 Scarlet Gallegos
Scarlet Gallegos Carla Pacheco
Carla Pacheco Christian Peters
Christian Peters Carlos M. Opazo
Carlos M. Opazo Luis G. Aguayo
Luis G. Aguayo- 1Laboratory of Neurophysiology, Department of Physiology, University of Concepcion, Concepcio, Chile
- 2Oxidation Biology Laboratory, The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, VIC, Australia
Alpha-synuclein is a presynaptic protein expressed throughout the central nervous system, and it is the main component of Lewy bodies, one of the histopathological features of Parkinson's disease (PD) which is a progressive and irreversible neurodegenerative disorder. The conformational flexibility of α-synuclein allows it to adopt different conformations, i.e., bound to membranes or form aggregates, the oligomers are believed to be the more toxic species. In this review, we will focus on two major features of α-synuclein, transmission and toxicity, that could help to understand the pathological characteristics of PD. One important feature of α-synuclein is its ability to be transmitted from neuron to neuron using mechanisms such as endocytosis, plasma membrane penetration or through exosomes, thus propagating the Lewy body pathology to different brain regions thereby contributing to the progressiveness of PD. The second feature of α-synuclein is that it confers cytotoxicity to recipient cells, principally when it is in an oligomeric state. This form causes mitochondrial dysfunction, endoplasmic reticulum stress, oxidative stress, proteasome impairment, disruption of plasma membrane and pore formation that lead to apoptosis pathway activation and consequent cell death. The complexity of α-synuclein oligomerization and formation of toxic species could be a major factor for the irreversibility of PD and could also explain the lack of successful therapies to halt the disease.
General Overview
Misfolded proteins are rich in β-sheet structure with a high tendency to form long fibrillar aggregates known as amyloid deposits (Soto, 2012). Amyloid is a generic term referring to organized protein aggregates with specific staining properties, higher resistance to proteolytic degradation and a fibrillar appearance when observed with electronic microscopy (Soto, 2003). β-structures are harder to degrade than α-helixes (main conformation of native proteins), which explains why amyloid deposits cannot be removed by the proteasome system (Lee and Yu, 2005). The aggregation of misfolded proteins is believed to occur when hydrophobic residues exposed at the surface of proteins interact with other misfolded proteins (Doyle et al., 2011).
There is a group of neurodegenerative diseases characterized by the misfolding of proteins into β-sheet aggregated structures that accumulate as amyloid deposits in affected tissues (Moreno-Gonzalez and Soto, 2011). These conformational disorders or protein misfolding diseases include Alzheimer's disease (AD) with extracellular plaques of amyloid-β protein (Aβ) and tangles of hyperphosphorylated tau protein in the cytoplasm of neurons; Parkinson's disease (PD) where the cytoplasm of neurons in the substantia nigra contains aggregates called Lewy bodies composed principally of the protein α-synuclein (α-syn); Huntington's disease; Amyotrophic Lateral Sclerosis; and transmissible spongiform encephalopathy characterized by an accumulation of aggregates of the prion protein (PrP) in the brain (Soto, 2003).
Prion proteins propagate by auto-catalytic conversion of native, nonpathogenic forms of the protein (PrPC) expressed in several types of human cells (Frost and Diamond, 2010), into misfolded pathological conformation (PrPSc) (Moreno-Gonzalez and Soto, 2011; Costanzo and Zurzolo, 2013). In prion disease, the pathological conformation is the primary infectious agent (Soto and Satani, 2011; Munch and Bertolotti, 2012) that propagates using a mechanism called “seeding/nucleation” where PrPSc acts like a seed that recruits and converts soluble protein into aggregates of PrPC which form polymers (Moreno-Gonzalez and Soto, 2011; Munch and Bertolotti, 2012). As prions grow and spread, they interfere with the function of the nervous system resulting in progression of the disease in affected patients (Jucker and Walker, 2013).
Accumulating experimental data indicate that the seeding principle, similar to the conversion of PrPC to PrPSc in prion diseases, also applies to other proteins associated to neurodegenerative diseases such as AD and PD (Soto, 2012; Costanzo and Zurzolo, 2013; Jucker and Walker, 2013). In this review, we will address features of the α-syn protein that propagates and causes toxicity in recipient neurons, which could explain the progressive and irreversible pathology of PD.
Parkinson's Disease
Parkinson's disease (PD) is the second most common neurodegenerative disease after AD (Feng et al., 2010). The two pathological hallmarks of PD are the presence of cytoplasmic inclusions termed Lewy bodies and Lewy neurites, and the selective degeneration and loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) (Smith et al., 2005; Luk et al., 2012a) leading to the main motor symptoms of PD, i.e., resting tremor, muscle rigidity, bradykinesia, and postural instability (Jankovic, 2008; Feng et al., 2010; Luk et al., 2012b). For this reason, PD is generally defined as a movement disorder associated with degeneration of neurons in the nigrostriatal system (Desplats et al., 2009). The diagnosis of PD relies mainly on the clinical detection of these motor symptoms, but there is no definitive diagnostic test. The symptoms are often accompanied by the appearance of autonomic, cognitive and psychiatric problems (Mercuri and Bernardi, 2005; Jankovic, 2008).
During the early stages of the disease, called the presymptomatic phase, PD patients develop non-motor deficits including olfaction impairment, vagal dysfunction and sleep disorders (Angot and Brundin, 2009). The typical motor symptoms appear when there is 50–60% of dopaminergic neuron loss and 70–80% of dopamine depletion (Schapira, 2009; Sato et al., 2013). The cognitive functions decline at more advanced stages (Angot and Brundin, 2009). As a result of these clinical symptoms, PD is now recognized as a complex clinicopathological entity. The presence of cognitive impairment or dementia in patients with PD is associated with loss of independence, a lower quality of life and a reduction in survival time (Irwin et al., 2013). The majority of PD cases are sporadic (Feng et al., 2010) and the etiology and pathogenesis remain enigmatic (Brundin et al., 2008). The origin and development of the disease seems to involve both genetic susceptibility and environmental factors such as oxidative stress, proteasome inhibition, and aging (Ross and Poirier, 2004; Smith et al., 2005).
Pathological Findings
The pathological hallmark of PD is the presence of intraneuronal proteinaceous inclusions called Lewy bodies (LBs) or Lewy neurites (LNs) depending if they are localized in the cell body or the processes, respectively (Angot and Brundin, 2009). LBs are spherical eosinophilic cytoplasmic protein aggregates that contain ubiquitin and fibrils of α-syn, located in the substantia nigra and in several central nervous system structures (Bisaglia et al., 2009; Soto, 2012). The main protein component of LBs is α-syn, a synaptic protein with the propensity to misfold and aggregate (Angot et al., 2012). Analysis of these inclusions with immunohistochemistry techniques revealed that they have affinity to specific dyes that identify misfolded α-syn, such as Thioflavin S that recognizes β-sheet conformations, and for α-syn phosphorylated on serine 129 (Ser129), which is a post-translational modification that only occurs in LBs (Angot and Brundin, 2009). In PD, the accumulation of Lewy bodies undergo an ascending pattern of progression, spreading from the lower brainstem and olfactory bulb into the limbic system and, eventually, to the neocortex, suggesting a propagation mechanism similar to prion diseases (Desplats et al., 2009).
PD is Progressive and Irreversible
There are two major features that identify PD: (1) progressive damage of dopaminergic neurons in the SNpc that leads to the depletion of dopamine (DA) release necessary to maintain essential functions (Mercuri and Bernardi, 2005), and (2) the appearance of non-motor symptoms associated to the degeneration of non-dopaminergic systems (Obeso et al., 2010) together with propagation and accumulation of Lewy bodies in different brain regions (Ross and Poirier, 2004; Desplats et al., 2009; Obeso et al., 2010) making PD an irreversible, and at the present, an incurable disorder (Singh et al., 2007).
Current pharmacological treatment for PD involves the use of the dopamine precursor L-3,4-dihydroxyphenylalanine (L-dopa) which alleviates bradykinesia, the increase in muscle tone and tremor, but does not reduce non-motor symptoms (Mercuri and Bernardi, 2005). DA agonists, COMT, and MAO-B inhibitors are also used as adjunctive treatment to L-dopa (Schapira, 2009; Tarazi et al., 2014). In addition, the efficiency of L-dopa decreases over time and many patients develop motor fluctuations (wearing-off and on–off phenomena), dyskinesias and behavioral abnormalities (Mercuri and Bernardi, 2005; Singh et al., 2007; Obeso et al., 2010). Newer non-pharmacological therapies include the use of viral vector genes to silence defective genes associated with PD. Surgical interventions such as deep brain stimulation, pallidotomy, and thalamotomy, or non-invasive procedures such as gamma knife radiation are also employed (Tarazi et al., 2014). The available pharmacological and non-pharmacological treatments focus primarily on motor symptoms, but do not modify the progression of DA neuronal degeneration in the SNpc or the other affected areas (Mercuri and Bernardi, 2005; Singh et al., 2007; Tarazi et al., 2014).
Alpha-Synuclein
Structure
Alpha-synuclein (α-syn) is a small, acidic protein of 14.5 kDa and 140 amino acids (Bisaglia et al., 2009) that is highly conserved in vertebrates and is expressed in presynaptic nerve terminals in several regions of the brain (Jain et al., 2013). The protein belongs to the synuclein family, α-, β- and γ-synuclein, from three highly expressed human genes (SNCA, SNCB, and SNCG) (Lavedan, 1998). The β-isoform also exhibits a presynaptic location and co-localizes with α-syn at many, but not all presynaptic terminals, whereas γ-synuclein is expressed by glia cells and specific neuronal populations, mainly dopamine neurons (Bendor et al., 2013). α-syn lacks a defined secondary structure and therefore belongs to the intrinsically unstructured protein family (Bisaglia et al., 2009; Deleersnijder et al., 2013). However, the remarkable conformational plasticity of α-syn allows it to adopt a wide range of dynamic structures depending on the environment and binding partners (Jain et al., 2013).
Figure 1A illustrates the three distinct regions of α-syn. (1) The amino-terminal sequence of α-syn (residues 1–60) contains variants of an imperfect 11 amino acid repeat with a highly conserved hexamer motif similar to that found in the amphipathic helices of apolipoproteins (Bellucci et al., 2012; Deleersnijder et al., 2013). This portion of the protein includes the A30P, A53T, and E46K mutation sites found in familial PD cases (Bisaglia et al., 2009). (2) The central region (residues 61–95) termed NAC (non-amyloid-β component) contains two additional motifs and is the most hydrophobic portion of the protein. This region can undergo a conformational change from a random coil to β-sheet structure and is able to form cylindrical β-sheets and amyloid-β-like fibrils (Bellucci et al., 2012; Deleersnijder et al., 2013). (3) The carboxy-terminal of α-syn (residue 96–140) is rich in acidic residues (Bisaglia et al., 2009) and is responsible for the intrinsically disordered nature of α-syn. This region also plays a regulatory role in the aggregation and fibril formation of the protein (Deleersnijder et al., 2013). Dopamine can interact nonspecifically with the C-terminal residues of α-syn, suggesting that inappropriate C-terminal cleavage of this protein, which is known to occur in PD brains, might affect DA homeostasis (Bisaglia et al., 2009; Bellucci et al., 2012).
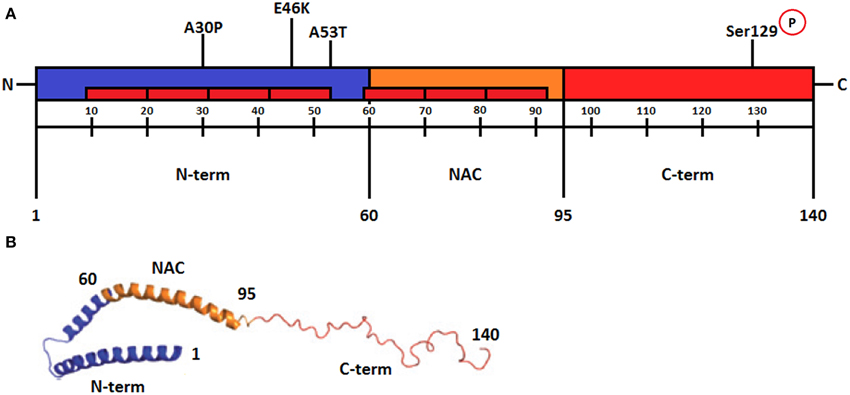
Figure 1. (A) Schematic representation of α-synuclein regions. The amino-terminal from amino acids 1–60 is an amphipathic region responsible for α-syn-membrane interactions. It contains repeats of hexamer motifs similar to apolipoproteins (represented in the figure as red rectangles). The point mutations of α-syn are located in this region (A30P, E46K, and A53T). The central region from amino acids 61-95, termed NAC (non-β amyloid component), is the most hydrophobic portion of the protein and is required for the aggregation process. This region folds into a β-sheet secondary structure and forms amyloid fibrils. The C-terminal from amino acids 96–140 is characterized by the presence of acidic residues and several negative charges. The residue serine 129 in this region is phosphorylated in Lewy bodies (Modified from Plotegher et al., 2014). (B) Schematic representation of micelle-bound α-synuclein. The N-terminal region with antiparallel α-helices is shown in blue, the NAC region is also an α-helix and is shown in orange, and the unstructured C-terminal part is shown in red. Numbers refer to amino acid residues. (Modified from Lashuel et al., 2013). Reprinted by permission from MacMillan Publishers Ltd (Nature Reviews Neuroscience).
Phosphorylation is one of the most common posttranslational modifications and has a role on the function of numerous target proteins (Sato et al., 2013). Thus, α-syn phosphorylated in Ser129 (pS129-α-syn) may have some functions in the normal brain. Residue Ser129 in the C-terminal region of α-syn is approximately 90% phosphorylated in Lewy Bodies (Figure 1A). This form is biochemically detectable in cerebral cortex, substantia nigra and nucleus basalis of Meynert in normal human subjects (Walker et al., 2013). Interestingly, no more than 4% of α-syn is phosphorylated at this residue in normal brain suggesting a strict control. The phosphorylation of Ser129 is controlled by kinases, phosphatases, and degradation pathways (Hara et al., 2013; Sato et al., 2013). Several studies identified the kinases responsible for the phosphorylation of α-syn in vitro and in vivo, such as Polo-like kinase PLK2 and PLK3, casein kinase CK1 and CK2 and members of the G protein-coupled receptor kinase family GRK2, GRK3, GRK5, and GRK6 (Hara et al., 2013; Sato et al., 2013; Kosten et al., 2014). Phosphoprotein phosphatase 2A (PPA2) might be responsible for dephosphorylation of α-syn, and stimulating the activity of PP2A resulted in reduced levels of pS129-α-syn and aggregated α-syn (Sato et al., 2013; Walker et al., 2013).
In vitro studies suggest that α-syn interacts with lipid membranes through its repeat motifs and with small unilamellar vesicles and micelles preferentially containing negatively charged head groups (Ulmer et al., 2005). When α-syn interacts with vesicles or membranes, the N-terminal of the protein acquires an α-helical conformation that can be an extended helix or antiparallel helices connected by a short linker, depending on the membrane properties. The C-terminal of the protein remains unstructured, as observed in Figure 1B (Ulmer et al., 2005; Plotegher et al., 2014). The binding of α-syn to phospholipid micelles produces a flattening in their surface curvature. In addition, the α-syn mutants A53T and E46K exhibit increased membrane-binding affinity and also flatten micelle surface curvature, whereas the A30P α-syn mutant has a decreased membrane affinity and does not alter the surface curvature of the micelles (Auluck et al., 2010).
Physiological Roles
Studies suggest that α-synuclein is involved in the control of synaptic membrane processes (Bellucci et al., 2012) and participates in the control of neurotransmitter release via interactions with members of the SNARE family (Tsigelny et al., 2012). More specifically, α-syn promotes SNARE-complex assembly through a non-enzymatic mechanism, binding to phospholipids via its N-terminal and to synaptobrevin-2 via its C-terminal (Burre et al., 2010). However, the precise physiological functions of α-syn remain uncertain and studies are often conflicting (Luk et al., 2009). Individual synuclein knockout animals (α-, β- or γ-syn) are viable (Lashuel et al., 2013) and results obtained in α-syn knockout mice suggest that α-syn is not essential for synapse formation or cell survival (Bisaglia et al., 2009) as supported by the relatively late translocation of α-syn into presynaptic terminals during synaptogenesis (Chandra et al., 2004).
The existence of three synuclein isoforms and the high degree of co-expression of α- and β-syn raises the possibility of redundant synaptic functions (Chandra et al., 2004; Greten-Harrison et al., 2010). Analysis of α-/β-synuclein double knockout mice did not show major changes in synaptic functions such as neurotransmitter release, synaptic vesicle numbers, or synaptic plasticity, and caused no structural abnormalities in the overall brain morphology. However, the α-/β- double knockout did show a modest reduction in striatal DA levels (Chandra et al., 2004; Bendor et al., 2013), and it has been reported that α-syn binds to C-terminal of the DA transporter increasing the DA uptake (Hara et al., 2013). The triple synuclein knockout mice (TKO) were viable and fertile, but developed severe neurological impairments resulting in a striking age-dependent survival deficit ending in premature death (Burre et al., 2010; Greten-Harrison et al., 2010). Furthermore, the TKO mice exhibited an age-dependent decrease in SNARE-complex assembly. Thus, synucleins are required for maintaining normal SNARE-complex assembly during aging in mice (Burre et al., 2010), and they are also relevant for long-term survival (Greten-Harrison et al., 2010).
Pathological Roles
The pathological roles of α-syn arise from the intracellular accumulation of α-syn amyloid fibrils that define a family of neurological disorders termed α-synucleinopathies in which PD is included (Luk et al., 2009; Danzer et al., 2012). α-syn is able to form oligomers, fibrils and large aggregates following overexpression, exposure to changes in pH, oxidative stress, or by interaction with dopamine (Feng et al., 2010). Unlike α-syn, β-syn does not fibrillize and both β- and γ-syn can inhibit the aggregation of α-syn in vitro and in vivo (Bendor et al., 2013). Several post-translational covalent modifications of α-syn have been described to promote the pathological changes in the protein, including serine and tyrosine phosphorylation, ubiquitination, nitration and C-terminal truncation (Ross and Poirier, 2004; Deleersnijder et al., 2013). The predominant modification of α-syn in Lewy bodies is single phosphorylation at Ser129. However, the relationship between Ser129 phosphorylation and α-syn aggregation remains unclear (Sato et al., 2013). Histopathological studies of human brain show that increased levels of pS129-α-syn are present in soluble and detergent insoluble brain fractions of Lewy Body pathology. Thus, phosphorylation and ubiquitination presumably increase aggregation and formation of an insoluble fraction. As the α-syn solubility decreases, phosphorylation seems to increase even more (Walker et al., 2013).
Mutations in the gene encoding α-syn (SNCA) directly link α-syn with the onset of PD (Plotegher et al., 2014). The overexpression of α-syn due to duplication or triplication of the SNCA gene causes rare familial forms of parkinsonism, and single nucleotide polymorphisms in the SNCA gene (Ala53Thr, Ala30Pro, and Glu46Lys) are linked to sporadic PD (Angot et al., 2012). Recently, new mutations have also been found in the N-terminal part of the protein (Deleersnijder et al., 2013). The three most common PD-related point mutations have shown to accelerate α-syn aggregation (but not necessarily fibril formation) in vitro. These effects can be explained by changes in net charge, hydrophobicity, and secondary structure propensity (Deleersnijder et al., 2013). This suggests that α-syn is heavily implicated in the pathogenesis of PD, both familial and sporadic cases (Smith et al., 2005; Angot et al., 2012).
Critical Features of α-Syn Favoring its Transmission and Neurotoxicity
Cellular transmission
Autopsy reports of PD patients who received bilateral intrastriatal grafts from healthy embryonic mesencephalic dopaminergic neurons, 10–22 years before the postmortem analysis (Li et al., 2008), showed that some of the grafted neurons contained pathological α-syn or Lewy bodies similar to those observed in the host brain (Li et al., 2008; Angot et al., 2012). These studies reported that the LB and LN developed in the grafted neurons were ubiquitinated, positive for Thioflavin S staining, and that α-syn was phosphorylated on Ser129 indicating that it was aggregated, post-translationally modified and disease-related (Brundin et al., 2008; Li et al., 2008; Angot and Brundin, 2009). It was also found that the α-syn accumulation in grafted neurons was time-dependent (Brundin et al., 2008). The presence of pathology in the grafted neurons could have been triggered by misfolded α-syn in the host brain, which was transmitted into grafted cells with subsequent seeding of aggregates in the recipient cells (Li et al., 2008). These findings suggest that α-syn pathology propagates by a mechanism similar to prion diseases (Hansen et al., 2011). For this reason, numerous studies have attempted to replicate and explain these findings in cell and animal models (Angot et al., 2012).
The introduction of exogenously assembled α-syn fibrils to various cells overexpressing α-syn catalyzed intracellular α-syn aggregation. The α-syn fibrils recruited endogenous soluble α-syn, converting them into misfolded detergent-insoluble inclusions which were hyperphosphorylated and ubiquitinated, resembling Lewy bodies (Luk et al., 2009). In another study, a specific type of α-syn oligomer was capable of inducing α-syn seeding and aggregation after exogenous application to a neuroblastoma cell line and primary cortical neurons. This aggregation process was dose- and time-dependent (Danzer et al., 2009). An in vivo study showed that human α-syn expressed in mice was transmitted from cells in the brain to dopaminergic neurons grafted into the striatum, in analogy to the mechanism suggested to take place in the grafted PD cases previously mentioned (Angot et al., 2012). The presence of Lewy bodies in grafted neurons was a time-dependent process, with a higher percentage of neurons displaying Lewy bodies in older grafts than in younger transplants (Angot et al., 2012).
Studies with injected homogenates prepared from brainstem and spinal cord of aged symptomatic animals that contained abundant LB-like pathology, or with preformed fibrils (PFF) assembled from human α-syn, into the neocortex and striatum (Luk et al., 2012a) showed that despite the fact that inoculations were unilateral, intraneuronal α-syn deposits were widely distributed bilaterally and present throughout the CNS. This indicated that α-syn pathology expanded through the CNS in a time-dependent manner and accelerated the disease in vivo (Luk et al., 2012b). Another study with non-transgenic mice (WT) that were injected unilaterally with PFFs of α-syn in the striatum showed that deposits of hyperphosphorylated α-syn were visible at the injection site and several areas directly interconnected to the striatum (Luk et al., 2012b). The contralateral neocortex also developed LBs, revealing once again a time-dependent dissemination of these inclusions. The striatal inoculation led to abundant α-syn pathology in neurons located in the SNpc. This was accompanied by the gradual loss of tyrosine hydroxylase immunoreactivity, suggesting that intraneuronal α-syn inclusions lead to neurodegeneration and DA neuronal loss. In summary, this study demonstrated that a single intrastriatal injection of synthetic misfolded α-syn into WT mice induced a neurodegenerative process, with accumulation of intracellular LB/LN pathology, selective loss of SNpc neurons, and impaired motor coordination (Luk et al., 2012a).
Cellular transmission mechanisms
Lewy Body propagation within the CNS could be associated with cell-to-cell transmission that involves the release of misfolded α-syn from donor cells into the extracellular space. Recipient cells could then take up the misfolded α-syn which recruits native unfolded α-syn from the cytosol of the recipient cell and acts as a template for the development of aggregates that eventually lead to the formation of Lewy Bodies (Angot and Brundin, 2009; Steiner et al., 2011). Studies have shown that cells can release α-syn into their surroundings. For example, nanomolar concentrations of extracellular monomeric and oligomeric α-syn have been detected in cerebrospinal fluid and plasma samples from both PD patients and controls (El-Agnaf et al., 2006; Bellucci et al., 2012; Costanzo and Zurzolo, 2013), with a significantly higher concentration in PD patients (El-Agnaf et al., 2006) supporting the occurrence of a secretory process for α-syn (Bellucci et al., 2012).
Several transmission mechanisms have been reported for α-syn (see Figure 2). For example, SH-SY5Y cells release α-syn and its mutant variants A53T and A30P into the extracellular space. The secretion is considerably reduced at 18°C, suggesting that α-syn is released from these cells via exocytosis (Lee et al., 2005). However, Brefeldin A, a classical inhibitor of ER/Golgi-dependent secretion, does not block α-syn release indicating that α-syn exocytosis relies on an unconventional secretory pathway (Lee et al., 2005; Angot and Brundin, 2009; Costanzo and Zurzolo, 2013). Neuronal cells subjected to various stress conditions including oxidative stress and proteolytic impairment increase the translocation of α-syn into vesicles and the release of monomeric and aggregated α-syn (Jang et al., 2010). Failure of intracellular clearance pathways, such as autophagy, could also contribute to the pathological release of α-syn increasing its transfer to other cells (Lee et al., 2013). The release of pathogenic α-syn from dying cells [see Figure 2(5)] could also contribute to the extracellular pool of α-syn in the brain (Angot and Brundin, 2009). It has been reported that α-syn moves antero- and retrogradely along axons (Steiner et al., 2011). For example, α-syn fibrils could be internalized, anterogradely transported within axons, released, and subsequently taken up by additional neurons (Freundt et al., 2012). Recently, tunneling nanotubes (TNTs) have been proposed to be involved in intercellular propagation of PrPSc. As observed in [Figure 2(4)], TNTs are actin-containing membrane bridges between cells (Angot and Brundin, 2009) acting as conduits for the exchange of cytosolic and membrane-bound molecules and organelles, as well as for the spreading of pathogens (Costanzo and Zurzolo, 2013). TNTs have not yet been implicated in other neurodegenerative diseases, or the spreading of other misfolded proteins such as α-syn (Angot and Brundin, 2009).
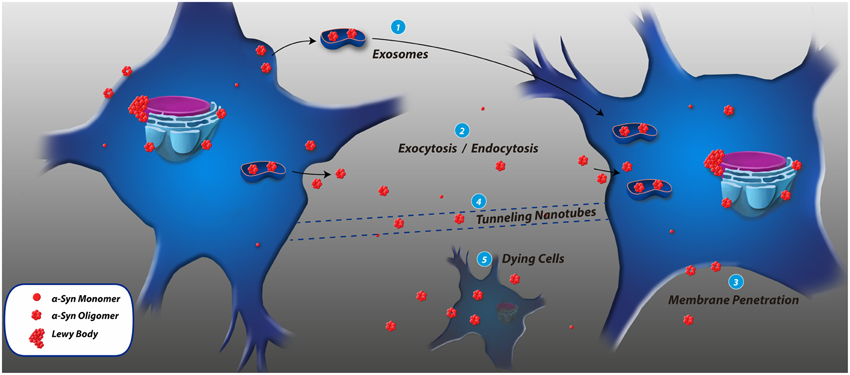
Figure 2. Cell-to-cell transmission mechanisms of α-synuclein. (1) Exosomes are small membrane vesicles derived from the endocytic pathway, and the presence of α-syn inside exosomes and its transmission to recipient cells has been reported. (2) α-syn could be released through an exocytosis process and accumulate in the extracellular space and be taken up by other cells through endocytosis. (3) α-syn associates to biological membranes by its N-terminal, penetrates the membrane and gains access to the cytosol. (4) Tunneling nanotubes are membrane bridges of actin between cells, but this mechanism has not been identified to participate in α-syn transmission. (5) Dying cells could be an important reservoir of pathological α-syn when they eventually expel their contents after being lysed.
Membrane penetration. Several studies have shown that the 11-amino acid imperfect repeats of α-syn play a critical role in the cell membrane translocation of α-syn. This uptake process is not inhibited when Chinese hamster ovary cells were incubated at a low temperature (4°C), suggesting that receptor-mediated endocytosis is not involved. In addition, the uptake of α-syn is insensitive to treatment with the general exo/endocytosis inhibitors Brefeldin A and Cytochalasin D (Ahn et al., 2006). A study using computer modeling and membrane simulations investigated the course of penetration of WT and A53T mutant α-syn in the membranes and pore formation activity. Their data showed that the N-terminal initially makes contact with the surface of the membrane, undergoing changes in secondary structure, and that the penetration of the A53T mutant α-syn across the membrane was 20% faster than WT α-syn (Tsigelny et al., 2012).
Exosomes. Exosomes have been proposed to participate in the spreading of disease-related proteins within the brain, such as PrPSc in prion diseases and β-amyloid peptide (Aβ) in AD. Exosomes are small membrane vesicles derived from the endocytic pathway and are released from cells into the surroundings. A wide range of cells secrete exosomes in vitro, including neurons and astrocytes (Angot and Brundin, 2009). For this reason, Danzer et al. (2012) investigated whether oligomeric species of α-syn are present in exosomes. The authors provided evidence that α-syn oligomers are indeed present in the exosomal fractions from both neuronal and non-neuronal cells. According to their results, exosome-associated α-syn oligomers are more prone to being taken up by cells than exosome-free α-syn oligomers and confer more cytotoxicity when compared to the increase in Caspase 3/7 activation (Danzer et al., 2012).
In another study, α-syn was also detected in exosomes of cells overexpressing α-syn and transmitted to normal SH-SY5Y cells. It has been reported that lysosomal function is decreased in PD patients, and α-syn requires the lysosome for its degradation. Thus, the investigators inhibited lysosomal function in the exosome donor cells and the exosomal α-syn level increased significantly, as well as the release of α-syn which led to greater transmission to recipient cells and a greater number of cells containing α-syn inclusions. The hypothesis of this group is that lysosomal dysfunction can accelerate exosomal α-syn release and propagation to neighboring cells with associated increase in α-syn inclusion formation (Alvarez-Erviti et al., 2011).
Endocytosis. Another transmission mechanism of α-syn was described in a study that used human dopaminergic neuronal cells treated with α-syn. The cells take up α-syn and approximately half of the inclusions formed in these cells displayed ubiquitin immunoreactivity and were stained with thioflavin S, resembling Lewy Bodies. Dynamin-1 K44A, a dominant negative mutant that blocks endocytic vesicle formation, reduced significantly the transmission of α-syn, as compared to cells not expressing this inhibitor. In addition, 90% of the transmitted α-syn was colocalized with endosomal GTPases rab5a and rab7 to further support endocytosis as a possible transmission mechanism (Desplats et al., 2009).
A similar study reported the entry of α-syn from the conditioned medium into SH-SY5Y cells. The α-syn uptake was observed in cells incubated at 37°C, but not in cells incubated at 4°C suggesting that α-syn could be taken up by endocytosis. To validate this observation, cells were co-cultured with the endocytosis inhibitors monodansylcadaverine or dynasore. These inhibitors decreased the number of cells with α-syn inclusions, indicating that endocytosis plays an important role in the α-syn intercellular transfer. To determine whether endocytic mechanisms were also involved in the uptake of α-syn in vivo, dynasore was coinjected with α-syn monomers in the right neocortex of rats. The data revealed that the α-syn signal was reduced by 40% in the right cortex where the inhibitor was coinjected with α-syn, suggesting that endocytosis contributes significantly to neuronal uptake of α-syn both in vitro and in vivo (Hansen et al., 2011).
PFFs of α-syn also enter inside neurons and seed recruitment of endogenous α-syn to initiate accumulation of pathologic hyperphosphorylated α-syn. Treatment of neurons with α-syn PFFs in the presence of wheat germ agglutinin (WGA) induced adsorptive mediated endocytosis, which increased the extent of α-syn pathology in a dose-dependent manner. The addition of a competitive inhibitor of WGA reduced its effects on α-syn-induced aggregate formation. Taken together, these findings indicate that α-syn PFFs could gain access to the neuronal cytoplasm by adsorptive endocytosis (Volpicelli-Daley et al., 2011).
Mechanisms of neurotoxicity of α-synuclein
Under pathological conditions, progressive accumulation of α-syn and the formation of oligomers have been proposed to play a critical role in the pathogenesis of PD and other α-synucleinopathies (Tsigelny et al., 2012). However, there are still unresolved issues according to the precise mechanisms through which α-syn aggregation contributes to neurodegeneration, the nature of the toxic forms of α-syn and the cellular pathways that are affected by α-syn (Tsigelny et al., 2012; Lashuel et al., 2013).
The multifunctional properties of α-syn may lie in its conformational flexibility, which allows the protein to change its conformation upon interaction with biological membranes of different compositions, other proteins or protein complexes (Lashuel et al., 2013; Plotegher et al., 2014). Several factors including oxidative stress, long incubations at 37°C, post-translational modifications, the concentrations of fatty acids, phospholipids, and metal ions (Fe2+, Cu2+, and Zn2+), or application of various ligands such as dopamine have shown to induce and/or modulate α-syn structure and oligomerization in vitro (Lashuel et al., 2013). Numerous studies show that the soluble oligomers have a higher cytotoxicity compared to the fibrillar form of the protein (Stockl et al., 2013).
Several different toxic mechanisms have been associated to the α-syn aggregation process and to its oligomeric and fibrillar products. Due to the heterogeneity of the aggregation products, it is reasonable to find a large variety of noxious effects attributed to α-syn (Plotegher et al., 2014). Similar to transmission, α-syn cytotoxicity may occur through different mechanisms. See Figure 3.
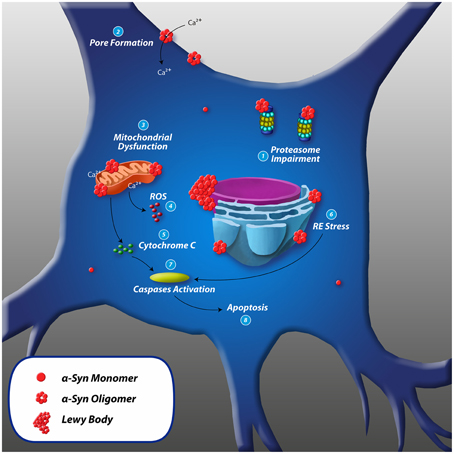
Figure 3. Toxic mechanisms of α-synuclein and cell death. (1) Proteasome impairment: inhibition of chymotryptic, tryptic and post-acidic proteasome activity leading to further intracellular accumulation of misfolded proteins such as α-syn. (2) Pore formation: α-syn oligomers penetrate cellular membranes increasing the conductance by forming pore-like structures that could act as non-selective channels, resulting in abnormal calcium influx. (3) Mitochondrial dysfunction: α-syn associates to the mitochondrial inner and outer membrane and (4) Increases mitochondrial and intracellular ROS levels. (5) Release of cytochrome C: accumulation of intramitochondrial ROS and Ca2+ leads to reduction in mitochondrial membrane potential and opening of permeability transition pores that could cause the release of cytochrome C to the cytosol. (6) RE stress: cellular accumulation of misfolded proteins can lead to chronic endoplasmic reticulum stress. α-syn associates to ER membrane and causes morphologic dysfunction such as dilated cisternae and increases the level of ER chaperones. (7) Cytochrome C leads to activation of caspase-3 and -9, and ER stress leads to activation of caspase-12. (8) Caspases initiate apoptosis leading to cell death.
Proteasome impairment and oxidative stress. One of the studies that evaluated α-syn toxicity found that the induction of A53T α-syn expression in PC12 cells increased cell death in a time-dependent manner (Smith et al., 2005). The expression of A53T α-syn inhibited chymotryptic, tryptic, and post-acidic proteasome activity. Additionally, the expression of A53T α-syn significantly increased the intracellular level of reactive oxygen species (ROS). These results indicate that the expression of A53T α-syn in PC12 cells caused early biochemical changes leading to proteasome inhibition and oxidative stress (Smith et al., 2005). The generation of ROS may cause oxidative neuronal damage. The main source of ROS in nigral neurons is thought to be the metabolism of dopamine itself. Another study using SH-SY5Y cells over-expressing human α-syn or mutant A53T or A30P showed increased generation of ROS that was greater in cells expressing α-syn mutants. Furthermore, exposure of all three α-syn-engineered cells to dopamine resulted in decreased cell viability (Junn and Mouradian, 2002).
Mitochondrial dysfunction. Mitochondria are membranous organelles that also have been proposed as cellular targets for α-syn neurotoxicity. Release of cytochrome c from mitochondria is a key event inducing apoptosis in many cells including neurons. One study showed that both A53T mutant and WT α-syn were localized at the mitochondria in human neuroblastoma dopaminergic SHSY cells and that overexpression of α-syn A53T or WT in these cells caused the release of cytochrome c from mitochondria (Parihar et al., 2008). The same phenomenon was observed with the induction of A53T α-syn expression in PC12 cells, where the increased release of cytochrome c from the mitochondria and its accumulation in the cytosol was also associated with increased caspase-3 and -9 activities over time [see Figure 3(3)] (Smith et al., 2005).
Mitochondria are main cellular calcium stores and play a crucial role for cellular calcium homeostasis. It is known that alterations in cellular calcium levels contribute to apoptosis of DA neurons in PD. The expression of α-syn A53T or WT is associated with higher concentrations of mitochondrial Ca2+, and excess Ca2+ initiates an apoptotic pathway. Thus, α-syn induces apoptosis via mitochondria-dependent pathways, including activation of caspases (Parihar et al., 2008).
Another study showed that aggregated and un-aggregated α-syn binds to mitochondrial membranes of human dopaminergic neuroblastoma SHSY cells (Parihar et al., 2009). Significantly higher mitochondrial ROS was observed in cells overexpressing α-syn, known to cause mitochondrial and cellular damage. The level of ROS in A53T α-syn expressing cells was higher than in A30P and WT expressing cells, suggesting greater neuronal toxicity of the A53T mutant. In addition, the mitochondrial membrane potential (Δψ) was measured as an indicator of mitochondrial function. A decrease in Δψ was observed in α-syn overexpressing cells in which A53T expressing cells had the lowest Δψ. These findings suggest that the decreased Δψ and consequent mitochondrial dysfunction are more likely to be a result of mitochondrial damage by elevated mitochondrial ROS and Ca2+ (Parihar et al., 2009).
ER stress. Cellular accumulation of misfolded proteins can lead to chronic endoplasmic reticulum stress (ERS) and trigger an integrated cellular response called unfolded protein response (UPR) (Doyle et al., 2011) that protects cells from accumulation of toxic misfolded proteins. However, persistent ERS leads to the activation of cell death cascade (Colla et al., 2012a).
A study found an increase in ER chaperones such as grp94, grp78, and PDI (widely used markers of ERS/UPR activation) in affected regions with α-syn pathology using a transgenic (Tg) mouse model expressing human α-syn (WT or mutant). The investigators identified a subset of phosphorylated α-syn reactivity that was localized on the ER membranes. The ER morphology in these neurons was highly abnormal with severely dilated ER cisternae, an ultrastructural indication of ER dysfunction in the A53T α-syn Tg mice (Colla et al., 2012a). Another study examined postmortem tissues of human PD patients and mouse cases of α-synucleinopathy and found selective accumulation of toxic α-syn oligomers within the ER/microsome compartment, supporting a direct pathological link between α-syn oligomers and ER stress in vivo (Colla et al., 2012b). The α-synucleinopathy and ER stress in the Tg mice model was also associated with the increased cleavage of caspase-12 and other downstream caspases (Colla et al., 2012a). The cleavage and activation of procaspase-12 is the hallmark of ER stress-induced apoptosis. In PC12 cells, the induction of A53T expression and the activity of caspase-12 was also significantly increased, suggesting that the expression of A53T α-syn causes ER stress mediated apoptosis in vitro (Smith et al., 2005).
Membrane disruption and pore formation. Another vastly studied cellular toxicity pathway of α-syn is the permeabilization of cellular membranes by oligomers (Stockl et al., 2013). As seen in [Figure 3(2)], the oligomers might interfere with the normal functions of cellular membranes and form pore-like structures, resulting in abnormal calcium influx (or other ions) with consequent neurodegeneration (Tsigelny et al., 2012).
Several studies indicate that α-syn oligomers are able to interact with lipid membranes, increase the conductance and form a pore complex in planar lipid bilayers (Kim et al., 2009; Schmidt et al., 2012; Tosatto et al., 2012). The same effect was observed in HEK293T cells over-expressing α-syn (Tsigelny et al., 2007). It has been reported that mutant α-syn (A53T or A30P) causes higher membrane permeability and induces the formation of pores in the plasma membrane of SH-SY5Y cells, which allows Ca2+ influx, and therefore plays an important role in cell degeneration (Furukawa et al., 2006). Similarly, a study by Feng et al. (2010) in a dopaminergic-like cell model demonstrated that the over-expression of WT α-syn caused the formation of oligomeric α-syn pore-like structures. Using whole-cell patch-clamp recordings, an increase in membrane conductance was found indicating the presence of opened membrane channels. These findings were associated with a modest but significant time-dependent increase in cell death and the authors hypothesized that pores formed by α-syn would act as non-selective channels and contribute to α-syn-induced toxicity (Feng et al., 2010).
According to molecular dynamic simulations done by Tsigelny et al. (2012), α-syn monomers penetrate the membrane and via attractive energies of intermolecular contact can develop a ring structure (α-syn octamer). This assembly possesses an intrinsic opening diameter of around 35Å in the middle of the oligomer and an external diameter of 130Å. The investigators observed that the accumulation of α-syn in neuronal membranes altered cellular function. Neurons overexpressing α-syn displayed increased permeability and this effect was more apparent in the cells expressing A53T α-syn. Consistent with this increased permeability, neuronal cell cultures expressing either WT or A53T α-syn displayed increased levels of intracellular calcium. In summary, these results suggest a model where penetration of membranes by α-syn gives rise to the formation of annular pore-like oligomeric structures with the ability to increase cell permeability and calcium influx (Tsigelny et al., 2012).
Conclusions
Accumulation of misfolded proteins have been linked to the generation and propagation of neurodegenerative diseases such as PD which is characterized by the degeneration and loss of DA neurons in the SNpc that leads to the main motor symptoms of the disease, as well as the degeneration of other non-dopaminergic brain regions. Protein inclusions called Lewy Bodies of which the main component is alpha-synuclein, a pre-synaptic unfolded protein that can adopt different conformations and form aggregates, are found in the cytoplasm of neurons. Mutations in the gene that encodes α-syn (A53T, A30P or E46K) result in proteins with a high tendency to misfold and form fibrillar aggregates rich in β-sheet structure. These mutations highlight the relevance and involvement of α-syn in the onset of PD cases.
Throughout this review we have referred to two important properties of alpha-synuclein. First, the ability of this protein to move from neuron to neuron, propagating the pathology by using a seeding/nucleation mechanism similar to prion proteins where misfolded α-syn recruits endogenous native α-syn to misfold and aggregate. Interestingly, the α-syn transmission occurs through different mechanisms such as exocytosis/endocytosis, by exosomes or directly by penetrating the plasma membrane. Second, the ability of α-syn to cause cellular toxicity, mainly in its oligomeric state. Studies have reported that α-syn oligomers affect several cellular organelles by binding to them and impairing their physiological functions. The most investigated mechanisms of WT and mutated α-syn toxicity include proteasome inhibition, oxidative stress, endoplasmic reticulum stress that leads to the activation of caspases, mitochondrial dysfunction and the eventual release of cytochrome c that also activates caspase pathways. One relevant mechanism of α-syn cytotoxicity is the formation of pore-like structures in different kinds of cellular membranes that act as a non-selective channel allowing the passage of small molecules and cations like Ca2+ that are well known to be involved in neurodegeneration. A recent study demonstrated that Aβ also forms pore-like structures and allows the entry of molecules such as Ca2+ that could cause neurotoxicity in brains of AD patients (Sepulveda et al., 2014). Table 1 shows the similarities and differences between α-syn and Aβ peptide.
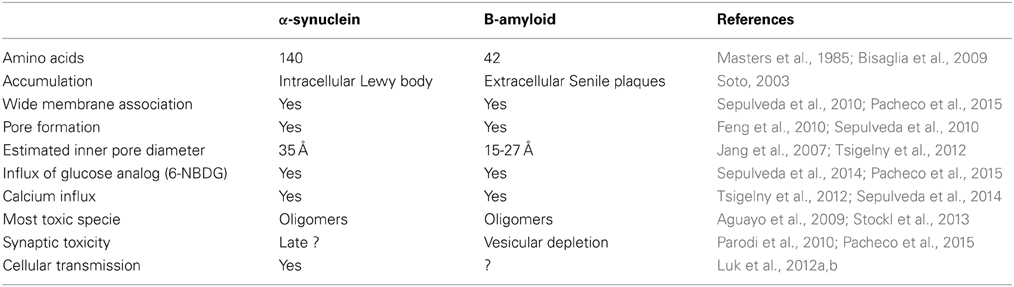
Table 1. Comparison between α-synuclein and β-amyloid characteristics.
The reason why α-syn is able to propagate and cause toxicity is not fully understood, but may rely on its conformational plasticity which allows the protein to adopt a secondary structure under different conditions. A study by Danzer et al. (2007) identified diverse types of α-syn oligomers using distinct preparation protocols. One oligomer type increased the intracellular calcium level, provoked increase membrane permeability and triggered cell death. Other oligomers were able to enter cells directly and seed intracellular α-syn aggregation. The conclusion of this study was that depending on the brain milieu conditions, heterogeneous populations of α-syn oligomers could be formed that have different biophysical properties and cellular effects (Danzer et al., 2007). Therefore, some important questions would be, what makes α-syn able to form different aggregated structures that differ in number of monomers and cellular effects? Why does α-syn use a transmission mechanism or cause a toxic effect in one type of cell, and in other cells uses another transmission mechanism and causes other toxic effects? These are questions that would be relevant to discuss in further studies. The properties of α-syn discussed in this review could explain why PD is a progressive and irreversible disease. However, it is possible that α-syn is not the only agent participating in the development and progression of PD, but it does indeed have a strong influence on the complexity of developing an effective therapy for PD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work supported by FONDECYT 1140473 (LGA). The authors thank Mrs. Lauren Aguayo for revising the article, and Ms. Cecilia Gonzalez for drawing Figures 2, 3.
Abbreviations
Aβ, amyloid beta; α-syn, alpha synuclein; A30P, alanine 30 proline; A53T, alanine 53 threonine; AD, Alzheimer's disease; DA, dopamine; E46K, glutamic acid 46 lysine; ER, endoplasmic reticulum; ERS, endoplasmic reticulum stress; Grp 78, glucose-regulated protein 78; Grp 94, glucose-regulated protein 94; LB, Lewy body; L-dopa, L-dihydroxyphenylalanine; LN, Lewy neurite; PD, Parkinson's disease; PDI, protein disulfide isomerase; PFF, preformed fibrils; PrP, prion protein; PrPC, cellular prion protein; PrPSC, scrapie prion protein; pS129-α-syn, α-syn phosphorylated at serine 129; ROS, reactive oxygen species; Ser129, serine 129; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SNpc, substantia nigra pars compacta; Tg, transgenic; TKO, triple knock-out; TNT, tunneling nanotubes; UPR, unfolded protein response; WGA, wheat germ agglutinin; WT, wild type.
References
Aguayo, L. G., Parodi, J., Sepúlveda, F. J., and Opazo, C. (2009). “Pore-forming neurotoxin-like mechanism for Aβ oligomer-induced synaptic failure,” in Pore-Forming Neurotoxin-Like Mechanism for Aβ Oligomer-Induced Synaptic Failure, eds G. Perry and R. B. Maccioni (New York, NY: Springer), 13–21. doi: 10.1007/978-0-387-87995-6
Ahn, K. J., Paik, S. R., Chung, K. C., and Kim, J. (2006). Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 97:265279. doi: 10.1111/j.1471-4159.2006.03731.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angot, E., and Brundin, P. (2009). Dissecting the potential molecular mechanisms underlying α-synuclein cell-to-cell transfer in Parkinson's disease. Parkinsonism Relat. Disord. 15, S143–S147. doi: 10.1016/S1353-8020(09)70802-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angot, E., Steiner, J. A., Lema Tome, C. M., Ekstrom, P., Mattsson, B., Bjorklund, A., et al. (2012). Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PloS ONE 7:e39465. doi: 10.1371/journal.pone.0039465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). alpha-Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bellucci, A., Zaltieri, M., Navarria, L., Grigoletto, J., Missale, C., and Spano, P. (2012). From α-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson's disease. Brain Res. 1476, 183–202. doi: 10.1016/j.brainres.2012.04.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bisaglia, M., Mammi, S., and Bubacco, L. (2009). Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 23, 329–340. doi: 10.1096/fj.08-119784
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brundin, P., Li, J. Y., Holton, J. L., Lindvall, O., and Revesz, T. (2008). Research in motion: the enigma of Parkinson's disease pathology spread. Nat. Rev. Neurosci. 9, 741–745. doi: 10.1038/nrn2477
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., et al. (2004). Double-knockout mice for α- and β-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. U.S.A. 101, 14966–14971. doi: 10.1073/pnas.0406283101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012a). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M., K. (2012b). Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Costanzo, M., and Zurzolo, C. (2013). The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17. doi: 10.1042/BJ20121898
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of -synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegen. 7:42. doi: 10.1186/1750-1326-7-42
Danzer, K. M., Krebs, S. K., Wolff, M., Birk, G., and Hengerer, B. (2009). Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J. Neurochem. 111, 192–203. doi: 10.1111/j.1471-4159.2009.06324.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deleersnijder, A., Gerard, M., Debyser, Z., and Baekelandt, V. (2013). The remarkable conformational plasticity of alpha-synuclein: blessing or curse? Trends Mol. Med. 19, 368–377. doi: 10.1016/j.molmed.2013.04.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Desplats, P. A., Lee, H., Eun-Jin, B., Christina, P., Edward, R., Leslie, C., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doyle, K. M., Kennedy, D., Gorman, A. M., Gupta, S., Healy, S. J., and Samali, A. (2011). Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 15, 2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
El-Agnaf, O. M., Salem, S. A., Paleologou, K. E., Curran, M. D., Gibson, M. J., Court, J. A., et al. (2006). Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. FASEB J. 20, 419–425. doi: 10.1096/fj.03-1449com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feng, L. R., Federoff, H. J., Vicini, S., and Maguire-Zeiss, K. A. (2010). Alpha-synuclein mediates alterations in membrane conductance: a potential role for alpha-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 32, 10–17. doi: 10.1111/j.1460-9568.2010.07266.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frost, B., and Diamond, M. I. (2010). Prion-like mechanisms in neurodegnerative diseases. Nat. Rev. Neurosci. 11, 155–159. doi: 10.1038/nrn2786
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Furukawa, K., Matsuzaki-Kobayashi, M., Hasegawa, T., Kikuchi, A., Sugeno, N., Itoyama, Y., et al. (2006). Plasma membrane ion permeability induced by mutant α−synuclein contributes to the degeneration of neural cells. J. Neurochem. 97, 1071–1077. doi: 10.1111/j.1471-4159.2006.03803.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). αβ γ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U.S. A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hara, S., Arawaka, S., Sato, H., Machiya, Y., Cui, C., Sasaki, A., et al. (2013). Serine 129 phosphorylation of membrane-associated α-synuclein modulates dopamine transporter function in a G protein-coupled receptor kinase-dependent manner. Mol. Biol. Cell 24:1649. doi: 10.1091/mbc.E12-12-0903
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Irwin, D. J., Lee, V. M., and Trojanowski, J. Q. (2013). Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636. doi: 10.1038/nrn3549
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jain, N., Bhasne, K., Hemaswasthi, M., and Mukhopadhyay, S. (2013). Structural and dynamical insights into the membrane-bound α-Synuclein. PLoS ONE 8:e83752. doi: 10.1371/journal.pone.0083752
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jang, A., Lee, H., Suk, J., Jung, J., Kim, K., and Lee, S. (2010). Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274.
Jang, H., Zheng, J., and Nussinov, R. (2007). Models of β-Amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 93, 19381949. doi: 10.1529/biophysj.107.110148
Jankovic, J. (2008). Parkinson's disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Junn, E., and Mouradian, M., M. (2002). Human α-Synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 320, 146150. doi: 10.1016/S0304-3940(02)00016-2
Kim, H. Y., Cho, M. K., Kumar, A., Maier, E., Siebenhaar, C., Becker, S., et al. (2009). Structural properties of pore-forming oligomers of alpha-synuclein. J. Am. Chem. Soc. 131, 17482–17489. doi: 10.1021/ja9077599
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kosten, J., Binolfi, A., Stuiver, M., Verzini, S., Theillet, F. X., Bekei, B., et al. (2014). Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosc. 5:12031208. doi: 10.1021/cn5002254
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, C., and Yu, M.-H. H. (2005). Protein folding and diseases. J. Biochem. Mol. Biol. 38, 275–280. doi: 10.5483/BMBRep.2005.38.3.275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, H. J., Cho, E. D., Lee, K. W., Kim, J. H., Cho, S. G., and Lee, S. J. (2013). Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 45:e22. doi: 10.1038/emm.2013.45
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O'Brien, P., Trojanowski, J. Q., et al. (2012a). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luk, K. C., Kehm, V. M., Zhang, B., O'Brien, P., Trojanowski, J. Q., and Lee, V. M. (2012b). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp.Med. 209, 975–986. doi: 10.1084/jem.20112457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luk, K. C., Song, C., O'Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056. doi: 10.1073/pnas.0908005106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Masters, C. S. G., Weiman, N. A., Multhaup, G., McDonald, D. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mercuri, N. B., and Bernardi, G. (2005). The ‘magic’ of -dopa: why is it the gold standard Parkinson's disease therapy? Trends Pharmacol. Sci. 26, 341344. doi: 10.1016/j.tips.2005.05.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moreno-Gonzalez, I., and Soto, C. (2011). Misfolded protein aggregates: mechanisms, structures and potential for disease transmission. Semin. Cell Dev.Biol. 22, 482–487. doi: 10.1016/j.semcdb.2011.04.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munch, C., and Bertolotti, A. (2012). Propagation of the prion phenomenon: beyond the seeding principle. J. Mol. Biol. 421, 491–498. doi: 10.1016/j.jmb.2011.12.061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pacheco, C., Morales, C., Ramirez, E. A., Muñoz, F. J., Gallegos, S., Caviedes, P., et al. (2015). Extracellular α-synuclein oligomers alter synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J. Neurochem. doi: 10.1111/jnc.13060. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2009). Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int. J. Biochem. Cell Biol. 41, 2015–2024. doi: 10.1016/j.biocel.2009.05.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parodi, J., Sepúlveda, F. J., Roa, J., Opazo, C., Inestrosa, N. C., and Aguayo, L. G. (2010). Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 285, 2506–2514. doi: 10.1074/jbc.M109.030023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plotegher, N., Greggio, E., Bisaglia, M., and Bubacco, L. (2014). Biophysical groundwork as a hinge to unravel the biology of α-synuclein aggregation and toxicity. Q. Rev. Biophys. 47:148. doi: 10.1017/S0033583513000097
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17. doi: 10.1038/nm1066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sato, H., Kato, T., and Arawaka, S. (2013). The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson's disease: a review of in vivo models. Rev. Neurosci. 24, 115–123. doi: 10.1515/revneuro-2012-0071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schapira, A. H. (2009). Neurobiology and treatment of Parkinson's disease. Trends Pharmacol. Sci. 30, 41–47. doi: 10.1016/j.tips.2008.10.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmidt, F., Levin, J., Kamp, F., Kretzschmar, H., Giese, A., and Botzel, K. (2012). Single-channel electrophysiology reveals a distinct and uniform pore complex formed by α-synuclein oligomers in lipid membranes. PLoS ONE 7:e42545. doi: 10.1371/journal.pone.0042545
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sepulveda, F. J., Fierro, H., Fernandez, E., Castillo, C., Peoples, R. W., Opazo, C., et al. (2014). Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer's disease. Neurobiol. Aging 35, 472481. doi: 10.1016/j.neurobiolaging.2013.08.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sepulveda, F. J., Parodi, J., Peoples, R. W., Opazo, C., and Aguayo, L. G. (2010). Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 5:e11820. doi: 10.1371/journal.pone.0011820
Singh, N., Pillay, V., and Choonara, Y. E. (2007). Advances in the treatment of Parkinson's disease. Prog. Neurobiol. 81:2944. doi: 10.1016/j.pneurobio.2006.11.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811. doi: 10.1093/hmg/ddi396
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. doi: 10.1038/nrn1007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Soto, C. (2012). Transmissible proteins: expanding the prion heresy. Cell 149, 968–977. doi: 10.1016/j.cell.2012.05.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Soto, C., and Satani, N. (2011). The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 17:1424. doi: 10.1016/j.molmed.2010.09.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steiner, J. A., Angot, E., and Brundin, P. (2011). A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Diff. 18, 1425–1433. doi: 10.1038/cdd.2011.53
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stockl, M. T., Zijlstra, N., and Subramaniam, V. (2013). α-Synuclein oligomers: an amyloid pore? Insights into mechanisms of α-synuclein oligomer-lipid interactions. Mol. Neurobiol. 47, 613–621. doi: 10.1007/s12035-012-8331-4
Tarazi, F. I., Sahli, Z. T., Wolny, M., and Mousa, S. A. (2014). Emerging therapies for Parkinson's disease: from bench to bedside. Pharmacol. Ther. 144, 123–133. doi: 10.1016/j.pharmthera.2014.05.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tosatto, L., Andrighetti, A. O., Plotegher, N., Antonini, V., Tessari, I., Ricci, L., et al. (2012). Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta 1818, 2876–2883. doi: 10.1016/j.bbamem.2012.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsigelny, I. F., Bar-On, P., Sharikov, Y., Crews, L., Hashimoto, M., Miller, M. A., et al. (2007). Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-synuclein. FEBS J. 274, 18621877. doi: 10.1111/j.1742-4658.2007.05733.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsigelny, I. F., Sharikov, Y., Wrasidlo, W., Gonzalez, T., Desplats, P. A., Crews, L., et al. (2012). Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 279, 1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005). “Structure and Dynamics of Micelle-bound Human α-Synuclein.” J. Biol. Chem. 280, 9595–9603. doi: 10.1074/jbc.M411805200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walker, D. G., Lue, L. F., Adler, C. H., Shill, H. A., Caviness, J. N., Sabbagh, M. N., et al. (2013). Changes in properties of serine 129 phosphorylated α-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 240, 190–204. doi: 10.1016/j.expneurol.2012.11.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Parkinson's disease, alpha-synuclein, transmission, toxicity, neurodegeneration
Citation: Gallegos S, Pacheco C, Peters C, Opazo CM and Aguayo LG (2015) Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson's disease. Front. Neurosci. 9:59. doi: 10.3389/fnins.2015.00059
Received: 09 December 2014; Paper pending published: 15 January 2015;
Accepted: 10 February 2015; Published online: 09 March 2015.
Edited by:
Ritchie Williamson, University of Dundee, UKReviewed by:
Christian Gonzalez-Billault, Universidad de Chile, ChileEfthimios M. C. Skoulakis, Biomedical Sciences Research Centre “Alexander Fleming,” Greece
Copyright © 2015 Gallegos, Pacheco, Peters, Opazo and Aguayo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis G. Aguayo, Department of Physiology, University of Concepción, PO Box 160-C, Concepción, Chile e-mail:bHVpc2FndWFAZ21haWwuY29t