 Ingrid Kratzer
Ingrid Kratzer Sophorn Chip
Sophorn Chip Zinaida S. Vexler
Zinaida S. Vexler- Department of Neurology, University of California San Francisco, San Francisco, CA, USA
Clinical data continue to reveal that the incidence of perinatal stroke is high, similar to that in the elderly. Perinatal stroke leads to significant morbidity and severe long-term neurological and cognitive deficits, including cerebral palsy. Experimental models of cerebral ischemia in neonatal rodents have shown that the pathophysiology of perinatal brain damage is multifactorial. Cerebral vasculature undergoes substantial structural and functional changes during early postnatal brain development. Thus, the state of the vasculature could affect susceptibility of the neonatal brain to cerebral ischemia. In this review, we discuss some of the most recent findings regarding the neurovascular responses of the immature brain to focal arterial stroke in relation to neuroinflammation. We also discuss a possible role of the neonatal blood-CSF barrier in modulating inflammation and the long-term effects of early neurovascular integrity after neonatal stroke on angiogenesis and neurogenesis.
Introduction
The blood-brain barrier (BBB) protects the central nervous system (CNS) and prevents non-specific leakage of molecules and cells from the blood into the brain. Stroke can disintegrate the BBB in many ways by disrupting cell-cell communication between endothelial cells (ECs), pericytes, and astrocytes, cumulatively referred to as the “neurovascular unit” (NVU), and by affecting neurons and microglial cells, thereby enhancing injury. Reperfusion and re-oxygenation of previously ischemic brain regions further affect BBB function, restoring or disrupting, depending on various factors such as the extent, length of initial cerebral blood flow (CBF) disruption, genetic background, sex, age, and the presence of other confounding factors.
The timing of injury during brain development has a major impact on determining the pathophysiology of ischemic brain damage. Maturation of individual cell types, individual components of the NVU and of the extracellular matrix (ECM), and of individual brain regions is not synchronized, contributing to the existence of “windows of susceptibility” to ischemia during particular fetal and postnatal periods. Here we discuss some of the most recent findings regarding the neurovascular responses of the immature brain to focal arterial stroke in relation to neuroinflammation and long-term effects on repair.
Age at the Time of Cerebral Ischemia as a Determinant of the Pathophysiology of Brain Damage: Preterm vs. Term
In preterm human babies (23–32-weeks of gestation) intracerebral hemorrhage (ICH), intraventricular hemorrhage (IVH), and periventricular white matter injury (PWMI) are the most common types of ischemia-related injuries (Volpe, 2009). Clinical aspects of perinatal stroke have been extensively discussed (Ferriero, 2004; Nelson and Lynch, 2004). Vulnerability of oligodendrocyte progenitor cells (OPCs) to ischemia and hypoxia including a concomitant loss of subplate neurons, a transient neuronal subpopulation important in corticogenesis and proper wiring of the developing brain, contribute to PWMI (McQuillen et al., 2003; Volpe, 2009). The germinal matrix with its weak, leaky vasculature, high local production of vascular endothelial growth factor (VEGF), angiopoietin-2 (Angpt2), and matrix metalloproteinases (MMPs) also make the preterm brain prone to ICH and IVH (Ballabh, 2010). Low pericyte coverage and ensheathement of astrocytic endfeets along blood vessels, together with immaturity of the basal membrane (BM), also make the preterm brain susceptible to ischemia-related injury (El-Khoury et al., 2006; Braun et al., 2007). At term (between birth and 28 days of life), ischemia-related white matter injury does occur (Rothstein and Levison, 2005; Van Den Broeck et al., 2008), but injury predominantly affects gray matter (Ferriero, 2004).
Several models were developed to understand the pathophysiological mechanisms of hypoxic-ischemic encephalopathy (HIE) and focal arterial stroke at term (Yager and Ashwal, 2009). A model of hypoxia-ischemia (HI) consists of a unilateral ligation of the common carotid artery (CCA) followed by a variable duration of exposure to 8% O2 in postnatal day 7 (P7) rats (Rice et al., 1981), and P9 mice and mimics HIE, whereas transient middle cerebral artery occlusion (tMCAO) model in P7 rats (Derugin et al., 1998), P10 rats (Mu et al., 2003), and P9 mice (Woo et al., 2012), and a combined permanent MCAO and transient CCA occlusion in P7 rats (Renolleau et al., 1998) mimic focal arterial stroke. The HI is associated with increased CBF during systemic hypoxia whereas CBF is disrupted after MCAO. Models of HI were also developed in rabbit and sheep (Marks et al., 1999; Derrick et al., 2007).
Studies in these age-appropriate ischemic models revealed several features unique to neonatal brain injury. First, excitotoxicity-induced neuronal death is a significant injury component (Ikonomidou et al., 1989). Second, neonatal brain is prone to reactive oxygen species (ROS) after HI (Sheldon et al., 2004). Third, apoptosis, a major neuronal cell death mode during this period, coexists with necrosis and necroptosis, creating a “continuum” with features of both cell death types (Blomgren et al., 2007; Northington et al., 2011). Finally, inflammation associated with failure to complete apoptosis is pivotal to ischemia-induced injury (Vexler and Yenari, 2009). Figure 1 outlines identified differences in ischemic injury mechanisms between adults and neonates.
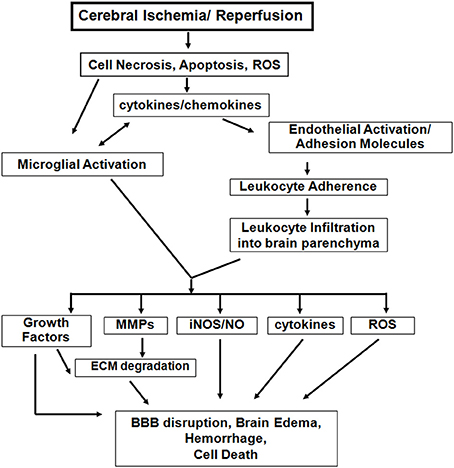
Figure 1. Inflammatory mechanisms following acute cerebral ischemia-reperfusion injury. There are both common and distinct features of the inflammatory response to cerebral ischemia between adults and neonates. However, neonatal brain is more susceptible to excitotoxic-damage and oxidative injury by ROS, resulting in necrosis and apoptosis continuum. The induction of cytokine/chemokine production, activation of microglial cells, and the systemic inflammatory response lead to neuroinflammation. Differences of adhesion molecule expression on ECs and on peripheral leukocytes exist between the injured adult and neonatal brain. Among the inflammatory mediators, activated matrix metalloproteinases (MMPs), inducible nitric oxide synthase (iNOS), and further cytokine and ROS accumulation also contribute to the variance in magnitude and spatial distribution of BBB disruption, brain edema and injury between neonatal and adult stroke. (modified from Vexler et al., 2006).
Brain Maturation-Dependent Susceptibility of the BBB After Stroke
The BBB undergoes major disruption early after adult stroke. Depending on initial stroke severity BBB opening can be bi-phasic or gradual (Knowland et al., 2014). The timing, extent of leukocyte-mediated BBB disturbances and redistribution of tight junctions (TJs) can vary. Degradation of TJ proteins occludin, claudin-5, and of a TJ-associated protein, zonula occludens 1 (ZO-1), is increased after stroke in part due to activation and/or de novo synthesis of MMPs (Yang et al., 2007a; Liu et al., 2012). TJ proteins can also internalize into the cytosol by caveolin-1-mediated endocytosis or redistribute to other membrane domains after cerebral ischemia, coinciding with the early post-ischemic BBB opening. Adherens junction proteins support BBB properties (Petty and Lo, 2002) and their altered composition after stroke changes TJ stability, affecting BBB permeability (Dejana and Giampietro, 2012; Wacker et al., 2012).
Surprisingly little is known about BBB function after neonatal ischemic injury. Major differences in functional BBB response to acute ischemia-reperfusion between neonates and adults have been recently identified (Fernandez-Lopez et al., 2012). While BBB permeability to albumin or intravascular tracers of a similar size is significantly increased after acute tMCAO in adult rats, BBB permeability remains low in injured neonatal rats. Gene and protein expression of occludin, claudin-5 and ZO-1 are better preserved in injured neonatal brain than in injured adult brain, whereas gene expression of the efflux transporters ATP-binding cassette, subfamily G2 (Abcg2) and P-glycoprotein (P-gp) is reduced in both ages 24 h after reperfusion (Fernandez-Lopez et al., 2012). Transcript levels of several adhesion molecules and ECM components are differentially affected by injury in immature and adult brain, including E-selectin and P-selectin. Gene expression of Mmp-9 is significantly upregulated in injured adults and, while high transcript levels of collagen type IV α1 (Col4a1), and Col4a2 remain unaltered in neonates, a significant increase of these two genes is evident in injured adult rats. Interestingly, transcripts of angiogenic regulators Vegfr-2, and Angpt2 are increased after stroke in adults but not in neonates (Fernandez-Lopez et al., 2012). Figure 2 summarizes these findings. In contrast, a transient leakage of much smaller tracers, sucrose and inulin, was observed in a mouse HI model, with the peak at 6 h and normalization by 24 h (D'Angelo et al., 2012), conveying that size and chemical structure of molecules affect leakage and highlighting the need for future BBB studies in injured immature brain.
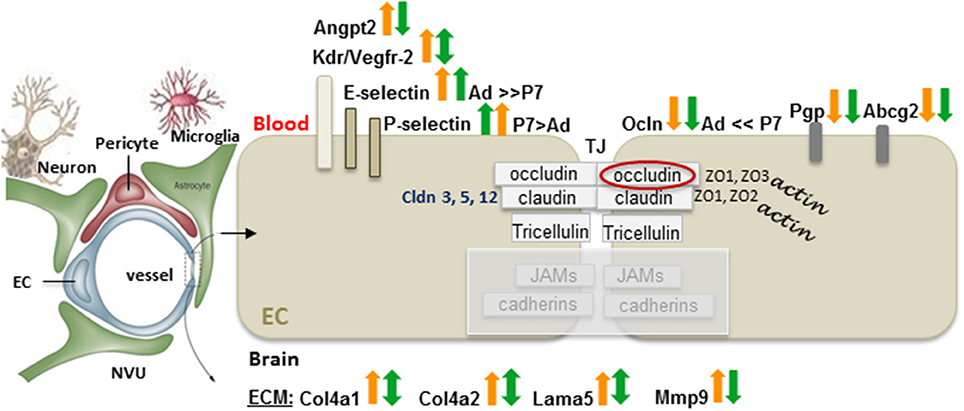
Figure 2. Differences in the gene expression of the BBB components between adults and neonates 24 h after tMCAO. On a diagram of the NVU, arrows demarcate the gene expression changes in injured vs. matching contralateral region (ratio, ipsilateral/contralateral) in adult (orange arrows), and neonatal rats (green arrows) subjected to 3 h of MCAO followed by 24 h of reperfusion. Arrows pointing upwards demarcate increased gene expression in injured regions vs. matching contralateral regions (statistically significant > 2-fold change). Downward arrows show reduced expression in injured regions vs. matching contralateral regions (statistically significant > 2-fold change). Double-sided arrows indicate no changes between injured and contralateral regions. Abbreviations: Ad: adult, P7: postnatal 7-days-old, EC, endothelial cell; angiopoetin 2, Angpt2; Kdr, Vegfr-2; Cldn, claudin; Ocln, occludin; ZO, zonula occludens; JAM, junctional adhesion molecules; TJ, tight junction; Pgp, P-glycoprotein; Abcg2, ATP-binding cassette transporter, subfamily G 2; Col4a1, collagen, type IV, alpha 1; Col4a2, collagen, type IV, alpha 2; Lama5, laminin, alpha 5; Mmp-9, matrix metalloproteinase 9. Original data are published in Fernandez-Lopez et al. (2012). The NVU contains fragments from Eichler et al. (2011).
The ECM proteins and their corresponding receptors on ECs and astrocytes provide both physical and biochemical glial-vascular “scaffolding” while BM components laminin, collagen IV (Col-IV), fibronectin, and perlecan provide proper cell-cell interactions. Endothelial-ECM interaction via β 1 integrins regulates the expression of claudin-5 and BBB tightness whereas other ECM proteins, like galectin-3 mediate integrin-induced stabilization of focal adhesions, and activate cytokine receptors to enhance actions of growth factors (Goetz et al., 2008). Homozygous mutations in Col-IV are lethal in mid-gestation due to blockage of capillary bed development, and mutations in the COL4A1 gene cause ICH both in newborn mouse and human (Gould et al., 2005; Labelle-Dumais et al., 2011). Laminin degradation after focal stroke in adults causes detachment of astrocytic endfeet, disrupts BBB and induces ICH (Fukuda et al., 2004), while in neonates expression of this ECM protein is not reduced acutely (Fernandez-Lopez et al., 2012). The role of other ECM proteins in injured neonates is less studied but opposite effects of galectin-3 in adult stroke and HI has been demonstrated (Doverhag et al., 2010; Lalancette-Hebert et al., 2012).
Parenchymal Brain Cells as Modulators of BBB Integrity After Neonatal Stroke
Neuroinflammation is a characteristic feature of stroke progression and a major contributor to brain injury (Iadecola and Anrather, 2011). Multiple cell types (i.e. neurons, astrocytes, ECs, and microglia) increase production of inflammatory mediators post-ischemia, which can adversely affect BBB integrity and propagate injury.
ECs are sensitive to oxidative stress (Freeman and Keller, 2012). Excessive ROS accumulation generated by ischemia/reperfusion contributes to damage of TJs and other EC components, and promotes activation of cell death pathways (Rizzo and Leaver, 2010). Although less sensitive to cerebral ischemia than neurons, ECs also undergo cell death via several mechanisms (Rizzo and Leaver, 2010). Our comparison of gene expression in ECs from injured and contralateral cortex in neonatal and adult rats after tMCAO (endothelial transcriptome) revealed a markedly distinct signature of up-regulated and down-regulated transcripts in injured regions between two ages (Fernandez-Lopez et al., 2012).
Astrocytes are rather resistant to ischemic injury but impairment of water fluxes through astrocytic swelling and increased expression of aquaporin 4 (AQP4) leads to edema after adult focal ischemia (Loreto and Reggio, 2010). No edema was observed in injured P10 pups in brain regions with greatest AQP4 expression (Badaut et al., 2007), suggesting that enhanced water clearance at border regions can protect.
Microglial cells have been considered toxic after cerebral ischemia due to production of inflammatory mediators. However, phagocytotic ability and production of growth factors by microglia can be beneficial via several mechanisms, including support of neuronal and endothelial survival (Baburamani et al., 2012; Luo and Chen, 2012). Microglial cells have several direct modulatory effects on the vasculature. During brain development they mediate vasculogenesis (Arnold and Betsholtz, 2013), and act as a physical bridge that guides vascular anastomosis, facilitating normal angiogenesis, and vascular sprouting. Microglia patrol the vasculature in the naïve brain (Davalos et al., 2012) and, upon BBB disruption, rapidly extend their processes, shielding injured sites (Nimmerjahn et al., 2005). Depletion of microglia worsens parenchymal injury in neonatal rats after tMCAO, increases levels of inflammatory mediators in acutely injured regions, and induces hemorrhagic transformation (Faustino et al., 2011; Fernandez Lopez et al., 2014) perhaps due to differing cell phenotypes in the immature brain.
Pericytes are important regulators of vessel contractility. Pericyte loss or dissociation from vessels lead to edema (Peppiatt et al., 2006) and impaired reflow (Yemisci et al., 2009). Lower than in adult pericyte coverage in the neonatal brain (Daneman et al., 2010) may differentially affect CBF regulation and BBB function after injury but this is yet to be demonstrated.
OPCs support EC survival but they are sensitive to ROS and can adversely affect BBB integrity via release of inflammatory mediators. Mast cells contribute to early ischemic brain swelling, BBB leakage and neutrophil infiltration (Strbian et al., 2006). Their stabilization protects and reduces hemorrhage formation (Strbian et al., 2007). Degranulation of mast cells is injurious in neonatal HI (Jin et al., 2007) and stroke (Biran et al., 2008) but direct effects on BBB have not been studied.
Systemic Inflammation Alters BBB Permeability After Neonatal Stroke
Parenchymal, perivascular and peripheral circulating cells independently and in concert contribute to stroke-induced production of inflammatory mediators, upregulation of integrins and adhesion molecules on ECs and leukocytes (Iadecola and Anrather, 2011).
Neutrophils rapidly, often transiently infiltrate ischemic tissue and exacerbate reperfusion injury in adults by priming the endothelium, causing “no-reflow” phenomenon, releasing ROS and proteolytic enzymes, and stimulating cytokine release (Mori et al., 1992; Amantea et al., 2009; Engelhardt and Liebner, 2014). Both loosely attached and infiltrated leukocytes can contribute to generation of pro-inflammatory molecules in the CNS (Denes et al., 2010). Neutropenia or treatments that prevent leukocyte adhesion and infiltration are neuroprotective (Kochanek and Hallenbeck, 1992; Yamasaki et al., 1995). Compared to adults, in neonates neutrophil infiltration is markedly lower in response to HI (Hudome et al., 1997; Bona et al., 1999), and tMCAO (Fernandez-Lopez et al., 2012). Neutralization of cytokine-induced neutrophil chemoattractant 1 protein (CINC-1) following tMCAO in adults holts neutrophil transmigration, reduces brain edema and protects (Yamasaki et al., 1997) whereas it promotes neutrophil infiltration following tMCAO and disrupts the BBB in neonates (Fernandez-Lopez et al., 2012).
Monocytes have been recently shown to have a dual role, elicit both inflammatory effects and maintain NVU integrity following cerebral ischemia. Monocyte depletion, chemokine (C-C motif) receptor 2 (CCR2) knockout, and bone marrow chimeric approach in murine stroke models demonstrated that CCR2 in bone marrow-derived cells alters injury and hemorrhagic transformation (Gliem et al., 2012). The stabilizing effects of monocytes are transforming growth factor beta 1 (TGF-β 1)-dependent (Gliem et al., 2012). Compared to adult stroke, infiltration of circulating monocytes across the BBB is relatively low during the acute injury phase in neonates (Denker et al., 2007). It remains poorly understood whether leukocyte immaturity at the time of insult or a distinct gene expression pattern of selectins, and cytokines/chemokines account for the difference.
T and B cell infiltration may be less profound (Bona et al., 1999) or transient (Benjelloun et al., 1999) in injured neonates than in adults (Catania and Lipton, 1998; Chu et al., 2014).
Individual Inflammatory Signaling Mechanisms
Evidence is growing that the inflammatory responses after stroke are different in neonates and adults (Vexler and Yenari, 2009). Genetic deletion of various inflammatory mediators, including NADPH oxidase (Doverhag et al., 2008), or interleukin 1 (IL-1) β, IL-1α, or both αβ (Hedtjarn et al., 2005) are not neuroprotective in the neonatal brain compared to adults. Some mediators that are upregulated and injurious during the acute injury phase, such as nitric oxide (NO), MMPs, macrophage inflammatory protein 1 alpha (MIP-1α), monocyte chemoattractant protein 1 (MCP-1), and complement, may be beneficial and mediate repair (Fernandez-Lopez et al., 2014).
MMPs degrade TJ and BM proteins, including collagen, laminin and fibronectin, thereby leading to brain edema, BBB leakage and leukocyte infiltration. MMP upregulation after stroke contributes to ECM breakdown and acute brain damage (Rosenberg et al., 1998; Asahi et al., 2000). MMP-2 and MMP-9, the two most studied MMPs in stroke, play different roles in BBB disruption (Asahi et al., 2001). MMP-3 targets several ECM components, including laminin and proteoglycans, and propagates injury by mediating BBB opening by inflammatory mediators (Cunningham et al., 2005). While activated microglia, macrophages and infiltrating leukocytes are the major sources of MMPs early after injury (Gidday et al., 2005; McColl et al., 2008), over time activated astrocytes and neurons begin producing MMPs, enhancing repair (Zhao et al., 2006). MMP-9 was shown to predict HIE in human newborns (Bednarek et al., 2012). Cerebrovascular ECs from neonates in culture contain more tissue plasminogen activator (t-PA) and gelatinases upon glutamate challenge than adult cells (Omouendze et al., 2013). MMP inhibition is protective after HI (Chen et al., 2009) but long-term MMP inhibition may holt ECM remodeling, as shown in adult stroke (Zhao et al., 2006).
The patterns of monocyte and neutrophil recruitment are cytokine- and chemokine-specific. The multifaceted roles for α, β, and δ classes of chemokines were shown in adult stroke models (Yamasaki et al., 1995). Integrins are central for cell communication within the NVU and for leukocyte recruitment after stroke (Iadecola and Anrather, 2011) but information on the role of integrins in neonatal stroke is scant.
Blood-CSF Barrier (BCSFB)
The choroid plexuses (CPs), forming the BCSFB, are involved in immune cell entry after brain injury (Shechter et al., 2013). CPs express chemokines and support transepithelial trafficking of neutrophils, monocytes and T cells (Szmydynger-Chodobska et al., 2009, 2012; Kunis et al., 2013). Engraftment of CPs in adult stroke models reduced infarct size and improved neurological function, in part via secretion of glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), and nerve growth factor (NGF) (Borlongan et al., 2004). CPs have unique functions in the developing brain (Dziegielewska et al., 2001) but their role in protection of the immature brain after stroke is unknown.
Neurovascular Responses and Repair After Neonatal Stroke
Long-term neural repair is less studied after neonatal stroke than after adult stroke. Cell proliferation in the subventricular zone (SVZ) after ischemia is triggered in both adults (Ohab et al., 2006), and neonates (Plane et al., 2004; Yang et al., 2007b). The dynamic changes within the SVZ neurogenic niche permit neuroblast migration into the ischemic striatum (Young et al., 2011) in the adult, where they express phenotypic region-specific mature neuronal markers (Parent et al., 2002), and into peri-infarct striatum after neonatal HI (Plane et al., 2004; Yang et al., 2007b). Niche astrocytes and SVZ microglia are also involved in neuroblast migration (Young et al., 2011). The newborn SVZ contains numerous cell types, including unipotential astrocytes and OPCs as well as bipotential glial progenitors (Levison and Goldman, 1997).
Given the dynamic nature of postnatal brain development one would expect robust repair processes in neonatal stroke but a 1–2 week delay in induction of angiogenesis was shown after tMCAO in P7 and P10 rats (Shimotake et al., 2010; Dzietko et al., 2013; Fernandez-Lopez et al., 2013) despite induction of VEGF (Mu et al., 2003). Another obstacle for endogenous repair is that proliferating cells in the postnatal SVZ differentiate into astrocytes, rather than neurons or oligodendrocytes, and astrogliosis holts the repair (Spadafora et al., 2010; Gonzalez et al., 2013). Changing the neural stem cell fate to route the differentiation into neurons and oligodendrocytes after stroke, for example, with erythropoietin, enhances the repair (Iwai et al., 2010; Gonzalez et al., 2013). Cell based therapies, including mesenchymal stem cells, improve functional outcomes after neonatal HI (Van Velthoven et al., 2010) and arterial stroke (Van Velthoven et al., 2013) by changing the microenvironment and stimulating Angpt1 and VEGF signaling, which amplify angiogenesis and “loosen” the barrier, allowing vessel remodeling and neuroblast migration.
Translational Aspects and Future Directions
A broad range of therapeutic agents was used in neonatal ischemic brain injury models to target the excitotoxic, oxidative, and inflammatory injury components, but, as in adult stroke, studies revealed limits in protection. To date, hypothermia is the only neuroprotective treatment for perinatal HIE with efficacy limited to moderate injury (Azzopardi et al., 2009; Edwards et al., 2010).
Importantly, recent studies have improved our understanding of the events at the BBB after neonatal ischemia by revealing that the developmental stage of the BBB at the time of ischemic insult is of prime importance and that careful consideration should be given to whether the BBB is in fact disrupted or it limits therapies from reaching an injured neonatal brain. The role of local parenchymal cells, microglia, as modulators of neurovascular integrity after neonatal stroke was also uncovered. Future studies should shed light on relationships between neurovascular integrity and interaction with neuroprogenitors, endogenous, or engrafted, including the migration and differentiation of neural progenitors during stroke-induced neurogenesis.
Author Contributions
Ingrid Kratzer and Sophorn Chip wrote individual parts of the review and contributed equally. Together with Zinaida S. Vexler who provided conceptual framework and wrote parts of the review, they contributed to revisions and prepared artwork.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The following grants NS080015 (Zinaida S. Vexler), NS44025 (Zinaida S. Vexler), and NS76726 (Zinaida S. Vexler) were used as principal sources of funding.
References
Amantea, D., Nappi, G., Bernardi, G., Bagetta, G., and Corasaniti, M. T. (2009). Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 276, 13–26. doi: 10.1111/j.1742-4658.2008.06766.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arnold, T., and Betsholtz, C. (2013). The importance of microglia in the development of the vasculature in the central nervous system. Vasc. Cell 5, 4. doi: 10.1186/2045-824X-5-4
Asahi, M., Asahi, K., Jung, J. C., Del Zoppo, G. J., Fini, M. E., and Lo, E. H. (2000). Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 20, 1681–1689. doi: 10.1097/00004647-200012000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asahi, M., Sumii, T., Fini, M. E., Itohara, S., and Lo, E. H. (2001). Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport 12, 3003–3007. doi: 10.1097/00001756-200109170-00050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azzopardi, D. V., Strohm, B., Edwards, A. D., Dyet, L., Halliday, H. L., Juszczak, E., et al. (2009). Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 361, 1349–1358. doi: 10.1056/NEJMoa0900854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baburamani, A. A., Ek, C. J., Walker, D. W., and Castillo-Melendez, M. (2012). Vulnerability of the developing brain to hypoxic-ischemic damage: contribution of the cerebral vasculature to injury and repair? Front. Physiol. 3:424. doi: 10.3389/fphys.2012.00424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Badaut, J., Ashwal, S., Tone, B., Regli, L., Tian, H. R., and Obenaus, A. (2007). Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr. Res. 62, 248–254. doi: 10.1203/PDR.0b013e3180db291b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ballabh, P. (2010). Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr. Res. 67, 1–8. doi: 10.1203/PDR.0b013e3181c1b176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bednarek, N., Svedin, P., Garnotel, R., Favrais, G., Loron, G., Schwendiman, L., et al. (2012). Increased MMP-9 and TIMP-1 in mouse neonatal brain and plasma and in human neonatal plasma after hypoxia-ischemia: a potential marker of neonatal encephalopathy. Pediatr. Res. 71, 63–70. doi: 10.1038/pr.2011.3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benjelloun, N., Renolleau, S., Represa, A., Ben-Ari, Y., and Charriaut-Marlangue, C. (1999). Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal Rat. Stroke 30, 1916–1923. discussion 1923-4.
Biran, V., Cochois, V., Karroubi, A., Arrang, J. M., Charriaut-Marlangue, C., and Heron, A. (2008). Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol. 18, 1–9. doi: 10.1111/j.1750-3639.2007.00092.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blomgren, K., Leist, M., and Groc, L. (2007). Pathological apoptosis in the developing brain. Apoptosis 12, 993–1010. doi: 10.1007/s10495-007-0754-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bona, E., Andersson, A. L., Blomgren, K., Gilland, E., Puka-Sundvall, M., Gustafson, K., et al. (1999). Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res. 45, 500–509. doi: 10.1203/00006450-199904010-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borlongan, C. V., Skinner, S. J., Geaney, M., Vasconcellos, A. V., Elliott, R. B., and Emerich, D. F. (2004). Intracerebral transplantation of porcine choroid plexus provides structural and functional neuroprotection in a rodent model of stroke. Stroke 35, 2206–2210. doi: 10.1161/01.STR.0000138954.25825.0b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braun, A., Xu, H., Hu, F., Kocherlakota, P., Siegel, D., Chander, P., et al. (2007). Paucity of pericytes in germinal matrix vasculature of premature infants. J. Neurosci. 27, 12012–12024. doi: 10.1523/JNEUROSCI.3281-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catania, A., and Lipton, J. M. (1998). Peptide modulation of fever and inflammation within the brain. Ann. N.Y. Acad. Sci. 856, 62–68. doi: 10.1111/j.1749-6632.1998.tb08313.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, W., Hartman, R., Ayer, R., Marcantonio, S., Kamper, J., Tang, J., et al. (2009). Matrix metalloproteinases inhibition provides neuroprotection against hypoxia-ischemia in the developing brain. J. Neurochem. 111, 726–736. doi: 10.1111/j.1471-4159.2009.06362.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, H. X., Kim, H. A., Lee, S., Moore, J. P., Chan, C. T., Vinh, A., et al. (2014). Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 34, 450–459. doi: 10.1038/jcbfm.2013.217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cunningham, L. A., Wetzel, M., and Rosenberg, G. A. (2005). Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 50, 329–339. doi: 10.1002/glia.20169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Angelo, B. D., Baburamani, A., Hagberg, H., and Mallard, C. (2012). “Role of blood-brain barrier in perinatal brain injury,” in Abstract Presented at the SfN in a Nanosymposium, New Orleans, LA, USA. Abstract 831.0399. Available online at: http://am2012.sfn.org/am2012/pdf/final_program/final_program_b6.pdf
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davalos, D., Ryu, J. K., Merlini, M., Baeten, K. M., Le Moan, N., Petersen, M. A., et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3, 1227. doi: 10.1038/ncomms2230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dejana, E., and Giampietro, C. (2012). Vascular endothelial-cadherin and vascular stability. Curr. Opin. Hematol. 19, 218–223. doi: 10.1097/MOH.0b013e3283523e1c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denes, A., Thornton, P., Rothwell, N. J., and Allan, S. M. (2010). Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav. Immun. 24, 708–723. doi: 10.1016/j.bbi.2009.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denker, S., Ji, S., Lee, S. Y., Dingman, A., Derugin, N., Wendland, M., et al. (2007). Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem. 100, 893–904. doi: 10.1111/j.1471-4159.2006.04162.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derrick, M., Drobyshevsky, A., Ji, X., and Tan, S. (2007). A model of cerebral palsy from fetal hypoxia-ischemia. Stroke 38, 731–735. doi: 10.1161/01.STR.0000251445.94697.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derugin, N., Ferriero, D. M., and Vexler, Z. S. (1998). Neonatal reversible focal cerebral ischemia: a new model. Neurosci. Res. 32, 349–353. doi: 10.1016/S0168-0102(98)00096-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doverhag, C., Hedtjarn, M., Poirier, F., Mallard, C., Hagberg, H., Karlsson, A., et al. (2010). Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 38, 36–46. doi: 10.1016/j.nbd.2009.12.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doverhag, C., Keller, M., Karlsson, A., Hedtjarn, M., Nilsson, U., Kapeller, E., et al. (2008). Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol. Dis. 31, 133–144. doi: 10.1016/j.nbd.2008.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dziegielewska, K. M., Ek, J., Habgood, M. D., and Saunders, N. R. (2001). Development of the choroid plexus. Microsc. Res. Tech. 52, 5–20. doi: 10.1002/1097-0029(20010101)52:1<5::AID-JEMT3>3.0.CO;2-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dzietko, M., Derugin, N., Wendland, M. F., Vexler, Z. S., and Ferriero, D. M. (2013). Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl. Stroke Res. 4, 189–200. doi: 10.1007/s12975-012-0221-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edwards, A. D., Brocklehurst, P., Gunn, A. J., Halliday, H., Juszczak, E., Levene, M., et al. (2010). Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 340, c363. doi: 10.1136/bmj.c363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eichler, A. F., Chung, E., Kodack, D. P., Loeffler, J. S., Fukumura, D., and Jain, R. K. (2011). The biology of brain metastases-translation to new therapies. Nat. Rev. Clin. Oncol. 8, 344–356. doi: 10.1038/nrclinonc.2011.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El-Khoury, N., Braun, A., Hu, F., Pandey, M., Nedergaard, M., Lagamma, E. F., et al. (2006). Astrocyte end-feet in germinal matrix, cerebral cortex, and white matter in developing infants. Pediatr. Res. 59, 673–679. doi: 10.1203/01.pdr.0000214975.85311.9c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Engelhardt, B., and Liebner, S. (2014). Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res. 355, 687–699. doi: 10.1007/s00441-014-1811-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faustino, J., Wang, X., Jonhson, C., Klibanov, A., Derugin, N., Wendland, M., et al. (2011). Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 31, 12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez-Lopez, D., Faustino, J., Daneman, R., Zhou, L., Lee, S. Y., Derugin, N., et al. (2012). Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke. J. Neurosci. 32, 9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez-Lopez, D., Faustino, J., Derugin, N., and Vexler, Z. S. (2013). Acute and chronic vascular responses to experimental focal arterial stroke in the neonate rat. Transl. Stroke Res. 4, 179–188. doi: 10.1007/s12975-012-0214-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez Lopez, D., Faustino, J., Klibanov, A., Derugin, N., Akassoglou, K., and Vexler, Z. S. (2014). “Activated microglia contribute to neurovascular integrity and limit brain injury after acute neonatal focal stroke,” in International Stroke Meeting (San Diego, CA).
Fernandez-Lopez, D., Natarajan, N., Ashwal, S., and Vexler, Z. S. (2014). Mechanisms of perinatal arterial ischemic stroke. J. Cereb. Blood Flow Metab. 34, 921–932. doi: 10.1038/jcbfm.2014.41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferriero, D. M. (2004). Neonatal brain injury. N. Engl. J. Med. 351, 1985–1995. doi: 10.1056/NEJMra041996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freeman, L. R., and Keller, J. N. (2012). Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 1822, 822–829. doi: 10.1016/j.bbadis.2011.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fukuda, S., Fini, C. A., Mabuchi, T., Koziol, J. A., Eggleston, L. L. Jr., and Del Zoppo, G. J. (2004). Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke 35, 998–1004. doi: 10.1161/01.STR.0000119383.76447.05
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gidday, J. M., Gasche, Y. G., Copin, J. C., Shah, A. R., Perez, R. S., Shapiro, S. D., et al. (2005). Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 289, H558–H568. doi: 10.1152/ajpheart.01275.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gliem, M., Mausberg, A. K., Lee, J. I., Simiantonakis, I., Van Rooijen, N., Hartung, H. P., et al. (2012). Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752. doi: 10.1002/ana.23529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goetz, J. G., Joshi, B., Lajoie, P., Strugnell, S. S., Scudamore, T., Kojic, L. D., et al. (2008). Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J. Cell Biol. 180, 1261–1275. doi: 10.1083/jcb.200709019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, F. F., Larpthaveesarp, A., McQuillen, P., Derugin, N., Wendland, M., Spadafora, R., et al. (2013). Erythropoietin increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke 44, 753–758. doi: 10.1161/STROKEAHA.111.000104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gould, D. B., Phalan, F. C., Breedveld, G. J., Van Mil, S. E., Smith, R. S., Schimenti, J. C., et al. (2005). Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308, 1167–1171. doi: 10.1126/science.1109418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hedtjarn, M., Mallard, C., Iwakura, Y., and Hagberg, H. (2005). Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia-ischemia in the immature brain. Dev. Neurosci. 27, 143–148. doi: 10.1159/000085986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hudome, S., Palmer, C., Roberts, R. L., Mauger, D., Housman, C., and Towfighi, J. (1997). The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 41, 607–616. doi: 10.1203/00006450-199705000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., and Anrather, J. (2011). The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808. doi: 10.1038/nm.2399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikonomidou, C., Mosinger, J. L., Salles, K. S., Labruyere, J., and Olney, J. W. (1989). Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J. Neurosci. 9, 2809–2818.
Iwai, M., Stetler, R. A., Xing, J., Hu, X., Gao, Y., Zhang, W., et al. (2010). Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 41, 1032–1037. doi: 10.1161/STROKEAHA.109.570325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, Y., Silverman, A. J., and Vannucci, S. J. (2007). Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev. Neurosci. 29, 373–384. doi: 10.1159/000105478
Knowland, D., Arac, A., Sekiguchi, K. J., Hsu, M., Lutz, S. E., Perrino, J., et al. (2014). Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617. doi: 10.1016/j.neuron.2014.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kochanek, P. M., and Hallenbeck, J. M. (1992). Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke 23, 1367–1379. doi: 10.1161/01.STR.23.9.1367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kunis, G., Baruch, K., Rosenzweig, N., Kertser, A., Miller, O., Berkutzki, T., et al. (2013). IFN-gamma-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain 136, 3427–3440. doi: 10.1093/brain/awt259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Labelle-Dumais, C., Dilworth, D. J., Harrington, E. P., De Leau, M., Lyons, D., Kabaeva, Z., et al. (2011). COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 7, e1002062. doi: 10.1371/journal.pgen.1002062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lalancette-Hebert, M., Swarup, V., Beaulieu, J. M., Bohacek, I., Abdelhamid, E., Weng, Y. C., et al. (2012). Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32, 10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levison, S. W., and Goldman, J. E. (1997). Multipotential and lineage restricted precursors coexist in the mammalian perinatal subventricular zone. J. Neurosci. Res. 48, 83–94.
Liu, J., Jin, X., Liu, K. J., and Liu, W. (2012). Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci. 32, 3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loreto, C., and Reggio, E. (2010). Aquaporin and vascular diseases. Curr. Neuropharmacol. 8, 105–111. doi: 10.2174/157015910791233196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, X. G., and Chen, S. D. (2012). The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 1, 9. doi: 10.1186/2047-9158-1-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marks, K. A., Mallard, C. E., Roberts, I., Williams, C. E., Gluckman, P. D., and Edwards, A. D. (1999). Nitric oxide synthase inhibition and delayed cerebral injury after severe cerebral ischemia in fetal sheep. Pediatr. Res. 46, 8–13. doi: 10.1203/00006450-199907000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McColl, B. W., Rothwell, N. J., and Allan, S. M. (2008). Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J. Neurosci. 28, 9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McQuillen, P. S., Sheldon, R. A., Shatz, C. J., and Ferriero, D. M. (2003). Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J. Neurosci. 23, 3308–3315.
Mori, E., Del Zoppo, G. J., Chambers, J. D., Copeland, B. R., and Arfors, K. E. (1992). Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 23, 712–718. doi: 10.1161/01.STR.23.5.712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mu, D., Jiang, X., Sheldon, R. A., Fox, C. K., Hamrick, S. E., Vexler, Z. S., et al. (2003). Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol. Dis. 14, 524–534. doi: 10.1016/j.nbd.2003.08.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nelson, K. B., and Lynch, J. K. (2004). Stroke in newborn infants. Lancet Neurol. 3, 150–158. doi: 10.1016/S1474-4422(04)00679-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Northington, F. J., Chavez-Valdez, R., and Martin, L. J. (2011). Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 69, 743–758. doi: 10.1002/ana.22419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohab, J. J., Fleming, S., Blesch, A., and Carmichael, S. T. (2006). A neurovascular niche for neurogenesis after stroke. J. Neurosci. 26, 13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Omouendze, P. L., Henry, V. J., Porte, B., Dupre, N., Carmeliet, P., Gonzalez, B. J., et al. (2013). Hypoxia-ischemia or excitotoxin-induced tissue plasminogen activator- dependent gelatinase activation in mice neonate brain microvessels. PLoS ONE 8:e71263. doi: 10.1371/journal.pone.0071263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parent, J. M., Vexler, Z. S., Gong, C., Derugin, N., and Ferriero, D. M. (2002). Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 52, 802–813. doi: 10.1002/ana.10393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peppiatt, C. M., Howarth, C., Mobbs, P., and Attwell, D. (2006). Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704. doi: 10.1038/nature05193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petty, M. A., and Lo, E. H. (2002). Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog. Neurobiol. 68, 311–323. doi: 10.1016/S0301-0082(02)00128-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Plane, J. M., Liu, R., Wang, T. W., Silverstein, F. S., and Parent, J. M. (2004). Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol. Dis. 16, 585–595. doi: 10.1016/j.nbd.2004.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renolleau, S., Aggoun-Zouaoui, D., Ben-Ari, Y., and Charriaut-Marlangue, C. (1998). A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: morphological changes indicative of apoptosis. Stroke 29, 1454–1460. discussion: 1461. doi: 10.1161/01.STR.29.7.1454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rice, J. E. D., Vannucci, R. C., and Brierley, J. B. (1981). The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 9, 131–141. doi: 10.1002/ana.410090206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rizzo, M. T., and Leaver, H. A. (2010). Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 42, 52–63. doi: 10.1007/s12035-010-8132-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosenberg, G. A., Estrada, E. Y., and Dencoff, J. E. (1998). Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 29, 2189–2195. doi: 10.1161/01.STR.29.10.2189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rothstein, R. P., and Levison, S. W. (2005). Gray matter oligodendrocyte progenitors and neurons die caspase-3 mediated deaths subsequent to mild perinatal hypoxic/ischemic insults. Dev. Neurosci. 27, 149–159. doi: 10.1159/000085987
Shechter, R., Miller, O., Yovel, G., Rosenzweig, N., London, A., Ruckh, J., et al. (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38, 555–569. doi: 10.1016/j.immuni.2013.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheldon, R. A., Jiang, X., Francisco, C., Christen, S., Vexler, Z. S., Tauber, M. G., et al. (2004). Manipulation of antioxidant pathways in neonatal murine brain. Pediatr. Res. 56, 656–662. doi: 10.1203/01.PDR.0000139413.27864.50
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimotake, J., Derugin, N., Wendland, M., Vexler, Z. S., and Ferriero, D. M. (2010). Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke 41, 343–349. doi: 10.1161/STROKEAHA.109.564229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spadafora, R., Gonzalez, F. F., Derugin, N., Wendland, M., Ferriero, D., and McQuillen, P. (2010). Altered fate of subventricular zone progenitor cells and reduced neurogenesis following neonatal stroke. Dev. Neurosci. 32, 101–113. doi: 10.1159/000279654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strbian, D., Karjalainen-Lindsberg, M. L., Kovanen, P. T., Tatlisumak, T., and Lindsberg, P. J. (2007). Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation 116, 411–418. doi: 10.1161/CIRCULATIONAHA.106.655423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strbian, D., Karjalainen-Lindsberg, M. L., Tatlisumak, T., and Lindsberg, P. J. (2006). Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J. Cereb. Blood Flow Metab. 26, 605–612. doi: 10.1038/sj.jcbfm.9600228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szmydynger-Chodobska, J., Strazielle, N., Gandy, J. R., Keefe, T. H., Zink, B. J., Ghersi-Egea, J. F., et al. (2012). Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J. Cereb. Blood Flow Metab. 32, 93–104. doi: 10.1038/jcbfm.2011.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szmydynger-Chodobska, J., Strazielle, N., Zink, B. J., Ghersi-Egea, J. F., and Chodobski, A. (2009). The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J. Cereb. Blood Flow Metab. 29, 1503–1516. doi: 10.1038/jcbfm.2009.71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Den Broeck, C., Himpens, E., Vanhaesebrouck, P., Calders, P., and Oostra, A. (2008). Influence of gestational age on the type of brain injury and neuromotor outcome in high-risk neonates. Eur. J. Pediatr. 167, 1005–1009. doi: 10.1007/s00431-007-0629-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Velthoven, C. T., Kavelaars, A., Van Bel, F., and Heijnen, C. J. (2010). Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 24, 387–393. doi: 10.1016/j.bbi.2009.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Velthoven, C. T., Sheldon, R. A., Kavelaars, A., Derugin, N., Vexler, Z. S., Willemen, H. L., et al. (2013). Mesenchymal stem cell transplantation attenuates brain injury after neonatal stroke. Stroke 44, 1426–1432. doi: 10.1161/STROKEAHA.111.000326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vexler, Z. S., and Yenari, M. A. (2009). Does inflammation after stroke affect the developing brain differently than adult brain? Dev. Neurosci. 31, 378–393. doi: 10.1159/000232556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vexler, Z. S., Tang, X. N., and Yenari, M. A. (2006). Inflammation in adult and neonatal stroke. Clin. Neurosci. Res. 6, 293–313. doi: 10.1016/j.cnr.2006.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volpe, J. J. (2009). The encephalopathy of prematurity–brain injury and impaired brain development inextricably intertwined. Semin. Pediatr. Neurol. 16, 167–178. doi: 10.1016/j.spen.2009.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wacker, B. K., Freie, A. B., Perfater, J. L., and Gidday, J. M. (2012). Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J. Cereb. Blood Flow Metab. 32, 1014–1023. doi: 10.1038/jcbfm.2012.3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woo, M. S., Wang, X., Faustino, J., Derugin, N., Wendland, M. F., Zhou, P., et al. (2012). Genetic deletion of CD36 enhances injury after acute neonatal stroke. Ann. Neurol. 72, 961–970. doi: 10.1002/ana.23727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yager, J. Y., and Ashwal, S. (2009). Animal models of perinatal hypoxic-ischemic brain damage. Pediatr. Neurol. 40, 156–167. doi: 10.1016/j.pediatrneurol.2008.10.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamasaki, Y., Matsuo, Y., Matsuura, N., Onodera, H., Itoyama, Y., and Kogure, K. (1995). Transient increase of cytokine-induced neutrophil chemoattractant, a member of the interleukin-8 family, in ischemic brain areas after focal ischemia in rats. Stroke 26, 318–22; discussion 322-3. doi: 10.1161/01.STR.26.2.318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamasaki, Y., Matsuo, Y., Zagorski, J., Matsuura, N., Onodera, H., Itoyama, Y., et al. (1997). New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Res. 759, 103–111. doi: 10.1016/S0006-8993(97)00251-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Estrada, E. Y., Thompson, J. F., Liu, W., and Rosenberg, G. A. (2007a). Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 27, 697–709. doi: 10.1038/sj.jcbfm.9600375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Covey, M. V., Bitel, C. L., Ni, L., Jonakait, G. M., and Levison, S. W. (2007b). Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann. Neurol. 61, 199–208. doi: 10.1002/ana.21068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yemisci, M., Gursoy-Ozdemir, Y., Vural, A., Can, A., Topalkara, K., and Dalkara, T. (2009). Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037. doi: 10.1038/nm.2022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Young, C. C., Brooks, K. J., Buchan, A. M., and Szele, F. G. (2011). Cellular and molecular determinants of stroke-induced changes in subventricular zone cell migration. Antioxid. Redox Signal. 14, 1877–1888. doi: 10.1089/ars.2010.3435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, B. Q., Wang, S., Kim, H. Y., Storrie, H., Rosen, B. R., Mooney, D. J., et al. (2006). Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 12, 441–445. doi: 10.1038/nm1387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: vascular permeability, microglia, leukocyte, inflammation, CSF-brain barrier, neonatal ischemia
Citation: Kratzer I, Chip S and Vexler ZS (2014) Barrier mechanisms in neonatal stroke. Front. Neurosci. 8:359. doi: 10.3389/fnins.2014.00359
Received: 26 August 2014; Accepted: 20 October 2014;
Published online: 07 November 2014.
Edited by:
Helen B. Stolp, King's College London, UKReviewed by:
Jun Zhang, Texas Tech University Health Sciences Center, USAAna A. Baburamani, King's College London, UK
Copyright © 2014 Kratzer, Chip and Vexler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zinaida S. Vexler, Department of Neurology, University of California San Francisco, 675 Nelson Rising Lane, San Francisco, CA 94158-0663, USA e-mail:emVuYS52ZXhsZXJAdWNzZi5lZHU=
†These authors have contributed equally to this work.