 Sherry-Ann Brown
Sherry-Ann Brown Leslie M. Loew
Leslie M. Loew- 1Department of Medicine, Mayo Clinic, Rochester, MN, USA
- 2Richard D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, Farmington, CT, USA
A suite of models was developed to study the role of inositol 1,4,5-trisphosphate receptor 1 (IP3R1) in spinocerebellar ataxias (SCAs). Several SCAs are linked to reduced abundance of IP3R1 or to supranormal sensitivity of the receptor to activation by its ligand inositol 1,4,5-trisphosphate (IP3). Detailed multidimensional models have been created to simulate biochemical calcium signaling and membrane electrophysiology in cerebellar Purkinje neurons. In these models, IP3R1-mediated calcium release is allowed to interact with ion channel response on the cell membrane. Experimental findings in mice and clinical observations in humans provide data input for the models. The SCA modeling suite helps interpret experimental results and provides suggestions to guide experiments. The models predict IP3R1 supersensitivity in SCA1 and compensatory mechanisms in SCA1, SCA2, and SCA3. Simulations explain the impact of calcium buffer proteins. Results show that IP3R1-mediated calcium release activates voltage-gated calcium-activated potassium channels in the plasma membrane. The SCA modeling suite unifies observations from experiments in a number of SCAs. The cadre of simulations demonstrates the central role of IP3R1.
Introduction
Several classes of spinocerebellar ataxia in humans and mice are associated with reduced IP3R1 levels (Matsumoto et al., 1996; Street et al., 1997; Zecevic et al., 1999; Lin et al., 2000; Ogura et al., 2001; Serra et al., 2004; Kurnellas et al., 2007; van de Leemput et al., 2007; Chou et al., 2008; Novak et al., 2010b; Castrioto et al., 2011; Marelli et al., 2011; Huang et al., 2012; Obayashi et al., 2012; Hansen et al., 2013; Sugawara et al., 2013) or increased sensitivity of the receptor to IP3 (Inoue et al., 2001; Chen et al., 2008; Liu et al., 2009), or both. Such disorders may be termed “IP3R1-associated ataxias” (Brown and Loew, 2009, 2012), which also encompasses ataxias with yet undetermined IP3R1 characteristics (e.g., Huang et al., 2012; Conroy et al., 2014). Beyond IP3R1-associated ataxias, a vast majority of ataxias converge on IP3R1-dependent signaling (Figure 1). Schorge et al. argue that the unifying feature of many cerebellar ataxias is their impact on IP3R1 (Schorge et al., 2010).
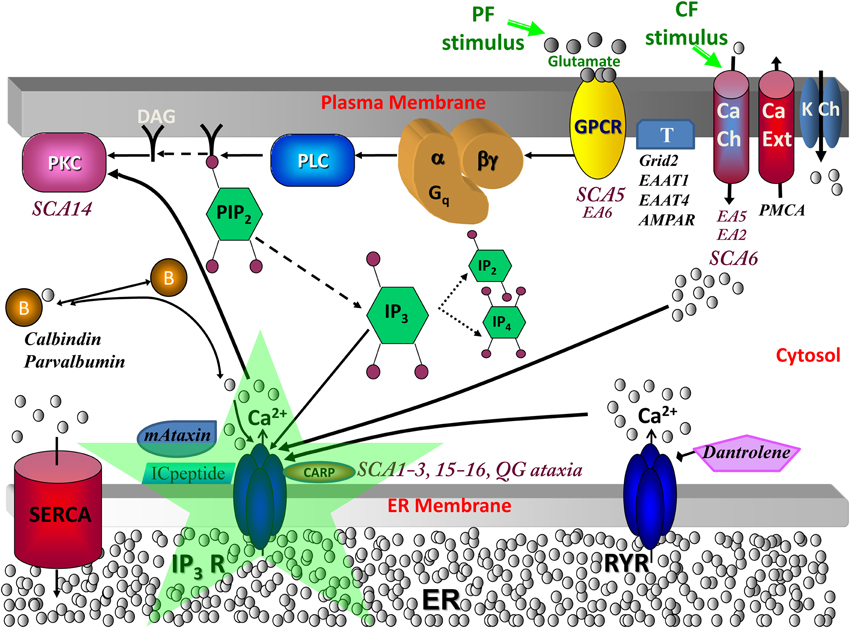
Figure 1. Calcium signaling in cerebellar Purkinje neuron spiny dendrites with various molecules involved in spinocerebellar ataxias. When a parallel fiber (PF) stimulates a distal dendrite of the Purkinje neuron, the neurotransmitter glutamate is released and binds its receptor, metabotropic glutamate receptor (mGluR; from the group C family of G protein-coupled receptors, GPCR), on the plasma membrane of the Purkinje dendrite (Finch and Augustine, 1998; Takechi et al., 1998). This leads to the activation of phospholipase C (PLC), a hydrolyzing enzyme that cleaves PIP2 in the plasma membrane to produce diacylglycerol (DAG) and IP3 (Cohen, 2003). DAG stays in the membrane, where it can activate cytosolic molecules close to the membrane, such as PKC (Nishizuka, 1984). IP3 diffuses away from the reaction site (the plasma membrane) to its receptor on the endoplasmic reticulum (ER) (Berridge et al., 2000). IP3 is also free to diffuse out of the spine head, through the spine neck, into the adjacent dendrite, and along the length of dendrite nearby, part of the dendritic reservoir. At the ER, IP3 binds its receptor, IP3R; the channel opens, releasing calcium from the internal store into the cytosol. This rise in calcium can activate calcium-dependent proteins, including calmodulin, various kinases (including PKC), and other enzymes. The climbing fiber (CF) stimulus depolarizes the entire neuron membrane, opening the voltage-gated calcium channels (Ito et al., 1982). The calcium that flows into the Purkinje neuron spine can also bind IP3R1, along with IP3 from parallel fiber activation of the spine, leading to supralinear calcium release. SCA, spinocerebellar ataxia; EA, episodic ataxia; QG, quadrupedal gait ataxia; K Ch, potassium channels including Kv1.1 (mutated in Episodic Ataxia type 1; Imbrici et al., 2003), Kv3.3 (mutated in SCA13; Waters et al., 2006), Kv4.3 (mutated in SCA1; Duarri et al., 2012 and SCA22 Lee et al., 2012), BK and SK, which are respectively, the large (big) conductance calcium-activated voltage-gated potassium channel which is the potassium channel involved in Purkinje membrane repolarization, and the small conductance calcium-activated voltage-independent potassium channel; DAG, diacylglycerol, which is a product of PLC hydrolysis that along with calcium co-activates PKC; PIP2, Phosphatidylinositol 4,5-bisphosphate, a plasma membrane phospholipid of the inner leaflet that gives rise to DAG and IP3 when hydrolyzed; PLC, phospholipase C, an enzyme that hydrolyzes PIP2 when activated by G-βγ from mGluR; mGluR or Grm1, metabotropic glutamate receptor type 1 (Guergueltcheva et al., 2012); T, other glutamate transporters and receptors including Grid2, Excitatory amino-acid transporter type 1 (EAAT1; mutated in Episodic Ataxia type 6, de Vries et al., 2009), Excitatory amino-acid transporter type 4 (EAAT4; Spectrin β, an anchor for EAAT4 and Grm1 is mutated in SCA5, Ikeda et al., 2006), and AMPAR; B, calcium-binding proteins or buffers including calbindin and parvalbumin (Supplemental Material, S1 Calcium buffers in SCA and Supplemental Figure S2); mAtaxin, mutant ataxin proteins including Ataxin-1 through Ataxin-7; ICpeptide, peptides that resemble the tip of IP3R1 and thereby competitively bind mAtaxin; SERCA, sarcoendoplasmic reticulum calcium ATPase, a transporter for calcium entry from the cytosol to the smooth endoplasmic reticulum (ER); CARP, Carbonic anhydrase-related protein (particularly CARP VIII), an IP3R1 antagonist (Türkmen et al., 2009) (Supplemental Material, S4 IP3R1 suppression by CARP); RYR, ryanodine receptor, a transporter of calcium from the ER to the cytosol in dendrites (but not present in spines) in response to binding of specific ligands such as ryanodine; Dantrolene, a drug that inhibits RYR (Supplemental Material, S2 Calcium-induced calcium release crosstalk); PMCA, Plasma membrane calcium ATP-ase transports calcium out of the cell; Ca Ch, calcium channels including store-operate channels (SOC) for store-operated calcium entry and Cav2.1, which is the main P-type calcium channel in PCs with nonsense/missense mutations causing episodic ataxia type 2, expansion of CAG repeats causing SCA6, and mutations in CavB4 an accessory subunit for Cav2.1 causing EA5 (Escayg et al., 2000); IP3R, inositol trisphosphate receptor (mutated in SCA15/16 and altered sensitivity in SCA1-3, antagonized in QG ataxia), intracellular calcium release channel on the endoplasmic reticulum gated by IP3; PKC, protein kinase C (mutated in SCA14) expressed in Purkinje neurons helps control expression of surface molecules including AMPAR. Adapted from Hernjak et al. (2005).
The SCA modeling suite is a collection of well-mixed (compartmental) and spatial (1D, 3D) computational models that simulate the biochemical and electrophysiological properties of the cerebellar Purkinje neuron involving various calcium signaling and ion channel molecules in constructed or experimentally derived geometries (Supplemental Material, Supplemental Figure S1 and Supplemental Table S1). The SCA suite examines the role of IP3R1 in SCA pathophysiology (Brown and Loew, 2012), with potential for translational studies.
Novel Predictions from the SCA Modeling Suite
Coincidence Detection Time Window at Spine IP3R1
Cerebellar Purkinje neurons receive input from more than 150,000 granule cell axons (parallel fibers), leading to hydrolysis of PIP2 and subsequent IP3-mediated calcium release from the endoplasmic reticulum (ER) (Finch and Augustine, 1998; Takechi et al., 1998; Berridge et al., 2000; Cohen, 2003) (Figure 1). Models 1–7 explored PIP2 signaling and IP3 production (Xu et al., 2003; Hernjak et al., 2005; Brown et al., 2008). Results from Models 4–7 indicated that baseline PIP2 levels are insufficient for requisite IP3 production in the spine, even with apparent anomalous lateral diffusion of PIP2 from the neighboring dendrite (Brown and Loew, 2012). On average, the Purkinje neuron has approximately 14 spines per micron of dendrite (Harris and Stevens, 1988). Each spine is attached to the dendrite branchlet by a neck with diverse morphology (Harris and Stevens, 1989) (see Supplemental Material, S3 IP3R1 in dendritic formation and spine morphology). Model 6 results showed that spine necks of varying radii and lengths also restricted diffusion of produced IP3 out of the spine head (Brown et al., 2008). This supported experimental results from Santamaria et al. with IP3 diffusing more slowly in spiny dendrites than in aspiny dendrites (Santamaria et al., 2006). This suggested that spines might compartmentalize IP3 via spine necks. Simulation results from Model 2 suggested local PIP2 sequestration as a likely source of sufficient PIP2, to fine-tune an experimentally observed (Wang et al., 2000; Sarkisov and Wang, 2008) time window between PF and climbing fiber (CF; from the inferior olive) activation of the Purkinje neuron spine. Stimulation from a single CF innervating the Purkinje neuron cell body and proximal dendrites leads to calcium influx across the plasma membrane, through voltage-gated calcium channels (Ito et al., 1982) (Figure 1). Calcium binding of IP3R1 increases open probability of the receptor (Fiala et al., 1996). IP3R1 serves as the gate-keeper for IP3-induced calcium release. Thus, coincidence detection at IP3R1 leads to more calcium release than with activation of IP3R1 by IP3 or calcium alone (Wang et al., 2000; Hernjak et al., 2005; Ogasawara et al., 2008; Sarkisov and Wang, 2008; Brown et al., 2011).
“Compensatory Pathology” in a Mouse Model of Human SCA1
Mutant Ataxin-1, Ataxin-2, and Ataxin-3 have multiple polyglutamine (CAG, polyQ) repeats (Orr et al., 1993; Kawaguchi et al., 1994; Pulst et al., 1996). Corresponding mouse models exhibit complex behavior with reduced IP3R1 abundance (Lin et al., 2000; Serra et al., 2004; Chou et al., 2008; Hansen et al., 2013) juxtaposed with IP3R1 supersensitivity (Inoue et al., 2001; Serra et al., 2004; Chen et al., 2008; Chou et al., 2008; Brown and Loew, 2012). IP3R1 supersensitivity has been directly shown in SCA2 and SCA3 (Chen et al., 2008; Liu et al., 2009), and speculated in SCA1 (Liu et al., 2009; Kasumu and Bezprozvanny, 2012) (Table 1). All three ataxic mouse models associate with supranormal calcium release (Inoue et al., 2001; Chen et al., 2008; Liu et al., 2009). Models 11 and 12 in the SCA modeling suite support the plausibility of IP3R1 supersensitivity as a necessary component to the Purkinje neuron dysfunction observed in SCA1 (Brown and Loew, 2012).
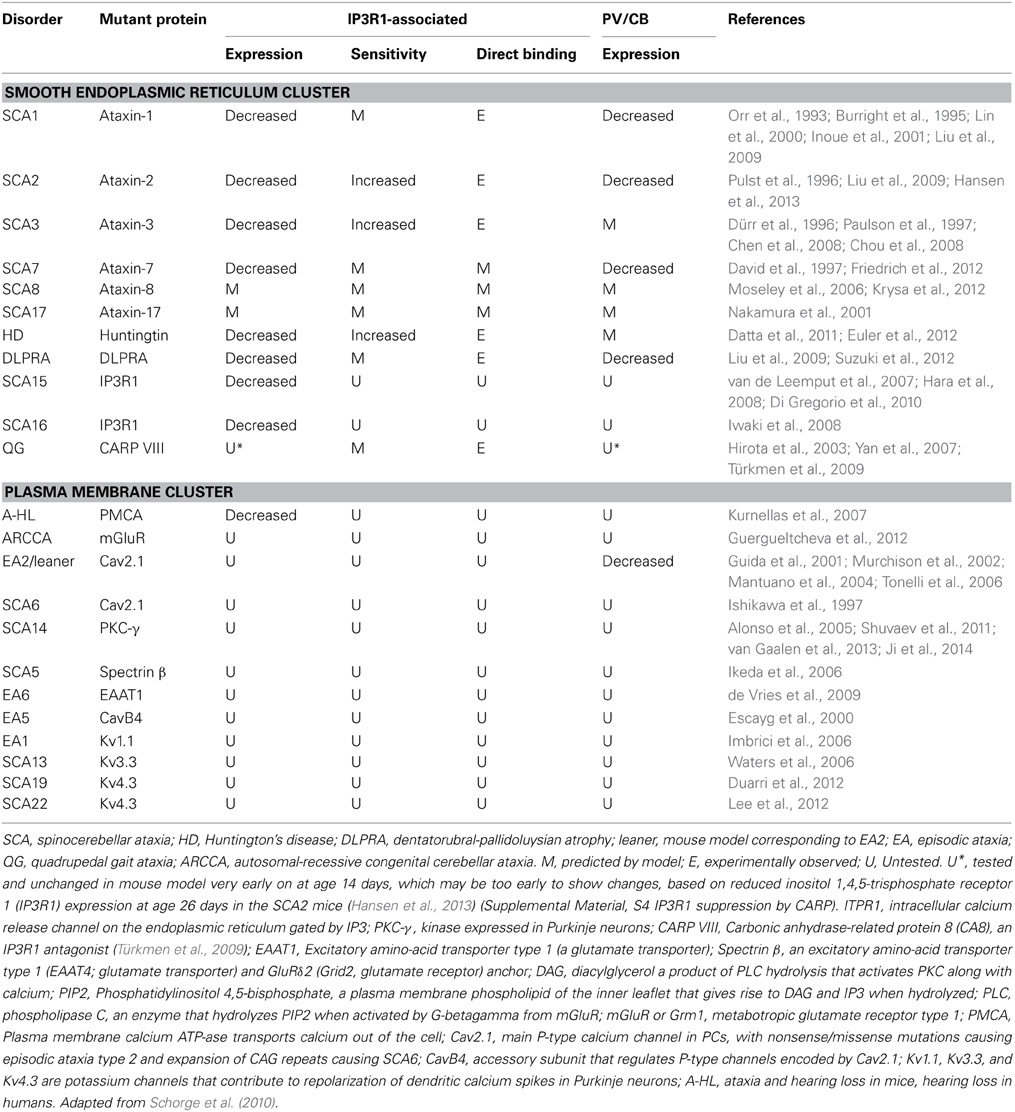
Table 1. Neurological disorders in humans and mice involving IP3R1, calbindin (CB), parvalbumin (PV), and other calcium signaling proteins.
One of many common denominators for downregulation of calcium homeostasis and glutamatergic signaling proteins in polyQ disorders (e.g., Orr et al., 1993; Kawaguchi et al., 1994; Koide et al., 1994, 1999; Trottier et al., 1994; Pulst et al., 1996; David et al., 1997; Nakamura et al., 2001) is transcription factor retinoid acid receptor-related orphan receptor alpha (RORα) (Serra et al., 2006; Gehrking et al., 2011; Euler et al., 2012). Model 12 results suggest that downregulation of IP3R1, sarcoendoplasmic reticulum calcium ATPase (SERCA), homer, and various other glutamatergic signaling proteins (Serra et al., 2004; Chou et al., 2008) in SCA1, SCA2, and SCA3 may compensate for IP3R1 supersensitivity (Brown and Loew, 2012). IP3R1 expression and sensitivity therefore cannot be considered independently in these polyQ ataxias, but interdependently. Huntington's disease and dentatorubral-pallidoluysian atrophy (DLPRA) are related polyQ disorders with increased IP3R1 sensitivity and reduced IP3R1 abundance (Liu et al., 2009; Datta et al., 2011; Euler et al., 2012; Suzuki et al., 2012). Although the phenotype in these disorders is very different, Model 12 predicts that downregulation of IP3R1 should compensate for supersensitivity. Conversely, Model 12 suggests that downregulation of calcium buffers such as calbindin and parvalbumin worsen pathology with potential feedback and feedforward loops or network motifs (Brown and Loew, 2012) (Supplemental Material, S1 Calcium buffers in SCA and Supplemental Figure S2). This is supported by experimental observations (Vig et al., 1998, 2001). Upregulation of any endogenous inhibitors of IP3R1 should also assist with homeostasis (Supplemental Material, S4 IP3R1 suppression by CARP).
BK Channel in IP3R1-Associated Ataxia Biochemicoelectrophysiological Model
IP3R1 interacts closely with the large conductance calcium-activated voltage-gated potassium (BK) channel in glioma cells (Weaver et al., 2007). BK channels appear in lipid rafts in the plasma membrane apposed to the smooth ER (sER). IP3R1 also functionally activates the BK channel in arterial smooth muscle cells (Zhao et al., 2010). It is thought that in other cell types, including neurons, BK channels may form physical complexes with various plasma membrane calcium channels, resulting in a proximity of only a few nanometers from the calcium channel pores (Dai et al., 2009).
In Purkinje neurons, BK channels contribute to repolarization of membrane potential transients in dendrites (Miyasho et al., 2001) and afterhyperpolarization of action potentials at the soma (Sausbier et al., 2004). BK channels are involved in several ataxias that converge on IP3R1-dependent signaling. BK knockout mice are ataxic and show markedly decreased spontaneous firing (tonic and bursting) of Purkinje neurons, with longer interspike intervals due to lack of BK contribution to afterhyperpolarization of the sodium (action potential) spikes which would normally help to reset the sodium channels in wild type mice (Sausbier et al., 2004; Brown and Loew, 2012). BK channels are activated by the P/Q-type calcium channels (Walter et al., 2006), which are mutated in episodic ataxia 2 (EA2) (Guida et al., 2001; Mantuano et al., 2004; Tonelli et al., 2006; Walter et al., 2006) and spinocerebellar ataxia 6 (SCA6) (Ishikawa et al., 1997; Bürk et al., 2014). In the SCA modeling suite (Models 8–9, 13–15), BK channels plays a key role in mediating the effects of IP3R1-mediated calcium release on electrophysiological signals (Brown and Loew, 2012). Combining electrophysiology with detailed biochemistry leads to emergent properties (altered firing of the Purkinje neuron in Models 13–15) that are not possible to simulate in purely electrophysiological or biochemical models (Brown and Loew, 2012).
Expanding the SCA Modeling Suite
Biochemical-Electrophysiological Modeling
There are other calcium channels that functionally couple with IP3R1. The small conductance calcium-activated potassium (SK) channels are not voltage-gated, but contribute to precision timing (Womack and Khodakhah, 2003; Womack et al., 2004; Walter et al., 2006; Alviña and Khodakhah, 2010a,b). Targeted overactivation of SK channels in SCA2 mice restores regular pacemaking activity (Kasumu et al., 2012). Isolated underactivation of SK channels without a counteracting mutation also yields ataxic mice (Alviña and Khodakhah, 2010a). Addition of this channel to the SCA models (Models 8–9, 13–15) will help mediate the influence of biochemical calcium release on electrophysiology.
Glutamate Receptor Modulation by PKC
With insufficient IP3R1, reduced calcium release should result in decreased activation of conventional PKC isoforms. Normally, cytosolic calcium and DAG together activate PKC (Nishizuka, 1984) (Figure 1). PKC phosphorylates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), another glutamate receptor, and other molecules that are recruited to the plasma membrane and induce AMPAR internalization (Chung et al., 2000, 2003). This depresses Purkinje neuron dendritic spine response to PF stimuli (Figure 1). Human SCA14 involve PKC mutations (Alonso et al., 2005; Shuvaev et al., 2011; van Gaalen et al., 2013; Ji et al., 2014) (Figure 1) (Table 1), which can lead to aberrant AMPAR modulation. Model 16 (“Brown et al., 2012—AMPAR.PKC.PP2A.NO” at www.vcell.org) simulates PKC-AMPAR signaling and can be incorporated into other SCA models (Models 10–15) to investigate SCA14. This PKC-AMPAR sub-model is adapted from simulations by Ogasawara et al. (2007, 2008).
PKC Impact on BK Channel Activity
Phosphorylation by PKC also inhibits neuronal BK channel activity (Shipston and Armstrong, 1996). Decreased PKC levels could therefore attenuate BK inhibition. This would balance suppression of BK channel activation by lower calcium transients in IP3R1-deficient Purkinje spines. Merging Models 13 and14 with Model 16 could illustrate contributions of BK regulation by PKC phosphorylation to SCAs. Protein kinase A (PKA) (Hall and Armstrong, 2000; Widmer et al., 2003) and PIP2 activation of BK and other potassium channels (Hilgemann et al., 2001; Falkenburger et al., 2010; Zhang et al., 2010) could also be added to these models.
Potential Confirmatory Studies to test SCA Modeling Suite Predictions
SCA Mouse Models
In addition to SCA1, SCA2, and SCA3 mice (Colomer Gould, 2012; Hansen et al., 2013; Hearst et al., 2014; Switonski et al., 2014), there are other mouse models available for testing SCA modeling suite predictions. The IP3R1+/− mice can most be likened to IP3R1 haploinsufficiency in humans with SCA15/SCA16 (Ogura et al., 2001; van de Leemput et al., 2007; Hara et al., 2008; Iwaki et al., 2008; Di Gregorio et al., 2010; Novak et al., 2010b; Castrioto et al., 2011; Marelli et al., 2011; Obayashi et al., 2012) (Model 10). The ITPR1opt/opt mice also have reduced IP3R1. However, IP3R1 is likely misregulated in these mice, as evidenced by IP3R1-mediated calcium transients that paradoxically show less attenuation to repeated stimulation than wild type mice (Street et al., 1997). If IP3R1 sensitivity is increased in ITPR1opt/opt mice, then these mice could serve as an additional candidate model for polyQ ataxias or other ataxias with decreased expression of supersensitive IP3R1 (see Model 11). Similarly, the reported SCA15 mouse model ITPR1Δ18/Δ18 shows reduced levels of IP3R1, but the 18 bp mutation is in the regulatory region of IP3R1 (van de Leemput et al., 2007). Calcium release and membrane electrophysiology need to be probed in these mice to ascertain whether they match the anticipated physiology of SCA15. Human SCA29 has missense mutations in the regulatory domain of IP3R1 (Huang et al., 2012) and would also need such studies in any corresponding mouse model.
Experimentally Available ICpeptides
A number of synthetic experimental peptides resembling sections of the C-terminal of IP3R1 are available for competitive binding in SCA mouse models. The IC4 peptide (also reported as IC1, Q2714-A2749; Tang et al., 2003; Tu et al., 2004) is available for competitive inhibition of PP1α in ataxias with reduced levels of IP3R1 (simulated in Model 10). IP3R1 dephosphorylation by protein phosphatase alpha (PP1α) decreases IP3R1 sensitivity to IP3 (Tang et al., 2003). IC4 (ICpeptide, Figure 1) resembles the tip of the C-terminal of IP3R1 that encodes the PP1α-binding domain. All these peptides can be used to validate and confirm predictions from the SCA modeling suite. The IC-G2736X and IC-10 peptides (Tang et al., 2009) are available for competitive inhibition of mutant Ataxin in ataxias with supersensitive IP3R1 (IC-G2736X simulated in Model 11). Peptide-based therapeutic approaches (Lucchese and Kanduc, 2014) could use viral vectors, as explored for Huntington's disease (HD) (Tang et al., 2009).
Clinical Translation
The SCA modeling suite is poised for continued use in translational studies (Brown et al., 2015). Cerebellar IP3R1 levels (Ogura et al., 2001; van de Leemput et al., 2007) in various SCA mouse models could be experimentally correlated with levels of peripheral lymphocyte IP3R1 from the same mice. The two sets of values could be plotted against each other. Levels of peripheral lymphocyte IP3R1 from ataxic individuals (van de Leemput et al., 2007) could then potentially be compared with corresponding levels in mice to estimate cerebellar levels in humans. In one study of SCA15, Western blot showed variably reduced IP3R1 levels in peripheral lymphocytes from three affected members of the same family relative to an unaffected family member (van de Leemput et al., 2007). Measuring peripheral blood lymphocyte levels of IP3R1 would be relatively noninvasive for humans. Correlated estimates of cerebellar IP3R1 would be useful to help guide therapy, particularly in presymptomatic patients who have undergone genetic testing and counseling (Supplemental Material, S1.6 Presymptomatic staging to consider calbindin modulation).
Functional Consideration of IP3R1 in Ataxia
IP3R-mediated calcium release occurs in various tissues (see deSouza et al., 2007; Mondin et al., 2009; Cárdenas et al., 2010; Chen et al., 2010; Ehrlich et al., 2010; Healy et al., 2010; Mandal et al., 2010; Park et al., 2010) including peripheral lymphocytes, but IP3R1 mutations in mice and humans (Matsumoto et al., 1996; Street et al., 1997; Ogura et al., 2001; van de Leemput et al., 2007) (van de Leemput et al., 2007; Hara et al., 2008; Iwaki et al., 2008; Di Gregorio et al., 2010; Huang et al., 2012) lead to primarily cerebellar defects. Nevertheless, the IP3R1−/− (Matsumoto et al., 1996) and IP3R1opt/opt (Street et al., 1997) mice also present with epileptic symptoms, and the conditional cerebellum/brainstem IP3R1 knockout mice and waddles mice present with dystonia (Jiao et al., 2005; Hisatsune et al., 2013) (Supplemental Material, S5 IP3R1 in basal ganglia-independent dystonia).
Most tissues have similar concentrations of all three IP3R isoforms or favor high levels of IP3R2 or IP3R3 over IP3R1 (De Smedt et al., 1997; Tu et al., 2005). Yet, 90% of IP3R in the cerebellar Purkinje neuron is IP3R1 (De Smedt et al., 1997). Insufficient levels of IP3R1 remarkably disrupt Purkinje neuron function, as observed in ataxia (SCA15 van de Leemput et al., 2007; Hara et al., 2008; Iwaki et al., 2008; Di Gregorio et al., 2010; Novak et al., 2010a,b; Marelli et al., 2011).
Other tissues such as smooth muscle, which has 75% of IP3R as IP3R1 (De Smedt et al., 1997), and peripheral lymphocytes, in which the major IP3R isoform is also type 1 (deSouza et al., 2007), likely have compensatory mechanisms involving 25% of IP3R as IP3R2 and IP3R3 to overcome IP3R1 deficits. In addition, there are two regulatory domain phosphorylation site splice variants of IP3R1 (Tu et al., 2002; Wagner et al., 2003). S(II) is favored in the brain (Wagner et al., 2003). Other tissues, such as smooth muscle and peripheral lymphocytes, may differentially phosphorylate their IP3R1 at S(I) (Tang et al., 2003) in response to insufficient levels of the receptor.
Further, there is a high density of sER containing IP3R1 in Purkinje spines (calculated average of ~15% of spine volume from Harris and Stevens, 1988), relative to hippocampal spines (reported as <5% of spine volume from Harris and Stevens, 1989) (Supplemental Material, S6 IP3R1 in hippocampal spines), which are important for synaptic plasticity involved in cognitive learning and memory. This suggests that IP3R1 on sER preferentially serves particular functions in Purkinje spines that may manifest differently in other cell types.
All of these reasons underlie the observation that in IP3R1 mutations, and in several human ataxias with biochemical and electrophysiological signals that converge on IP3R1-dependent signaling (Mikoshiba, 2007; Schorge et al., 2010), the primary clinical manifestation is spinocerebellar ataxia.
SCA in Computational Systems Neurobiology
Spatial quantitative models have given some insight into how cerebellar Purkinje neuron intracellular processes work together as an efficient system. A computational foundation for studying a wide array of spinocerebellar ataxias that involve mutations in various calcium and potassium channels, kinases, and other molecules, including IP3R1 was developed (Supplemental Material, Supplemental Figure S1). The result is a practical application of Computational Systems Neurobiology (Brown et al., 2012). Using these models to study various ataxias will help us to explain a wide array of experimental observations, elucidate cellular causes of these ataxias in mice and humans, and further understand the relationship between cytosolic calcium and membrane electrophysiology. The SCA modeling suite can help characterize the cellular pathophysiology of IP3R1-associated ataxia. That will help us to understand the biochemical and electrophysiological coupling in excitable membranes, since IP3R1 is highly expressed in the brain, and enriched in the cerebellum (Furuichi et al., 1989; De Smedt et al., 1997; Mikoshiba, 2007).
Author Contributions
Sherry-Ann Brown conceived of, analyzed, designed, drafted, critically revised, approved, and agreed to be accountable for this submitted work. Leslie M. Loew analyzed, designed, drafted, critically revised, approved, and agreed to be accountable for this submitted work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We are grateful to Dr. Louise McCullough for reading and editing this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fnins.2014.00453/abstract
References
Alonso, I., Costa, C., Gomes, A., Ferro, A., Seixas, A., Silva, S., et al. (2005). A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J. Hum. Genet. 50, 523–529. doi: 10.1007/s10038-005-0287-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alviña, K., and Khodakhah, K. (2010a). KCa channels as therapeutic targets in episodic ataxia type-2. J. Neurosci. 30, 7249–7257. doi: 10.1523/JNEUROSCI.6341-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alviña, K., and Khodakhah, K. (2010b). The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 30, 7258–7268. doi: 10.1523/JNEUROSCI.3582-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berridge, M., Lipp, P., and Bootman, M. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S.-A., Holmes, R., and Loew, L. M. (2012). “Spatial organization and diffusion in neuronal signaling” in Computational Systems Neurobiology, ed N. L. Novere (Dordrecht: Springer), 133–161.
Brown, S.-A., and Loew, L. M. (2009). Toward a computational model of IP3R1-associated ataxia. Biophys. J. 96(Suppl. 1), 96a. doi: 10.1016/j.bpj.2008.12.405
Brown, S. A., and Loew, L. M. (2012). Computational analysis of calcium signaling and membrane electrophysiology in cerebellar Purkinje neurons associated with ataxia. BMC Syst. Biol. 6:70. doi: 10.1186/1752-0509-6-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S.-A., McCullough, L. D., and Loew, L. M. (2015). Computational neurobiology is a useful tool in translational neurology: the example of ataxia. Front. Neurosci. 9:1. doi: 10.3389/fnins.2015.00001
Brown, S.-A., Moraru, I., Schaff, J., and Loew, L. M. (2011). Virtual NEURON: a strategy for merged biochemical and electrophysiological modeling. J. Comput. Neurosci. 31, 385–400. doi: 10.1007/s10827-011-0317-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S-A., Morgan, F., Watras, J., and Loew, L. (2008). Analysis of phosphatidylinositol-4,5-bisphosphate signaling in cerebellar Purkinje spines. Biophys. J. 95, 1795–1812. doi: 10.1529/biophysj.108.130195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bürk, K., Kaiser, F. J., Tennstedt, S., Schöls, L., Kreuz, F. R., Wieland, T. et al. (2014). A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non-episodic spinocerebellar ataxia with slow progression. Eur. J. Med. Genet. 57, 207–211. doi: 10.1016/j.ejmg.2014.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burright, E., Clark, H., Servadio, A., Matilla, T., Feddersen, R., Yunis, W., et al. (1995). SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82, 937–948. doi: 10.1016/0092-8674(95)90273-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cárdenas, C., Juretic, N., Bevilacqua, J., García, I., Figueroa, R., Hartley, R., et al. (2010). Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 24, 3210–3221. doi: 10.1096/fj.09-152017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castrioto, A., Prontera, P., Di Gregorio, E., Rossi, V., Parnetti, L., Rossi, A., et al. (2011). A novel spinocerebellar ataxia type 15 family with involuntary movements and cognitive decline. Eur. J. Neurol. 18, 1263–1265. doi: 10.1111/j.1468-1331.2011.03366.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., Tao, R., Sun, H., Tse, H., Lau, C., and Li, G. (2010). Multiple Ca2+ signaling pathways regulate intracellular Ca2+ activity in human cardiac fibroblasts. J. Cell. Physiol. 223, 68–75. doi: 10.1002/jcp.22010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X., Tang, T., Tu, H., Nelson, O., Pook, M., Hammer, R., et al. (2008). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 28, 12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chou, A., Yeh, T., Ouyang, P., Chen, Y., Chen, S., and Wang, H. (2008). Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 31, 89–101. doi: 10.1016/j.nbd.2008.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, H., Steinberg, J., Huganir, R., and Linden, D. (2003). Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300, 1751–1755. doi: 10.1126/science.1082915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, H., Xia, J., Scannevin, R., Zhang, X., and Huganir, R. (2000). Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci. 20, 7258–7267. Available online at: http://www.jneurosci.org/content/20/19/7258.long
Cohen, D. (2003). Of rafts and moving water. Sci. STKE 2003:pe36. doi: 10.1126/stke.2003.199.pe36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colomer Gould, V. F. (2012). Mouse models of spinocerebellar ataxia type 3 (Machado-Joseph disease). Neurotherapeutics 9, 285–296. doi: 10.1007/s13311-012-0117-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conroy, J., McGettigan, P., Murphy, R., Webb, D., Murphy, S. M., McCoy, B., et al. (2014). A novel locus for episodic ataxia:UBR4 the likely candidate. Eur. J. Hum. Genet. 22, 505–510. doi: 10.1038/ejhg.2013.173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dai, S., Hall, D., and Hell, J. (2009). Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev. 89, 411–452. doi: 10.1152/physrev.00029.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Datta, M., Choudhury, A., Lahiri, A., and Bhattacharyya, N. P. (2011). Genome wide gene expression regulation by HIP1 Protein Interactor, HIPPI: prediction and validation. BMC Genomics 12:463. doi: 10.1186/1471-2164-12-463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, G., Abbas, N., Stevanin, G., Dürr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi: 10.1038/ng0997-65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Smedt, H., Missiaen, L., Parys, J., Henning, R., Sienaert, I., Vanlingen, S., et al. (1997). Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem. J. 322(Pt 2), 575–583.
de Vries, B., Mamsa, H., Stam, A. H., Wan, J., Bakker, S. L., Vanmolkot, K. R. et al. (2009). Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch. Neurol. 66, 97–101. doi: 10.1001/archneurol.2008.53
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
deSouza, N., Cui, J., Dura, M., McDonald, T., and Marks, A. (2007). A function for tyrosine phosphorylation of type 1 inositol 1,4,5-trisphosphate receptor in lymphocyte activation. J. Cell Biol. 179, 923–934. doi: 10.1083/jcb.200708200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Gregorio, E., Orsi, L., Godani, M., Vaula, G., Jensen, S., Salmon, E., et al. (2010). Two Italian families with ITPR1 gene deletion presenting a broader phenotype of SCA15. Cerebellum 9, 115–123. doi: 10.1007/s12311-009-0154-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duarri, A., Jezierska, J., Fokkens, M., Meijer, M., Schelhaas, H. J., den Dunnen, W. F., et al. (2012). Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 72, 870–880. doi: 10.1002/ana.23700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dürr, A., Stevanin, G., Cancel, G., Duyckaerts, C., Abbas, N., Didierjean, O., et al. (1996). Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann. Neurol. 39, 490–499. doi: 10.1002/ana.410390411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ehrlich, L., Medina, G., Khan, M., Powell, M., Mikoshiba, K., and Carter, C. (2010). Activation of the inositol (1,4,5)-triphosphate calcium gate receptor is required for HIV-1 Gag Release. J. Virol. 84, 6438–6451. doi: 10.1128/JVI.01588-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Escayg, A., De Waard, M., Lee, D. D., Bichet, D., Wolf, P., Mayer, T., et al. (2000). Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet. 66, 1531–1539. doi: 10.1086/302909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Euler, P., Friedrich, B., Ziegler, R., Kuhn, A., Lindenberg, K. S., Weiller, C., et al. (2012). Gene expression analysis on a single cell level in Purkinje cells of Huntington's disease transgenic mice. Neurosci. Lett. 517, 7–12. doi: 10.1016/j.neulet.2012.03.080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falkenburger, B., Jensen, J., and Hille, B. (2010). Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 99–114. doi: 10.1085/jgp.200910345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fiala, J., Grossberg, S., and Bullock, D. (1996). Metabotropic glutamate receptor activation in cerebellar Purkinje cells as substrate for adaptive timing of the classically conditioned eye-blink response. J. Neurosci. 16, 3760–3774.
Finch, E., and Augustine, G. (1998). Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature 396, 753–756. doi: 10.1038/25541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedrich, B., Euler, P., Ziegler, R., Kuhn, A., Landwehrmeyer, B. G., Luthi-Carter, R., et al. (2012). Comparative analyses of Purkinje cell gene expression profiles reveal shared molecular abnormalities in models of different polyglutamine diseases. Brain Res. 1481, 37–48. doi: 10.1016/j.brainres.2012.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Furuichi, T., Yoshikawa, S., Miyawaki, A., Wada, K., Maeda, N., and Mikoshiba, K. (1989). Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 342, 32–38. doi: 10.1038/342032a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gehrking, K. M., Andresen, J. M., Duvick, L., Lough, J., Zoghbi, H. Y., and Orr, H. T. (2011). Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum. Mol. Genet. 20, 2204–2212. doi: 10.1093/hmg/ddr108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guergueltcheva, V., Azmanov, D. N., Angelicheva, D., Smith, K. R., Chamova, T., Florez, L., et al. (2012). Autosomal-recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am. J. Hum. Genet. 91, 553–564. doi: 10.1016/j.ajhg.2012.07.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guida, S., Trettel, F., Pagnutti, S., Mantuano, E., Tottene, A., Veneziano, L., et al. (2001). Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet. 68, 759–764. doi: 10.1086/318804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, S., and Armstrong, D. (2000). Conditional and unconditional inhibition of calcium-activated potassium channels by reversible protein phosphorylation. J. Biol. Chem. 275, 3749–3754. doi: 10.1074/jbc.275.6.3749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hansen, S. T., Meera, P., Otis, T. S., and Pulst, S. M. (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22, 271–283. doi: 10.1093/hmg/dds427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hara, K., Shiga, A., Nozaki, H., Mitsui, J., Takahashi, Y., Ishiguro, H., et al. (2008). Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 71, 547–551. doi: 10.1212/01.wnl.0000311277.71046.a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, K., and Stevens, J. (1988). Dendritic spines of rat cerebellar Purkinje cells: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 8, 4455–4469.
Harris, K., and Stevens, J. (1989). Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 2982–2997.
Healy, J., Nilsson, K., Hohmeier, H., Berglund, J., Davis, J., Hoffman, J., et al. (2010). Cholinergic augmentation of insulin release requires ankyrin-B. Sci. Signal. 3, ra19. doi: 10.1126/scisignal.2000771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hearst, S. M., Shao, Q., Lopez, M., Raucher, D., and Vig, P. J. (2014). Focused cerebellar laser light induced hyperthermia improves symptoms and pathology of polyglutamine disease sca1 in a mouse model. Cerebellum 13, 596–606. doi: 10.1007/s12311-014-0576-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernjak, N., Slepchenko, B., Fernald, K., Fink, C., Fortin, D., Moraru, I., et al. (2005). Modeling and analysis of calcium signaling events leading to long-term depression in cerebellar Purkinje cells. Biophys. J. 89, 3790–3806. doi: 10.1529/biophysj.105.065771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hilgemann, D., Feng, S., and Nasuhoglu, C. (2001). The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001:re19. doi: 10.1126/stke.2001.111.re19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirota, J., Ando, H., Hamada, K., and Mikoshiba, K. (2003). Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem. J. 372(Pt 2), 435–441. doi: 10.1042/BJ20030110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hisatsune, C., Miyamoto, H., Hirono, M., Yamaguchi, N., Sugawara, T., Ogawa, N., et al. (2013). IP3R1 deficiency in the cerebellum/brainstem causes basal ganglia-independent dystonia by triggering tonic Purkinje cell firings in mice. Front. Neural Circuits 7:156. doi: 10.3389/fncir.2013.00156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, L., Chardon, J. W., Carter, M. T., Friend, K. L., Dudding, T. E., Schwartzentruber, J., et al. (2012). Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 7:67. doi: 10.1186/1750-1172-7-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikeda, Y., Dick, K. A., Weatherspoon, M. R., Gincel, D., Armbrust, K. R., Dalton, J. C., et al. (2006). Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 38, 184–190. doi: 10.1038/ng1728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., Cusimano, A., D'Adamo, M., De Curtis, A., and Pessia, M. (2003). Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels. Pflugers Arch. 446, 373–379. doi: 10.1007/s00424-002-0962-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., D'Adamo, M., Kullmann, D., and Pessia, M. (2006). Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur. J. Neurosci. 24, 3073–3083. doi: 10.1111/j.1460-9568.2006.05186.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, T., Lin, X., Kohlmeier, K., Orr, H., Zoghbi, H., and Ross, W. (2001). Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J. Neurophysiol. 85, 1750–1760. Available online at: http://jn.physiology.org/content/85/4/1750.long
Ishikawa, K., Tanaka, H., Saito, M., Ohkoshi, N., Fujita, T., Yoshizawa, K., et al. (1997). Japanese families with autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1-p13.2 and are strongly associated with mild CAG expansions in the spinocerebellar ataxia type 6 gene in chromosome 19p13.1. Am. J. Hum. Genet. 61, 336–346. doi: 10.1086/514867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, M., Sakurai, M., and Tongroach, P. (1982). Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 324, 113–134. doi: 10.1113/jphysiol.1982.sp014103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwaki, A., Kawano, Y., Miura, S., Shibata, H., Matsuse, D., Li, W., et al. (2008). Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J. Med. Genet. 45, 32–35. doi: 10.1136/jmg.2007.053942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ji, J., Hassler, M. L., Shimobayashi, E., Paka, N., Streit, R., and Kapfhammer, J. P. (2014). Increased protein kinase C gamma activity induces Purkinje cell pathology in a mouse model of spinocerebellar ataxia 14. Neurobiol. Dis. 70C, 1–11. doi: 10.1016/j.nbd.2014.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiao, Y., Yan, J., Zhao, Y., Donahue, L. R., Beamer, W. G., Li, X., et al. (2005). Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics 171, 1239–1246. doi: 10.1534/genetics.105.044487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasumu, A., and Bezprozvanny, I. (2012). Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum 11, 630–639. doi: 10.1007/s12311-010-0182-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasumu, A. W., Hougaard, C., Rode, F., Jacobsen, T. A., Sabatier, J. M., Eriksen, B. L., et al. (2012). Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 19, 1340–1353. doi: 10.1016/j.chembiol.2012.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawaguchi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S., et al. (1994). CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228. doi: 10.1038/ng1194-221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koide, R., Ikeuchi, T., Onodera, O., Tanaka, H., Igarashi, S., Endo, K., et al. (1994). Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 6, 9–13. doi: 10.1038/ng0194-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koide, R., Kobayashi, S., Shimohata, T., Ikeuchi, T., Maruyama, M., Saito, M., et al. (1999). A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum. Mol. Genet. 8, 2047–2053. doi: 10.1093/hmg/8.11.2047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krysa, W., Rajkiewicz, M., and Sułek, A. (2012). Rapid detection of large expansions in progressive myoclonus epilepsy type 1, myotonic dystrophy type 2 and spinocerebellar ataxia type 8. Neurol. Neurochir. Pol. 46, 113–120. doi: 10.5114/ninp.2012.28253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurnellas, M., Lee, A., Li, H., Deng, L., Ehrlich, D., and Elkabes, S. (2007). Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons. Mol. Cell. Neurosci. 34, 178–188. doi: 10.1016/j.mcn.2006.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, Y. C., Durr, A., Majczenko, K., Huang, Y. H., Liu, Y. C., Lien, C. C., et al. (2012). Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 72, 859–869. doi: 10.1002/ana.23701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, X., Antalffy, B., Kang, D., Orr, H., and Zoghbi, H. (2000). Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 3, 157–163. doi: 10.1038/72101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., Tang, T., Tu, H., Nelson, O., Herndon, E., Huynh, D., et al. (2009). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 29, 9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucchese, G., and Kanduc, D. (2014). Single amino acid repeats connect viruses to neurodegeneration. Curr. Drug Discov. Technol. 11, 214–219. doi: 10.2174/1570163811666140212112300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mandal, A., Shahidullah, M., and Delamere, N. (2010). Hydrostatic pressure-induced release of stored calcium in cultured rat optic nerve head astrocytes. Invest. Ophthalmol. Vis. Sci. 51, 3129–3138. doi: 10.1167/iovs.09-4614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mantuano, E., Veneziano, L., Spadaro, M., Giunti, P., Guida, S., Leggio, M., et al. (2004). Clusters of non-truncating mutations of P/Q type Ca2+ channel subunit Ca(v)2.1 causing episodic ataxia 2. J. Med. Genet. 41, e82. doi: 10.1136/jmg.2003.015396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marelli, C., van de Leemput, J., Johnson, J. O., Tison, F., Thauvin-Robinet, C., Picard, F., et al. (2011). SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia. Arch. Neurol. 68, 637–643. doi: 10.1001/archneurol.2011.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsumoto, M., Nakagawa, T., Inoue, T., Nagata, E., Tanaka, K., Takano, H., et al. (1996). Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature 379, 168–171. doi: 10.1038/379168a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mikoshiba, K. (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem. 102, 1426–1446. doi: 10.1111/j.1471-4159.2007.04825.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyasho, T., Takagi, H., Suzuki, H., Watanabe, S., Inoue, M., Kudo, Y., et al. (2001). Low-threshold potassium channels and a low-threshold calcium channel regulate Ca2+ spike firing in the dendrites of cerebellar Purkinje neurons: a modeling study. Brain Res. 891, 106–115. doi: 10.1016/S0006-8993(00)03206-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mondin, L., Balghi, H., Constantin, B., Cognard, C., and Sebille, S. (2009). Negative modulation of inositol 1,4,5-trisphosphate type 1 receptor expression prevents dystrophin-deficient muscle cells death. Am. J. Physiol. Cell Physiol. 297, C1133–C1145. doi: 10.1152/ajpcell.00048.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moseley, M. L., Zu, T., Ikeda, Y., Gao, W., Mosemiller, A. K., Daughters, R. S., et al. (2006). Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 38, 758–769. doi: 10.1038/ng1827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murchison, D., Dove, L., Abbott, L., and Griffith, W. (2002). Homeostatic compensation maintains Ca2+ signaling functions in Purkinje neurons in the leaner mutant mouse. Cerebellum 1, 119–127. doi: 10.1080/147342202753671259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakamura, K., Jeong, S., Uchihara, T., Anno, M., Nagashima, K., Nagashima, T., et al. (2001). SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 10, 1441–1448. doi: 10.1093/hmg/10.14.1441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishizuka, Y. (1984). The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 308, 693–698. doi: 10.1038/308693a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Novak, M., Davis, M., Li, A., Goold, R., Tabrizi, S., Sweeney, M., et al. (2010a). PAW32 ITPR1 gene deletion causes spinocerebellar ataxia 15/16: a genetic, clinical and radiological description of a novel kindred. J. Neurol. Neurosurg. Psychiatr. 81, e32. doi: 10.1136/jnnp.2010.226340.60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Novak, M. J., Sweeney, M. G., Li, A., Treacy, C., Chandrashekar, H. S., Giunti, P., et al. (2010b). An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: a genetic, clinical and radiological description. Mov. Disord. 25, 2176–2182. doi: 10.1002/mds.23223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Obayashi, M., Ishikawa, K., Izumi, Y., Takahashi, M., Niimi, Y., Sato, N., et al. (2012). Prevalence of inositol 1, 4, 5-triphosphate receptor type 1 gene deletion, the mutation for spinocerebellar ataxia type 15, in Japan screened by gene dosage. J. Hum. Genet. 57, 202–206. doi: 10.1038/jhg.2012.5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogasawara, H., Doi, T., Doya, K., and Kawato, M. (2007). Nitric oxide regulates input specificity of long-term depression and context dependence of cerebellar learning. PLoS Comput. Biol. 3:e179. doi: 10.1371/journal.pcbi.0020179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogasawara, H., Doi, T., and Kawato, M. (2008). Systems biology perspectives on cerebellar long-term depression. Neurosignals 16, 300–317. doi: 10.1159/000123040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogura, H., Matsumoto, M., and Mikoshiba, K. (2001). Motor discoordination in mutant mice heterozygous for the type 1 inositol 1,4,5-trisphosphate receptor. Behav. Brain Res. 122, 215–219. doi: 10.1016/S0166-4328(01)00187-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orr, H., Chung, M., Banfi, S., Kwiatkowski, T. J., Servadio, A., Beaudet, A., et al. (1993). Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 4, 221–226. doi: 10.1038/ng0793-221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, K., Yule, D., and Bowers, W. (2010). Impaired TNF-alpha control of IP3R-mediated Ca2+ release in Alzheimer's disease mouse neurons. Cell. Signal. 22, 519–526. doi: 10.1016/j.cellsig.2009.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paulson, H., Perez, M., Trottier, Y., Trojanowski, J., Subramony, S., Das, S., et al. (1997). Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–344. doi: 10.1016/S0896-6273(00)80943-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pulst, S., Nechiporuk, A., Nechiporuk, T., Gispert, S., Chen, X., Lopes-Cendes, I., et al. (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276. doi: 10.1038/ng1196-269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santamaria, F., Wils, S., De Schutter, E., and Augustine, G. (2006). Anomalous diffusion in Purkinje cell dendrites caused by spines. Neuron 52, 635–648. doi: 10.1016/j.neuron.2006.10.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarkisov, D., and Wang, S. (2008). Order-dependent coincidence detection in cerebellar Purkinje neurons at the inositol trisphosphate receptor. J. Neurosci. 28, 133–142. doi: 10.1523/JNEUROSCI.1729-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sausbier, M., Hu, H., Arntz, C., Feil, S., Kamm, S., Adelsberger, H., et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 9474–9478. doi: 10.1073/pnas.0401702101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schorge, S., van de Leemput, J., Singleton, A., Houlden, H., and Hardy, J. (2010). Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 33, 211–219. doi: 10.1016/j.tins.2010.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serra, H., Byam, C., Lande, J., Tousey, S., Zoghbi, H., and Orr, H. (2004). Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 13, 2535–2543. doi: 10.1093/hmg/ddh268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serra, H., Duvick, L., Zu, T., Carlson, K., Stevens, S., Jorgensen, N., et al. (2006). RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708. doi: 10.1016/j.cell.2006.09.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shipston, M., and Armstrong, D. (1996). Activation of protein kinase C inhibits calcium-activated potassium channels in rat pituitary tumour cells. J Physiol 493(Pt 3), 665–672. doi: 10.1113/jphysiol.1996.sp021413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shuvaev, A. N., Horiuchi, H., Seki, T., Goenawan, H., Irie, T., Iizuka, A., et al. (2011). Mutant PKCgamma in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J. Neurosci. 31, 14324–14334. doi: 10.1523/JNEUROSCI.5530-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Street, V., Bosma, M., Demas, V., Regan, M., Lin, D., Robinson, L., et al. (1997). The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J. Neurosci. 17, 635–645.
Sugawara, T., Hisatsune, C., Le, T. D., Hashikawa, T., Hirono, M., Hattori, M., et al. (2013). Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintaining the spine morphology of purkinje cells in adult mice. J. Neurosci. 33, 12186–12196. doi: 10.1523/JNEUROSCI.0545-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, K., Zhou, J., Sato, T., Takao, K., Miyagawa, T., Oyake, M., et al. (2012). DRPLA transgenic mouse substrains carrying single copy of full-length mutant human DRPLA gene with variable sizes of expanded CAG repeats exhibit CAG repeat length- and age-dependent changes in behavioral abnormalities and gene expression profiles. Neurobiol. Dis. 46, 336–350. doi: 10.1016/j.nbd.2012.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Switonski, P. M., Szlachcic, W. J., Krzyzosiak, W. J., and Figiel, M. (2014). A new humanized ataxin-3 knock-in mouse model combines the genetic features, pathogenesis of neurons and glia and late disease onset of SCA3/MJD. Neurobiol. Dis. 73, 174–188. doi: 10.1016/j.nbd.2014.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takechi, H., Eilers, J., and Konnerth, A. (1998). A new class of synaptic response involving calcium release in dendritic spines. Nature 396, 757–760. doi: 10.1038/25547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T., Guo, C., Wang, H., Chen, X., and Bezprozvanny, I. (2009). Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J. Neurosci. 29, 1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T., Tu, H., Wang, Z., and Bezprozvanny, I. (2003). Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase a and protein phosphatase 1alpha. J. Neurosci. 23, 403–415. Available online at: http://www.jneurosci.org/content/23/2/403.long
Tonelli, A., D'Angelo, M., Salati, R., Villa, L., Germinasi, C., Frattini, T., et al. (2006). Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 241, 13–17. doi: 10.1016/j.jns.2005.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trottier, Y., Biancalana, V., and Mandel, J. (1994). Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J. Med. Genet. 31, 377–382. doi: 10.1136/jmg.31.5.377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, H., Miyakawa, T., Wang, Z., Glouchankova, L., Iino, M., and Bezprozvanny, I. (2002). Functional characterization of the type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/-) splice variants and the Opisthotonos mutant form. Biophys. J. 82, 1995–2004. doi: 10.1016/S0006-3495(02)75548-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, H., Tang, T., Wang, Z., and Bezprozvanny, I. (2004). Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J. Biol. Chem. 279, 19375–19382. doi: 10.1074/jbc.M313476200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, H., Wang, Z., Nosyreva, E., De Smedt, H., and Bezprozvanny, I. (2005). Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys. J. 88, 1046–1055. doi: 10.1529/biophysj.104.049593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Türkmen, S., Guo, G., Garshasbi, M., Hoffmann, K., Alshalah, A. J., Mischung, C., et al. (2009). CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 5:e1000487. doi: 10.1371/journal.pgen.1000487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van de Leemput, J., Chandran, J., Knight, M., Holtzclaw, L., Scholz, S., Cookson, M., et al. (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 3:e108. doi: 10.1371/journal.pgen.0030108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Gaalen, J., Vermeer, S., van Veluw, M., van de Warrenburg, B. P., and Dooijes, D. (2013). A de novo SCA14 mutation in an isolated case of late-onset cerebellar ataxia. Mov. Disord. 28, 1902–1903. doi: 10.1002/mds.25572
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vig, P., Subramony, S., Burright, E., Fratkin, J., McDaniel, D., Desaiah, D., et al. (1998). Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1 transgenic mice. Neurology 50, 106–113. doi: 10.1212/WNL.50.1.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vig, P., Subramony, S., and McDaniel, D. (2001). Calcium homeostasis and spinocerebellar ataxia-1 (SCA-1). Brain Res. Bull. 56, 221–225. doi: 10.1016/S0361-9230(01)00595-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagner, L. N., Li, W., and Yule, D. (2003). Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2- splice variants. J. Biol. Chem. 278, 45811–45817. doi: 10.1074/jbc.M306270200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walter, J., Alviña, K., Womack, M., Chevez, C., and Khodakhah, K. (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 9, 389–397. doi: 10.1038/nn1648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Denk, W., and Häusser, M. (2000). Coincidence detection in single dendritic spines mediated by calcium release. Nat. Neurosci. 3, 1266–1273. doi: 10.1038/81792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weaver, A., Olsen, M., McFerrin, M., and Sontheimer, H. (2007). BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: a novel mechanism for coupling [Ca(2+)](i) to ion channel activation. J. Biol. Chem. 282, 31558–31568. doi: 10.1074/jbc.M702866200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Widmer, H., Rowe, I., and Shipston, M. (2003). Conditional protein phosphorylation regulates BK channel activity in rat cerebellar Purkinje neurons. J. Physiol. 552(Pt 2), 379–391. doi: 10.1113/jphysiol.2003.046441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Womack, M., Chevez, C., and Khodakhah, K. (2004). Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J. Neurosci. 24, 8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Womack, M., and Khodakhah, K. (2003). Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J. Neurosci. 23, 2600–2607. doi: 10.1523/JNEUROSCI.2915-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, C., Watras, J., and Loew, L. (2003). Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 161, 779–791. doi: 10.1083/jcb.200301070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, J., Jiao, Y., Jiao, F., Stuart, J., Donahue, L. R., Beamer, W. G., et al. (2007). Effects of carbonic anhydrase VIII deficiency on cerebellar gene expression profiles in the wdl mouse. Neurosci. Lett. 413, 196–201. doi: 10.1016/j.neulet.2006.11.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zecevic, N., Milosevic, A., and Ehrlich, B. (1999). Calcium signaling molecules in human cerebellum at midgestation and in ataxia. Early Hum. Dev. 54, 103–116. doi: 10.1016/S0378-3782(98)00090-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Chen, X., Jia, C., Geng, X., Du, X., and Zhang, H. (2010). Depolarization increases phosphatidylinositol (PI) 4,5-bisphosphate level and KCNQ currents through PI 4-kinase mechanisms. J. Biol. Chem. 285, 9402–9409. doi: 10.1074/jbc.M109.068205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, G., Neeb, Z. P., Leo, M. D., Pachuau, J., Adebiyi, A., Ouyang, K. et al. (2010). Type 1 IP3 receptors activate BKCa channels via local molecular coupling in arterial smooth muscle cells. J. Gen. Physiol. 136, 283–291. doi: 10.1085/jgp.201010453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: spinocerebellar ataxia, translational, model, computational, inositol 1,4,5-trisphosphate receptor 1, Purkinje, carbonic anhydrase related proteins, homer
Citation: Brown S-A and Loew LM (2015) Integration of modeling with experimental and clinical findings synthesizes and refines the central role of inositol 1,4,5-trisphosphate receptor 1 in spinocerebellar ataxia. Front. Neurosci. 8:453. doi: 10.3389/fnins.2014.00453
Received: 04 November 2014; Accepted: 22 December 2014;
Published online: 21 January 2015.
Edited by:
Firas H. Kobeissy, University of Florida, USAReviewed by:
Katsuhiko Mikoshiba, RIKEN Brain Science Institute, JapanIlya Bezprozvanny, University of Texas Southwestern Medical Center at Dallas, USA
Copyright © 2015 Brown and Loew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sherry-Ann Brown, Department of Medicine, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA e-mail:YnJvd24uc2hlcnJ5YW5uQG1heW8uZWR1