 René Günther1†
René Günther1† Kim-Ann Saal1Martin Suhr1David Scheer1
Kim-Ann Saal1Martin Suhr1David Scheer1 Jan Christoph Koch1
Jan Christoph Koch1 Mathias Bähr1,2
Mathias Bähr1,2 Paul Lingor1,2
Paul Lingor1,2 Lars Tönges1,2*
Lars Tönges1,2*- 1Department of Neurology, University Medicine Göttingen, Göttingen, Germany
- 2Nanoscale Microscopy and Molecular Physiology of the Brain, Cluster of Excellence 171–DFG Research Center 103 (CNMPB), Göttingen, Germany
Disease progression in amyotrophic lateral sclerosis (ALS) is characterized by degeneration of motoneurons and their axons which results in a progressive muscle weakness and ultimately death from respiratory failure. The only approved drug, riluzole, lacks clinical efficacy so that more potent treatment options are needed. We have identified rho kinase (ROCK) as a target, which can be manipulated to beneficially influence disease progression in models of ALS. Here, we examined the therapeutic potential of the ROCK inhibitor Y-27632 in both an in vitro and in an in vivo paradigm of motoneuron disease. Application of Y-27632 to primary motoneurons in vitro increased survival and promoted neurite outgrowth. In vivo, SOD1G93A mice were orally treated with 2 or 30 mg/kg body weight of Y-27632. The 2 mg/kg group did not benefit from Y-27632 treatment, whereas treatment with 30 mg/kg resulted in improved motor function in male mice. Female mice showed only limited improvement and overall survival was not modified in both 2 and 30 mg/kg Y-27632 groups. In conclusion, we provide evidence that inhibition of ROCK by Y-27632 is neuroprotective in vitro but has limited beneficial effects in vivo being restricted to male mice. Therefore, the evaluation of ROCK inhibitors in preclinical models of ALS should always take gender differences into account.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neuromuscular disease which is characterized by degeneration of the first and second motoneuron resulting in severe progressive muscle weakness and most often death from respiratory insufficiency within only a few years after initial diagnosis. It is known to be a neurodegenerative process that comprises the loss of motoneuronal cells, of their axons and neuromuscular junctions but also non-neuronal cell populations contribute to the progression of disease. Because first pathological signs of motoneuron degeneration are observed at the distal end of its axonal projections some authors consider ALS to be a distal axonopathy (Fischer and Glass, 2007). The molecular and pathophysiological mechanisms of the disorder are multifactorial and include protein aggregation, oxidative stress, excitotoxicity, mitochondrial dysfunction, and impaired axonal transport (Ferraiuolo et al., 2011). Moreover, there is a strong involvement of glial cells such as astrocytes, microglia and oligodendrocytes, which may determine the onset and the dynamics of disease progression (Philips and Robberecht, 2011).
Apart from the majority of sporadic forms, there exist at least 10% of familial forms of ALS (fALS). Among these, genetic mutations in the super oxide dismutase gene 1 (SOD1) were identified first and today represent about 20% of all familial forms (Rosen et al., 1993). Recently, many other familial cases have been identified which exhibit mutations e.g., in Fused in sarcoma (FUS), TAR DNA-binding protein 43 (TDP-43) or exhibit an intronic hexanucleotide repeat in the gene C9orf72. The C9orf72 repeat expansion is considered to be the most frequent genetic alteration in fALS patients but has also been identified in many patients with sporadic ALS (sALS). Therefore, longstanding concepts of the cause and molecular pathology in ALS are currently re-evaluated (Turner et al., 2013).
The fALS gene mutations have permitted the generation of transgenic mouse models of ALS. Because it closely recapitulates human disease progression the SOD1G93A mouse model has evolved as a standard model for the evaluation of therapeutic effects in preclinical studies (Turner and Talbot, 2008). A variety of neuroprotective agents have been examined to both delay the onset of clinical disease and to prolong the disease course ultimately extending life span in animal models, but only the glutamate antagonist riluzole has reached clinical use so far (Benatar, 2007). However, this agent has very limited benefits in ALS patients (Stewart et al., 2001).
Rho kinases (ROCK) are serine/threonine kinases that regulate cytoskeleton structure through phosphorylation of LIM kinases, myosin light chain or the ezrin/radixin/moesin protein complex (Tonges et al., 2011). Our group and others have shown that inhibition of ROCK with pharmacological small molecule inhibitors is not only able to improve the regenerative response in the lesioned CNS (Lingor et al., 2008; Planchamp et al., 2008; Bermel et al., 2009), but also exerts neuroprotection via activation of intracellular cell survival signaling such as the Akt pathway (Tonges et al., 2012; Takata et al., 2013). In addition, we have recently demonstrated that the use of fasudil, a small-molecule ROCK inhibitor of the isoquinoline series, strongly modulates microglial function in SOD1G93A female mice which may be a major component of the therapeutic effect (Tonges et al., 2014).
Based on these results, we have designed both an in vitro and an in vivo study to evaluate the neuroprotective potential of ROCK inhibition with a more specific ROCK inhibitor, the 4-aminopyridine derivative Y-27632.
Materials and Methods
Motoneuron Preparation
Lumbar spinal cord mouse motoneurons (MN) of E11.5–E13.5 embryos of wild type (WT) mice (B6/SJL background) were generated applying a preparation technique adapted from Wiese et al. (2010). In the case of later immunoblot and cell toxicity analysis the panning step was omitted in order to obtain a higher cell yield. In all cases, cells were collected and seeded on poly-L-ornithine/laminin coated cover slips in MN complete medium containing Neurobasal medium without (2)L-Glutamine (Invitrogen, Darmstadt, Germany), supplemented with 2% horse serum (Linaris, Wertheim, Germany), B27-supplement (Invitrogen, Darmstadt, Germany), BDNF (final concentration 10 ng/mL; Tebu Bio, Offenbach, Germany), and CNTF (final concentration 10 ng/mL; Tebu Bio, Offenbach, Germany) at a density of 25,000 cells/cm2.
Motoneuron Culture, Quantification of Cellular Survival, Cytotoxicity and Neurite Length
MN were cultured in MN complete medium and supplemented with Y-27632 (final concentration 10 μM; Sigma-Aldrich, St. Louis, Mo) or the respective amount of vehicle every second day. Additionally, BDNF and CNTF (final concentration 10 ng/mL, both Tebu Bio) were supplemented every second day. Motoneuron survival was assessed by counting ChAT-immunopositive cells after fixation and immunocytochemistry on DIV4.
Cytotoxicity assays were also done on DIV4. Here, a bioluminescence-based assay for the release of adenylate kinase (AK) from lesioned cells was applied according to the manufacturer's instructions (ToxiLight®, Lonza, Wakersville, USA). Briefly, the amount of adenylate kinase (AK) was determined in the culture medium by measuring the AK-dependent conversion of ADP to ATP and subsequent light emission by luciferase with a luminometer (Wallac 1450 MicroBeta Trilux, PerkinElmer, Shelton, USA).
The length of all neurites of ChAT-immunopositive cells was evaluated semi-automatically using the axon tracing module of “Image J” (Free Java software provided by the National Institutes of Health, Bethesda, Maryland, USA) and was divided by the numbers of ChAT-immunopositive cells in order to obtain neurite length/cell. Results were expressed in relation to vehicle treated cells. The immunofluorescence-based quantification of intracellular ROCK2 protein was done by measuring mean fluorescence intensity values in MN perikarya with “ImageJ.” For MN survival at least three and for neurite outgrowth at least two independent experiments were evaluated. The quantification of perikaryal ROCK2 was done in at least 6 MN per treatment condition.
Immunocytochemistry
For MN immunolabeling, cells were fixed in PFA 4% for 10 min at room temperature (RT, 22°C), permeabilized with 100% ice-cold acetone (AppliChem, Darmstadt, Germany) 10 min at −20°C, washed twice with PBS and blocked with 10% normalized goat serum 10 min at RT. Probes were incubated with the primary antibodies (rabbit anti ChAT 1:50, Millipore, Schwalbach, Germany; goat anti-ROCK2 1:50, Santa Cruz Biotechnology Inc., Heidelberg, Germany) for 1 h at 37°C or were fixed in PFA 4% for 10 min at room temperature (RT, 22°C), washed twice with PBS, incubated 30 min in 25 mM Glycine in PBS (Applichem, Darmstadt, Germany), permeabilized and blocked with 10% horse serum, 5% BSA, 0,3% Triton, 25 mM Glycine in PBS at RT for 1 h and then incubated with primary antibodies (rabbit anti ChAT 1:50, Millipore, Schwalbach, Germany; mouse anti-MAP2 1:500 Chemicon/Millipore, Schwalbach, Germany) over night at 4°C.
Following three PBS washes, Cy3- or Cy2-labeled secondary antibodies (1:250, Dianova, Hamburg, Germany) were applied for 1 h at room temperature. After another three PBS washes, cells were then nuclear counter-stained with DAPI (4,6-diamidino-2-phenylindole) (Sigma, Taufkirchen, Germany), optionally incubated with additional Rhodamine-Phalloidin 1:500 in PBS (Invitrogen, Eugene, Oregon, USA) and mounted in Mowiol (Hoechst, Frankfurt, Germany). Immunolabeled fluorescent cells were imaged on a Zeiss Axioplan 2 fluorescence microscope equipped with a CCD camera and AxioVision software (Zeiss, Göttingen, Germany). For evaluation of survival and neurite outgrowth of ChAT-immunopositive cells, micrographs were taken with a 10× objective of at least four random visual fields per culture well and of at least two wells per condition.
Motoneuron Cell Culture Lysis and Immunoblotting
Motoneuron cultures were plated at a density of 25,000 cells/cm2, cultured in MN complete medium and supplemented with Y-27632 (final concentration 10 μM; Sigma-Aldrich, St. Louis, Mo) or the respective amount of vehicle every second day. After 5 days cells were lysed with a cell lysis buffer (Thermo Scientific™ Pierce™ RIPA Buffer, Thermo Fisher Scientific Inc., Waltham, MA USA) plus protease inhibitors (“Complete tablets,” Roche, Basel, Switzerland) and phosphatase inhibitor (“PhosSTOP,” Roche, Basel, Switzerland). The protein content of all cell samples was determined using Bradford (Biorad, Munich, Germany) and equal amounts of protein (20 μg) were separated on a sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Proteins were then electrotransferred onto a PVDF membrane (Applichem, Darmstadt, Germany) and blocked with 5% milk in Tris-buffered saline/Tween-20 (TBS-T) for 1 h. Membranes were then incubated with the primary antibodies goat anti-ROCK2 1:50 (Santa Cruz Biotechnology Inc., Heidelberg, Germany) and mouse anti-GAPDH 1:1000 (HyTest Ltd, Turku, Finland) in 5% milk TBS-T or 5% BSA TBS-T over night at 4°C. This was followed by incubation with corresponding horseradish peroxidase-coupled secondary antibodies (1:1000 for 1 h at room temperature; Dianova, Hamburg, Germany). ECL-Plus reagent (Amersham, Arlington Heights, IL, USA) was applied on the membrane and the chemiluminescence was visualized on an Amersham Hyperfilm ECL (GE Healthcare, Chalfont St Giles, GB).
Animal Housing, Breeding, Genotyping and Application of Y-27632
All animal experiments were carried out according to the regulations of the local animal research council and legislation of the State of Lower Saxony. High-copy number B6/SJL-Tg(SOD1*G93A)1Gur/J (labeled as SOD1G93A in the following text) (Gurney et al., 1994) were obtained from Jackson Labs (Stock Number 002726; Bar Harbor, USA). The colony was maintained by crossing B6/SJL males harboring the transgene with wild-type B6/SJL females. Housing of animals was performed under a 12 h light/12 h dark cycle with free access to food and water.
For genotyping, tail biopsies of 14-day-old pups were subjected to a standardized PCR protocol (Jackson Labs). Probe sequences were: hSOD1-forward, CATCAGCCCTAATCCATCTGA; hSOD1-reverse, CGCGACTAACAATCAAAGTGA; Interleukin 2-forward, CTAGGCCACAGAATTGAAAGATCT, Interleukin 2-reverse, GTAGGTGGAAATTCTAGCATCATCC).
Y-27632 was administered at a concentration of 2 mg/kg body weight per day (subsequently termed as Y2) or at a concentration of 30 mg/kg body weight per day (Y30) in drinking water according to previous studies with Y-27632 in animal disease models (Nagaoka et al., 2005). Control groups received drinking water without supplementation (Veh).
Clinical and Behavioral Animal Testing
Experimental groups
At day of life 50 (d50) mice were allocated to the different treatment groups. In a first trial a Y-27632 dose of 2 mg/kg (Y2) was compared with vehicle (Veh). In a second trial a Y-27632 dose of 30 mg/kg (Y30) was compared with vehicle (Veh). Both trials were performed in an observer-blinded fashion and groups were constituted to minimize inter-group variability by matching of animals with respect to body weight, age and litter.
Assessment of neurological score, body weight and survival
Neurological scores and body weight were assessed every 3 days for each mouse from 50 days of age. The neurological score employed a scale of 0 (worst) to 4 (best) (Weydt et al., 2003). Mice showing an inability to right themselves 30 s after being placed on a side or having a 25% loss of their initial body weight were scored as “dead,” and were euthanized using carbon dioxide.
Rotarod test
After training for three consecutive times of at least 180 s at a constant speed of 15 r.p.m. the time for which an animal could remain on the rotating rod (Ugo-Basile, Comerio, Italy) was measured (Gunther et al., 2012). The mean of three trials was recorded for each animal.
Hanging wire test
Animals were placed on a custom-made wire lid (0.8 cm spacing) and cautiously turned upside down, 60 cm above a straw-covered bottom (Gunther et al., 2012). After training for three consecutive times of at least 180 s the latency to fall was measured. The mean of three trials was recorded.
Statistical Analysis
For in vitro experiments, the comparison between two groups was carried out using an unpaired Student's t-test. Behavioral data were subjected to a multifactorial ANOVA analysis and post hoc Fisher's test. Survival data were analyzed using Kaplan-Meier survival fit analysis with Log-Rank tests for statistical significance. Statistical analyses were performed using Statistica 10 (StatSoft, Hamburg, Germany) and Kyplot 2.0 (KyensLab Inc, Tokyo, Japan). Data are represented as mean ± standard error of the mean. Significances are *P < 0.05; **P < 0.01; ***P < 0.001.
Results
Pharmacological Inhibition of ROCK with Y-27632 Increases Survival and Promotes Neurite Outgrowth of Motoneurons in vitro
Based on findings that ROCK inhibition is able to attenuate cell death in CNS neurons we examined the neuroprotective potential of Y-27632 in primary MN in dissociated cell culture. In comparison to the number of surviving MN after 4 days in vitro in the vehicle treated culture which was set to 100% (100.0 ± 5.3%), the Y-27632 treated culture showed significantly higher numbers of MN (123.7 ± 9.5%) (Figure 1A). This protective effect was also observed in a cytotoxicity assay in which the release of adenylate kinase was reduced after Y-27632 treatment (Supplementary Figure 1).
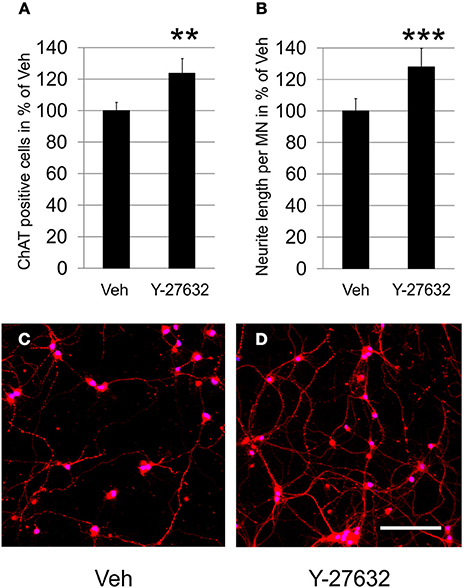
Figure 1. Y-27632 increases survival of dissociated MN and promotes neurite outgrowth. (A) Relative numbers of ChAT-immunopositive MN per visual field on DIV4 in cultures treated with Veh or with Y-27632 (10 μM) (n = 3 MN cultures, bars represent means ± s.e.m.; **P < 0.005, according to unpaired Student's t-test). (B) Relative neurite length per ChAT-immunopositive MN treated as in (A) (n = 2 MN cultures, bars represent means ± s.e.m.; ***P < 0.001, according to unpaired Student's t-test). (C,D) Representative photomicrographs of MN immunolabeled with ChAT (Cy3, red) and treated with Veh (C) or Y-27632 (10 μM) (D) (nuclear counter-stain with DAPI, blue). Scale bar, 200 μm.
In the same cultures we measured the total neurite net and calculated neurite length per MN in order to remain independent from MN cell numbers. Compared to the vehicle control treatment, which was set to 100% (100.0 ± 7.7%), application of 10 μM Y-27632 resulted in a significantly increased neurite length of ChAT-immunopositive motoneurons (128.0 ± 11.8%) (Figures 1B–D). In a dose-escalating experiment we observed that a 10 μM final dose of Y-27632 was superior to 5-fold lower or higher doses, which were not significantly promoting neurite outgrowth (Supplementary Figure 2).
The presence of ROCK2 in motoneurons was demonstrated both immunocytochemically in ChAT immunopositive cells and by immunoblot of cell culture lysates. Here, treatment with Y-27632 was not found to influence the amount of ROCK2 protein being expressed. However, the application of Y-27632 resulted in altered growth cone morphology with prolonged processes if compared to vehicle treated cells (Figure 2).
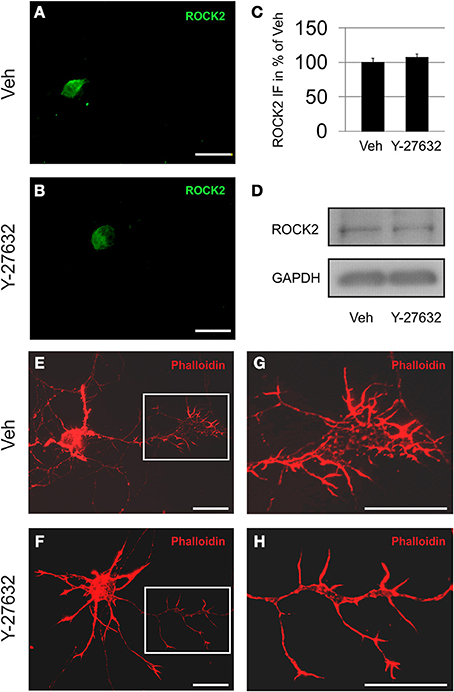
Figure 2. Motoneurons express ROCK2 protein. Depicted are MN treated with vehicle (A,E,G) or Y-27632 (10 μM) (B,F,H). (A,B) ROCK2 is localized primarily in the cytoplasm in both vehicle and Y-27632-treated MN. (C) Quantification of perikaryal ROCK2 immunofluorescence (IF) in MN (n = 6 cells, bars represent means ± s.e.m., according to unpaired Student's t-test). (D) Immunoblots of motoneuron cell cultures for ROCK2 and GAPDH. (E,F) MN harbor high amounts of f-actin (rhodamine-phalloidin, red). (G,H) Magnifications of growth cone areas as depicted in (E,F). Scale bar, 25 μm.
Weight Dynamics and Survival of Y-27632 Treated Mice
In a proof-of-principle approach, we used the SOD1G93A mouse model to assess the effects of oral Y-27632 treatment with 2 mg/kg body weight (Y2) and 30 mg/kg body weight (Y30) compared against vehicle (Veh) in a presymptomatic trial design starting at day of life 50 (d50).
Female mice, which have been treated with Y30, had a slightly higher weight in an advanced age, which was, however, not reaching significance. The Y2 female treatment group was not different from vehicle control (Figures 3A–D). Male animals treated with Y2 did not gain as much maximal weight as controls. The Y30 male treatment group was not different from the control (Figures 3E–H).
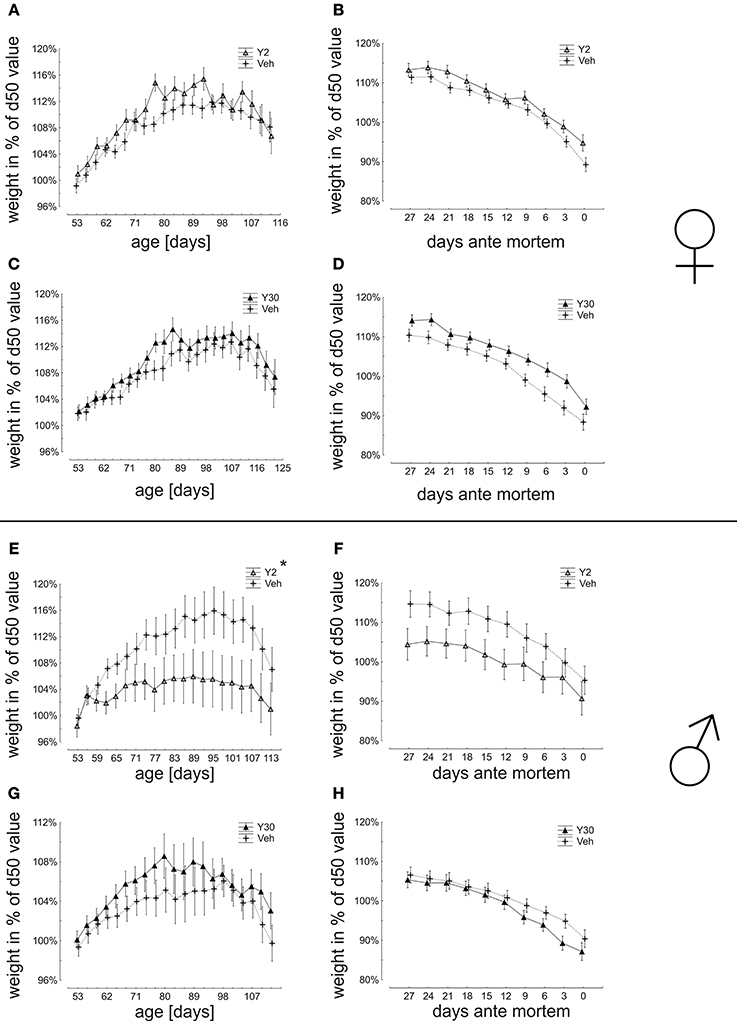
Figure 3. Weight dynamics in relation to body weight at day 50 (start of presymptomatic treatment) in SOD1G93A mice treated with 2 or 30 mg/kg body weight Y-27632. (A–D) Weight dynamics in female mice treated with Y-27632 from d50 in a dosage of 2 mg/kg (A,B) (Y2 n = 13; Veh n = 18) or 30 mg/kg (C,D) (Y30 n = 13; Veh n = 12). (E–H) Weight dynamics in male mice treated with Y-27632 from d50 in a dosage of 2 mg/kg (E,F) (Y2 n = 10; Veh n = 14) or 30 mg/kg (G,H) (Y30 n = 8; Veh n = 8). Statistics was done applying multifactorial ANOVA and post-hoc Fisher's test. *P < 0.05.
The analysis of mean survival times did not show a significant improvement by Y-27632 treatment for any group (females: Veh 132,8 ± 1,9 days vs. Y2 130,8 ± 2,4 days; Veh 141,2 ± 2,5 days vs. Y30 143,9 ± 3,4 days; males: Veh 125,9 ± 1,9 days vs. Y2 125,5 ± 2,2 days; Veh 132 ± 3,5 days vs. Y30 133,5 ± 2,6 days) (Figure 4).
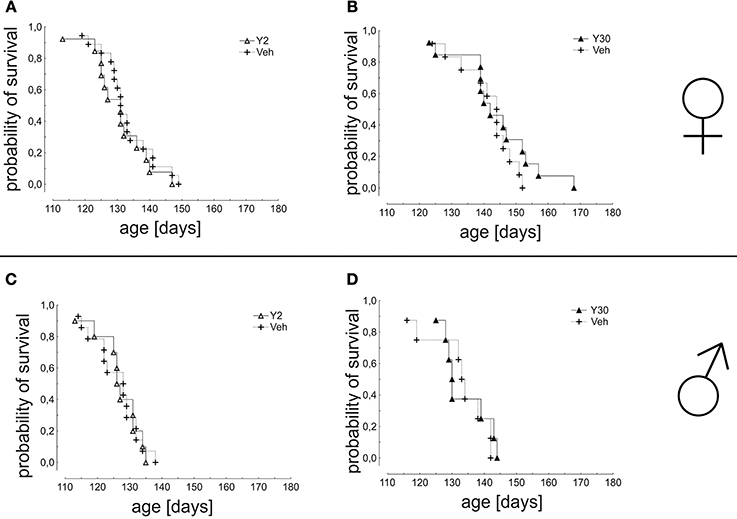
Figure 4. Survival in SOD1G93A mice treated with 2 or 30 mg/kg body weight Y-27632. Depicted is a Kaplan-Meier curve for cumulative probability of survival for female mice treated with 2 mg/kg (A) (Y2 n = 13; Veh n = 18) or 30 mg/kg Y-27632 (B) (Y30 n = 13; Veh n = 12) and for male mice treated with 2 mg/kg (C) (Y2 n = 10; Veh n = 14) or 30 mg/kg Y-27632 (D) (Y30 n = 8; Veh n = 8). Days ante mortem refers to the time point before death of an individual animal measured in days. Statistics was done applying Kaplan-Meier survival fit analysis with Log-Rank test.
Neurological Scoring and Motor Behavior are Improved in Male Mice Treated with High Dose of Y-27632
In a detailed analysis of disease progression based on the neurological score we found nearly no significant differences between Y-27632 and Veh-treated groups. A significant effect was, however, seen in Y30 treated male mice which showed improved values for the neurological score from d107 until d122 (Figure 5).
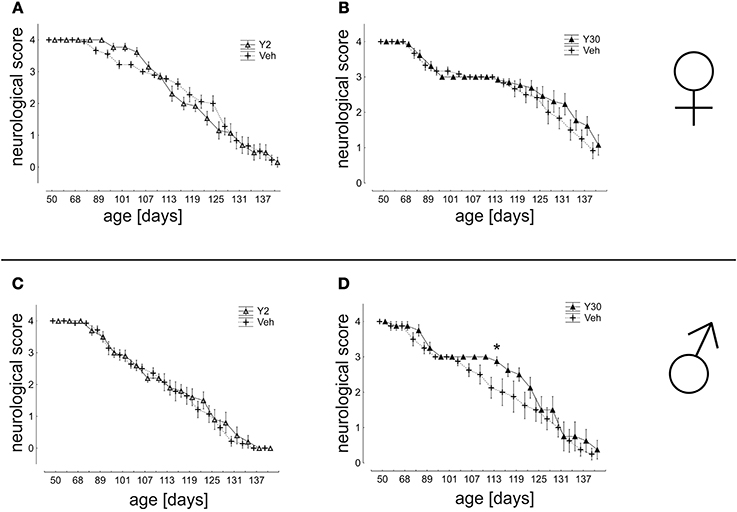
Figure 5. Neurological score in SOD1G93A mice treated with 2 or 30 mg/kg body weight Y-27632. (A,B) Depicted are scores for female mice treated with Y-27632 from d50 in a final dosage of 2 mg/kg (A) (Y2 n = 13; Veh n = 18) or 30 mg/kg (B) (Y30 n = 13; Veh n = 12). (C,D) Scores for male mice treated with Y-27632 from d50 in a final dosage of 2 mg/kg (C) (Y2 n = 10; Veh n = 14) or 30 mg/kg (D) (Y30 n = 8; Veh n = 8). Statistics was done applying multifactorial ANOVA and post-hoc Fisher's test. *P < 0.05.
In the analysis of motor coordination and muscle strength as assessed by the ability to remain on a rotating rod, a significant advantage was again detected for male mice treated with Y30 in the time period from d80 until d113 (d104: Veh 114,04 ± 30,24 s; Y30 168,17 ± 11,83 s; d107: Veh 117,13 ± 26,94 s; Y30 173,31 ± 6,69 s; d110: Veh 70,67 ± 22,90 s; Y30 145,79 ± 16,97 s; d113 Veh 50,29 ± 21,19 s; Y30 105,56 ± 20,31 s). Similarly, in the analysis of muscle strength measured by hanging wire the vehicle group performed worse in comparison to the Y30 male group, however, without reaching significance. Animals treated with Y2 did not exhibit improved motor behavior nor did any of the female cohorts (Figure 6).
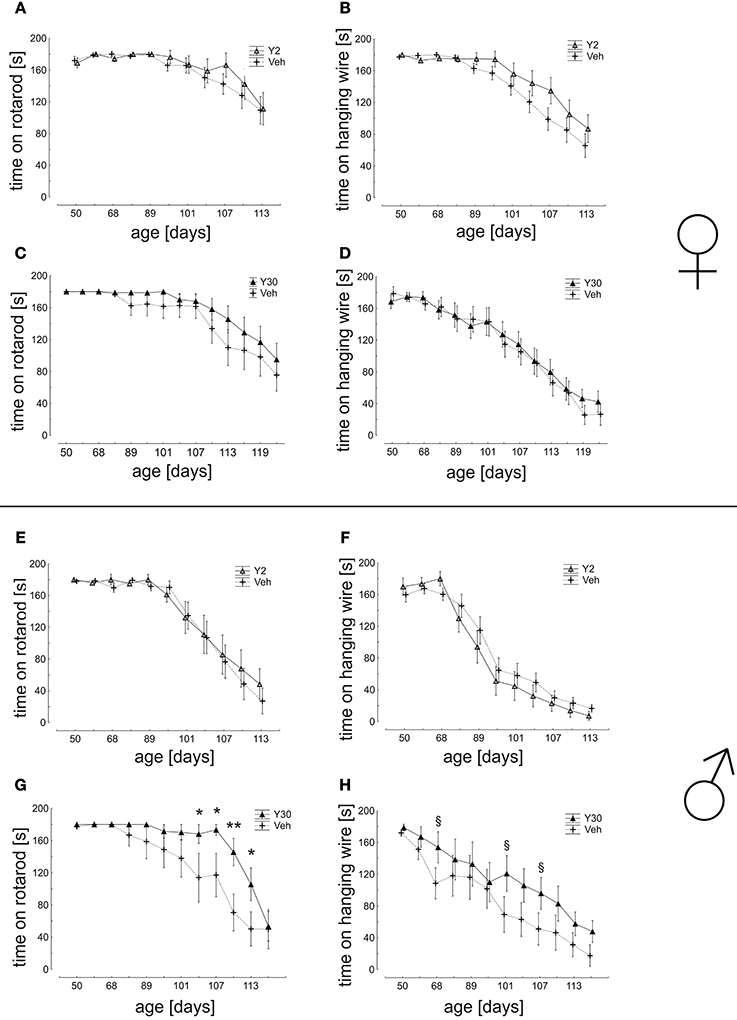
Figure 6. Motor behavior in SOD1G93A mice treated with 2 or 30 mg/kg body weight Y-27632. (A–D) Mean time of endurance on the rotarod and in the hanging wire test for female mice treated with Y-27632 from d50 in a dosage of 2 mg/kg (A,B) (Y2 n = 13; Veh n = 18) or 30 mg/kg (C,D) (Y30 n = 13; Veh n = 12). (E–H) Scores for male mice treated with Y-27632 from d50 in a dosage of 2 mg/kg (E,F) (Y2 n = 10; Veh n = 14) or 30 mg/kg (G,H) (Y30 n = 8; Veh n = 8). Statistics was done applying multifactorial ANOVA and post-hoc Fisher's test; *P < 0.05; **P < 0.01; in H: § d68 p = 0.12; § d101 p = 0.08; § d107 p = 0.12.
Discussion
Motoneuron death and axonal degeneration are critical pathogenic features in the development of human ALS (Fischer et al., 2004). ROCK inhibition by fasudil was previously shown to exert neuroprotective, but also pro-regenerative and axon-stabilizing effects, but fasudil is a rather unspecific drug in comparison to the more specific ROCK inhibitor Y-27632. Therefore, we have now evaluated the therapeutic potential of orally applied Y-27632 in the SOD1G93A mouse model of ALS.
Pharmacological Inhibition of ROCK Protects Motoneurons in Culture
ROCK inhibitors have been applied successfully in a multitude of cell death paradigms. They have shown to attenuate apoptotic cell death of retinal ganglion cells (Lingor et al., 2008; Tura et al., 2009) and prevent dopaminergic cell loss in toxin-based models of Parkinson's disease (Tonges et al., 2012). Recently, Y-27632 was also shown to protect hippocampal neurons against neuronal cell death induced by glutamate in vitro and by kainic acid in vivo (Jeon et al., 2013).
In a model of kainate-induced glutamate excitotoxicity, our group was able to demonstrate that the ROCK inhibitor fasudil significantly increased the number of surviving MN (Tonges et al., 2014). This neuroprotective effect was now also observed for Y-27632, which reduced the loss of motoneurons in dissociated cell culture. Furthermore, neurite outgrowth was promoted on a permissive substrate—an effect, which is well known for ROCK inhibitors if applied to neuronal cells (Lingor et al., 2008; Tonges et al., 2012).
However, it has to be taken into account that there seems to be an optimal dosage window for Y-27632 in vitro—as is known for other ROCK inhibitors, too—because final concentrations of 2 or 50 μM, being fivefold lower or higher than the optimal 10 μM, were not able to elicit a significant outgrowth promoting effect of MN anymore. This could be due to the inhibition of protein kinases other than ROCK which are known for both Y-27632 and fasudil—however, to a much lower extent for Y-27632. In fact, a concentration of 20 μM fasudil is needed to reduce ROCK-II activity in vitro to 7% of control and a concentration of 10 μM Y-27632 is needed to reduce ROCK-II activity in vitro to 13%. The concentrations of both compounds required for 50% inhibition (IC50) of ROCK2 are 1900 nM for fasudil and 800 nM for Y-27632. Most importantly, the activities of several other kinases are much stronger reduced with fasudil (20 μM) (MAPKAP-K1b 37%, MSK1: 19%, PKA: 35%, S6K1 32%) than with Y-27632 (10 μM) (MAPKAP-K1b: 72%, MSK1: 57%; PKA: 91%, S6K1 94%), which may elicit neurobiological side effects albeit being difficult to predict.
The Disease Course in SOD1G93A Mice is only Transiently Improved by Y-27632 Treatment
In the evaluation of clinical symptoms of disease, we found only a transient improvement for male mice with the high-dose Y30 treatment, which correlated with the transient improvement in motor performance. In contrast, survival of SOD1G93A mice was not significantly prolonged in this study in any group.
In a previous study of our group, we had orally applied the more unspecific ROCK inhibitor fasudil to SOD1G93A mice starting at postnatal day 50 (d50) (Tonges et al., 2014). Here, survival was significantly prolonged with fasudil 30 mg/kg and by trend with fasudil 100 mg/kg. The smaller effect by fasudil 100 mg/kg could be due to a U-shaped dose-response relationship, which we have observed before in vitro (Lingor et al., 2007; Tonges et al., 2012). Similar effects of fasudil on survival in the SOD1G93A model have been reported by another group who found a prosurvival response, which was attributed to an attenuation of ROCK activity and an increased level of phospho-Akt (Takata et al., 2013).
Other trials with Y-27632 in models of neurodegenerative disease had shown more favorable results. In a model of Huntington's disease, the ROCK inhibitor Y-27632 improved rotarod performance without influencing survival time at a dosage of 100 mg/kg (Li et al., 2009). The two ROCK inhibitors Y-27632 (30 mg/kg) and fasudil (30 mg/kg) were also beneficially applied in mouse models of SMA, in which they prolonged survival and preserved the integrity of the neuromuscular junction (NMJ). Interestingly, the effect on survival was more pronounced with fasudil than with Y-27632 (Bowerman et al., 2010, 2012).
Overall, the application of Y-27632 was less successful in the SOD1G93A mouse model than that of fasudil. Since 30 mg/kg body weight showed only significant effects on motor function, we cannot exclude that an even higher dosage would be more beneficial. It could also be that the differential inhibitory profile toward other kinases is responsible for the lack of efficacy (Davies et al., 2000). Thus, some of these “off-target” effects on other kinases by fasudil may have been even beneficial to a certain extent in this ALS model.
Gender-Differences of ROCK Inhibition with Y-27632 in the SOD1G93A Model
It is known that female mice have longer survival times in the SOD1G93A mouse model than males. This fact has also been observed in our study. However, a minor trend to improved survival with Y-27632 is only seen in females treated with 30 mg/kg Y-27632 but not in male mice. Interestingly, the transient improvement of motor function was detected only in male mice on a significant level and was observed only by trend in female mice. This could be due to a more aggressive disease course associated with increased ROCK activity in intermediate stages in male mice, which then benefit more from the Y-27632 application. Supportive of this hypothesis is a study that has been performed in the MPTP model of Parkinson's disease in female mice. If these mice were ovariectomized, an increased loss of dopaminergic neurons after MPTP toxicity was observed, which was shown to be associated with an increase of ROCK activity. Thus, higher ROCK activity correlated to increased vulnerability of dopaminergic neurons to MPTP. When Y-27632 was applied in this model, the increased loss of dopaminergic cells was reversed being as effective as an estrogen-replacement therapy (Rodriguez-Perez et al., 2013).
Conclusion
The role of ROCK and the therapeutic potential of its inhibition are increasingly studied in models of neurodegenerative disease. However, findings obtained with one ROCK inhibitor may not be generalized to the entire class of ROCK inhibitors, which show high chemical heterogeneity. Whereas the application of fasudil was successfully promoting survival of SOD1G93A mice in two independent studies, this was not found for Y-27632 in two different dosages in the present paradigm. Suboptimal targeting of the therapeutic window as well as the differential inhibition of other protein kinases may contribute to these differences. As most studies have been performed in female mice, a gender-dependent responsiveness to ROCK inhibition may play an additional role. Our study thus supports the need for thorough evaluation of ROCK inhibitors in models of neurodegenerative disease in order to better characterize their effects and thereby optimally prepare their translation into human clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants of the Research Support Program of the University Medicine Göttingen to Lars Tönges. and the Else Kröner Fresenius-Stiftung to Paul Lingor. Paul Lingor and Mathias Bähr were supported by the DFG-Cluster of Excellence Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), Göttingen. The authors thank Elisa Barski and Vivian Dambeck for expert technical assistance. We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fnins.2014.00304/abstract
Abbreviations
ANOVA, analysis of variance; MAPKAP-K1b, MAPK-activated protein kinase-1b; MN, motoneuron; MSK1, mitogen- and stress-activated protein kinase 1; PKA, cAMP-dependent protein kinase; ROCK, rho kinase; S6K1, p70 ribosomal protein S6 kinase; SMA, spinal muscular atrophy; SOD1G93A, superoxide dismutase 1 G93A transgenic; Veh, vehicle; Y2, Y-27632 2 mg/kg body weight; Y30, Y-27632 30 mg/kg body weight.
References
Benatar, M. (2007). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13. doi: 10.1016/j.nbd.2006.12.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bermel, C., Tonges, L., Planchamp, V., Gillardon, F., Weishaupt, J. H., Dietz, G. P., et al. (2009). Combined inhibition of Cdk5 and ROCK additively increase cell survival, but not the regenerative response in regenerating retinal ganglion cells. Mol. Cell. Neurosci. 42, 427–437. doi: 10.1016/j.mcn.2009.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bowerman, M., Beauvais, A., Anderson, C. L., and Kothary, R. (2010). Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 19, 1468–1478. doi: 10.1093/hmg/ddq021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bowerman, M., Murray, L. M., Boyer, J. G., Anderson, C. L., and Kothary, R. (2012). Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 10:24. doi: 10.1186/1741-7015-10-24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. doi: 10.1042/0264-6021:3510095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, L. R., and Glass, J. D. (2007). Axonal degeneration in motor neuron disease. Neurodegener. Dis. 4, 431–442. doi: 10.1159/000107704
Gunther, R., Suhr, M., Koch, J. C., Bahr, M., Lingor, P., and Tonges, L. (2012). Clinical testing and spinal cord removal in a mouse model for amyotrophic lateral sclerosis (ALS). J. Vis. Exp. 61:e3936. doi: 10.3791/3936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeon, B. T., Jeong, E. A., Park, S. Y., Son, H., Shin, H. J., Lee, D. H., et al. (2013). The Rho-kinase (ROCK) inhibitor Y-27632 protects against excitotoxicity-induced neuronal death in vivo and in vitro. Neurotox. Res. 23, 238–248. doi: 10.1007/s12640-012-9339-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., Huang, Y., Ma, A. A., Lin, E., and Diamond, M. I. (2009). Y-27632 improves rotarod performance and reduces huntingtin levels in R6/2 mice. Neurobiol. Dis. 36, 413–420. doi: 10.1016/j.nbd.2009.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lingor, P., Teusch, N., Schwarz, K., Mueller, R., Mack, H., Bahr, M., et al. (2007). Inhibition of rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J. Neurochem. 103, 181–189. doi: 10.1111/j.1471-4159.2007.04756.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lingor, P., Tonges, L., Pieper, N., Bermel, C., Barski, E., Planchamp, V., et al. (2008). ROCK inhibition and CNTF interact on intrinsic signalling pathways and differentially regulate survival and regeneration in retinal ganglion cells. Brain 131, 250–263. doi: 10.1093/brain/awm284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagaoka, T., Fagan, K. A., Gebb, S. A., Morris, K. G., Suzuki, T., Shimokawa, H., et al. (2005). Inhaled rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am. J. Respir. Crit. Care Med. 171, 494–499. doi: 10.1164/rccm.200405-637OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Planchamp, V., Bermel, C., Tonges, L., Ostendorf, T., Kugler, S., Reed, J. C., et al. (2008). BAG1 promotes axonal outgrowth and regeneration in vivo via Raf-1 and reduction of ROCK activity. Brain 131, 2606–2619. doi: 10.1093/brain/awn196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Perez, A. I., Dominguez-Meijide, A., Lanciego, J. L., Guerra, M. J., and Labandeira-Garcia, J. L. (2013). Inhibition of rho kinase mediates the neuroprotective effects of estrogen in the MPTP model of Parkinson's disease. Neurobiol. Dis. 58, 209–219. doi: 10.1016/j.nbd.2013.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stewart, A., Sandercock, J., Bryan, S., Hyde, C., Barton, P. M., Fry-Smith, A., et al. (2001). The clinical effectiveness and cost-effectiveness of riluzole for motor neurone disease: a rapid and systematic review. Health Technol. Assess. 5, 1–97.
Takata, M., Tanaka, H., Kimura, M., Nagahara, Y., Tanaka, K., Kawasaki, K., et al. (2013). Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental amyotrophic lateral sclerosis models. Br. J. Pharmacol. 170, 341–351. doi: 10.1111/bph.12277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonges, L., Frank, T., Tatenhorst, L., Saal, K. A., Koch, J. C., Szego, E. M., et al. (2012). Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain 135, 3355–3370. doi: 10.1093/brain/aws254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonges, L., Gunther, R., Suhr, M., Jansen, J., Balck, A., Saal, K. A., et al. (2014). Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 62, 217–232. doi: 10.1002/glia.22601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonges, L., Koch, J. C., Bahr, M., and Lingor, P. (2011). ROCKing regeneration: rho kinase inhibition as molecular target for neurorestoration. Front. Mol. Neurosci. 4:39. doi: 10.3389/fnmol.2011.00039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tura, A., Schuettauf, F., Monnier, P. P., Bartz-Schmidt, K. U., and Henke-Fahle, S. (2009). Efficacy of Rho-kinase inhibition in promoting cell survival and reducing reactive gliosis in the rodent retina. Invest. Ophthalmol. Vis. Sci. 50, 452–461. doi: 10.1167/iovs.08-1973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turner, B. J., and Talbot, K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. doi: 10.1016/j.pneurobio.2008.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., De Carvalho, M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322. doi: 10.1016/S1474-4422(13)70036-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weydt, P., Hong, S. Y., Kliot, M., and Moller, T. (2003). Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport 14, 1051–1054. doi: 10.1097/00001756-200305230-00029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiese, S., Herrmann, T., Drepper, C., Jablonka, S., Funk, N., Klausmeyer, A., et al. (2010). Isolation and enrichment of embryonic mouse motoneurons from the lumbar spinal cord of individual mouse embryos. Nat. Protoc. 5, 31–38. doi: 10.1038/nprot.2009.193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ROCK, Y-27632, SOD1G93A, ALS, neuroprotection
Citation: Günther R, Saal K-A, Suhr M, Scheer D, Koch JC, Bähr M, Lingor P and Tönges L (2014) The rho kinase inhibitor Y-27632 improves motor performance in male SOD1G93A mice. Front. Neurosci. 8:304. doi: 10.3389/fnins.2014.00304
Received: 15 July 2014; Accepted: 09 September 2014;
Published online: 07 October 2014.
Edited by:
Nick Andrews, Harvard Medical School, USAReviewed by:
David Tattersall, GlaxoSmithCline, ChinaYung-Chih Cheng, Boston Children's Hospital/Harvard Medical School, USA
Copyright © 2014 Günther, Saal, Suhr, Scheer, Koch, Bähr, Lingor and Tönges. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Tönges MD, Department of Neurology, University Medicine Göttingen, Robert-Koch-Str. 40, 37075 Göttingen, Germany e-mail:bHRvZW5nZUBnd2RnLmRl
†Present address: René Günther, Department of Neurology, Dresden University of Technology, Dresden, Germany