 Esperanza Fernández
Esperanza Fernández Nicholas Rajan
Nicholas Rajan Claudia Bagni
Claudia Bagni- 1Center for the Biology of Disease, Vlaams Institut voor Biotechnologie, Leuven, Belgium
- 2Center for Human Genetics, Leuven Institute for Neuroscience and Disease, KU Leuven, Leuven, Belgium
- 3Department of Biomedicine and Prevention, University “Tor Vergata”, Rome, Italy
The fragile X mental retardation protein (FMRP) is an RNA-binding protein that regulates mRNA metabolism. FMRP has been largely studied in the brain, where the absence of this protein leads to fragile X syndrome, the most frequent form of inherited intellectual disability. Since the identification of the FMRP gene in 1991, many studies have primarily focused on understanding the function/s of this protein. Hundreds of potential FMRP mRNA targets and several interacting proteins have been identified. Here, we report the identification of FMRP mRNA targets in the mammalian brain that support the key role of this protein during brain development and in regulating synaptic plasticity. We compared the genes from databases and genome-wide association studies with the brain FMRP transcriptome, and identified several FMRP mRNA targets associated with autism spectrum disorders, mood disorders and schizophrenia, showing a potential common pathway/s for these apparently different disorders.
Introduction
Protein synthesis at subcellular sites is a well-conserved mechanism that allows the rapid expression of specific genes in response to localized cues (Xing and Bassell, 2013). During transport, mRNAs are stabilized via association with multiple and different trans-acting factors, such as RNA-binding proteins (RBPs) and non-coding RNAs, forming ribonucleoparticles (RNPs) that vary in size and composition during cell cycle and development.
In highly polarized cells, such as neurons, mRNAs are transported from the nucleus to dendrites and axons where these molecules undergo local translation and degradation (Steward and Schuman, 2003; Bramham, 2008; Cajigas et al., 2010; Doyle and Kiebler, 2012; Hornberg and Holt, 2013) according to their subcellular localization and cellular inputs (Bramham, 2008).
RNA-binding proteins recognize and bind mRNA targets through regulatory elements in the 5′ and 3′ untranslated regions (UTRs) (Pichon et al., 2012), and in some cases the coding regions are also involved in these interactions (Anko and Neugebauer, 2012). Binding to mRNAs is mediated through well-known RNA-binding motifs, which are often present in multiple copies (Clery et al., 2008) and typically bind short RNA sequences (Anko and Neugebauer, 2012). Several RBPs cooperate for the binding of mRNA, thereby increasing the specificity of this interaction (Matlin et al., 2005; Ule and Darnell, 2006). The actin cytoskeleton might well facilitate RNA recognition, as this structure associates with RBPs and coordinates the binding of these proteins to mRNA (Percipalle, 2009). However, individual RBPs bind to several mRNAs. The multi-targeted binding property of RBPs has led to a model of regulated gene expression in eukaryotes termed “the post-transcriptional operon” (Keene, 2007).
The fragile X mental retardation protein (FMRP) is a widely studied RBP in the brain. Silencing of the FMR1 gene encoding FMRP leads to fragile X mental retardation syndrome (FXS), the most common cause of inherited intellectual disability (Bagni et al., 2012). A majority of the clinical cases of FXS reflect a lack of FMRP due to a large trinucleotide CGG-repeat expansion in the 5′ UTR of the gene, resulting in FMR1 gene silencing. Rare cases have been reported to carry partially deleted or mutated FMRP (De Boulle et al., 1993; Mila et al., 2000; Coffee et al., 2008; Collins et al., 2010). The FMR1 gene and FMRP have also been associated with the pathogenesis of other disorders, such as fragile X-associated tremor ataxia syndrome (FXTAS), premature ovarian failure (POF), and autism spectrum disorder (ASD) (Bagni et al., 2012).
Here, we briefly reviewed the structure and function of FMRP, a multifunctional RBP that regulates the transport, stability and local protein synthesis of hundreds of RNAs in the brain. We further discuss how anomalies in the expression of FMRP alter the condition of its targets and ultimately, highlight a subset of FMRP target mRNAs dysregulated in autism spectrum disorders (ASDs), mood disorders (MDs) including bipolar disorder (BD), major depressive disorder (MDD), attention deficit hyperactive disorder (ADHD), and schizophrenia (SCZ).
FMRP Structure, RNA Targets and Protein Partners
The human FMR1 gene is ubiquitously expressed (https://www.genevestigator.com/gv/), with higher abundance in some tissues (Kaufmann et al., 2002; Xie et al., 2009). The gene comprises 17 exons spanning 38 kb of Xq27.3 (Eichler et al., 1993). Alternative splicing of the gene results in the generation of 12 protein isoforms (De Boulle et al., 1993; Brackett et al., 2013).
In the mammalian brain, FMRP targets hundreds of mRNAs (Miyashiro et al., 2003; Darnell et al., 2011; Bagni et al., 2012; Gross et al., 2012; Wang et al., 2012) and non-coding RNAs, such as the brain cytoplasmic RNA BC1/BC200 in vitro and in vivo (Zalfa et al., 2003, 2005; Gabus et al., 2004; Johnson et al., 2006; Lacoux et al., 2012) and a few microRNAs (Jin et al., 2004; Edbauer et al., 2010; Gessert et al., 2010; Muddashetty et al., 2011; Tian et al., 2013).
Structural studies of the FMRP domain have contributed to the understanding of the molecular function/s of this protein. The N-terminal region, characterized by the presence of two Tudor domains (TD)(Ramos et al., 2006), binds in vitro RNA homopolymers and the small non-coding BC1 RNA (Gabus et al., 2004; Zalfa et al., 2005; Lacoux et al., 2012) (Figure 1). The central region contains two K homology domains (KH) and a nuclear export signal (NES) (Valverde et al., 2008). The most severe single point mutation identified in a patient with FXS is an lle367Asn, located on helix α2 of the KH2 domain (De Boulle et al., 1993). The murine FMRP, carrying the corresponding mutation (Ile304Asn), loses the ability to bind RNA (Zang et al., 2009), likely reflecting the destabilization of the hydrophobic core, which partially unfolds the domain (Di Marino et al., 2013). A recent study in non-neuronal cells has shown that the FMRP Ile304Asn mutation reduces the binding affinity of a subset of mRNAs, such as neurofibromatosis type 1 (NF1), FMR1, bifunctional glutamate/proline-tRNA ligase (EPRS), serine/threonine-protein phosphatase 2A catalytic subunit alpha isoform (PPP2CA), ubiquitin-protein ligase E3A (UBE3A), structural maintenance of chromosomes protein 1A (SMC1A) and cohesin subunit SA-2 (STAG2) (Ascano et al., 2012).
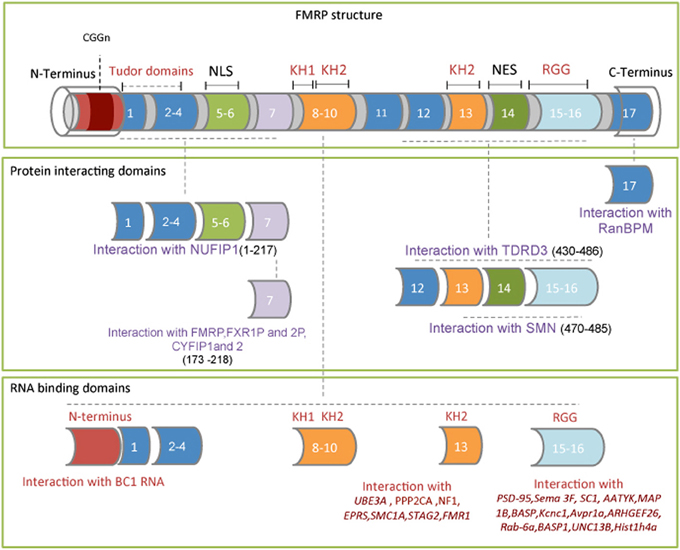
Figure 1. FMRP exon structure comprising its functional domains. Upper frame: The red box at the N-terminus of exon 1 indicates the location of the CGG triplet repeat within the 5′ UTR of the mRNA. The four RNA binding domains are: the N-terminus, the two K homology domains (KH1 and KH2) and the RGG box. Middle frame: FMRP domains interacting with NUFIP1, CYFIP1, CYFIP2, FXR1P, FXR2P, TDRD3, and SMN proteins. The FMRP amino acid sequence involved in these interactions is shown between the brackets. The nuclear localization signal (NLS) and the nuclear export signal (NES) are also indicated. Lower frame: The FMRP RNA binding domains and the RNA/mRNA targets directly bound are indicated.
The C-terminal region, containing an RGG box, is involved in the interaction of well-characterized FMRP mRNA targets (Darnell et al., 2001; Menon and Mihailescu, 2007; Westermark and Malter, 2007; Zalfa et al., 2007; Menon et al., 2008; Blackwell et al., 2010). The C-terminal region of FMRP binds post synaptic protein-95 (PSD-95) mRNA (Zalfa et al., 2007), microtubule associated protein 1B (MAP1B) (Darnell et al., 2001; Zalfa et al., 2003), semaphorin 3F (SEMA3F) (Menon and Mihailescu, 2007), extracellular matrix protein 2 (SC1), brain acid soluble protein 1 (NAP22) (Darnell et al., 2001) and serine/threonine-protein kinase LMTK1 (AATYK) (Blackwell et al., 2010) mRNAs, a few of which are depicted in Figure 1.
FMRP homodimerises and interacts with several cytoplasmic and nuclear proteins involved in mRNA metabolism and cytoskeleton-remodeling proteins (Bagni and Klann, 2012). Among the best characterized FMRP-interacting proteins are protein argonaute-2 (Ago2) (Muddashetty et al., 2011), 58 kDa microspherule protein (MSP58) (Davidovic et al., 2006), nuclear fragile X mental retardation-interacting proteins 1 and 2 (NUFIP1 and 2) (Bardoni et al., 2003), the survival of motor neuron (SMN) (Piazzon et al., 2008), the Tudor domain-containing protein 3 (Linder et al., 2008), nuclear export factor 2 (NXF2) (Zhang et al., 2007), dicer (Cheever and Ceman, 2009), cytoplasmic interacting protein CYFIP1 (Schenck et al., 2001, 2003; Napoli et al., 2008; De Rubeis et al., 2013) and the two paralogs, fragile X-related proteins 1 and 2 (FXRP1 and FXRP2) (Tamanini et al., 1999) (Figure 1).
Cellular and Molecular Functions of FMRP
Although in neurons, FMRP has been localized in the nucleus, cell body and dendrites (Willemsen et al., 1996), the cytoplasmic function of FMRP has been the most studied. FMRP forms large cytoplasmic RNPs containing several proteins and RNAs, and this protein is involved in the transport, stability and translation of several mRNAs (Bagni et al., 2012). One report suggested FMRP might also function as splicing enhancer (Didiot et al., 2008). Additionally, Drosophila FMRP has been related to the RNA-editing pathway (Bhogal et al., 2011).
Regulation of mRNA Transport
FMRP transports RNA/mRNAs from the cell body to synapses in an activity-dependent manner and through a dynamic association with microtubule motors (Kanai et al., 2004; Antar et al., 2005; Ferrari et al., 2007; Dictenberg et al., 2008; Charalambous et al., 2013). FMRP granules transport mRNA including its own (Antar et al., 2004; Ferrari et al., 2007; Kao et al., 2010), and the absence of FMRP impairs the localization of Map1b and SAP90/PSD-95-associated protein 4 (Sapap4) mRNAs, thus altering the proper synthesis of these proteins at synapses (Dictenberg et al., 2008; Kao et al., 2010).
Regulation of mRNA Stability
Initial studies performed in Fmr1 KO mice have revealed that the absence of FMRP alters the abundance of hundreds of mRNAs in the brain (Brown et al., 2001; Miyashiro et al., 2003; Gantois et al., 2006); a few mRNAs were found to be down regulated in all three studies. Further analyses on specific mRNAs showed that dysregulation occurred in specific brain areas and/or subcellular compartments, suggesting that FMRP might regulate the same mRNA in multiple ways (Miyashiro et al., 2003). FMRP modulates the stability of certain mRNAs by preventing or sustaining mRNA decay (De Rubeis and Bagni, 2010). As an example of the two opposite activities on different mRNAs, it has been shown that hippocampal FMRP protects PSD-95 mRNA from decay (Zalfa et al., 2007) in an activity-dependent manner; however, FMRP protein also facilitates the decay of nuclear RNA export factor 1 (NXF1) mRNA in mouse neuroblastoma (N2a) cells (Zhang et al., 2007). Furthermore, FMRP regulates PSD-95 mRNA stability in the hippocampus (Zalfa et al., 2007) and regulates translation at cortical synapses (Muddashetty et al., 2007). PSD-95 mRNA is an important player in synaptic plasticity and is affected in ASD (Feyder et al., 2010) and SCZ (Toro and Deakin, 2005).
The cortical region of the Fmr1 KO mouse brain shows the reduced expression of different GABAA receptor subunits (El Idrissi et al., 2005; Gantois et al., 2006), consistent with evidence of imbalanced GABAergic signaling in FXS patients. Taken together, FMRP-RNPs might play different roles in several brain regions and regulate mRNAs through different mechanisms according to the developmental stage and subcellular localization.
Regulation of mRNA Translation
The translational dysregulation of FMRP mRNA targets significantly contributes to the FXS phenotype (Bagni et al., 2012; Darnell and Klann, 2013). Initial studies performed in lymphoblastoid cells derived from FXS individuals showed an increased translation rate in several FMRP targets (Brown et al., 2001). The increased translation of FMRP mRNA targets was also observed in Fmr1 KO mice specifically at synapses, consistent with the idea that FMRP functions as a repressor of translation (Muddashetty et al., 2007; Narayanan et al., 2007; Napoli et al., 2008; De Rubeis et al., 2013).
FMRP activity is regulated in response to different receptor signaling cascades, i.e., type I metabotropic glutamate receptors (mGluRs) (Huber et al., 2002), the 2-amino-3-(5-methyl-3-oxo-1,2-ox-azol-4-yl) propanoic acid (AMPA) receptors (Nakamoto et al., 2007), the γ-aminobutyric acid (GABA) receptors (Centonze et al., 2008; Curia et al., 2009; Shang et al., 2009), the N-methyl-D-aspartate (NMDA) receptors (Suvrathan et al., 2010; Yun and Trommer, 2011; Eadie et al., 2012), the tyrosine kinase or BDNF/NT-3 growth factor (TrkB) receptors (Napoli et al., 2008; Louhivuori et al., 2011; De Rubeis et al., 2013), the dopamine (DA) receptors (Wang et al., 2008) and recently the cannabinoid receptors (Maccarrone et al., 2010; Busquets-Garcia et al., 2013).
One of the most affected and best characterized signaling cascades in fragile X is the mGluR (Bear et al., 2004). Upon mGluR receptor activation, FMRP-mediated translational block is released and protein synthesis can ensue. In the absence of FMRP, the increase in protein synthesis results in a receptor imbalance; an increase in the mGluR1 and mGluR5 activity and the reduced insertion of AMPA receptors at the surface that leads to enhanced mGluR long-term depression (mGluR-LTD) (Bear et al., 2004).
mGluR-LTD is a form of synaptic plasticity that involves mRNA targeting and local protein synthesis and degradation (Bear and Malenka, 1994), and this condition can be induced through the application of (S)-3,5-dihydroxyphenylglycine (DHPG) (Wisniewski and Car, 2002) in a protein synthesis-independent manner (Huber et al., 2002). In Fmr1 KO mice, DHPG-induced LTD is strongly increased and these electrophysiological phenotypes established the “mGluR theory” in FXS (Bear et al., 2004).
FMRP activity is regulated through posttranslational modifications. DHPG-induced LTD also activates FMRP synthesis at synapses (Antar et al., 2004; Ferrari et al., 2007; Kao et al., 2010), which in turn is quickly degraded through the ubiquitin-proteasome system (Hou et al., 2006). The effect of FMRP on protein synthesis is influenced by the phosphorylation status of FMRP (Ceman et al., 2003), via the mTOR pathway (Narayanan et al., 2007): phosphorylated FMRP represses translation, while dephosphorylated FMRP releases the inhibition, allowing protein synthesis to ensue, a mechanism similarly shown for previously characterized eukaryotic initiation factor 4E binding proteins (eIF4E-BPs) in non-neuronal cells (Richter and Klann, 2009).
FMRP has also been detected in P bodies (PB), stress granules (SG) (Kedersha et al., 2005), and cytoplasmic structures, containing translationally silent pre-initiation complexes. FMRP is part of mRNPs (Siomi et al., 1996; Laggerbauer et al., 2001; Ishizuka et al., 2002; Zalfa et al., 2003; Anderson and Kedersha, 2006; Monzo et al., 2006; Papoulas et al., 2010; Charalambous et al., 2013), supporting the function of FMRP as a translational repressor at the initiation level, as observed at synapses both in vitro (Laggerbauer et al., 2001) and in vivo (Napoli et al., 2008; De Rubeis et al., 2013).
We have shown that FMRP represses translation through its binding to CYFIP1, a neuronal eIF4E-BP (Napoli et al., 2008). CYFIP1 binds to eIF4E, blocking the initiation of translation. Subsequently, the synaptic stimuli CYFIP1-FMRP complex is released from eIF4E and translation ensues (Napoli et al., 2008). Notably, CYFIP1 is also implicated in actin cytoskeleton remodeling (Kobayashi et al., 1998; Eden et al., 2002; Schenck et al., 2003; Stradal et al., 2004; Chen et al., 2010). We have recently shown that CYFIP1 links local protein synthesis and actin dynamics (De Rubeis et al., 2013). FMRP has also been proposed to regulate mRNA elongation (Darnell et al., 2011).
FXS and Commonalities with Other Diseases
FXS is the most common monogenic cause of ASD, and 30% of patients with FXS present autistic behaviors (Bagni et al., 2012). Early studies performed on heterozygous females carrying the fragile X mutant gene showed a greater frequency of psychopathologies associated with schizophrenia spectrum diagnoses (Reiss et al., 1988). Furthermore, carriers of premutated FMR1 alleles (reduced FMRP levels) have been associated with a significant degree of psychiatric disorders (Bourgeois et al., 2009). Recently, low FMRP levels have been detected in the postmortem brain from subjects with SCZ, BD and MDD (Fatemi et al., 2010; Kelemen et al., 2013; Kovacs et al., 2013) and in blood samples from schizophrenia patients (Kovacs et al., 2013). Some individuals that display psychoses also carry FMR1 full and pre-mutations (Jonsson et al., 1995; Ashworth et al., 1996; Khin et al., 1998).
It is not known whether decreased levels of FMRP are the cause or the consequence of the development of these disorders. However, it is tempting to speculate that the loss or reduced function of FMRP might lead to a dysregulation of particular FMRP target genes associated with ASD, SCZ, and MD, suggesting the correlation of certain FXS features with these neuronal disorders. Because the GABAergic system is dysfunctional in these disorders (Kelemen et al., 2013) and the lack of FMRP affects the expression of some GABA receptor subunits (D'Hulst and Kooy, 2007), it is reasonable to hypothesize that FMRP reduction might explain the alterations of proteins associated with the GABAergic system in these different neurological diseases. Indeed, recent findings showed that a selective activator of GABAB receptor reversed some FXS associated pathologies (Henderson et al., 2012).
It cannot be ruled out that certain proteins, which are risk factors for ASD, SCZ and/or MD, work together with FMRP and might disrupt the function of this protein in a disease context. Recently, it has been observed that topoisomerase Top3β, a risk gene for SCZ and ASD (Iossifov et al., 2012; Xu et al., 2012; Stoll et al., 2013), binds to FMRP and modifies the function of this protein in vitro, thereby supporting normal neurodevelopment and averting mental disorders (Xu et al., 2013). In addition, the authors observed that the disruption of either Top3β or Fmr1 genes in Drosophila led to a dysregulation of ptk2, which is genetically associated with SCZ (Walsh et al., 2008). Notably, CYFIP1 has been associated with ASD (Sahoo et al., 2006) (Doornbos et al., 2009; Van Der Zwaag et al., 2010; Von Der Lippe et al., 2010; Talebizadeh et al., 2013), SCZ and epilepsy (Sahoo et al., 2006; Tam et al., 2010; Zhao et al., 2012). In addition, we have recently shown that the CYFIP1 interactome contains many novel proteins associated with ASD, SCZ, and MDD, providing new perspectives to define the regulatory pathways shared by neurological disabilities characterized by spine dysmorphogenesis (De Rubeis et al., 2013), a common feature of several neuropsychiatric disorders (Penzes et al., 2011).
Over the last 10 years, several hundred putative FMRP mRNA targets have been identified in the brain (Brown et al., 2001; Chen et al., 2003; Miyashiro et al., 2003; Zalfa et al., 2003, 2007; Muddashetty et al., 2007; Darnell et al., 2011), and more than 6000 targets have been identified in non-neuronal cells (Ascano et al., 2012). While these analyses have expanded the number of FMRP targets, further studies are required to elucidate the extent to which each mRNA contributes to the FXS clinical phenotype/s. There is substantial evidence that individuals with intellectual disabilities are prone to psychological profiles independently of the genetic and/or environmental cause (Turk, 2011).
Based on several large-scale studies, the number of FMRP neuronal target mRNAs is approximately 1,400 (Brown et al., 2001; Chen et al., 2003; Miyashiro et al., 2003; Darnell et al., 2011). We compared 1,169 unique (non-overlapping in the mentioned studies) FMRP mRNA targets with de novo ASD associated genes identified through recent genome-wide association studies (GWAS)(Iossifov et al., 2012; Neale et al., 2012; O'roak et al., 2012a,b; Sanders et al., 2012), obtained from the SFARI (http://gene.sfari.org) and the Autism databases (AutDB (http://www.mindspec.org/autdb.html).
As represented in Figure 2, according to the GWAS, 35 FMRP target mRNAs are associated with ASD (in red), while the SFARI and AutDB databases have revealed that 100 FMRP target mRNAs are candidate genes for ASD (in black). Fifteen genes overlap between the results obtained in the GWAS and the SFARI and AutDB databases (in black/red color). This analysis shows that approximately 10% of the neuronal FMRP targets identified, in the above-mentioned studies, overlap with the genes associated with ASDs (120 out of 1169).
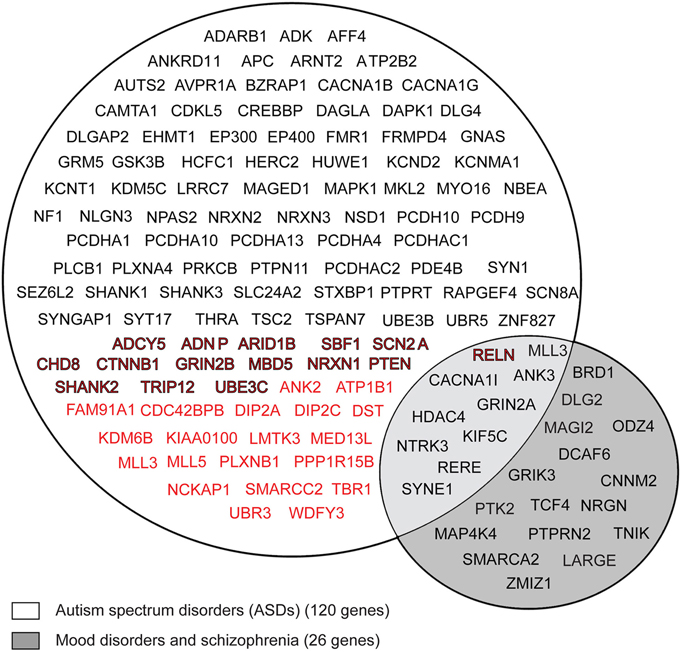
Figure 2. Venn diagram of FMRP mRNA targets associated with autistic spectrum disorder (ASD), schizophrenia (SCZ) and mood disorder (MD). FMRP neuronal target genes (1169) were compared with de novo gene disruptions (nonsense, splice and frameshifts) reported in GWAS are associated with ASD, as previously described (Iossifov et al., 2012; Neale et al., 2012; O'roak et al., 2012b; Sanders et al., 2012), the 528 genes from the SFARI (https://sfari.org/resources/sfari-base) and the 304 genes from the AutDB databases (http://autism.mindspec.org/autdb/Welcome.do). Approximately 120 FMRP target genes were associated with ASD (in black). Among the 120 target genes, included 35 genes were identified from the GWAS (in red) and 100 genes were identified from the two databases (in black). Fifteen genes showed overlap between the GWAS, the SFARI, and AutDB databases (in black/red color). The 1169 FMRP target genes were compared with the genes associated with SCZ and MD in the NHGRI GWAS database (http://www.genome.gov/gwastudies/) and 26 common FMRP targets were shown.
We also compared the 1,169 FMRP target mRNAs with 176 genes associated with BD, attention deficit-hyperactivity disorder (ADHD), mood disorder (MD), and SCZ (GWAS compiled by the National Human Genome Research Institute catalog http://www.genome.gov/). Twenty-six (out of 176) FMRP target mRNAs were also identified in this cohort (Figure 2, in gray). Because a few genes in this group (10) were also detected among the FMRP targets in the ASD group, it is reasonable to hypothesize that ASD, SCZ, and mood disorders (BD, MDD, ADHD) share certain common signaling pathways.
Future Perspectives
Post-transcriptional studies have revealed that the FMRP regulon controls disease-related proteins that affect both neurodevelopment and adult brain plasticity and homeostasis. The emerging wave of genetic association studies has revealed a large number of risk genes for several neurodegenerative diseases and neurodevelopmental disorders, such as SCZ, ASD, and BD (http://www.genome.gov/gwastudies).
The risk genes for neurodevelopmental disorders, identified through GWAS, were compared with the list of the FMRP targets, and the results suggest that several pathways are dysregulated in FXS and might account for specific FXS phenotypes.
As the FMRP acts as a protein synthesis repressor, it is reasonable to propose that the FXS phenotype might reflect the overexpression of specific genes. However, FMRP not only regulates gene expression at the translational level, but it also influences the stability of several mRNAs. Furthermore, to determine the functional association of the FMRP regulon with the repertoire of genes altered in individuals carrying ASD, SCZ, and MD, it is important to investigate the dosage of these genes in individuals with FXS. Moreover, FXS is a neurodevelopmental disorder, and the absence of FMRP could affect the expression of specific targets at different developmental stages and in different brain areas. Further studies on FMRP targets and the FMRP interactome at specific developmental stages would help to determine the cause of these disorders and develop further strategies to ameliorate FXS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported through funding from VIB (Flemish Institute for Biotechnology), Associazione Italiana Sindrome X Fragile, FWO (Fonds Wetenschappelijk Onderzoek Vlaanderen, G.0705.11), Telethon (GGP10150), The Queen Elisabeth Foundation of Belgium, and a HEALTH-2009-2.1.2-1 EU-FP7 (European Union–Frame Programe 7) “SynSys” grant to Claudia Bagni. Nicholas Rajan was supported through funding from a Brain Train EU-FP7 grant to Claudia Bagni. Esperanza Fernández received an Intra-European Marie Curie Fellowship FP7 for career development.
References
Anderson, P., and Kedersha, N. (2006). RNA granules. J. Cell Biol. 172, 803–808. doi: 10.1083/jcb.200512082
Anko, M. L., and Neugebauer, K. M. (2012). RNA-protein interactions in vivo: global gets specific. Trends Biochem. Sci. 37, 255–262. doi: 10.1016/j.tibs.2012.02.005
Antar, L. N., Afroz, R., Dictenberg, J. B., Carroll, R. C., and Bassell, G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004
Antar, L. N., Dictenberg, J. B., Plociniak, M., Afroz, R., and Bassell, G. J. (2005). Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350–359. doi: 10.1111/j.1601-183X.2005.00128.x
Ascano, M. Jr., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., Corcoran, D. L., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Ashworth, A., Abusaad, I., Walsh, C., Nanko, S., Murray, R. M., Asherson, P., et al. (1996). Linkage analysis of the fragile X gene FMR-1 and schizophrenia: no evidence for linkage but report of a family with schizophrenia and an unstable triplet repeat. Psychiatr. Genet. 6, 81–86. doi: 10.1097/00041444-199622000-00008
Bagni, C., and Klann, E. (2012). Molecular functions of the mammalian fragile X mental retardation protein: insights into mental retardation and synaptic plasticity. Autisms 126–139. doi: 10.1093/med/9780199744312.003.0008
Bagni, C., Tassone, F., Neri, G., and Hagerman, R. (2012). Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J. Clin. Invest. 122, 4314–4322. doi: 10.1172/JCI63141
Bardoni, B., Castets, M., Huot, M. E., Schenck, A., Adinolfi, S., Corbin, F., et al. (2003). 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum. Mol. Genet. 12, 1689–1698. doi: 10.1093/hmg/ddg181
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Bear, M. F., and Malenka, R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 4, 389–399. doi: 10.1016/0959-4388(94)90101-5
Bhogal, B., Jepson, J. E., Savva, Y. A., Pepper, A. S., Reenan, R. A., and Jongens, T. A. (2011). Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci. 14, 1517–1524. doi: 10.1038/nn.2950
Blackwell, E., Zhang, X., and Ceman, S. (2010). Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum. Mol. Genet. 19, 1314–1323. doi: 10.1093/hmg/ddq007
Bourgeois, J. A., Coffey, S. M., Rivera, S. M., Hessl, D., Gane, L. W., Tassone, F., et al. (2009). A review of fragile X premutation disorders: expanding the psychiatric perspective. J. Clin. Psychiatry 70, 852–862. doi: 10.4088/JCP.08r04476
Brackett, D. M., Qing, F., Amieux, P. S., Sellers, D. L., Horner, P. J., and Morris, D. R. (2013). FMR1 transcript isoforms: association with polyribosomes; regional and developmental expression in mouse brain. PLoS ONE 8:e58296. doi: 10.1371/journal.pone.0058296
Bramham, C. R. (2008). Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol. 18, 524–531. doi: 10.1016/j.conb.2008.09.013
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O'donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/S0092-8674(01)00568-2
Busquets-Garcia, A., Gomis-Gonzalez, M., Guegan, T., Agustin-Pavon, C., Pastor, A., Mato, S., et al. (2013). Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat. Med. 19, 603–607. doi: 10.1038/nm.3127
Cajigas, I. J., Will, T., and Schuman, E. M. (2010). Protein homeostasis and synaptic plasticity. EMBO J. 29, 2746–2752. doi: 10.1038/emboj.2010.173
Ceman, S., O'donnell, W. T., Reed, M., Patton, S., Pohl, J., and Warren, S. T. (2003). Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum. Mol. Genet. 12, 3295–3305. doi: 10.1093/hmg/ddg350
Centonze, D., Rossi, S., Mercaldo, V., Napoli, I., Ciotti, M. T., De Chiara, V., et al. (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 63, 963–973. doi: 10.1016/j.biopsych.2007.09.008
Charalambous, D. C., Pasciuto, E., Mercaldo, V., Pilo Boyl, P., Munck, S., Bagni, C., et al. (2013). KIF1Bbeta transports dendritically localized mRNPs in neurons and is recruited to synapses in an activity-dependent manner. Cell. Mol. Life Sci. 70, 335–356. doi: 10.1007/s00018-012-1108-0
Cheever, A., and Ceman, S. (2009). Phosphorylation of FMRP inhibits association with Dicer. RNA 15, 362–366. doi: 10.1261/rna.1500809
Chen, L., Yun, S. W., Seto, J., Liu, W., and Toth, M. (2003). The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 120, 1005–1017. doi: 10.1016/S0306-4522(03)00406-8
Chen, Z., Borek, D., Padrick, S. B., Gomez, T. S., Metlagel, Z., Ismail, A. M., et al. (2010). Structure and control of the actin regulatory WAVE complex. Nature 468, 533–538. doi: 10.1038/nature09623
Clery, A., Blatter, M., and Allain, F. H. (2008). RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 18, 290–298. doi: 10.1016/j.sbi.2008.04.002
Coffee, B., Ikeda, M., Budimirovic, D. B., Hjelm, L. N., Kaufmann, W. E., and Warren, S. T. (2008). Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis. Am. J. Med. Genet. A. 146A, 1358–1367. doi: 10.1002/ajmg.a.32261
Collins, S. C., Bray, S. M., Suhl, J. A., Cutler, D. J., Coffee, B., Zwick, M. E., et al. (2010). Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am. J. Med. Genet. A 152A, 2512–2520. doi: 10.1002/ajmg.a.33626
Curia, G., Papouin, T., Seguela, P., and Avoli, M. (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520. doi: 10.1093/cercor/bhn159
D'Hulst, C., and Kooy, R. F. (2007). The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 30, 425–431. doi: 10.1016/j.tins.2007.06.003
Darnell, J. C., Jensen, K. B., Jin, P., Brown, V., Warren, S. T., and Darnell, R. B. (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499. doi: 10.1016/S0092-8674(01)00566-9
Darnell, J. C., and Klann, E. (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1–7. doi: 10.1038/nn.3379
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Davidovic, L., Bechara, E., Gravel, M., Jaglin, X. H., Tremblay, S., Sik, A., et al. (2006). The nuclear microspherule protein 58 is a novel RNA-binding protein that interacts with fragile X mental retardation protein in polyribosomal mRNPs from neurons. Hum. Mol. Genet. 15, 1525–1538. doi: 10.1093/hmg/ddl074
De Boulle, K., Verkerk, A. J., Reyniers, E., Vits, L., Hendrickx, J., Van Roy, B., et al. (1993). A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 3, 31–35. doi: 10.1038/ng0193-31
De Rubeis, S., and Bagni, C. (2010). Fragile X mental retardation protein control of neuronal mRNA metabolism: insights into mRNA stability. Mol. Cell. Neurosci. 43, 43–50. doi: 10.1016/j.mcn.2009.09.013
De Rubeis, S., Pasciuto, E., Li, K. W., Fernández, E., Di Marino, D., Buzzi, A., et al. (2013). CYFIP1 co-ordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182. doi: 10.1016/j.neuron.2013.06.039
Di Marino, D., Achsel, T., Lacoux, C., Falconi, M., and Bagni, C. (2013). Molecular dynamics simulations show how the FMRP Ile304Asn mutation destabilizes the KH2 domain structure and affects its function. J. Biomol. Struct. Dyn. doi: 10.1080/07391102.2013.768552. [Epub ahead of print].
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
Didiot, M. C., Tian, Z., Schaeffer, C., Subramanian, M., Mandel, J. L., and Moine, H. (2008). The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 36, 4902–4912. doi: 10.1093/nar/gkn472
Doornbos, M., Sikkema-Raddatz, B., Ruijvenkamp, C. A., Dijkhuizen, T., Bijlsma, E. K., Gijsbers, A. C., et al. (2009). Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur. J. Med. Genet. 52, 108–115. doi: 10.1016/j.ejmg.2009.03.010
Doyle, M., and Kiebler, M. A. (2012). A numbers game underpins cytoplasmic mRNA transport. Nat. Cell Biol. 14, 333–335. doi: 10.1038/ncb2475
Eadie, B. D., Cushman, J., Kannangara, T. S., Fanselow, M. S., and Christie, B. R. (2012). NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 22, 241–254. doi: 10.1002/hipo.20890
Edbauer, D., Neilson, J. R., Foster, K. A., Wang, C. F., Seeburg, D. P., Batterton, M. N., et al. (2010). Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65, 373–384. doi: 10.1016/j.neuron.2010.01.005
Eden, S., Rohatgi, R., Podtelejnikov, A. V., Mann, M., and Kirschner, M. W. (2002). Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418, 790–793. doi: 10.1038/nature00859
Eichler, E. E., Richards, S., Gibbs, R. A., and Nelson, D. L. (1993). Fine structure of the human FMR1 gene. Hum. Mol. Genet. 2, 1147–1153. doi: 10.1093/hmg/2.8.1147
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146. doi: 10.1016/j.neulet.2004.11.087
Fatemi, S. H., Kneeland, R. E., Liesch, S. B., and Folsom, T. D. (2010). Fragile X mental retardation protein levels are decreased in major psychiatric disorders. Schizophr. Res. 124, 246–247. doi: 10.1016/j.schres.2010.07.017
Ferrari, F., Mercaldo, V., Piccoli, G., Sala, C., Cannata, S., Achsel, T., et al. (2007). The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol. Cell. Neurosci. 34, 343–354. doi: 10.1016/j.mcn.2006.11.015
Feyder, M., Karlsson, R. M., Mathur, P., Lyman, M., Bock, R., Momenan, R., et al. (2010). Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams' syndrome. Am. J. Psychiatry 167, 1508–1517. doi: 10.1176/appi.ajp.2010.10040484
Gabus, C., Mazroui, R., Tremblay, S., Khandjian, E. W., and Darlix, J. L. (2004). The fragile X mental retardation protein has nucleic acid chaperone properties. Nucleic Acids Res. 32, 2129–2137. doi: 10.1093/nar/gkh535
Gantois, I., Vandesompele, J., Speleman, F., Reyniers, E., D'Hooge, R., Severijnen, L. A., et al. (2006). Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 21, 346–357. doi: 10.1016/j.nbd.2005.07.017
Gessert, S., Bugner, V., Tecza, A., Pinker, M., and Kuhl, M. (2010). FMR1/FXR1 and the miRNA pathway are required for eye and neural crest development. Dev. Biol. 341, 222–235. doi: 10.1016/j.ydbio.2010.02.031
Gross, C., Berry-Kravis, E. M., and Bassell, G. J. (2012). Therapeutic strategies in fragile X syndrome: dysregulated mglur signaling and beyond. Neuropsychopharmacology 37, 178–195. doi: 10.1038/npp.2011.137
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128. doi: 10.1126/scitranslmed.3004218
Hornberg, H., and Holt, C. (2013). RNA-binding proteins and translational regulation in axons and growth cones. Front. Neurosci. 7:81. doi: 10.3389/fnins.2013.00081
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. doi: 10.1016/j.neuron.2006.07.005
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Ishizuka, A., Siomi, M. C., and Siomi, H. (2002). A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 16, 2497–2508. doi: 10.1101/gad.1022002
Jin, P., Zarnescu, D. C., Ceman, S., Nakamoto, M., Mowrey, J., Jongens, T. A., et al. (2004). Biochemical and genetic interaction between the fragile X mental retardation protein and themicroRNA pathway. Nat. Neurosci. 7, 113–117. doi: 10.1038/nn1174
Jonsson, E., Bjorck, E., Wahlstrom, J., Gustavsson, P., and Sedvall, G. (1995). Screening for CGG trinucleotide repeat expansion in the fragile X mental retardation 1 gene in schizophrenic patients. Psychiatr. Genet. 5, 157–160. doi: 10.1097/00041444-199524000-00002
Johnson, E. M., Kinoshita, Y., Weinreb, D. B., Wortman, M. J., Simon, R., Khalili, K., et al. (2006). Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J. Neurosci. Res. 83, 929–943. doi: 10.1002/jnr.20806
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Kao, D. I., Aldridge, G. M., Weiler, I. J., and Greenough, W. T. (2010). Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc. Natl. Acad. Sci. U.S.A. 107, 15601–15606. doi: 10.1073/pnas.1010564107
Kaufmann, W. E., Cohen, S., Sun, H. T., and Ho, G. (2002). Molecular phenotype of Fragile X syndrome: FMRP, FXRPs, and protein targets. Microsc. Res. Tech. 57, 135–144. doi: 10.1002/jemt.10066
Kedersha, N., Stoecklin, G., Ayodele, M., Yacono, P., Lykke-Andersen, J., Fritzler, M. J., et al. (2005). Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884. doi: 10.1083/jcb.200502088
Keene, J. D. (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543. doi: 10.1038/nrg2111
Kelemen, O., Kovacs, T., and Keri, S. (2013). Contrast, motion, perceptual integration, and neurocognition in schizophrenia: the role of fragile-X related mechanisms. Prog. Neuropsychopharmacol. Biol. Psychiatry 46C, 92–97. doi: 10.1016/j.pnpbp.2013.06.017
Khin, N. A., Tarleton, J., Raghu, B., and Park, S. K. (1998). Clinical description of an adult male with psychosis who showed FMR1 gene methylation mosaicism. Am. J. Med. Genet. 81, 222–224.
Kobayashi, K., Kuroda, S., Fukata, M., Nakamura, T., Nagase, T., Nomura, N., et al. (1998). p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J. Biol. Chem. 273, 291–295. doi: 10.1074/jbc.273.1.291
Kovacs, T., Kelemen, O., and Keri, S. (2013). Decreased fragile X mental retardation protein (FMRP) is associated with lower IQ and earlier illness onset in patients with schizophrenia. Psychiatry Res. doi: 10.1016/j.psychres.2012.12.022. [Epub ahead of print].
Lacoux, C., Di Marino, D., Boyl, P. P., Zalfa, F., Yan, B., Ciotti, M. T., et al. (2012). BC1-FMRP interaction is modulated by 2'-O-methylation: RNA-binding activity of the tudor domain and translational regulation at synapses. Nucleic Acids Res. 40, 4086–4096. doi: 10.1093/nar/gkr1254
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A., and Fischer, U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338. doi: 10.1093/hmg/10.4.329
Linder, B., Plottner, O., Kroiss, M., Hartmann, E., Laggerbauer, B., Meister, G., et al. (2008). Tdrd3 is a novel stress granule-associated protein interacting with the Fragile-X syndrome protein FMRP. Hum. Mol. Genet. 17, 3236–3246. doi: 10.1093/hmg/ddn219
Louhivuori, V., Vicario, A., Uutela, M., Rantamaki, T., Louhivuori, L. M., Castren, E., et al. (2011). BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol. Dis. 41, 469–480. doi: 10.1016/j.nbd.2010.10.018
Maccarrone, M., Rossi, S., Bari, M., De Chiara, V., Rapino, C., Musella, A., et al. (2010). Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology 35, 1500–1509. doi: 10.1038/npp.2010.19
Matlin, A. J., Clark, F., and Smith, C. W. (2005). Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 6, 386–398. doi: 10.1038/nrm1645
Menon, L., Mader, S. A., and Mihailescu, M. R. (2008). Fragile X mental retardation protein interactions with the microtubule associated protein 1B RNA. RNA 14, 1644–1655. doi: 10.1261/rna.1100708
Menon, L., and Mihailescu, M. R. (2007). Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res. 35, 5379–5392. doi: 10.1093/nar/gkm581
Mila, M., Castellvi-Bel, S., Sanchez, A., Barcelo, A., Badenas, C., Mallolas, J., et al. (2000). Rare variants in the promoter of the fragile X syndrome gene (FMR1). Mol. Cell. Probes 14, 115–119. doi: 10.1006/mcpr.2000.0293
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/S0896-6273(03)00034-5
Monzo, K., Papoulas, O., Cantin, G. T., Wang, Y., Yates, J. R. 3rd., and Sisson, J. C. (2006). Fragile X mental retardation protein controls trailer hitch expression and cleavage furrow formation in Drosophila embryos. Proc. Natl. Acad. Sci. U.S.A. 103, 18160–18165. doi: 10.1073/pnas.0606508103
Muddashetty, R. S., Kelic, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007
Muddashetty, R. S., Nalavadi, V. C., Gross, C., Yao, X., Xing, L., Laur, O., et al. (2011). Reversible Inhibition of PSD-95 mRNA Translation by miR-125a, FMRP Phosphorylation, and mGluR Signaling. Mol. Cell 42, 673–688. doi: 10.1016/j.molcel.2011.05.006
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 15537–15542. doi: 10.1073/pnas.0707484104
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. doi: 10.1016/j.cell.2008.07.031
Narayanan, U., Nalavadi, V., Nakamoto, M., Pallas, D. C., Ceman, S., Bassell, G. J., et al. (2007). FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neurosci. 27, 14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
O'roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O'roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Papoulas, O., Monzo, K. F., Cantin, G. T., Ruse, C., Yates, J. R. 3rd., Ryu, Y. H., et al. (2010). dFMRP and Caprin, translational regulators of synaptic plasticity, control the cell cycle at the Drosophila mid-blastula transition. Development 137, 4201–4209. doi: 10.1242/dev.055046
Penzes, P., Cahill, M. E., Jones, K. A., Vanleeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Percipalle, P. (2009). The long journey of actin and actin-associated proteins from genes to polysomes. Cell. Mol. Life Sci. 66, 2151–2165. doi: 10.1007/s00018-009-0012-8
Piazzon, N., Rage, F., Schlotter, F., Moine, H., Branlant, C., and Massenet, S. (2008). In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J. Biol. Chem. 283, 5598–5610. doi: 10.1074/jbc.M707304200
Pichon, X., Wilson, L. A., Stoneley, M., Bastide, A., King, H. A., Somers, J., et al. (2012). RNA binding protein/RNA element interactions and the control of translation. Curr. Protein Pept. Sci. 13, 294–304. doi: 10.2174/138920312801619475
Ramos, A., Hollingworth, D., Adinolfi, S., Castets, M., Kelly, G., Frenkiel, T. A., et al. (2006). The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein-protein interaction. Structure 14, 21–31. doi: 10.1016/j.str.2005.09.018
Reiss, A. L., Hagerman, R. J., Vinogradov, S., Abrams, M., and King, R. J. (1988). Psychiatric disability in female carriers of the fragile X chromosome. Arch. Gen. Psychiatry 45, 25–30. doi: 10.1001/archpsyc.1988.01800250029005
Richter, J. D., and Klann, E. (2009). Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 23, 1–11. doi: 10.1101/gad.1735809
Sahoo, T., Peters, S. U., Madduri, N. S., Glaze, D. G., German, J. R., Bird, L. M., et al. (2006). Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations. J. Med. Genet. 43, 512–516. doi: 10.1136/jmg.2005.036913
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Schenck, A., Bardoni, B., Langmann, C., Harden, N., Mandel, J. L., and Giangrande, A. (2003). CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38, 887–898. doi: 10.1016/S0896-6273(03)00354-4
Schenck, A., Bardoni, B., Moro, A., Bagni, C., and Mandel, J. L. (2001). A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. U.S.A. 98, 8844–8849. doi: 10.1073/pnas.151231598
Shang, Y., Wang, H., Mercaldo, V., Li, X., Chen, T., and Zhuo, M. (2009). Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J. Neurochem. 111, 635–646. doi: 10.1111/j.1471-4159.2009.06314.x
Siomi, M. C., Zhang, Y., Siomi, H., and Dreyfuss, G. (1996). Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol. Cell. Biol. 16, 3825–3832.
Steward, O., and Schuman, E. M. (2003). Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40, 347–359. doi: 10.1016/S0896-6273(03)00635-4
Stoll, G., Pietilainen, O. P., Linder, B., Suvisaari, J., Brosi, C., Hennah, W., et al. (2013). Deletion of TOP3beta, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 16, 1228–1237. doi: 10.1038/nn.3484
Stradal, T. E., Rottner, K., Disanza, A., Confalonieri, S., Innocenti, M., and Scita, G. (2004). Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 14, 303–311. doi: 10.1016/j.tcb.2004.04.007
Suvrathan, A., Hoeffer, C. A., Wong, H., Klann, E., and Chattarji, S. (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 11591–11596. doi: 10.1073/pnas.1002262107
Talebizadeh, Z., Aldenderfer, R., and Wen Chen, X. (2013). A proof-of-concept study: exon-level expression profiling and alternative splicing in autism using lymphoblastoid cell lines. Psychiatr. Genet. doi: 10.1097/YPG.0b013e3283635526. [Epub ahead of print].
Tam, G. W., Van De Lagemaat, L. N., Redon, R., Strathdee, K. E., Croning, M. D., Malloy, M. P., et al. (2010). Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem. Soc. Trans. 38, 445–451. doi: 10.1042/BST0380445
Tamanini, F., Van Unen, L., Bakker, C., Sacchi, N., Galjaard, H., Oostra, B. A., et al. (1999). Oligomerization properties of fragile-X mental-retardation protein (FMRP) and the fragile-X-related proteins FXR1P and FXR2P. Biochem. J. 343(Pt 3), 517–523. doi: 10.1042/0264-6021:3430517
Tian, H., Cao, Y. X., Zhang, X. S., Liao, W. P., Yi, Y. H., Lian, J., et al. (2013). The targeting and functions of miRNA-383 are mediated by FMRP during spermatogenesis. Cell Death Dis. 4, e617. doi: 10.1038/cddis.2013.138
Toro, C., and Deakin, J. F. (2005). NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 80, 323–330. doi: 10.1016/j.schres.2005.07.003
Turk, J. (2011). Fragile X syndrome: lifespan developmental implications for those without as well as with intellectual disability. Curr. Opin. Psychiatry 24, 387–397. doi: 10.1097/YCO.0b013e328349bb77
Ule, J., and Darnell, R. B. (2006). RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr. Opin. Neurobiol. 16, 102–110. doi: 10.1016/j.conb.2006.01.003
Valverde, R., Edwards, L., and Regan, L. (2008). Structure and function of KH domains. FEBS J. 275, 2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x
Van Der Zwaag, B., Staal, W. G., Hochstenbach, R., Poot, M., Spierenburg, H. A., De Jonge, M. V., et al. (2010). A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 960–966. doi: 10.1002/ajmg.b.3/055
Von Der Lippe, C., Rustad, C., Heimdal, K., and Rodningen, O. K. (2010). 15q11.2 microdeletion–seven new patients with delayed development and/or behavioural problems. Eur. J. Med. Genet. 54, 357–360. doi: 10.1016/j.ejmg.2010.12.008
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Wang, H., Wu, L. J., Kim, S. S., Lee, F. J., Gong, B., Toyoda, H., et al. (2008). FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59, 634–647. doi: 10.1016/j.neuron.2008.06.027
Wang, T., Bray, S. M., and Warren, S. T. (2012). New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 22, 256–263. doi: 10.1016/j.gde.2012.02.002
Westermark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5:e52. doi: 10.1371/journal.pbio.0050052
Willemsen, R., Bontekoe, C., Tamanini, F., Galjaard, H., Hoogeveen, A., and Oostra, B. (1996). Association of FMRP with ribosomal precursor particles in the nucleolus. Biochem. Biophys. Res. Commun. 225, 27–33. doi: 10.1006/bbrc.1996.1126
Wisniewski, K., and Car, H. (2002). (S)-3,5-DHPG: a review. CNS Drug Rev. 8, 101–116. doi: 10.1111/j.1527-3458.2002.tb00218.x
Xie, W., Dolzhanskaya, N., Lafauci, G., Dobkin, C., and Denman, R. B. (2009). Tissue and developmental regulation of fragile X mental retardation 1 exon 12 and 15 isoforms. Neurobiol. Dis. 35, 52–62. doi: 10.1016/j.nbd.2009.03.015
Xing, L., and Bassell, G. J. (2013). mRNA localization: an orchestration of assembly, traffic and synthesis. Traffic 14, 2–14. doi: 10.1111/tra.12004
Xu, B., Ionita-Laza, I., Roos, J. L., Boone, B., Woodrick, S., Sun, Y., et al. (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369. doi: 10.1038/ng.2446
Xu, D., Shen, W., Guo, R., Xue, Y., Peng, W., Sima, J., et al. (2013). Top3beta is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci. 16, 1238–1247. doi: 10.1038/nn.3479
Yun, S. H., and Trommer, B. L. (2011). Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J. Neurosci. Res. 89, 176–182. doi: 10.1002/jnr.22546
Zalfa, F., Adinolfi, S., Napoli, I., Kuhn-Holsken, E., Urlaub, H., Achsel, T., et al. (2005). Fragile X mental retardation protein (FMRP) binds specifically to the brain cytoplasmic RNAs BC1/BC200 via a novel RNA-binding motif. J. Biol. Chem. 280, 33403–33410. doi: 10.1074/jbc.M504286200
Zalfa, F., Eleuteri, B., Dickson, K. S., Mercaldo, V., De Rubeis, S., Di Penta, A., et al. (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 10, 578–587. doi: 10.1038/nn1893
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., et al. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. doi: 10.1016/S0092-8674(03)00079-5
Zang, J. B., Nosyreva, E. D., Spencer, C. M., Volk, L. J., Musunuru, K., Zhong, R., et al. (2009). A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 5:e1000758. doi: 10.1371/journal.pgen.1000758
Zhang, M., Wang, Q., and Huang, Y. (2007). Fragile X mental retardation protein FMRP and the RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10057–10062. doi: 10.1073/pnas.0700169104
Keywords: fragile X syndrome, autism, schizophrenia, major depressive disorders, FMRP, RNA-binding proteins, synaptic plasticity, local protein synthesis
Citation: Fernández E, Rajan N and Bagni C (2013) The FMRP regulon: from targets to disease convergence. Front. Neurosci. 7:191. doi: 10.3389/fnins.2013.00191
Received: 02 July 2013; Paper pending published: 30 July 2013;
Accepted: 04 October 2013; Published online: 24 October 2013.
Edited by:
Jernej Ule, University College London, UKReviewed by:
Scott E. Hemby, Wake Forest University School of Medicine, USAKa Wan Li, VU University, Netherlands
Copyright © 2013 Fernández, Rajan and Bagni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esperanza Fernández, Center for the Biology of Disease, Vlaams Institut voor Biotechnologie, O&N IV Herestraat 49 - box 602, 3000 Leuven, Belgium e-mail:ZXNwZXJhbnphLmZlcm5hbmRlekBjbWUudmliLWt1bGV1dmVuLmJl;
Claudia Bagni, Center for Human Genetics, Leuven Institute for Neuroscience and Disease, KU Leuven, O&N IV Herestraat 49 - box 602, 3000 Leuven, Belgium e-mail:Y2xhdWRpYS5iYWduaUB1bmlyb21hMi5pdA==;Y2xhdWRpYS5iYWduaUBjbWUudmliLWt1bGV1dmVuLmJl