 Joshua R. Gold
Joshua R. Gold Victoria M. Bajo
Victoria M. Bajo- Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK
The brain displays a remarkable capacity for both widespread and region-specific modifications in response to environmental challenges, with adaptive processes bringing about the reweighing of connections in neural networks putatively required for optimizing performance and behavior. As an avenue for investigation, studies centered around changes in the mammalian auditory system, extending from the brainstem to the cortex, have revealed a plethora of mechanisms that operate in the context of sensory disruption after insult, be it lesion-, noise trauma, drug-, or age-related. Of particular interest in recent work are those aspects of auditory processing which, after sensory disruption, change at multiple—if not all—levels of the auditory hierarchy. These include changes in excitatory, inhibitory and neuromodulatory networks, consistent with theories of homeostatic plasticity; functional alterations in gene expression and in protein levels; as well as broader network processing effects with cognitive and behavioral implications. Nevertheless, there abounds substantial debate regarding which of these processes may only be sequelae of the original insult, and which may, in fact, be maladaptively compelling further degradation of the organism's competence to cope with its disrupted sensory context. In this review, we aim to examine how the mammalian auditory system responds in the wake of particular insults, and to disambiguate how the changes that develop might underlie a correlated class of phantom disorders, including tinnitus and hyperacusis, which putatively are brought about through maladaptive neuroplastic disruptions to auditory networks governing the spatial and temporal processing of acoustic sensory information.
The adult mammalian auditory system shows a remarkable degree of plasticity in a variety of contexts, and at a number of processing levels, manifesting as changes in the central representation of acoustic stimuli. These modulatory processes in the adult auditory brain are thought to be crucial to the performance and learning of ecologically relevant behaviors (Froemke and Martins, 2011; King et al., 2011), as well as being differentially affected during active and passive listening (Pienkowski and Eggermont, 2011). The types of changes observed in these contexts tend to be adaptive if behavior is assessed. However, as previously reviewed (Salvi et al., 2000; Syka, 2002), the mechanisms that develop following a sensorineural auditory insult may represent a unique assemblage, specific to abnormal or damaged sensory input. For example, cholinergic modulation of auditory cortical plasticity plays a key role in driving tonotopic reorganization (Kilgard and Merzenich, 1998) or training-related plasticity (Reed et al., 2011), including regaining acoustic spatial localization after unilateral conductive hearing loss (Leach et al., 2013). However, the cholinergic system is seemingly extraneous to cortical tonotopic plasticity following permanent cochlear damage (Kamke et al., 2005). Therefore, the various outcomes of auditory insults that affect normal cochlear function may constitute an important route for investigating the capacity of the mammalian brain for dealing with disrupted sensory inputs.
The burden posed by hearing-related trauma represents a significant challenge to healthcare services globally, with the rate of hearing impairment approaching ~40% in some adult populations (Agrawal et al., 2008; Nondahl et al., 2011), while the risk of hearing impairment appears to be climbing amongst younger cohorts (Niskar et al., 2001). Each of these epidemiological observations suggest that hearing loss and auditory trauma-related symptoms are liable to affect larger proportions of the future population. There is thus impetus motivating the investigation of the underlying mechanisms that might be responsible for peripheral and the central changes that cause abnormalities in spectrotemporal processing, which in day-to-day life can have a substantial impact on speech perception and auditory scene analysis.
Furthermore, a link between auditory disruption and phantom percept generation has been appreciated for years (e.g., Axelsson and Sandh, 1985). More recently, interest in the possible central origins of such phantoms has been sparked by suggestions that maladaptive neuroplasticity may be responsible for these percepts (Eggermont and Roberts, 2004), even in the absence of any audiometrically identifiable hearing loss (Schaette and McAlpine, 2011). Clearly, the instantiation of tinnitus represents a problem: both for its human sufferers, substantially decreasing quality of life in some patients, particularly given the dearth of effective, broadly applicable treatments available (Baguley et al., 2013; Langguth et al., 2013); but also for researchers investigating the neurobiological mechanisms of the disease, particularly given the little consensus regarding the factors that induce and then maintain the disease chronically (Kaltenbach, 2011; Knipper et al., 2013; Noreña and Farley 2013; Roberts et al., 2013).
To this end, the current review will seek to examine the confluence of factors related to auditory insults from a bottom-up perspective, addressing changes in peripheral function associated with mechanically-, acoustically-, pharmacologically-, and aging-mediated disturbances. The central consequences of these changes to hearing receptor function will then be evaluated: at the level of the single neuron, its local circuitry, as well as the global systemic effects likely to be responsible for behavioral changes that have been categorized in animal models of auditory trauma (Table 1). By occupying a non-hypothesis-driven position in considering the available data across a variety of fields, certain important functional commonalities and/or exceptions will hopefully come to light.
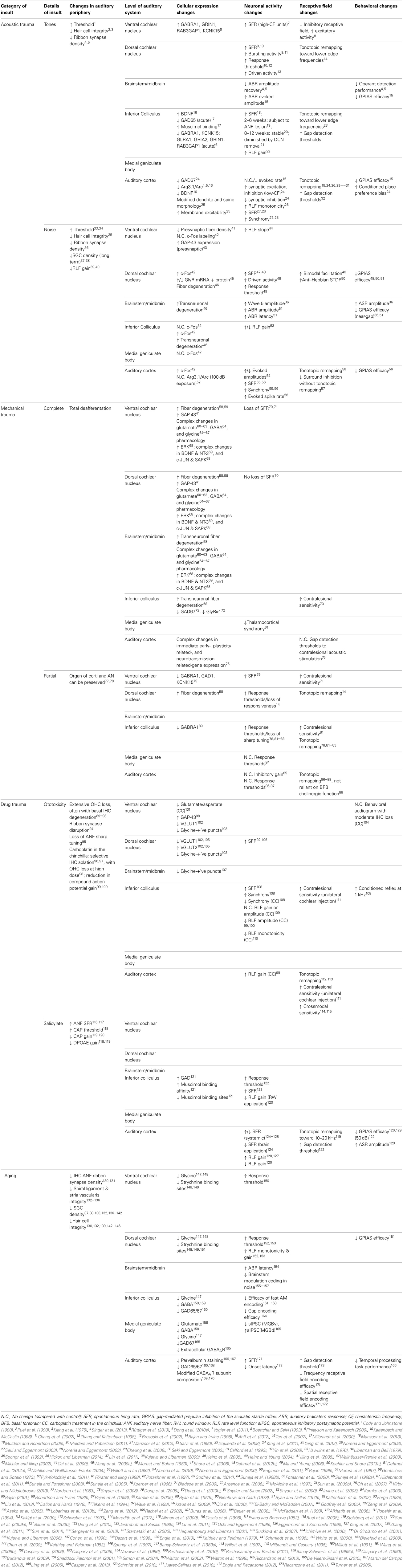
Table 1. Summary of the changes observed at the various levels of the auditory pathway after different types of auditory insults.
Changes in Cochlear Function Following Peripheral Insult
Studies of hearing loss predominantly focus on damage inflicted upon the organ of Corti, with the outcomes of direct mechanical (e.g., Robertson and Irvine, 1989; Rajan et al., 1993) or thermal (e.g., Snyder et al., 2008) lesions of the cochlea and its afferents clear in terms of the extent of damage induced. The time scale is also variable, depending upon the idiosyncrasies of other deafening protocols, leading to temporary- (e.g., Calford et al., 1993; Syka and Rybalko, 2000; Dehmel et al., 2012b; but see Kujawa and Liberman, 2009), or permanent threshold shifts, or indeed related to natural aging. Nonetheless, different experimental approaches appear to share certain commonalities that may be responsible for the induction of stereotypical responses in central processing stations.
The Anatomy of Cochlear Injury
Following an acutely induced acoustic overexposure, the degree of hair cell loss generally scales with the amplitude and duration of the insult. This usually manifests as extensive outer hair cell (OHC) death, with frequency-delimited loss of inner hair cells (IHCs) scaling with the trauma severity (Spongr et al., 1992). Lesion patterns following similar exposure protocols are variable, however. Descriptions have been made of equivalent outer and inner hair cell degeneration (Dolan et al., 1975; Kiang et al., 1975), as well as consistent damage to basally-located OHCs and IHCs regardless of the spectral content or duration of the acoustic trauma (Hawkins et al., 1976; Mulders et al., 2011).
By comparison, disruption of hair cell stereocilia has been observed to occur without inducing apoptosis of the affected hair cells (Liberman and Beil, 1979). This heterogeneous susceptibility to trauma appears to be conserved in certain models of ototoxicity, such as aminoglycoside antibiotic exposure (Forge, 1985), or that induced by platinum-derived cancer treatment drugs (specifically cisplatin and carboplatin) (Yorgason et al., 2006). Certainly, these treatments are recognized for their potentially devastating side-effects in human patients. Interestingly, while each of these latter, platinum-based agents leads primarily to OHC destruction, a lack of dose dependency has been reported, often leading to variable degrees of OHC damage for a range of exposure concentrations (Kaltenbach et al., 2002). Notable is the unique case of the chinchilla, which suffers a severe, and often, complete, ablation of the IHC population by carboplatin (Wake et al., 1993; Takeno et al., 1994), affecting OHCs only when doses verge on systemic toxicity, at which point total hair cell losses are substantial (Kraus et al., 2009).
It is constructive to highlight an equivalency between losses developed following acute trauma, and those acquired during aging. Age-related changes affect the cochlea in a stereotyped, multifactorial manner (Schuknecht, 1964; Schuknecht and Gacek, 1993; Ohlemiller, 2004), with hearing deterioration being characterized according to the underlying pathology of the receptor organ. According to Schuknecht's revised categories of the pathology, specific cochlear components may undergo degradation. The types of presbyacusis typically referred to include sensory presbyacusis—a loss of hair cell or organ of Corti integrity; metabolic presbyacusis, indicative of aberrant strial physiology; neural presbyacusis, involving atrophy and/or apoptosis of the afferent spiral ganglion cell fibers in the cochlea; or some combination thereof.
In models of presbyacusis, the relative distribution of hair cell damage appears to vary between species and is often defined by greater OHC impairment (Keithley and Feldman, 1982; Spongr et al., 1997). During the onset of age-related peripheral pathology, progressive deterioration of a number of cochlear structures has also been described: in the stria vascularis (Di Girolamo et al., 2001; Hequembourg and Liberman, 2001) and in the spiral ligament (Ichimiya et al., 2000), as well as a metabolic deterioration of the organ of Corti, which may exacerbate the functional consequences of OHC prestin reduction (Buckiova et al., 2007; Bielefeld et al., 2008; Chen et al., 2009). Ultimately, each of these various cochlear pathologies seems to converge upon auditory neuropathy (Keithley and Feldman, 1979; Cohen et al., 1990; Dazert et al., 1996; White et al., 2000; Engle et al., 2013). This putatively common endpoint, involving complete deafferentation, is likely preceded by functional disturbance of the synaptic interface between spiral ganglion afferents and IHCs (Stamataki et al., 2006; Sergeyenko et al., 2013).
A similar template of neuropathy—in particular concerning damage at the IHC-ANF interface—has been observed in acute experimentally-induced hearing loss, with regions of IHC loss correlated with reactive swelling and peripheral deafferentation of an equivalent spiral ganglion cell (SGC) population (Feng et al., 2012). Akin to the description above, degeneration of SGC perikarya occurs over the weeks to months following trauma, as does selective stria vascularis pathology (Wang et al., 2002). Similarly, inspection of drug ototoxicity has unveiled the development of afferent fiber terminal vacuolization in advance of any discernible IHC damage (Wang et al., 2003). Systemic toxicity is probably responsible for certain aspects of peripheral trauma, although the retraction of peripheral afferents following trauma-mediated terminal swelling has been postulated to be associated with disruption of the IHC ribbon synapse, a specialized glutamatergic structure necessary for synchronized auditory nerve activity (Buran et al., 2010). Importantly, under traumatic conditions, the ribbon synapse structure is capable of mediating an excitotoxic injury of afferent dendrites (Puel et al., 1998; Pujol and Puel, 1999).
Ribbon synaptopathy has been noted in the absence of overt cellular morphological disruption (Liu et al., 2013), with recent evidence indicating that it may well be a predictable consequence of cochlear trauma (Rüttiger et al., 2013; Singer et al., 2013). Observations of ribbon synaptopathy following even mild acoustic exposure (Maison et al., 2013) are particularly compromising from a functional perspective, since cochlear degeneration may have unwanted consequences at much later time points after the initial insult (Kujawa and Liberman, 2006, 2009; Sergeyenko et al., 2013) (Figure 1). How, then, are the various structural consequences of cochlear trauma to be reconciled with functional changes occurring in auditory nerve afferent signaling?
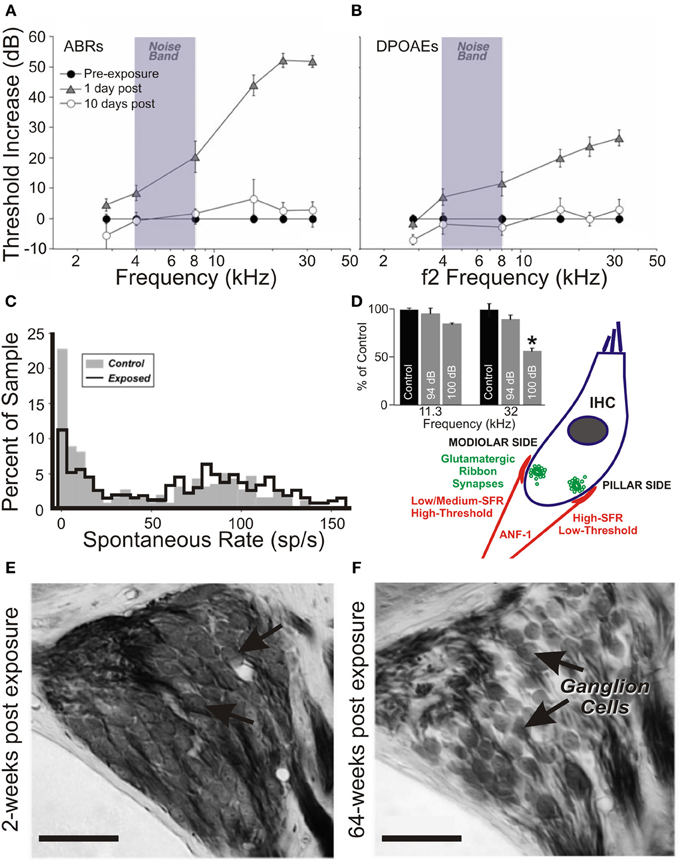
Figure 1. Insult-induced changes to auditory nerve physiology can occur in the absence of audiometric hearing loss. Peripheral thresholds measured using auditory brainstem responses (A) and distortion product otoacoustic emissions (B) following exposure to moderate narrowband noise trauma. Both are elevated in the short term following overexposure (24 h; triangles), however by >7 days thresholds have returned to normal (open circles). Modified with permission from Lin et al. (2011). (C) There is a significant shift in the normally bimodal distribution of spontaneous rate in the auditory nerve of exposed animals over a similar time course, leading to underrepresentation of type-1 auditory nerve fibers, coding for frequencies at and above the trauma high-pass corner, with spontaneous rates <20 spikes/s. Modified with permission from Furman et al. (2013). (D) Anatomically, such fibers synapse on the modiolar side of inner hair cells, and typically have a wide dynamic range of response, allowing effective rate coding up to high stimulus intensities. In a mouse model of exposure (inset), these high-threshold type-1 ANF responses are selectively reduced in the acoustic trauma region in the short term following traumatic exposure (8–16 kHz octave-band noise, 100 dB SPL), but not for exposure to a non-traumatic stimulus (94 dB SPL). Modified with permission from Hickox and Liberman (2014). (E,F) Histological comparison between the spiral ganglia basal turns of mice in the short- (E) and long- (F) terms following trauma finds that this pattern of high-threshold response loss precipitates ongoing SGC apoptosis. Scale bars 50 μm. Modified with permission from Kujawa and Liberman (2009). *p ≤ 0.01.
The Assessment and Outcomes of Peripheral Trauma
Audiogram-based assessment is often capable of detecting hearing loss defined in terms of OHC dysfunction and/or threshold elevation, and is used extensively to define the consequences of cochlear trauma as “permanent” or “temporary” threshold shifts. Typically, this is via inspection of waveforms of the auditory brainstem response (ABR) or auditory nerve fiber compound action potential (CAP), in conjunction with distortion product otoacoustic emissions (DPOAEs), or simply via self-reporting in human patients. By contrast with the threshold losses mediated by mixed OHC/IHC lesions (Cody and Johnstone, 1980; Liberman and Dodds, 1984a) or OHC loss alone (Dallos and Harris, 1978), the pattern of extensive cochlear damage induced by carboplatin administration in the chinchilla manifests as a time-dependent reduction in the CAP amplitude, often without pathological modulation of DPOAEs or ABR synchrony (El-Badry and McFadden, 2009), and only minor elevation (~9 dB) of electrophysiological thresholds (El-Badry and McFadden, 2007). This is reflected functionally in the preservation of behavioral audiograms collected from carboplatin-exposed chinchillas (Lobarinas et al., 2013b). It is certainly concerning that IHC pathology can extend to include up to 80% of the cochlear population, without similarly drastic changes in measures provided by commonly-used diagnostic tools.
Similarly, long-term degeneration of SGCs has been noted in aged mice with intact IHCs and permanently raised ABR thresholds (Kujawa and Liberman, 2006). More recently, a type of insidious pathology has been characterized following an apparently limited, acute hearing loss, previously thought to have minimal ongoing ramifications at the periphery (Figure 1). Inspection of mouse cochleae without evidence of raised thresholds (on ABRs or DPOAEs), nor with any overt structural damage to OHC or IHC populations, revealed IHC-specific ribbon synaptopathy within 24 h of acoustic trauma, manifesting as suprathreshold reductions in ABR amplitude at the affected cochleotopic frequencies (Kujawa and Liberman, 2009). More surprisingly, late-stage (>2 years) cochleography exposed a reduction by ≤50% of SGC density, regardless of the apparent health of the organ of Corti. In addition to these findings being replicated in the guinea pig (Lin et al., 2011), afferent degeneration coming about in this “silent” deafness seems only to affect those auditory nerve fibers encoding high-intensity stimuli (Furman et al., 2013).
Diagnostic issues would undoubtedly be associated with detecting this type of selective insult to high-threshold, low-spontaneous-rate fibers, especially since classical studies have correlated hearing loss with reductions in afferent spontaneous and evoked firing rates (Lonsbury-Martin and Meikle, 1978; Liberman and Dodds, 1984b). Recent demonstration that the phase-locking capacity of the auditory nerve fibers is diminished in the presence of low-level noise following acoustic trauma (Henry and Heinz, 2012, 2013) suggests that the functional ramifications of high-threshold fiber pathology are liable to be complex and context-specific. Given the multifaceted peripheral sequelae of cochlear trauma, it is important that comprehensive evaluation of auditory afferent function is performed to fully characterize the extent of cochlear pathology following auditory trauma, both clinically and experimentally (e.g., Ahlf et al., 2012; Dehmel et al., 2012a; Gu et al., 2012; Rüttiger et al., 2013).
Of particular use would be the routine implementation of suprathreshold evoked potential-based measurements of peripheral and brainstem auditory function. The collection of ABR data at multiple intensities for each subject (prior to and following trauma in animal experiments) would allow auditory thresholds to be established (either generally, in response to broadband click-train stimuli; or at greater spectral resolution by using tonal or narrowband-passed noise stimuli). Moreover, possible sites of lesion-mediated plasticity could be ascertained by evaluating differences in waveform amplitudes and latencies (either between the trauma population and matched controls, or in repeat measurements of the same subjects under different treatment conditions). Given that ABRs can be obtained repeatedly, they represent a convenient way of tracking, longitudinally, the immediate and longer term effects of commonly-implemented trauma protocols. The collection of such data would greatly improve our understanding of the impact of trauma on brainstem neural populations. For example, data of this kind have been reported in human tinnitus patients with and without audiometric threshold elevations, and have been integral to our ongoing understanding of that pathology (Attias et al., 1996; Kehrle et al., 2008; Schaette and McAlpine, 2011; Gu et al., 2012) (see below).
Neurochemical and Structural Changes Induced by Auditory Damage
Induced damage to the peripheral hearing organ causes physiological disruption of neurons at various levels along the auditory neuraxis, affecting not only neurotransmitter communication mechanisms, but also intracellular signaling pathways and metabolism. Although the precise functional impact is yet to be fully elucidated, given the complexity of the central auditory system, plus the extent and temporal features of the various insults examined experimentally, examining how cellular-level processes are affected may enhance our understanding of the higher-order systemic or perceptual outcomes of auditory trauma.
Activity-Related Gene Modulation
The expression of certain immediate early genes (IEGs), as a surrogate for neuronal metabolic state, has revealed time-varying modulation patterns in the auditory cortex following bilateral cochlear ablation that indicate the short time course necessary for insult-induced genetic mobilization in auditory neurons (Oh et al., 2007). c-fos is an IEG induced by stimulus novelty (e.g., Ehret and Fischer, 1991; Rouiller et al., 1992), thought to link neuronal activity with gene transduction associated with plasticity and learning (Carretta et al., 1999; Tischmeyer and Grimm, 1999). In the auditory cortex, c-fos is expressed within 1 h following acoustic trauma (Wallhäusser-Franke et al., 2003), in a diffuse manner indicating an acute, widespread (non-tonotopic) hyperreactivity within the cortex (Mahlke and Wallhäusser-Franke, 2004). Enhancements of c-fos expression levels were also detected over similar time frames at key subcortical auditory centers (including IC and DCN) following acoustic trauma, suggesting a feedforward upregulation of neural activity throughout the central auditory system.
As an alternative method of modifying cochlear activity, salicylate administration at high doses has been noted to affect hearing thresholds (Cazals, 2000), simultaneously elevating spontaneous activity (Evans and Borerwe, 1982) and diminishing driven response rates (Sun et al., 2009a; Stolzberg et al., 2011) within the auditory nerve. Appropriately, c-fos labeling after systemic salicylate injection yielded limited labeling in the non-lemniscal dorsal cortex and external nucleus of the IC (Wallhäusser-Franke, 1997; Mahlke and Wallhäusser-Franke, 2004), with more consistent, dose-dependent expression found at the primary auditory cortex (A1) and anterior auditory field (AAF) (Wallhäusser-Franke et al., 2003).
More recently, region-specific insult-induced changes in the expression of another IEG, Arg3.1/Arc (activity-regulated gene/activity-regulated cytoskeleton-associated protein), have been investigated. Arg3.1/Arc mobilization putatively occurs through brain-derived neurotrophic factor (BDNF)-mediated activation of the MEK-ERK signaling pathway (Ying et al., 2002). Components of this pathway are upregulated in the auditory brainstem following cochlear ablation (Suneja and Potashner, 2003; Suneja et al., 2005) and acoustic trauma (Tan et al., 2007). On examining the effects of salicylate and acoustic trauma on Arg3.1/Arc expression in the adult gerbil, marked divergence from the pattern of c-fos labeling is seen, with Arg3.1/Arc upregulation occurring in cortex only (Mahlke and Wallhäusser-Franke, 2004). Furthermore, dissimilarities between the tonotopic and laminar arrangements of Arg3.1/Arc expression were qualitatively related to the type of insult. Systemic salicylate injection enhanced Arg3.1/Arc expression in the high-frequency domain of A1, but predominantly outside of layer IV, thereby suggesting its effects are less likely to be mediated by substantial thalamic input. By contrast, moderate acoustic trauma (at an intensity sufficient to produce long-term cochlear synaptic and afferent dysfunction) yielded a non-layer-specific Arg3.1/Arc expression pattern in a tonotopically constrained manner, effectively matching the cochlear trauma profile (Mahlke and Wallhäusser-Franke, 2004).
In more recent studies of acoustic trauma, it was intriguing that those exposed subjects with demonstrable IHC ribbon synapse degradation displayed Arg3.1/Arc mobilization that appeared to be down, not up (Tan et al., 2007; Rüttiger et al., 2013; Singer et al., 2013) (Figure 2). Indeed, this bidirectional Arg3.1/Arc modulation is inversely correlated with the degree of BDNF and c-fos expression in the spiral ganglion, for periods spanning 3 h to 6 days after acoustic exposure (Tan et al., 2007); Arg3.1/Arc was downregulated in the cerebral cortex for at least 14–30 days thereafter (Rüttiger et al., 2013; Singer et al., 2013). Although the functional consequences of long-term up- or downregulation following auditory trauma remain uncertain, Arg3.1/Arc is thought to play a key role in controlling AMPA-type glutamate receptor distribution during homeostatic plasticity (e.g., Shepherd et al., 2006; Bramham et al., 2010; Gao et al., 2010; Béïque et al., 2011).
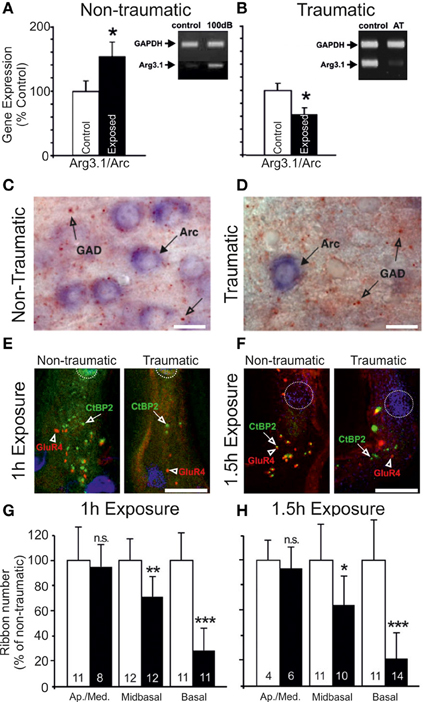
Figure 2. Changes in the auditory cortex expression of Arg3.1/Arc and cochlear ribbon synapse density related to the degree of acoustic trauma. (A) Following exposure to acoustic trauma that renders non-permanent threshold elevation (100 dB SPL, 2 h), reverse-transcriptase polymerase chain reaction (RT-PCR) analysis reveals Arg3.1/Arc expression is significantly upregulated in the auditory cortex; (B) by contrast, animals overexposed at 115–119 dB SPL for 2 h displayed marked downregulation of Arg3.1/Arc transcipts. Modified with permission from Tan et al. (2007). *p ≤ 0.05. (C,D) This relative downregulation is illustrated by immunohistochemical labeling of Arg3.1/Arc mRNA in the auditory cortex of animals exposed to a similar trauma (120 dB SPL, 2 h). Glutamate decarboxylase puncta are also indicated. Modified with permission from Singer et al. (2013). When animals were acoustically overexposed for either (E) 1 h or (F) 1.5 h, staining in the cochlea for IHC ribbon synapses (CtBP2, green, open arrows) indicated a subset of animals with marked reduction in ribbon synapse density, illustrated here for the mid-basal turn of the cochlea. IHC nuclei are labeled with DAPI (blue, circled); glutamate receptor subunit GluR4 protein labeled (red, open arrowheads). Scale bars 10 μm. (G,H) For each exposure protocol, a subset of animals could be categorized according to significant proportional reductions in midbasal and basal cochlear IHC ribbon synapse densities, relative to exposed animals without this pathology. Open bars = controls; filled bars = exposed. Numbers in each bar correspond to respective n. Modified with permission from Rüttiger et al. (2013). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Excitatory Signaling Function
On the basis of those data, it might be expected that significant alterations in glutamatergic function would be observed after the auditory insult—however, characterization of glutamate receptor pharmacology in the deafferented auditory cortex remains conspicuously underrepresented in the literature. As such, the observed modulations in Arg3.1/Arc mobilization (reviewed in Knipper et al., 2010, 2013) might be functionally linked with those changes in cortical neurotransmission and neuronal response properties under similar exposure conditions, or rather might simply correlated with the induction of certain forms of hearing loss, without attendant regulation of homeostatic plasticity mechanisms (Bramham et al., 2010).
Changes in glutamatergic metabolism have been better documented subcortically, such as the small, non-significant reduction of glutamate concentrations in the medial geniculate body (MGB), inferior colliculus (IC) and cochlear nucleus (CN) of a rodent model of presbyacusis (Banay-Schwartz et al., 1989b). Cochlear ablation, by contrast, induces enhanced glutamate metabolism, manifesting as elevated release and uptake, and concomitant with a putative compensatory strengthening of excitatory processing following extensive afferent fiber death (Potashner et al., 1997; Illing et al., 2005), as levels of glutamate and aspartate in the CN correlate with the degree of cochlear damage (Godfrey et al., 2005, 2014). While the proportional expression of glutamate vesicular transporter (VGLUT) subtypes has been reported to undergo adjustment in the insult-exposed CN (Fyk-Kolodziej et al., 2011), work specifically directed at such changes in the dorsal cochlear nucleus (DCN) has unveiled marked crossmodal reweighing. By assigning VGLUT subtypes 1 and 2 to auditory and non-auditory inputs, respectively (Zhou et al., 2007), the upregulated representation of non-auditory afferents in the fusiform layer of the DCN following cochlear trauma was revealed (Zeng et al., 2009, 2012).
Furthermore, in the short term, mRNAs encoding glutamatergic AMPA and NMDA-type receptor subunits are reduced in the central nucleus of the IC (CNIC) contralateral to a partial mechanical lesion of the cochlea (Dong et al., 2009, 2010a). Although these changes are only transient, they may be sufficient to more chronically disrupt cellular excitability elsewhere in the central auditory system. Such an outcome might follow in parallel within the auditory brainstem, given the reported remodeling of AMPA binding and NMDA-receptor subunit expression in the ventral cochlear nucleus (VCN) (Förster and Illing, 1998; Suneja et al., 2000). In spite of these efforts, many details of post-traumatic glutamate pharmacology remain to be elucidated.
Inhibitory Signaling Function
A more comprehensive understanding has been gained of inhibitory changes, which, in addition to the reactive changes in glutamate release and signaling subcortically, may precipitate a disruption of the balance of excitation and inhibition. Even at the earliest stages of auditory processing, inhibitory inputs are affected, failing to recover to pre-lesion levels in the ventral CN (Hildebrandt et al., 2011). In line with these disruptions, reductions in the density of glycine-immunoreactive puncta (Asako et al., 2005) and glycine receptor subunit protein (Wang et al., 2009a) have each been reported throughout the cochlear nucleus (CN). Such changes develop alongside altered strychnine binding and glycine metabolism in the ipsilateral CN and superior olivary complex, earlier described in the deafened guinea pig (Suneja et al., 1998a,b) and rat (Buras et al., 2006), suggesting a sustained post-deafening hyperexcitability (Potashner et al., 2000). These inhibitory disruptions occurring in a number of auditory brainstem nuclei are, perhaps, not unexpected, since previous work has demonstrated significant reductions in both the levels of glycine (Banay-Schwartz et al., 1989a; Willott et al., 1997; Wang et al., 2009b) and glycine/strychnine binding sites (Milbrandt and Caspary, 1995; Willott et al., 1997) in rodent models of presbyacusis.
Possibly the most heavily scrutinized locus for such representative disruptions to the auditory pathway is the inferior colliculus (reviewed in Caspary et al., 2008; Ouda and Syka, 2012). Aged rodents display decreases in free GABA concentrations throughout the IC (Banay-Schwartz et al., 1989b), as well as in evoked GABA release and GABAergic cell density (without modification of GABAergic cellular morphology) (Caspary et al., 1990), and reduced labeling for GAD65 and GAD67 (Burianova et al., 2009). GAD67 is also reduced following acute, complete deafferentation (Argence et al., 2006), although when the extent of damage is limited to certain frequency domains, the downregulation of transcripts coding for GABAA receptor subunit 1 and GAD1, respectively, were matched bilaterally by an effective reduction in GABAergic receptor protein expression in a tonotopically limited fashion (Dong et al., 2010b). It is noteworthy that GAD disruptions may occur only transiently (Milbrandt et al., 2000), indicating the possibility for the metabolic recovery of GABAergic signaling if trauma severity is limited. Indeed, enhanced collicular GABA immunoreactivity (Tan et al., 2007) (Figure 2) and GAD65 levels (Bauer et al., 2000) each have been described, possibly compensating for elevated network activity following auditory trauma.
A comparable GABAergic dysregulation is also seen cortically. A reduction in free GABA concentration is found in aged rat cortex (Banay-Schwartz et al., 1989b), and is possibly related to reductions in the levels of GAD65/67 mRNA, and GAD67 protein across cortical layers. Those observations were reported in two different rat strains that, importantly, display divergent peripheral symptoms as a function of aging but shared central inhibitory sequelae (Ling et al., 2005; Burianova et al., 2009). Indeed, lower GABA density throughout the auditory cortex appears to be co-determined by age and hearing loss, since GAD65 expression is also reduced after acoustic trauma in younger adult animals (Yang et al., 2011). Moreover, the receptor subunit composition of GABAA receptors is heterogeneously modified in aged rats (Schmidt et al., 2010), with a 30% reduction in the wild-type α1 subunit compared with young controls, as well as in certain subtypes of β- and γ-subunits (Caspary et al., 2013). In total, then, despite the picture of post-traumatic auditory neurochemistry standing broadly incomplete, certain characteristic changes appear to occur regardless of insult type. The extent to which these affect physiological function and neural response properties will therefore require examination. Additionally, an improved understanding of the neuropharmacological correlates of auditory trauma would be beneficial in the development of patient treatment related to the compensation for those changes (Schreiber et al., 2010; Mukherjea et al., 2011).
Neuronal Spontaneous and Evoked Response Properties Undergo Plastic Modulation During and After Auditory Insults
It is worthwhile considering that, even in the absence of acoustic stimulation, the net firing rate of the auditory nerve is particularly high in healthy animals (e.g., Kiang et al., 1975; Liberman and Dodds, 1984b); as such, cochlear exposure to various insults affecting peripheral and/or central function (particularly pharmacological and age-related) might be predicted to have profound effects on baseline levels of activity throughout the auditory system. Consequently, it is important to evaluate how single cells in the auditory system, when faced with abnormal synaptic inputs, may dynamically modify their activity and, by association, their properties related to the processing of acoustic stimuli.
Auditory Nerve
The activity of auditory nerve fibers (ANFs) changes in response to various experimental insults. Interpretation of data concerning post-traumatic amplitude transfer functions and temporal coding under diminished hearing conditions has been informed by classical physiological studies. In early experiments, wholesale mechanical cochlear ablation was shown to yield a complete loss of spontaneous activity within the auditory nerve and its afferent terminals within the ventral cochlear nucleus (VCN) (Koerber et al., 1966). This ultimately produced substantial fiber loss and density reduction throughout the VCN (Gentschev and Sotelo, 1973; Morest and Bohne, 1983; Morest et al., 1997). Under such circumstances, it is apparent that some ANFs remain functionally normal, even if the unlikelihood of recording from an active fiber scales with the degree of IHC damage (Wang et al., 1997). In such cases, functional normality is apparently contingent upon modification of the electrical properties of individual axons (Fryatt et al., 2011), and the survival of the OHC population. The latter is particularly necessary in maintaining sharp, low threshold tuning at the fibers' respective characteristic frequencies (CF) (Kiang et al., 1975; Dallos and Harris, 1978).
Nerve response properties may nevertheless be affected in other ways. By chronically monitoring the CAP over a period of weeks, long term (≤5 weeks post-exposure) reductions in the maximum evoked CAP magnitude can be observed for chinchillas with selective IHC ablation (Qiu et al., 2000) (Figure 3). Since the CAP represents the summed activity of multiple fibers, reduction in the steepness (or gain) of the nerve's rate-level function (RLF) may ostensibly derive from either, or each, of two sources: a similar reduction in gain of the RLFs of individual fibers; or a loss of fibers encoding high intensity sounds (Liberman, 1982; Taberner and Liberman, 2005). Each is likely, particularly in conditions of permanent threshold elevation and the associated cochlear pathophysiology. Certainly, acoustic exposure, yielding a range of threshold elevations, was reported to mediate a reduction in the gain of individual fiber responses to a spectrally diverse range of stimuli, regardless of the fiber's baseline spontaneous firing rate (SFR) (Heinz and Young, 2004; Heinz et al., 2005).
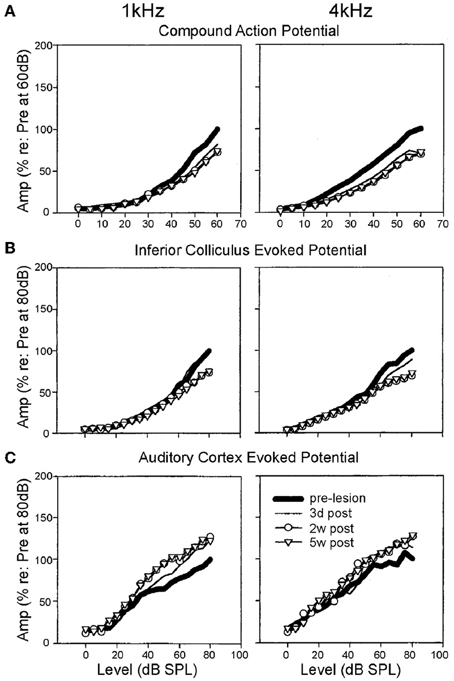
Figure 3. Specific elevation of auditory evoked rate level functions after selective inner hair cell ablation. Following systemic injection of carboplatin (various doses) into chinchillas, the selective IHC lesion that develops was evaluated by chronic electrode implantation with measurement of evoked field response functions from the cochlear compound action potential (A), the inferior colliculus (B) and the auditory cortex (C) in the awake animal in response to 1 kHz (left column) and 4 kHz (right column) tonal acoustic stimulation. By recording prior to lesion induction (thick lines), 3 days (thin lines), 2 weeks (circles) and 5 weeks (triangles) after lesioning, there was found to be a maintained suppression of evoked response amplitudes in both the cochlea and the inferior colliculus; paradoxically, IHC ablation yielded a marked enhancement of evoked response functions recorded from the auditory cortex. Modified with permission from Qiu et al. (2000).
By contrast, even if cochlear trauma is minimal, such as following recovery from an acoustically-induced “temporary” hearing loss, a frequency-selective deactivation of ANFs responsive up to high intensities may occur, causing a shift in the distribution of ensemble spontaneous activity toward higher firing rates (>20 sp/s) (Furman et al., 2013) (Figure 1). This pattern of deafferentation has been associated with a depression in IHC ribbon synapse density (see above), and has been observed to arise in presbyacusis (Schmiedt et al., 1996). Thus, cochlear trauma is potentially linked with a degradation of high intensity coding beginning at the level of the auditory nerve, in spite of possible threshold preservation. As a matter of comparison, although the ototoxic and threshold-elevating aspects of salicylate administration are well-documented (Cazals, 2000), its effects on spontaneous rate are ambiguous. Auditory nerve hyperactivity has been reported following salicylate infusion (e.g., Ruel et al., 2008) or systemic treatment (Evans and Borerwe, 1982; Cazals et al., 1998). From the available data, however, this phenomenon may simply be a by-product of extremely high systemic concentrations, far beyond those generally observed in human patients (Stolzberg et al., 2012).
Ventral and Dorsal Cochlear Nucleus
As a major site of ANF afferent termination, neurons in the VCN would be expected to display profound changes in activity, putatively compensating for the proposed reduction in peripheral drive. Given the high SFRs of ANFs, even the “silent” loss of high-threshold (low spontaneous rate) fibers might have profound effects on the baseline activity of VCN cells. SFRs of cells in the VCN appear to be fundamentally determined by ANF activity (Koerber et al., 1966). Although it is marked by a frequency-specific increase in threshold to tonal stimulation and apparent loss of evoked sensitivity in mouse presbyacusis (Willott et al., 1991), the VCN nevertheless may display an increase in contralaterally driven activity (Bledsoe et al., 2009).
The idea that the VCN expresses post-lesion response enhancement through plastic remodeling is intriguing. In the case of a partial cochlear lesion achieved mechanically or acoustically, a significant elevation of spontaneous discharge developed (Vogler et al., 2011). Interestingly, only in the former case was this increase limited cochleotopically, and in terms of the cell subpopulations concerned, with primary-like and onset response-type neurons displaying the largest enhancements (Vogler et al., 2011). This seems to confirm the reported enhancement of tone-evoked activity by transient acoustic trauma in those neural types (Boettcher and Salvi, 1993). By comparison, when identified units of the VCN were recorded following permanent hearing loss, primary-like neurons in particular displayed diminished rate-level function gain (RLF), whereas non-primary chopper type cells consistently exhibited monotonic steep RLFs (Cai et al., 2009). This remodeling of neuronal input/output functions is potentially a consequence of diminished inhibitory function, in conjunction with adaptive hyperactivity after diminished afferent drive. If this is the case, the VCN may be a major station for providing an augmented feedforward representation of cochlear activity to higher levels of the auditory pathway (Noreña, 2011; Noreña and Farley, 2013).
As the other major subdivision of the CN, and thus in receipt of ANF afferent terminals, neurons in the dorsal cochlear nucleus (DCN) have been extensively investigated for changes in excitability under a variety of traumatic conditions. A significant SFR increase in DCN cells develops within a few (2–5) days following cochlear trauma, and persists for weeks thereafter (Kaltenbach and McCaslin, 1996; Zhang and Kaltenbach, 1998; Kaltenbach and Afman, 2000; Kaltenbach et al., 2000; Chang et al., 2002; Finlayson and Kaltenbach, 2009; but see Ma and Young, 2006). The intrinsic excitability of fusiform cells (the major excitatory output from the DCN) is elevated bilaterally even after unilateral noise exposure (Brozoski et al., 2002). This activity change mirrors those seen in the response profiles of fusiform cells (Caspary et al., 2005) and glycinergic cartwheel cells (Caspary et al., 2006) during presbyacusis onset in rats. Although amplified responses may be derived from systemic inhibitory dysfunction (Middleton et al., 2011), the existence of a correlation between OHC disruption and DCN spontaneous discharge indicates that the onset of hyperactivity within the DCN is probably the result of a confluence of factors (Kaltenbach et al., 2002; Rachel et al., 2002).
Inferior Colliculus
At the level of the inferior colliculus (IC) in the auditory midbrain, the predictability of trauma-related neuronal excitability modulations becomes far less concrete, with outcomes varying as a function of exposure mechanism and recovery time. Spontaneous firing rate (SFR) elevations have been characterized in the days-to-weeks following an acoustic trauma (Dong et al., 2009; Mulders and Robertson, 2009; Manzoor et al., 2013; Robertson et al., 2013), complete cochlear deafferentation (Moore et al., 1997) and acutely after high-dose salicylate injection (Jastreboff and Sasaki, 1986; but see Ma et al., 2006). Generally, SFR develops in a manner that is correlated with those cochlear regions displaying equivalent bandpassed CAP threshold increases, with IHC and/or OHC loss (Willott et al., 1988a; Mulders et al., 2011). The origin/s of this hyperexcitability remains under debate, however. First, the peripheral origin of spontaneous rate enhancement appears to be contingent on some degree of OHC ablation only. This is suggested by carboplatin injection in chinchillas—selectively lesioning the IHC population—which had no effect on SFR of IC neurons 7–9 months after trauma. By contrast, such elevations did emerge following acoustic or cisplatin trauma, each of which produces some degree of OHC damage (Bauer et al., 2008).
Moreover, sectioning of the cochlear nerve has been reported to normalize trauma-related hyperactivity in the IC central nucleus (CNIC) (Mulders and Robertson, 2009), as has lesion of the DCN (Manzoor et al., 2012). These data suggest that the IC may chiefly adopt the evolved, persistent hyperactivity of the DCN (Zacharek et al., 2002; Zhang et al., 2006). Nevertheless, longer term observations have revealed that the IC may transition through a period of SFR lability prior to a more permanent acquisition of hyperactivity. By contrast with ipsilateral cochlear nerve lesions performed at 2–6 weeks after acoustic trauma, when identical auditory neurectomy was repeated at 8–12 weeks, IC hyperactivity remained (Mulders and Robertson, 2011). Evidently, then, the IC represents another auditory centre that displays compensatory upregulation of resting activity in response to trauma (Robertson et al., 2013).
Available data remain controversial regarding the evoked input/output gain function changes in IC neurons. In the extreme case of complete cochlear ablation, little-to-no effect upon the response properties of the contralateral CNIC during acoustic stimulation of the intact ear was initially reported (Nordeen et al., 1983). Strong response enhancement has, however, more recently been demonstrated, when the impacts of similar cochlear lesion models were investigated acutely and after three months (Popelár et al., 1994; McAlpine et al., 1997). In addition to an enhancement of the CNIC neural responses to ipsilateral acoustic stimulation, an increase in evoked activity on the contralateral side develops in the next three months after the cochlear insult. The dynamic range of recorded units were also found to be selectively broadened. This adaptive reweighing of collicular responses to favor input from the remaining ear is thought to be motivated by contralateral deafferentation removing the afferent drive to which these cells are predominantly responsive.
How, then, do the collicular cells respond to acoustic stimulation following an incomplete lesion? While response thresholds may be transiently elevated, largely as a function of OHC damage or apoptosis (Salvi et al., 1990), in the case of OHC preservation, threshold shifts are undetectable (McFadden et al., 1998; Alkhatib et al., 2006). When suprathreshold stimulation levels are considered, whereas maximum response amplitude enhancement has been documented (Willott and Lu, 1982; Salvi et al., 1990; McFadden et al., 1998), more often the loss of IHC integrity appears to engender a reduction in neuronal suprathreshold responsivity (McFadden et al., 1998; Qiu et al., 2000; El-Badry and McFadden, 2007) (Figure 3). This is akin to the effects of cochlear administration of salicylate (Sun et al., 2009a), which is thought to be active against inhibitory pharmacology (Su et al., 2009). In addition, there is a reduction in the proportion of non-monotonic-type rate level functions (Alkhatib et al., 2006), a response pattern that is characteristically reliant upon normal inhibitory activity. In presbyacusis-affected C57Bl/6 mice, similar effects were dependent on cochlear threshold elevation (Willott et al., 1988b), since rate-level function steepening did not emerge in aged CBA/J mice that displayed less marked cochlear degeneration (Willott et al., 1988a). This correlation between peripheral disruption and alterations of rate-level function statistics, plus the preponderance of depressed suprathreshold evoked potential amplitudes, seems to support the conclusion that loss of peripheral normality may selectively depress inhibitory function within the IC.
Auditory Thalamus
The auditory thalamus (medial geniculate body, MGB) is a major lynchpin in the transmission of sensory information to the auditory cortex, and a key mediator of auditory receptive field properties of cortical neurons. Nevertheless the effects of trauma upon MGB neurons have been little explored thus far, particularly in terms of categorical shifts in response properties following auditory insult.
Suprathreshold neural response latencies are predominantly retained across a range of characteristic tuning frequencies following partial cochlear ablation (Kamke et al., 2003). Yet, indications of excitability shifts were provided by evaluating shifts in the strength of synchronous activation, specifically in the ventral division of the MGB (Sun et al., 2009b). These latter recordings were only reported for a single animal at each of 4 and 24 h after trauma. Nevertheless, they are congruent with the only comprehensive evaluation of insult-related response plasticity in the auditory thalamus (Richardson et al., 2012, 2013). By performing patch recordings made in the MGB of aged rats, substantial reductions in the amplitude of the evoked phasic and tonic GABAergic current density throughout the MGB were seen. Moreover, region-specific modulations of spontaneous inhibitory activity appeared to develop during the aging process. This was seen in comparisons of the dorsal (non-lemniscal) division of the MGB, which underwent significant reductions in spontaneous inhibitory postsynaptic current, and the ventral (lemniscal) MGB, where a paradoxical enhancement developed in parallel (Richardson et al., 2013). On the basis of these limited but intriguing observations, it appears that the MGB, as a whole, displays the characteristic perturbation of GABAergic inhibitory signaling seen in other auditory centers.
Auditory Cortex
By the level of primary auditory cortex (A1), basal activity and suprathreshold responses might each be conceivably elevated, based on its afferent inputs. Certainly, aging has been demonstrated to bring about spontaneous rate increases in cortical units in a number of species, including rat (Hughes et al., 2010) and rhesus macaque (Juarez-Salinas et al., 2010). A similar effect appears to manifest after acoustic overexposure (Noreña and Eggermont, 2003, 2006), though at a delay of a few hours after trauma, leading to sustained augmentation across A1 for weeks to months (Seki and Eggermont, 2003; Engineer et al., 2011).
In the case of acoustic trauma, cross-correlation analysis revealed coincident enhancement of neural spiking synchrony. This elevation in synchrony was particularly prevalent in those regions that displayed some amount of threshold elevation post-traumatically (Noreña and Eggermont, 2006). Based on the observation that this aspect of neural activity was enhanced almost instantaneously following trauma (Noreña and Eggermont, 2003), and prior to the onset of spontaneous rate enhancements, correlated network spiking may be acting as a precursor to the ongoing plastic modulation of single cell and network response properties.
In each of the studies to have evaluated the effects of salicylate administration on auditory cortex basal activity, the route of administration appears to affect the outcome, even if salicylate may qualitatively raise thresholds, like certain other forms of trauma. Depression of neural activity ensues following direct application of salicylate to the cortical surface (Lu et al., 2011). Systemic administration has, by contrast, been associated with wholesale depressions in spontaneous rate (Yang et al., 2007; Zhang et al., 2011) or simultaneous elevations and depressions in spontaneous discharge (Ochi and Eggermont, 1996; Lu et al., 2011). This latter phenomenon appears to indicate a process of normalizing the mean ensemble discharge by augmenting the spontaneous rate of minimally active units, and depressing the basal discharge of those units with high spontaneous activity. These shifts in spontaneous rate develop in the absence of short-term modulation of intracortical spike synchrony (Ochi and Eggermont, 1996). This is certainly intriguing, given that synchrony is, apparently, enhanced almost immediately after (non-salicylate-based) cochlear trauma.
The idiosyncratic nature of these perturbations, relative to other experimentally induced insults, is further evidence that salicylate is likely to have complex, multifactorial effects throughout the auditory system, both peripheral and central. For example, enhanced driven neural activity is consistently observed across the tonotopic extent of the cortical surface even when salicylate is administered by different routes, such as systemically (Qiu et al., 2000; Yang et al., 2007; Sun et al., 2009a; Deng et al., 2010; Noreña et al., 2010; Lu et al., 2011; Zhang et al., 2011) (Figure 3), or directly to the cortical surface (Lu et al., 2011)—the latter approach being unlikely to bring about changes within the cochlea itself. The likelihood that this stably inducible effect is mediated via a suppression of GABAergic signaling efficacy is strongly suggested by the absence of any such enhancement when agents that positively modulate GABA pharmacology were coadministered (Sun et al., 2009a; Lu et al., 2011).
If, on the other hand, cortical evoked activity patterns are contrasted with those in the cochlear nuclei or the inferior colliculi induced by peripheral pathology, A1 appears to display changes that are uniquely indicative of early adaptation to deprived or modulated sensory input. In one case, recordings from cortical neurons were collected during the 50 h following cochlear ablation (Moore et al., 1997). In that experiment, there was a gradual decline in response thresholds to best-frequency stimulation of the ipsilateral (unlesioned) ear, suggesting adaptive reweighing to favor conserved inputs was occurring.
The evolution of this reweighing appears to vary according to the cortical region affected most severely by deafferentation, leading to cases in which driven activity may simultaneously increase and decrease within the same animal (Noreña et al., 2010). Still, a greater proportion of monotonic rate-level functions are seen in neurons tuned to frequencies outside of the region of cortex responding to the lesioned cochlea (Noreña and Eggermont, 2003) (this region of the auditory lemniscal pathway has been dubbed the “lesion projection zone,” or LPZ Schmid et al., 1996; Calford, 2002). This observation, alongside that of enhanced evoked (Engineer et al., 2011) or maximum firing rates, may well be illustrative of enhanced excitatory activity as well as reduced inhibitory regulation (e.g., Rajan, 2001).
Evidence that the mechanistic underpinnings of this putatively homeostatic reweighing may operate for both excitatory and inhibitory synaptic signaling has been suggested in single cell recordings from animals with noise-induced threshold elevations. Whereas cells outside of the LPZ displayed proportional enhancement of excitatory and inhibitory activity, those neurons that underwent peripheral deafferentation instead displayed reweighing toward more excitatory responses (Yang et al., 2011) (Figure 4). This effect was correlated with reduced tonic GABAA signaling and GAD65 expression in the LPZ. Reduced inhibitory signaling efficacy is common to a rodent presbyacusis model (Llano et al., 2012), as is the description of significantly depressed tonic GABAA activity at the level of the ventral MGB (Richardson et al., 2013). As such, the elevation of response magnitudes in the auditory cortex, rather than simply being the result of passive unmasking of pre-existing excitatory inputs, may be the result of homeostatic reweighing of physiological excitatory/inhibitory balance toward network hyperexcitability. In future work, it will be important to disentangle the degree to which this homeostatic plasticity is endogenous to each auditory centre recorded from in a trauma model, or if the apparent excitability changes are simply inherited from elsewhere. It is likely that a combination of each is the case, though recent data nevertheless indicate the existence of categorical differences between centers with respect to neuronal excitability (e.g., Cai et al., 2014).
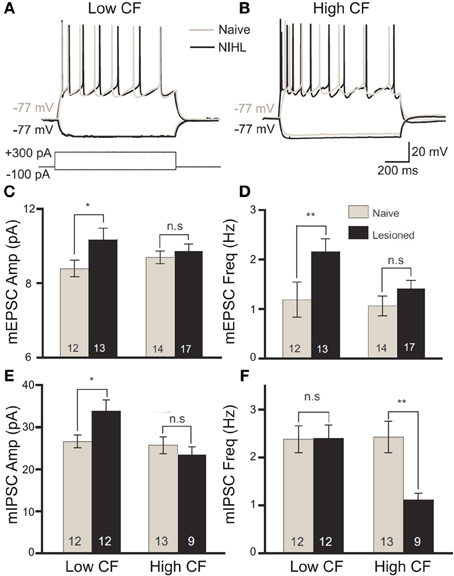
Figure 4. The balance of excitation and inhibition is differentially modified by band-limited acoustic trauma according to the frequency tuning of cortical neurons. Following acoustic trauma (123 dB SPL, 4 kHz, 7 h) of adult rats, in vitro patch recordings from primary auditory cortex layer II/III pyramidal neurons were performed to evaluate relative changes in excitability between neurons with low- or high-categorized characteristic frequency tuning. In response to square current pulse injection, low-CF neurons showed no enhancement in the number of spikes fired (A), whereas high-CF cells displayed significantly elevated excitability (B). Modified with permission from Yang et al. (2012). On recording miniature excitatory postsynaptic potentials (mEPSCs), there was a significant increase in the amplitude (C) and frequency (D) of mEPSCs in low-CF neurons only following trauma. Similar analysis of miniature inhibitory postsynaptic potentials (mIPSCs) revealed a significant trauma-driven elevation of mIPSC amplitude in low-CF neurons (E), while the frequency of mIPSCs was significantly depressed in high-CF cells (F). Modified with permission from Yang et al. (2011). *p ≤ 0.05; **p ≤ 0.01.
Insult-Mediated Changes in Neural Spectrotemporal Receptive Fields and Networks
Insults to the auditory system are capable of disrupting or even abrogating the network response features to stimuli that vary in spectral and temporal content. Efforts at characterizing these network-level modulations have typically taken two approaches: defining the spectral bandwidth characteristics of auditory cells in the tonotopically organized centers of the lemniscal auditory pathway, and their temporal tuning characteristics in response to more complex, time-varying stimuli, such as amplitude- or frequency-modulated sounds.
Frequency Receptive Field Remapping
Among the earlier explorations of ensemble responses to deafferentation and trauma are the systematic characterizations conducted of the tonotopic remapping that develops following the insult. This phenomenon (reviewed in Calford, 2002; Irvine, 2007; Kilgard, 2012) concerns the representation of the cochlear frequency domain along an axis of a lemniscal auditory centre that is modified to produce an enhanced sensitivity to one or more frequency bands—effectively to the exclusion of others. Pioneering work from Irvine and colleagues in the guinea pig (Robertson and Irvine, 1989), later extended to the adult cat (Rajan et al., 1993), unveiled a systematic flattening of the primary auditory cortex tonotopic map that was contralateral to partial cochlear damage (Figure 5). This damage was characterized by complete hair cell destruction with limited (<5%) retention of spiral ganglion afferents. In these experiments, a notable change in neural frequency receptive field properties emerged upon comparing acoustic stimulation to the ipsilateral and contralateral ears: whereas in the naive animal the monaural tonotopic maps were effectively in register for each form of tonal stimulation (within ±0.03 kHz), an asymmetry of responsiveness following peripheral lesion developed such that only the contralateral frequency response map was flattened.
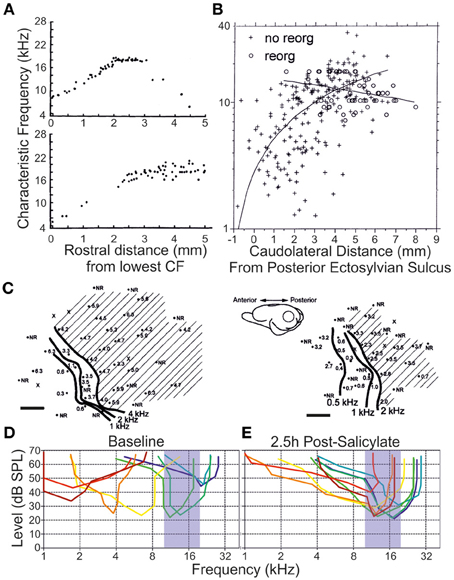
Figure 5. Remapping of the tonotopy in the primary auditory cortex following a variety of peripheral insults. (A) A flattening of the linear progression of characteristic frequency (CF) tuning when units were recorded along the dorsoventral axis of primary auditory cortex is observed following partial mechanical lesion of the cochlea in adult cats (individual subjects, upper and lower. Each datapoint represents a single unit CF. Modified with permission from Rajan et al. (1993). (B) A similar flattening of tonotopic progression is demonstrated as a function of distance along the cortical surface dorsoventrally in adult cats exposed to narrowband-passed acoustic trauma (crosses), compared with naive controls (circles). Modified with permission from Seki and Eggermont (2002). (C) In individual adult chinchillas (left, right) exposed to amikacin-induced basal cochlear hair cell lesions, there is a rearrangement of cortical responses to preference low characteristic frequency responses at regions of the cortex normally responsive at threshold to higher frequency stimuli (shaded region). Scale bars 1 mm. Modified with permission from Kakigi et al. (2000). (D) When individual multiunits were isolated in the adult rat primary auditory cortex (each unit is a different color), together spanning the hearing range of the animal, (E) systemic injection of salicylate (250 mg/kg intraperitoneal) produced a dynamic retuning of multiunits toward a CF range of 10–20 kHz by 2.5 h after injection. Modified with permission from Stolzberg et al. (2011).
Similar, high-frequency, chemical spiral ganglion lesions may also induce tonotopic remapping process, being refined over the course of months to yield enhanced cortical representation of low frequency sounds (Schwaber et al., 1993; Kakigi et al., 2000) (Figure 5). Notably, however, the phenomenon may be observed within days to weeks following selective deafferentation (Seki and Eggermont, 2002; Noreña et al., 2003; Engineer et al., 2011; Yang et al., 2011). Frequency reweighing in the cortex following salicylate has also been described (Stolzberg et al., 2011). Rather than displaying shifts toward the low-frequency edge of the retained normal inputs, instead neurons became abnormally sensitive to stimuli between 10 and 20 kHz at physiological thresholds. In these experiments, recorded units displayed substantially broadened bandwidths, implying that a large degree of their sensitivity to pre-exposure CFs was presumably retained (Stolzberg et al., 2011). It is also rather striking that both salicylate exposure (Deng et al., 2010) and acoustic trauma (Yin et al., 2008) each yield an apparently equivalent reduction in the cortical gap-detection threshold for stimulation off-channel, i.e., completely outside of the region displaying peripheral threshold elevation. This might suggest that each insult commonly disrupts a frequency-insensitive mechanism, or mechanisms, critical to the precise encoding of temporally salient information.
In cases of acute remapping like those described above, the loss of feedforward drive may simply intensify the relative expression of certain frequencies that had previously occupied peripheral regions of each unit's frequency response area. While evoked response thresholds are elevated, albeit moderately, by the induction of trauma, the overexpression of particular bandwidths tends to occur outside of the LPZ. Elevated driven response magnitudes, in addition, would imply the existence of a short-term plastic unmasking of afferent inputs that affects the relative interaction of excitatory and inhibitory receptive fields (Rajan, 2001). This phenomenon may develop even in the absence of CF shift (Rajan, 1998). In the chronic phase, however, the redevelopment of normalized peak thresholds and sharp tuning, with little variance of frequency receptive field width across the cortical surface, advocates for intrinsic plastic phenomena, adaptively reweighing neural sensitivity in favor of the remaining inputs.
Furthermore, there exists the possibility that cortical remapping may be inherited, in part, from plasticity in subcortical centers. Recordings made in the ventral division of the MGB contralateral to the cochlear lesion indicate the existence of putative tonotopic remapping in that centre (Kamke et al., 2003). The importance of the thalamocortical circuitry in potentially mediating high-level remodeling of receptive fields after peripheral receptor organ damage, as distinct from simple rostral transmission of deafferented, occasionally silenced (Rajan and Irvine, 1998), projections can be hypothesized.
As a point for comparison, the smooth, linear progression of tonotopy along the dorsoventral axis of the CNIC was predictably flattened after a cochlear lesion, producing an expanded region in which the characteristic frequency of collicular cells matched that of the lower edge of the LPZ (Irvine et al., 2003). However, there was a pronounced dichotomy among those neural units measured, in terms of the stimulus intensity required to drive activity. Although many neurons displayed markedly raised thresholds, akin to those observed in the VCN, others presented driven onset activity at ecologically relevant sound levels. While there exists the possibility of selective remodeling of neural connectivity, the fact that similar tuning patterns were observed in the acute phase following high-frequency spiral ganglion lesions (Snyder et al., 2000, 2008; Snyder and Sinex, 2002) might suggest a release from inhibition of previously subthreshold inputs coding for infra-LPZ frequencies. Indeed, these inputs are highly convergent, and partially comprise suprathreshold ipsilateral projections, which in the lesioned animal are enhanced approximately 5-fold (Irvine et al., 2003; Izquierdo et al., 2008).
Interestingly, such adaptive remodeling may not be universal. In recordings from A1 in the short term, tonotopic remapping has been found to normalize over the course of a week post-trauma (Ahlf et al., 2012). Moreover, following a low-frequency permanent deafferentation, ipsi- and contralateral cortical tuning were found, by 6 months post-insult, to be brought back into register (in contrast with the conserved tuning described above) (Cheung et al., 2009). The mechanism by which this occurred involved an elevation of the thresholds and reweighing of the receptive field characteristics of the cortex ipsilateral to the lesion. This is of particular concern for studies into the longer-term perceptual effects of monaurally limited hearing loss, given that this process develops despite the apparent disadvantages involved with accommodating for the lesioned ear bilaterally, and may be important in considering the utility of fitting hearing aids as early as possible in unilaterally impaired audiological patients. It is, also notable that the frequency receptive fields of infragranular cortical neurons were acutely unstable in aged rats (Turner et al., 2005). Concurrent with a remapping of frequency representations at the cortical surface associated with presbyacusis-mediated cochlear threshold shifts (Willott et al., 1993), an age-related change to the distribution of receptive field shapes of infragranular cortical neurons appears to develop, with overrepresentation of “complex,” non-V-shaped fields (Turner et al., 2005). How this may affect frequency-dependent perception remains to be seen.
Changes in Properties of Network Responses to Temporally Complex Stimuli
In the context of complex stimulus processing, it is possible that more fundamental shifts in neural output may distort the representation or encoding of stimuli in “higher” cortical centers. Recordings in the rhesus monkey have highlighted an age-related loss of hierarchical abstraction across cortical fields, such that in young monkeys the spatial, directional tuning acuity of neuronal responses is amplified from A1 to the more secondary caudolateral field (area CL), whereas such refinement is absent in older animals (Juarez-Salinas et al., 2010). These effects derive from reductions in inhibition to off-target locations in both CL and A1 (yielding broader receptive fields), as well as a reduction in onset latency in CL, suggesting that plasticity may selectively arise in the primate corticothalamic system, thus enhancing the representation of primary afferent stimuli in non-primary cortical fields (Engle and Recanzone, 2012). To the extent that such cortical remodeling may have the capacity for normalization, recent experiments have demonstrated that following auditory-driven behavioral training, aged rats displayed rectification of the tonotopic map found in A1, as well as in neuronal spectrotemporal response properties (De Villers-Sidani et al., 2010). The correlation between these functional improvements in A1 network activity with a post-training enhancement in parvalbumin levels—a marker of fast spiking inhibitory neurons important to perception- and learning-derived network plasticity (e.g., Donato et al., 2013), which undergoes downregulation following age-related hearing loss (Martin del Campo et al., 2012)—is further indicative of the possible maladaptive effects generated by inhibitory dysregulation.
The suggestion that temporal processing deficits may endure at the level of the IC, having also been documented in the cochlear nuclei, is apparent even from ABR analysis, which provides a surrogate for synchronous network activation (Buchwald and Huang, 1975). In long-latency ABR waveforms, which correlate with auditory midbrain activity (Melcher and Kiang, 1996; Melcher et al., 1996), clear age-dependent modulations in network response timing emerged among older animals (Nozawa et al., 1996). Although no such age-related differences were found during frequency-modulated stimulation of collicular cells (Lee et al., 2002), markedly worsened synchrony to modulated stimulation in aged animals was effected by the addition of background noise (Parthasarathy et al., 2010) and by varying the stimulus modulation depth (Parthasarathy and Bartlett, 2011). Regarding modulation rate, substantial deficits are evidently present at higher modulation rates (Parthasarathy and Bartlett, 2012), and indeed for collicular multi-units, gap-detection thresholds were significantly elevated in aged CBA/CaJ mice (Walton et al., 1998).
These temporal processing deficits align conceptually with observations from a two-tone suppression protocol that indicated significant differences in post-stimulus suppression and facilitation among collicular neurons (Finlayson, 2002). Here, some aged cells displayed abnormally long suppression time constants, indicative of altered encoding of temporally precise stimuli. Since a reduction in rise times to sinusoidally amplitude-modulated (SAM) stimuli also developed in aged CBA mice (Simon et al., 2004), inhibitory dysregulation affecting IC information processing may be a factor common to auditory trauma phenotypes.
It is, however, important to note that specific changes appear to develop across the different IC subdivisions. External cortex (ECIC) neurons in F344 rats displayed a proportional shift toward non-monotonic response when presented with contralateral stimuli (Palombi and Caspary, 1996a), although response bandwidths and net dynamic range of ECIC and CNIC cells failed to change over time with aging. This absence of effective age-related response modulation in F344 rats was also reported during binaural stimulation, typically associated with ipsilaterally-derived suppression of contralaterally-derived excitation (Palombi and Caspary, 1996b). However, when the same authors investigated responses to temporally-complex SAM stimuli, both ECIC and CNIC neurons showed clear divergence from those response distributions recorded in young rats (Shaddock Palombi et al., 2001). Such data concur with experiments in the mouse, in which dramatic differences in preferred modulation rate emerged in aged animals, reducing from 200 to 70 Hz in the upper quartile of units recorded (Walton et al., 2002). Despite failing to express consistent tonotopic changes, used elsewhere as a metric for neuroplastic rearrangement, the clear changes to the temporal coding operations of the IC, which may have behavior- and context-specific impacts with age (Harrison, 1981; Brown, 1984), are strong evidence of cellular- and network-level imbalances with functional consequences.
Altered Cross-Modal Sensitivity of the Auditory System Following Trauma
The occurrence of receptive field expansion in the trauma-exposed auditory system is therefore interesting, since integration across neuronal computational modes may have unexpected network or perceptual consequences. Indeed, aside from displaying compromised acoustic responsivity, the bilaterally deafferented auditory cortex comprised a significant proportion of neurons that exhibited salient responses to somatosensory inputs (Allman et al., 2009). However, the proportion displaying verifiable multisensory integration, under conditions of selectively preserved auditory input, is in fact diminished relative to control animals (Meredith et al., 2012).
In related work from Shore and colleagues, an enhancement of somatosensory input into the DCN driven by traumatic threshold elevations has been described (Shore et al., 2008). This modified sensory afferent input in the DCN is accompanied by alterations of the typical stimulus timing-dependent plasticity rules for bimodal integration during successive auditory/somatosensory stimulation (Dehmel et al., 2012b; Koehler and Shore, 2013a,b) (Figure 6), generally producing a long-term enhancement of unimodal acoustically evoked activity. Given that earlier descriptions of the effects of cochlear trauma concluded the absence of plastic remapping in the DCN (Kaltenbach et al., 1992; Rajan and Irvine, 1998), it is worthwhile, in future work, to consider alternative plasticity mechanisms, including cross-modal reorganization, which may develop post-traumatically throughout the auditory neuraxis.
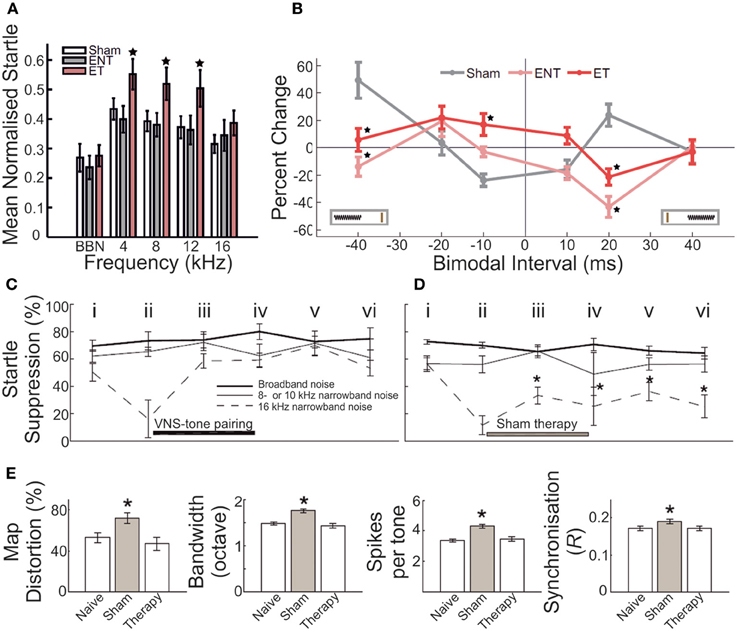
Figure 6. Gap-prepulse inhibition of the acoustic startle reflex (GPIAS) in rodents acts as a marker for underlying neurobiological changes following auditory insult. (A) In a cohort of adult guinea pigs, acoustic overexposure (1/4 octave band noise centered at 7 kHz, 93 dB SPL, 2 h; twice-exposed, separated by 6–8 weeks) yielded a subset of animals that displayed significantly decreased normalized startle response inhibition for background masking in the 4–16 kHz bandwidths. Sham controls = white bars; GPIAS-unimpaired animals = gray bars; GPIAS-impaired animals = pink bars. Star = significantly different (p < 0.05) from other bars in the same frequency band (B) On the basis of categorization according to the presence of GPIAS deficits, this subset of animals displayed significantly different stimulus-timing dependent plasticity functions for bimodal somatosensory-auditory stimulation compared with both sham controls, and exposed but unimpaired animals. Sham = gray; GPIAS-unimpaired = pink; GPIAS-impaired = red. Modified with permission from Koehler and Shore (2013a). (C–E) Following exposure to acoustic trauma (octave band noise centered at 16 kHz, 115 dB SPL, 1 h), adult rats underwent daily pairing of tonal stimuli outside the 8–10 kHz frequency bandwidth, either with vagal nerve stimulation (VNS) (C) or sham stimulation (D). VNS-tone pairing was found to produce rapid and long-lasting remediation of GPIAS impairments for 8–10 kHz background masking; sham-tone pairing produced no such improvement, with startle suppression ratios remaining depressed throughout the testing period following trauma. Time points: i, before trauma; ii, 4 weeks after exposure; iii, 10 days after initiation of VNS-tone or sham-tone pairing therapy; iv, 20 days after therapy initiation; v, 1 week after conclusion of therapy; vi, 3 weeks after conclusion of therapy. Broadband noise (thick line), non-tinnitus frequency (thin line), putative tinnitus frequency (dashed line). (E) In addition, VNS-tone pairing was found to remediate and normalize with respect to control measurements, aspects of primary auditory cortex neuronal operation which had been significantly enhanced following trauma, including tonotopic map distortion, frequency receptive field bandwidth, evoked spike response number, and neural spike-timing synchrony. For each bar plot, leftmost bar = naive; middle bar (gray) = sham-tone pairing therapy; rightmost bar = VNS-tone pairing therapy. Asterisk = significantly different (p < 0.05) compared with controls. Modified with permission from Engineer et al. (2011).
The Functional and Behavioral Implications of Trauma-Driven Auditory Plasticity
A constellation of neural changes evolves in parallel throughout the auditory system in response to the challenge posed by the introduction of mechanically-, pharmacologically-, acoustically-, or aging-derived insults (Table 1). These changes can be classed as dynamic modulations of spontaneous and driven neural activity, the underpinnings of which appear to rely upon putatively homeostatic gain-modulation mechanisms that affect the extent of neural excitability, and selectively modulate the balance of excitation and inhibition. Changes at the single neuron and network processing levels can be demonstrated in vivo (e.g., Noreña, 2011; Yang et al., 2011, 2012) and by in silico modeling (Dominguez et al., 2006; Schaette and Kempter, 2006, 2008, 2012; Chrostowski et al., 2011; Tass and Popovych, 2012; Schaette, 2013). Although these neurophysiological sequelae are becoming better understood, it remains an ongoing problem as to whether the various components of central modulation, at different levels of the auditory pathway, might translate into perceptual abnormalities, like tinnitus, that could interrogated by behavioral testing.
A major point of inquiry in current auditory neuroscience thus seeks to explain how it is that (subjective) tinnitus, being the perception of an auditory stimulus in the absence of an environmental equivalent, may come about in the wake of auditory trauma of a variety of forms, and manifest in only a proportion of any tested cohort (e.g., Eggermont and Roberts, 2004; Roberts et al., 2010; Noreña and Farley, 2013). In addition, concerns have recently been raised regarding the capacity to behaviorally test for the presence/absence of some abnormal positive percept. A possible division might be drawn between tinnitus as distinct from other correlated perceptual abnormalities, such as hyperacusis (Baguley, 2003), or from the predicted outcomes of hearing loss in the absence of any positively generative perceptual changes (Eggermont, 2013).
To that extent, a number of questions ought to be addressed: what classes of behavioral changes have been conclusively shown to develop following the introduction of some specific auditory insult (in the absence of a priori assumptions regarding the perceptual correlates of these behavioral data)? What are the neurobiological changes that have been shown to develop in parallel with these behavioral changes? And is it possible to conclude some effective functional link between certain aspect/s of the underlying insult-affected neurobiology and a categorical class of phantom percept?
Cochlear Lesion-Induced Behavioral Threshold Modulations
Of the behavioral studies that have been conducted related to sensorineural auditory insults in adult mammals, the range of observed effects correlates well with predictions made according to early physiological studies. In particular, those studies aimed at disambiguating the changes that develop in the periphery and at the level of the auditory nerve have been informative. Some insults have little effect on detection thresholds—for example, partial section of the auditory nerve in cats was found to have no impact on intensity thresholds except in the most severe of cases (Neff, 1947; Schuknecht and Woellner, 1953, 1955). Clearly, in the absence of complete destruction of afferent cochlear transmission, a small population of surviving neurons is sufficient to provide a basis for accurate and sensitive detection profiles. Conversely, damage at the cochlea is associated with behavioral threshold elevation in a manner related primarily to the extent of OHC loss (Ryan and Dallos, 1975; Hawkins et al., 1976; Ryan et al., 1979). After that kind of trauma, auditory nerve fiber tuning curves displaying marked loss of both sharp peak tuning and sensitivity by approximately 40 dB SPL at maximum.
In lieu of OHC loss, there is relatively good preservation of audiometric thresholds (Lobarinas et al., 2013b), whereas frequency discrimination is apparently reliant on effective preservation of the IHC population (Nienhuys and Clark, 1978). It is likely that the majority of peripherally-mediated insults affect one or both subtypes of hair cell population, even in cases in which the animal's electrophysiologically characterized thresholds are normalized following trauma (Kujawa and Liberman, 2009). As such, it might be predicted that the perceptual experience of an adult mammal subject would be affected in some fashion.
More recently, the focus of behavioral characterization has included the evaluation of changes in temporal processing ability, primarily (though not exclusively) in the form of testing gap-detection thresholds following insult. Electrophysiological data have demonstrated that neurometric gap detection thresholds deteriorate in the awake animal with aging (e.g., Recanzone et al., 2011) or acoustic trauma (e.g., Yin et al., 2008) (c.f. unilateral cochlear ablation, in which acoustic stimulation of the intact ear produced normal neurometric functions Kirby and Middlebrooks, 2010). Such neural changes appear to be behaviorally reflected by reductions in psychometric discrimination performance using gap-in-noise (Giraudi-Perry et al., 1982; Salvi and Arehole, 1985; Rybalko and Syka, 2005; Gold et al., 2013) or amplitude-modulated stimuli (Henderson et al., 1984). Performance of these operant behavioral paradigms is certainly reliant upon the normal function of the central auditory system for effective fine-scale perceptual discrimination of temporally-modulated stimuli. This can be reasoned from the effects of severe bilateral decortication/auditory cortical lesioning, which do not abolish task performance, but nonetheless significantly raises the detection thresholds of adult animals with otherwise normal hearing function (Ison et al., 1991; Kelly et al., 1996; Threlkeld et al., 2008). On the basis of these ablation/inactivation studies, it is not unreasonable to conclude that the accurate perception of the temporal fine structure of the stimuli, and so the performance of operant tasks reliant upon that perception, is expected to be affected following sensorineural insults that perturb normal auditory functionality.
As an additional measure of temporal processing abnormalities, a number of studies have leveraged various paradigms concerning the prepulse inhibition (PPI) of the acoustic startle response in small mammals. The behavior is reflexive, and thus can be recorded in the absence of prior training. It is notable, however, that there are indications of species-related efficacy, or rather lack thereof, of the response (Gruner, 1989; Pilz and Leaton, 1999). Nevertheless, it can be optimized for investigating aspects of an animal's perceptual experience. In particular, suppression of the acoustic startle using a gap-in-noise prepulse (gap-mediated prepulse inhibition of the acoustic startle, or GPIAS) can be modulated according to the specific experimental conditions imposed. These include the temporal interval between the prepulse and the startle stimulus, the amplitude of the startle stimulus, and the spectral content of the background noise masker (Longenecker and Galazyuk, 2012; Lobarinas et al., 2013a; Hickox and Liberman, 2014).
The introduction of an insult akin to those described that affected operant conditioning-derived deficits of gap detection may be interrogated by way of the acoustic startle response. From recordings performed either in the same subjects, or subjects exposed to identical stimuli which impaired GPIAS, aspects of subcortical temporal processing plasticity were found to be selectively affected in animals with gap-detection impairments, as distinct from subjects which have undergone comparable insult exposure, or naive controls (Koehler and Shore, 2013a) (see Figures 6A,B). Also, recordings in the auditory cortex have suggested that GPIAS may be a useful metric for evaluating insult-mediated changes in cortical activity. In particular, there was correlation between levels of neural synchrony and frequency tuning, and remediation of behavioral deficits observed to occur following a paired vagal nerve stimulation protocol (Engineer et al., 2011) (see Figures 6C,D).
Recent data have suggested that the susceptibility of subjects to impaired gap-mediated startle suppression following acoustic trauma might be related to baseline levels of excitability of the auditory neuraxis. Differential outcomes were found among subjects with variable levels of resting cortical firing rates and evoked activity, manifesting in addition as instability of cortical tonotopy (Ahlf et al., 2012). This variable expression of neural excitability is almost certainly related to the plasticity phenotype of animals in various insult conditions. Indeed, systemic salicylate injection, which is understood to affect neural excitability (see above), has been recognized as effective in similarly modulating the suppression efficacy, largely in parallel with upregulation of neural gain in the auditory cortex (Deng et al., 2010; Sun et al., 2014).
Given the extensive excitatory/inhibitory remodeling that is associated with aging (Caspary et al., 2008), it is possibly unsurprising that GPIAS impairment has also been demonstrated in a mouse model of presbyacusis. In this model, behavioral dysfunction was correlated with cortical inhibitory dysfunction demonstrated using novel functional imaging techniques (Llano et al., 2012). GPIAS behavioral impairment is undoubtedly a useful indicator of certain aspects of the possible underlying pathophysiology that affects neuroplastic changes after trauma. It may not be premature to suggest that the GPIAS represents an effective metric of auditory temporal processing deficits that occur subcortically. Indeed, cortical deficits may also modulate subcortical processing of the reflex circuitry by way of strong corticofugal projections (Bajo and King, 2012), which may also undergo insult-driven plastic remodeling. The role of these projections in the context of insult-mediated plasticity is largely unknown, and requires exploration in future experiments.
Perceptual Phantoms and Proposed Behavioral Correlates
The development of methods for reliably demonstrating a tinnitus-like percept in experimental animals represents another major research topic still in progress. The existence of tinnitus is, by convention, categorically different from the perception of silence. It is thus not unreasonable that a popular approach has sought to detect the phantom percept using operant conditioning paradigms to test perception. Often—but not always—under negative motivation, these require the animal to choose between a background noise stimulus, and either the absence of an acoustic stimulus, or the presence of some acoustic stimulus whose spectral content is deemed similar to the tinnitus percept (Jastreboff et al., 1988; Bauer and Brozoski, 2001; Heffner and Harrington, 2002; Guitton et al., 2003; Rüttiger et al., 2003; Yang et al., 2007, 2011; Sederholm and Swedberg, 2013).
Intuitively, experimental approaches of this kind are likely to be successful in revealing a subjective abnormality in the animal's perceptual state. The evaluation of the decision criterion, which is contingent upon the subject's integration of stimulus- and network-driven auditory activity, is likely to take into account the possible existence of a phantom percept. Indeed, the various demonstrations that long-known tinnitus inducers in human patients, such as high-dose salicylate (Cazals, 2000), bring about behavioral disruptions under such operant conditioning paradigms, indicate that the pursuit of phantom percepts in animals may not be wholly intractable.
More recently, the GPIAS paradigm has been proffered as a potential tool for behavioral interrogation of the phantom percept. Under the working framework that the presence of the tinnitus will internally mask, or “fill-in,” the presence of the gap in noise acting to suppress the startle response, a cohort of animals with diminished startle suppression as a function of some auditory insult might be revealed (Turner et al., 2006, 2012). The paradigm has definitely yielded interesting and promising data regarding the behavioral correlates of neurophysiological modulation—related to changes in tuning, evoked and spontaneous single unit activity, and broader network correlational aspects of auditory function—and has been validated in relation to other reported tinnitus detectors (Bauer and Brozoski, 2001; Yang et al., 2007). However, the likelihood that the technique acts as a reliable indicator of tinnitus proper remains under debate (Eggermont, 2013).
If indeed tinnitus “fills in the gap,” it is puzzling how it is that modifying the prepulse-startle inter-stimulus interval can renormalize GPIAS activity in animals whose suppression behaviors are selectively compromised (Hickox and Liberman, 2014). In those cases, IHC ribbon synapse pathology was comparable with the pathology displayed by subjects with operant-demonstrated behavioral deficits (Rüttiger et al., 2013; Singer et al., 2013). Moreover, gap-mediated startle suppression was absent during salicylate overdose when the gap-in-noise contained slowly ramped offset windows (Sun et al., 2014). This is despite the same animals displaying effective startle suppression when the gap on/offset ramp characteristics are otherwise modified (Sun et al., 2014). Observations of this kind may indicate that tinnitus, if present, fails to fill in the silent period consistently in the manner suggested in other reports of tinnitus-like behavior.
A further worry is the apparent susceptibility of GPIAS to modulation by unilateral earplug insertion (Lobarinas et al., 2013a) (Figure 7). This result has been interpreted as a false-positive detection of tinnitus in non-tinnitus animals, on the grounds that transient earplugging failed to reveal a tinnitus-like behavior under operant conditioning detection (Bauer and Brozoski, 2001). According to recent human data, chronic (>7 days) unilateral earplug insertion can induce positive phantom percepts in a majority of young, healthy listeners (Schaette et al., 2012). Animal behavioral studies have thus far only investigated transient earplugging effects, and so are not wholly comparable to the chronic earplugging condition of Schaette and colleagues. Yet, it is notable that enhanced neural synchrony has been postulated as a potential correlate of tinnitus in the auditory cortex (Noreña et al., 2003; Eggermont and Roberts, 2004; Roberts et al., 2010), developing more or less instantaneously following sensorineural auditory trauma (but not, it seems, following salicylate Ochi and Eggermont, 1996). If the phantom perceptual outcome of plugging operates by way of a similar functional framework as is to blame in “sensorineural” tinnitus (Schaette and Kempter, 2006; Schaette et al., 2012), it is not unlikely that transient tinnitus-like percepts may be inducible in a subset of subjects with a unilateral earplug. This may (Lobarinas et al., 2013a) or may not (Bauer and Brozoski, 2001; Turner et al., 2006) manifest during “tinnitus-sensitive” behavioral testing.
Figure 7. Unilateral conductive hearing loss yields conflicting reports of tinnitus generation in adult human and non-human normal-hearing subjects. (A) Chronic (>7 days) insertion of an earplug into one ear produced subjective reports of tinnitus in 10/18 human listeners, evaluated on a tinnitus similarity likeness scale; the group average tinnitus likeness indicated the development of a low-pass sensation peaking between 8 and 12 kHz. Inset is the attenuation function for earplug insertion, indicating a sloping attenuation of high-frequency stimuli up to ~30 dB SPL for stimuli >3–4 kHz. Modified with permission from Schaette et al. (2012). (B) Using GPIAS as a surrogate reporter of tinnitus presence in adult normal-hearing rats, unilateral earplug insertion also revealed tinnitus-like symptoms, as a reduction in startle-suppression efficacy for gaps inserted into carrier noise (60 dB SPL) narrow-bandpassed at 12, 16, 20, and 24 kHz. No such reduction developed for a 6 kHz narrowband carrier. Gray bars = baseline startle suppression ratio; black bars = plug startle suppression ratio; asterisk = significant difference within frequency band compared to baseline (p < 0.05). Modified with permission from Lobarinas et al. (2013a). (C) Conversely, testing of the effects of unilateral plugging on an operant sound detection task for stimuli presented at 10 kHz (upper) and lower (20 kHz) in adult rats showed there to be no effect upon detection functions for either stimulus bandwidth; by contrast, animals that underwent acoustic overexposure (narrowband noise centered at 16 kHz, 105 dB SPL, 1 h) displayed marked reduction in detection functions at 20 kHz, interpreted as the presence of tinnitus whose spectral content matched that frequency band. Control = open circles; normal-hearing with plug = filled squares; acoustic trauma without plug = open squares. Modified with permission from Bauer and Brozoski (2001). (D) A similar evaluation of the effects of unilateral earplug in adult rats found no difference between GPIAS functions in the same subjects prior to and following earplug insertion. Background noise was centered at 10 kHz, at which plug insertion produced a mean attenuation of 22 dB SPL. Modified with permission from Turner et al. (2006).
The emergence of non-specific gap-detection deficits has recently been demonstrated among a subset of tinnitus patients (Fournier and Hébert, 2013; but see Campolo et al., 2013), and thus such perceptual abnormalities may well form part of the tinnitus syndrome in animal models thus far reported. It is, however, insufficient to rely on deficits of this kind to categorically define the presence of tinnitus, even in the context of physiological and anatomical features that have been related to the disease in the past. What is required is the development of behavioral paradigms that more specifically disentangle the presence of a phantom percept from the spatiotemporal processing deficits that may arise from auditory trauma, neuroplastic changes, or otherwise. Almost certainly, the implementation of these paradigms would rely upon baseline measurements being obtained in the same animal population as is subsequently exposed to trauma, enabling repeated-measures statistical tests to be leveraged. In addition, the exploration of novel behavioral and pathological models will expand our capacity to interrogate the perceptual effects of central auditory changes which develop following trauma. Prior to causally relating behavioral phenotypes to particular neurophysiological modulations to that have been labeled “tinnitus-like,” it would be constructive to develop a fuller appreciation of the neurobiological changes that consistently develop in the wake of tinnitus-related effectors, irrespective of the proposed instantiation of the percept itself.
The Future of Insult-Related Plasticity and Perceptual Changes
Recent circumspection has called into question the proposed relationship between certain neurophysiological changes and the development of auditory phantoms (Eggermont, 2013). Contrasting changes to spontaneous discharge rates were found following different insults that each yielded equivalent behavioral results (e.g., Bauer et al., 2008). There is a lack of correspondence between remediation of behavioral changes and spontaneous rate elevations, despite other neurophysiological features undergoing normalization (Engineer et al., 2011), and an apparently paradoxical depression or amplification of spontaneous rate in certain studies of salicylate administration (Eggermont and Kenmochi, 1998; Yang et al., 2007; Zhang et al., 2011). Similarly, the use of tonotopic remapping as a tinnitus-specific metric can be problematic due to the contrast between short term reversals in tonotopic map plasticity (Ahlf et al., 2012) compared with therapy-related tonotopy restoration (Engineer et al., 2011) in animals with “tinnitus-like” behavior. Indeed, in human patients, both the presence (Mühlnickel et al., 1998; Weisz et al., 2005) and the absence (Langers et al., 2012) of tonotopic remapping have been reported, further frustrating the interpretation of animal data.
While historically less focused upon in research than tinnitus, hyperacusis is gaining traction as a potential sequela of the trauma-induced maladaptive plasticity syndrome. Recent reports have linked the state of enhanced auditory sensitivity to the development of tinnitus in human patients (Dauman and Bouscau-Faure, 2005; Fournier and Hébert, 2013; Hébert et al., 2013). Behavioral data have suggested that the possibility for detection of a hyperacusis-like state in animal models may exist via the emergence of enhanced gain of the acoustic startle reflex, and its attendant prepulse suppression. This type of behavior has been noted following acoustic trauma (Dehmel et al., 2012a; Sun et al., 2012; Chen et al., 2013; Pace and Zhang, 2013; Hickox and Liberman, 2014), salicylate treatment (Turner and Parrish, 2008; Sun et al., 2009a), or aging (Ison et al., 2007).
It is possible that the neurobiological factors mediating the maintenance of hyperacusis are distinct from, though likely related to, those inducing tinnitus. Computational (Zeng, 2013) and molecular models (Knipper et al., 2013) each have sought to assign specific network modulation patterns, particularly those associated with aberrant central signaling gain changes, to the occurrence of post-traumatic hyperacusis. Certainly, evidence of enhanced driven neural activity has been noted subcortically (Cai et al., 2009) and cortically (Qiu et al., 2000; Yang et al., 2007; Sun et al., 2009a, 2012) following various forms of auditory overexposure (see above). It is intriguing to ask how it is that the auditory system's innate gain control systems may be perturbed to potentially generate an abnormal perceptual experience. Indeed, the observed disparities between cortical and subcortical gain functions may in part be explained by hierarchically distinct, physiological differences in gain control (Rabinowitz et al., 2011, 2012). As lines of inquiry, these certainly warrant further examination in integrated behavioral and neurophysiological models, chiefly to answer key concerns regarding the development of the condition, observed with (Dauman and Bouscau-Faure, 2005) and without (Gu et al., 2010) peripheral threshold elevations.
Fruitful investigation is likely to be achieved by evaluating the causes underlying changes to neural gain and network spiking synchrony following some tinnitus-related insult, particularly with respect to rebalancing of excitatory/inhibitory signaling mechanisms (Knipper et al., 2013). A broader consideration of multiregional network effects throughout the auditory pathway is required. It is necessary to highlight the temporally-defined differential effects of post-insult regional disruption, particularly at the level of the auditory brainstem and midbrain (Brozoski and Bauer, 2005; Mulders and Robertson, 2009, 2011; Brozoski et al., 2012). In addition, the necessary involvement of ascending and descending projections throughout the auditory neuraxis in mediating physiological and behavioral changes following auditory insult remain underexplored (Bajo et al., 2010; Bajo and King, 2012). In the absence of any effective and reliable therapeutic measures being available for tinnitus treatment in human sufferers (Baguley et al., 2013; Langguth et al., 2013), it is clear that the insult-related plastic processes related to, and distinct from, perceptual phantom generation must be unraveled for clinical progress to proceed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. F. R. Nodal and Prof. A. J. King for fruitful comments on a previous version of this review. Our research is supported by the Wellcome Trust (WT07650AIA to A. J. King) and by Action on Hearing Loss (S72_Bajo).
References
Agrawal, Y., Platz, E. A., and Niparko, J. K. (2008). Prevalence of hearing loss and differences by demographic characteristics among US adults: data from the National Health and Nutrition Examination Survey, 1999-2004. Arch. Intern. Med. 168, 1522–1530. doi: 10.1001/archinte.168.14.1522
Ahlf, S., Tziridis, K., Korn, S., Strohmeyer, I., and Schulze, H. (2012). Predisposition for and prevention of subjective tinnitus development. PLoS ONE 7:e44519. doi: 10.1371/journal.pone.0044519
Alkhatib, A., Biebel, U. W., and Smolders, J. W. T. (2006). Reduction of inhibition in the inferior colliculus after inner hair cell loss. Neuroreport 17, 1493–1497. doi: 10.1097/01.wnr.0000234754.11431.ee
Allman, B. L., Keniston, L. P., and Meredith, M. A. (2009). Adult deafness induces somatosensory conversion of ferret auditory cortex. Proc. Natl. Acad. Sci. U.S.A. 106, 5925–5930. doi: 10.1073/pnas.0809483106
Argence, M., Saez, I., Sassu, R., Vassias, I., Vidal, P. P., and de Waele, C. (2006). Modulation of inhibitory and excitatory synaptic transmission in rat inferior colliculus after unilateral cochleectomy: an in situ and immunofluorescence study. Neuroscience 141, 1193–1207. doi: 10.1016/j.neuroscience.2006.04.058
Asako, M., Holt, A. G., Griffith, R. D., Buras, E. D., and Altschuler, R. A. (2005). Deafness-related decreases in glycine-immunoreactive labeling in the rat cochlear nucleus. J. Neurosci. Res. 81, 102–109. doi: 10.1002/jnr.20542
Attias, J., Pratt, H., Reshef, I., Bresloff, I., Horowitz, G., Polyakov, A., et al. (1996). Detailed analysis of auditory brainstem responses in patients with noise-induced tinnitus. Audiology 35, 259–270. doi: 10.3109/00206099609071946
Axelsson, A., and Sandh, A. (1985). Tinnitus in noise-induced hearing loss. Br. J. Audiol. 19, 271–276. doi: 10.3109/03005368509078983
Baguley, D., McFerran, D., and Hall, D. (2013). Tinnitus. Lancet 382, 1600–1607. doi: 10.1016/S0140-6736(13)60142-7
Bajo, V. M., and King, A. J. (2012). Cortical modulation of auditory processing in the midbrain. Front. Neural Circuits 6:114. doi: 10.3389/fncir.2012.00114
Bajo, V. M., Nodal, F. R., Moore, D. R., and King, A. J. (2010). The descending corticocollicular pathway mediates learning-induced auditory plasticity. Nat. Neurosci. 13, 253–260. doi: 10.1038/nn.2466
Banay-Schwartz, M., Lajtha, A., and Palkovits, M. (1989a). Changes with aging in the levels of amino acids in rat CNS structural elements. II. Taurine and small neutral amino acids. Neurochem. Res. 14, 563–570. doi: 10.1007/BF00964919
Banay-Schwartz, M., Lajtha, A., and Palkovits, M. (1989b). Changes with aging in the levels of amino acids in rat CNS structural elements. I. Glutamate and related amino acids. Neurochem. Res. 14, 555–562. doi: 10.1007/BF00964918
Bauer, C. A., and Brozoski, T. J. (2001). Assessing tinnitus and prospective tinnitus therapeutics using a psychophysical animal model. J. Assoc. Res. Otolaryngol. 2, 54–64. doi: 10.1007/s101620010030
Bauer, C. A., Brozoski, T. J., Holder, T. M., and Caspary, D. M. (2000). Effects of chronic salicylate on GABAergic activity in rat inferior colliculus. Hear Res. 147, 175–182. doi: 10.1016/S0378-5955(00)00130-1
Bauer, C. A., Turner, J. G., Caspary, D. M., Myers, K. S., and Brozoski, T. J. (2008). Tinnitus and inferior colliculus activity in chinchillas related to three distinct patterns of cochlear trauma. J. Neurosci. Res. 86, 2564–2578. doi: 10.1002/jnr.21699
Béïque, J., Na, Y., Kuhl, D., Worley, P. F., and Huganir, R. L. (2011). Arc-dependent synapse-specific homeostatic plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 816–821. doi: 10.1073/pnas.1017914108
Bielefeld, E. C., Coling, D., Chen, G.-D., Li, M., Tanaka, C., Hu, B.-H., et al. (2008). Age-related hearing loss in the Fischer 344/NHsd rat substrain. Hear Res. 241, 26–33. doi: 10.1016/j.heares.2008.04.006
Bledsoe, S. C., Koehler, S., Tucci, D. L., Zhou, J., Le Prell, C., and Shore, S. E. (2009). Ventral cochlear nucleus responses to contralateral sound are mediated by commissural and olivocochlear pathways. J. Neurophysiol. 102, 886–900. doi: 10.1152/jn.91003.2008
Boettcher, F. A., and Salvi, R. J. (1993). Functional changes in the ventral cochlear nucleus following acute acoustic overstimulation. J. Acoust. Soc. Am. 94, 2123–2134. doi: 10.1121/1.407484
Bramham, C. R., Alme, M. N., Bittins, M., Kuipers, S. D., Nair, R. R., Pai, B., et al. (2010). The Arc of synaptic memory. Exp. Brain Res. 200, 125–140. doi: 10.1007/s00221-009-1959-2
Brown, C. H. (1984). Directional hearing in aging rats. Exp. Aging Res. 10, 35–38. doi: 10.1080/03610738408258539
Brozoski, T. J., and Bauer, C. A. (2005). The effect of dorsal cochlear nucleus ablation on tinnitus in rats. Hear Res. 206, 227–236. doi: 10.1016/j.heares.2004.12.013
Brozoski, T. J., Bauer, C. A., and Caspary, D. M. (2002). Elevated fusiform cell activity in the dorsal cochlear nucleus of chinchillas with psychophysical evidence of tinnitus. J. Neurosci. 22, 2383–2390.
Brozoski, T. J., Wisner, K. W., Sybert, L. T., and Bauer, C. A. (2012). Bilateral dorsal cochlear nucleus lesions prevent acoustic-trauma induced tinnitus in an animal model. J. Assoc. Res. Otolaryngol. 13, 55–66. doi: 10.1007/s10162-011-0290-3
Buchwald, J. S., and Huang, C. (1975). Far-field acoustic response: origins in the cat. Science 189, 382–384. doi: 10.1126/science.1145206
Buckiova, D., Popelar, J., and Syka, J. (2007). Aging cochleas in the F344 rat: morphological and functional changes. Exp. Gerontol. 42, 629–638. doi: 10.1016/j.exger.2007.02.007
Buran, B. N., Strenzke, N., Neef, A., Gundelfinger, E. D., Moser, T., and Liberman, M. C. (2010). Onset coding is degraded in auditory nerve fibers from mutant mice lacking synaptic ribbons. J. Neurosci. 30, 7587–7597. doi: 10.1523/JNEUROSCI.0389-10.2010
Buras, E. D., Holt, A. G., Griffith, R. D., Asako, M., and Altschuler, R. A. (2006). Changes in glycine immunoreactivity in the rat superior olivary complex following deafness. J. Comp. Neurol. 494, 179–189. doi: 10.1002/cne.20795
Burianova, J., Ouda, L., Profant, O., and Syka, J. (2009). Age-related changes in GAD levels in the central auditory system of the rat. Exp. Gerontol. 44, 161–169. doi: 10.1016/j.exger.2008.09.012
Cai, R., Kalappa, B. I., Brozoski, T. J., Ling, L. L., and Caspary, D. M. (2014). Is GABA neurotransmission enhanced in auditory thalamus relative to inferior colliculus? J. Neurophysiol. 111, 229–238. doi: 10.1152/jn.00556.2013
Cai, S., Ma, W.-L. D., and Young, E. D. (2009). Encoding intensity in ventral cochlear nucleus following acoustic trauma: implications for loudness recruitment. J. Assoc. Res. Otolaryngol. 10, 5–22. doi: 10.1007/s10162-008-0142-y
Calford, M. B. (2002). Dynamic representational plasticity in sensory cortex. Neuroscience 111, 709–738. doi: 10.1016/S0306-4522(02)00022-2
Calford, M. B., Rajan, R., and Irvine, D. R. (1993). Rapid changes in the frequency tuning of neurons in cat auditory cortex resulting from pure-tone-induced temporary threshold shift. Neuroscience 55, 953–964. doi: 10.1016/0306-4522(93)90310-C
Campolo, J., Lobarinas, E., and Salvi, R. (2013). Does tinnitus “fill in” the silent gaps? Noise Health 15, 398–405. doi: 10.4103/1463-1741.121232
Carretta, D., Hervé-Minvielle, A., Bajo, V. M., Villa, A. E., and Rouiller, E. M. (1999). c-Fos expression in the auditory pathways related to the significance of acoustic signals in rats performing a sensory-motor task. Brain Res. 841, 170–183. doi: 10.1016/S0006-8993(99)01840-5
Caspary, D. M., Hughes, L. F., and Ling, L. L. (2013). Age-related GABAA receptor changes in rat auditory cortex. Neurobiol. Aging 34, 1486–1496. doi: 10.1016/j.neurobiolaging.2012.11.009
Caspary, D. M., Hughes, L. F., Schatteman, T. A., and Turner, J. G. (2006). Age-related changes in the response properties of cartwheel cells in rat dorsal cochlear nucleus. Hear Res. 216–217, 207–215. doi: 10.1016/j.heares.2006.03.005
Caspary, D. M., Ling, L., Turner, J. G., and Hughes, L. F. (2008). Inhibitory neurotransmission, plasticity and aging in the mammalian central auditory system. J. Exp. Biol. 211, 1781–1791. doi: 10.1242/jeb.013581
Caspary, D. M., Raza, A., Lawhorn Armour, B. A., Pippin, J., and Arneriæ, S. P. (1990). Immunocytochemical and neurochemical evidence for age-related loss of GABA in the inferior colliculus: implications for neural presbycusis. J. Neurosci. 10, 2363–2372.
Caspary, D. M., Schatteman, T. A., and Hughes, L. F. (2005). Age-related changes in the inhibitory response properties of dorsal cochlear nucleus output neurons: role of inhibitory inputs. J. Neurosci. 25, 10952–10959. doi: 10.1523/JNEUROSCI.2451-05.2005
Cazals, Y. (2000). Auditory sensori-neural alterations induced by salicylate. Prog. Neurobiol. 62, 583–631. doi: 10.1016/S0301-0082(00)00027-7
Cazals, Y., Horner, K. C., and Huang, Z. W. (1998). Alterations in average spectrum of cochleoneural activity by long-term salicylate treatment in the guinea pig: a plausible index of tinnitus. J. Neurophysiol. 80, 2113–2120.
Chang, H., Chen, K., Kaltenbach, J. A., Zhang, J., and Godfrey, D. A. (2002). Effects of acoustic trauma on dorsal cochlear nucleus neuron activity in slices. Hear Res. 164, 59–68. doi: 10.1016/S0378-5955(01)00410-5
Chen, G.-D., Li, M., Tanaka, C., Bielefeld, E. C., Hu, B.-H., Kermany, M. H., et al. (2009). Aging outer hair cells (OHCs) in the Fischer 344 rat cochlea: function and morphology. Hear Res. 248, 39–47. doi: 10.1016/j.heares.2008.11.010
Chen, G., Lee, C., Sandridge, S. A., Butler, H. M., Manzoor, N. F., and Kaltenbach, J. A. (2013). Behavioral evidence for possible simultaneous induction of hyperacusis and tinnitus following intense sound exposure. J. Assoc. Res. Otolaryngol. 14, 413–424. doi: 10.1007/s10162-013-0375-2
Cheung, S. W., Bonham, B. H., Schreiner, C. E., Godey, B., and Copenhaver, D. A. (2009). Realignment of interaural cortical maps in asymmetric hearing loss. J. Neurosci. 29, 7065–7078. doi: 10.1523/JNEUROSCI.6072-08.2009
Chrostowski, M., Yang, L., Wilson, H. R., Bruce, I. C., and Becker, S. (2011). Can homeostatic plasticity in deafferented primary auditory cortex lead to travelling waves of excitation? J. Comput. Neurosci. 30, 279–299. doi: 10.1007/s10827-010-0256-1
Cody, A. R., and Johnstone, B. M. (1980). Single auditory neuron response during acute acoustic trauma. Hear Res. 3, 3–16. doi: 10.1016/0378-5955(80)90004-0
Cohen, G. M., Park, J. C., and Grasso, J. S. (1990). Comparison of demyelination and neural degeneration in spiral and Scarpa's ganglia of C57BL/6 mice. J. Electron Microsc. Tech. 15, 165–172. doi: 10.1002/jemt.1060150208
Dallos, P., and Harris, D. (1978). Properties of auditory nerve responses in absence of outer hair cells. J. Neurophysiol. 41, 365–383.
Dauman, R., and Bouscau-Faure, F. (2005). Assessment and amelioration of hyperacusis in tinnitus patients. Acta Otolaryngol. 125, 503–509.
Dazert, S., Feldman, M. L., and Keithley, E. M. (1996). Cochlear spiral ganglion cell degeneration in wild-caught mice as a function of age. Hear Res. 100, 101–106. doi: 10.1016/0378-5955(96)00100-1
De Villers-Sidani, E., Alzghoul, L., Zhou, X., Simpson, K. L., Lin, R. C. S., and Merzenich, M. M. (2010). Recovery of functional and structural age-related changes in the rat primary auditory cortex with operant training. Proc. Natl. Acad. Sci. U.S.A. 107, 13900–13905. doi: 10.1073/pnas.1007885107
Dehmel, S., Eisinger, D., and Shore, S. E. (2012a). Gap prepulse inhibition and auditory brainstem-evoked potentials as objective measures for tinnitus in guinea pigs. Front. Syst. Neurosci. 6:42. doi: 10.3389/fnsys.2012.00042
Dehmel, S., Pradhan, S., Koehler, S., Bledsoe, S., and Shore, S. (2012b). Noise overexposure alters long-term somatosensory-auditory processing in the dorsal cochlear nucleus–possible basis for tinnitus-related hyperactivity? J. Neurosci. 32, 1660–1671. doi: 10.1523/JNEUROSCI.4608-11.2012
Deng, A., Lu, J., and Sun, W. (2010). Temporal processing in inferior colliculus and auditory cortex affected by high doses of salicylate. Brain Res. 1344, 96–103. doi: 10.1016/j.brainres.2010.04.077
Di Girolamo, S., Quaranta, N., Picciotti, P., Torsello, A., and Wolf, F. (2001). Age-related histopathological changes of the stria vascularis: an experimental model. Audiology 40, 322–326. doi: 10.3109/00206090109073129
Dolan, T. R., Ades, H. W., Bredberg, G., and Neff, W. D. (1975). Inner ear damage and hearing loss after exposure to tones of high intensity. Acta Otolaryngol. 80, 343–352. doi: 10.3109/00016487509121336
Dominguez, M., Becker, S., Bruce, I., and Read, H. (2006). A spiking neuron model of cortical correlates of sensorineural hearing loss: spontaneous firing, synchrony, and tinnitus. Neural Comput. 18, 2942–2958. doi: 10.1162/neco.2006.18.12.2942
Donato, F., Rompani, S. B., and Caroni, P. (2013). Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504, 272–276. doi: 10.1038/nature12866
Dong, S., Mulders, W. H., Rodger, J., and Robertson, D. (2009). Changes in neuronal activity and gene expression in guinea-pig auditory brainstem after unilateral partial hearing loss. Neuroscience 159, 1164–1174. doi: 10.1016/j.neuroscience.2009.01.043
Dong, S., Mulders, W. H., Rodger, J., Woo, S., and Robertson, D. (2010a). Acoustic trauma evokes hyperactivity and changes in gene expression in guinea-pig auditory brainstem. Eur. J. Neurosci. 31, 1616–1628. doi: 10.1111/j.1460-9568.2010.07183.x
Dong, S., Rodger, J., Mulders, W. H., and Robertson, D. (2010b). Tonotopic changes in GABA receptor expression in guinea pig inferior colliculus after partial unilateral hearing loss. Brain Res. 1342, 24–32. doi: 10.1016/j.brainres.2010.04.067
Eggermont, J. J. (2013). Hearing loss, hyperacusis, or tinnitus: what is modeled in animal research? Hear Res. 295, 140–149. doi: 10.1016/j.heares.2012.01.005
Eggermont, J. J., and Roberts, L. E. (2004). The neuroscience of tinnitus. Trends Neurosci. 27, 676–682. doi: 10.1016/j.tins.2004.08.010
Eggermont, J., and Kenmochi, M. (1998). Salicylate and quinine selectively increase spontaneous firing rates in secondary auditory cortex. Hear Res. 117, 149–160. doi: 10.1016/S0378-5955(98)00008-2
Ehret, G., and Fischer, R. (1991). Neuronal activity and tonotopy in the auditory system visualized by c-fos gene expression. Brain Res. 567, 350–354. doi: 10.1016/0006-8993(91)90819-H
El-Badry, M. M., and McFadden, S. L. (2007). Electrophysiological correlates of progressive sensorineural pathology in carboplatin-treated chinchillas. Brain Res. 1134, 122–130. doi: 10.1016/j.brainres.2006.11.078
El-Badry, M. M., and McFadden, S. L. (2009). Evaluation of inner hair cell and nerve fiber loss as sufficient pathologies underlying auditory neuropathy. Hear Res. 255, 84–90. doi: 10.1016/j.heares.2009.06.003
Engineer, N. D., Riley, J. R., Seale, J. D., Vrana, W. A., Shetake, J. A., Sudanagunta, S. P., et al. (2011). Reversing pathological neural activity using targeted plasticity. Nature 470, 101–104. doi: 10.1038/nature09656
Engle, J. R., and Recanzone, G. H. (2012). Characterizing spatial tuning functions of neurons in the auditory cortex of young and aged monkeys: a new perspective on old data. Front. Aging Neurosci. 4:36. doi: 10.3389/fnagi.2012.00036
Engle, J. R., Tinling, S., and Recanzone, G. H. (2013). Age-related hearing loss in rhesus monkeys is correlated with cochlear histopathologies. PLoS ONE 8:e55092. doi: 10.1371/journal.pone.0055092
Evans, E. F., and Borerwe, T. A. (1982). Ototoxic effects of salicylates on the responses of single cochlear nerve fibres and on cochlear potentials. Br. J. Audiol. 16, 101–108. doi: 10.3109/03005368209081454
Feng, J., Bendiske, J., and Morest, D. K. (2012). Degeneration in the ventral cochlear nucleus after severe noise damage in mice. J. Neurosci. Res. 90, 831–841. doi: 10.1002/jnr.22793
Finlayson, P. G. (2002). Paired-tone stimuli reveal reductions and alterations in temporal processing in inferior colliculus neurons of aged animals. J. Assoc. Res. Otolaryngol. 3, 321–331. doi: 10.1007/s101620020038
Finlayson, P. G., and Kaltenbach, J. A. (2009). Alterations in the spontaneous discharge patterns of single units in the dorsal cochlear nucleus following intense sound exposure. Hear Res. 256, 104–117. doi: 10.1016/j.heares.2009.07.006
Forge, A. (1985). Outer hair cell loss and supporting cell expansion following chronic gentamicin treatment. Hear Res. 19, 171–182. doi: 10.1016/0378-5955(85)90121-2
Förster, C. R., and Illing, R. B. (1998). Redistribution of NMDA receptors in the cochlear nucleus following cochleotomy. Neuroreport 9, 3531–3535. doi: 10.1097/00001756-199810260-00036
Fournier, P., and Hébert, S. (2013). Gap detection deficits in humans with tinnitus as assessed with the acoustic startle paradigm: does tinnitus fill in the gap? Hear Res. 295, 16–23. doi: 10.1016/j.heares.2012.05.011
Froemke, R. C., and Martins, A. R. O. (2011). Spectrotemporal dynamics of auditory cortical synaptic receptive field plasticity. Hear Res. 279, 149–161. doi: 10.1016/j.heares.2011.03.005
Fryatt, A. G., Mulheran, M., Egerton, J., Gunthorpe, M. J., and Grubb, B. D. (2011). Ototrauma induces sodium channel plasticity in auditory afferent neurons. Mol. Cell Neurosci. 48, 51–61. doi: 10.1016/j.mcn.2011.06.005
Furman, A. C., Kujawa, S. G., and Liberman, M. C. (2013). Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. J. Neurophysiol. 110, 577–586. doi: 10.1152/jn.00164.2013
Fyk-Kolodziej, B., Shimano, T., Gong, T.-W., and Holt, A. G. (2011). Vesicular glutamate transporters: spatio-temporal plasticity following hearing loss. Neuroscience 178, 218–239. doi: 10.1016/j.neuroscience.2010.12.059
Gao, M., Sossa, K., Song, L., Errington, L., Cummings, L., Hwang, H., et al. (2010). A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci. 30, 7168–7178. doi: 10.1523/JNEUROSCI.1067-10.2010
Gentschev, T., and Sotelo, C. (1973). Degenerative patterns in the ventral cochlear nucleus of the rat after primary deafferentation. An ultra-structural study. Brain Res. 62, 37–60. doi: 10.1016/0006-8993(73)90618-5
Giraudi-Perry, D. M., Salvi, R. J., and Henderson, D. (1982). Gap detection in hearing-impaired chinchillas. J. Acoust. Soc. Am. 72, 1387–1393. doi: 10.1121/1.388444
Godfrey, D. A., Godfrey, M. A., Ding, D.-L., Chen, K., and Salvi, R. J. (2005). Amino acid concentrations in chinchilla cochlear nucleus at different times after carboplatin treatment. Hear Res. 206, 64–73. doi: 10.1016/j.heares.2005.03.004
Godfrey, D. A., Jin, Y.-M., Liu, X., and Godfrey, M. A. (2014). Effects of cochlear ablation on amino acid levels in the rat cochlear nucleus and superior olive. Hear. Res. 309, 44–54. doi: 10.1016/j.heares.2013.11.005
Gold, J. R., Peters, F., Nodal, F. R., King, A. J., and Bajo, V. M. (2013). “Towards a new animal model of tinnitus: gap detection behaviour in adult ferrets,” in 7th International TRI Tinnitus Conference (Valencia).
Gruner, J. A. (1989). Comparison of vestibular and auditory startle responses in the rat and cat. J. Neurosci. Methods 27, 13–23. doi: 10.1016/0165-0270(89)90049-6
Gu, J. W., Halpin, C. F., Nam, E., Levine, R. A., and Melcher, J. R. (2010). Tinnitus, diminished sound-level tolerance, and elevated auditory activity in humans with clinically normal hearing sensitivity. J. Neurophysiol. 104, 3361–3370. doi: 10.1152/jn.00226.2010
Gu, J. W., Herrmann, B. S., Levine, R. A., and Melcher, J. R. (2012). Brainstem auditory evoked potentials suggest a role for the ventral cochlear nucleus in tinnitus. J. Assoc. Res. Otolaryngol. 13, 819–833. doi: 10.1007/s10162-012-0344-1
Guitton, M. J., Caston, J., Ruel, J., Johnson, R. M., Pujol, R., and Puel, J.-L. (2003). Salicylate induces tinnitus through activation of cochlear NMDA receptors. J. Neurosci. 23, 3944–3952.
Harrison, J. M. (1981). Effects of age on acquisition and maintenance of a location discrimination in rats. Exp. Aging Res. 7, 467–476. doi: 10.1080/03610738108259825
Hawkins, J. E., Johnsson, L. G., Stebbins, W. C., Moody, D. B., and Coombs, S. L. (1976). Hearing loss and cochlear pathology in monkeys after noise exposure. Acta Otolaryngol. 81, 337–343. doi: 10.3109/00016487609119971
Hébert, S., Fournier, P., and Noreña, A. (2013). The auditory sensitivity is increased in tinnitus ears. J. Neurosci. 33, 2356–2364. doi: 10.1523/JNEUROSCI.3461-12.2013
Heffner, H. E., and Harrington, I. A. (2002). Tinnitus in hamsters following exposure to intense sound. Hear Res. 170, 83–95. doi: 10.1016/S0378-5955(02)00343-X
Heinz, M. G., Issa, J. B., and Young, E. D. (2005). Auditory-nerve rate responses are inconsistent with common hypotheses for the neural correlates of loudness recruitment. J. Assoc. Res. Otolaryngol. 6, 91–105. doi: 10.1007/s10162-004-5043-0
Heinz, M. G., and Young, E. D. (2004). Response growth with sound level in auditory-nerve fibers after noise-induced hearing loss. J. Neurophysiol. 91, 784–795. doi: 10.1152/jn.00776.2003
Henderson, D., Salvi, R., Pavek, G., and Hamernik, R. (1984). Amplitude modulation thresholds in chinchillas with high-frequency hearing loss. J. Acoust. Soc. Am. 75, 1177–1183. doi: 10.1121/1.390767
Henry, K. S., and Heinz, M. G. (2012). Diminished temporal coding with sensorineural hearing loss emerges in background noise. Nat. Neurosci. 15, 1362–1364. doi: 10.1038/nn.3216
Henry, K. S., and Heinz, M. G. (2013). Effects of sensorineural hearing loss on temporal coding of narrowband and broadband signals in the auditory periphery. Hear Res. 303, 39–47. doi: 10.1016/j.heares.2013.01.014
Hequembourg, S., and Liberman, M. C. (2001). Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J. Assoc. Res. Otolaryngol. 2, 118–129. doi: 10.1007/s101620010075
Hickox, A. E., and Liberman, M. C. (2014). Is noise-induced cochlear neuropathy key to the generation of hyperacusis or tinnitus? J. Neurophysiol. 111, 552–564. doi: 10.1152/jn.00184.2013
Hildebrandt, H., Hoffmann, N. A., and Illing, R. B. (2011). Synaptic reorganization in the adult rat's ventral cochlear nucleus following its total sensory deafferentation. PLoS ONE 6:e23686. doi: 10.1371/journal.pone.0023686
Hughes, L. F., Turner, J. G., Parrish, J. L., and Caspary, D. M. (2010). Processing of broadband stimuli across A1 layers in young and aged rats. Hear Res. 264, 79–85. doi: 10.1016/j.heares.2009.09.005
Ichimiya, I., Suzuki, M., and Mogi, G. (2000). Age-related changes in the murine cochlear lateral wall. Hear Res. 139, 116–122. doi: 10.1016/S0378-5955(99)00170-7
Illing, R.-B., Kraus, K. S., and Meidinger, M. A. (2005). Reconnecting neuronal networks in the auditory brainstem following unilateral deafening. Hear Res. 206, 185–199. doi: 10.1016/j.heares.2005.01.016
Irvine, D. R. F. (2007). Auditory cortical plasticity: does it provide evidence for cognitive processing in the auditory cortex? Hear Res. 229, 158–170. doi: 10.1016/j.heares.2007.01.006
Irvine, D. R. F., Rajan, R., and Smith, S. (2003). Effects of restricted cochlear lesions in adult cats on the frequency organization of the inferior colliculus. J. Comp. Neurol. 467, 354–374. doi: 10.1002/cne.10921
Ison, J. R., Allen, P. D., and O'Neill, W. E. (2007). Age-related hearing loss in C57BL/6J mice has both frequency-specific and non-frequency-specific components that produce a hyperacusis-like exaggeration of the acoustic startle reflex. J. Assoc. Res. Otolaryngol. 8, 539–550. doi: 10.1007/s10162-007-0098-3
Ison, J. R., O'Connor, K., Bowen, G. P., and Bocirnea, A. (1991). Temporal resolution of gaps in noise by the rat is lost with functional decortication. Behav. Neurosci. 105, 33–40. doi: 10.1037//0735-7044.105.1.33
Izquierdo, M. A., Gutiérrez-Conde, P. M., Merchán, M. A., and Malmierca, M. S. (2008). Non-plastic reorganization of frequency coding in the inferior colliculus of the rat following noise-induced hearing loss. Neuroscience 154, 355–369. doi: 10.1016/j.neuroscience.2008.01.057
Jastreboff, P. J., Brennan, J. F., Coleman, J. K., and Sasaki, C. T. (1988). Phantom auditory sensation in rats: an animal model for tinnitus. Behav. Neurosci. 102, 811–822. doi: 10.1037/0735-7044.102.6.811
Jastreboff, P. J., and Sasaki, C. T. (1986). Salicylate-induced changes in spontaneous activity of single units in the inferior colliculus of the guinea pig. J. Acoust. Soc. Am. 80, 1384–1391. doi: 10.1121/1.394391
Juarez-Salinas, D. L., Engle, J. R., Navarro, X. O., and Recanzone, G. H. (2010). Hierarchical and serial processing in the spatial auditory cortical pathway is degraded by natural aging. J. Neurosci. 30, 14795–14804. doi: 10.1523/JNEUROSCI.3393-10.2010
Kakigi, A., Hirakawa, H., Harel, N., Mount, R. J., and Harrison, R. V. (2000). Tonotopic mapping in auditory cortex of the adult chinchilla with amikacin-induced cochlear lesions. Audiology 39, 153–160. doi: 10.3109/00206090009073068
Kaltenbach, J. A. (2011). Tinnitus: models and mechanisms. Hear Res. 276, 52–60. doi: 10.1016/j.heares.2010.12.003
Kaltenbach, J. A., and Afman, C. E. (2000). Hyperactivity in the dorsal cochlear nucleus after intense sound exposure and its resemblance to tone-evoked activity: a physiological model for tinnitus. Hear Res. 140, 165–172. doi: 10.1016/S0378-5955(99)00197-5
Kaltenbach, J. A., Czaja, J. M., and Kaplan, C. R. (1992). Changes in the tonotopic map of the dorsal cochlear nucleus following induction of cochlear lesions by exposure to intense sound. Hear Res. 59, 213–223. doi: 10.1016/0378-5955(92)90118-7
Kaltenbach, J. A., and McCaslin, D. L. (1996). Increases in spontaneous activity in the dorsal cochlear nucleus following exposure to high intensity sound: a possible neural correlate of tinnitus. Audit. Neurosci. 3, 57–78.
Kaltenbach, J. A., Rachel, J. D., Mathog, T. A., Zhang, J., Falzarano, P. R., and Lewandowski, M. (2002). Cisplatin-induced hyperactivity in the dorsal cochlear nucleus and its relation to outer hair cell loss: relevance to tinnitus. J. Neurophysiol. 88, 699–714.
Kaltenbach, J. A., Zhang, J., and Afman, C. E. (2000). Plasticity of spontaneous neural activity in the dorsal cochlear nucleus after intense sound exposure. Hear Res. 147, 282–292. doi: 10.1016/S0378-5955(00)00138-6
Kamke, M. R., Brown, M., and Irvine, D. R. F. (2003). Plasticity in the tonotopic organization of the medial geniculate body in adult cats following restricted unilateral cochlear lesions. J. Comp. Neurol. 459, 355–367. doi: 10.1002/cne.10586
Kamke, M. R., Brown, M., and Irvine, D. R. F. (2005). Basal forebrain cholinergic input is not essential for lesion-induced plasticity in mature auditory cortex. Neuron 48, 675–686. doi: 10.1016/j.neuron.2005.09.014
Kehrle, H. M., Granjeiro, R. C., Sampaio, A. L. L., Bezerra, R., Almeida, V. F., and Oliveira, C. A. (2008). Comparison of auditory brainstem response results in normal-hearing patients with and without tinnitus. Arch. Otolaryngol. Head Neck Surg. 134, 647–651. doi: 10.1001/archotol.134.6.647
Keithley, E. M., and Feldman, M. L. (1979). Spiral ganglion cell counts in an age-graded series of rat cochleas. J. Comp. Neurol. 188, 429–442. doi: 10.1002/cne.901880306
Keithley, E. M., and Feldman, M. L. (1982). Hair cell counts in an age-graded series of rat cochleas. Hear Res. 8, 249–262. doi: 10.1016/0378-5955(82)90017-X
Kelly, J. B., Rooney, B. J., and Phillips, D. P. (1996). Effects of bilateral auditory cortical lesions on gap-detection thresholds in the ferret (Mustela putorius). Behav. Neurosci. 110, 542–550. doi: 10.1037/0735-7044.110.3.542
Kiang, N. Y., Liberman, M. C., and Levine, R. A. (1975). Auditory-nerve activity in cats exposed to ototoxic drugs and high-intensity sounds. Ann. Otol. Rhinol. Laryngol. 85, 752–768.
Kilgard, M. P. (2012). Harnessing plasticity to understand learning and treat disease. Trends Neurosci. 35, 715–722. doi: 10.1016/j.tins.2012.09.002
Kilgard, M. P., and Merzenich, M. M. (1998). Plasticity of temporal information processing in the primary auditory cortex. Nat. Neurosci. 1, 727–731. doi: 10.1038/3729
King, A. J., Dahmen, J. C., Keating, P., Leach, N. D., Nodal, F. R., and Bajo, V. M. (2011). Neural circuits underlying adaptation and learning in the perception of auditory space. Neurosci. Biobehav. Rev. 35, 2129–2139. doi: 10.1016/j.neubiorev.2011.03.008
Kirby, A. E., and Middlebrooks, J. C. (2010). Auditory temporal acuity probed with cochlear implant stimulation and cortical recording. J. Neurophysiol. 103, 531–542. doi: 10.1152/jn.00794.2009
Knipper, M., Van Dijk, P., Nunes, I., Rüttiger, L., and Zimmermann, U. (2013). Advances in the neurobiology of hearing disorders: recent developments regarding the basis of tinnitus and hyperacusis. Prog. Neurobiol. 111, 17–33. doi: 10.1016/j.pneurobio.2013.08.002
Knipper, M., Zimmermann, U., and Müller, M. (2010). Molecular aspects of tinnitus. Hear Res. 266, 60–69. doi: 10.1016/j.heares.2009.07.013
Koehler, S. D., and Shore, S. E. (2013a). Stimulus timing-dependent plasticity in dorsal cochlear nucleus is altered in tinnitus. J. Neurosci. 33, 19647–19656. doi: 10.1523/JNEUROSCI.2788-13.2013
Koehler, S. D., and Shore, S. E. (2013b). Stimulus-timing dependent multisensory plasticity in the guinea pig dorsal cochlear nucleus. PLoS ONE 8:e59828. doi: 10.1371/journal.pone.0059828
Koerber, K. C., Pfeiffer, R. R., Warr, W. B., and Kiang, N. Y. (1966). Spontaneous spike discharges from single units in the cochlear nucleus after destruction of the cochlea. Exp. Neurol. 16, 119–130. doi: 10.1016/0014-4886(66)90091-4
Kraus, K. S., Ding, D., Zhou, Y., and Salvi, R. J. (2009). Central auditory plasticity after carboplatin-induced unilateral inner ear damage in the chinchilla: up-regulation of GAP-43 in the ventral cochlear nucleus. Hear Res. 255, 33–43. doi: 10.1016/j.heares.2009.05.001
Kujawa, S. G., and Liberman, M. C. (2006). Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J. Neurosci. 26, 2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009
Langers, D. R. M., de Kleine, E., and van Dijk, P. (2012). Tinnitus does not require macroscopic tonotopic map reorganization. Front. Syst. Neurosci. 6:2. doi: 10.3389/fnsys.2012.00002
Langguth, B., Kreuzer, P. M., Kleinjung, T., and De Ridder, D. (2013). Tinnitus: causes and clinical management. Lancet Neurol. 12, 920–930. doi: 10.1016/S1474-4422(13)70160-1
Leach, N. D., Nodal, F. R., Cordery, P. M., King, A. J., and Bajo, V. M. (2013). Cortical cholinergic input is required for normal auditory perception and experience-dependent plasticity in adult ferrets. J. Neurosci. 33, 6659–6671. doi: 10.1523/JNEUROSCI.5039-12.2013
Lee, H. J., Wallani, T., and Mendelson, J. R. (2002). Temporal processing speed in the inferior colliculus of young and aged rats. Hear Res. 174, 64–74. doi: 10.1016/S0378-5955(02)00639-1
Liberman, M. C. (1982). Single-neuron labeling in the cat auditory nerve. Science 216, 1239–1241. doi: 10.1126/science.7079757
Liberman, M. C., and Beil, D. G. (1979). Hair cell condition and auditory nerve response in normal and noise-damaged cochleas. Acta Otolaryngol. 88, 161–176. doi: 10.3109/00016487909137156
Liberman, M. C., and Dodds, L. W. (1984a). Single-neuron labeling and chronic cochlear pathology. III. Stereocilia damage and alterations of threshold tuning curves. Hear Res. 16, 55–74. doi: 10.1016/0378-5955(84)90025-X
Liberman, M. C., and Dodds, L. W. (1984b). Single-neuron labeling and chronic cochlear pathology. I. I. Stereocilia damage and alterations of spontaneous discharge rates. Hear Res. 16, 43–53. doi: 10.1016/0378-5955(84)90024-8
Lin, H. W., Furman, A. C., Kujawa, S. G., and Liberman, M. C. (2011). Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J. Assoc. Res. Otolaryngol. 12, 605–616. doi: 10.1007/s10162-011-0277-0
Ling, L. L., Hughes, L. F., and Caspary, D. M. (2005). Age-related loss of the GABA synthetic enzyme glutamic acid decarboxylase in rat primary auditory cortex. Neuroscience 132, 1103–1113. doi: 10.1016/j.neuroscience.2004.12.043
Liu, K., Jiang, X., Shi, C., Shi, L., Yang, B., Shi, L., et al. (2013). Cochlear inner hair cell Ribbon Synapse is the primary target of ototoxic aminoglycoside stimuli. Mol. Neurobiol. 48, 647–654. doi: 10.1007/s12035-013-8454-2
Llano, D. A., Turner, J., and Caspary, D. M. (2012). Diminished cortical inhibition in an aging mouse model of chronic tinnitus. J. Neurosci. 32, 16141–16148. doi: 10.1523/JNEUROSCI.2499-12.2012
Lobarinas, E., Hayes, S. H., and Allman, B. L. (2013a). The gap-startle paradigm for tinnitus screening in animal models: limitations and optimization. Hear Res. 295, 150–160. doi: 10.1016/j.heares.2012.06.001
Lobarinas, E., Salvi, R., and Ding, D. (2013b). Insensitivity of the audiogram to carboplatin induced inner hair cell loss in chinchillas. Hear Res. 302, 113–120. doi: 10.1016/j.heares.2013.03.012
Longenecker, R. J., and Galazyuk, A. V. (2012). Methodological optimization of tinnitus assessment using prepulse inhibition of the acoustic startle reflex. Brain Res. 1485, 54–62. doi: 10.1016/j.brainres.2012.02.067
Lonsbury-Martin, B. L., and Meikle, M. B. (1978). Neural correlates of auditory fatigue: frequency-dependent changes in activity of single cochlear nerve fibers. J. Neurophysiol. 41, 987–1006.
Lu, J., Lobarinas, E., Deng, A., Goodey, R., Stolzberg, D., Salvi, R. J., et al. (2011). GABAergic neural activity involved in salicylate-induced auditory cortex gain enhancement. Neuroscience 189, 187–198. doi: 10.1016/j.neuroscience.2011.04.073
Ma, W.-L. D., Hidaka, H., and May, B. J. (2006). Spontaneous activity in the inferior colliculus of CBA/J mice after manipulations that induce tinnitus. Hear. Res. 212, 9–21. doi: 10.1016/j.heares.2005.10.003
Ma, W.-L. D., and Young, E. D. (2006). Dorsal cochlear nucleus response properties following acoustic trauma: response maps and spontaneous activity. Hear Res. 216–217, 176–188. doi: 10.1016/j.heares.2006.03.011
Mahlke, C., and Wallhäusser-Franke, E. (2004). Evidence for tinnitus-related plasticity in the auditory and limbic system, demonstrated by arg3.1 and c-fos immunocytochemistry. Hear Res. 195, 17–34. doi: 10.1016/j.heares.2004.03.005
Maison, S. F., Usubuchi, H., and Liberman, M. C. (2013). Efferent feedback minimizes cochlear neuropathy from moderate noise exposure. J. Neurosci. 33, 5542–5552. doi: 10.1523/JNEUROSCI.5027-12.2013
Manzoor, N. F., Gao, Y., Licari, F., and Kaltenbach, J. A. (2013). Comparison and contrast of noise-induced hyperactivity in the dorsal cochlear nucleus and inferior colliculus. Hear Res. 295, 114–123. doi: 10.1016/j.heares.2012.04.003
Manzoor, N. F., Licari, F. G., Klapchar, M., Elkin, R. L., Gao, Y., Chen, G., et al. (2012). Noise-induced hyperactivity in the inferior colliculus: its relationship with hyperactivity in the dorsal cochlear nucleus. J. Neurophysiol. 108, 976–988. doi: 10.1152/jn.00833.2011
Martin del Campo, H. N., Measor, K. R., and Razak, K. A. (2012). Parvalbumin immunoreactivity in the auditory cortex of a mouse model of presbycusis. Hear Res. 294, 31–39. doi: 10.1016/j.heares.2012.08.017
McAlpine, D., Martin, R. L., Mossop, J. E., and Moore, D. R. (1997). Response properties of neurons in the inferior colliculus of the monaurally deafened ferret to acoustic stimulation of the intact ear. J. Neurophysiol. 78, 767–779.
McFadden, S. L., Kasper, C., Ostrowski, J., Ding, D., and Salvi, R. J. (1998). Effects of inner hair cell loss on inferior colliculus evoked potential thresholds, amplitudes and forward masking functions in chinchillas. Hear Res. 120, 121–132. doi: 10.1016/S0378-5955(98)00052-5
Melcher, J. R., Guinan, J. J., Knudson, I. M., and Kiang, N. Y. (1996). Generators of the brainstem auditory evoked potential in cat. II. Correlating lesion sites with waveform changes. Hear Res. 93, 28–51. doi: 10.1016/0378-5955(95)00179-4
Melcher, J. R., and Kiang, N. Y. (1996). Generators of the brainstem auditory evoked potential in cat. III: identified cell populations. Hear Res. 93, 52–71. doi: 10.1016/0378-5955(95)00200-6
Meredith, M. A., Keniston, L. P., and Allman, B. L. (2012). Multisensory dysfunction accompanies crossmodal plasticity following adult hearing impairment. Neuroscience 214, 136–148. doi: 10.1016/j.neuroscience.2012.04.001
Middleton, J. W., Kiritani, T., Pedersen, C., Turner, J. G., Shepherd, G. M. G., and Tzounopoulos, T. (2011). Mice with behavioral evidence of tinnitus exhibit dorsal cochlear nucleus hyperactivity because of decreased GABAergic inhibition. Proc. Natl. Acad. Sci. U.S.A. 108, 7601–7606. doi: 10.1073/pnas.1100223108
Milbrandt, J. C., and Caspary, D. M. (1995). Age-related reduction of [3H]strychnine binding sites in the cochlear nucleus of the Fischer 344 rat. Neuroscience 67, 713–719. doi: 10.1016/0306-4522(95)00082-T
Milbrandt, J. C., Holder, T. M., Wilson, M. C., Salvi, R. J., and Caspary, D. M. (2000). GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hear Res. 147, 251–260. doi: 10.1016/S0378-5955(00)00135-0
Michler, S. A., and Illing, R. B. (2002). Acoustic trauma induces reemergence of the growth- and plasticity-associated protein GAP-43 in the rat auditory brainstem. J. Comp. Neurol. 451, 250–266. doi: 10.1002/cne.10348
Moore, D. R., France, S. J., McAlpine, D., Mossop, J. E., and Versnel, H. (1997). “Plasticity of inferior colliculus and auditory cortex following unilateral deafening in adult ferrets,” in Acoustical Signal Processing in the Central Auditory System, ed J. Syka (New York, NY: Plenum), 489–499. doi: 10.1007/978-1-4419-8712-9_45
Morest, D. K., and Bohne, B. A. (1983). Noise-induced degeneration in the brain and representation of inner and outer hair cells. Hear Res. 9, 145–151. doi: 10.1016/0378-5955(83)90024-2
Morest, D. K., Kim, J., and Bohne, B. A. (1997). Neuronal and transneuronal degeneration of auditory axons in the brainstem after cochlear lesions in the chinchilla: cochleotopic and non-cochleotopic patterns. Hear Res. 103, 151–168. doi: 10.1016/S0378-5955(96)00172-4
Mühlnickel, W., Elbert, T., Taub, E., and Flor, H. (1998). Reorganization of auditory cortex in tinnitus. Proc. Natl. Acad. Sci. U.S.A. 95, 10340–10343. doi: 10.1073/pnas.95.17.10340
Mukherjea, D., Rybak, L. P., Sheehan, K. E., Kaur, T., Ramkumar, V., Jajoo, S., et al. (2011). The design and screening of drugs to prevent acquired sensorineural hearing loss. Expert Opin. Drug Discov. 6, 491–505. doi: 10.1517/17460441.2011.562887
Mulders, W. H., Ding, D., Salvi, R., and Robertson, D. (2011). Relationship between auditory thresholds, central spontaneous activity, and hair cell loss after acoustic trauma. J. Comp. Neurol. 519, 2637–2647. doi: 10.1002/cne.22644
Mulders, W. H., and Robertson, D. (2009). Hyperactivity in the auditory midbrain after acoustic trauma: dependence on cochlear activity. Neuroscience 164, 733–746. doi: 10.1016/j.neuroscience.2009.08.036
Mulders, W. H., and Robertson, D. (2011). Progressive centralization of midbrain hyperactivity after acoustic trauma. Neuroscience 192, 753–760.
Neff, W. D. (1947). The effects of partial section of the auditory nerve. J. Comp. Physiol. Psychol. 40, 203–215. doi: 10.1037/h0056931
Nienhuys, T. G., and Clark, G. M. (1978). Frequency discrimination following the selective destruction of cochlear inner and outer hair cells. Science 199, 1356–1357. doi: 10.1126/science.628846
Niskar, A. S, Kieszak, S. M., Holmes, A. E, Esteban, E., Rubin, C., and Brody, D. J. (2001). Estimated prevalence of noise-induced hearing threshold shifts among children 6 to 19 years of age: the third national health and nutrition examination survey, 1988-1994, United States. Pediatrics 108, 40–43. doi: 10.1542/peds.108.1.40
Nondahl, D. M., Cruickshanks, K. J., Huang, G.-H., Klein, B. E. K., Klein, R., Nieto, F. J., et al. (2011). Tinnitus and its risk factors in the Beaver Dam offspring study. Int. J. Audiol. 50, 313–320. doi: 10.3109/14992027.2010.551220
Nordeen, K. W., Killackey, H. P., and Kitzes, L. M. (1983). Ascending projections to the inferior colliculus following unilateral cochlear ablation in the neonatal gerbil, Meriones unguiculatus. J. Comp. Neurol. 214, 144–153. doi: 10.1002/cne.902140204
Noreña, A. J. (2011). An integrative model of tinnitus based on a central gain controlling neural sensitivity. Neurosci. Biobehav. Rev. 35, 1089–1109. doi: 10.1016/j.neubiorev.2010.11.003
Noreña, A. J., and Eggermont, J. J. (2003). Changes in spontaneous neural activity immediately after an acoustic trauma: implications for neural correlates of tinnitus. Hear Res. 183, 137–153. doi: 10.1016/S0378-5955(03)00225-9
Noreña, A. J., and Eggermont, J. J. (2006). Enriched acoustic environment after noise trauma abolishes neural signs of tinnitus. Neuroreport 17, 559–563.
Noreña, A. J., and Farley, B. J. (2013). Tinnitus-related neural activity: theories of generation, propagation, and centralization. Hear Res. 295, 161–171. doi: 10.1016/j.heares.2012.09.010
Noreña, A. J., Moffat, G., Blanc, J. L., Pezard, L., and Cazals, Y. (2010). Neural changes in the auditory cortex of awake guinea pigs after two tinnitus inducers: salicylate and acoustic trauma. Neuroscience 166, 1194–1209. doi: 10.1016/j.neuroscience.2009.12.063
Noreña, A. J., Tomita, M., and Eggermont, J. J. (2003). Neural changes in cat auditory cortex after a transient pure-tone trauma. J. Neurophysiol. 90, 2387–2401. doi: 10.1152/jn.00139.2003
Nozawa, I., Imamura, S., Fujimori, I., Hashimoto, K., Shimomura, S., Hisamatsu, K., et al. (1996). Age-related alterations in the auditory brainstem responses and the compound action potentials in guinea pigs. Laryngoscope 106, 1034–1039. doi: 10.1097/00005537-199608000-00024
Ochi, K., and Eggermont, J. J. (1996). Effects of salicylate on neural activity in cat primary auditory cortex. Hear Res. 95, 63–76. doi: 10.1016/0378-5955(96)00019-6
Oh, S.-H., Kim, C.-S., and Song, J.-J. (2007). Gene expression and plasticity in the rat auditory cortex after bilateral cochlear ablation. Acta Otolaryngol. 127, 341–350. doi: 10.1080/00016480701275246
Ohlemiller, K. K. (2004). Age-related hearing loss: the status of Schuknecht's typology. Curr. Opin. Otolaryngol. Head Neck Surg. 12, 439–443. doi: 10.1097/01.moo.0000134450.99615.22
Ouda, L., and Syka, J. (2012). Immunocytochemical profiles of inferior colliculus neurons in the rat and their changes with aging. Front. Neural Circuits 6:68. doi: 10.3389/fncir.2012.00068
Pace, E., and Zhang, J. (2013). Noise-induced tinnitus using individualized gap detection analysis and its relationship with hyperacusis, anxiety, and spatial cognition. PLoS ONE 8:e75011. doi: 10.1371/journal.pone.0075011
Palombi, P. S., and Caspary, D. M. (1996a). Physiology of the aged Fischer 344 rat inferior colliculus: responses to contralateral monaural stimuli. J. Neurophysiol. 76, 3114–3125.
Palombi, P. S., and Caspary, D. M. (1996b). Responses of young and aged Fischer 344 rat inferior colliculus neurons to binaural tonal stimuli. Hear Res. 100, 59–67. doi: 10.1016/0378-5955(96)00113-X
Parthasarathy, A., and Bartlett, E. (2012). Two-channel recording of auditory-evoked potentials to detect age-related deficits in temporal processing. Hear Res. 289, 52–62. doi: 10.1016/j.heares.2012.04.014
Parthasarathy, A., and Bartlett, E. L. (2011). Age-related auditory deficits in temporal processing in F-344 rats. Neuroscience 192, 619–630. doi: 10.1016/j.neuroscience.2011.06.042
Parthasarathy, A., Cunningham, P. A., and Bartlett, E. L. (2010). Age-related differences in auditory processing as assessed by amplitude-modulation following responses in quiet and in noise. Front. Aging Neurosci. 2:152. doi: 10.3389/fnagi.2010.00152
Pienkowski, M., and Eggermont, J. J. (2011). Cortical tonotopic map plasticity and behavior. Neurosci. Biobehav. Rev. 35, 2117–2128. doi: 10.1016/j.neubiorev.2011.02.002
Pilz, P. K. D., and Leaton, R. N. (1999). Short-term and long-term habituation of the acoustic startle response as a function of stimulus rise time in rats. Psychobiology 27, 402–414.
Popelár, J., Erre, J. P., Aran, J. M., and Cazals, Y. (1994). Plastic changes in ipsi-contralateral differences of auditory cortex and inferior colliculus evoked potentials after injury to one ear in the adult guinea pig. Hear Res. 72, 125–134. doi: 10.1016/0378-5955(94)90212-7
Potashner, S. J., Suneja, S. K., and Benson, C. G. (1997). Regulation of D-aspartate release and uptake in adult brain stem auditory nuclei after unilateral middle ear ossicle removal and cochlear ablation. Exp. Neurol. 148, 222–235. doi: 10.1006/exnr.1997.6641
Potashner, S. J., Suneja, S. K., and Benson, C. G. (2000). Altered glycinergic synaptic activities in guinea pig brain stem auditory nuclei after unilateral cochlear ablation. Hear Res. 147, 125–136. doi: 10.1016/S0378-5955(00)00126-X
Puel, J. L., Ruel, J. Gervais d'Aldin, C., and Pujol, R. (1998). Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport 9, 2109–2114. doi: 10.1097/00001756-199806220-00037
Pujol, R., and Puel, J. L. (1999). Excitotoxicity, synaptic repair, and functional recovery in the mammalian cochlea: a review of recent findings. Ann. N.Y. Acad. Sci. 884, 249–254. doi: 10.1111/j.1749-6632.1999.tb08646.x
Qiu, C., Salvi, R., Ding, D., and Burkard, R. (2000). Inner hair cell loss leads to enhanced response amplitudes in auditory cortex of unanesthetized chinchillas: evidence for increased system gain. Hear Res. 139, 153–171. doi: 10.1016/S0378-5955(99)00171-9
Rabinowitz, N. C., Willmore, B. D. B., Schnupp, J. W. H., and King, A. J. (2011). Contrast gain control in auditory cortex. Neuron 70, 1178–1191. doi: 10.1016/j.neuron.2011.04.030
Rabinowitz, N. C., Willmore, B. D. B., Schnupp, J. W. H., and King, A. J. (2012). Spectrotemporal contrast kernels for neurons in primary auditory cortex. J. Neurosci. 32, 11271–11284. doi: 10.1523/JNEUROSCI.1715-12.2012
Rachel, J. D., Kaltenbach, J. A., and Janisse, J. (2002). Increases in spontaneous neural activity in the hamster dorsal cochlear nucleus following cisplatin treatment: a possible basis for cisplatin-induced tinnitus. Hear Res. 164, 206–214. doi: 10.1016/S0378-5955(02)00287-3
Rajan, R. (1998). Receptor organ damage causes loss of cortical surround inhibition without topographic map plasticity. Nat. Neurosci. 1, 138–143. doi: 10.1038/388
Rajan, R. (2001). Plasticity of excitation and inhibition in the receptive field of primary auditory cortical neurons after limited receptor organ damage. Cereb. Cortex 11, 171–182. doi: 10.1093/cercor/11.2.171
Rajan, R., and Irvine, D. R. (1998). Absence of plasticity of the frequency map in dorsal cochlear nucleus of adult cats after unilateral partial cochlear lesions. J. Comp. Neurol. 399, 35–46. doi: 10.1002/(SICI)1096-9861(19980914)399:1%3C35::AID-CNE3%3E3.3.CO;2-D
Rajan, R., Irvine, D. R., Wise, L. Z., and Heil, P. (1993). Effect of unilateral partial cochlear lesions in adult cats on the representation of lesioned and unlesioned cochleas in primary auditory cortex. J. Comp. Neurol. 338, 17–49. doi: 10.1002/cne.903380104
Recanzone, G. H., Engle, J. R., and Juarez-Salinas, D. L. (2011). Spatial and temporal processing of single auditory cortical neurons and populations of neurons in the macaque monkey. Hear Res. 271, 115–122. doi: 10.1016/j.heares.2010.03.084
Reed, A., Riley, J., Carraway, R., Carrasco, A., Perez, C., Jakkamsetti, V., et al. (2011). Cortical map plasticity improves learning but is not necessary for improved performance. Neuron 70, 121–131. doi: 10.1016/j.neuron.2011.02.038
Richardson, B. D., Brozoski, T. J., Ling, L. L., and Caspary, D. M. (2012). Targeting inhibitory neurotransmission in tinnitus. Brain Res. 1485, 77–87. doi: 10.1016/j.brainres.2012.02.014
Richardson, B. D., Ling, L. L., Uteshev, V. V., and Caspary, D. M. (2013). Reduced GABA(A) receptor-mediated tonic inhibition in aged rat auditory thalamus. J. Neurosci. 33, 1218–1227. doi: 10.1523/JNEUROSCI.3277-12.2013
Roberts, L. E., Eggermont, J. J., Caspary, D. M., Shore, S. E., Melcher, J. R., and Kaltenbach, J. A. (2010). Ringing ears: the neuroscience of tinnitus. J. Neurosci. 30, 14972–14979. doi: 10.1523/JNEUROSCI.4028-10.2010
Roberts, L. E., Husain, F. T., and Eggermont, J. J. (2013). Role of attention in the generation and modulation of tinnitus. Neurosci. Biobehav. Rev. 37, 1754–1773. doi: 10.1016/j.neubiorev.2013.07.007
Robertson, D., Bester, C., Vogler, D., and Mulders, W. H. (2013). Spontaneous hyperactivity in the auditory midbrain: relationship to afferent input. Hear Res. 295, 124–129. doi: 10.1016/j.heares.2012.02.002
Robertson, D., and Irvine, D. R. (1989). Plasticity of frequency organization in auditory cortex of guinea pigs with partial unilateral deafness. J. Comp. Neurol. 282, 456–471. doi: 10.1002/cne.902820311
Rouiller, E. M., Wan, X. S., Moret, V., and Liang, F. (1992). Mapping of c-fos expression elicited by pure tones stimulation in the auditory pathways of the rat, with emphasis on the cochlear nucleus. Neurosci. Lett. 144, 19–24. doi: 10.1016/0304-3940(92)90706-D
Ruel, J., Chabbert, C., Nouvian, R., Bendris, R., Eybalin, M., Leger, C. L., et al. (2008). Salicylate enables cochlear arachidonic-acid-sensitive NMDA receptor responses. J. Neurosci. 28, 7313–7323. doi: 10.1523/JNEUROSCI.5335-07.2008
Rüttiger, L., Ciuffani, J., Zenner, H.-P., and Knipper, M. (2003). A behavioral paradigm to judge acute sodium salicylate-induced sound experience in rats: a new approach for an animal model on tinnitus. Hear Res. 180, 39–50. doi: 10.1016/S0378-5955(03)00075-3
Rüttiger, L., Singer, W., Panford-Walsh, R., Matsumoto, M., Lee, S. C., Zuccotti, A., et al. (2013). The reduced cochlear output and the failure to adapt the central auditory response causes tinnitus in noise exposed rats. PLoS ONE 8:e57247. doi: 10.1371/journal.pone.0057247
Ryan, A., and Dallos, P. (1975). Effect of absence of cochlear outer hair cells on behavioural auditory threshold. Nature 253, 44–46. doi: 10.1038/253044a0
Ryan, A., Dallos, P., and McGee, T. (1979). Psychophysical tuning curves and auditory thresholds after hair cell damage in the chinchilla. J. Acoust. Soc. Am. 66, 370–378. doi: 10.1121/1.383194
Rybalko, N., and Syka, J. (2005). Effect of noise exposure on gap detection in rats. Hear Res. 200, 63–72. doi: 10.1016/j.heares.2004.08.014
Salvi, R. J., and Arehole, S. (1985). Gap detection in chinchillas with temporary high-frequency hearing loss. J. Acoust. Soc. Am. 77, 1173–1177. doi: 10.1121/1.392181
Salvi, R. J., Saunders, S. S., Gratton, M. A., Arehole, S., and Powers, N. (1990). Enhanced evoked response amplitudes in the inferior colliculus of the chinchilla following acoustic trauma. Hear Res. 50, 245–257. doi: 10.1016/0378-5955(90)90049-U
Salvi, R. J., Wang, J., and Ding, D. (2000). Auditory plasticity and hyperactivity following cochlear damage. Hear Res. 147, 261–274. doi: 10.1016/S0378-5955(00)00136-2
Schaette, R. (2013). Tinnitus in men, mice (as well as other Rodents), and machines. Hear. Res. doi: 10.1016/j.heares.2013.12.004. [Epub ahead of print].
Schaette, R., and Kempter, R. (2006). Development of tinnitus-related neuronal hyperactivity through homeostatic plasticity after hearing loss: a computational model. Eur. J. Neurosci. 23, 3124–3138. doi: 10.1111/j.1460-9568.2006.04774.x
Schaette, R., and Kempter, R. (2008). Development of hyperactivity after hearing loss in a computational model of the dorsal cochlear nucleus depends on neuron response type. Hear Res. 240, 57–72. doi: 10.1016/j.heares.2008.02.006
Schaette, R., and Kempter, R. (2012). Computational models of neurophysiological correlates of tinnitus. Front. Syst. Neurosci. 6:34. doi: 10.3389/fnsys.2012.00034
Schaette, R., and McAlpine, D. (2011). Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J. Neurosci. 31, 13452–13457. doi: 10.1523/JNEUROSCI.2156-11.2011
Schaette, R., Turtle, C., and Munro, K. J. (2012). Reversible induction of phantom auditory sensations through simulated unilateral hearing loss. PLoS ONE 7:e35238. doi: 10.1371/journal.pone.0035238
Schmid, L. M., Rosa, M. G., Calford, M. B., and Ambler, J. S. (1996). Visuotopic reorganization in the primary visual cortex of adult cats following monocular and binocular retinal lesions. Cereb. Cortex 6, 388–405. doi: 10.1093/cercor/6.3.388
Schmidt, S., Redecker, C., Bruehl, C., and Witte, O. W. (2010). Age-related decline of functional inhibition in rat cortex. Neurobiol. Aging 31, 504–511. doi: 10.1016/j.neurobiolaging.2008.04.006
Schmiedt, R. A., Mills, J. H., and Boettcher, F. A. (1996). Age-related loss of activity of auditory-nerve fibers. J. Neurophysiol. 76, 2799–2803.
Schreiber, B. E., Agrup, C., Haskard, D. O., and Luxon, L. M. (2010). Sudden sensorineural hearing loss. Lancet 375, 1203–1211. doi: 10.1016/S0140-6736(09)62071-7
Schuknecht, H. F. (1964). Further observations on the pathology of presbycusis. Arch. Otolaryngol. 80, 369–382. doi: 10.1001/archotol.1964.00750040381003
Schuknecht, H. F., and Gacek, M. R. (1993). Cochlear pathology in presbycusis. Ann. Otol. Rhinol. Laryngol. 102, 1–16.
Schuknecht, H. F., and Woellner, R. C. (1953). Hearing losses following partial section of the cochlear nerve. Laryngoscope 63, 441–465. doi: 10.1288/00005537-195306000-00001
Schuknecht, H. F., and Woellner, R. C. (1955). An experimental and clinical study of deafness from lesions of the cochlear nerve. J. Laryngol. Otol. 69, 75–97. doi: 10.1017/S0022215100050465
Schwaber, M. K., Garraghty, P. E., and Kaas, J. H. (1993). Neuroplasticity of the adult primate auditory cortex following cochlear hearing loss. Am. J. Otol. 14, 252–258.
Sederholm, F., and Swedberg, M. D. B. (2013). Establishment of auditory discrimination and detection of tinnitus induced by salicylic acid and intense tone exposure in the rat. Brain Res. 1510, 48–62. doi: 10.1016/j.brainres.2013.03.013
Seki, S., and Eggermont, J. J. (2002). Changes in cat primary auditory cortex after minor-to-moderate pure-tone induced hearing loss. Hear Res. 173, 172–186. doi: 10.1016/S0378-5955(02)00518-X
Seki, S., and Eggermont, J. J. (2003). Changes in spontaneous firing rate and neural synchrony in cat primary auditory cortex after localized tone-induced hearing loss. Hear Res. 180, 28–38. doi: 10.1016/S0378-5955(03)00074-1
Sergeyenko, Y., Lall, K., Liberman, M. C., and Kujawa, S. G. (2013). Age-related cochlear synaptopathy: an early-onset contributor to auditory functional decline. J. Neurosci. 33, 13686–13694. doi: 10.1523/JNEUROSCI.1783-13.2013
Shaddock Palombi, P., Backoff, P. M., and Caspary, D. M. (2001). Responses of young and aged rat inferior colliculus neurons to sinusoidally amplitude modulated stimuli. Hear Res. 153, 174–180. doi: 10.1016/S0378-5955(00)00264-1
Shepherd, J. D., Rumbaugh, G., Wu, J., Chowdhury, S., Plath, N., Kuhl, D., et al. (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484. doi: 10.1016/j.neuron.2006.08.034
Shore, S. E., Koehler, S., Oldakowski, M., Hughes, L. F., and Syed, S. (2008). Dorsal cochlear nucleus responses to somatosensory stimulation are enhanced after noise-induced hearing loss. Eur. J. Neurosci. 27, 155–168. doi: 10.1111/j.1460-9568.2007.05983.x
Simon, H., Frisina, R. D., and Walton, J. P. (2004). Age reduces response latency of mouse inferior colliculus neurons to AM sounds. J. Acoust. Soc. Am. 116, 469. doi: 10.1121/1.1760796
Singer, W., Zuccotti, A., Jaumann, M., Lee, S. C., Panford-Walsh, R., Xiong, H., et al. (2013). Noise-induced inner hair cell ribbon loss disturbs central arc mobilization: a novel molecular paradigm for understanding tinnitus. Mol. Neurobiol. 47, 261–279. doi: 10.1007/s12035-012-8372-8
Snyder, R. L., Bonham, B. H., and Sinex, D. G. (2008). Acute changes in frequency responses of inferior colliculus central nucleus (ICC) neurons following progressively enlarged restricted spiral ganglion lesions. Hear Res. 246, 59–78. doi: 10.1016/j.heares.2008.09.010
Snyder, R. L., and Sinex, D. G. (2002). Immediate changes in tuning of inferior colliculus neurons following acute lesions of cat spiral ganglion. J. Neurophysiol. 87, 434–452.
Snyder, R. L., Sinex, D. G., McGee, J. D., and Walsh, E. W. (2000). Acute spiral ganglion lesions change the tuning and tonotopic organization of cat inferior colliculus neurons. Hear Res. 147, 200–220. doi: 10.1016/S0378-5955(00)00132-5
Spongr, V. P., Boettcher, F. A., Saunders, S. S., and Salvi, R. J. (1992). Effects of noise and salicylate on hair cell loss in the chinchilla cochlea. Arch. Otolaryngol. Head Neck Surg. 118, 157–164. doi: 10.1001/archotol.1992.01880020051015
Spongr, V. P., Flood, D. G., Frisina, R. D., and Salvi, R. J. (1997). Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J. Acoust. Soc. Am. 101, 3546–3553. doi: 10.1121/1.418315
Stamataki, S., Francis, H. W., Lehar, M., May, B. J., and Ryugo, D. K. (2006). Synaptic alterations at inner hair cells precede spiral ganglion cell loss in aging C57BL/6J mice. Hear Res. 221, 104–118. doi: 10.1016/j.heares.2006.07.014
Stolzberg, D., Chen, G.-D., Allman, B. L., and Salvi, R. J. (2011). Salicylate-induced peripheral auditory changes and tonotopic reorganization of auditory cortex. Neuroscience 180, 157–164. doi: 10.1016/j.neuroscience.2011.02.005
Stolzberg, D., Salvi, R. J., and Allman, B. L. (2012). Salicylate toxicity model of tinnitus. Front. Syst. Neurosci. 6:28. doi: 10.3389/fnsys.2012.00028
Su, Y.-Y., Luo, B., Wang, H.-T., and Chen, L. (2009). Differential effects of sodium salicylate on current-evoked firing of pyramidal neurons and fast-spiking interneurons in slices of rat auditory cortex. Hear Res. 253, 60–66. doi: 10.1016/j.heares.2009.03.007
Sun, W., Deng, A., Jayaram, A., and Gibson, B. (2012). Noise exposure enhances auditory cortex responses related to hyperacusis behavior. Brain Res. 1485, 108–116. doi: 10.1016/j.brainres.2012.02.008
Sun, W., Doolittle, L., Flowers, E., Zhang, C., and Wang, Q. (2014). High doses of salicylate causes prepulse facilitation of onset-gap induced acoustic startle response. Behav. Brain Res. 258, 187–192. doi: 10.1016/j.bbr.2013.10.024
Sun, W., Lu, J., Stolzberg, D., Gray, L., Deng, A., Lobarinas, E., et al. (2009a). Salicylate increases the gain of the central auditory system. Neuroscience 159, 325–334. doi: 10.1016/j.neuroscience.2008.12.024
Sun, X., Guo, Y. P., Shum, D. K.-Y., Chan, Y.-S., and He, J. (2009b). Time course of cortically induced fos expression in auditory thalamus and midbrain after bilateral cochlear ablation. Neuroscience 160, 186–197. doi: 10.1016/j.neuroscience.2009.02.020
Suneja, S. K., Benson, C. G., and Potashner, S. J. (1998a). Glycine receptors in adult guinea pig brain stem auditory nuclei: regulation after unilateral cochlear ablation. Exp. Neurol. 154, 473–488. doi: 10.1006/exnr.1998.6946
Suneja, S. K., and Potashner, S. J. (2003). ERK and SAPK signaling in auditory brainstem neurons after unilateral cochlear ablation. J. Neurosci. Res. 73, 235–245. doi: 10.1002/jnr.10644
Suneja, S. K., Potashner, S. J., and Benson, C. G. (1998b). Plastic changes in glycine and GABA release and uptake in adult brain stem auditory nuclei after unilateral middle ear ossicle removal and cochlear ablation. Exp. Neurol. 151, 273–288. doi: 10.1006/exnr.1998.6812
Suneja, S. K., Potashner, S. J., and Benson, C. G. (2000). AMPA receptor binding in adult guinea pig brain stem auditory nuclei after unilateral cochlear ablation. Exp. Neurol. 165, 355–369. doi: 10.1006/exnr.2000.7471
Suneja, S. K., Yan, L., and Potashner, S. J. (2005). Regulation of NT-3 and BDNF levels in guinea pig auditory brain stem nuclei after unilateral cochlear ablation. J. Neurosci. Res. 80, 381–390. doi: 10.1002/jnr.20457
Syka, J. (2002). Plastic changes in the central auditory system after hearing loss, restoration of function, and during learning. Physiol. Rev. 82, 601–636. doi: 10.1152/physrev.00002.2002
Syka, J., and Rybalko, N. (2000). Threshold shifts and enhancement of cortical evoked responses after noise exposure in rats. Hear Res. 139, 59–68. doi: 10.1016/S0378-5955(99)00175-6
Taberner, A. M., and Liberman, M. C. (2005). Response properties of single auditory nerve fibers in the mouse. J. Neurophysiol. 93, 557–569. doi: 10.1152/jn.00574.2004
Takeno, S., Harrison, R. V., Ibrahim, D., Wake, M., and Mount, R. J. (1994). Cochlear function after selective inner hair cell degeneration induced by carboplatin. Hear Res. 75, 93–102. doi: 10.1016/0378-5955(94)90060-4
Tan, J., Rüttiger, L., Panford-Walsh, R., Singer, W., Schulze, H., Kilian, S. B., et al. (2007). Tinnitus behavior and hearing function correlate with the reciprocal expression patterns of BDNF and Arg3.1/arc in auditory neurons following acoustic trauma. Neuroscience 145, 715–726. doi: 10.1016/j.neuroscience.2006.11.067
Tass, P. A., and Popovych, O. V. (2012). Unlearning tinnitus-related cerebral synchrony with acoustic coordinated reset stimulation: theoretical concept and modelling. Biol. Cybern. 106, 27–36. doi: 10.1007/s00422-012-0479-5
Threlkeld, S. W., Penley, S. C., Rosen, G. D., and Fitch, R. H. (2008). Detection of silent gaps in white noise following cortical deactivation in rats. Neuroreport 19, 893–898. doi: 10.1097/WNR.0b013e3283013d7e
Tischmeyer, W., and Grimm, R. (1999). Activation of immediate early genes and memory formation. Cell Mol. Life Sci. 55, 564–574. doi: 10.1007/s000180050315
Turner, J. G., Brozoski, T. J., Bauer, C. A., Parrish, J. L., Myers, K., Hughes, L. F., et al. (2006). Gap detection deficits in rats with tinnitus: a potential novel screening tool. Behav. Neurosci. 120, 188–195. doi: 10.1037/0735-7044.120.1.188
Turner, J., Larsen, D., Hughes, L., Moechars, D., and Shore, S. (2012). Time course of tinnitus development following noise exposure in mice. J. Neurosci. Res. 90, 1480–1488. doi: 10.1002/jnr.22827
Turner, J. G., Hughes, L. F., and Caspary, D. M. (2005). Affects of aging on receptive fields in rat primary auditory cortex layer V neurons. J. Neurophysiol. 94, 2738–2747. doi: 10.1152/jn.00362.2005
Turner, J. G., and Parrish, J. (2008). Gap detection methods for assessing salicylate-induced tinnitus and hyperacusis in rats. Am. J. Audiol. 17, S185–S192. doi: 10.1044/1059-0889(2008/08-0006)
Vogler, D. P., Robertson, D., and Mulders, W. H. (2011). Hyperactivity in the ventral cochlear nucleus after cochlear trauma. J. Neurosci. 31, 6639–6645. doi: 10.1523/JNEUROSCI.6538-10.2011
Wake, M., Takeno, S., Ibrahim, D., Harrison, R., and Mount, R. (1993). Carboplatin ototoxicity: an animal model. J. Laryngol. Otol. 107, 585–589. doi: 10.1017/S0022215100123771
Wallhäusser-Franke, E. (1997). Salicylate evokes c-fos expression in the brain stem: implications for tinnitus. Neuroreport 8, 725–728. doi: 10.1097/00001756-199702100-00029
Wallhäusser-Franke, E., Mahlke, C., Oliva, R., Braun, S., Wenz, G., and Langner, G. (2003). Expression of c-fos in auditory and non-auditory brain regions of the gerbil after manipulations that induce tinnitus. Exp. Brain Res. 153, 649–654. doi: 10.1007/s00221-003-1614-2
Walton, J. P., Frisina, R. D., and O'Neill, W. E. (1998). Age-related alteration in processing of temporal sound features in the auditory midbrain of the CBA mouse. J. Neurosci. 18, 2764–2776.
Walton, J. P., Simon, H., and Frisina, R. D. (2002). Age-related alterations in the neural coding of envelope periodicities. J. Neurophysiol. 88, 565–578.
Wang, H., Brozoski, T. J., Turner, J. G., Ling, L., Parrish, J. L., Hughes, L. F., et al. (2009a) Plasticity at glycinergic synapses in dorsal cochlear nucleus of rats with behavioral evidence of tinnitus. Neuroscience 164, 747–759. doi: 10.1016/j.neuroscience.2009.08.026
Wang, H., Turner, J. G., Ling, L., Parrish, J. L., Hughes, L. F., and Caspary, D. M. (2009b) Age-related changes in glycine receptor subunit composition and binding in dorsal cochlear nucleus. Neuroscience 160, 227–239. doi: 10.1016/j.neuroscience.2009.01.079
Wang, J., Ding, D., and Salvi, R. J. (2003). Carboplatin-induced early cochlear lesion in chinchillas. Hear Res. 181, 65–72. doi: 10.1016/S0378-5955(03)00176-X
Wang, J., Powers, N. L., Hofstetter, P., Trautwein, P., Ding, D., and Salvi, R. (1997). Effects of selective inner hair cell loss on auditory nerve fiber threshold, tuning and spontaneous and driven discharge rate. Hear Res. 107, 67–82. doi: 10.1016/S0378-5955(97)00020-8
Wang, Y., Hirose, K., and Liberman, M. C. (2002). Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J. Assoc. Res. Otolaryngol. 3, 248–268. doi: 10.1007/s101620020028
Weisz, N., Wienbruch, C., Dohrmann, K., and Elbert, T. (2005). Neuromagnetic indicators of auditory cortical reorganization of tinnitus. Brain 128, 2722–2731. doi: 10.1093/brain/awh588
White, J. A., Burgess, B. J., Hall, R. D., and Nadol, J. B. (2000). Pattern of degeneration of the spiral ganglion cell and its processes in the C57BL/6J mouse. Hear Res. 141, 12–18. doi: 10.1016/S0378-5955(99)00204-X
Willott, J. F., Aitkin, L. M., and McFadden, S. L. (1993). Plasticity of auditory cortex associated with sensorineural hearing loss in adult C57BL/6J mice. J. Comp. Neurol. 329, 402–411. doi: 10.1002/cne.903290310
Willott, J. F., and Lu, S. M. (1982). Noise-induced hearing loss can alter neural coding and increase excitability in the central nervous system. Science 216, 1331–1334. doi: 10.1126/science.7079767
Willott, J. F., Milbrandt, J. C., Bross, L. S., and Caspary, D. M. (1997). Glycine immunoreactivity and receptor binding in the cochlear nucleus of C57BL/6J and CBA/CaJ mice: effects of cochlear impairment and aging. J. Comp. Neurol. 385, 405–414. doi: 10.1002/(SICI)1096-9861(19970901)385:3%3C405::AID-CNE5%3E3.3.CO;2-%23
Willott, J. F., Parham, K., and Hunter, K. P. (1988a). Response properties of inferior colliculus neurons in young and very old CBA/J mice. Hear Res. 37, 1–14. doi: 10.1016/0378-5955(88)90073-1
Willott, J. F., Parham, K., and Hunter, K. P. (1988b). Response properties of inferior colliculus neurons in middle-aged C57BL/6J mice with presbycusis. Hear Res. 37, 15–27. doi: 10.1016/0378-5955(88)90074-3
Willott, J. F., Parham, K., and Hunter, K. P. (1991). Comparison of the auditory sensitivity of neurons in the cochlear nucleus and inferior colliculus of young and aging C57BL/6J and CBA/J mice. Hear Res. 53, 78–94. doi: 10.1016/0378-5955(91)90215-U
Yang, G., Lobarinas, E., Zhang, L., Turner, J., Stolzberg, D., Salvi, R., et al. (2007). Salicylate induced tinnitus: behavioral measures and neural activity in auditory cortex of awake rats. Hear Res. 226, 244–253. doi: 10.1016/j.heares.2006.06.013
Yang, S., Su, W., and Bao, S. (2012). Long-term, but not transient, threshold shifts alter the morphology and increase the excitability of cortical pyramidal neurons. J. Neurophysiol. 108, 1567–1574. doi: 10.1152/jn.00371.2012
Yang, S., Weiner, B. D., Zhang, L. S., Cho, S.-J., and Bao, S. (2011). Homeostatic plasticity drives tinnitus perception in an animal model. Proc. Natl. Acad. Sci. U.S.A. 108, 14974–14979. doi: 10.1073/pnas.1107998108
Yin, S.-K., Feng, Y.-M., Chen, Z.-N., and Wang, J. (2008). The effect of noise-induced sloping high-frequency hearing loss on the gap-response in the inferior colliculus and auditory cortex of guinea pigs. Hear Res. 239, 126–140. doi: 10.1016/j.heares.2008.02.002
Ying, S., Futter, M., Rosenblum, K., Webber, M. J., Hunt, S. P., Bliss, T. V. P., et al. (2002). Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 22, 1532–1540.
Yorgason, J. G., Fayad, J. N., and Kalinec, F. (2006). Understanding drug ototoxicity: molecular insights for prevention and clinical management. Expert Opin. Drug Saf. 5, 383–399. doi: 10.1517/14740338.5.3.383
Zacharek, M. A., Kaltenbach, J. A., Mathog, T. A., and Zhang, J. (2002). Effects of cochlear ablation on noise induced hyperactivity in the hamster dorsal cochlear nucleus: implications for the origin of noise induced tinnitus. Hear Res. 172, 137–143. doi: 10.1016/S0378-5955(02)00575-0
Zeng, C., Nannapaneni, N., Zhou, J., Hughes, L. F., and Shore, S. (2009). Cochlear damage changes the distribution of vesicular glutamate transporters associated with auditory and nonauditory inputs to the cochlear nucleus. J. Neurosci. 29, 4210–4217. doi: 10.1523/JNEUROSCI.0208-09.2009
Zeng, C., Yang, Z., Shreve, L., Bledsoe, S., and Shore, S. (2012). Somatosensory projections to cochlear nucleus are upregulated after unilateral deafness. J. Neurosci. 32, 15791–15801. doi: 10.1523/JNEUROSCI.2598-12.2012
Zeng, F.-G. (2013). An active loudness model suggesting tinnitus as increased central noise and hyperacusis as increased nonlinear gain. Hear Res. 295, 172–179. doi: 10.1016/j.heares.2012.05.009
Zhang, J. S., and Kaltenbach, J. A. (1998). Increases in spontaneous activity in the dorsal cochlear nucleus of the rat following exposure to high-intensity sound. Neurosci. Lett. 250, 197–200. doi: 10.1016/S0304-3940(98)00482-0
Zhang, J. S., Kaltenbach, J. A., Godfrey, D. A., and Wang, J. (2006). Origin of hyperactivity in the hamster dorsal cochlear nucleus following intense sound exposure. J. Neurosci. Res. 84, 819–831. doi: 10.1002/jnr.20985
Zhang, X., Yang, P., Cao, Y., Qin, L., and Sato, Y. (2011). Salicylate induced neural changes in the primary auditory cortex of awake cats. Neuroscience 172, 232–245. doi: 10.1016/j.neuroscience.2010.10.073
Keywords: cochlea, auditory cortex, hearing loss, neural plasticity, tinnitus, peripheral insult
Citation: Gold JR and Bajo VM (2014) Insult-induced adaptive plasticity of the auditory system. Front. Neurosci. 8:110. doi: 10.3389/fnins.2014.00110
Received: 07 March 2014; Accepted: 28 April 2014;
Published online: 23 May 2014.
Edited by:
Yukiko Kikuchi, Newcastle University Medical School, UKReviewed by:
Gregg H. Recanzone, University of California, USAWilliam Sedley, Newcastle University, UK
Copyright © 2014 Gold and Bajo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria M. Bajo, Department of Physiology, Anatomy and Genetics, Sherrington Building, Parks Road, Oxford OX1 3PT, UK e-mail:dmljdG9yaWEuYmFqb0BkcGFnLm94LmFjLnVr