 Shelton T. Mariga
Shelton T. Mariga Miriam Kolko1,2
Miriam Kolko1,2 Albert Gjedde
Albert Gjedde Linda H. Bergersen
Linda H. Bergersen- 1Department of Neuroscience and Pharmacology, University of Copenhagen, Copenhagen, Denmark
- 2Department of Ophthalmology, Roskilde Hospital, Roskilde, Denmark
- 3The Brain and Muscle Energy Group and SN-Lab, Department of Anatomy and Department of Oral Biology, Institute of Basic Medical Sciences and Centre for Molecular Biology and Neuroscience/SERTA Healthy Brain Aging Centre, University of Oslo, Oslo, Norway
Cerebral malaria (CM), caused by Plasmodium falciparum infection, is a prevalent neurological disorder in the tropics. Most of the patients are children, typically with intractable seizures and high mortality. Current treatment is unsatisfactory. Understanding the pathogenesis of CM is required in order to identify therapeutic targets. Here, we argue that cerebral energy metabolic defects are probable etiological factors in CM pathogenesis, because malaria parasites consume large amounts of glucose metabolized mostly to lactate. Monocarboxylate transporters (MCTs) mediate facilitated transfer, which serves to equalize lactate concentrations across cell membranes in the direction of the concentration gradient. The equalizing action of MCTs is the basis for lactate’s role as a volume transmitter of metabolic signals in the brain. Lactate binds to the lactate receptor GPR81, recently discovered on brain cells and cerebral blood vessels, causing inhibition of adenylyl cyclase. High levels of lactate delivered by the parasite at the vascular endothelium may damage the blood–brain barrier, disrupt lactate homeostasis in the brain, and imply MCTs and the lactate receptor as novel therapeutic targets in CM.
Cerebral Malaria
Malaria is caused by the protozoan parasite Plasmodium. About two million children die each year world-wide (Breman, 2001), mainly due to severe complications, especially from cerebral malaria (CM), of Plasmodium falciparum infection, the majority in sub-Saharan Africa, but also in Southeast Asia and South America. In developed countries, CM affects mainly returning travelers. Brain edema, lactate accumulation, and intracranial hypertension are important characteristics of CM, and untreated CM often leads to coma and death within 24 h (Newton and Krishna, 1998). CM survivors sometimes suffer from general disorders such as acidosis, as well as more specific neurological disorders such as ataxia, epilepsy, and blindness. However, the pathogenesis of CM remains unclear. Despite intense research, no effective vaccine is yet available, and the problem of drug-resistant malaria is increasing (Mariga et al., 2004).
CM from Circulating Asexual Plasmodium falciparum
The Plasmodium parasite invades erythrocytes as part of its asexual life cycle within the human host. It matures within these blood cells (Plasmodium-invaded red blood cells, PRBC) to the trophozoite and schizont stages. It then disappears from the peripheral circulation and is sequestrated in the vascular beds of critical organs such as the brain (Kristensson et al., 2013). Progression to CM is closely linked to erythrocytes containing mature stages of P. falciparum confined in the brain microvasculature (Dorovini-Zis et al., 2011) and host immunocompentent cells targeting the malaria parasite (Idro et al., 2005). Mechanisms by wish an intraerythrocytic parasite lodged in the vascular space of the brain can elicit such dramatic neurological effects necessarily involve local and systemic metabolic intermediates, as well as the transport systems for water and metabolites that link the membranes of the PRBC to the blood–brain barrier (BBB) and the end-feet of astrocytes.
Dynamics of Blood and Brain Lactate in CM
Several factors contribute to the accumulation of lactate in brain tissue in CM. The lack of a functional citric acid (TCA) cycle in human and murine Plasmodium parasites makes them largely reliant on glycolysis to fulfill their very substantial energy requirements (Sherman, 1998). Erythrocytes infected with mature parasites at the trophozoite stage consume glucose two orders of magnitude faster than normal, uninfected erythrocytes (Scheibel et al., 1979; Roth et al., 1982) with commensurate generation of lactic acid (Pfaller et al., 1982) i.e., at 5–100 times the rates of uninfected erythrocytes (Zolg et al., 1984). The resulting quantities of lactate leave the cells as directed by the concentration gradient. At rest, lactate in brain also is transported from brain tissue to plasma, but given the high rate of release of lactate from Plasmodium infected erythrocytes sequestered on the endothelial cells, lactate generated by parasites moves from the plasma to brain tissue down a concentration gradient.
Cerebral Lactate Content and Transport
The quantities of lactate produced by the parasites compete with the lactate generated in the host’s own brain metabolism for transport to or from the brain tissue. A system of transporters of lactate is present at the parasite surface as H++-coupled monocarboxylate transporters (MCTs) with features in common with members of the MCT family of higher eukaryotes (Kanaani and Ginsburg, 1991, 1992; Elliot et al., 2001). The MCT family members are well characterized in human brain (Bergersen et al., 1999, 2001; Pierre and Pellerin, 2005; Bergersen, 2007; Lauritzen et al., 2013a,b). In general, MCT1 is at the vascular endothelium, MCT4 in astrocytes, and MCT2 on neurons. Microglia expresses MCT1 and MCT2 after cerebral ischemia (Moreira et al., 2009). Both the MCTs and the lactate receptor GPR81 display affinities for L-lactate in the low mM range (Cai et al., 2008) that are consistent with the range of extracellular lactate concentrations measured by microdialysis in the brain tissue in vivo in human subjects (Abi-Saab et al., 2002; Jalloh et al., 2013).
Lactate Flux Receptor Action at the Blood–Brain Barrier
The BBB is constituted by the tight junctions of the endothelium of cerebral microvessels and capillaries. Interaction between lactate and lactate transport systems is likely to be perturbed at the BBB that is an important site of CM pathogenesis according to data from human autopsies, animal models, and in vitro cell cultures (Tripathi et al., 2007). At the BBB interfaces between the brain parenchyma and the parasite infected erythrocytes, any impairment of the integrity of endothelial cells of the cerebral microvasculature will lead to cerebral edema and lactic acidosis by the disruption of ion and solute homeostasis that follows increased membrane permeability. Normally, lactate enters the brain by first being transported through the endothelial cells and then diffusing extracellularly in the brain interstitial space and intracellularly after transport by the MCTs. Lactate down-regulates cAMP by interacting with GRP81, which is exposed on the luminal as well as the abluminal membranes of the endothelial cells and to a lesser extent on the membranes of astrocytes (Lauritzen et al., 2013a,b).
CM Association with Elevated Lactate
Association of CM with increased CSF (cerebral spine fluid) lactate is now well established. As a signaling molecule as well as a substrate for energy metabolism, brain lactate is an important diagnostic marker in clinical settings. Lactate concentrations can be used to monitor disease progress and during trials of drug effects in CM. Results from clinical settings and animal models provide convincing evidence of increased lactate concentrations both in extracellular fluid and CSF in CM. Several investigators confirmed lactate as the single best indicator of prognosis (White et al., 1985; Krishna et al., 1994) when measured in CSF of patients infected with P. falciparum (Tosti et al., 2007). In the patients, CSF concentrations correlated with the state of the disease (White et al., 1985), with death observed at levels higher than 6–9 mM lactate. Survivors always had less than 3.4 mM.
More than 80% of children from Malawi, Kenya, and Gambia with CM (Waller et al., 1991) with elevated CSF pressure revealed 40% higher lactate concentrations compared to normal (Newton et al., 1994; Potchen et al., 2012). The combination of elevated pressure and elevated lactate often is fatal (Newton et al., 1994, 1997), with most autopsies revealing high brain lactate concentrations (SenGupta and Naraqi, 1992; Walker et al., 1992). In adult Thai patients, CNS lactate in CSF at twice the normal value was associated with 100% mortality, suggestive of metabolic disturbances in the brain, indicative of a fundamental pathological process (Dorovini-Zis et al., 2011). Declining lactate during CM is associated with recovery, confirming that severe lactacidosis is a reliable indicator of brain damage, (Newton, 2005). Similar lactate profiles are observed in murine CM models, when brain lactate increases a few days post-infection (Sanni et al., 2001).
Association with Malaria Retinopathy and Lactate
Malaria retinopathy occurs in approximately two-thirds of pediatric patients with CM (Beare et al., 2004, 2006). It is characterized by retinal whitening, vessel changes and retinal hemorrhages. Correlation between malaria retinopathy and the severity of clinical CM symptoms has consistently been shown, and examination of the retina by ophthalmoscopy is useful for diagnosing CM (Beare et al., 2011). Increased plasma lactate levels are associated with the severity of retinopathy and CM, suggesting that the microvascular obstruction observed in the retina reflects systemic and cerebral microvascular obstruction and focal ischemia (Maude et al., 2009a). The eye is an easily examined, and the retina is developmentally a part of the brain and its vasculature a continuation of the cerebral vasculature. Consequently there is increasing attention on retinal pathology in malaria, with the perspective of diagnosis as well as of understanding mechanisms in CM (Maude et al., 2009b; White et al., 2009; Beare et al., 2011). Yet, the cascade of reactions involving lactate and its relation to malaria retinopathy is not fully understood.
Mouse Models of CM
The present perspective focuses on the critical role of lactate in the etiology and development of CM, although CM likely has a multifactorial pathogenesis. Murine models are critical to the study of mechanisms of underlying symptoms observed in CM (Hunt and Grau, 2003; Rénia et al., 2010; Riley et al., 2010). In humans, the signs typically encountered with CM include lactacidosis, seizures, brain edema (Polder et al., 1992; Potchen et al., 2012), and coma, followed by death (Rest, 1982; Hunt and Grau, 2003; Medana and Turner, 2006). Lactate accumulation is associated with decreased brain perfusion and brain hypoxia in a mouse model (Promeneur et al., 2013). These observations indicate intravascular obstruction and reduced perfusion of brain microvessels in CM.
To resolve the mechanisms of these changes, it is necessary to determine the cellular expression, localization and potential redistribution of the brain MCTs (MCT1, MCT2, MCT4), as well as the lactate receptor (GPR81), to reveal the dynamics of lactate’s role in CM. This is possible with the mouse models of CM, established by infection with the rodent malaria parasite P. berghei ANKA in susceptible strains, C57BL/6 (Rest, 1982) or CBA/J (Penet et al., 2005). In vivo studies of the latter model demonstrated massive brain edema coupled to lactacidosis associated with compression of cerebral arteries, ultimately causing coma and death, but no attempt was made to study the brain lactate transport systems (Penet et al., 2005). Using the CM C57BL/6 mouse model, Promeneur and collaborators (Promeneur et al., 2013) found that the water channel aquaporine 4 (AQP4) was downregulated in CM. Also, knock-out C57BL/6 mice lacking AQP4 succumbed earlier to P. berghei ANKA infection and were more severely affected than wild type mice, indicating that the water channel is protective in CM. It is particularly important to repeat these experiments in GPR81 knock-out mice to determine whether volume transmission followed by remote action at the lactate receptor is an important factor in CM.
MCT and GPR81 in CM
In the brain and retina, MCTs, like AQP4, are highly expressed in ependymal cells and astrocytes with polarized distribution in end-feet membranes facing cerebral capillaries and pia mater (Bergersen, 2007). GPR81 has a similar distribution (Lauritzen et al., 2013a,b). MCTs have also been identified in retinal sections (Bergersen et al., 1999) and unpublished results reveal GPR81 staining in human retinal sections (Kolko and Bergersen, in preparation). In addition MCT1 and GPR81 are highly expressed in the endothelial cells. The strategic locations at the blood–brain and cerebrospinal fluid–brain interfaces indicate that MCTs and GPR81 are major factors in the regulation of lactate movement into and out of the brain. Such movements were suggested already by early in vivo work (Gjedde et al., 1975) and are now firmly established (van Hall et al., 2009). Lactate receptor actions have been demonstrated in brain, reducing cAMP (apparent IC50 29 mM; Lauritzen et al., 2013a,b) and neuronal calcium spiking (apparent IC50 4.2 mM; Bozzo et al., 2013) through GPR81 activation. In addition, lactate stimulates locus coeruleus neurons (apparent IC50 0.7 mM; Tang et al., 2014), ascribed to a receptor other than GPR81. These observations and the volume transmitter role of lactate (Bergersen and Gjedde, 2012), whereby lactate diffuses through large volumes of brain tissue to produce distant receptor effects, therefore imply actions of lactate produced by malaria parasites as important factors in CM.
Epileptic seizures occur in more than 70% of CM patients on admission (Newton et al., 2000; Idro et al., 2010). Elevated lactate concentrations in the brain are highly correlated with the severity of repeated seizures commonly observed in CM. Similarly, seizures in temporal lobe epilepsy (TLE) with hippocampal sclerosis, known as mesial temporal lobe epilepsy (MTLE), are correlated with high lactate levels as observed by microdialysis in epileptic patients (During et al., 1994). Brain MCTs expressions and distributions might be influenced by multiple forms of brain pathology including MTLE, stroke, meningitis, neuromyelitis optica, and brain tumors (Bergersen, 2007; Lauritzen et al., 2011, 2012, 2013a,b). Observations in MTLE and other neuropathological disorders resemble in some respects, the CM-related clinical and pathological status (i.e., seizures, lactacidosis). However, the role of lactate and the receptor action and transport in CM and other disease states need to be clarified.
Hypothesis of a New Factor in CM
Based on the extant evidence, we claim that the lactate transporters and the lactate receptor GPR81 together are an essential factor in the pathophysiology of CM. The most obvious scenario (Figure 1) under this hypothesis is that lactate, profusely released from Plasmodium infected erythrocytes sequestered at the capillary endothelium, excessively down-regulates cAMP via GPR81. High concentrations of lactate will reach receptors both at the luminal and abluminal surface of the endothelial cells, because of the presence of MCT1 (Lauritzen et al., 2011) as well as GPR81 (Lauritzen et al., 2013a,b) at both sides of the endothelium. Normally as well as in disease, cAMP is perhaps the most potent signaling molecule to stabilize the endothelial barrier, and also regulates inflammation response and vascular tone (Roberts and Dart, 2014; Schlegel and Waschke, 2014). It acts on three classes of proteins: protein kinase A (PKA), cyclic-nucleotide-gated ion channels, and Epacs (exchange proteins directly activated by cAMP). The Epacs constitute a family of guanine-nucleotide-exchange factors for the small Ras-related GTPases Rap1 and Rap2. Both PKA and Rap1 can cause Rac1-mediated strengthening of adherens junctions and of the actin cytoskeleton (Schlegel and Waschke, 2014).
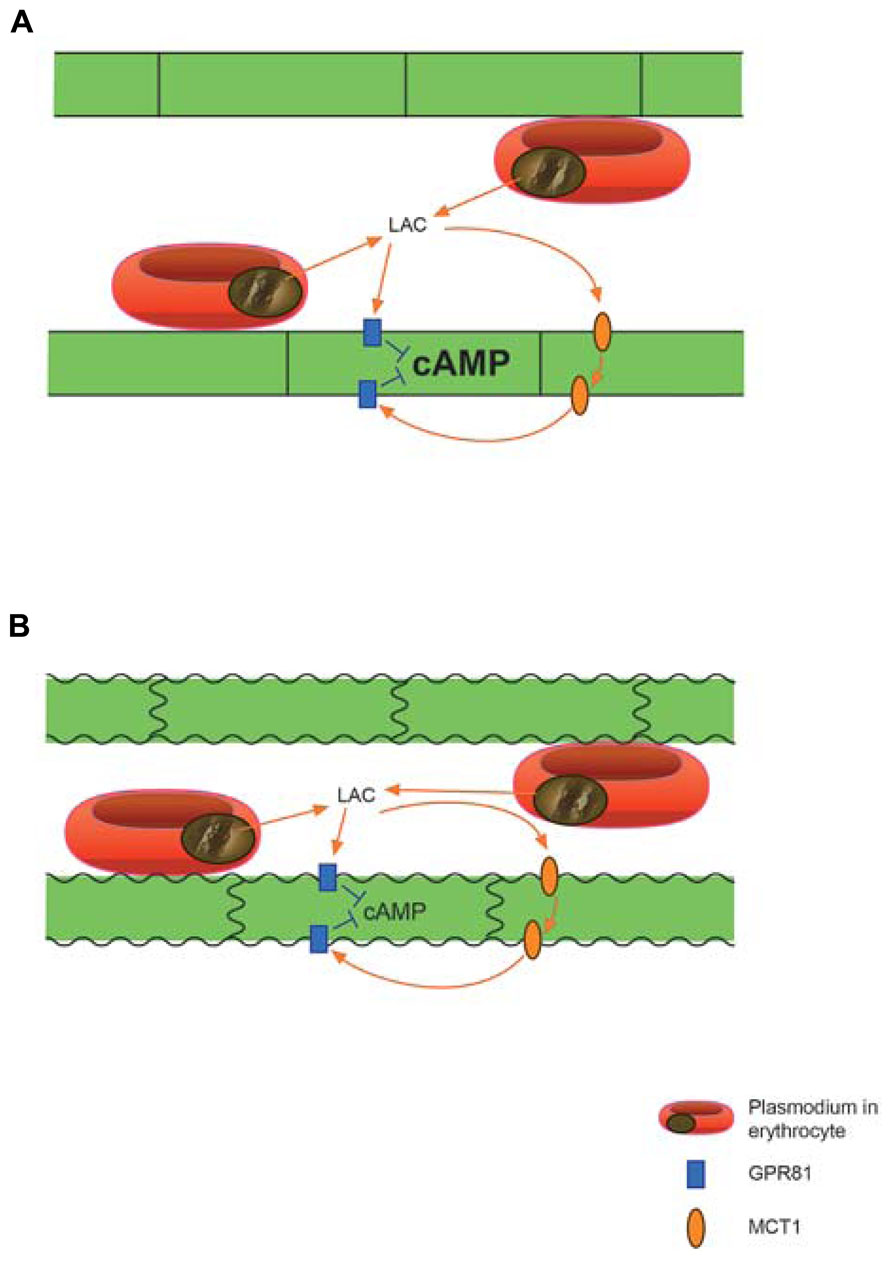
FIGURE 1. Proposed action of extreme quantities of Plasmodium derived lactic acid on brain vascular endothelium. (A) Lactate penetrating the endothelial cells through MCT1 activates the lactate receptor GPR81, which is situated on both sides of the endothelium, to cause down-regulation of cAMP by inhibiting adenylyl cyclase activity. (B) Reduced levels of cAMP can lead to vasoconstriction and endothelial dysfunction with weakening of cell–cell junctions ultimately causing break-down of the blood-brain-barrier. Not shown: the GPR81 mediated effects may act in concert with pH effects of lactic acid and with other deleterious agents from the protozoon. Lactate penetrating the endothelium may also act on GPR81 expressed on neurons and to a lesser degree on astrocytic perivascular end-feet.
Extreme down-regulation of cAMP can therefore be expected to cause deterioration of the endothelium, including break-down of regulated trans-endothelial transport and of the junctions between endothelial cells, i.e., break-down of the BBB (Figure 1). In addition, increased vascular tone (Roberts and Dart, 2014) may aggravate the hypoperfusion caused by clogging of microvessels by sequestered erythrocytes. The effects via GPR81, and possibly other lactate receptors (cf. Tang et al., 2014), may converge with effects of acidification caused by proton from the parasite produced lactic acid, as well as with other deleterious compounds originating in the Plasmodium infected erythrocytes (Aird et al., 2014).
Within the brain parenchyma, excessive lactate concentrations delivered by the parasites must be expected to perturb the volume transmitter function of lactate (Bergersen and Gjedde, 2012), although the concentration of lactate will decrease with the distance from the parasites, which are confined to the vasculature. The vascular end-feet processes of astrocytes have relatively low GPR81 concentrations, while the synaptic membranes of excitatory synapses have the highest concentrations of GPR81 (Lauritzen et al., 2013a,b), but are at a greater distance from the parasites. The effects of excessive lactate in the parenchyma are harder to predict than those on the BBB, but they are expected to be exacerbated in CM, because lactate induced down-regulation of cAMP will upregulate MCT1 at the endothelial plasma membrane (Smith et al., 2012).
Inflammatory responses through the innate immune system may serve to confine the infection or cause serious tissue damage, the relative weight of these effects depending on genetic characteristics of the Plasmodium and of the host (Wu et al., 2014). Careful dissection of the mechanisms may reveal ways to augment antiparasite mechanisms but suppress tissue damage. A recent paper (Hoque et al., 2014) found that lactate by activating GPR81 reduced tissue injury in disease models. The fact that this effect was mediated down stream through arrestin beta-2 rather than cAMP predicts the possibility of separately modifying deleterious and beneficial effects of GPR81 activation.
Our hypothesis may be tested in mouse models of CM. The role of GPR81 can be ascertained by inducing CM in GPR81 knock-out mice. This will reveal whether the lack of the lactate receptor confers resistance to CM and/or ameliorates the pathology and outcome, in which case GPR81 antagonists would offer novel treatment for CM. GPR81 antagonists are not available at present, but may soon be. Further, MCT1 blockers, which are presently introduced in cancer therapy (Parks et al., 2013; Draoui et al., 2014) may slow the release of lactate from the infected erythrocytes and its transfer across the endothelium. If so, they would offer another novel therapy against CM. The existence of MCT blockers that selectively inhibit lactate influx but not efflux (Draoui et al., 2014) would allow the action to be restricted to the luminal side of the endothelium, avoiding possibly deleterious actions of the drugs on the brain parenchyma, which may also be avoided by using GPR81 and MCT antagonists that are hydrophilic and do not penetrate the BBB. The hypothesis can be further tested by analysis of MCT and GPR81 mRNAs and protein expressions and by immunohistochemistry by light and electron microscopy of the brain in CM mice and non-CM controls. It is particularly important to examine the hippocampal formation, in view of its central role in epileptogenesis and in view of the demonstrated roles of MCTs in human epilepsy and animal models of TLE (Lauritzen et al., 2013a,b). In addition, retinal sections must be examined to identify the expression pattern of MCTs and GPR81 in CM mice. The possible impact of antimalarial drugs on lactate transport and receptor activity should be tested.
Conclusion
Studies on lactate transport and receptor action in CM are expected to provide important new insights into the pathogenesis of CM, and to identify novel therapeutic targets.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study has been supported by grants from the University of Copenhangen and the Lundbeck Foundation, Denmark, and from the University of Oslo and the Research Council of Norway.
References
Abi-Saab, W. M., Maggs, D. G., Jones, T., Jacob, R., Srihari, V., Thompson, J., et al. (2002). Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: effects of hyperglycemia and hypoglycemia. J. Cereb. Blood Flow Metab. 22, 271–279. doi: 10.1097/00004647-200203000-00004
Aird, W. C., Mosnier, L. O., and Fairhurst R. M. (2014). Plasmodium falciparum picks (on) EPCR. Blood 123, 163–167. doi: 10.1182/blood-2013-09-521005
Beare, N. A. V., Lewallen, S., Taylor, T. E., and Molyneux, M. E. (2011). Redefining cerebral malaria by including malaria retinopathy. Future Microbiol. 6, 349–355. doi: 10.2217/fmb.11.3
Beare, N. A. V., Southern, C., Chalira, C., and Taylor, T. E. (2004). Prognostic significance and course of retinopathy in children with severe malaria. Arch. Ophthalmol. 122, 1141–1147. doi: 10.1001/archopht.122.8.1141
Beare, N. A. V., Taylor, T. E., Harding, S. P., Lewallen, S., and Molyneux, M. E. (2006). Malarial retinopathy: a newly established diagnostic sign in severe malaria. Am. J. Trop. Med. Hyg. 75, 790–797.
Bergersen, L. H. (2007). Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 145, 11–19. doi: 10.1016/j.neuroscience.2006.11.062
Bergersen, L. H., and Gjedde, A. (2012). Is lactate a volume transmitter of metabolic states of the brain? Front. Neuroenergetics 4:5. doi: 10.3389/fnene.2012.00005
Bergersen, L., Jóhannsson, E., Veruki, M. L., Nagelhus, E. A., Halestrap, A., Sejersted, O. M., et al. (1999). Cellular and subcellular expression of monocarboxylate transporters in the pigment epithelium and retina of the rat. Neuroscience 90, 319–331. doi: 10.1016/S0306-4522(98)00427-8
Bergersen, L., Wærhaug, O., Helm, J., Thomas, M., Laake, P., Davies, A. J., et al. (2001). A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp. Brain Res. 136, 523–534. doi: 10.1007/s002210000600
Bozzo, L., Puyal, J., and Chatton, J. Y. (2013). Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE 8:e71721. doi: 10.1371/journal.pone.0071721
Breman, J. G. (2001). The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am. J. Trop. Med. Hyg. 64(Suppl. 12), 1–11.
Cai, T. Q., Ren, N., Jin, L., Cheng, K., Kash, S., Chen, R., et al. (2008). Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem. Biophys. Res. Commun. 377, 987–991. doi: 10.1016/j.bbrc.2008.10.088
Dorovini-Zis, K., Schmidt, K., Huynh, H., Fu, W., Whitten, R. O., Milner, D., et al. (2011). The neuropathology of fatal cerebral malaria in Malawian children. Am. J. Pathol. 178, 2146–2158. doi: 10.1016/j.ajpath.2011.01.016
Draoui, N., Schicke, O., Seront, E., Bouzin, C., Sonveaux, P., Riant, O., et al. (2014). Antitumor activity of 7-aminocarboxycoumarin derivatives, a new class of potent inhibitors of lactate influx but not efflux. Mol. Cancer Ther. doi: 10.1158/1535-7163.MCT-13-0653 [Epub ahead of print].
During, M. J., Fried, I., Leone, P., Katz, A., and Spencer, D. D. (1994). Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J. Neurochem. 62, 2356–2361. doi: 10.1046/j.1471-4159.1994.62062356.x
Elliot, J. L., Saliba, K. J., and Kirk, K. (2001). Transport of lactate and pyruvate in the intraerythrocytic malaria parasite, Plasmodium falciparum. Biochem. J. 355, 733–739.
Gjedde, A., Andersson, J., and Eklöf, B. (1975). Brain uptake of lactate, antipyrine, water and ethanol. Acta Physiol. Scand. 93, 145–149. doi: 10.1111/j.1748-1716.1975.tb05802.x
Hoque, R., Farooq, A., Ghani, A., Gorelick, F., and Mehal, W. Z. (2014). Lactate reduces organ injury in toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology doi: 10.1053/j.gastro.2014.03.014 [Epub ahead of print].
Hunt, N. H., and Grau, G. E. (2003). Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 24, 491–499. doi: 10.1016/S1471-4906(03)00229-1
Idro, R., Jenkins, N. E., and Newton, C. R. (2005). Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 4, 827–840. doi: 10.1016/S1474-4422(05)70247-7
Idro, R., Marsh, K., John, C. C., and Newton, C. R. J. (2010). Cerebral malaria; mechanisms of brain injury and strategies for improved neuro-cognitive outcome. Pediatr. Res. 68, 267–274. doi: 10.1203/PDR.0b013e3181eee738
Jalloh, I., Helmy, A., Shannon, R. J., Gallagher, C. N., Menon, D. K., Carpenter, K. L., et al. (2013). Lactate uptake by the injured human brain: evidence from anarteriovenous gradient and cerebral microdialysis study. J. Neurotrauma 30, 2031–2037. doi: 10.1089/neu.2013.2947
Kanaani, J., and Ginsburg, H. (1991). Transport of lactate in Plasmodium falciparum-infected human erythrocytes. J. Cell. Physiol. 149, 469–476. doi: 10.1002/jcp.1041490316
Kanaani, J., and Ginsburg, H. (1992). Effects of cinnamic acid derivatives on in vitro growth of Plasmodium falciparum and on the permeability of the membrane of malaria-infected erythrocytes. Antimicrob. Agents Chemother. 36, 1102–1108. doi: 10.1128/AAC.36.5.1102
Krishna, S., Waller, D. W., ter Kuile, F., Kwiatkowski, D., Crawley, J., Craddock, C. F., et al. (1994). Lactic acidosis and hypoglycaemia in children with severe malaria: pathophysiological and prognostic significance. Trans. R. Soc. Trop. Med. Hyg. 88, 67–73. doi: 10.1016/0035-9203(94)90504-5
Kristensson, K., Masocha, W., and Bentivoglio, M. (2013). Mechanisms of CNS invasion and damage by parasites. Handb. Clin. Neurol. 114, 11–22. doi: 10.1016/B978-0-444-53490-3.00002-9
Lauritzen, F., de Lanerolle, N. C., Lee, T. S., Spencer, D. D., Kim, J. H., Bergersen, L. H., et al. (2011). Monocarboxylate transporter 1 is deficient on microvessels in the human epileptogenic hippocampus. Neurobiol. Dis. 41, 577–584. doi: 10.1016/j.nbd.2010.11.005
Lauritzen, F., Heuser, K., de Lanerolle, N. C., Lee, T. W., Spencer, D. D., Kim, J. H., et al. (2012). Redistribution of monocarboxylate transporter 2 on the surface of astrocytes in the human epileptogenic hippocampus. Glia 60, 0894–1491.
Lauritzen, F., Eid, T., and Bergersen, L. H. (2013a). Monocarboxylate transporters in temporal lobe epilepsy: roles of lactate and ketogenic diet. Brain Struct. Funct. doi: 10.1007/s00429-013-0672-x [Epub ahead of print].
Lauritzen, K. H., Morland, C., Puchades, M., Holm-Hansen, S., Hagelin, E. M., Lauritzen F., et al. (2013b). Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex doi: 10.1093/cercor/bht136 [Epub ahead of print].
Mariga, S. T., Gil, J. P., Sisowath, C., Wernsdorfer, W. H., and Björkman, A. (2004). Synergism between amodiaquine and Its major metabolite, desethylamodiaquine, against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 48, 4089–4096. doi: 10.1128/AAC.48.11.4089-4096.2004
Maude, R. J., Beare, N. A. V., Abu, S. A., Chang, C. C., Charunwatthana, P., Faiz, M. A., et al. (2009a). The spectrum of retinopathy in adults with Plasodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 103, 665–671. doi: 10.1016/j.trstmh.2009.03.001
Maude, R. J., Dondorp, A. M., Abu, S. A., Day, N. P. J., White, N. J., and Beare, N. A. V. (2009b). The eye in cerebral malaria: what can it teach us? Trans. R. Soc. Trop. Med. Hyg. 103, 661–664. doi: 10.1016/j.trstmh.2008.11.003
Medana, I. M., and Turner, G. D. (2006). Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 36, 555–568. doi: 10.1016/j.ijpara.2006.02.004
Moreira, T. J., Pierre, K., Maekawa, F., Repond, C., Cebere, A., Liljequist, S., et al. (2009). Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J. Cereb. Blood Flow Metab. 29, 1273–1283. doi: 10.1038/jcbfm.2009.50
Newton, C. R. (2005). Interaction between Plasmodium falciparum and human immunodeficiency virus type 1 on the central nervous system of African children. J. Neurovirol. 3, 45–51. doi: 10.1080/13550280500511881
Newton, C. R., Crawley, J., Sowumni, A., Waruiru, C., Mwangi, I., English, M., et al. (1997). Intracranial hypertension in Africans with cerebral malaria. Arch. Dis. Child. 76, 219–226. doi: 10.1136/adc.76.3.219
Newton, C. R. J., Hien, T. T., and White, N. (2000). Neurological aspects of tropical disease Cerebral malaria J. Neurol. Neurosurg. Psychiatry 69, 433–441. doi: 10.1136/jnnp.69.4.433
Newton, C. R., and Krishna, S. (1998). Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol. Ther. 79, 1–53. doi: 10.1016/S0163-7258(98)00008-4
Newton, C. R., Peshu, N., Kendall, B., Kirkham, F. J., Sowunmi, A., Waruiru, C., et al. (1994). Brain swelling and ischaemia in Kenyans with cerebral malaria. Arch. Dis. Child. 70, 281–287. doi: 10.1136/adc.70.4.281
Parks, S. K., Chiche, J., and Pouysségur, J. (2013). Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 13, 611–623. doi: 10.1038/nrc3579
Pierre, K., and Pellerin, L. (2005). Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. 94, 1–14. doi: 10.1111/j.1471-4159.2005.03168.x
Penet, M. F., Viola, A., Confort-Gouny, S., Le Fur, Y., Duhamel, G., Kober, F., et al. (2005). Imaging experimental cerebral malaria in vivo: significant role of ischemic brain edema. J. Neurosci. 25, 7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005
Pfaller, M. A., Krogstad, D. J., Parquette, A. R., and Nguyen-Dinh, P. (1982). Plasmodium falciparum: stage-specific lactate production in synchronized cultures. Exp. Parasitol. 54, 391–396. doi: 10.1016/0014-4894(82)90048-0
Polder, T. W., Eling, W. M., Curfs, J. H., Jerusalem, C. R., and Wijers-Rouw, M. (1992). Ultrastructural changes in the blood-brain barrier of mice infected with Plasmodium berghei. Acta Leiden 60, 31–46.
Potchen, M. J., Kampondeni, S. D., Seydel, K. B., Birbeck, G. L., Hammond, C. A., Bradley, W. G., et al. (2012). Acute brain MRI findings in 120 malawian children with cerebral malaria: new insights into an ancient disease. AJNR Am. J. Neuroradiol. 33, 1740–1746. doi: 10.3174/ajnr.A3035
Promeneur, D., Lunde, L. K., Amiry-Moghaddam, M., and Agre, P. (2013). Protective role of brain water channel AQP4 in murine cerebral malaria. Proc. Natl. Acad. Sci. U.S.A. 110, 1035–1040. doi: 10.1073/pnas.1220566110
Rénia, L., Grüner, A. C., and Snounou, G. (2010). Cerebral malaria: In praise of epistemes. Trends Parasitol. 26, 275–277. doi: 10.1016/j.pt.2010.03.005
Rest, J. R. (1982). Cerebral malaria in inbred mice. a new model and its pathology. Trans. R. Soc. Trop. Med. Hyg. 76, 410–415. doi: 10.1016/0035-9203(82)90203-6
Riley, E. M., Couper, K. N., Helmby, H., Julius, C. R., Hafalla, J. C. R., de Souza, J. B., et al. (2010). Neuropathogenesis of human and murine malaria. Trends Parasitol. 26, 277–278. doi: 10.1016/j.pt.2010.03.002
Roth, E. F. Jr., Raventos-Suarez, C., Perkins, M., and Nagel, R. L. (1982). Glutathione stability and oxidative stress in P. falciparum infection in vitro: responses of normal and G6PD deficient cells. Biochem. Biophys. Res. Commun. 109, 355–362. doi: 10.1016/0006-291X(82)91728-4
Roberts, O. L., and Dart, C. (2014). cAMP signalling in the vasculature: the role of Epac (exchange protein directly activated by cAMP). Biochem. Soc. Trans. 42, 89–97. doi: 10.1042/BST20130253
Sanni, L. A., Rae, C., Maitland, A., Stocker, R., and Hunt, N. H. (2001). Is ischemia involved in the pathogenesis of murine cerebral malaria? Am. J. Pathol. 159, 1105–1112. doi: 10.1016/S0002-9440(10)61786-5
Scheibel, L. W., Adler, A., and Trager, W. (1979). Tetraethylthiuram disulfide (Antabuse) inhibits the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 76, 5303–5307. doi: 10.1073/pnas.76.10.5303
Schlegel, N., and Waschke, J. (2014). cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 355, 587–596. doi: 10.1007/s00441-013-1755-y
SenGupta, S. K., and Naraqi, S. (1992). The brain in cerebral malaria: a pathological study of 24 fatal cases in Papua New Guinea. P. N. G. Med. J. 35, 270–274.
Sherman, I. W. (1998). Malaria: Parasite Biology, Pathogenesis, and Protection. Washington, DC: ASM Press.
Smith, J. P., Uhernik, A. L., Li, L., Liu, Z., and Drewes, L. R. (2012). Regulation of Mct1 by cAMP-dependent internalization in rat brain endothelial cells. Brain Res. 1480, 1–11. doi: 10.1016/j.brainres.2012.08.026
Tang, F., Lane, S., Korsak, A., Paton, J. F., Gourine, A. V., Kasparov, S., et al. (2014). Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat. Commun. 5:3284. doi: 10.1038/ncomms4284
Tosti, C. L., Petersen, E. T., Laothamatas, J., Golay, X., Swaminathan, S. V., Van Cauteren, M., et al. (2007). Cerebrospinal fluid lactate in P. falciparum malaria: measurement by chemical shift imaging at 3 Tesla. Proc. Intl. Soc. Mag. Reson. Med. 15:2262.
Tripathi, A. K., Sullivan, D. J., and Stins, M. F. (2007). Plasmodium falciparum–infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J. Infect. Dis. 195, 942–950. doi: 10.1086/512083
van Hall, G., Strømstad, M., Rasmussen, P., Jans, Ø., Zaar, M., Gam, C., et al. (2009). Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 29, 1121–1129. doi: 10.1038/jcbfm.2009.35
Waller, D., Waller, D., Crawley, J., Nosten, F., Chapman, D., Krishna, S., et al. (1991). Intracranial pressure in childhood cerebral malaria. Trans. R. Soc. Trop. Med. Hyg. 85, 362–364. doi: 10.1016/0035-9203(91)90291-6
Walker, O., Salako, L. A., Sowunmi, A., Thomas, J. O., Sodeine, O., and Bondi, F. S. (1992). Prognostic risk factors and post mortem findings in cerebral malaria in children. Trans. R. Soc. Trop. Med. Hyg. 86, 491–493. doi: 10.1016/0035-9203(92)90082-N
White, N. J., Looareesuwan, S., Phillips, R. E., Warrell, D. A., Chanthavanich, P., and Pongpaew, P. (1985). Pathophysiological and prognostic significance of cerebrospinal fluid lactate in cerebral malaria. Lancet 325, 776–778. doi: 10.1016/S0140-6736(85)91445-X
White, V. A., Lewallen, S., Beare, N. A. V., Molyneux, M. E., and Taylor, T. E. (2009). Retinal pathology of pediatric cerebral malaria in Malawi. PLoS ONE 4:e4317. doi: 10.1371/journal.pone.0004317
Wu, J., Tian, L., Yu, X., Pattaradilokrat, S., Li, J., Wang, M., et al. (2014). Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. U.S.A. 111, E511–E520. doi: 10.1073/pnas.1316467111
Keywords: energy metabolism, cerebral malaria, lactate transport, lactate receptor, volume transmitter
Citation: Mariga ST, Kolko M, Gjedde A and Bergersen LH (2014) Lactate transport and receptor actions in cerebral malaria. Front. Neurosci. 8:2. doi: 10.3389/fnins.2014.00125
Received: 13 March 2014; Accepted: 22 April 2014;
Published online: 19 May 2014.
Edited by:
Avital Schurr, University of Louisville, USAReviewed by:
Oliver Kann, University of Heidelberg, GermanyJuan P. Bolanos, University of Salamanca, Spain
Leif Hertz, China Medical University, China
Copyright © 2014 Mariga, Kolko, Gjedde and Bergersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda H. Bergersen, The Brain and Muscle Energy Group and SN-Lab, Department of Anatomy and Department of Oral Biology, Institute of Basic Medical Sciences and Centre for Molecular Biology and Neuroscience/SERTA Healthy Brain Aging Centre, University of Oslo, N-0317 Oslo, Norway e-mail:bC5oLmJlcmdlcnNlbkBtZWRpc2luLnVpby5ubw==