
 David E. Hamilton2
David E. Hamilton2
- 1Neuroscience Graduate Program, University of Michigan, Ann Arbor, MI, USA
- 2Molecular and Behavioral Neuroscience Institute, University of Michigan, Ann Arbor, MI, USA
- 3Department of Psychiatry, University of Michigan, Ann Arbor, MI, USA
Previous studies have primarily interpreted gene expression regulation by glucocorticoids in the brain in terms of impact on neurons; however, less is known about the corresponding impact of glucocorticoids on glia and specifically astrocytes in vivo. Recent microarray experiments have identified glucocorticoid-sensitive mRNAs in primary astrocyte cell culture, including a number of mRNAs that have reported astrocyte-enriched expression patterns relative to other brain cell types. Here, we have tested whether elevations of glucocorticoids regulate a subset of these mRNAs in vivo following acute and chronic corticosterone exposure in adult mice. Acute corticosterone exposure was achieved by a single injection of 10 mg/kg corticosterone, and tissue samples were harvested 2 h post-injection. Chronic corticosterone exposure was achieved by administering 10 mg/mL corticosterone via drinking water for 2 weeks. Gene expression was then assessed in two brain regions associated with glucocorticoid action (prefrontal cortex and hippocampus) by qPCR and by in situ hybridization. The majority of measured mRNAs regulated by glucocorticoids in astrocytes in vitro were similarly regulated by acute and/or chronic glucocorticoid exposure in vivo. In addition, the expression levels for mRNAs regulated in at least one corticosterone exposure condition (acute/chronic) demonstrated moderate positive correlation between the two conditions by brain region. In situ hybridization analyses suggest that select mRNAs are regulated by chronic corticosterone exposure specifically in astroctyes based on (1) similar general expression patterns between corticosterone-treated and vehicle-treated animals and (2) similar expression patterns to the pan-astrocyte marker Aldh1l1. Our findings demonstrate that glucocorticoids regulate astrocyte-enriched mRNAs in vivo and suggest that glucocorticoids regulate gene expression in the brain in a cell type-dependent fashion.
Introduction
At the level of the organism, stress occurs via environmental or psychological stimuli (stressors) that disrupt homeostasis. The classic stress response involves communication between the brain and the rest of the body through the hypothalamic-pituitary-adrenal (HPA) axis. In response to stress, the brain integrates signals through the brainstem and the limbic system (e.g., prefrontal cortex, hippocampus, and amygdala), which in turn activates signaling cascades through the hypothalamus and pituitary gland that result in the stimulation of the adrenal glands and release of glucocorticoids into the circulatory system. Glucocorticoids are the major output hormones of the HPA axis and enable the organism to adapt to stressors by influencing many physiological pathways (e.g., modulate attention and appraisal, alter energy metabolism, regulate immune signaling, modulate memory formation, etc.) (De Kloet et al., 2005; Lupien et al., 2009). Glucocorticoids also act in negative feedback mechanisms at multiple levels of the HPA axis (e.g., limbic system, hypothalamus) to regulate stress response activation and return the system to baseline following stress. Appropriate stress response is thus critical for appropriate responsiveness to external stimuli. Chronic disruption of appropriate stress signaling (e.g., altered HPA axis signaling, chronic stress) can have drastic impact on well-being and has been linked to numerous disease states, including Cushing's disease [HPA hyperactivity (Newell-Price et al., 2006)], Addison's disease [HPA hypoactivity (Ten et al., 2001)], and multiple psychiatric disorders, including depression and bipolar disorder (McEwen, 2005). Given the importance of the stress response to health and disease, there is great interest in understanding the impact of glucocorticoids on the brain, both in terms of acute exposure (e.g., stress mechanisms) and chronic exposure (e.g., disease states).
At the cellular level, glucocorticoids are known to act as transcriptional regulators through interaction with their associated receptors (i.e., mineralocorticoid receptor, glucocorticoid receptor) (Strachan and Read, 1999). These receptors have unique neuroanatomical expression patterns but are both prominently expressed in limbic structures (e.g., prefrontal cortex, hippocampus) to facilitate HPA axis signaling. Glucocorticoids readily pass through the cell membrane and bind their inactive receptors that are complexed with chaperone proteins in the cytoplasm. Ligand-bound receptors are activated and translocate to the nucleus [mechanisms reviewed in Vandevyver et al. (2012)]. Activated receptors can bind to DNA sequences known as glucocorticoid response elements (GREs) and upregulate or downregulate target gene expression. Glucocorticoids are known to regulate many mRNAs in the brain. While numerous studies have reported in vitro findings of glucocorticoid regulation on a gene by gene basis, fewer studies have looked at glucocorticoid regulation in the brain at the level of the transcriptome in vivo. A limited number of gene expression profiling studies of glucocorticoid mRNA regulation using brain tissue have found that numerous mRNAs are regulated by glucocorticoids in hippocampal slices (Datson et al., 2001) and that the observed regulation varies by the duration of glucocorticoid exposure (Morsink et al., 2006).
The brain consists of many diverse cell types (e.g., neurons, astrocytes, oligodendrocytes, microglia) that vary significantly in function. Historically, investigations of the brain have focused on neurons, but there is increasing evidence that glial cells play an active role in numerous brain processes. Astrocytes participate in many functions, including neurotransmission regulation, electrical coupling and gap junctions, gliotransmission, metabolism and the blood brain barrier, and astrogliosis and immune function [reviewed in Maragakis and Rothstein (2006)]. In addition to the growing functional importance of glial cells, all of these cell types are known to express glucocorticoid receptors and are thus likely responsive to glucocorticoid signaling (McEwen et al., 1968; Vielkind et al., 1990; Sierra et al., 2008). Previous studies examining the impact of glucocorticoids upon brain tissues have routinely interpreted the action of these steroids in terms of neuronal physiology, but glucocorticoids are also known to impact glial cells. For example, glucocorticoids can inhibit astrocyte proliferation in vivo (Unemura et al., 2012). In terms of mRNA regulation, glucocorticoids have been shown to regulate a limited number of astrocyte mRNAs, including glutamine synthetase and glial fibrillary acidic protein (GFAP) (Nichols et al., 1990; Laping et al., 1994). Beyond these limited reports, the impact of glucocorticoids on global gene expression in cell types other than neurons in the brain is largely unknown.
There is also a clinical interest in understanding how glucocorticoids impact astrocytes due to their growing association with major depression, a disorder strongly associated with altered HPA axis signaling. Postmortem morphological studies have observed changes in astrocytes associated with depression (e.g., decreased astrocyte density and increased astrocyte size in prefrontal cortex) (Rajkowska et al., 1999; Miguel-Hidalgo et al., 2000), and postmortem brain transcriptional profiling studies have also identified astrocyte-associated mRNAs as being altered in depression (Barley et al., 2009; Bernard et al., 2010). However, how and even if glucocorticoids influence astrocytes in pathological conditions such as depression is currently speculative, and the extent of glucocorticoid regulation occurring in astrocytes in vivo under basal conditions is not well-understood. We recently characterized glucocorticoid receptor-mediated regulation of mRNAs in cortical and hippocampal astrocytes in vitro, defining glucocorticoid-regulated mRNA targets that included both astrocyte-specific mRNAs and mRNAs ubiquitously expressed among cell types in the brain (Carter et al., 2012). However, the physiological relevance of this glucocorticoid regulation observed in astrocyte cell culture has not been investigated in the intact brain. Here we have assessed whether a subset of mRNAs regulated by glucocorticoids in astrocytes in vitro are responsive to glucocorticoids in vivo following acute and chronic glucocorticoid exposure, including mRNAs reported to be enriched in astrocytes (Cahoy et al., 2008).
Materials and Methods
Animal Treatments and Brain Tissue Collection
All animal procedures at the University of Michigan were approved by UCUCA (University Committee on Use and Care of Animals) and monitored by ULAM (Unit for Laboratory Animal Medicine). Adult C57B/6 male mice (8 weeks of age) were obtained from Charles River. For all experiments, mice were group-housed (n = 3–5 per cage). Mice were housed for 1 week under basal conditions (14:10 light dark cycle) before experiments. To collect tissue samples, mice were euthanized using cervical dislocation and decapitation. For both the acute and chronic glucocorticoid exposure protocols, upon euthanasia, trunk blood was collected, and brains were removed and bisected along the mid-sagittal plane. One brain hemisphere was frozen in chilled isopentane (−35°C) and stored at −80°C for subsequent tissue sectioning and in situ hybridization analyses. The remaining brain hemisphere was further dissected on wet ice to collect hippocampi and prefrontal cortex tissues for quantitative PCR (qPCR) analyses. These tissue samples were frozen immediately on dry ice and stored at −80°C for subsequent RNA extraction.
Acute Glucocorticoid Mouse Model
Mice were injected (s.c.) with 10 mg/kg corticosterone or vehicle (1% ethanol solution) 1 h after lights on. Mice were returned to their home cages for specified time durations prior to euthanasia and sample collection. Corticosterone (Cort, Cat. #C2505) was obtained from Sigma Aldrich.
Chronic Glucocorticoid Mouse Model
Chronic corticosterone exposure was produced based on previously published protocols (Gourley et al., 2008; Karatsoreos et al., 2010; Lee et al., 2010). Mice were given access to drinking water containing 10 mg/mL corticosterone or vehicle for 2 weeks (n = 5/group). Drinking solutions were replaced 3 times over course of 2 weeks (every 4–5 days). On day 14, mice were weighed on an analytical balance. Mice were then euthanized 1 h after lights on, and brains were removed. Adrenal glands, spleen, and thymus tissues were dissected on ice and weighed on an analytical balance.
Plasma Corticosterone Assay
Immediately following decapitation, trunk blood was collected and mixed with 0.5 M EDTA (pH 8.0, final concentration in blood sample ~10 μM) and stored immediately on wet ice. Blood samples were subsequently spun in a table-top centrifuge (3500 × g for 10 min at 4°C). Blood plasma was transferred to a fresh 1.5 mL tube and stored at −80°C. Plasma corticosterone concentrations were measured using a corticosterone double antibody radioimmunoassay kit (MP Biomedicals, Cat. #07120102). Individual samples were measured in triplicate, including standard curve controls; the average value was used for subsequent calculations.
RNA Isolation
Total RNA samples were isolated from tissues using Trizol reagent per manufacturer's protocol (Invitrogen), and RNA concentrations were obtained using a Nanodrop spectrophotometer (Thermo Scientific).
Quantitative PCR (qPCR) Analysis
RNA samples (500 ng–1 μg) were converted to cDNA using Superscript II via random hexamer priming (Invitrogen). Approximately Fifty percentage of each cDNA reaction was used for Applied Biosystems (ABI) Taqman mRNA qPCR assays in custom Low-Density Array (TLDA) format (ABI #4346799) with Taqman reagents (ABI #4440048). The Taqman arrays were processed on an Applied Biosystems Viia7 instrument according to manufacturer protocols. mRNAs were selected for measurement based on combinations of the following of factors as defined previously (Carter et al., 2012); (1) glucocorticoid sensitivity vs. insensitivity in primary astrocyte cultures, (2) magnitude of glucocorticoid regulation, (3) previous reporting of glucocorticoid sensitivity vs. novel glucocorticoid regulation, and (4) previous association with astrocyte function and/or astrocyte-enrichment (Cahoy et al., 2008). Previously reported glucocorticoid regulation vs. novel glucocorticoid regulation was determined by literature analysis of each individual gene (i.e., using PubMed and Google Scholar search tools, terms used: gene symbol and/or probe ID + “glucocorticoids, corticosteroids, corticosterone, dexamethasone, prednisone”). mRNA expression was measured in technical triplicate per sample. Taqman mRNA assays were selected based on manufacturer recommendations; all but one assay spanned exon-exon junctions (Glul). Specific Taqman mRNA assays used in this analysis are: Actb (Mm01205647_g1), Adora2b (Mm00839292_m1), Aldh1l1 (Mm00550947_m1), Atp6v1b2 (Mm00431987_m1), Ch25h (Mm00515486_s1), Egr2 (Mm00456650_m1), Fgfr1 (Mm00438930_m1), Fgfr3 (Mm00433294_m1), Fkbp5 (Mm00487401_m1), Folh1 (Mm00489655_m1), Foxo1 (Mm00490672_m1), Gap43 (Mm00500404_m1), Gfap (Mm01253033_m1), Gja1 (Mm00439105_m1), Gjb6 (Mm01317508_m1), Glul (Mm00725701_s1), Hdac7 (Mm00469520_m1), Klf9 (Mm00495172_m1), Mapk4 (Mm00554001_m1), Mertk (Mm00434920_m1), Pdk4 (Mm01166879_m1), Per1 (Mm00501813_m1), Phlda1 (Mm00456345_g1), Prodh (Mm00448401_m1), Sgk1 (Mm00441380_m1), Slc1a2 (Mm00441457_m1), Slc1a3 (Mm00600697_m1), Sult1a1 (Mm01132072_m1), Syn2 (Mm00449780_m1), Txnip (Mm00452393_m1), Wnt7a (Mm00437354_m1). Gene symbols, definitions, and reported astrocyte fold-enrichment (Cahoy et al., 2008) for measured mRNAs are listed in Table 1.
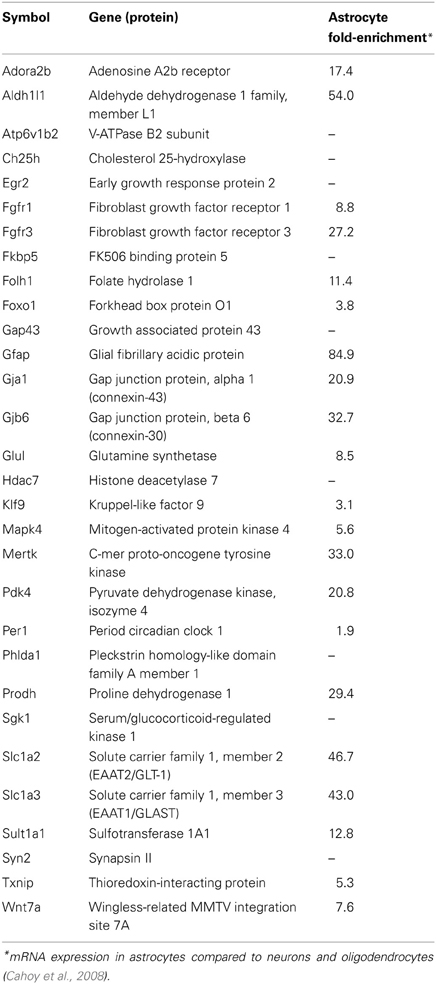
Table 1. mRNAs assessed for glucocorticoid regulation in the brain in vivo.
In situ Hybridization (ISH)
Brain hemisections were cut on a cryostat (10 μm thick), mounted on Superfrost microscope slides (2 sections per slide), and stored at −80°C prior to ISH experiments. To create ISH probes, specific mRNA domains that displayed low levels of nucleotide homology were amplified by PCR (Native Taq DNA polymerase, Invitrogen) using primers designed with NCBI Primer Blast software (Table 2). PCR amplicons were cloned into pCR-II-TOPO Vector; plasmids were transformed using TOP10 cells (Invitrogen). Plasmid DNA was isolated using a QIAprep Spin Miniprep Kit (Qiagen). The identity of all plasmid clones was verified by Sanger Sequencing (DNA sequencing core, University of Michigan). Plasmids were linearized using appropriate restriction enzymes (NEB). Linearized plasmids were used to create 35S-RNA probes using T7 or SP6 RNA polymerases (varied by plasmid). Briefly, linearized plasmid (100–500 ng) was combined with 1 μl each of ATP, GTP, and CTP (10 mM), 1 μl RNAse inhibitor, 1.66 μl DTT (100 μM), 4 μl 5 × transcription buffer (Promega) and 7.8 μl of 35S-UTP (12.5 μCi/ul, Perkin Elmer). Probes were purified using BioRad P-6 columns; the effluent was measured with a scintillation counter and then diluted with hybridization solution (~2 × 106 cpms of probe in 40 μl hybridization buffer per slide). All remaining tissue processing steps were performed according to published lab protocols (Carter et al., 2010). Probe hybridization specificity was defined in control experiments by sense probes which failed to yield autoradiographic signals above background. Sense probes demonstrate significantly lower signal compared to antisense probes for all mRNAs assessed (Figure 1).
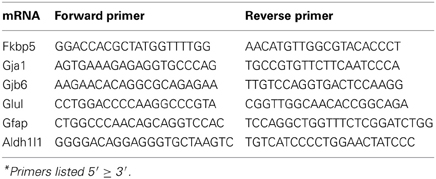
Table 2. Primers for in situ hybridization probes.
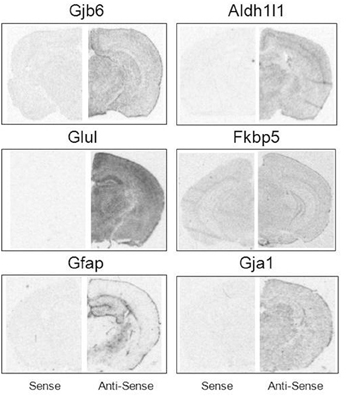
Figure 1. Specificity of mRNA in situ hybridization probes. Sense (Left) and antisense (Right) comparisons of ISH probe signal under matched exposure times per probe.
Densitometry Analysis
Anatomical boundaries of the prefrontal cortex and hippocampus where defined based upon reference to the mouse brain atlas of Paxinos and Franklin (2001). Autoradiograms from ISH were scanned with a ScanMaker 1000XL Pro Flatbed Scanner (Microtek, Carson, CA) using SilverFast Ai Imaging Software (LaserSoft Imaging, Sarasota, FL). The scanned images were analyzed based on optical density (OD) measurements using ImageJ software (Version 1.45S, NIH). For each probe, a normalized OD value for each section image was determined by subtracting a background value from the OD value (Background = background mean + 3.5 * standard deviation of background mean). Background measurements were taken from a non-tissue area of each film. Four section images were measured for each brain region per animal; an average normalized value of the 4 sections per probe was used for downstream analyses.
Statistical Analysis
For qPCR experiments, differential expression analysis was performed using the delta-delta-Ct method [(Livak and Schmittgen, 2001), β-actin as control reference] using Statminer software (Integromics). An average of Ct-values from technical replicates was taken as the Ct-value for each gene measurement; individual Ct-values identified as outliers via the Grubbs' outlier test were excluded from downstream analyses. For comparisons between groups in terms of qPCR analysis of mRNA differential expression, measurements were compared using a moderated t-test; statistical significance was defined as p < 0.05. The relationship between corticosterone levels over time due to corticosterone injection was analyzed using a Two-Way ANOVA with multiple replicates; comparison of groups at individual time points was performed post-hoc using Fisher's least-significant difference (LSD) test. For individual comparisons between groups in terms of organ weight, body weight, corticosterone levels, and ISH analysis, measurements were compared using a two-tailed student's t-test; statistical significance was defined as p < 0.05. The standard error of the mean (SEM) was used for error bars on graphs.
Results
Single Corticosterone Injection Results in Transient Rise of Corticosterone Plasma Levels
In order to determine the plasma concentrations of corticosterone in vivo following a single bolus injection of corticosterone, 3 sets of mice were injected with 10 mg/kg corticosterone or vehicle solution, and blood samples were then collected at 1, 2, and 4 h post-injection (n = 3 animals/group/time point, 18 animals total; Figure 2A). Two-Way ANOVA revealed a significant effect of treatment [F(1, 12) = 9.54, p = 0.009], time point [F(2, 12) = 7.08, p = 0.009] and a significant interaction between treatment and time point [F(2, 12) = 5.51, p = 0.02]. Post-hoc comparisons revealed a trend for a significant effect of treatment on corticosterone levels at 1 h (Fisher's LSD p = 0.06). Overall, corticosterone-treated animals tended to exhibit higher corticosterone levels at each time point compared with vehicle-treated animals, and the difference between treatment groups was dependent on the length of exposure. Specifically, a 10 mg/kg dose of corticosterone increased plasma corticosterone concentration to a supraphysiological level by 1 h. The plasma corticosterone level remained significantly elevated at 2 h at levels similar in magnitude to acute stress (Ma et al., 1997; McClennen et al., 1998) and then returned to near baseline by 4 h. Given the robust increase in plasma corticosterone levels during the initial hours post-injection, we chose to analyze gene expression 2 h post-injection in subsequent experiments, a time point temporally consistent with direct glucocorticoid-based transcriptional mechanisms. A replicate experiment yielded a corticosterone elevation of similar magnitude, and samples from this second experiment were used for subsequent qPCR experiments (n = 8 animals/group, Figure 2B).
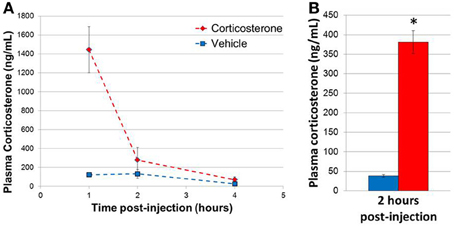
Figure 2. Single injection of 10 mg/kg corticosterone results in transient rise in plasma corticosterone levels in mice. (A) Time course of plasma corticosterone levels following injection (n = 3/treatment/time point). (B) Plasma corticosterone levels in mice (2 h post-injection exposure) used in qPCR experiments (n = 8/group). Error bars = SEM. *p < 0.05.
Single Injection of Corticosterone Results in mRNA Regulation in Brain in vivo of mRNAs Previously Reported to be Regulated by Glucocorticoids in Astrocytes in vitro
mRNA levels for select genes in RNA samples derived from two brain regions (prefrontal cortex and hippocampus) of mice injected with corticosterone or vehicle solution were measured by qPCR using Taqman Low Density Arrays (TLDAs). 18 mRNAs were measured based on robust glucocorticoid-sensitivity in astrocytes in vitro (Carter et al., 2012). Twelve of these 18 mRNAs were statistically regulated by acute corticosterone exposure in vivo in at least one of the brain regions tested (Figure 3). Among all mRNAs measured, 6 mRNAs were differentially expressed in both cortex and hippocampus following acute corticosterone treatment (upregulated; Fkbp5, Gjb6, Klf9, Pdk4, Sgk1, Sult1a1). 2 mRNAs were regulated in cortex but not in hippocampus (upregulated: Txnip; downregulated: Phlda1), and 6 mRNAs were regulated in hippocampus but not cortex (upregulated: Adora2b, Egr2; downregulated: Hdac7, Prodh, Slc1a2, Wnt7a). 15 mRNAs that have been reported to be regulated by glucocorticoids in vitro [listed in Carter et al. (2012)] were not statistically regulated in either brain region by acute corticosterone exposure in vivo (previously reported upregulation: Ch25h, Folh1, Foxo1, Gap43, Glul, Mapk4, Mertk, Per1, Syn2; previously reported downregulation: Atp6v1b2, Gfap). 3 mRNAs were regulated by acute corticosterone treatment in vivo that were not regulated or regulated in the opposite direction by glucocorticoids in astrocytes in vitro (downregulated: Hdac7, Slc1a2; downregulated instead of upregulated: Egr2). The magnitude of regulation induced by acute corticosterone exposure in this condition ranged from +3.15-fold regulation (Sgk1 in hippocampus) to +0.65/−1.55-fold regulation (Egr2 in hippocampus). Complete numerical values of mRNA regulation by acute corticosterone exposure in vivo and corresponding statistical p-values for all mRNAs measured are listed in Table 3.
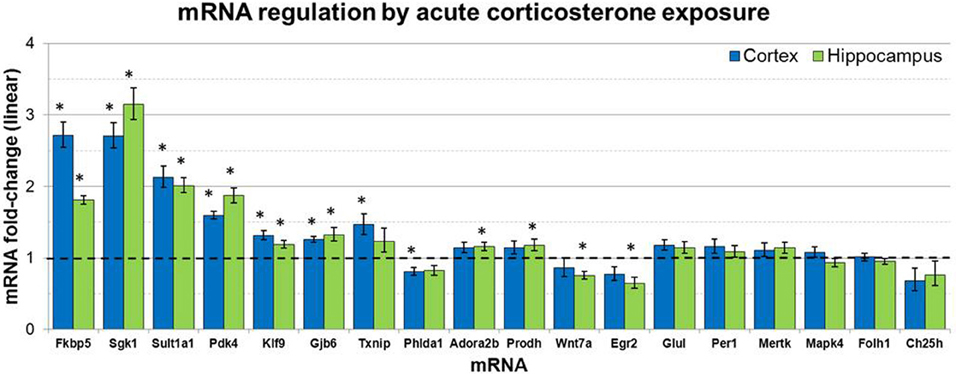
Figure 3. Acute corticosterone exposure regulates select mRNAs in hippocampus and prefrontal cortex. Gene expression fold-change ratios for mRNAs regulated by glucocorticoids in astrocytes in vitro (Carter et al., 2012) in prefrontal cortex and hippocampus following injection of 10 mg/kg corticosterone vs. saline control in vivo (2 h post-injection exposure, n 8 per group). Dashed line indicates no change compared to vehicle. Error bars = SEM. *p < 0.05.
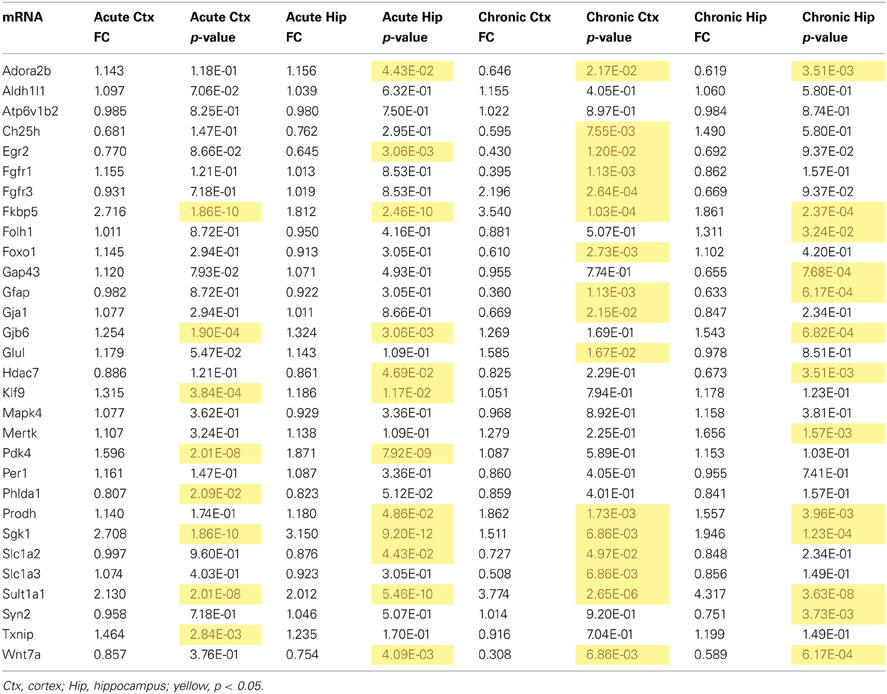
Table 3. qPCR data for all mRNAs measured for mRNA regulation by acute and chronic corticosterone exposure in the brain in vivo.
Mice Given Extended Access to Drinking Water Containing Corticosterone Demonstrate Hormonal and Organ Changes Consistent with Acute and Chronic Glucocorticoid Elevation
Mice given access to drinking water containing corticosterone for 14 days displayed statistically elevated corticosterone plasma levels relative to mice given access to vehicle drinking water (Figure 4A). Chronic corticosterone-treated mice also had significantly reduced thymus, adrenal, and spleen organ weights compared to vehicle-treated mice (Figure 4B), data consistent with chronic glucocorticoid exposure. There was no difference in the total body weight between treatment groups (Figure 4C).
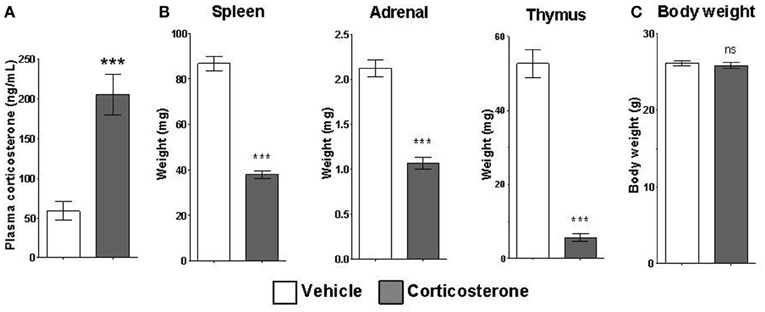
Figure 4. Chronic corticosterone administration via drinking water results in acute and chronic corticosterone elevations. (A) Plasma corticosterone levels after 2 weeks access to drinking water containing 10 mg/mL corticosterone vs. vehicle. (B) Weights of corticosterone-sensitive organs after chronic corticosterone administration. (C) Body weight of mice after chronic corticosterone administration. N = 5 per group. Error bars = SEM. ***p < 0.0001. ns = not significant.
Chronic Corticosterone Exposure Results in mRNA Regulation in Brain in vivo of mRNAs Previously Reported to be Regulated by Glucocorticoids in Astrocytes in vitro
mRNA levels for select genes in RNA samples derived from two brain regions (prefrontal cortex and hippocampus) of mice treated with chronic corticosterone solution or vehicle solution were measured by qPCR using TLDAs. Of the 18 mRNAs measured that were regulated by glucocorticoids in astrocytes in vitro (Carter et al., 2012), 12 of these mRNAs were statistically regulated by chronic corticosterone exposure in vivo in at least one of the brain regions tested (Figure 5). Among all mRNA measured, 7 mRNAs were regulated by chronic corticosterone exposure in both cortex and hippocampus (upregulated: Adora2b, Fkbp5, Prodh, Sgk1, Sult1a1; downregulated: Gfap, Wnt7a). 9 mRNAs were regulated by chronic corticosterone exposure in cortex but not in hippocampus (upregulated: Ch25h, Egr2, Fgfr3, Foxo1, Glul; downregulated: Ch25h, Fgfr3, Gja1, Slc1a2, Slc1a3); 6 mRNAs were regulated by chronic corticosterone exposure in hippocampus but not cortex (upregulated: Folh1, Gap43, Gjb6, Mertk, Syn2; downregulated: Hdac7). 7 mRNAs that have been reported to be regulated in vitro were not regulated in either region by chronic corticosterone exposure in vivo [previously reported upregulation: Klf9, Mapk4, Per1, Pdk4, Txnip; previously reported downregulation: Atp6v1b2, Phlda1; referenced in (Carter et al., 2012)]. 15 mRNAs were regulated in at least one brain region in a manner consistent with previously reported glucocorticoid regulation in vitro [listed in Carter et al. (2012)] (upregulated: Adora2b, Fkbp5, Folh1, Gap43, Gjb6, Glul, Mertk, Prodh, Sgk1, Sult1a1, Syn2; downregulated: Ch25h, Gfap, Gja1, Wnt7a) (Figure 5A). In addition, 12 mRNAs were regulated by chronic corticosterone treatment in vivo that were not regulated or regulated in the opposite direction by glucocorticoids in astrocytes in vitro (upregulated: Fgfr3; downregulated: Fgfr1, Foxo1, Gap43, Gfap, Gja1, Hdac7, Slc1a2, Slc1a3, Syn2; downregulated instead of upregulated: Egr2, Adora2b) (Figure 5B). The magnitude of regulation induced by chronic corticosterone exposure in this condition ranged from +4.38-fold regulation (Sult1a1 in hippocampus) to +0.31/−3.24-fold regulation (Wnt7a in frontal cortex). Complete numerical values of mRNA regulation by chronic corticosterone exposure in vivo and corresponding statistical p-values are for all mRNAs measured are listed in Table 3.
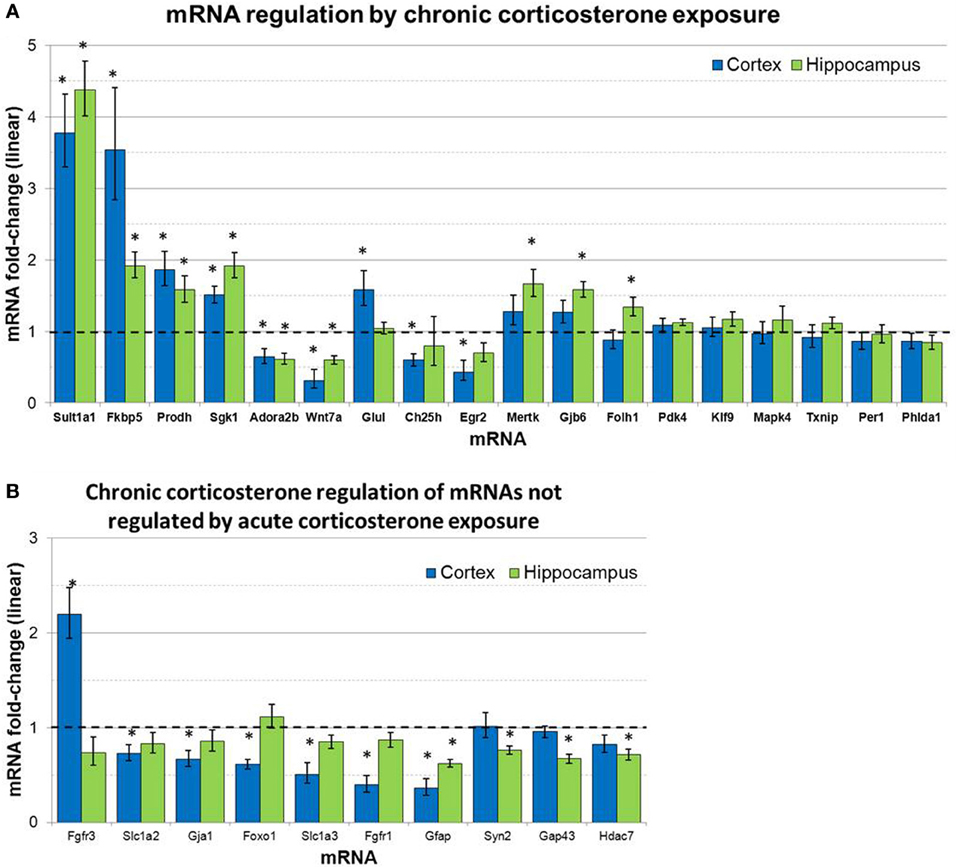
Figure 5. Chronic corticosterone exposure regulates select mRNAs in hippocampus and prefrontal cortex. (A) Gene expression fold-change ratios for mRNAs regulated by glucocorticoids in astrocytes in vitro (Carter et al., 2012) in prefrontal cortex and hippocampus following chronic corticosterone exposure for 2 weeks vs. vehicle control (n = 8 per group). (B) mRNA fold-changes of mRNAs regulated by chronic corticosterone exposure in vivo that were not regulated by glucocorticoids in astrocytes in vitro or acute corticosterone exposure in vivo. Dashed line indicates no change compared to vehicle. Error bars = SEM. *p < 0.05.
Select mRNAs Regulated by Chronic Corticosterone Exposure in vivo have Neuroanatomical Expression Patterns Consistent with Astrocyte Marker Aldh1L1
Regulation of select mRNAs by chronic corticosterone exposure was further assessed in the prefrontal cortex and hippocampus by semi-quantitative radioactive in situ hybridization (Figure 6). These mRNAs were selected based on previously reported glucocorticoid sensitivity (Fkbp5, Gfap, Glul), their regulation by chronic corticosterone exposure in cortex and/or hippocampus based on qPCR measurements (Fkbp5, Gfap, Gja1, Gjb6, Glul), and/or an established association with astrocyte localization and function (Aldh1l1, Gfap, Gja1, Gjb6, Glul). Of the 6 mRNAs examined via in situ hybridization, 3 mRNAs were statistically regulated in at least one brain region in the same direction as the qPCR data (upregulated: Fkbp5, Gfap, Gjb6). 2 mRNAs demonstrated non-significant trends in the same direction as the qPCR data (upregulated: Glul; downregulated: Gja1), and 1 mRNA demonstrated differential expression in at least one brain region that was not observed by qPCR (upregulated: Aldh1l1). In terms of hippocampal expression patterns between chronic corticosterone treatment and vehicle treatment, these mRNAs showed generally consistent expression patterns between treatment groups (Figure 6A). Fkbp5 appeared predominantly expressed in pyramidal cell hippocampal subfields, while expression of Gfap, Gja1, Gjb6, and Glul was noticeably lacking in the pyramidal cell regions but displayed diffuse levels of expression in molecular cell regions. This latter anatomical distribution was highly similar to the expression pattern of the pan-astrocytic marker Aldh1l1 (Figure 6A).
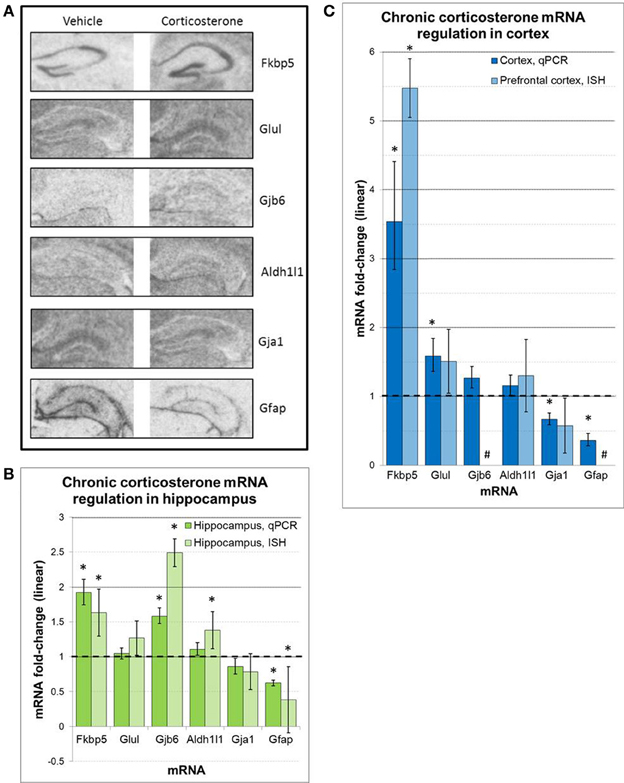
Figure 6. ISH measures of select mRNAs confirm chronic corticosterone regulation and reveal mRNA expression patterns consistent with pan-astrocyte marker Aldh1l1 (A). Representative images of in situ hybridization data for select mRNAs in hippocampus. (B,C) in situ hybridization densitometry measures of differential mRNA expression in whole hippocampus (B) and prefrontal cortex (C). n = 5/group. Dashed line = no change compared to vehicle. *p < 0.05. #sub-threshold ISH signal.
Acute Corticosterone Exposure in vivo and Chronic Corticosterone Exposure in vivo (1) Regulate Distinct Sets of mRNAs Among mRNAs Previously Reported to be Regulated by Glucocorticoids in Astrocytes in vitro and (2) Demonstrate Positive Correlation Among the Expression Levels of Regulated mRNAs Between Conditions
Comparison of select mRNAs measured by qPCR that were regulated by glucocorticoids in astrocytes in vitro [18 regulated mRNAs, (Carter et al., 2012)], acute corticosterone exposure in vivo (14 regulated mRNAs), or chronic corticosterone exposure in vivo (22 regulated mRNAs) revealed significant overlap yet also distinct patterns of mRNA regulation between conditions (in vitro + acute overlap: 10 mRNAs, in vitro + chronic overlap: 12 mRNAs, acute + chronic overlap: 10 mRNAs, Figure 7). For mRNAs regulated in multiple conditions, the directionality of glucocorticoid regulation was generally consistent across conditions (Figure 7A). 8 mRNAs were regulated in all three conditions, and all mRNAs regulated by acute corticosterone exposure in vivo were also regulated in one of the other conditions (Figure 7B). Among mRNAs regulated by glucocorticoids in at least one in vivo condition, there was a moderate, positive correlation among the measured mRNAs between mRNA expression changes between the two conditions (cortex correlation coefficient R = 0.641, hippocampus correlation coefficient R = 0.720) (Figure 7C).
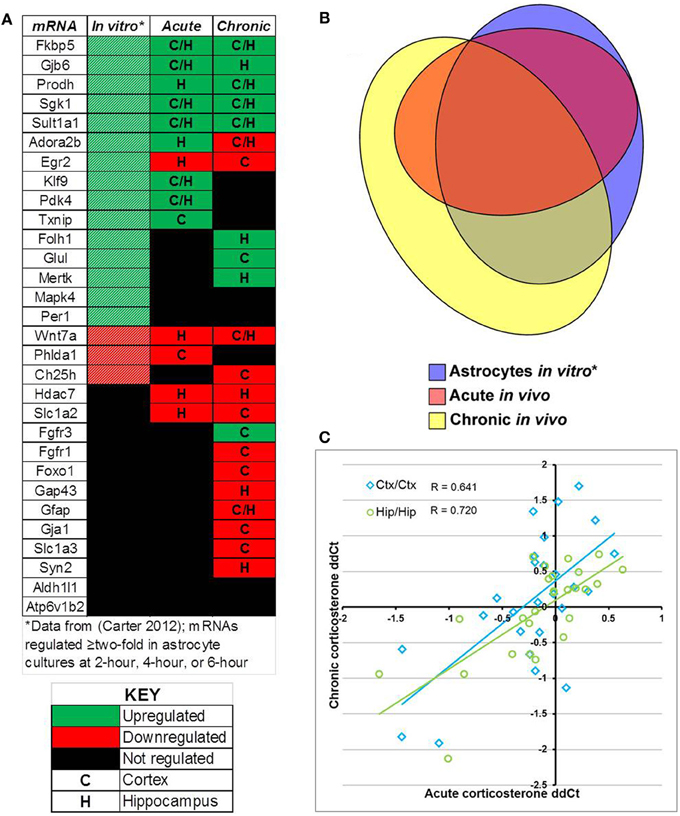
Figure 7. Summary of results for measured mRNAs regarding glucocorticoid regulation by acute corticosterone exposure and chronic corticosterone exposure in vivo. (A) Heatmap summarizing directionality of glucocorticoid regulation of mRNAs across conditions. Key summarizes table terminology. (B) Proportional Venn diagram comparing measured mRNAs regulated by glucocorticoid treatment among 3 experimental conditions (astrocytes in vitro, acute exposure in vivo, chronic exposure in vivo). (C) Plot of mRNA regulation by acute corticosterone exposure vs. mRNA regulation by chronic corticosterone exposure for all mRNAs regulated in at least one in vivo condition (log2 scale). Regulated = p < 0.05. *in vitro data from Carter et al. (2012).
Discussion
Glucocorticoids are important mediators of the classic stress response. Previous reports have characterized extensive glucocorticoid-mediated mRNA regulation in CNS tissue using in vitro systems and hippocampal slice models, demonstrating that stress hormones indeed have a broad impact on mRNA expression in the brain. Although many cell types in the brain express receptors for glucocorticoids [e.g., neurons (McEwen et al., 1968), astrocytes and oligodendrocytes (Vielkind et al., 1990), microglia (Sierra et al., 2008)] and are thus presumably responsive to glucocorticoids, most studies of glucocorticoid signaling in the brain have interpreted resulting data in terms of glucocorticoid regulation in neurons. Relatively little is known about the impact of glucocorticoids on other brain cell types (e.g., glia). We and others have recently investigated glucocorticoid regulation in astrocytes in vitro using primary cortical astrocyte cultures derived from newborn mice and reported extensive time-dependent gene expression changes (Carter et al., 2012; Slezak et al., 2013), including glucocorticoid regulation of astrocyte-enriched mRNAs (Cahoy et al., 2008). While these in vitro data are intriguing, physiological significance of these results would hinge upon in vivo evidence of similar glucocorticoid regulation of astrocyte-enriched mRNA expression. Here we have examined the effect of exposing the brain to acute and chronic glucocorticoid treatment in vivo on the expression of select mRNAs that are regulated by glucocorticoids in astrocytes in vitro.
We first determined if a subset of mRNAs robustly regulated by short-term glucocorticoid treatment in astrocytes in vitro [i.e., 18 mRNAs identified in Carter et al. (2012)] were also regulated by acute glucocorticoid exposure in the brain in vivo. We measured mRNA expression in prefrontal cortex and hippocampus, two regions of well-characterized glucocorticoid action in the brain. A majority of the measured mRNAs regulated by glucocorticoids in astrocytes in vitro were also regulated by acute corticosterone exposure in at least one brain region in vivo (12/18 regulated, Figure 3). Almost all of the observed mRNA regulation in vivo was directionally consistent with the regulation in astrocytes in vitro (upregulated: Fkbp5, Gjb6, Prodh, Sgk1, Sult1a1, Adora2b, Klf9, Pdk4, Txnip; downregulated: Wnt7a, Phlda1). A portion of the regulated mRNAs are known to be both enriched in astrocytes and involved in specific processes, such as cellular metabolism (Pdk4, Prodh, Sult1a1), intercellular signaling (Adora2b, Gjb6, Slc1a2), and transcriptional regulation (Klf9, Txnip). While these qPCR data do not identify the cell types in which these glucocorticoid-mediated mRNA changes take place, these data confirm that these mRNAs are glucocorticoid-sensitive in vivo. An intriguing subsequent question will be to determine if other physiological challenges known to acutely increase glucocorticoids (e.g., stress) similarly regulate these same mRNAs. Additionally, a portion of mRNAs regulated by glucocorticoids in astrocytes in vitro were not regulated by acute corticosterone treatment in the brain in vivo (Ch25h, Folh1, Glul, Mapk4, Mertk). This result may indicate these particular mRNAs are not regulated by elevated glucocorticoids in vivo or that the cellular environment of the intact brain is sufficiently different than in astrocytes in vitro and glucocorticoid regulation of these mRNAs may be delayed and/or less robust in vivo. If these mRNAs are indeed regulated in the brain by glucocorticoids, another possible explanation for no observed mRNA expression changes is differential regulation by cell types; for example, glucocorticoids has been shown to increase NGF mRNA expression in neurons but decrease NGF expression in astrocytes (Lindholm et al., 1992), a scenario that may result in no net change in tissue-level mRNA expression. Alternatively, this acute corticosterone exposure condition may not be sufficient to elicit regulation of these mRNAs based on condition-dependent features (e.g., corticosterone dosage, relatively short duration of exposure). Overall, given the previous reporting of the astrocyte-enrichment of many mRNAs measured (Cahoy et al., 2008), the observed regulation of many of these mRNAs suggests that astrocytes are acutely responsive to glucocorticoids in vivo.
In general, the absolute magnitude of the glucocorticoid-mediated changes observed in vivo was lower than mRNA expression differences observed following similar steroid treatment durations in astrocytes in vitro. One potential explanation for this difference may be due to the active clearance of glucocorticoids from the brain compared to cell culture. In cell culture, the glucocorticoid concentration remains high and relatively constant, whereas there is a more temporally limited elevation of corticosterone in the intact brain, likely resulting in less robust regulation. Consistent with this possibility, active clearance of plasma corticosterone was directly observed in the acute exposure condition, returning corticosterone concentrations to baseline within 4 h (Figure 2A). In general, the cellular environment in the brain constantly strives to maintain homeostasis; in a situation of elevated glucocorticoid levels, the brain would engage systems to limit responsiveness to continued steroid exposure by activating opposing regulatory mechanisms to control the response to changes induced by glucocorticoid signaling. There are also less intercellular interactions in the isolated astrocyte cell culture that may counteract or limit cell-type dependent glucocorticoid responses in vivo. Another possible mechanism influencing the observed glucocorticoid regulation is steroid metabolism. Neurons and glia contain enzymes that modify glucocorticoid structure (e.g., 5-alpha-reductase) (Melcangi et al., 1993). The models used in these experiments cannot distinguish between regulation by corticosterone and associated metabolites in these experiments; whether directly or indirectly, we can conclude that these mRNAs are responsive to corticosterone treatment, but changes in steroid metabolism may impact the magnitude of regulation [e.g., corticosterone metabolites have different affinities for glucocorticoid receptor (McInnes et al., 2004)]. The role of glucocorticoid metabolites in the regulation of these mRNAs in the brain in vivo remains unresolved, which may be as or more important as regulation by the natural ligand corticosterone. Future studies that specifically utilize glucocorticoid metabolites and/or include the use of glucocorticoid receptor antagonists (e.g., RU—486) would aid in addressing this important issue. Additionally, the previous in vitro astrocyte experiments were focused on glucocorticoid receptor-mediated mRNA regulation; given the presence of mineralocorticoid receptor in astrocytes and the limbic system, differential receptor binding may also influence the observed mRNA regulation. Taken together, although the magnitude of mRNA regulation was reduced in vivo relative to astrocytes in vitro, these data indicate that these mRNAs can indeed be regulated in the brain by acute glucocorticoid exposure in vivo.
Although these data involving acute glucocorticoid regulation are relevant for understanding physiological glucocorticoid signaling (e.g., stress response), clinical conditions involving alterations in glucocorticoid signaling are often chronic in nature. Given that our acute condition resulted in a temporary increase in glucocorticoid levels, we wanted to further investigate regulation of these mRNAs in response to chronically elevated glucocorticoid levels in the brain in vivo. We assessed the impact of chronic glucocorticoid exposure on mice using an established protocol for administering corticosterone via drinking water (Gourley et al., 2008; Karatsoreos et al., 2010) (Figure 4). Consistent with prior studies, 2 weeks of corticosterone treatment of mice lead to elevated plasma levels of corticosterone, indicating that all mice exposed to the corticosterone solution had consumed water containing corticosterone at least in the short term. Physiological indices of chronic corticosterone treatment were observed in the form of hypotrophy of several glucocorticoid-sensitive tissues (i.e., adrenal glands, thymus, and spleen). These indices were observed without changes in body weight, suggesting limited secondary metabolic impact of this protocol. Together, these data demonstrate that mice under this protocol show signs of acute and chronic corticosterone exposure.
With evidence of chronic corticosterone exposure, we then assessed glucocorticoid-mediated mRNA regulation in the brain. Among the mRNAs measured, the majority of the mRNAs regulated by glucocorticoids in astrocytes in vitro were also statistically regulated in at least one brain region by chronic corticosterone exposure in vivo (12/18 mRNAs, Figure 5). Many mRNAs were regulated by chronic corticosterone exposure in the same direction as the glucocorticoid regulation observed in astrocytes in vitro and acute corticosterone exposure in vivo (upregulated: Fkbp5, Folh1, Gjb6, Glul, Mertk, Prodh, Sgk1, Sult1a1; downregulated: Ch25h, Hdac7, Slc1a2, Wnt7a), displaying a consistent response to glucocorticoids independent of steroid treatment duration. In contrast, several other mRNAs in the chronic corticosterone experiment were either not regulated (Klf9, Pdk4, Txnip, Mapk4, Per1, Phlda1) or regulated in the opposite direction (Adora2b, Egr2) when compared with glucocorticoid regulation observed in acute corticosterone exposure in vivo and astrocyte cultures in vitro. These data may be a result of compensatory mechanisms that reverse or limit regulation of these mRNAs under chronic glucocorticoid stimulation. Unexpectedly, chronic corticosterone exposure in vivo also regulated mRNAs that were not regulated in either acute glucocorticoid exposure condition in vivo or in vitro (upregulated: Fgfr3; downregulated: Fgfr1, Foxo1, Gap43, Gfap, Gja1, Slc1a3, Syn2). These discrepancies may be due to indirect, compensatory or adaptive regulation to prolonged glucocorticoid exposure. In addition, since we assessed a single chronic corticosterone exposure condition, one caveat linked to interpreting these results is that we cannot determine if the mRNAs measured change in other contexts of chronic corticosterone manipulation (e.g., longer exposures, adrenalectomy/corticosterone replacement, chronic stress). We also do not know if these changes are transient (e.g., reversible with washout period), although a previous study found long-lasting behavioral effects of chronic corticosterone exposure administered via drinking water even after an abstinence period (Gourley et al., 2008). Overall, these data provide initial evidence that astrocytes respond to chronic elevations of glucocorticoids in vivo and potentially demonstrate secondary regulation compared with acute glucocorticoid exposure.
A summary of the results comparing glucocorticoid regulation based on glucocorticoid exposure in astrocytes in vitro, acute corticosterone exposure in the brain in vivo, and chronic corticosterone exposure in the brain in vivo are shown in Figure 7. The high concordance of in vivo regulation by corticosterone with glucocorticoid regulation in astrocytes in vitro is important in part because most of these mRNAs have not been reported as glucocorticoid-sensitive in the brain in vivo. A subset of measured mRNAs have been reported to be regulated in the brain in vivo by glucocorticoids or stress conditions [e.g., Fkbp5 (Scharf et al., 2011), Gfap (O'Callaghan et al., 1989), Glul (Patel et al., 1983), Sgk1 (Sarabdjitsingh et al., 2010)], but a number of astrocyte-enriched mRNAs demonstrate novel in vivo glucocorticoid regulation in the brain (e.g., Gjb6, Prodh, Adora2b, Klf9, Pdk4, Txnip, Folh1, Mertk, Wnt7a). Of the mRNAs with reported glucocorticoid regulation in the brain in vivo, the directionality matches the previous data, giving further confidence that our data documents authentic glucocorticoid regulation in the brain. The additional in vivo regulation observed in these experiments may not have been previously observed for multiple reasons; one possibility is that the magnitude of changes are small enough that they may not have been statistically significant if measured with techniques less sensitive than qPCR (e.g., microarray).
In addition, these results have implications for understanding glucocorticoid regulation in astrocytes by brain region. There is increasing evidence of astrocyte diversity based on neuroanatomical location; given the extensive differential mRNA regulation among neurons, astrocytes may also exhibit differential mRNA regulation by brain region. This point is also relevant given the data comparisons to cortical astrocyte cultures in vitro (Carter et al., 2012). Our results characterize glucocorticoid-mediated mRNA regulation in two brain regions known to be involved in glucocorticoid action, the prefrontal cortex and hippocampus; whether these mRNAs are regulated in a similar manner in other brain regions remains to be investigated. In response to acute or chronic corticosterone exposure, certain mRNAs were statistically regulated in one brain region but not the other (e.g., Hdac7 only in hippocampus). While there may indeed be differential glucocorticoid mRNA regulation by brain region, such results may be due to specific factors unique to these particular experimental conditions (e.g., exposure times, criteria used to define differential expression). Additional experiments showing consistent brain-region-dependent glucocorticoid regulation across multiple exposure times and conditions in vivo are needed to address this possibility.
We also noted a moderate positive correlation between the measured mRNA changes induced by acute corticosterone exposure and chronic corticosterone exposure by brain region (Acute-Cortex vs. Chronic-Cortex: r = 0.641, Acute-Hippocampus vs. Chronic-Hippocampus: r = 0.720). In other words, mRNAs regulated by glucocorticoids in one condition generally were regulated in the other condition with a similar directionality and magnitude. Given that the mRNAs in this study were selected based on their association with glucocorticoid regulation in astrocytes, perhaps this pattern would be applicable to other glucocorticoid-regulated mRNAs in astrocytes. Although we cannot determine with these data if the observed changes in the two conditions are occurring by similar mechanisms (e.g., direct regulation by glucocorticoid receptor), these correlations suggest that robust glucocorticoid sensitivity may be able to induce regulation of select mRNAs in the brain independent of exposure duration.
Although we have substantiated the notion of glucocorticoid regulation in vivo of mRNAs regulated by glucocorticoids in astrocytes in vitro via qPCR, we cannot localize mRNA regulation to specific cell types in a multicellular brain tissue sample using this technique. In order to examine the neuroanatomical nature of these glucocorticoid-regulated mRNAs in vivo, we performed in situ hybridization studies on brain tissue from the chronic corticosterone exposure experiments (e.g., mice chronically exposed to corticosterone solution or vehicle solution). Using semi-quantitative densitometry, we were able to assess the glucocorticoid regulation of a number of mRNAs in the prefrontal cortex and/or hippocampus by ISH (Figure 6). Although only a subset of differential expression as measured by qPCR were validated by ISH, the ISH data was directionally consistent with the qPCR data in almost all cases; the discrepancy of statistical significance may be in part due to a more robust sensitivity of qPCR measurements.
The hippocampus has a well-known cytoarchitecture that can readily associate cell types with specific locations in this brain region. Within the hippocampus, there are well-known layers such as the pyramidal cell layer in the hippocampal subfields (e.g., dentate gyrus, CA1, CA2, CA3), which is characterized by a higher density of neurons compared with other regions. In contrast, the molecular layer contains a relatively lower neuron density. We thus examined the neuroantomical expression patterns of these 6 mRNAs in an effort to associate the observed mRNA regulation with the location of specific cell types. Similarly general expression patterns of these mRNAs were observed by ISH in sections from both vehicle-treated animals and corticosterone-treated animals, suggesting that the observed changes are likely occurring in cells that express these mRNAs under basal conditions. Among mRNAs measured, we detected ISH signal for Fkbp5 predominately in the pyramidal cell layer throughout multiple hippocampal subfields (Figure 6A). Following chronic glucocorticoids, the hybridization signal appears to rise across the pyramidal cell layer of the hippocampal subfields without any noticeable increase in signal in the molecular layer. While additional high resolution images will be necessary to fully determine in which cell types Fkbp5 is being regulated, this expression pattern suggests the mRNA regulation occurs in pyramidal neuronal cell populations. In contrast, mRNAs regulated by chronic corticosterone but not expressed in pyramidal cell regions may be regulated in other cell types (e.g., astrocytes). Strikingly, all of the other corticosterone-regulated mRNAs analyzed by ISH in this study demonstrate very low or undetectable levels of expression in pyramidal neuron subfields (Gfap, Glul, Gja1, Gjb6). The gene expression patterns of these mRNAs appear much more homogeneous across the hippocampus, an anatomical distribution that is largely consistent with the reported distribution of astrocytes in this tissue (Ogata and Kosaka, 2002; de Vivo et al., 2010). Importantly, these mRNAs also show similar expression patterns as that of Aldh1l1, a mRNA previously shown to be substantially enriched in astrocytes and described as a pan-astrocytic marker (Cahoy et al., 2008). While further studies will be required to definitively identify the cell type of this differential glucocorticoid-sensitive gene expression, given the similar general expression patterns of these mRNAs between corticosterone-treated and vehicle treated animals along with the highly similar anatomical distribution of Gja1, Gbj6, and Glu1 the pan-astrocytic marker Aldh1l1, these results are consistent with the hypothesis that these mRNAs are regulated by chronic glucocorticoid exposure in astrocytes in vivo.
We also note a differential regulation of astrocyte cell markers by corticosterone treatment in vivo that may be important for future studies of glucocorticoid signaling involving astrocytes. Gfap, a cytoskeletal filament and a marker historically associated with astrocytes in the brain, yields a different expression pattern compared with the pan-astrocytic marker Aldh1l1 and the other astrocyte-enriched mRNAs in the hippocampus (Figure 6A). Gfap mRNA is detected at relatively lower levels throughout the hippocampus but at relatively higher levels at the boundary of the structure; this boundary expression is consistent with previously reported Gfap expression in ependymal cells (i.e., pial distribution) (Liu et al., 2006). Following chronic glucocorticoid-treatment, Gfap mRNA levels decrease across all areas (ependymal cells and non-pyramidal cell areas of the hippocampus), consistent with our qPCR data and previous reports (O'Callaghan et al., 1989). Recent studies on major depression have used Gfap in human postmortem experiments and animal models of depression to measure astrocyte cell number; multiple reports have reported decreased astrocyte cell density in these conditions based on Gfap protein measurements (e.g., Rajkowska et al., 1999; Miguel-Hidalgo et al., 2000). However, this interpretation may be confounded by glucocorticoid-induced downregulation of Gfap mRNA and corresponding protein in these conditions associated with hypercortisolemia. If astrocyte cell numbers are changing, then all astrocyte markers would also be expected to change in parallel. We have measured Aldh1l1, a more recently identified pan-astrocyte marker, and found that Aldh1l1 is not downregulated by glucocorticoids under any measured condition (in contrast, our ISH data suggest possible upregulation by chronic corticosterone exposure). These findings suggest that astrocyte numbers may not be decreasing but that the astrocyte cytoskeleton is dynamically regulated in conditions of hypercortisolemia. Additional cell counting studies using Gfap and Aldh1l1 are needed to clarify this issue. In general, future studies in astrocytes would benefit from using multiple cell markers or a marker known not to be affected by any component of the manipulation or disease of interest.
What are the functional consequences of glucocorticoid regulation in astrocytes in vivo? We can project functional alterations based on the known roles of the mRNAs measured. Two potential functional alterations involve gap junction coupling and glutamate signaling. Astrocytes express two main gap junction proteins, connexin-30 (Gjb6) and connexin-43 (Gja1). Gjb6 was strongly upregulated by both acute and chronic corticosterone exposure in vivo, while Gja1 was downregulated by chronic corticosterone exposure. Changes in these gap junctions would likely alter the permeability within astrocytes and between astrocytes and other cells [e.g., oligodendrocytes (Orthmann-Murphy et al., 2007)], a significant alteration given the important role of gap junctions in intercellular communication among astrocytes. Astrocytes also play an important role in glutamate signaling, both in terms of glutamate synthesis and glutamate transport. While glucocorticoids have been known to increase glutamine synthetase (Glul) expression, we observed that chronic glucocorticoid exposure decreased the expression of both astrocytic glutamate transporters, GLT-1 (Slc1a2) and GLAST (Slc1a3). Decreases in astrocyte glutamate uptake have been reported in chronic stress conditions (Almeida et al., 2010; de Vasconcellos-Bittencourt et al., 2011); our results suggest that impairment of glutamate transport could be mediated by chronic glucocorticoid exposure, a potential mechanism for the changes seen in chronic stress.
These data also have interesting parallels to clinical findings in postmortem studies of major depression, a condition associated with elevations of glucocorticoids. Multiple mRNAs regulated by chronic corticosterone exposure in this study are reported to be changed in the same direction in human depression (e.g., downregulation of Gja1, Slc1a2, and Slc1a3) (Bernard et al., 2010). While the data presented in these studies cannot be directly linked to depression biology, they do provide a potential mechanism (glucocorticoid regulation) by which these genes may be altered. Alterations of astrocyte-enriched fibroblast growth factor (FGF) receptors observed here (upregulation of Fgfr3, downregulation of Fgfr1) may also be relevant to depression biology given the findings of altered expression of FGF receptors and ligands in depression (Evans et al., 2004). We would be interested in determining if these same mRNAs are regulated in other animal models demonstrating chronically elevated corticosterone levels, such as animal models of depression. Given the associations of increased depressive-like behavior and astrocyte dysfunction (Banasr and Duman, 2008), perhaps these mRNAs are relevant to the underlying mechanisms of these observations. A caveat of such extrapolations is that we have documented glucocorticoid-mediated mRNA changes but not changes in corresponding proteins; further studies are needed to measure correlations between glucocorticoid-mediated changes in mRNA expression and any changes in corresponding protein levels.
In summary, our data demonstrate that select mRNAs regulated by glucocorticoids in astrocytes in vitro are also regulated by acute and/or chronic corticosterone exposure in vivo. A number of these mRNAs have been reported as astrocyte-enriched (Cahoy et al., 2008), and ISH data of anatomical expression patterns within the hippocampus suggest that certain mRNA changes occur within regions containing astrocytes. Together, these data suggest that the observed glucocorticoid-mediated mRNA regulation may be occurring in astrocytes. These data add physiological significance to parallel gene expression findings in astrocyte cell culture and suggest that additional findings of glucocorticoid regulation in astrocyte cultures in vitro may extend to the brain in vivo. Based on these results, we posit that differential responses to glucocorticoids by cell type is an important factor for fully understanding glucocorticoid-mediated mechanisms in the brain. The functional contribution of glucocorticoid regulation in astrocytes to stress signaling remains to be investigated and may be integral to both the classical stress response and pathological conditions associated with elevations in glucocorticoid levels in the brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grants T32-EY017878 (Bradley S. Carter), T32-MH014279 (Bradley S. Carter), and R01-DA025973 (Robert C. Thompson).
References
Almeida, R. F., Thomazi, A. P., Godinho, G. F., Saute, J. A., Wofchuk, S. T., Souza, D. O., et al. (2010). Effects of depressive-like behavior of rats on brain glutamate uptake. Neurochem. Res. 35, 1164–1171. doi: 10.1007/s11064-010-0169-4
Banasr, M., and Duman, R. S. (2008). Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 64, 863–870. doi: 10.1016/j.biopsych.2008.06.008
Barley, K., Dracheva, S., and Byne, W. (2009). Subcortical oligodendrocyte-and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr. Res. 112, 54–64. doi: 10.1016/j.schres.2009.04.019
Bernard, R., Kerman, I. A., Thompson, R. C., Jones, E. G., Bunney, W. E., Barchas, J. D., et al. (2010). Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 16, 634–646. doi: 10.1038/mp.2010.44
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Carter, B., Fletcher, J., and Thompson, R. (2010). Analysis of messenger RNA expression by in situ hybridization using RNA probes synthesized via in vitro transcription. Methods 52, 322. doi: 10.1016/j.ymeth.2010.08.001
Carter, B. S., Meng, F., and Thompson, R. C. (2012). Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol. Genomics 44, 1188–1200. doi: 10.1152/physiolgenomics.00097.2012
Datson, N., van der Perk, J., de Kloet, E., and Vreugdenhil, E. (2001). Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur. J. Neurosci. 14, 675–689. doi: 10.1046/j.0953-816x.2001.01685.x
De Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
de Vasconcellos-Bittencourt, A. P., Vendite, D. A., Nassif, M., Crema, L. M., Frozza, R., Thomazi, A. P., et al. (2011). Chronic stress and lithium treatments alter hippocampal glutamate uptake and release in the rat and potentiate necrotic cellular death after oxygen and glucose deprivation. Neurochem. Res. 36, 793–800. doi: 10.1007/s11064-011-0404-7
de Vivo, L., Melone, M., Rothstein, J. D., and Conti, F. (2010). GLT-1 promoter activity in astrocytes and neurons of mouse hippocampus and somatic sensory cortex. Front. Neuroanat. 3:31. doi: 10.3389/neuro.05.031.2009
Evans, S. J., Choudary, P. V., Neal, C. R., Li, J. Z., Vawter, M. P., Tomita, H., et al. (2004). Dysregulation of the fibroblast growth factor system in major depression. Proc. Natl. Acad. Sci. U.S.A 101, 15506–15511. doi: 10.1073/pnas.0406788101
Gourley, S. L., Wu, F. J., Kiraly, D. D., Ploski, J. E., Kedves, A. T., Duman, R. S., et al. (2008). Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol. Psychiatry 63, 353–359. doi: 10.1016/j.biopsych.2007.07.016
Karatsoreos, I. N., Bhagat, S. M., Bowles, N. P., Weil, Z. M., Pfaff, D. W., and McEwen, B. S. (2010). Endocrine and physiological changes in response to chronic corticosterone: a potential model of the metabolic syndrome in mouse. Endocrinology 151, 2117–2127. doi: 10.1210/en.2009-1436
Laping, N. J., Nichols, N. R., Day, J. R., Johnson, S. A., and Finch, C. E. (1994). Transcriptional control of glial fibrillary acidic protein and glutamine synthetase in vivo shows opposite responses to corticosterone in the hippocampus. Endocrinology 135, 1928–1933. doi: 10.1210/en.135.5.1928
Lee, R. S., Tamashiro, K. L., Yang, X., Purcell, R. H., Harvey, A., Willour, V. L., et al. (2010). Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151, 4332–4343. doi: 10.1210/en.2010-0225
Lindholm, D., Castren, E., Hengerer, B., Zafra, F., Berninger, B., and Thoenen, H. (1992). Differential regulation of Nerve Growth Factor (NGF) synthesis in neurons and astrocytes by glucocorticoid hormones. Eur. J. Neurosci. 4, 404–410. doi: 10.1111/j.1460-9568.1992.tb00889.x
Liu, X., Bolteus, A. J., Balkin, D. M., Henschel, O., and Bordey, A. (2006). GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54, 394–410. doi: 10.1002/glia.20392
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. doi: 10.1038/nrn2639
Ma, X., Levy, A., and Lightman, S. (1997). Rapid changes in heteronuclear RNA for corticotrophin-releasing hormone and arginine vasopressin in response to acute stress. J. Endocrinol. 152, 81–89. doi: 10.1677/joe.0.1520081
Maragakis, N. J., and Rothstein, J. D. (2006). Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689. doi: 10.1038/ncpneuro0355
McClennen, S. J., Cortright, D. N., and Seasholtz, A. F. (1998). Regulation of pituitary corticotropin-releasing hormone-binding protein messenger ribonucleic acid levels by restraint stress and adrenalectomy. Endocrinology 139, 4435–4441. doi: 10.1210/en.139.11.4435
McEwen, B. S. (2005). Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism 54, 20–23. doi: 10.1016/j.metabol.2005.01.008
McEwen, B. S., Weiss, J. M., and Schwartz, L. S. (1968). Selective retention of corticosterone by limbic structures in rat brain. Nature 220, 911–912. doi: 10.1038/220911a0
McInnes, K. J., Kenyon, C. J., Chapman, K. E., Livingstone, D. E., Macdonald, L. J., Walker, B. R., et al. (2004). 5alpha-reduced glucocorticoids, novel endogenous activators of the glucocorticoid receptor. J. Biol. Chem. 279, 22908–22912. doi: 10.1074/jbc.M402822200
Melcangi, R. C., Celotti, F., Castano, P., and Martini, L. (1993). Differential localization of the 5 alpha-reductase and the 3 alpha-hydroxysteroid dehydrogenase in neuronal and glial cultures. Endocrinology 132, 1252–1259. doi: 10.1210/en.132.3.1252
Miguel-Hidalgo, J. J., Baucom, C., Dilley, G., Overholser, J. C., Meltzer, H. Y., Stockmeier, C. A., et al. (2000). Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol. Psychiatry 48, 861–873. doi: 10.1016/S0006-3223(00)00999-9
Morsink, M., Steenbergen, P., Vos, J., Karst, H., Joels, M., Kloet, E. R., et al. (2006). Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J. Neuroendocrinol. 18, 239–252. doi: 10.1111/j.1365-2826.2006.01413.x
Newell-Price, J., Bertagna, X., Grossman, A. B., and Nieman, L. K. (2006). Cushing's syndrome. Lancet 367, 1605–1617. doi: 10.1016/S0140-6736(06)68699-6
Nichols, N. R., Osterburg, H. H., Masters, J. N., Millar, S. L., and Finch, C. E. (1990). Messenger RNA for glial fibrillary acidic protein is decreased in rat brain following acute and chronic corticosterone treatment. Brain Res. Mol. Brain Res. 7, 1–7. doi: 10.1016/0169-328X(90)90066-M
O'Callaghan, J. P., Brinton, R. E., and McEwen, B. S. (1989). Glucocorticoids regulate the concentration of glial fibrillary acidic protein throughout the brain. Brain Res. 494, 159–161.
Ogata, K., and Kosaka, T. (2002). Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113, 221–233. doi: 10.1016/S0306-4522(02)00041-6
Orthmann-Murphy, J. L., Freidin, M., Fischer, E., Scherer, S. S., and Abrams, C. K. (2007). Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27, 13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007
Patel, A. J., Hunt, A., and Tahourdin, C. S. (1983). Regulation of in vivo glutamine synthetase activity by glucocorticoids in the developing rat brain. Brain Res. 312, 83–91.
Paxinos, G., and Franklin, K. (2001). The Mouse Brain Atlas in Stereotaxic Coordinates. San Diego, CA: Academic.
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer, H. Y., et al. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085–1098. doi: 10.1016/S0006-3223(99)00041-4
Sarabdjitsingh, R. A., Isenia, S., Polman, A., Mijalkovic, J., Lachize, S., Datson, N., et al. (2010). Disrupted corticosterone pulsatile patterns attenuate responsiveness to glucocorticoid signaling in rat brain. Endocrinology 151, 1177–1186. doi: 10.1210/en.2009-1119
Scharf, S. H., Liebl, C., Binder, E. B., Schmidt, M. V., and Muller, M. B. (2011). Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS ONE 6:e16883. doi: 10.1371/journal.pone.0016883
Sierra, A., Gottfried-Blackmore, A., Milner, T. A., McEwen, B. S., and Bulloch, K. (2008). Steroid hormone receptor expression and function in microglia. Glia 56, 659–674. doi: 10.1002/glia.20644
Slezak, M., Korostynski, M., Gieryk, A., Golda, S., Dzbek, J., Piechota, M., et al. (2013). Astrocytes are a neural target of morphine action via glucocorticoid receptor-dependent signaling. Glia 61, 623–635. doi: 10.1002/glia.22460
Strachan, T., and Read, A. P. (eds.). (1999). Human Molecular Genetics, 2nd Edn. New York, NY: Wiley-Liss.
Ten, S., New, M., and Maclaren, N. (2001). Addison's disease 2001. J. Clin. Endocrinol. Metab. 86, 2909–2922. doi: 10.1210/jc.86.7.2909
Unemura, K., Kume, T., Kondo, M., Maeda, Y., Izumi, Y., and Akaike, A. (2012). Glucocorticoids decrease astrocyte numbers by reducing glucocorticoid receptor expression in vitro and in vivo. J. Pharmacol. Sci. 119, 30–39. doi: 10.1254/jphs.12047FP
Vandevyver, S., Dejager, L., and Libert, C. (2012). On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic 13, 364–374. doi: 10.1111/j.1600-0854.2011.01288.x
Keywords: glucocorticoids, corticosterone, RNA, messenger, brain, astrocytes, mice
Citation: Carter BS, Hamilton DE and Thompson RC (2013) Acute and chronic glucocorticoid treatments regulate astrocyte-enriched mRNAs in multiple brain regions in vivo. Front. Neurosci. 7:139. doi: 10.3389/fnins.2013.00139
Received: 14 May 2013; Accepted: 19 July 2013;
Published online: 07 August 2013.
Edited by:
François Pralong, Centre Hospitalier Universitaire Vaudois, SwitzerlandReviewed by:
Julie A. Chowen, Hospital Infantil Universitario Niño Jesús, SpainValerio Magnaghi, Università Degli Studi di Milano, Italy
Copyright © 2013 Carter, Hamilton and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert C. Thompson, Department of Psychiatry, Molecular and Behavioral Neuroscience Institute, University of Michigan, 5049 Biomedical Research Science Building, 109 Zina Pitcher Place, Ann Arbor, MI 48109, USA e-mail:bXV0YW50QHVtaWNoLmVkdQ==