 Hsiao-Jou Cortina Chen
Hsiao-Jou Cortina Chen Jereme G. Spiers
Jereme G. Spiers Conrad Sernia
Conrad Sernia Nickolas A. Lavidis
Nickolas A. Lavidis- School of Biomedical Sciences, The University of Queensland, Brisbane, QLD, Australia
Exposure to stressful stimuli causes activation of the hypothalamic-pituitary-adrenal axis which rapidly releases high concentrations of glucocorticoid stress hormones, resulting in increased cellular metabolism and spontaneous oxygen and nitrogen radical formation. High concentrations of nitrogen radicals, including nitric oxide, cause damage to cellular proteins in addition to inhibiting components of the mitochondrial transport chain, leading to cellular energy deficiency. During stress exposure, pharmacological inhibition of nitric oxide production reduces indicators of anxiety- and depressive-like behavior in animal models. Therefore, the purpose of this review is to present an overview of the current literature on stress-evoked changes in the nitrergic system, particularly within neural tissue.
Introduction
An acute stress response is mediated by the tripartite activation of the sympatho-adrenal-medullary (SAM), hypothalamic-spinal-adrenal (HSA), and hypothalamic-pituitary-adrenal (HPA) axes. The first of these axes to respond is the autonomic SAM system, consisting of several hypothalamic and brainstem nuclei, notably including the locus ceruleus (Jansen et al., 1995). The locus ceruleus is the primary source of central noradrenergic signaling, functioning via the ascending noradrenergic bundle and descending through preganglionic neurons in the intermediolateral cell column (IML) of the spinal cord to innervate the adrenal medulla (Sara, 2009; Ulrich-Lai and Herman, 2009). Through the combined action of catecholamines, this system promotes increased arousal and vigilance and is responsible for the rapid generation of the “fight-or-flight” response (Jansen et al., 1995). The paraventricular nucleus (PVN) of the hypothalamus is considered the apex of the HPA stress response as release of corticotropin-releasing hormone from the parvocellular neurosecretory neurons triggers anterior pituitary corticotrophs to release the pro-opiomelanocortin fragment, adrenocorticotropic hormone (ACTH), into the circulation. However, the PVN also facilitates corticosterone release directly through the HSA stress axis via adrenocortical innervation from the IML, and indirectly via an alternative stress pathway involving prolactin release (Buijs et al., 1999; Lowry, 2002; Ulrich-Lai et al., 2006; Jaroenporn et al., 2009). This ultimately sensitizes the adrenal gland to ACTH, resulting in corticosterone release from the zona fasciculata of the adrenal cortex thereby exerting the characteristics downstream cellular and metabolic effects of stress (Buijs et al., 1999; Lowry, 2002; Weiser et al., 2011). Adrenal glucocorticoids accelerate cellular metabolism to increase available energy which consequently increases free radical formation in specific regions of the central nervous system (Spiers et al., 2013). This stress-induced increase in radical production, including nitric oxide (NO) formation, leads to oxidative and nitrosative stress (Chen et al., 2014). Furthermore, the toxic metabolite of NO, peroxynitrite, is capable of inhibiting components of the mitochondrial respiratory chain, leading to cellular energy deficiency (Sarti et al., 2012). Since dysfunction of the nitrergic system has been implicated in the neuropathogenesis of several stress-related disease states, the present review summarizes our current understanding and advances relating to the impact of stress on the nitrergic system.
Nitric Oxide Biosynthesis and Functions
Nitric oxide, a gaseous free radical belonging to the family of reactive nitrogen species (RNS), is synthesized through the conversion of L-arginine to L-citrulline by nitric oxide synthase (NOS) in the presence of oxygen, NADPH, and cofactors such as tetrahydrobiopterin (Andrew and Mayer, 1999). There are three main isoforms, each with a specific distribution profile; neuronal NOS (nNOS, type I), inducible NOS (iNOS, type II), and endothelial NOS (eNOS, type III) (Stuehr, 1999). Though nNOS is predominantly active in the cytosol of central and peripheral neurons for signaling and regulation, it has also been found in the sarcolemma and cytoplasm of all muscle fibers (Frandsen et al., 1996). Interestingly, nNOS is present in the hippocampus, hypothalamus, pituitary, and adrenal gland, suggesting co-localization with the HPA axis (Lai et al., 2005; Gadek-Michalska et al., 2012). Furthermore, several studies have demonstrated transcriptional regulation of nNOS by glucocorticoids in the hippocampus, implicating its importance in the stress response, although the upstream promoter of NOS1 does not carry a glucocorticoid responsive element (López-Figueroa et al., 1998; Reagan et al., 1999; Zhou et al., 2011). There are four nNOS splice variants, α, β, γ, and μ, with nNOSα being the most dominant and therefore being physically and functionally coupled to the glutamate receptors of the N-methyl-D-aspartate (NMDA) subtype through their mutual post-synaptic density-95/discs-large/zona occludens-1 (PDZ) binding motif (Eliasson et al., 1997). Within the hippocampus, local calcium influx through NMDA receptors can trigger the production of NO, which subsequently activates its receptor, soluble guanylyl cyclase, leading to release of second messenger cyclic guanosine monophosphate (cGMP) (Figure 1). This NO-cGMP signaling has been implicated in the induction of hippocampal long-term potentiation which is known to be one of the principal mechanisms in learning and memory (Schuman and Madison, 1991; Arancio et al., 1996; Kelley et al., 2010). The nNOSμ mainly localizes in the skeletal muscles, with nNOSμ-deficient muscles being myopathic (Percival et al., 2008). The β variant lacks the PDZ domain while nNOSγ has very little to no enzymatic activity (Eliasson et al., 1997). Endothelial NOS contains a putative shear stress responsive element in the promoter region of the NOS3 gene while the protein is membrane-bound to the golgi apparatus and caveolae, producing NO mainly in the endothelium of blood vessels responsible for vasodilation and smooth muscle relaxation (Smith et al., 2006). The inducible form of NOS responds at the transcriptional level to inflammatory factors (Zamora et al., 2000; Aktan, 2004). Within the central nervous system, the iNOS-mediated release of NO by astrocytes and microglia has a major role in antimicrobial and tumoricidal activity in response to various inflammatory signals (Hua et al., 2002; Brantley et al., 2010). Moreover, upon transcriptional activation, this soluble subtype can produce micromolar levels of NO and is known to be associated with diseases such as artherosclerosis, rheumatoid arthritis, diabetes, septic shock, and multiple sclerosis (Kuhlencordt et al., 2001; Hill et al., 2004; Maki-Petaja et al., 2008; Heemskerk et al., 2009; Soskic et al., 2011). Both nNOS and eNOS are constitutively active isoforms producing low concentrations of NO (in the nanomolar range) over long periods and are activated by calcium ions though transient binding to the calcium-binding protein, calmodulin (Knott and Bossy-Wetzel, 2009). Comparatively, the inducible form of NOS can produce high concentrations of NO in relatively short periods and is calcium independent due to a high binding affinity to calmodulin (Aktan, 2004). The inorganic ions, nitrate and nitrite (NOx), were previously thought to be the end products of NO metabolism. However, recent studies have demonstrated a NOS-independent pathway in which NO can be produced by reducing NOx, a reaction catalyzed by xanthine reductase under low oxygen tension and low pH environment. The NO produced by this nitrate-nitrite-NO pathway may have similar roles to NO generated from the L-arginine-NOS pathway representing an important secondary pool (see review by Lundberg et al., 2008).
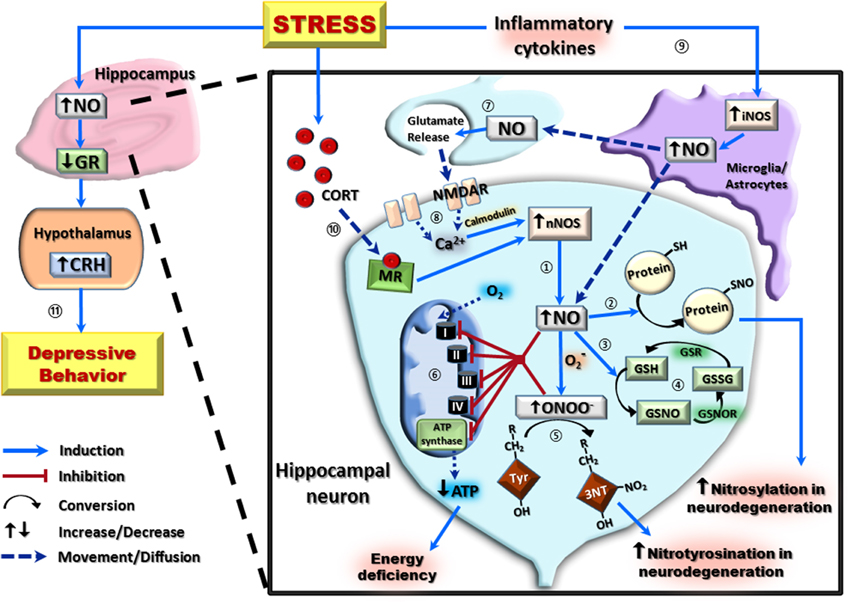
Figure 1. A schematic representation of the nitrergic system and its downstream effects in hippocampal neurons following stress exposure. In hippocampal neurons, the majority of nitric oxide (NO) production occurs via the conversion of L-arginine to L-citrulline by the neuronal isoform of nitric oxide synthase (nNOS) ①. High concentrations of NO can then covalently bond with protein thiol groups (protein-SH) to form S-nitroso-proteins (protein-SNO) ② or interact with the reduced form of glutathione (GSH) forming S-nitrosoglutathione (GSNO) ③. This can be regenerated back to GSH via an initial conversion to oxidized glutathione (GSSG) by S-nitrosoglutathione reductase (GSNOR), and subsequent reduction of GSSG by glutathione reductase (GSR) ④. Interaction of NO with the superoxide radical (O2−) results in the formation of the neurotoxic radical, peroxynitrite (ONOO−) which irreversibly reacts with protein tyrosine (Tyr) residues to form 3-nitrotyrosine (3-NT) ⑤. Increased NO and ONOO− are capable of causing cellular energy deficiency by inhibition of all components of the electron transport chain (Complex I–IV), including ATP synthase, resulting in decreased ATP production ⑥. Both post-synaptically produced NO, and NO produced by the inducible isoform of nitric oxide synthase (iNOS), can act as a neurotransmitter on pre-synaptic neurons ⑦. This pre-synaptic NO causes glutamate release, which activates post-synaptic NMDA receptors (NMDAR) to increase calcium (Ca2+) concentration and, in the presence of calmodulin, further potentiate nNOS-derived NO ⑧. Stress exposure increases NO by activating inflammatory cytokines to potentiate glial/astrocyte iNOS activity ⑨, and by increasing circulating corticosterone (CORT) which induces nNOS activity via a mineralocorticoid receptor (MR)-mediated pathway ⑩. This increase in NO results in downregulation of hippocampal glucocorticoid receptors (GR) and subsequently increases hypothalamic corticotropin-releasing hormone (CRH) to induce depressive-like behaviors ⑪.
Nitrosative Stress
High levels of NO and its derivatives are destructive to cellular components such as proteins, lipids and DNA. Nitric oxide can react directly with molecular oxygen to produce two relatively strong oxidants, nitrogen dioxide and dinitrogen trioxide. However, at physiological levels of NO these reactions are relatively slow. A primary reaction in the production of RNS is the combination of NO and superoxide anions to form the highly reactive metabolite, peroxynitrite, a potent neurotoxin (Lipton et al., 1993). It has been suggested that NO and peroxynitrite can disrupt adenosine 5′-triphosphate (ATP) synthase and almost all components of the mitochondrial respiratory chain (Almeida and Bolanos, 2001; Sarti et al., 2012). These RNS reversibly or irreversibly inhibit mitochondrial oxygen consumption, particularly at complex IV (also known as cytochrome c oxidase), and may lead to cellular energy deficiency and ultimately cell death in pathological conditions (Sarti et al., 2012). Inhibition of cytochrome c oxidase by NO and peroxynitrite causes neuronal dysfunction and, in addition to high iNOS expression, has been observed in the cortex of Alzheimer's patients (Mutisya et al., 1994; Haas et al., 2002).
S-nitrosylation is the covalent attachment of NO to the thiol side chain of the amino acid cysteine, forming other NO derivatives termed S-nitroso-proteins. Under physiological conditions, it has been demonstrated that NO is converted to the nitrosonium ion which subsequently S-nitrosylates the NMDA receptor, thereby preventing glutamate excitotoxicity by blocking calcium influx, promoting cell survival (Lipton and Stamler, 1994). Excessive production of NO can be counteracted by conjugation with reduced glutathione, forming the stable adduct S-nitrosoglutathione which has important role in signal transduction and regulation of a variety of protein functions (Klatt and Lamas, 2000; Anand and Stamler, 2012). Abnormal S-nitrosylation to proteins such as apolipoprotein E, cyclin-dependent kinase 5, dynamin-related protein 1, parkin, peroxiredoxin 2, protein disulfide isomerase, heat-shock protein 90, and X-linked inhibitor of apoptosis have all being linked to neurodegenerative conditions such as Alzheimer's and Parkinson's diseases (Anand and Stamler, 2012). Lastly, peroxynitrite provokes protein nitrotyrosination, an irreversible chemical addition of a nitro group to the tyrosine residue in target proteins generating 3-nitrotyrosine. This post-translational modification usually impairs the normal physiological function of the proteins and therefore nitrotyrosination has been used as a marker in several neurodegenerative conditions such as amyotrophic lateral sclerosis (Peluffo et al., 2004). These aspects of the nitrergic system have been summarized in Figure 1.
Stress-Evoked Modulation of the Nitrergic System
It has been generally accepted that psychophysiological stress is associated with upregulation of NOS mRNA expression and enzymatic activity. For example, a single 6 h acute immobilization stress induces upregulation of iNOS expression and activity in the cerebral cortex which is mediated by the NMDA receptor and subsequent activation of the transcriptional factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Madrigal et al., 2001). The acute stress-induced activation of the NMDA receptor also increases tumor necrosis factor-alpha (TNFα) via upregulation of TNFα-convertase. Antagonism of TNFα-convertase prevents the stress-induced translocation of NF-κB and subsequent iNOS expression, thus confirming the involvement of TNFα (Madrigal et al., 2002). This is also supported by Shirakawa et al. (2004) who demonstrated glutamatergic activation and not catecholaminergic drive of the hypothalamic paraventricular nucleus to be responsible for the acute stress-induced increase in NO metabolites. Interestingly, biting activity is capable of suppressing the stress-induced increase in hypothalamic nNOS mRNA expression in rats (Hori et al., 2005). A single 2 h acute restraint stress significantly increases the density of neurons expressing nNOS visualized by nicotinamide adenine dinucleotide phosphate-diaphorase (NADPH-d) histochemistry in the amygdaloid nucleus, an effect delayed by 5 days in the hippocampus and entorhinal cortex (Echeverry et al., 2004). Predator-induced post-traumatic stress significantly increases nNOS positive neurons and total NOx in the medial prefrontal cortex 7 days after the 10 min predator stress treatment (Campos et al., 2013). Conversely, Chakraborti et al. (2014) demonstrated that acute restraint stress causes a reduction in total NOx and an increase in the major endogenous NOS inhibitor, asymmetric dimethylarginine, in whole brain homogenates. This suggests that the stress-induced NOx increases in regions such as the hippocampus and hypothalamus may hold a high degree of functional significance. These biochemical changes in NOx and asymmetric dimethylarginine were observed alongside anxiety-like behavior and were more pronounced in male compared to female rats. The pharmacological blockade of estrogen biosynthesis exacerbated these biochemical and behavioral changes in females, suggesting that the observed sex differences are due to a protective role of estrogen. Interestingly, bilateral injection of an NMDA receptor antagonist, NOS inhibitor, or NO scavenger into the dorsal hippocampus attenuated autonomic responses such as hypertension and tachycardia following a 60 min acute restraint stress, suggesting that NMDA/NOS activation within the hippocampus plays a role in autonomic modulation during stress (Moraes-Neto et al., 2014). Another study from the same group proposed a glutamatergic NMDA receptor-NO-cGMP signaling pathway in modulating contextual fear conditioning within the dorsal hippocampus, where intra-hippocampal injection of NMDA receptor antagonist DL-AP7, NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO), and cGMP inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), attenuated the fear-conditioned response (Fabri et al., 2014).
Chronic immobilization stress has been shown to increase NOx, iNOS activity, and peroxynitrite-induced 3-nitrotyrosine accumulation in cortical neurons (Olivenza et al., 2000). Notably, de Pablos et al. (2014) recently found a degree of regional specificity associated with this chronic stress-induced iNOS expression, with little to no constitutive expression in the substantia nigra following 9 days of unpredictable stress exposure. However, this same unpredictable stress model potentiates iNOS expression following exposure to exogenous immunostimulatory stressors such as lipopolysaccharides. Recent studies in several animal paradigms have demonstrated that inhibitors of NOS significantly modulate stress-related behaviors. In support of these findings, the commercially available antidepressant paroxetine, a selective serotonin reuptake inhibitor, also possesses NOS inhibition capability (Finkel et al., 1996). Wegener and Volke (2010) have reviewed and summarized these studies including data on each of the NOS inhibitor's specificity and potency, and their anxiolytic- and antidepressant-like properties. Chronic unpredictable mild stress increases plasma nitrite levels and iNOS mRNA expression in the cortex, in addition to damaging cortical neurons and inducing depressive-like behavior (Wang et al., 2008; Peng et al., 2012). These effects can be attenuated or prevented using NOS inhibitors, which was demonstrated by intra-hippocampal injection of the selective iNOS inhibitor, aminoguanidine, resulting in suppression of the chronic unpredictable mild stress-induced depressive-like behavior in rats (Wang et al., 2008). Regional infusion of a selective nNOS inhibitor 7-nitroindazole (7-NI) into the hippocampus showed antidepressant-like effects similar to those with the iNOS inhibitor, aminoguanidine (Joca and Guimaraes, 2006). Likewise, the anxiogenic-like behavior observed in rats during ethanol withdrawal is inhibited by administration of the selective iNOS inhibitor, 1400W, into the dorsolateral periaqueductal gray (Bonassoli et al., 2013). The data with intra-cerebral NOS inhibition is further supported by studies using systemic treatment. Intraperitoneal injection of 1400W increases survival of cortical neurons and decreases the depressive-like behavior in mice (Peng et al., 2012). The nNOS inhibitor 1-(-2-trifluoromethylphenyl)-imidazole (TRIM) given systemically 30 min prior to testing induces anxiolytic-like behavior shown by increased time spent in the light compartment of a light-dark compartment test (Volke et al., 2003). Furthermore, TRIM administration decreased the immobility time in the forced swimming test, demonstrating an antidepressant-like effect comparable to the tricyclic antidepressant imipramine. In agreement with these observations, Ulak et al. (2008) injected TRIM intraperitoneally 50 min before a forced swim test and showed the involvement of the serotonergic system in the antidepressant-like actions of TRIM. This was further clarified in a later study in which the serotonin type II receptors were found to be responsible for this effect (Ulak et al., 2010). Furthermore, Joung et al. (2012) demonstrated that following a 2 h immobilization stress, the selective inhibitor 7-NI produced its anxiolytic-like effects shown by an increase in the time spent on the open arms of the elevated plus-maze through the direct reduction of NO metabolites in the PVN and locus ceruleus. A less specific NOS inhibitor, L-NG-Nitroarginine methyl ester (L-NAME), injected systemically 30 min prior to testing shows protective effects against chronic swim stress-induced impairment of passive avoidance learning and hyperalgesia in rats (Nazeri et al., 2014). In a similar vein, Ferreira et al. (2012) performed behavioral, genomic, and proteomic analyses in rats and suggested that the antidepressant-like effects of NOS inhibition may involve the expression of additional factors including members of the glutathione redox system.
Genetic animal models have also contributed to the current understanding of nitrergic changes in stress. Thus, inhibition of NO production by nNOS gene deletion in mice suppressed hippocampal neurogenesis and exhibited antidepressant-like properties while nNOS over-expression in the hippocampus was essential for chronic stress-induced depression (Zhou et al., 2007). Recently, a number of studies have proposed a regulatory role of NO on the limbic HPA stress axis. Zhang et al. (2010) used mice lacking the nNOS gene to demonstrate an anxiolytic-like phenotype when tested using an elevated plus-maze, similar to normal mice treated with intra-hippocampal microinjection of the selective nNOS inhibitor 7-NI. The authors proposed a signaling pathway involving the activation of serotonin type IA receptors which mediate, via an unknown mechanism, the downregulation of hippocampal nNOS, leading to a decrease in NO and subsequent inhibition of cAMP response element-binding (CREB) protein phosphorylation. A follow up study elucidated further the link between NO and the HPA axis by showing that chronic mild stress and glucocorticoid exposure lead to hippocampal nNOS overexpression via activating hippocampal mineralocorticoid receptor (MR) (Zhou et al., 2011). The excessive nNOS-derived NO significantly downregulated local glucocorticoid receptor (GR) expression through either the soluble guanylyl cyclase/cGMP or peroxynitrite/extracellular signal-regulated kinase (ERK) signaling pathways. The significant downregulation of GR in the hippocampus leads to an elevation in hypothalamic corticotropin-releasing hormone and the depressive-like behaviors in mice as illustrated in Figure 1. It is important to note that nNOS deletion, infusion of intrahippocampal nNOS inhibitor, and NO-cGMP signaling blockade prevented the chronic mild stress-evoked behavioral modification. Interestingly, this chronic glucocorticoid-induced MR-nNOS-NO pathway is exclusive to the MR-rich hippocampus and drives HPA axis hyperactivity through impaired negative feedback (Zhu et al., 2014).
The considerable body of evidence from animal models is progressively expanding and supported by modest but significant clinical studies. Several reports have shown that increased levels of NO metabolites are present in depressed and autistic patients (Suzuki et al., 2001; Sogut et al., 2003; Lee et al., 2006). Patients with recurrent depressive behavior displayed higher plasma NOx concentrations which were associated with cognitive impairment (Talarowska et al., 2012). Galecki et al. (2010, 2011) discovered single nucleotide polymorphisms in exon 22 of the NOS2A gene (iNOS) and exon 29 of the NOSI gene (nNOS) in depressed Caucasian individuals. Furthermore, three single nucleotide polymorphisms located at the regulatory region of NOSI gene are responsible for the susceptibility of an individual to depressive disorders (Sarginson et al., 2014).
Summary
A growing body of evidence suggests that the etiology of anxiety and depression-related conditions can be derived from the sensitization of particular stress-related circuits that are “primed” following exposure to a short-term stressor. The duration for stress-related circuitry priming far exceeds responses to adrenergic and glucocorticoid-mediated stress responses. Understanding the mechanisms underlying the induction of this long latency will provide a significant link between stress and the pathogenesis of anxiety and depressive disorders. The nitrergic system has been implicated in regulating both short and long-term activation of the stress response, with a variety of NOS inhibitors demonstrating potent anxiolytic and antidepressant activity. The intrinsic cross talk between neuroendocrine stress and nitrergic system activation is now an important physiological consideration. Further understanding the role of this system is important in identifying early players in stress-induced pathological conditions.
Author Contributions
Author Hsiao-Jou Cortina Chen managed the literature searches, wrote the first draft of the manuscript, and produced the graphic. Author Jereme G. Spiers, Conrad Sernia and Nickolas A. Lavidis critically revised the manuscript. All authors have approved the final version of the manuscript for journal submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Szu-Ju Patricia Chen for assistance in graphical design and production.
References
Aktan, F. (2004). iNOS-mediated nitric oxide production and its regulation. Life Sci. 75, 639–653. doi: 10.1016/j.lfs.2003.10.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Almeida, A., and Bolanos, J. P. (2001). A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J. Neurochem. 77, 676–690. doi: 10.1046/j.1471-4159.2001.00276.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anand, P., and Stamler, J. S. (2012). Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. 90, 233–244. doi: 10.1007/s00109-012-0878-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andrew, P. J., and Mayer, B. (1999). Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 43, 521–531. doi: 10.1016/S0008-6363(99)00115-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arancio, O., Kiebler, M., Lee, C. J., Lev-Ram, V., Tsien, R. Y., Kandel, E. R., et al. (1996). Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87, 1025–1035. doi: 10.1016/S0092-8674(00)81797-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonassoli, V. T., Contardi, E. B., Milani, H., and de Oliveira, R. M. (2013). Effects of nitric oxide synthase inhibition in the dorsolateral periaqueductal gray matter on ethanol withdrawal-induced anxiety-like behavior in rats. Psychopharmacology (Berl). 228, 487–498. doi: 10.1007/s00213-013-3049-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brantley, E. C., Guo, L., Zhang, C., Lin, Q., Yokoi, K., Langley, R. R., et al. (2010). Nitric oxide-mediated tumoricidal activity of murine microglial cells. Transl. Oncol. 3, 380–388. doi: 10.1593/tlo.10208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buijs, R. M., Wortel, J., Van Heerikhuize, J. J., Feenstra, M. G., Ter Horst, G. J., Romijn, H. J., et al. (1999). Anatomical and functional demonstration of a multisynaptic suprachiasmatic nucleus adrenal (cortex) pathway. Eur. J. Neurosci. 11, 1535–1544. doi: 10.1046/j.1460-9568.1999.00575.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campos, A. C., Piorino, E. M., Ferreira, F. R., and Guimaraes, F. S. (2013). Increased nitric oxide-mediated neurotransmission in the medial prefrontal cortex is associated with the long lasting anxiogenic-like effect of predator exposure. Behav. Brain Res. 256, 391–397. doi: 10.1016/j.bbr.2013.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chakraborti, A., Gulati, K., and Ray, A. (2014). Possible role of nitric oxide (NO) in the regulation of gender related differences in stress induced anxiogenesis in rats. Nitric Oxide 43, 74–80. doi: 10.1016/j.niox.2014.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, H. J. C., Spiers, J. G., Sernia, C., Anderson, S. T., and Lavidis, N. A. (2014). Reactive nitrogen species contribute to the rapid onset of redox changes induced by acute immobilization stress in rats. Stress 17, 520–527. doi: 10.3109/10253890.2014.966264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Pablos, R. M., Herrera, A. J., Espinosa-Oliva, A. M., Sarmiento, M., Munoz, M. F., Machado, A., et al. (2014). Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J. Neuroinflammation 11, 34. doi: 10.1186/1742-2094-11-34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Echeverry, M. B., Guimaraes, F. S., and Del Bel, E. A. (2004). Acute and delayed restraint stress-induced changes in nitric oxide producing neurons in limbic regions. Neuroscience 125, 981–993. doi: 10.1016/j.neuroscience.2003.12.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eliasson, M. J., Blackshaw, S., Schell, M. J., and Snyder, S. H. (1997). Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc. Natl. Acad. Sci. U.S.A. 94, 3396–3401. doi: 10.1073/pnas.94.7.3396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fabri, D. R. S., Hott, S. C., Reis, D. G., Biojone, C., Corrêa, F. M. A., and Resstel, L. B. M. (2014). The expression of contextual fear conditioning involves activation of a NMDA receptor-nitric oxide-cGMP pathway in the dorsal hippocampus of rats. Eur. Neuropsychopharmacol. 24, 1676–1686. doi: 10.1016/j.euroneuro.2014.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferreira, F. R., Oliveira, A. M., Dinarte, A. R., Pinheiro, D. G., Greene, L. J., Silva, W. A., et al. (2012). Changes in hippocampal gene expression by 7-nitroindazole in rats submitted to forced swimming stress. Genes Brain Behav. 11, 303–313. doi: 10.1111/j.1601-183X.2011.00757.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finkel, M. S., Laghrissi-Thode, F., Pollock, B. G., and Rong, J. (1996). Paroxetine is a novel nitric oxide synthase inhibitor. Psychopharmacol. Bull. 32, 653–658.
Frandsen, U., Lopez-Figueroa, M., and Hellsten, Y. (1996). Localization of nitric oxide synthase in human skeletal muscle. Biochem. Biophys. Res. Commun. 227, 88–93. doi: 10.1006/bbrc.1996.1472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gadek-Michalska, A., Tadeusz, J., Rachwalska, P., Spyrka, J., and Bugajski, J. (2012). Effect of repeated restraint on homotypic stress-induced nitric oxide synthases expression in brain structures regulating HPA axis. Pharmacol. Rep. 64, 1381–1390. doi: 10.1016/S1734-1140(12)70935-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galecki, P., Maes, M., Florkowski, A., Lewinski, A., Galecka, E., Bienkiewicz, M., et al. (2010). An inducible nitric oxide synthase polymorphism is associated with the risk of recurrent depressive disorder. Neurosci. Lett. 486, 184–187. doi: 10.1016/j.neulet.2010.09.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galecki, P., Maes, M., Florkowski, A., Lewinski, A., Galecka, E., Bienkiewicz, M., et al. (2011). Association between inducible and neuronal nitric oxide synthase polymorphisms and recurrent depressive disorder. J. Affect. Disord. 129, 175–182. doi: 10.1016/j.jad.2010.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haas, J., Storch-Hagenlocher, B., Biessmann, A., and Wildemann, B. (2002). Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer's dementia and following stimulation with beta-amyloid 1-42 in vitro. Neurosci. Lett. 322, 121–125. doi: 10.1016/S0304-3940(02)00095-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heemskerk, S., Masereeuw, R., Russel, F. G., and Pickkers, P. (2009). Selective iNOS inhibition for the treatment of sepsis-induced acute kidney injury. Nat. Rev. Nephrol. 5, 629–640. doi: 10.1038/nrneph.2009.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hill, K. E., Zollinger, L. V., Watt, H. E., Carlson, N. G., and Rose, J. W. (2004). Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: distribution, cellular expression and association with myelin damage. J. Neuroimmunol. 151, 171–179. doi: 10.1016/j.jneuroim.2004.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hori, N., Lee, M. C., Sasaguri, K., Ishii, H., Kamei, M., Kimoto, K., et al. (2005). Suppression of stress-induced nNOS expression in the rat hypothalamus by biting. J. Dent. Res. 84, 624–628. doi: 10.1177/154405910508400708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hua, L. L., Kim, M. O., Brosnan, C. F., and Lee, S. C. (2002). Modulation of astrocyte inducible nitric oxide synthase and cytokine expression by interferon beta is associated with induction and inhibition of interferon gamma-activated sequence binding activity. J. Neurochem. 83, 1120–1128. doi: 10.1046/j.1471-4159.2002.01226.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jansen, A. S., Nguyen, X. V., Karpitskiy, V., Mettenleiter, T. C., and Loewy, A. D. (1995). Central command neurons of the sympathetic nervous system: basis of the fight-or-flight response. Science 270, 644–646. doi: 10.1126/science.270.5236.644
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jaroenporn, S., Nagaoka, K., Ohta, R., Shirota, M., Watanabe, G., and Taya, K. (2009). Differences in adrenocortical secretory and gene expression responses to stimulation in vitro by ACTH or prolactin between high- and low-avoidance Hatano rats. Stress 12, 22–29. doi: 10.1080/10253890801976652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joca, S. R., and Guimaraes, F. S. (2006). Inhibition of neuronal nitric oxide synthase in the rat hippocampus induces antidepressant-like effects. Psychopharmacology (Berl). 185, 298–305. doi: 10.1007/s00213-006-0326-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joung, H. Y., Jung, E. Y., Kim, K., Lee, M. S., Her, S., and Shim, I. (2012). The differential role of NOS inhibitors on stress-induced anxiety and neuroendocrine alterations in the rat. Behav. Brain Res. 235, 176–181. doi: 10.1016/j.bbr.2012.07.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelley, J. B., Anderson, K. L., and Itzhak, Y. (2010). Pharmacological modulators of nitric oxide signaling and contextual fear conditioning in mice. Psychopharmacology (Berl). 210, 65–74. doi: 10.1007/s00213-010-1817-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klatt, P., and Lamas, S. (2000). Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 267, 4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knott, A. B., and Bossy-Wetzel, E. (2009). Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 11, 541–554. doi: 10.1089/ars.2008.2234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuhlencordt, P. J., Chen, J., Han, F., Astern, J., and Huang, P. L. (2001). Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation 103, 3099–3104. doi: 10.1161/01.CIR.103.25.3099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, F. J., Huang, S. S., Hsieh, M. C., Hsin, S. C., Wu, C. H., Hsin, Y. C., et al. (2005). Upregulation of neuronal nitric oxide synthase mRNA and protein in adrenal medulla of water-deprived rats. J. Histochem. Cytochem. 53, 45–53. doi: 10.1177/002215540505300106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, B. H., Lee, S. W., Yoon, D., Lee, H. J., Yang, J. C., Shim, S. H., et al. (2006). Increased plasma nitric oxide metabolites in suicide attempters. Neuropsychobiology 53, 127–132. doi: 10.1159/000092542
Lipton, S. A., Choi, Y. B., Pan, Z. H., Lei, S. Z., Chen, H. S., Sucher, N. J., et al. (1993). A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626–632. doi: 10.1038/364626a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipton, S. A., and Stamler, J. S. (1994). Actions of redox-related congeners of nitric oxide at the NMDA receptor. Neuropharmacology 33, 1229–1233. doi: 10.1016/0028-3908(94)90021-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
López-Figueroa, M. O., Itoi, K., and Watson, S. J. (1998). Regulation of nitric oxide synthase messenger RNA expression in the rat hippocampus by glucocorticoids. Neuroscience 87, 439–446. doi: 10.1016/S0306-4522(98)00075-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lowry, C. A. (2002). Functional subsets of serotonergic neurones: implications for control of the hypothalamic-pituitary-adrenal axis. J. Neuroendocrinol. 14, 911–923. doi: 10.1046/j.1365-2826.2002.00861.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lundberg, J. O., Weitzberg, E., and Gladwin, M. T. (2008). The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 7, 156–167. doi: 10.1038/nrd2466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madrigal, J. L., Hurtado, O., Moro, M. A., Lizasoain, I., Lorenzo, P., Castrillo, A., et al. (2002). The increase in TNF-alpha levels is implicated in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology 26, 155–163. doi: 10.1016/S0893-133X(01)00292-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madrigal, J. L., Moro, M. A., Lizasoain, I., Lorenzo, P., Castrillo, A., Bosca, L., et al. (2001). Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J. Neurochem. 76, 532–538. doi: 10.1046/j.1471-4159.2001.00108.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maki-Petaja, K. M., Cheriyan, J., Booth, A. D., Hall, F. C., Brown, J., Wallace, S. M., et al. (2008). Inducible nitric oxide synthase activity is increased in patients with rheumatoid arthritis and contributes to endothelial dysfunction. Int. J. Cardiol. 129, 399–405. doi: 10.1016/j.ijcard.2008.02.011
Moraes-Neto, T. B., Scopinho, A. A., Biojone, C., Correa, F. M., and Resstel, L. B. (2014). Involvement of dorsal hippocampus glutamatergic and nitrergic neurotransmission in autonomic responses evoked by acute restraint stress in rats. Neuroscience 258, 364–373. doi: 10.1016/j.neuroscience.2013.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mutisya, E. M., Bowling, A. C., and Beal, M. F. (1994). Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J. Neurochem. 63, 2179–2184. doi: 10.1046/j.1471-4159.1994.63062179.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nazeri, M., Razavinasab, M., Abareghi, F., and Shabani, M. (2014). Role of nitric oxide in altered nociception and memory following chronic stress. Physiol. Behav. 129, 214–220. doi: 10.1016/j.physbeh.2014.02.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olivenza, R., Moro, M. A., Lizasoain, I., Lorenzo, P., Fernandez, A. P., Rodrigo, J., et al. (2000). Chronic stress induces the expression of inducible nitric oxide synthase in rat brain cortex. J. Neurochem. 74, 785–791. doi: 10.1046/j.1471-4159.2000.740785.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peluffo, H., Shacka, J. J., Ricart, K., Bisig, C. G., Martìnez-Palma, L., Pritsch, O., et al. (2004). Induction of motor neuron apoptosis by free 3-nitro-l-tyrosine. J. Neurochem. 89, 602–612. doi: 10.1046/j.1471-4159.2004.02363.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peng, Y.-L., Liu, Y.-N., Liu, L., Wang, X., Jiang, C.-L., and Wang, Y.-X. (2012). Inducible nitric oxide synthase is involved in the modulation of depressive behaviors induced by unpredictable chronic mild stress. J. Neuroinflammation 9:75. doi: 10.1186/1742-2094-9-75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Percival, J. M., Anderson, K. N., Gregorevic, P., Chamberlain, J. S., and Froehner, S. C. (2008). Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS ONE 3:e3387. doi: 10.1371/journal.pone.0003387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reagan, L. P., McKittrick, C. R., and McEwen, B. S. (1999). Corticosterone and phenytoin reduce neuronal nitric oxide synthase messenger RNA expression in rat hippocampus. Neuroscience 91, 211–219. doi: 10.1016/S0306-4522(98)00615-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sara, S. J. (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 10, 211–223. doi: 10.1038/nrn2573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarginson, J. E., Deakin, J. W., Anderson, I. M., Downey, D., Thomas, E., Elliott, R., et al. (2014). Neuronal Nitric Oxide Synthase (NOS1) polymorphisms interact with financial hardship to affect depression risk. Neuropsychopharmacology 39, 2857–2866. doi: 10.1038/npp.2014.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarti, P., Forte, E., Mastronicola, D., Giuffre, A., and Arese, M. (2012). Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim. Biophys. Acta 1817, 610–619. doi: 10.1016/j.bbabio.2011.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuman, E. M., and Madison, D. V. (1991). A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506. doi: 10.1126/science.1720572
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shirakawa, T., Mitome, M., Kikuiri, T., Nakamura, W., Oshima, S., Hasegawa, T., et al. (2004). Immobilization induces acute nitric oxide production in the rat hypothalamus: a role of ionotropic glutamate receptors in the paraventricular nucleus. Endocrinology 145, 3603–3607. doi: 10.1210/en.2004-0068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, A. R., Visioli, F., and Hagen, T. M. (2006). Plasma membrane-associated endothelial nitric oxide synthase and activity in aging rat aortic vascular endothelia markedly decline with age. Arch. Biochem. Biophys. 454, 100–105. doi: 10.1016/j.abb.2006.02.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sogut, S., Zoroglu, S. S., Ozyurt, H., Yilmaz, H. R., Ozugurlu, F., Sivasli, E., et al. (2003). Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. Acta 331, 111–117. doi: 10.1016/S0009-8981(03)00119-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soskic, S. S., Dobutovic, B. D., Sudar, E. M., Obradovic, M. M., Nikolic, D. M., Djordjevic, J. D., et al. (2011). Regulation of Inducible Nitric Oxide Synthase (iNOS) and its potential role in insulin resistance, diabetes and heart failure. Open Cardiovasc. Med. J. 5, 153–163. doi: 10.2174/1874192401105010153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiers, J. G., Chen, H. J., Bradley, A. J., Anderson, S. T., Sernia, C., and Lavidis, N. A. (2013). Acute restraint stress induces rapid and prolonged changes in erythrocyte and hippocampal redox status. Psychoneuroendocrinology 38, 2511–2519. doi: 10.1016/j.psyneuen.2013.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stuehr, D. J. (1999). Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1411, 217–230. doi: 10.1016/S0005-2728(99)00016-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, E., Yagi, G., Nakaki, T., Kanba, S., and Asai, M. (2001). Elevated plasma nitrate levels in depressive states. J. Affect. Disord. 63, 221–224. doi: 10.1016/S0165-0327(00)00164-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Talarowska, M., Galecki, P., Maes, M., Orzechowska, A., Chamielec, M., Bartosz, G., et al. (2012). Nitric oxide plasma concentration associated with cognitive impairment in patients with recurrent depressive disorder. Neurosci. Lett. 510, 127–131. doi: 10.1016/j.neulet.2012.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulak, G., Mutlu, O., Akar, F. Y., Komsuoglu, F. I., Tanyeri, P., and Erden, B. F. (2008). Neuronal NOS inhibitor 1-(2-trifluoromethylphenyl)-imidazole augment the effects of antidepressants acting via serotonergic system in the forced swimming test in rats. Pharmacol. Biochem. Behav. 90, 563–568. doi: 10.1016/j.pbb.2008.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulak, G., Mutlu, O., Tanyeri, P., Komsuoglu, F. I., Akar, F. Y., and Erden, B. F. (2010). Involvement of serotonin receptor subtypes in the antidepressant-like effect of TRIM in the rat forced swimming test. Pharmacol. Biochem. Behav. 95, 308–314. doi: 10.1016/j.pbb.2010.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulrich-Lai, Y. M., Arnhold, M. M., and Engeland, W. C. (2006). Adrenal splanchnic innervation contributes to the diurnal rhythm of plasma corticosterone in rats by modulating adrenal sensitivity to ACTH. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1128–R1135. doi: 10.1152/ajpregu.00042.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulrich-Lai, Y. M., and Herman, J. P. (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 10, 397–409. doi: 10.1038/nrn2647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volke, V., Wegener, G., Bourin, M., and Vasar, E. (2003). Antidepressant- and anxiolytic-like effects of selective neuronal NOS inhibitor 1-(2-trifluoromethylphenyl)-imidazole in mice. Behav. Brain Res. 140, 141–147. doi: 10.1016/S0166-4328(02)00312-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, D., An, S. C., and Zhang, X. (2008). Prevention of chronic stress-induced depression-like behavior by inducible nitric oxide inhibitor. Neurosci. Lett. 433, 59–64. doi: 10.1016/j.neulet.2007.12.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wegener, G., and Volke, V. (2010). Nitric oxide synthase inhibitors as antidepressants. Pharmaceuticals 3, 273–299. doi: 10.3390/ph3010273
Weiser, M. J., Osterlund, C., and Spencer, R. L. (2011). Inhibitory effects of corticosterone in the hypothalamic paraventricular nucleus (PVN) on stress-induced adrenocorticotrophic hormone secretion and gene expression in the PVN and anterior pituitary. J. Neuroendocrinol. 23, 1231–1240. doi: 10.1111/j.1365-2826.2011.02217.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zamora, R., Vodovotz, Y., and Billiar, T. R. (2000). Inducible nitric oxide synthase and inflammatory diseases. Mol. Med. 6, 347–373.
Zhang, J., Huang, X. Y., Ye, M. L., Luo, C. X., Wu, H. Y., Hu, Y., et al. (2010). Neuronal nitric oxide synthase alteration accounts for the role of 5-HT1A receptor in modulating anxiety-related behaviors. J. Neurosci. 30, 2433–2441. doi: 10.1523/JNEUROSCI.5880-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Q. G., Hu, Y., Hua, Y., Hu, M., Luo, C. X., Han, X., et al. (2007). Neuronal nitric oxide synthase contributes to chronic stress-induced depression by suppressing hippocampal neurogenesis. J. Neurochem. 103, 1843–1854. doi: 10.1111/j.1471-4159.2007.04914.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Q. G., Zhu, L. J., Chen, C., Wu, H. Y., Luo, C. X., Chang, L., et al. (2011). Hippocampal neuronal nitric oxide synthase mediates the stress-related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J. Neurosci. 31, 7579–7590. doi: 10.1523/JNEUROSCI.0004-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, L. J., Liu, M. Y., Li, H., Liu, X., Chen, C., Han, Z., et al. (2014). The different roles of glucocorticoids in the hippocampus and hypothalamus in chronic stress-induced HPA axis hyperactivity. PLoS ONE 9:e97689. doi: 10.1371/journal.pone.0097689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: anxiety, depression, hypothalamic-pituitary-adrenal axis, glucocorticoids, nitrergic system, nitric oxide, peroxynitrite, reactive nitrogen species
Citation: Chen H-JC, Spiers JG, Sernia C and Lavidis NA (2015) Response of the nitrergic system to activation of the neuroendocrine stress axis. Front. Neurosci. 9:3. doi: 10.3389/fnins.2015.00003
Received: 30 October 2014; Accepted: 05 January 2015;
Published online: 21 January 2015.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Charles W. Wilkinson, University of Washington, USAAlline C. Campos, Max-Planck-Institute for Experimental Medicine, Germany
Copyright © 2015 Chen, Spiers, Sernia and Lavidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hsiao-Jou Cortina Chen, School of Biomedical Sciences, The University of Queensland, St. Lucia, Brisbane, QLD 4072, Australia e-mail:aC5jaGVuNkB1cS5lZHUuYXU=