 Sahar Al-Mahdawi
Sahar Al-Mahdawi Sara Anjomani Virmouni
Sara Anjomani Virmouni Mark A. Pook
Mark A. Pook- 1Ataxia Research Group, Division of Biosciences, Department of Life Sciences, College of Health and Life Sciences, Brunel University London, Uxbridge, UK
- 2Synthetic Biology Theme, Institute of Environment, Health and Societies, Brunel University London, Uxbridge, UK
DNA methylation primarily occurs within human cells as a 5-methylcytosine (5mC) modification of the cytosine bases in CpG dinucleotides. 5mC has proven to be an important epigenetic mark that is involved in the control of gene transcription for processes such as development and differentiation. However, recent studies have identified an alternative modification, 5-hydroxymethylcytosine (5hmC), which is formed by oxidation of 5mC by ten-eleven translocation (TET) enzymes. The overall levels of 5hmC in the mammalian genome are approximately 10% of 5mC levels, although higher levels have been detected in tissues of the central nervous system (CNS). The functions of 5hmC are not yet fully known, but evidence suggests that 5hmC may be both an intermediate product during the removal of 5mC by passive or active demethylation processes and also an epigenetic modification in its own right, regulating chromatin or transcriptional factors involved in processes such as neurodevelopment or environmental stress response. This review highlights our current understanding of the role that 5hmC plays in neurodegenerative diseases, including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), fragile X-associated tremor/ataxia syndrome (FXTAS), Friedreich ataxia (FRDA), Huntington's disease (HD), and Parkinson's disease (PD).
Introduction
Methylation of mammalian DNA occurs by DNA methyltransferase (DNMT) enzymatic conversion of cytosine residues in CpG dinucleotides to 5-methylcytosine (5mC) (Robertson, 2001). CpG sites are clustered together as CpG islands (CGIs), which locate to distinct gene regions, and the DNA methylation profile of a gene has been shown to have a significant impact upon its level of expression (Bird and Wolffe, 1999). Furthermore, aberrant DNA methylation profiles are known to be associated with many different human diseases, including Rett syndrome (Amir et al., 1999) and cancer (Kulis and Esteller, 2010), where there are alterations of global DNA methylation patterns, and Fragile X syndrome (FXS), where there is specific methylation of the CCG repeat mutation in the fragile X mental retardation-1 (FMR1) gene (Naumann et al., 2009). Abnormal global or localized DNA methylation patterns have also been associated with certain neurodegenerative diseases (Pook, 2012; Lu et al., 2013).
In 2009, two independent studies simultaneously reported the existence of an alternative modification, 5-hydroxymethylcytosine (5hmC), formed due to oxidation of 5mC by ten-eleven translocation (TET) enzymes, a family of 2-oxoglutarate- and Fe(II)-dependent dioxygenases, consisting of three proteins, TET1, TET2, and TET3 (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). Highly conserved homologous proteins have also been identified in mouse, designated Tet1, Tet2, and Tet3. Initial studies demonstrated roles for Tet enzymes, converting 5mC to 5hmC, in pluripotency and developmental reprogramming (Ito et al., 2010; Branco et al., 2011; Ficz et al., 2011; Gu et al., 2011; Inoue and Zhang, 2011). Subsequent studies in mouse embryonic stem (ES) cells suggested that Tet enzymes could control DNA methylation both by the conversion of 5mC to 5hmC and by binding to CpG rich regions to prevent DNMT activity (Xu et al., 2011). More recently, outside of developmental reprogramming, TET1 has been shown to act in differentiated cells as a maintenance demethylase that prevents aberrant methylation spreading into unmethylated or hypomethylated CGIs (Jin et al., 2014). The overall levels of 5hmC in the mammalian genome have been reported to be approximately 10% of 5mC levels (Branco et al., 2011), although higher levels have been detected in tissues of the CNS (Globisch et al., 2010). For example, 5hmC is approximately 40% as abundant as 5mC in the DNA of Purkinje cells of the cerebellum (Kriaucionis and Heintz, 2009). Subsequent to the identification of 5hmC, two other modifications of cytosine have been discovered, 5-formylcytosine (5fC) and 5-carbamylcytosine (5caC). These are less abundant than 5hmC and they are recognized as intermediates generated by TET enzyme activity in the 5hmC to cytosine conversion pathway (Ito et al., 2011).
Several research groups have studied the distribution of 5hmC throughout the genome. In brain tissue, 5hmC is particularly enriched within synaptic genes, exhibiting tissue-specific differences at exon-intron boundaries, suggesting a potential role for 5hmC in the differential splicing of these genes (Khare et al., 2012). Conversely, in human and mouse ES cells, 5hmC is enriched at specific gene bodies, promoters and enhancers, particularly at promoters marked with H3K4me3 and H3K27me3 and at enhancers marked with H3K4me1 and H3K27ac, suggesting a role for 5hmC in the epigenetic regulation of transcription (Pastor et al., 2011; Stroud et al., 2011; Szulwach et al., 2011a). Furthermore, enrichment of 5hmC at both gene bodies of actively transcribed genes and promoter regions of Polycomb-repressed developmental regulator genes has provided evidence of a dual role for 5hmC in pluripotent stem cells (Wu et al., 2011). 5hmC is also found in mitochondrial DNA (Iacobazzi et al., 2013), which has implications for disorders of mitochondrial dysfunction, including neurodegenerative diseases. Due to the CNS-selective tissue distribution of 5hmC and its involvement in epigenetic gene regulation during neurodevelopment, several recent investigations have focused on uncovering potential roles for 5hmC in neurodegenerative diseases. Here we summarize the findings of this recent epigenetic-based neurological research.
The Functions of 5hmC
Although the functions of 5hmC within the cell are not yet fully known, evidence emerging from recent 5hmC protein binding studies supports the existence of multiple roles (Figure 1). Several proteins have now been shown to bind to 5hmC, including UHRF1 (ubiquitin-like, containing PHD, and RING finger domain 1) (Frauer et al., 2011), MBD3 (methyl-CpG binding domain protein 3) (Yildirim et al., 2011), MeCP2 (methyl-CpG binding protein 2) (Mellen et al., 2012), UHRF2 (ubiquitin-like, containing PHD and RING finger domain 2) and a number of other proteins identified by proteomics analysis (Spruijt et al., 2013). These studies suggest that 5hmC is involved, both indirectly and directly, in the dynamic interplay between DNA methylation status and gene transcription.
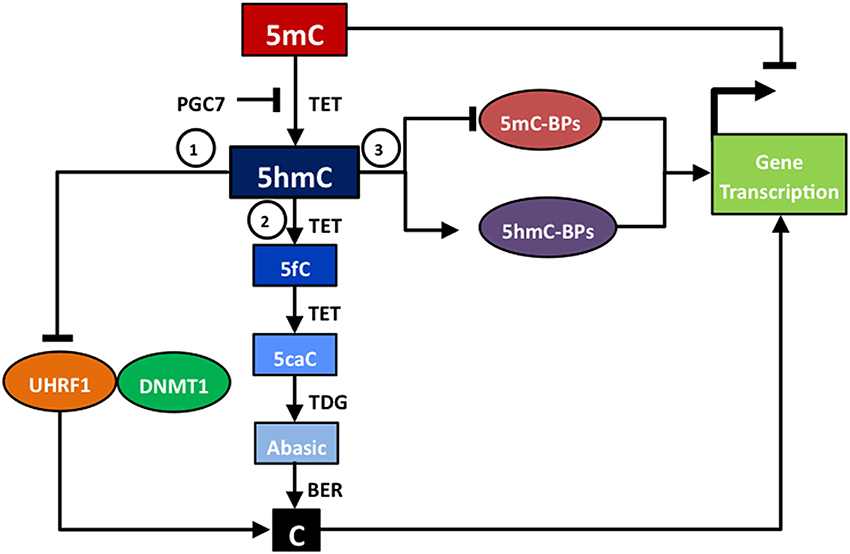
Figure 1. Functions of 5hmC. 5hmC has several different functions that impact upon gene transcription: (1) acting as an intermediate in passive DNA demethylation due to poor binding between 5hmC and UHRF1, the partner of DNMT1; (2) acting as an intermediate in TET/TDG/BER-based active DNA methylation; (3) altering the ratio of 5mC-binding proteins (5mC-BPs) to 5hmC-binding proteins (5hmC-BPs) that impair or activate gene transcription.
Firstly, 5hmC acts as an intermediate in both the passive and the active DNA demethylation conversion of 5mC to cytosine, and thus is indirectly involved in regulating gene transcription. Passive DNA demethylation occurs due to poor binding of UHRF1 to 5hmC. Usually UHRF1 and its partner, DNMT1, act together to bind to 5mC and carry out maintenance DNA methylation (Bostick et al., 2007). However, UHRF1 has a reduced binding affinity for 5hmC compared to 5mC (Frauer et al., 2011), and therefore DNMT1 may not be recruited to maintain levels of DNA methylation and hence passive demethylation occurs. Shortly after the discovery that TET enzymes mediated the conversion of 5mC to 5hmC, other studies revealed that TET enzymes could further oxidize 5hmC to 5fC and then 5caC (Ito et al., 2011). This led to the understanding that active DNA demethylation can occur through sequential stages of TET enzyme-mediated oxidation, 5mC to 5hmC to 5fC to 5caC, followed by the action of TDG to form an abasic site, which is then repaired to a cytosine residue by BER activity (Guo et al., 2011; He et al., 2011; Maiti and Drohat, 2011). An alternative active DNA demethylation pathway that involves deamination of 5hmC to a hydroxyuracil (5hmU) intermediate has also been proposed (Cortellino et al., 2011). In addition, there is preliminary in vitro evidence that the de novo DNA methyltransferases DNMT3A and DNMT3B can act as DNA dehydroxymethylases, which may be able to directly convert 5hmC to cytosine (Chen et al., 2012). Therefore, changes in 5hmC status may simply reflect changes in the biological processes that require DNA demethylation, such as the development of pre-implantation embryos or the reprogramming of primordial germ cells (PGCs) (Kohli and Zhang, 2013). Global DNA demethylation occurs during two stages of embryogenesis: (i) in zygotes where there is preferential DNA demethylation of the parental genome, (ii) in PGCs during the establishment of gender-specific DNA methylation patterns (Inoue and Zhang, 2011). Tet1 is not responsible for global demethylation in PGCs, but has been shown to mediate locus-specific demethylation of a subset of meiotic genes (Yamaguchi et al., 2012) and to have a critical function in the erasure of genomic imprinting (Yamaguchi et al., 2013).
Secondly, 5hmC binds chromatin regulator proteins, which suggests that it is not merely an intermediate in DNA demethylation, but that it can more directly influence the regulation of gene transcription in processes such as neurodevelopment (Szulwach et al., 2011b) or cellular responses to oxidative stress (Chia et al., 2011). For example, 5hmC may modulate the relative binding of methyl-CpG-binding domain proteins, such as MeCP2 and MBD3, to produce a more open chromatin state and activation of gene transcription (Yildirim et al., 2011; Mellen et al., 2012). Furthermore, 5hmC can be associated with, or affected by, particular histone modifications that influence gene transcription. For example, tight correlations of 5hmC localization have been reported with both histone H3K4me2, an epigenetic mark of euchromatin, and H3K27me3, an epigenetic mark of heterochromatin, across a variety of somatic tissues (Haffner et al., 2013; Chen et al., 2014). In addition, recent studies have shown that the conversion of 5mC to 5hmC can be prevented by binding of PGC7 (also known as Dppa3 or Stella) to histone H3K9me2 (Nakamura et al., 2012). Furthermore, as with 5hmC, it is possible that 5fC and 5caC may also have independent functions in the regulation of gene transcription (Raiber et al., 2012).
5hmC, Neurodevelopment and Neurodegenerative Diseases
Several studies have suggested a role for 5hmC in the epigenetic regulation of transcription, mediating brain development and functional maintenance of the adult brain. At the outset, comparatively high levels of 5hmC were detected in CNS tissues, which contain predominantly non-proliferating cells (Globisch et al., 2010). Thus, 5hmC was found to be approximately 40% as abundant as 5mC in the DNA of Purkinje cells of the cerebellum (Kriaucionis and Heintz, 2009). In contrast, loss of global 5hmC has been associated with cancer, suggesting that 5hmC cannot be well maintained in highly proliferating cells (Pfeifer et al., 2013). Throughout the stages of mouse neurodevelopment from embryonic to adult brain, 5hmC has been shown to be not merely an intermediate metabolite of DNA demethylation, but a long-lasting but dynamic epigenetic mark that is distinct from 5mC. Thus, while 5mC differentially binds MBD1 and MeCP2, and recruits H3K9me3 and H3K27me3, 5hmC progressively co-localizes with MBD3 and recruits H3K4me2 (Chen et al., 2014). Furthermore, a positive correlation has been reported between 5hmC levels and human cerebellum development (Wang et al., 2012) and 5hmC has been reported to regulate transcriptional factors involved in neurodevelopment (Szulwach et al., 2011b). Finally, alterations of 5hmC have been implicated in a number of neurodevelopmental diseases, including Rett syndrome, autism spectrum disorders, schizophrenia and fetal alcohol syndrome (Cheng et al., 2014).
Such growing evidence clearly indicates that 5hmC has an important role to play in normal neurodevelopment and maintenance of adult CNS function. Thus, it is intuitive that abnormalities of 5hmC distribution or function may also be important factors for neurodegenerative diseases. Indeed, a genome-wide study of 5hmC distribution in mouse cerebellum has revealed an age-related gene expression level-dependent enrichment of 5hmC in specific gene bodies that are linked to neurodegenerative diseases in mice and humans, including ataxia and disorders of Purkinje cell degeneration (Song et al., 2011b). The following sections describe the specific 5hmC studies that have been performed in relation to individual neurodegenerative diseases.
AD
Alzheimer's disease (AD) is the most common neurodegenerative disorder, characterized by progressive decline of cognitive functions, neuronal cell loss, and two hallmarks of pathology, extracellular amyloid beta plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein (Tanzi, 2012). The causes of AD are unknown, but some evidence has been gained from identifying abnormalities in selected genes, including the beta-amyloid precursor protein gene, APP, and the presenilin genes, PSEN1 and PSEN2. In addition, it has been suggested that there may be alterations of epigenetic factors due to aging or in response to environmental stresses (Wang et al., 2008; Coppieters and Dragunow, 2011; Bihaqi et al., 2012). Several studies have recently investigated the global levels of DNA methylation-based enzymes and chromatin marks, DNMT1, TET1, 5mC, 5hmC, 5fC, and 5caC, in AD brain tissues using a variety of immunohistochemical detection methods, with somewhat contradictory results (Table 1). Initial studies of human brain samples, specifically the entorhinal cortex layer II of the medial temporal lobe, revealed evidence of decreased levels of 5mC and DNMT1 in neurons of AD patients (Mastroeni et al., 2010). Similar decreases in 5mC, together with decreased levels of 5hmC, were subsequently identified in the hippocampal region of AD brains (Chouliaras et al., 2013). However, further studies have shown exactly the opposite effects, reporting increased levels of both 5mC and 5hmC in AD brains (Table 1). Firstly, increased levels of 5mC have been detected in frontal cortex of AD patients (Rao et al., 2012). Secondly, increased levels of TET1, 5mC, and 5hmC, accompanied by decreases in the levels of 5fC and 5caC, have been detected in the hippocampus of AD patients, while no changes were detected in cerebellum tissues (Bradley-Whitman and Lovell, 2013). Finally, increased levels of both 5mC and 5hmC have been detected in the frontal and temporal cortex of AD patients (Coppieters et al., 2014). The reasons for the discrepancies between the different studies will require further investigation, but they may be due to the analysis of different regions of the brain and the use of different immunohistochemical quantification techniques. It will also be interesting to investigate levels of locus-specific 5hmC at specific genes related to AD pathology, such as the APP and PSEN1 genes, which have previously shown AD-related changes in 5mC levels (Rogaev et al., 1994; Tohgi et al., 1999; Barrachina and Ferrer, 2009). Only after such studies will the role of 5hmC in either the causes or consequences of AD pathogenesis become apparent.

Table 1. Alterations of 5hmC in neurodegenerative diseases.
ALS
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by selective loss of motor neurons within the brain and spinal cord (Calvo et al., 2014). The cause of ALS is largely unknown, although defective genes have been identified in a small percentage of familial ALS cases, including those encoding superoxide dismutase 1 (SOD1), TAR DNA-Binding Protein (TARDBP), fused in sarcoma (FUS), Ubiquilin2 (UBQLN2) and C9ORF72 (Chen et al., 2013). For the much larger percentage of sporadic ALS cases, it has been suggested that environmental factors, such as exposure to toxins or dietary factors, may be driving more global epigenetic changes (Ahmed and Wicklund, 2011). Therefore, it is interesting to note that one recent study has reported global increases in both 5mC and 5hmC levels in postmortem sporadic ALS spinal cord, but not in blood samples (Figueroa-Romero et al., 2012) (Table 1). The differences between spinal cord and blood samples suggest that neither 5mC nor 5hmC would currently be suitable as biomarkers of ALS, but further studies will no doubt follow to shed further light onto the role of 5hmC in ALS.
FXTAS
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disease caused by CGG repeat expansion mutations within the 5' untranslated region (UTR) of the FMR1 gene (Hagerman et al., 2001). FXTAS patients carry 55–200 CGG repeats, regarded as premutation alleles, and although they do not exhibit signs of disease in early life, they generally develop severe tremor, ataxia and progressive cognitive decline in the fifth decades of life. In contrast, individuals who carry over 200 CGG repeats, regarded as full mutations, develop FXS, which is the commonest form of inherited mental retardation (Bagni and Oostra, 2013). In FXS, the CGG repeat expansion mutation becomes hyermethylated, as does the CpG island within the FMR1 promoter region, resulting in reduced expression of FMR1 (Naumann et al., 2009). However, in FXTAS there is increased expression of FMR1, and a toxic RNA gain of function is considered to be the primary disease mechanism (Jacquemont et al., 2003; Jin et al., 2003). To the best of our knowledge, there have not yet been any reports describing the levels of 5hmC at the FMR1 locus in either FXS or FXTAS. However, one study has investigated global levels of 5hmC in the rCGG mouse model of FXTAS, which is characterized by overexpression of human CGG repeats within the 5 UTR of the FMR1 gene in Purkinje neuronal cells, leading to cell death and subsequent behavioral deficits (Hashem et al., 2009). The results showed there to be a genome-wide decrease in 5hmC levels in the cerebellum tissues of rCGG mice compared with age-matched wild-type controls, mainly within gene bodies and CGIs (Yao et al., 2014). However, there were also increases of 5hmC levels in repetitive elements and cerebellum-specific enhancers that correlated with genes and transcription factors known to be involved in neurodevelopment (Table 1). These findings strongly suggest a potential function for 5hmC as part of the disease mechanism for FXTAS.
FRDA
Friedreich ataxia (FRDA) is a rare autosomal recessive neurodegenerative disorder caused by GAA repeat expansion mutation within intron 1 of the FXN gene, leading to decreased expression of the essential mitochondrial protein frataxin (Campuzano et al., 1996). The main sites of pathology are the large sensory neurons of the dorsal root ganglia and the dentate nucleus of the cerebellum (Koeppen, 2013), although non-CNS pathologies are also evident, including hypertrophic cardiomyopathy (Weidemann et al., 2013) and diabetes (Cnop et al., 2013). Several studies have identified FRDA-related epigenetic changes, including alterations of DNA methylation status, in the immediate vicinity of the expanded GAA repeats of the FXN gene (Evans-Galea et al., 2013; Sandi et al., 2014). An initial investigation of DNA methylation revealed hypermethylation of specific CpG sites upstream of the GAA repeat in FRDA patient lymphoblastoid cells compared to unaffected controls (Greene et al., 2007). Our group subsequently identified hypermethylation at the upstream GAA repeat region in FRDA postmortem cerebellum and heart tissues (Al-Mahdawi et al., 2008). Interestingly, we also identified reduced hypomethylation in the downstream GAA repeat region in FRDA patient tissues compared with controls. These findings were confirmed in blood and buccal cell samples from a large cohort of FRDA patients, where a significant inverse correlation was also detected between the level of DNA methylation in the upstream GAA region and the level of FXN expression (Evans-Galea et al., 2012). Yet another study has shown that the degree of DNA methylation in the upstream GAA repeat region in FRDA patients correlates with the length of the GAA repeats and inversely correlates with the age of disease onset (Castaldo et al., 2008). Therefore, there is good evidence that DNA methylation may have an, as yet unknown, role to play in the molecular mechanism of FRDA. With this in mind, our group have recently analyzed the 5hmC status of one of the FXN upstream GAA CpG sites in FRDA cerebellum and heart tissues using a restriction enzyme-based procedure that allowed distinction between 5hmC and 5mC and we found that the majority of the hypermethylated DNA at this CpG residue comprises 5hmC rather than 5mC (Al-Mahdawi et al., 2013) (Table 1). It is possible that raised 5hmC levels reflect an attempt to reverse GAA repeat-induced FXN gene silencing, which is marked by increased DNA methylation at the upstream GAA repeat region, rather than indicating any involvement in the cause of disease. Therefore, it will be interesting to see if there are further 5hmC alterations of the FXN gene, or indeed other genes, associated with FRDA.
HD
Huntington's disease (HD) is an autosomal dominant progressive neurodegenerative disease, characterized by chorea, dystonia, and cognitive decline, with the main site of pathology being the striatum. HD is caused by CAG repeat expansion mutation within exon 1 of the HTT gene, leading to abnormal polyglutamine formation within the amino-terminus of the HTT protein (MacDonald et al., 2003). The mechanism of disease is unknown, although many studies have provided evidence for polyglutamine or RNA toxic gains of function as well as haploinsufficiency or alternative splicing of the HTT gene as potential causes (Landles and Bates, 2004; Sathasivam et al., 2013). More recently, evidence has been put forward from two studies to suggest the potential involvement of 5hmC in HD by two distinct mechanisms (Table 1). Firstly, increased levels of 5mC and decreased levels of 5hmC were identified in the 5' UTR of the ADORA2A gene in the striatum (specifically the putamen) of HD patients compared with age-matched controls (Villar-Menendez et al., 2013). The ADORA2A gene encodes the adenosine A2A receptor, a G-protein-coupled receptor that is normally highly expressed in the basal ganglia, but severely reduced in HD (Glass et al., 2000). Secondly, genome-wide loss of 5hmC has been reported in YAC128 HD mouse striatum and cortex brain tissues compared with age-matched wild-type controls (Wang et al., 2013). A closer inspection of this data revealed 747 differentially hydroxymethylated regions in the striatum of which 49 showed HD-related increases of 5hmC, enriched in gene bodies and positively correlated with gene transcription, and 698 showed HD-related decreases of 5hmC. The authors speculate that 5hmC is a novel epigenetic feature in HD, involved in neurogenesis, neuronal function and survival in HD brain.
PD
Parkinson's disease (PD) is the second most common neurodegenerative disease after AD, characterized by the progressive loss of substantia nigra dopaminergic neurons, resulting in muscle rigidity, bradykinesia, tremor, and instability. The causes of PD are unknown, but mutations in several genes have now been identified in rare cases of inherited PD, including the genes SNCA (alpha-synuclein), PARK2 (parkin), PTEN induced Putative Kinase 1 (PINK1), PARK7 (DJ-1), Leucine Rich Repeat Kinase 2 (LRRK2) and ATP13A2 (Yang et al., 2009). Previous studies have suggested the potential involvement of DNA methylation in PD, particularly with regards to regulation of SNCA gene expression, but without solid conclusions (Lu et al., 2013). More recently, 5hmC levels have been studied in striatal brain tissues of the 6-OHDA induced rat model of PD, but while 5hmC content generally increased with age, no changes in 5hmC levels were detected compared with controls (Zhang et al., 2013). Therefore, any potential involvement of 5hmC in the causes or consequences of PD remain to be identified.
Conclusions: Implications for Diagnosis and Therapy of Neurodegenerative Diseases
To date, the investigations of 5hmC in relation to neurodegenerative diseases can be considered to be within their early days. Of the few studies that have been performed, no common features have emerged regarding 5hmC alterations and neurodegenerative diseases; both global increases and decreases of 5hmC levels have been identified in different diseases, and in the case of AD, within the same disease. It is likely that poorly reproducible techniques of immunohistochemical quantification have contributed to this lack of clarity. One impetus behind the recent studies of 5hmC is the potential to identify changes in 5hmC as a potential biomarker of neurodegenerative disease. Alterations of 5hmC levels may be useful, irrespective of whether they are considered to be a cause or a consequence of the disease process. However, initial studies of sporadic ALS have revealed no correlation between alterations of 5hmC, or indeed in 5mC, in spinal cord and blood, suggesting that these would not be suitable as biomarkers of this neurodegenerative disease (Figueroa-Romero et al., 2012). Another driver behind 5hmC studies of neurodegenerative disease is the potential to identify novel targets for therapy. In this case, alterations of 5hmC levels would have to be involved in the molecular disease process rather than being a consequence of disease. However, if 5hmC does indeed prove to be a significant epigenetic mark involved in the causation of neurodegenerative disease, then it may be possible to develop drugs that modify the 5hmC status, either to decrease 5hmC levels by inhibition of TET enzyme activity (Xiao et al., 2012) or to increase 5hmC levels by enhancement of TET enzyme activity (Yin et al., 2013). However, further investigations of 5hmC alterations are first required for all neurodegenerative diseases, both at global and locus-specific levels. Such studies will be enhanced by the development of state of the art technologies, such as the recently described third-generation sequencing and oxidative bisulfite (oxBs) BeadChip platforms (Song et al., 2011a; Stewart et al., 2014).
Author Contributions
All authors contributed to draft the manuscript and all authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sara Anjomani Virmouni was supported by funding to Mark A. Pook from the Friedreich's Ataxia Research Alliance (FARA).
References
Ahmed, A., and Wicklund, M. P. (2011). Amyotrophic lateral sclerosis: what role does environment play? Neurol. Clin. 29, 689–711. doi: 10.1016/j.ncl.2011.06.001
Al-Mahdawi, S., Pinto, R. M., Ismail, O., Varshney, D., Lymperi, S., Sandi, C., et al. (2008). The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 17, 735–746. doi: 10.1093/hmg/ddm346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Al-Mahdawi, S., Sandi, C., Mouro Pinto, R., and Pook, M. A. (2013). Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS ONE 8:e74956. doi: 10.1371/journal.pone.0074956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amir, R. E., Van den Veyver, I.B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bagni, C., and Oostra, B. A. (2013). Fragile X syndrome: from protein function to therapy. Am. J. Med. Genet. A 161A, 2809–2821. doi: 10.1002/ajmg.a.36241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrachina, M., and Ferrer, I. (2009). DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 68, 880–891. doi: 10.1097/NEN.0b013e3181af2e46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bihaqi, S. W., Schumacher, A., Maloney, B., Lahiri, D. K., and Zawia, N. H. (2012). Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): towards a new paradigm. Curr. Alzheimer Res. 9, 574–588. doi: 10.2174/156720512800617982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bird, A. P., and Wolffe, A. P. (1999). Methylation-induced repression–belts, braces, and chromatin. Cell 99, 451–454 doi: 10.1016/S0092-8674(00)81532-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bostick, M., Kim, J. K., Esteve, P. O., Clark, A., Pradhan, S., and Jacobsen, S. E. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764. doi: 10.1126/science.1147939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bradley-Whitman, M. A., and Lovell, M. A. (2013). Epigenetic changes in the progression of Alzheimer's disease. Mech. Ageing Dev. 134, 486–495. doi: 10.1016/j.mad.2013.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Branco, M. R., Ficz, G., and Reik, W. (2011). Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 13, 7–13. doi: 10.1038/nrg3080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calvo, A. C., Manzano, R., Mendonca, D. M., Munoz, M. J., Zaragoza, P., and Osta, R. (2014). Amyotrophic lateral sclerosis: a focus on disease progression. Biomed Res. Int. 2014:925101. doi: 10.1155/2014/925101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campuzano, V., Montermini, L., Molto, M. D., Pianese, L., Cossee, M., Cavalcanti, F., et al. (1996). Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 doi: 10.1126/science.271.5254.1423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castaldo, I., Pinelli, M., Monticelli, A., Acquaviva, F., Giacchetti, M., Filla, A., et al. (2008). DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 45, 808–812. doi: 10.1136/jmg.2008.058594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C. C., Wang, K. Y., and Shen, C. K. (2012). The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J. Biol. Chem. 287, 33116–33121. doi: 10.1074/jbc.C112.406975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., Sayana, P., Zhang, X., and Le, W. (2013). Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener. 8:28. doi: 10.1186/1750-1326-8-28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Damayanti, N. P., Irudayaraj, J., Dunn, K., and Zhou, F. C. (2014). Diversity of two forms of DNA methylation in the brain. Front. Genet. 5:46. doi: 10.3389/fgene.2014.00046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, Y., Bernstein, A., Chen, D., and Jin, P. (2014). 5-Hydroxymethylcytosine: a new player in brain disorders? Exp. Neurol. doi: 10.1016/j.expneurol.2014.05.008. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chia, N., Wang, L., Lu, X., Senut, M. C., Brenner, C., and Ruden, D. M. (2011). Hypothesis: environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 6, 853–856. doi: 10.4161/epi.6.7.16461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cnop, M., Mulder, H., and Igoillo-Esteve, M. (2013). Diabetes in Friedreich ataxia. J. Neurochem. 126(Suppl. 1), 94–102. doi: 10.1111/jnc.12216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coppieters, N., Dieriks, B. V., Lill, C., Faull, R. L., Curtis, M. A., and Dragunow, M. (2014). Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol. Aging 35, 1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coppieters, N., and Dragunow, M. (2011). Epigenetics in Alzheimer's disease: a focus on DNA modifications. Curr. Pharm. Des. 17, 3398–3412. doi: 10.2174/138161211798072544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortellino, S., Xu, J., Sannai, M., Moore, R., Caretti, E., Cigliano, A., et al. (2011). Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79. doi: 10.1016/j.cell.2011.06.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans-Galea, M. V., Carrodus, N., Rowley, S. M., Corben, L. A., Tai, G., Saffery, R., et al. (2012). FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 71, 487–497. doi: 10.1002/ana.22671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans-Galea, M. V., Hannan, A. J., Carrodus, N., Delatycki, M. B., and Saffery, R. (2013). Epigenetic modifications in trinucleotide repeat diseases. Trends Mol. Med. 19, 655–663. doi: 10.1016/j.molmed.2013.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ficz, G., Branco, M. R., Seisenberger, S., Santos, F., Krueger, F., Hore, T. A., et al. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473, 398–402. doi: 10.1038/nature10008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Figueroa-Romero, C., Hur, J., Bender, D. E., Delaney, C. E., Cataldo, M. D., Smith, A. L., et al. (2012). Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 7:e52672. doi: 10.1371/journal.pone.0052672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frauer, C., Hoffmann, T., Bultmann, S., Casa, V., Cardoso, M. C., Antes, I., et al. (2011). Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE 6:e21306. doi: 10.1371/journal.pone.0021306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glass, M., Dragunow, M., and Faull, R. L. (2000). The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 97, 505–519. doi: 10.1016/S0306-4522(00)00008-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Globisch, D., Munzel, M., Muller, M., Michalakis, S., Wagner, M., Koch, S., et al. (2010). Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5:e15367. doi: 10.1371/journal.pone.0015367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greene, E., Mahishi, L., Entezam, A., Kumari, D., and Usdin, K. (2007). Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 35, 3383–3390. doi: 10.1093/nar/gkm271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, T. P., Guo, F., Yang, H., Wu, H. P., Xu, G. F., Liu, W., et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. doi: 10.1038/nature10443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haffner, M. C., Pellakuru, L. G., Ghosh, S., Lotan, T. L., Nelson, W. G., De Marzo, A. M., et al. (2013). Tight correlation of 5-hydroxymethylcytosine and Polycomb marks in health and disease. Cell Cycle 12, 1835–1841. doi: 10.4161/cc.25010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, R. J., Leehey, M., Heinrichs, W., Tassone, F., Wilson, R., Hills, J., et al. (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130 doi: 10.1212/WNL.57.1.127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashem, V., Galloway, J. N., Mori, M., Willemsen, R., Oostra, B. A., Paylor, R., et al. (2009). Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet. 18, 2443–2451. doi: 10.1093/hmg/ddp182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iacobazzi, V., Castegna, A., Infantino, V., and Andria, G. (2013). Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 110, 25–34. doi: 10.1016/j.ymgme.2013.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, A., and Zhang, Y. (2011). Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334, 194. doi: 10.1126/science.1212483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, S., D'Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. doi: 10.1038/nature09303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi: 10.1126/science.1210597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquemont, S., Hagerman, R. J., Leehey, M., Grigsby, J., Zhang, L., Brunberg, J. A., et al. (2003). Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 72, 869–878. doi: 10.1086/374321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, C., Lu, Y., Jelinek, J., Liang, S., Estecio, M. R., Barton, M. C., et al. (2014). TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 42, 6956–6971. doi: 10.1093/nar/gku372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, P., Zarnescu, D. C., Zhang, F., Pearson, C. E., Lucchesi, J. C., Moses, K., et al. (2003). RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39, 739–747. doi: 10.1016/S0896-6273(03)00533-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khare, T., Pai, S., Koncevicius, K., Pal, M., Kriukiene, E., Liutkeviciute, Z., et al. (2012). 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 19, 1037–1043. doi: 10.1038/nsmb.2372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koeppen, A. H. (2013). Nikolaus Friedreich and degenerative atrophy of the dorsal columns of the spinal cord. J. Neurochem. 126(Suppl. 1), 4. doi: 10.1111/jnc.12218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kulis, M., and Esteller, M. (2010). DNA methylation and cancer. Adv. Genet. 70, 27–56. doi: 10.1016/B978-0-12-380866-0.60002-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landles, C., and Bates, G. P. (2004). Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 5, 958–963. doi: 10.1038/sj.embor.7400250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, H., Liu, X., Deng, Y., and Qing, H. (2013). DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 5:85. doi: 10.3389/fnagi.2013.00085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacDonald, M. E., Gines, S., Gusella, J. F., and Wheeler, V. C. (2003). Huntington's disease. Neuromolecular Med. 4, 7–20. doi: 10.1385/NMM:4:1-2:7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maiti, A., and Drohat, A. C. (2011). Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338. doi: 10.1074/jbc.C111.284620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., and Rogers, J. (2010). Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol. Aging 31, 2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mellen, M., Ayata, P., Dewell, S., Kriaucionis, S., and Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. doi: 10.1016/j.cell.2012.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., Liu, Y. J., Nakashima, H., Umehara, H., Inoue, K., Matoba, S., et al. (2012). PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 486, 415–419. doi: 10.1038/nature11093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Naumann, A., Hochstein, N., Weber, S., Fanning, E., and Doerfler, W. (2009). A distinct DNA-methylation boundary in the 5'- upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am. J. Hum. Genet. 85, 606–616. doi: 10.1016/j.ajhg.2009.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397. doi: 10.1038/nature10102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeifer, G. P., Kadam, S., and Jin, S. G. (2013). 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin. 6:10. doi: 10.1186/1756-8935-6-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pook, M. A. (2012). “DNA methylation and trinucleotide repeat expansion diseases,” in DNA Methylation - From Genomics to Technology, ed T. Tatarinova (InTech.). doi: 10.5772/34169
Raiber, E. A., Beraldi, D., Ficz, G., Burgess, H. E., Branco, M. R., Murat, P., et al. (2012). Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 13:R69. doi: 10.1186/gb-2012-13-8-r69
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, J. S., Keleshian, V. L., Klein, S., and Rapoport, S. I. (2012). Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients. Transl. Psychiatry 2, e132. doi: 10.1038/tp.2012.55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, K. D. (2001). DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155. doi: 10.1038/sj.onc.1204341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogaev, E. I., Lukiw, W. J., Lavrushina, O., Rogaeva, E. A., and St. George-Hyslop, P. H. (1994). The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics 22, 340–347. doi: 10.1006/geno.1994.1393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sandi, C., Sandi, M., Anjomani Virmouni, S., Al-Mahdawi, S., and Pook, M. A. (2014). Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 5:165. doi: 10.3389/fgene.2014.00165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sathasivam, K., Neueder, A., Gipson, T. A., Landles, C., Benjamin, A. C., Bondulich, M. K., et al. (2013). Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370. doi: 10.1073/pnas.1221891110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, C. X., Clark, T. A., Lu, X. Y., Kislyuk, A., Dai, Q., Turner, S. W., et al. (2011a). Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods 9, 75–77. doi: 10.1038/nmeth.1779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011b). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72. doi: 10.1038/nbt.1732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spruijt, C. G., Gnerlich, F., Smits, A. H., Pfaffeneder, T., Jansen, P. W., Bauer, C., et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159. doi: 10.1016/j.cell.2013.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stewart, S. K., Morris, T. J., Guilhamon, P., Bulstrode, H., Bachman, M., Balasubramanian, S., et al. (2014). oxBS-450K: a method for analysing hydroxymethylation using 450K BeadChips. Methods. doi: 10.1016/j.ymeth.2014.08.009. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stroud, H., Feng, S., Morey Kinney, S., Pradhan, S., and Jacobsen, S. E. (2011). 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 12:R54. doi: 10.1186/gb-2011-12-6-r54
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Han, J. W., Kim, S., et al. (2011a). Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 7:e1002154. doi: 10.1371/journal.pgen.1002154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011b). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, 1–10. doi: 10.1101/cshperspect.a006296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tohgi, H., Utsugisawa, K., Nagane, Y., Yoshimura, M., Genda, Y., and Ukitsu, M. (1999). Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 70, 288–292. doi: 10.1016/S0169-328X(99)00163-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villar-Menendez, I., Blanch, M., Tyebji, S., Pereira-Veiga, T., Albasanz, J. L., Martin, M., et al. (2013). Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. Neuromolecular Med. 15, 295–309. doi: 10.1007/s12017-013-8219-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, F., Yang, Y., Lin, X., Wang, J. Q., Wu, Y. S., Xie, W., et al. (2013). Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington's disease. Hum. Mol. Genet. 22, 3641–3653. doi: 10.1093/hmg/ddt214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S. C., Oelze, B., and Schumacher, A. (2008). Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS ONE 3:e2698. doi: 10.1371/journal.pone.0002698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Pan, Q., Lin, L., Szulwach, K. E., Song, C. X., He, C., et al. (2012). Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet. 21, 5500–5510. doi: 10.1093/hmg/dds394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weidemann, F., Stork, S., Liu, D., Hu, K., Herrmann, S., Ertl, G., et al. (2013). Cardiomyopathy of Friedreich ataxia. J. Neurochem. 126(Suppl. 1), 88–93. doi: 10.1111/jnc.12217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, H., D'Alessio, A. C., Ito, S., Wang, Z., Cui, K., Zhao, K., et al. (2011). Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 25, 679–684. doi: 10.1101/gad.2036011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. doi: 10.1101/gad.191056.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Wu, F., Tan, L., Kong, L., Xiong, L., Deng, J., et al. (2011). Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell 42, 451–464. doi: 10.1016/j.molcel.2011.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, S., Hong, K., Liu, R., Shen, L., Inoue, A., Diep, D., et al. (2012). Tet1 controls meiosis by regulating meiotic gene expression. Nature 492, 443–447. doi: 10.1038/nature11709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, S., Shen, L., Liu, Y., Sendler, D., and Zhang, Y. (2013). Role of Tet1 in erasure of genomic imprinting. Nature 504, 460–464. doi: 10.1038/nature12805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Y. X., Wood, N. W., and Latchman, D. S. (2009). Molecular basis of Parkinson's disease. Neuroreport 20, 150–156. doi: 10.1097/WNR.0b013e32831c50df
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yao, B., Lin, L., Street, R. C., Zalewski, Z. A., Galloway, J. N., Wu, H., et al. (2014). Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 23, 1095–1107. doi: 10.1093/hmg/ddt504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yildirim, O., Li, R., Hung, J. H., Chen, P. B., Dong, X., Ee, L. S., et al. (2011). Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510. doi: 10.1016/j.cell.2011.11.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, R., Mao, S. Q., Zhao, B., Chong, Z., Yang, Y., Zhao, C., et al. (2013). Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 135, 10396–10403. doi: 10.1021/ja4028346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: 5-hydroxymethylcytosine, Alzheimer's disease, amyotrophic lateral sclerosis, fragile X-associated tremor/ataxia syndrome, Friedreich ataxia, Huntington's disease, Parkinson's disease
Citation: Al-Mahdawi S, Anjomani Virmouni S and Pook MA (2014) The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front. Neurosci. 8:397. doi: 10.3389/fnins.2014.00397
Received: 01 October 2014; Accepted: 18 November 2014;
Published online: 05 December 2014.
Edited by:
Marguerite Evans-Galea, Murdoch Childrens Research Institute, AustraliaReviewed by:
Beth Wilmot, Oregon Health & Science University, USAFang He, University of Michigan, USA
Copyright © 2014 Al-Mahdawi, Anjomani Virmouni and Pook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark A. Pook, Ataxia Research Group, Division of Biosciences, Department of Life Sciences, College of Health and Life Sciences, Brunel University London, Kingston Lane, Uxbridge, Middlesex UB8 3PH, UK e-mail:bWFyay5wb29rQGJydW5lbC5hYy51aw==