 Sherry-Ann Brown
Sherry-Ann Brown Louise D. McCullough
Louise D. McCullough Leslie M. Loew
Leslie M. Loew- 1Department of Medicine, Mayo Clinic, Rochester, MN, USA
- 2Departments of Neurology and Neuroscience, University of Connecticut Health Center, Farmington, CT, USA
- 3Richard D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, Farmington, CT, USA
Hereditary ataxia, or motor incoordination, affects approximately 150,000 Americans and hundreds of thousands of individuals worldwide with onset from as early as mid-childhood. Affected individuals exhibit dysarthria, dysmetria, action tremor, and diadochokinesia. In this review, we consider an array of computational studies derived from experimental observations relevant to human neuropathology. A survey of related studies illustrates the impact of integrating clinical evidence with data from mouse models and computational simulations. Results from these studies may help explain findings in mice, and after extensive laboratory study, may ultimately be translated to ataxic individuals. This inquiry lays a foundation for using computation to understand neurobiochemical and electrophysiological pathophysiology of spinocerebellar ataxias and may contribute to development of therapeutics. The interdisciplinary analysis suggests that computational neurobiology can be an important tool for translational neurology.
Introduction
Computational systems neurobiology (Brown et al., 2012) can be used to understand neuronal systems, based on utilizing information garnered from clinical reports, animal studies and in vitro modeling. Results from computational neurobiology can be used to develop additional animal and cellular experiments that may ultimately be translated to clinical practice, i.e., translational neurology. One clinical condition poised to benefit from this marriage is spinocerebellar ataxia (SCA) (Figure 1). Ataxia refers to lack of motor coordination (Goetz, 2003). In this paper, we use SCA as an example to demonstrate how computation and translation can potentially be woven together to enhance our knowledge of cell function.
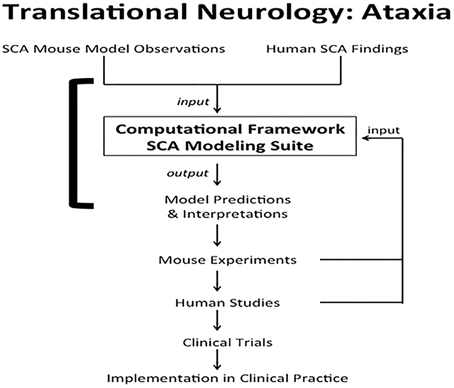
Figure 1. Translational neurology: ataxia. SCA mouse observations and human SCA findings are incorporated into the computational framework SCA modeling suite. The models help interpret experimental and clinical findings. The models also predict interactions between proteins and emergent properties that can be borne out in novel mouse experiments. Results from the laboratory and clinical observations can be used to validate, disprove, or tweak the computational models. Findings from mouse experiments can also ultimately be translated to human studies, leading to clinical trials to test therapeutics. The final step in translational neurology with the example of ataxia is implementation of the iterative findings in patient care. The solid square bracket highlights the components addressed directly by computational systems neurobiology (Brown et al., 2012; Brown and Loew, 2012).
The most common SCAs are spinocerebellar ataxia 1 (SCA1), spinocerebellar ataxia 2 (SCA2), spinocerebellar ataxia 3 (SCA3), and Spinocerebellar Ataxia 6 (SCA6) (Supplementary Material, Table S1) (Jacobi et al., 2012; Orr, 2012; Musova et al., 2013). They are caused by expanded polyglutamine (polyQ; CAG) repeat mutations in genes that code for ataxin 1, ataxin 2, ataxin 3, and the CACNA1A calcium channel, respectively (Supplementary Material, Table S1) (Gispert et al., 1993; Orr et al., 1993; Kawaguchi et al., 1994; Pulst et al., 1996; Zhuchenko et al., 1997; Tonelli et al., 2006; Bürk et al., 2014).
A number of mutations that cause SCA or episodic spinocerebellar ataxia (EA) also occur in genes directly involved in calcium signaling and plasma membrane excitability, which are critical for cerebellar Purkinje neuron function (Kim et al., 1997; Yue et al., 1997; Zhuchenko et al., 1997; Guida et al., 2001; Alonso et al., 2005; Iwaki et al., 2008; Becker et al., 2009; Alviña and Khodakhah, 2010; Kasumu and Bezprozvanny, 2012) (Supplementary Material, Table S1). A number of these genes impact the phosphoinositol signaling pathway (Supplementary Material, Figure S1A; Table S1) (Aiba et al., 1994; Kim et al., 1997; Alonso et al., 2005; van de Leemput et al., 2007). This pathway is important for calcium release from the smooth endoplasmic reticulum (sER) into the cytoplasm of cerebellar Purkinje neurons. Many other mutations affect calcium and potassium ion channels (EA2, SCA6, EA1, EA5, SCA13, SCA19, SCA22) (Supplementary Material, Table S1) (Yue et al., 1997; Zhuchenko et al., 1997; Lin et al., 2000; Guida et al., 2001; Imbrici et al., 2003; Sausbier et al., 2004; Tonelli et al., 2006; Bürk et al., 2014) that are important for regulating the rate of calcium influx into cells. spinocerebellar ataxia 14 (SCA14) involves a mutation in the gene encoding protein kinase C (PKC) that is also important for calcium homeostasis (Supplementary Material, Figure S1A; Table S1) (Alonso et al., 2005; Ueda et al., 2013; van Gaalen et al., 2013; Ji et al., 2014). spinocerebellar ataxia 15 (SCA15) and spinocerebellar ataxia 16 (SCA16) in humans and in mice are caused by deletion and missense mutations in the gene for inositol-1,4,5-trisphosphate receptor type 1 (IP3R1), a calcium channel on the sER (Desaiah et al., 1991; Street et al., 1997; Zecevic et al., 1999; Lin et al., 2000; Storey et al., 2001; Serra et al., 2004; van de Leemput et al., 2007; Chen et al., 2008; Chou et al., 2008; Hara et al., 2008; Iwaki et al., 2008; Liu et al., 2009; Di Gregorio et al., 2010; Novak et al., 2010a,b; Huang et al., 2012).
Data from mouse experiments and clinical observations have been incorporated into SCA computational models (Brown and Loew, 2015). The models are developed using computer engineering tools and software, such as Virtual Cell (Moraru et al., 2008; Slepchenko and Loew, 2010) and NEURON (Hines and Carnevale, 2001). These modeling platforms are based on mathematical equations for physics principles, such as reaction, diffusion, flux, and concentration gradients (Hines and Carnevale, 1997; Schaff et al., 2000; Cowan et al., 2012). The models simulate physiological interactions among intracellular reactions, molecular diffusion, and cell geometry and give insight into how these cellular processes work together as an efficient system (Brown et al., 2008; Brown and Loew, 2012). Simulations help clarify observed phenomena and suggest future experiments to help elucidate disease mechanisms and potential therapeutics.
Experiments Confirm Model Predictions
There are a handful of neurobiological and electrophysiological computational models relevant to IP3R1-associated ataxias (Brown and Loew, 2012), detailed in the following subsections.
Neurobiology Modeling and Simulation
PIP2 signaling upstream of IP3R1
First, a model of phosphatidylinositol-4,5-bisphosphate (PIP2) signaling upstream of IP3R1 in neuroblastoma cells was developed (Xu et al., 2003) (Supplementary Material, Figure S1A). The model predicted stimulated PIP2 synthesis in addition to PIP2 hydrolysis when the Purkinje neuron spine is activated by parallel fibers, to produce IP3R1-mediated calcium release. Subsequently, bench experiments confirmed simulation results in a mouse neuroblastoma cell line (Xu et al., 2003).
IP3R1 signaling downstream of PIP2
Second, models of IP3R1-related signaling downstream of PIP2 in the cerebellar Purkinje neuron were created (Doi et al., 2003; Hernjak et al., 2005). One model considered the high abundance and low sensitivity of IP3R1 in Purkinje cells (Hernjak et al., 2005). This model qualitatively reproduced experimentally observed calcium transients during coincident activation of the Purkinje spiny dendrite (Wang et al., 2000).
Sources of sufficient PIP2 for IP3R1-mediated signaling
Third, quantitative models of Purkinje neurons spiny dendrites were developed (Brown et al., 2008, 2011; Brown and Loew, 2012). Local sequestration of PIP2 (with a lower diffusion coefficient than unbound PIP2) on the inner leaflet of cerebellar Purkinje neuron spines (McLaughlin et al., 2002; Golebiewska et al., 2008) (Supplementary Material, Figure S1B) was assessed. Model results supported the efficacy of local sequestration as a means of providing sufficient PIP2 for IP3R1-mediate calcium release. Findings correlated with prior experimental results (Wang et al., 2000) (Table 1). The computational simulations predicted a time window during which coincident activation of the Purkinje spine by other cell types could occur (Brown et al., 2008). This time window was independently borne out in bench experiments in rat cerebellar brain slices (Sarkisov and Wang, 2008) (Table 1).
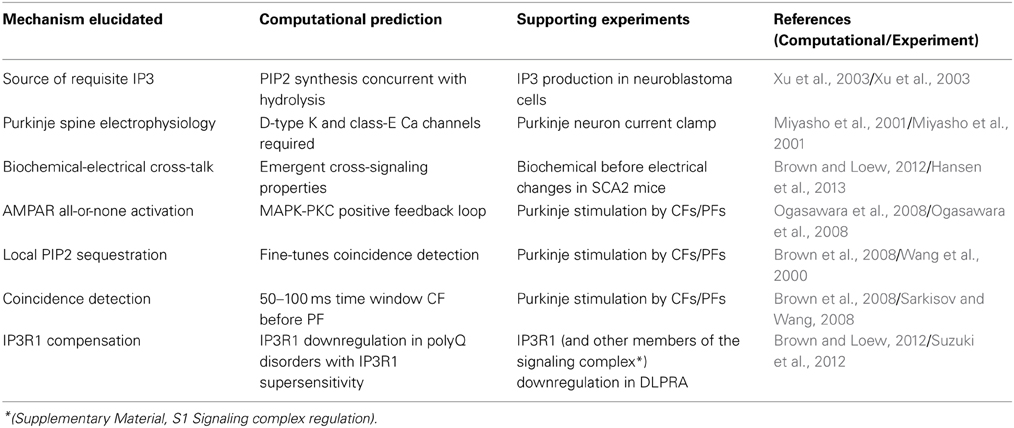
Table 1. Examples of contributions of computational systems neurobiology to translational neurology.
Signaling downstream of IP3R1
Fourth, predictions were generated about kinetic interactions between PKC and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptors (AMPAR) in Purkinje neurons that were experimentally verified (Ogasawara et al., 2008) (Table 1). These molecules are downstream of IP3R1-mediated calcium release, as calcium co-activates PKC.
Potential use of neurobiological model results
Using results from these computational models, drugs could be developed to interfere with local sequestration or other steps in the phosphoinositol signaling pathway in mice or rats and could potentially be translated to humans with supersensitive IP3R1 to help treat SCA1-3 and SCA14 (Supplementary Material, Table S1).
Neuroelectrophysiology Modeling
Prediction of purkinje neuron calcium and potassium channels
Normal electrophysiology of the Purkinje neuron was also modeled (De Schutter and Bower, 1994a,b; Miyasho et al., 2001). New properties of Purkinje neuron electrophysiology were predicted, requiring contributions of D-type potassium channels and class-E calcium channels that were not known previously to influence Purkinje neuron electrophysiology (Supplementary Material, Figure S1A). The predictions were confirmed with rat model experiments (Miyasho et al., 2001) (Table 1).
Reduction of purkinje neurons
A method to map realistic neurons into equivalent reduced models while maintaining high accuracy membrane potential changes during synaptic inputs, with direct links to experimental observables was subsequently developed (Marasco et al., 2012).
Potential use of neuroelectrophysiological model results
The examples provided suggest that iterative computational modeling can provide insight into normal and pathological neurophysiology. The neuroelectrophysiology models could be used to economically assess the impact of new therapeutics in research and development prior to studies in mice or rats. Manipulation in the virtual system would allow for precise input control and real-time output review with changes in firing dynamics of the Purkinje neuron. This would facilitate discovery of determinant and feedback loops (Brown and Loew, 2015) and other interactions that would otherwise be impossible to monitor in brain slices in the same time frame. Insights could potentially be translated to humans with various SCAs, particularly those such as SCA6, SCA13, SCA19, and SCA22 that involve disruption of membrane electrophysiology (Supplementary Material, Table S1).
Simulations Interpret Observed Phenotypes
Supranormal IP3R1 Sensitivity in SCA 1-3
A source of pathology caused by the polyQ repeats (Orr et al., 1993; Kawaguchi et al., 1994; Koide et al., 1994, 1999; Trottier et al., 1994; Pulst et al., 1996; David et al., 1997; Nakamura et al., 2001) is due to interaction of the mutant protein with IP3R1 (Bezprozvanny, 2011). In SCA2 (Liu et al., 2009) and SCA3 (Chen et al., 2008), mutant Ataxin-2 and Ataxin-3, respectively, directly bind to the C-terminal of IP3R1 and make it easier to solicit IP3-induced calcium responses. Association of mutant Ataxin-1 with IP3R1 has been reported (Liu et al., 2009), but supersensitivity in these mice has not yet been tested. SCA modeling results suggests that IP3R1 supersensitivity in SCA1 is necessary (Brown and Loew, 2012) to elicit observed supranormal calcium transients (Inoue et al., 2001).
IP3R1 Downregulation Provides Partial Compensation in SCA 1-3
Studies in SCA1 and SCA2 mouse models, as well as mice and humans with SCA3, found reduced levels of IP3R1, metabotropic glutamate receptor (mGluR), and other calcium signaling and glutamatergic proteins (Lin et al., 2000; Vig et al., 2001; Serra et al., 2004; Chou et al., 2008; Hansen et al., 2013) (Supplementary Material, Figure S1). Decreased expression of IP3R1 and sarcoendoplasmic reticulum calcium ATPase (SERCA) was also confirmed in SCA1 patients (Lin et al., 2000). Such findings were also discovered in Purkinje neurons from mouse models of HD (Datta et al., 2011; Euler et al., 2012). SCA modeling interprets downregulation of these key calcium signaling proteins as serving to partially compensate for supersensitive IP3R1 (Brown and Loew, 2012). Further downregulation of these glutamatergic signaling proteins could be manipulated to delay symptomatic disease in mouse models and in the long run in presymptomatic humans (Brown and Loew, 2015).
Homer and Myosin VA Association with IP3R1 in SCAs
Homer 3 is part of a signaling complex with reduced expression in SCA1 mice (Serra et al., 2004) (Supplementary Material, S1 Signaling complex regulation). Homer 3 localizes predominantly to Purkinje neuron spines (Shiraishi et al., 2004) and may associate with mGluR and IP3R1 in vivo (Tu et al., 1998; Sandonà et al., 2003). Myosin Va levels are also decreased in SCA1 (Serra et al., 2004). Both Homer and Myosin Va have been proposed to guide sER (sER) into spines as spines are being formed from dendritic shafts (Wagner and Hammer, 2003). Accordingly, Myosin Va knockout mice are ataxic with spines devoid of sER and IP3R1 (Takagishi et al., 1996). SCA modeling results suggest that reduced spine sER volume due to downregulation of Homer 3 and Myosin Va in SCA1 partially compensates for IP3R1 supersensitivity (Brown and Loew, 2012). This supports findings from an experiment in which downregulation of Homer 1b/c attenuated IP3R1-mediated calcium release in rat cortical neurons (Chen et al., 2012). Expression of Homer could potentially be manipulated to further compensate for IP3R1 supersensitivity in polyQ ataxias, which also include SCA7 (David et al., 1997), SCA17 (Nakamura et al., 2001), and dentatorubral-pallidoluysian atrophy (DLPRA) (Koide et al., 1994). SCA simulations could determine a therapeutic window for Homer expression to avoid overcompensation. As an example, although a very different mechanism, Homer 3 scaffolding protein has been implicated as an autoimmune target in subacute cerebellar ataxia. This ataxia is not hereditary and occurs post-infection or as a paraneoplastic process in some patients with Hodgkin's lymphoma (Zuliani et al., 2007). Presumably, disruption of Homer 3 scaffolding beyond a therapeutic window, or in the absence of supersensitive IP3R1 as in autoimmune subacute cerebellar ataxia, interrupts signaling complex formation and related cellular processes (Supplementary Material, S1 Signaling complex regulation).
Cross-Signaling between Biochemical and Electrophysiological Dysregulation in SCAs
These and other forms of biochemical dysregulation precede electrophysiological impairment in an SCA2 mouse model, with Purkinje neuron firing frequency decreased at 6 weeks compared to wild type (Hansen et al., 2013) and worsening as the mice age (Kasumu et al., 2012b). This is consistent with timing of electrophysiological changes in an SCA1 mouse model (Hourez et al., 2011) and in the large conductance calcium-activated voltage-gated potassium channel (BK) knockout mice (Sausbier et al., 2004). These changes are then followed by onset of motor discoordination at 8 weeks in the SCA2 mice (Hansen et al., 2013) and 6–8 weeks in SCA3 mice (Shakkottai et al., 2011). The time of ataxia onset for these mice is identical to that for mice heterozygous for IP3R1 deletion (Ogura et al., 2001). These findings imply shared pathophysiology leading to similar phenotype: biochemical dysfunction and subsequent electrophysiological aberrations leading to ataxia. The SCA modeling suite has also predicted altered Purkinje neuron firing arising from cross-talk between calcium signaling and membrane electrophysiology (Brown and Loew, 2012) (Table 1). Future iterations of the modeling suite could additionally include the small conductance calcium-activated potassium channels (SK), which has been shown to help mediate the influence of calcium signaling on membrane electrophysiology in SCA2 and EA, and has also been proposed as a potential therapeutic targets (Alviña and Khodakhah, 2010; Kasumu et al., 2012a).
Clinical Translation
Computational Models are Clinically Informative for SCAs
The cerebellum is conserved across all vertebrate species (Kandel et al., 2000). Thus, a combination of computational models and mouse models is clinically informative for human SCAs. Several SCA mouse models have been developed (Burright et al., 1995; Huynh et al., 2000; van de Leemput et al., 2007; Colomer Gould, 2012; Kelp et al., 2013). As virtual model neurons are created, details of the computational models are validated by comparison with experimental data in these mice (Xu et al., 2003; Marasco et al., 2012), with an end to translation to humans (Figure 1).
ICpeptide Application in vivo for SCAs and Other polyQ Diseases
Peptides resembling portions of the IP3R1 C-terminal (ICpeptides) (Supplementary Material, Figure S1B) have been created (Tang et al., 2003b, 2009; Tu et al., 2004). SCA model results suggest that application of IC-G2736X (IP3R1 base pairs D2590-G2736) (Supplementary Material, Figure S1B) restores normal calcium transients in polyQ ataxias (Brown and Loew, 2012). ICpeptides could be used to develop more selective therapeutics that can then be tested in animals and, if promising, in patients. Simulations also showed that treating SCA15/16 mice with IC4 (IP3R1 base pairs Q2714-A2749) normalizes calcium release by therapeutically increasing IP3R1 sensitivity to IP3 to counteract IP3R1 haploinsufficiency (Brown and Loew, 2012). IC4 competitively binds protein phosphatase 1 alpha (PP1α; a phosphatase that decreases IP3R1 sensitivity).
A different polyQ disorder, Huntington's disease (HD), is sometimes phenotypically confused with SCA if ataxia is prominent (Tang et al., 2003a; Bezprozvanny and Hayden, 2004; Bezprozvanny, 2007; Zhang et al., 2008; Dong et al., 2013; Rodríguez-Quiroga et al., 2013). Whereas ataxias primarily exhibit motor discoordination, HD is a neurodegenerative hyperkinetic movement disorder affecting the basal ganglia (Bezprozvanny, 2011). Although molecular interactions in Purkinje neurons are different from those in medial striatal neurons, the polyQ SCAs share underlying pathophysiology with HD involving supersensitive IP3R1 (Bezprozvanny, 2011). Application of the IC10 peptide (IP3R1 base pairs F2627-A2749) (Supplementary Material, Figure S1B) in medial striatal neurons in HD mice restored normal calcium response (Tang et al., 2009). These mice were largely spared from neurotoxicity, with improved motor coordination (Tang et al., 2009). IC-peptides therefore represent an alternative step toward thinking about new therapeutics for polyQ disorders.
IP3 Suppression in SCAs
Subsequent to these simulations (Brown and Loew, 2012), it was demonstrated that overexpressing inositol 1,4,5-phosphatase (5PP) to chronically suppress IP3R1-mediated calcium release improved motor coordination in SCA2 mice (Kasumu et al., 2012b). The enzyme 5PP converts IP3 to the inactive form inositol 1,4-bisphosphatase (Supplementary Material, Figure S1A), decreasing the overall amount of IP3 sensed by IP3R1. This demonstrated that suppression of IP3R1-mediated calcium release could be of therapeutic benefit for SCAs, as supported by SCA modeling predictions (Figure 1).
Anticipatory Therapeutics for SCAs
There is currently no direct way of treating hereditary ataxias. Patients can be cared for symptomatically, with physical and speech therapy and walking aids (Schöls et al., 2004). If SCA modeling can help us understand how alterations in calcium signaling and membrane electrophysiology can be restored in SCAs, this can enhance our chance at engineering therapeutics for ataxia. In the era of SCA genomic testing (Smeets and Verbeek, 2014), anticipatory therapeutics would be useful preferentially in presymptomatic patients (Brown and Loew, 2015). For example, if ICpeptides are applied before mice become symptomatic, this could reduce symptom occurrence, delay onset, slow progression, and maximize output from undamaged tissue. SCA modeling would therefore be integrated with genomic testing, family history considerations, and presymptomatic investigation and treatment. Presymptomatic testing is often pursued for severe late-onset neurodegenerative diseases, including SCAs (Guimarães et al., 2013; Schuler-Faccini et al., 2014). Even without genetic testing, other early clinical disease features (prodromes) are detectable prior to onset of ataxia and can be used to determine treatment timing (Velázquez-Pérez et al., 2014a,b). Such translation, after extensive lab research, could improve quality of life and alleviate economical, social, and occupational strains on patients, as well as family members and caregivers.
Uses for SCA Modeling Exemplified
In computational models, various parameters in the cerebellar Purkinje neuron may be deliberately perturbed, with results examined to see if any of these perturbations match those found in individuals with various cerebellar disorders. This can assist with making connections between subcellular dysfunction and phenotypic manifestations. In addition to understanding pathophysiology, the models could be used to screen drugs, investigate adverse effects, and examine ramifications of genetic replacements and knockouts.
There are other computational models that can be used to exemplify the potential of SCA models. Experiment-based computational modeling similar to SCA, including the use of individualized 3D reconstruction of in vivo-acquired computed tomography (CT) images from several patients, showed that peak wall stress calculated in vivo for abdominal aortic aneurysm (AAA) near the time of rupture was more predictive of rupture than the conventional assessment of aortic diameter (Fillinger et al., 2002; Raut et al., 2013; Soudah et al., 2013). Similarly, Caroli et al used computational modeling to create patient-specific computational vascular network models (Caroli et al., 2013). These models predicted blood flow 6 weeks after surgical creation of arteriovenous fistulas (AVF) for long-term hemodialysis (Caroli et al., 2013). The model was validated in a multicenter, prospective clinical study, and is expected to reduce AVF failure or dysfunction and related patient morbidity. A line of computational models were also created to compare various modes of pharmacologic delivery of Doxorubicin, a chemotherapeutic drug used for a variety of cancers. The models predicted optimal antitumor efficacy with protection from adverse effects with thermosensitive liposomes or with an administration protocol using increased duration of infusion with higher doses, compared to the conventional bolus injection (Reich et al., 1979; El-Kareh and Secomb, 2000, 2005; Evans et al., 2009; Zhan and Xu, 2013).
Systems Biology in Translational Medicine
A number of scientific contributions from computational modeling (Table 1) that enhance our understanding of SCA have been presented. The computational models are more economical, reproducible, and expedient than animal experiments. However, the computer models depend on and integrate information from animal studies and clinical observations. As a result, Figure 1 shows that animal experiments and SCA models work together in concert to iteratively explain and predict pathophysiology. In addition, computational predictions were made via a bioinformatics screen to identify transcripts that interact with and have functions relevant to polyQ SCAs (Spence and Wallihan, 2012). Next steps in translational systems biology will include mining and modeling network motifs in ataxia and considering their functional implications (Alon, 2007; Brown and Loew, 2015). Findings from such translational studies could be used to develop new drugs or treatment strategies (Matilla-Dueñas et al., 2014), suggesting that computational neurobiology plays a role in translational neurology (Figure 1). Expanding interactions between these related research and medical communities (De Schutter, 2008) will usher in an era that may create advanced support for Translational Medicine and, in particular, Neurology.
Author Contributions
Sherry-Ann Brown conceived of, analyzed, designed, drafted, critically revised, approved, and agreed to be accountable for this submitted work. Louise D. McCullough conceived of, critically revised, approved, and agreed to be accountable for this submitted work. Leslie M. Loew analyzed, critically revised, approved, and agreed to be accountable for this submitted work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fnins.2015.00001/abstract
References
Aiba, A., Kano, M., Chen, C., Stanton, M., Fox, G., Herrup, K., et al. (1994). Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79, 377–388. doi: 10.1016/0092-8674(94)90205-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alonso, I., Costa, C., Gomes, A., Ferro, A., Seixas, A., Silva, S., et al. (2005). A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J. Hum. Genet. 50, 523–529. doi: 10.1007/s10038-005-0287-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alviña, K., and Khodakhah, K. (2010). KCa channels as therapeutic targets in episodic ataxia Type-2. J. Neurosci. 30, 7249–7257. doi: 10.1523/JNEUROSCI.6341-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becker, E., Oliver, P., Glitsch, M., Banks, G., Achilli, F., Hardy, A., et al. (2009). A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. U.S.A. 106, 6706–6711. doi: 10.1073/pnas.0810599106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bezprozvanny, I. (2007). Inositol 1,4,5-tripshosphate receptor, calcium signalling and Huntington's disease. Subcell. Biochem. 45, 323–335. doi: 10.1007/978-1-4020-6191-2_11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bezprozvanny, I. (2011). Role of inositol 1,4,5-trishosphate receptors in pathogenesis of Huntington's disease and spinocerebellar ataxias. Neurochem. Res. 36, 1186–1197. doi: 10.1007/s11064-010-0393-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bezprozvanny, I., and Hayden, M. (2004). Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 322, 1310–1317. doi: 10.1016/j.bbrc.2004.08.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S.-A., Holmes, R., and Loew, L. M. (2012). “Spatial organization and diffusion in neuronal signaling,” in Computational Systems Neurobiology, ed N. L. Novere (Dordrecht: Springer), 133–161.
Brown, S. A., and Loew, L. M. (2012). Computational analysis of calcium signaling and membrane electrophysiology in cerebellar Purkinje neurons associated with ataxia. BMC Syst. Biol. 6:70. doi: 10.1186/1752-0509-6-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S.-A., and Loew, L. M. (2015). Inositol 1,4,5-trisphosphate receptor 1 is essential to spinocerebellar ataxia modeling. Front. Neurosci. 8:453. doi: 10.3389/fnins.2014.00453
Brown, S. A., Moraru, I. I., Schaff, J. C., and Loew, L. M. (2011). Virtual NEURON: a strategy for merged biochemical and electrophysiological modeling. J. Comput. Neurosci. 31, 385–400. doi: 10.1007/s10827-011-0317-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, S., Morgan, F., Watras, J., and Loew, L. (2008). Analysis of phosphatidylinositol-4,5-bisphosphate signaling in cerebellar Purkinje spines. Biophys. J. 95, 1795–1812. doi: 10.1529/biophysj.108.130195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bürk, K., Kaiser, F. J., Tennstedt, S., Schöls, L., Kreuz, F. R., Wieland, T., et al. (2014). A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non-episodic spinocerebellar ataxia with slow progression. Eur. J. Med. Genet. 57, 207–211. doi: 10.1016/j.ejmg.2014.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burright, E., Clark, H., Servadio, A., Matilla, T., Feddersen, R., Yunis, W., et al. (1995). SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82, 937–948. doi: 10.1016/0092-8674(95)90273-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caroli, A., Manini, S., Antiga, L., Passera, K., Ene-Iordache, B., Rota, S., et al. (2013). Validation of a patient-specific hemodynamic computational model for surgical planning of vascular access in hemodialysis patients. Kidney Int. 84, 1237–1245. doi: 10.1038/ki.2013.188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, T., Fei, F., Jiang, X. F., Zhang, L., Qu, Y., Huo, K., et al. (2012). Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic. Biol. Med. 52, 208–217. doi: 10.1016/j.freeradbiomed.2011.10.451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X., Tang, T., Tu, H., Nelson, O., Pook, M., Hammer, R., et al. (2008). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 28, 12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chou, A., Yeh, T., Ouyang, P., Chen, Y., Chen, S., and Wang, H. (2008). Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 31, 89–101. doi: 10.1016/j.nbd.2008.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colomer Gould, V. F. (2012). Mouse models of spinocerebellar ataxia type 3 (Machado-Joseph disease). Neurotherapeutics 9, 285–296. doi: 10.1007/s13311-012-0117-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cowan, A. E., Moraru, I. I., Schaff, J. C., Slepchenko, B. M., and Loew, L. M. (2012). Spatial modeling of cell signaling networks. Methods Cell Biol. 110, 195–221. doi: 10.1016/B978-0-12-388403-9.00008-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Datta, M., Choudhury, A., Lahiri, A., and Bhattacharyya, N. P. (2011). Genome wide gene expression regulation by HIP1 Protein Interactor, HIPPI: prediction and validation. BMC Genomics 12:463. doi: 10.1186/1471-2164-12-463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, G., Abbas, N., Stevanin, G., Dürr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi: 10.1038/ng0997-65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desaiah, D., Vig, P., Subramony, S., and Currier, R. (1991). Inositol 1,4,5-trisphosphate receptors and protein kinase C in olivopontocerebellar atrophy. Brain Res. 552, 36–40. doi: 10.1016/0006-8993(91)90656-G
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Schutter, E. (2008). Why are computational neuroscience and systems biology so separate? PLoS Comput. Biol. 4:e1000078. doi: 10.1371/journal.pcbi.1000078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Schutter, E., and Bower, J. (1994a). An active membrane model of the cerebellar Purkinje cell II. Simulation of synaptic responses. J. Neurophysiol. 71, 401–419.
De Schutter, E., and Bower, J. (1994b). An active membrane model of the cerebellar Purkinje cell. I. Simulation of current clamps in slice. J. Neurophysiol. 71, 375–400.
Di Gregorio, E., Orsi, L., Godani, M., Vaula, G., Jensen, S., Salmon, E., et al. (2010). Two Italian families with ITPR1 gene deletion presenting a broader phenotype of SCA15. Cerebellum. 9, 115–123. doi: 10.1007/s12311-009-0154-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doi, T., Kuroda, S., Michikawa, T., and Kawato, M. (2003). [Simulation of IP3/Ca2+ signaling pathway in neurons: the system of coincidence detection of input timing in cerebellar Purkinje cells]. Tanpakushitsu Kakusan Koso 48, 817–822.
Dong, Y., Sun, Y. M., Liu, Z. J., Ni, W., Shi, S. S., and Wu, Z. Y. (2013). Chinese patients with Huntington's disease initially presenting with spinocerebellar ataxia. Clin. Genet. 83, 380–383. doi: 10.1111/j.1399-0004.2012.01927.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El-Kareh, A. W., and Secomb, T. W. (2000). A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia 2, 325–338. doi: 10.1038/sj.neo.7900096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El-Kareh, A. W., and Secomb, T. W. (2005). Two-mechanism peak concentration model for cellular pharmacodynamics of Doxorubicin. Neoplasia 7, 705–713. doi: 10.1593/neo.05118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Euler, P., Friedrich, B., Ziegler, R., Kuhn, A., Lindenberg, K. S., Weiller, C., et al. (2012). Gene expression analysis on a single cell level in Purkinje cells of Huntington's disease transgenic mice. Neurosci. Lett. 517, 7–12. doi: 10.1016/j.neulet.2012.03.080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans, C. J., Phillips, R. M., Jones, P. F., Loadman, P. M., Sleeman, B. D., Twelves, C., et al. (2009). A mathematical model of doxorubicin penetration through multicellular layers. J. Theor. Biol. 257, 598–608. doi: 10.1016/j.jtbi.2008.11.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fillinger, M. F., Raghavan, M. L., Marra, S. P., Cronenwett, J. L., and Kennedy, F. E. (2002). In vivo analysis of mechanical wall stress and abdominal aortic aneurysm rupture risk. J. Vasc. Surg. 36, 589–597. doi: 10.1067/mva.2002.125478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gispert, S., Twells, R., Orozco, G., Brice, A., Weber, J., Heredero, L., et al. (1993). Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat. Genet. 4, 295–299. doi: 10.1038/ng0793-295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Golebiewska, U., Nyako, M., Woturski, W., Zaitseva, I., and McLaughlin, S. (2008). Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 19, 1663–1669. doi: 10.1091/mbc.E07-12-1208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guida, S., Trettel, F., Pagnutti, S., Mantuano, E., Tottene, A., Veneziano, L., et al. (2001). Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet. 68, 759–764. doi: 10.1086/318804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guimarães, L., Sequeiros, J., Skirton, H., and Paneque, M. (2013). What counts as effective genetic counselling for presymptomatic testing in late-onset disorders? A study of the consultand's perspective. J. Genet. Couns. 22, 437–447. doi: 10.1007/s10897-012-9561-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hansen, S. T., Meera, P., Otis, T. S., and Pulst, S. M. (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22, 271–283. doi: 10.1093/hmg/dds427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hara, K., Shiga, A., Nozaki, H., Mitsui, J., Takahashi, Y., Ishiguro, H., et al. (2008). Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 71, 547–551. doi: 10.1212/01.wnl.0000311277.71046.a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernjak, N., Slepchenko, B., Fernald, K., Fink, C., Fortin, D., Moraru, I., et al. (2005). Modeling and analysis of calcium signaling events leading to long-term depression in cerebellar Purkinje cells. Biophys. J. 89, 3790–3806. doi: 10.1529/biophysj.105.065771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hines, M., and Carnevale, N. (1997). The NEURON simulation environment. Neural Comput. 9, 1179–1209. doi: 10.1162/neco.1997.9.6.1179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hines, M., and Carnevale, N. (2001). NEURON: a tool for neuroscientists. Neuroscientist 7, 123–135. doi: 10.1177/107385840100700207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hourez, R., Servais, L., Orduz, D., Gall, D., Millard, I., de Kerchove d'Exaerde, A., et al. (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J. Neurosci. 31, 11795–11807. doi: 10.1523/JNEUROSCI.0905-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, L., Chardon, J. W., Carter, M. T., Friend, K. L., Dudding, T. E., Schwartzentruber, J., et al. (2012). Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 7:67. doi: 10.1186/1750-1172-7-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huynh, D., Figueroa, K., Hoang, N., and Pulst, S. (2000). Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26, 44–50. doi: 10.1038/79162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., Cusimano, A., D'Adamo, M., De Curtis, A., and Pessia, M. (2003). Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels. Pflugers Arch. 446, 373–379.
Inoue, T., Lin, X., Kohlmeier, K., Orr, H., Zoghbi, H., and Ross, W. (2001). Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J. Neurophysiol. 85, 1750–1760.
Iwaki, A., Kawano, Y., Miura, S., Shibata, H., Matsuse, D., Li, W., et al. (2008). Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J. Med. Genet. 45, 32–35. doi: 10.1136/jmg.2007.053942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacobi, H., Hauser, T. K., Giunti, P., Globas, C., Bauer, P., Schmitz-Hübsch, T., et al. (2012). Spinocerebellar ataxia types 1, 2, 3 and 6: the clinical spectrum of ataxia and morphometric brainstem and cerebellar findings. Cerebellum 11, 155–166. doi: 10.1007/s12311-011-0292-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ji, J., Hassler, M. L., Shimobayashi, E., Paka, N., Streit, R., and Kapfhammer, J. P. (2014). Increased protein kinase C gamma activity induces Purkinje cell pathology in a mouse model of spinocerebellar ataxia 14. Neurobiol. Dis. 70C, 1–11. doi: 10.1016/j.nbd.2014.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kandel, E., Schwartz, J., and Jessell, T. (2000). ‘The Cerebellum,’ Principles of Neuroscience. New York, NY: McGraw Hill.
Kasumu, A., and Bezprozvanny, I. (2012). Deranged calcium signaling in purkinje cells and pathogenesis in spinocerebellar Ataxia 2 (SCA2) and other Ataxias. Cerebellum 11, 630–639. doi: 10.1007/s12311-010-0182-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasumu, A. W., Hougaard, C., Rode, F., Jacobsen, T. A., Sabatier, J. M., Eriksen, B. L., et al. (2012a). Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 19, 1340–1353. doi: 10.1016/j.chembiol.2012.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasumu, A. W., Liang, X., Egorova, P., Vorontsova, D., and Bezprozvanny, I. (2012b). Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J. Neurosci. 32, 12786–12796. doi: 10.1523/JNEUROSCI.1643-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawaguchi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S., et al. (1994). CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228. doi: 10.1038/ng1194-221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelp, A., Koeppen, A. H., Petrasch-Parwez, E., Calaminus, C., Bauer, C., Portal, E., et al. (2013). A novel transgenic rat model for spinocerebellar ataxia type 17 recapitulates neuropathological changes and supplies in vivo imaging biomarkers. J. Neurosci. 33, 9068–9081. doi: 10.1523/JNEUROSCI.5622-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, D., Jun, K., Lee, S., Kang, N., Min, D., Kim, Y., et al. (1997). Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature 389, 290–293. doi: 10.1038/38508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koide, R., Ikeuchi, T., Onodera, O., Tanaka, H., Igarashi, S., Endo, K., et al. (1994). Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 6, 9–13. doi: 10.1038/ng0194-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koide, R., Kobayashi, S., Shimohata, T., Ikeuchi, T., Maruyama, M., Saito, M., et al. (1999). A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum. Mol. Genet. 8, 2047–2053. doi: 10.1093/hmg/8.11.2047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, X., Antalffy, B., Kang, D., Orr, H., and Zoghbi, H. (2000). Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 3, 157–163. doi: 10.1038/72101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., Tang, T., Tu, H., Nelson, O., Herndon, E., Huynh, D., et al. (2009). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 29, 9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marasco, A., Limongiello, A., and Migliore, M. (2012). Fast and accurate low-dimensional reduction of biophysically detailed neuron models. Sci. Rep. 2:928. doi: 10.1038/srep00928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matilla-Dueñas, A., Ashizawa, T., Brice, A., Magri, S., McFarland, K. N., Pandolfo, M., et al. (2014). Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 13, 269–302. doi: 10.1007/s12311-013-0539-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McLaughlin, S., Wang, J., Gambhir, A., and Murray, D. (2002). PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175. doi: 10.1146/annurev.biophys.31.082901.134259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyasho, T., Takagi, H., Suzuki, H., Watanabe, S., Inoue, M., Kudo, Y., et al. (2001). Low-threshold potassium channels and a low-threshold calcium channel regulate Ca2+ spike firing in the dendrites of cerebellar Purkinje neurons: a modeling study. Brain Res. 891, 106–115. doi: 10.1016/S0006-8993(00)03206-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moraru, I. I., Schaff, J. C., Slepchenko, B. M., Blinov, M. L., Morgan, F., Lakshminarayana, A., et al. (2008). Virtual Cell modelling and simulation software environment. IET Syst. Biol. 2, 352–362. doi: 10.1049/iet-syb:20080102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Musova, Z., Sedlacek, Z., Mazanec, R., Klempir, J., Roth, J., Plevova, P., et al. (2013). Spinocerebellar ataxias type 8, 12, and 17 and dentatorubro-pallidoluysian atrophy in Czech ataxic patients. Cerebellum 12, 155–161. doi: 10.1007/s12311-012-0403-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakamura, K., Jeong, S., Uchihara, T., Anno, M., Nagashima, K., Nagashima, T., et al. (2001). SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 10, 1441–1448. doi: 10.1093/hmg/10.14.1441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Novak, M., Davis, M., Li, A., Goold, R., Tabrizi, S., Sweeney, M., et al. (2010a). PAW32 ITPR1 gene deletion causes spinocerebellar ataxia 15/16: a genetic, clinical and radiological description of a novel kindred. J. Neurol. Neurosurg. Psychiatr. 81, e32. doi: 10.1136/jnnp.2010.226340.60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Novak, M., Sweeney, M., Li, A., Treacy, C., Chandrashekar, H., Giunti, P., et al. (2010b). An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: a genetic, clinical and radiological description. Mov. Disord. 25, 2176–2182. doi: 10.1002/mds.23223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogasawara, H., Doi, T., and Kawato, M. (2008). Systems biology perspectives on cerebellar long-term depression. Neurosignals 16, 300–317. doi: 10.1159/000123040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogura, H., Matsumoto, M., and Mikoshiba, K. (2001). Motor discoordination in mutant mice heterozygous for the type 1 inositol 1,4,5-trisphosphate receptor. Behav. Brain Res. 122, 215–219. doi: 10.1016/S0166-4328(01)00187-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orr, H., Chung, M., Banfi, S., Kwiatkowski, T. J., Servadio, A., Beaudet, A., et al. (1993). Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 4, 221–226. doi: 10.1038/ng0793-221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orr, H. T. (2012). Cell biology of spinocerebellar ataxia. J. Cell Biol. 197, 167–177. doi: 10.1083/jcb.201105092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pulst, S., Nechiporuk, A., Nechiporuk, T., Gispert, S., Chen, X., Lopes-Cendes, I., et al. (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276. doi: 10.1038/ng1196-269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raut, S. S., Chandra, S., Shum, J., and Finol, E. A. (2013). The role of geometric and biomechanical factors in abdominal aortic aneurysm rupture risk assessment. Ann. Biomed. Eng. 41, 1459–1477. doi: 10.1007/s10439-013-0786-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reich, S. D., Steinberg, F., Bachur, N. R., Riggs, C. E., Goebel, R., and Berman, M. (1979). Mathematical model for adriamycin (doxorubicin) pharmacokinetics. Cancer Chemother. Pharmacol. 3, 125–131. doi: 10.1007/BF00254984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodríguez-Quiroga, S. A., Gonzalez-Morón, D., Garretto, N., and Kauffman, M. A. (2013). Huntington's disease masquerading as spinocerebellar ataxia. BMJ Case Rep. 2013:bcr2012008380. doi: 10.1136/bcr-2012-008380
Sandonà, D., Scolari, A., Mikoshiba, K., and Volpe, P. (2003). Subcellular distribution of Homer 1b/c in relation to endoplasmic reticulum and plasma membrane proteins in Purkinje neurons. Neurochem. Res. 28, 1151–1158. doi: 10.1023/A:1024264025401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarkisov, D., and Wang, S. (2008). Order-dependent coincidence detection in cerebellar Purkinje neurons at the inositol trisphosphate receptor. J. Neurosci. 28, 133–142. doi: 10.1523/JNEUROSCI.1729-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sausbier, M., Hu, H., Arntz, C., Feil, S., Kamm, S., Adelsberger, H., et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 9474–9478. doi: 10.1073/pnas.0401702101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaff, J., Slepchenko, B., and Loew, L. (2000). Physiological modeling with virtual cell framework. Meth. Enzymol. 321, 1–23. doi: 10.1016/S0076-6879(00)21184-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schöls, L., Bauer, P., Schmidt, T., Schulte, T., and Riess, O. (2004). Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 3, 291–304. doi: 10.1016/S1474-4422(04)00737-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuler-Faccini, L., Osorio, C. M., Romariz, F., Paneque, M., Sequeiros, J., and Jardim, L. B. (2014). Genetic counseling and presymptomatic testing programs for Machado-Joseph Disease: lessons from Brazil and Portugal. Genet. Mol. Biol. 37(Suppl. 1), 263–270. doi: 10.1590/S1415-47572014000200012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serra, H., Byam, C., Lande, J., Tousey, S., Zoghbi, H., and Orr, H. (2004). Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 13, 2535–2543. doi: 10.1093/hmg/ddh268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shakkottai, V. G., do Carmo Costa, M., Dell'Orco, J. M., Sankaranarayanan, A., Wulff, H., and Paulson, H. L. (2011). Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. 31, 13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shiraishi, Y., Mizutani, A., Yuasa, S., Mikoshiba, K., and Furuichi, T. (2004). Differential expression of Homer family proteins in the developing mouse brain. J. Comp. Neurol. 473, 582–599. doi: 10.1002/cne.20116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slepchenko, B. M., and Loew, L. M. (2010). Use of virtual cell in studies of cellular dynamics. Int. Rev. Cell Mol. Biol. 283, 1–56. doi: 10.1016/S1937-6448(10)83001-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smeets, C. J., and Verbeek, D. S. (2014). Cerebellar ataxia and functional genomics: identifying the routes to cerebellar neurodegeneration. Biochim. Biophys. Acta. 1842, 2030–2038. doi: 10.1016/j.bbadis.2014.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soudah, E., Ng, E. Y., Loong, T. H., Bordone, M., Pua, U., and Narayanan, S. (2013). CFD modelling of abdominal aortic aneurysm on hemodynamic loads using a realistic geometry with CT. Comput. Math. Methods Med. 2013:472564. doi: 10.1155/2013/472564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spence, J. L., and Wallihan, S. (2012). Computational prediction of the polyQ and CAG repeat spinocerebellar ataxia network based on sequence identity to untranslated regions. Gene 509, 273–281. doi: 10.1016/j.gene.2012.07.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Storey, E., Gardner, R., Knight, M., Kennerson, M., Tuck, R., Forrest, S., et al. (2001). A new autosomal dominant pure cerebellar ataxia. Neurology 57, 1913–1915. doi: 10.1212/WNL.57.10.1913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Street, V., Bosma, M., Demas, V., Regan, M., Lin, D., Robinson, L., et al. (1997). The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J. Neurosci. 17, 635–645.
Suzuki, K., Zhou, J., Sato, T., Takao, K., Miyagawa, T., Oyake, M., et al. (2012). DRPLA transgenic mouse substrains carrying single copy of full-length mutant human DRPLA gene with variable sizes of expanded CAG repeats exhibit CAG repeat length- and age-dependent changes in behavioral abnormalities and gene expression profiles. Neurobiol. Dis. 46, 336–350. doi: 10.1016/j.nbd.2012.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takagishi, Y., Oda, S., Hayasaka, S., Dekker-Ohno, K., Shikata, T., Inouye, M., et al. (1996). The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci. Lett. 215, 169–172. doi: 10.1016/0304-3940(96)12967-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T., Guo, C., Wang, H., Chen, X., and Bezprozvanny, I. (2009). Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J. Neurosci. 29, 1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T., Tu, H., Chan, E., Maximov, A., Wang, Z., Wellington, C., et al. (2003a). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, T., Tu, H., Wang, Z., and Bezprozvanny, I. (2003b). Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase a and protein phosphatase 1alpha. J. Neurosci. 23, 403–415.
Tonelli, A., D'Angelo, M., Salati, R., Villa, L., Germinasi, C., Frattini, T., et al. (2006). Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 241, 13–17. doi: 10.1016/j.jns.2005.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trottier, Y., Biancalana, V., and Mandel, J. (1994). Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J. Med. Genet. 31, 377–382. doi: 10.1136/jmg.31.5.377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, H., Tang, T., Wang, Z., and Bezprozvanny, I. (2004). Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J. Biol. Chem. 279, 19375–19382. doi: 10.1074/jbc.M313476200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, J., Xiao, B., Yuan, J., Lanahan, A., Leoffert, K., Li, M., et al. (1998). Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21, 717–726. doi: 10.1016/S0896-6273(00)80589-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ueda, T., Seki, T., Katanazaka, K., Sekiguchi, K., Kobayashi, K., Kanda, F., et al. (2013). A novel mutation in the C2 domain of protein kinase C gamma associated with spinocerebellar ataxia type 14. J. Neurol. 260, 1664–1666. doi: 10.1007/s00415-013-6916-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van de Leemput, J., Chandran, J., Knight, M., Holtzclaw, L., Scholz, S., Cookson, M., et al. (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 3:e108. doi: 10.1371/journal.pgen.0030108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Gaalen, J., Vermeer, S., van Veluw, M., van de Warrenburg, B. P., and Dooijes, D. (2013). A de novo SCA14 mutation in an isolated case of late-onset cerebellar ataxia. Mov. Disord. 28, 1902–1903. doi: 10.1002/mds.25572
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Velázquez-Pérez, L., Rodríguez-Labrada, R., Canales-Ochoa, N., Montero, J. M., Sánchez-Cruz, G., Aguilera-Rodríguez, R., et al. (2014a). Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol. 13, 482–489. doi: 10.1016/S1474-4422(14)70027-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Velázquez-Pérez, L., Rodríguez-Labrada, R., Cruz-Rivas, E. M., Fernández-Ruiz, J., Vaca-Palomares, I., et al. (2014b). Comprehensive study of early features in spinocerebellar Ataxia 2: delineating the prodromal stage of the disease. Cerebellum. 13, 568–579. doi: 10.1007/s12311-014-0574-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vig, P., Subramony, S., and McDaniel, D. (2001). Calcium homeostasis and spinocerebellar ataxia-1 (SCA-1). Brain Res. Bull. 56, 221–225. doi: 10.1016/S0361-9230(01)00595-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagner, W., and Hammer, J. R. (2003). Myosin V and the endoplasmic reticulum: the connection grows. J. Cell Biol. 163, 1193–1196. doi: 10.1083/jcb.200311077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Denk, W., and Häusser, M. (2000). Coincidence detection in single dendritic spines mediated by calcium release. Nat. Neurosci. 3, 1266–1273. doi: 10.1038/81792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, C., Watras, J., and Loew, L. (2003). Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 161, 779–791. doi: 10.1083/jcb.200301070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yue, Q., Jen, J., Nelson, S., and Baloh, R. (1997). Progressive ataxia due to a missense mutation in a calcium-channel gene. Am. J. Hum. Genet. 61, 1078–1087. doi: 10.1086/301613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zecevic, N., Milosevic, A., and Ehrlich, B. (1999). Calcium signaling molecules in human cerebellum at midgestation and in ataxia. Early Hum. Dev. 54, 103–116. doi: 10.1016/S0378-3782(98)00090-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhan, W., and Xu, X. Y. (2013). A mathematical model for thermosensitive liposomal delivery of Doxorubicin to solid tumour. J. Drug Deliv. 2013:172529. doi: 10.1155/2013/172529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Li, Q., Graham, R., Slow, E., Hayden, M., and Bezprozvanny, I. (2008). Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol. Dis. 31, 80–88. doi: 10.1016/j.nbd.2008.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhuchenko, O., Bailey, J., Bonnen, P., Ashizawa, T., Stockton, D., Amos, C., et al. (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69. doi: 10.1038/ng0197-62
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuliani, L., Sabater, L., Saiz, A., Baiges, J., Giometto, B., and Graus, F. (2007). Homer 3 autoimmunity in subacute idiopathic cerebellar ataxia. Neurology 68, 239–240. doi: 10.1212/01.wnl.0000251308.79366.f9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: spinocerebellar ataxia, translational, model, computational, neurology, inositol 1,4,5-triphosphate receptor 1, Purkinje, homer
Citation: Brown S-A, McCullough LD and Loew LM (2015) Computational neurobiology is a useful tool in translational neurology: the example of ataxia. Front. Neurosci. 9:1. doi: 10.3389/fnins.2015.00001
Received: 14 November 2014; Paper pending published: 20 December 2014;
Accepted: 02 January 2015; Published online: 21 January 2015.
Edited by:
Firas H. Kobeissy, University of Florida, USAReviewed by:
Tianxiao Huan, Framingham Heart Study, USAZahraa Naji Sabra, American University of Beirut, Lebanon
Georges Nemer, American University of Beirut, Lebanon
Ilya Bezprozvanny, University of Texas, Southwestern Medical Center at Dallas, USA
Copyright © 2015 Brown, McCullough and Loew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sherry-Ann Brown, Department of Medicine, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA e-mail:YnJvd24uc2hlcnJ5YW5uQG1heW8uZWR1