 Kazue Muraki
Kazue Muraki Kenji Tanigaki
Kenji Tanigaki- Shiga Medical Center, Research Institute, Moriyama, Shiga, Japan
Schizophrenia is a complex mental disorder that displays behavioral deficits such as decreased sensory gating, reduced social interaction and working memory deficits. The neurodevelopmental model is one of the widely accepted hypotheses of the etiology of schizophrenia. Subtle developmental abnormalities of the brain which stated long before the onset of clinical symptoms are thought to lead to the emergence of illness. Schizophrenia has strong genetic components but its underlying molecular pathogenesis is still poorly understood. Genetic linkage and association studies have identified several genes involved in neuronal migrations as candidate susceptibility genes for schizophrenia, although their effect size is small. Recent progress in copy number variation studies also has identified much higher risk loci such as 22q11. Based on these genetic findings, we are now able to utilize genetically-defined animal models. Here we summarize the results of neurodevelopmental and behavioral analysis of genetically-defined animal models. Furthermore, animal model experiments have demonstrated that embryonic and perinatal neurodevelopmental insults in neurogenesis and neuronal migrations cause neuronal functional and behavioral deficits in affected adult animals, which are similar to those of schizophrenic patients. However, these findings do not establish causative relationship. Genetically-defined animal models are a critical approach to explore the relationship between neuronal migration abnormalities and behavioral abnormalities relevant to schizophrenia.
Introduction
Schizophrenia is a chronic psychiatric disorder with a strong genetic component. Twin studies indicated that the heritability for schizophrenia is estimated to be 0.81 (Sullivan et al., 2012). Most of genetic linkage studies failed to identify highly-shared risk alleles due to the complexity of genetic architecture of schizophrenia except for DISC1 (Millar et al., 2000). Many combinations of different gene variants cause genetic risk of schizophrenia. Genome-wide association studies have identified many schizophrenia susceptibility candidate genes. Most of such common variants confer only slight increase in risk for schizophrenia (odds ratio < 1.2) (Ripke et al., 2013), and often failed to be replicated. Some of them, Neuregulin1, ErbB4, and Reelin are involved in the regulation of neuronal migration. On the other hand, rare and de novo chromosomal microdeletion or microduplication [copy number variations (CNVs)] have been implicated in schizophrenia (Levinson et al., 2011; Rees et al., 2014). Such CNVs are from thousands to millions nucleotides and contain many genes and their odds ratios are high (2~20) compared with common variants. 22q11.2 deletion syndrome (22q11DS) is the most frequent known genetic cause of schizophrenia (Pulver et al., 1994). However, it remains to be elucidated how combinations of these genetic variants play pathogenic roles of schizophrenia.
Schizophrenia is believed to result from embryonic developmental abnormalities not from neuronal degenerations (Weinberger, 1987; O'Connell et al., 1997). Cytoarchitectural abnormalities were reported in the entorhinal cortex (Jakob and Beckmann, 1986; Arnold et al., 1991, 1997; Falkai et al., 2000) and the subcortical white matters in schizophrenia (Akbarian et al., 1993a,b, 1996). Decreased neuronal density in the superficial white matter and increased density in the deep white matter suggest neuronal migration defects in schizophrenia. However, other studies failed to replicate these findings (Akil and Lewis, 1997; Krimer et al., 1997; Bernstein et al., 1998; Beasley et al., 2002, 2009), suggesting these abnormalities might be too subtle to be detected without special methods. Prior human brain imaging studies also have indicated reduced cerebral volume, ventricular enlargement, and reduced hippocampal volume in schizophrenia (Shenton et al., 2001).
The development of the human cerebral cortex is similar to that in the mouse (Rakic, 2009; Hansen et al., 2013; Ma et al., 2013), which enabled investigation of the functions of schizophrenia susceptibility candidate genes in neuronal developments. The mammalian cerebral cortex consists mainly of excitatory glutametergic and inhibitory GABAergic neurons. Glutamatergic neurons are generated from neural progenitors in the dorsal forebrain (Glover et al., 2009; Rakic, 2009), whereas most of inhibitory neurons are thought to derive from the ventral pallium: medial, lateral and caudate ganglionic eminence (MGE, LGE, and CGE) (Hansen et al., 2013; Ma et al., 2013). Two types of migration are observed in the cortex. One is radial migration of glutamatergic neurons from the underlying ventricular zone along the radial glial fiber, while the other is tangential migration from the ventral forebrain of GABAergic interneurons (Corbin et al., 2001; Marin and Rubenstein, 2003). However, the differences between the human and mouse cortices are also reported. In the mouse, about 70% of cortical interneurons generate from the MGE and ~30% are from the CGE (Miyoshi et al., 2010). In contrast, more than half of interneurons derive from the CGE in the human (Hansen et al., 2013). The human cortex showed a much higher diversity in the interneuron types compared with that of the rodents (Feldman and Peters, 1978). In spite of the limitations caused by these differences, animal models are still valuable to elucidate the roles of neuronal migration deficits in the pathogenesis of schizophrenia. Many genetically-modified animal models with construct validity and cell-specific gene modification technique are available. The great advantage of rodent model to study schizophrenia is that we can establish a causal relationship between genetic abnormalities, neuronal developments, and behavioral abnormalities. Here we review a group of studies using rodent models which give insights into the pathogenesis of schizophrenia.
Developmental Neuronal Disruption Model of Schizophrenia
Perinatal insult of neuronal development can cause anatomical and behavioral deficits similar to human schizophrenic patients. One of the examples is a gestational day 17 (GD17) methylazoxymethanol acetate (MAM) administration rat model (Grace and Moore, 1998; Flagstad et al., 2004; Gourevitch et al., 2004; Paredes et al., 2006). MAM is a mitotic toxin and MAM administration specifically disrupts proliferating region. GD17 MAM treatment results in specific subtle reductions in the volume of prefrontal cortex (PFC) and hippocampus (HP), heterotopias in the HP resulting from neuronal migration deficits (Le Pen et al., 2006; Moore et al., 2006), which are characteristics of schizophrenia (Kovelman and Scheibel, 1984; Shenton et al., 2001; Heckers, 2004; Honea et al., 2005). GD17 MAM-treated rats also display decreased density of parvalbumin (PV)-positive interneurons in medial PFC and HP (Lodge et al., 2009). Interestingly, postmortem studies of human schizophrenia have shown decreased expression of PV and the 67 KDa isoform of glutamic acid decarboxylase (GAD67), which is an enzyme responsible for GABA synthesis, in PFC of schizophrenia subjects (Akbarian et al., 1995; Volk et al., 2000; Hashimoto et al., 2003; Fung et al., 2010). However, no difference is observed in the density of PV-positive interneurons in schizophrenia (Hashimoto et al., 2003). PV-positive interneurons are known to be indispensable for synchronized firing of excitatory pyramidal neurons in gamma frequencies (30–80 Hz), which plays essential roles for cognitive functions (Howard et al., 2003). Altered gamma oscillation activity and cognitive deficits have been reported in schizophrenia (Cho et al., 2006; Minzenberg et al., 2010). Consequently, PV-positive interneuron deficits are thought to be the cause of impairments of gamma oscillation and cognition in individuals with schizophrenia (Lewis et al., 2012). MAM-treated rats also show behavioral deficits in prepulse inhibition (PPI), which reflects an inability to filter out irrelevant sensory information, and working memory task (Flagstad et al., 2005; Le Pen et al., 2006; Moore et al., 2006), which are typical symptoms of schizophrenia in humans (Braff and Geyer, 1990; Liddle and Morris, 1991; Goldman-Rakic, 1994; Swerdlow et al., 1994; Nuechterlein et al., 2004). Furthermore, electrophysiological studies have shown that enhanced activity of ventral HP leads to dopaminergic neuronal activation in MAM-treated rats (Lodge and Grace, 2007). Again, these altered hippocampal activities are also observed in human schizophrenic patients (Medoff et al., 2001; Schobel et al., 2009). These abnormalities can be normalized by administration of α5GABA A receptor positive allosteric modulator, SH-053-2′F-R-CH3 (Gill et al., 2011), suggesting the involvement of GABA in embryonic MAM treatment-induced deficits. The MAM model provides a direct evidence that subtle embryonic disruptions of neuronal development result in behavioral alterations disorders, although the etiology is absolutely different from that of schizophrenia in humans.
Neuregulin-ERBB Signaling
Neuregulins are a large family of epidermal growth factor (EGF)-like proteins and play divergent roles both in neuronal development and in the neuronal activity homeostasis in the mature central nervous system. Several genetic linkage studies have shown Neuregulin1 (NRG1) as a strong candidate gene for schizophrenia (Badner and Gershon, 2002; Stefansson et al., 2002; Lewis et al., 2003). Some GWASs also support the hypothesis (Li et al., 2006; Munafo et al., 2006; Shi et al., 2009; Agim et al., 2013), although it has not been confirmed by a recent mega-analysis in which international consortia combined the resources to maximize the sample size and identified more than 100 candidate genes for schizophrenia with high levels of statistical significance (Schizophrenia Working Group of the Psychiztric Genomics Consortium, 2014). Most of schizophrenia-associated single nucleotide polymorphisms (SNPs) in NRG1 are localized in the 5′ and 3′ region of the gene. Some of them are associated with the expression level of NRG1 (Law et al., 2006; Weickert et al., 2012). A receptor of NRG1, ERBB4 is also associated with schizophrenia (Benzel et al., 2007; Law et al., 2007; Shi et al., 2009; Agim et al., 2013). It has been reported that a rare chromosome micro-deletion in a schizophrenic patient disrupts this gene, resulting in a truncated protein similar to dominant-negative ERBB4 (Walsh et al., 2008).
Nrg1 generates six types and at least 30 isoforms owing to multiple promoters and alternative splicing (Mei and Nave, 2014). Most pro-Nrg1 isoforms are transmembrane proteins and N-terminal domains containing EGF-like domain are released out of the cell after undergoing proteolytic processing except for TypeIII Nrg1 (cysteine-rich-domain containing Nrg1 (CRD-Nrg1)). This released mature Nrg1 activates ErbB receptor tyrosine kinase such as ErbB2/ErbB3 heterodimer and ErbB4 homodimers. Nrg1 regulates migration of excitatory glutamatergic neurons and γ-aminobutyric acid (GABA)-producing interneurons in the embryonic cortex. Nrg1 promotes the maintenance of radial glial cells in the cortex and induces elongation of radial fiber, which are essential for the radial migration of cortical excitatory neurons and cerebellar granule cells (Anton et al., 1997; Rio et al., 1997). NRG1 is also critical for interneuronal tangential migration (Flames et al., 2004; Li et al., 2012). ErbB4 is expressed in interneuronal progenitors migrating from the MGE to the cortex (Yau et al., 2003; Flames et al., 2004). Type III Nrg1 is expressed in lateral ganglionic eminence, and Type I and II Nrg1 (immunoglobulin (Ig)-domain containing Nrg1 (Ig-Nrg1)) are expressed in the embryonic cortex (Flames et al., 2004). Diffusible Type I and II Nrg1 in the cortex are thought to attract ErbB4-expressing interneurons along a permissive corridor of Type III Nrg1 (Flames et al., 2004), although this model is challenged. In another model, Nrg1, and Nrg3 have been proposed to be repellants for migrating interneurons (Li et al., 2012). Loss of ErbB4 cuases embryonic lethality due to failed development of myocardial trabeculae, which made it difficult to characterize the functions of ErbB4 signaling in interneuronal migration (Gassmann et al., 1995; Kramer et al., 1996). However, heart-rescued ErbB4 knockout mice with cardiac-specific ErbB4 transgene displayed decreased number of GABAergic interneurons in the postnatal cortex (Flames et al., 2004; Fisahn et al., 2009; Li et al., 2012), which clearly showed the essential roles of Nrg1/ErbB4 signaling in interneuronal migration.
Cell-specific gene modification techniques are now starting to elucidate a link between Nrg1/ErbB4 signaling and pathophysiology of schizophrenia. The gain and loss of function of Nrg1/ErbB4 signaling were examined because postmortem studies of schizophrenia reported both increased and decreased NRG1/ERBB4 signaling in schizophrenic patients (Silberberg et al., 2006; Law et al., 2007; Weickert et al., 2012; Joshi et al., 2014). Transgenic mice overexpressing Type I Nrg1 showed deficits in PPI and contextual fear conditioning, and hyperlocomotion (Deakin et al., 2009, 2012; Yin et al., 2013; Luo et al., 2014). If the overexpression of Nrg1 was switched off in adult mice, its effects were reversible (Yin et al., 2013; Luo et al., 2014). The influence of Nrg1 overexpression on neuronal development remains to be elucidated. Nrg1 or ErbB4 heterozygous mice and conditional knockout mice also displayed various behavioral abnormalities: locomotor hyperactivity in open field (OF), impairment in Prepulse inhibition (PPI), and fear conditioning (Stefansson et al., 2002; Golub et al., 2004; Boucher et al., 2007; O'Tuathaigh et al., 2007, 2010; Chen et al., 2008, 2010; Duffy et al., 2008; Ehrlichman et al., 2009; Shamir et al., 2012; Del Pino et al., 2013; Pei et al., 2014)(Table 1). Nrg1 heterozygous mice and heart-rescued ErbB4 knockout (KO) mice showed decreased number of cortical PV interneurons (Fisahn et al., 2009; Neddens and Buonanno, 2010; Shamir et al., 2012; Pei et al., 2014) (Table 1). However, PV interneuron-specific deletion of ErbB4 did not affect the number of cortical interneurons, which might be due to the slow turnover of ErbB4 (Fazzari et al., 2010; Shamir et al., 2012) (Table 1). A comparative behavioral analysis of ErbB4 KO and PV interneuron-specific ErbB4 KO mice demonstrated that PV interneuron-specific deletion is sufficient for hyperactivity and deficits in PPI. The only difference is that ErbB4 KO mice but not PV interneuron-specific ErbB4 KO mice exhibit reduced anxiety-like behaviors and deficits in cued and contextual fear conditioning (Shamir et al., 2012), which might be caused by developmental disorders in ErbB4-deficient interneurons.
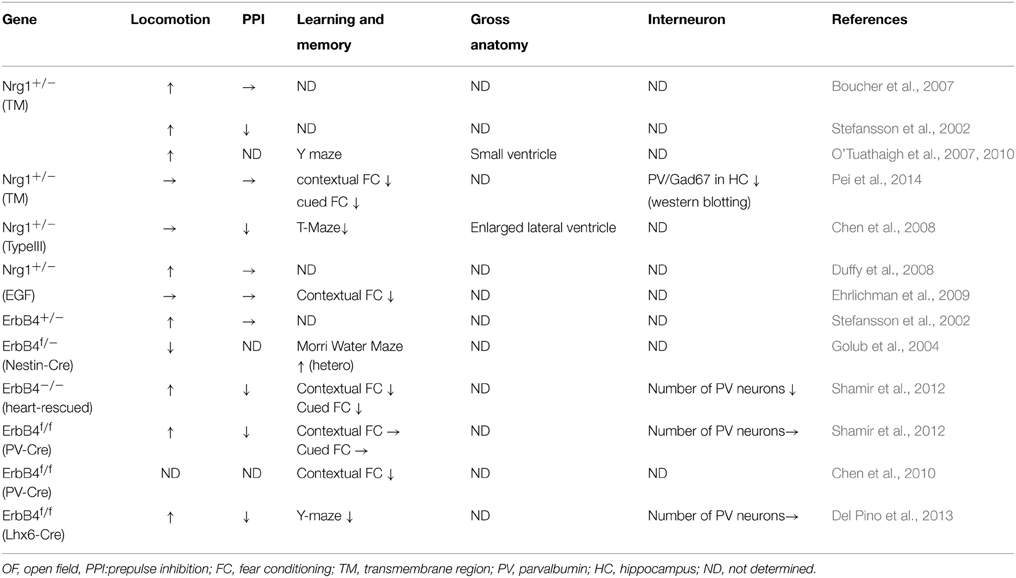
Table 1. Summary of mutant Nrg1/ErbB4 mouse models.
Disrupted-in Schizophrenia 1
The disrupted-in schizophrenia 1 (DISC1) gene was discovered at the breakpoint of inherited balanced chromosomal translocation in a Scottish family suffering from major depression, schizophrenia, and bipolar disorder (St Clair et al., 1990; Millar et al., 2000). Following linkage analysis and association studies demonstrated that DISC1 is significantly associated with schizophrenia, bipolar disorder and major depression (Ekelund et al., 2001, 2004; Macgregor et al., 2004; Hamshere et al., 2005; Hashimoto et al., 2006; Liu et al., 2006; Thomson et al., 2014), although it has not been confirmed by a recent mega-analysis of GWASs (Schizophrenia Working Group of the Psychiztric Genomics Consortium, 2014).
DISC1 is a scaffolding protein interacting with multiple proteins: nuclear distribution gene E homolog-like 1 (NDEL1), lissencephaly-1 (LIS1), phosphodiesterase 4B (PDE4B), glycogen synthase kinase 3β (GSK3β) and DIX domain-containing 1 (DIXDC1) (Ozeki et al., 2003; Millar et al., 2005; Taya et al., 2007; Mao et al., 2009). DISC1/DIXDC1 functions as a switch between neuronal proliferation and migration. DISC1/DIXDC1 binds to GSK3β and inhibits its activity leading to proliferation of neural progenitors through the inhibition of β-catenin degradation (Mao et al., 2009). CDK5 phosphorylation of DISC1 at S710 and DIXDC1 facilitates neuronal migration by dissociating DISC1 from GSK3β and promoting its binding with NDEL1 (Singh et al., 2010; Ishizuka et al., 2011). DISC1 variants associating with human brain structures and psychiatric phenotypes have been reported to impair this switching mechanism (Singh et al., 2011). Furthermore, knockdown of DISC1 also impairs interneuronal tangential migrations (Steinecke et al., 2012, 2014).
Two hypotheses are proposed on the pathophysiology of the disruption of the DISC1 gene: that the Scottish mutation decreases DISC1 expression and leads to haploinsufficiency; or that the Scottish mutation results in production of carboxy-terminal-truncated DISC1 (amino acids 1-598). This C-terminal truncated DISC1 functions as a dominant negative protein, and impairs microtubule dynamics by blocking interaction between DISC1 and dynein complex (Kamiya et al., 2005). Dynein complex contains LIS1 and NDEL1, and regulates coupling of the nucleus and centrosome, which is indispensable for radial migration of cortical excitatory neurons (Sasaki et al., 2000). Knockdown of DISC1 inhibits cortical neuronal cell migration (Kamiya et al., 2005; Kubo et al., 2010).
Acute knockdown of DISC1 using RNAi leads to drastic neuronal migration deficits. However, DISC1 KO mice (Disc1Δ2−3/Δ2−3) astonishingly showed almost normal cytoarchitectures of the cerebral cortex and the HP (Kuroda et al., 2011), whereas the number of PV-positive interneurons reduced in female Disc1Δ2−3/Δ2−3 mice (Nakai et al., 2014). The phenotypes in the proliferation of neuronal progenitors have not been examined in DISC1 KO mice, which will provide important insight into the pathogenesis of DISC1 deficiency. Furthermore, Disc1Δ2−3/Δ2−3 mice did not show schizophrenia-like phenotype but exhibited lower anxiety and higher impulsivity (Kuroda et al., 2011) (Table 2). Only female Disc1Δ2−3/Δ2−3 mice exhibited enhanced responsiveness to methamphetamine and deficits in PPI (Kuroda et al., 2011). These milder phenotypes of Disc1Δ2−3/Δ2−3 mice might be explained by a compensation mechanism after chronic loss of DISC1. N-nitroso-N-ethylurea (ENU) mutagenesis was utilized to generate missense mutations of Disc1. L100P mutant (Disc1L100P/L100P) mice showed reduced brain volume, reduced number of cortical neurons, altered distribution of cortical neurons, interneuonal migration deficits, and schizophrenia-like behavioral abnormalities, although the behavioral phenotypes were not confirmed by another group due to the difference in the genetic background (Clapcote et al., 2007; Lee et al., 2011, 2013; Shoji et al., 2012). 129S6/SvEv 25-bp deletion variant results in the production of a truncated isoform of DISC1 (amino acids 1-542) (Koike et al., 2006). C57BL/6J mice carrying the Disc1 gene from the 129S6/SvEv strain (Disc1Δ25 bp/Δ25 bpmice) exhibited enlarged ventricle and working memory deficits (Koike et al., 2006; Juan et al., 2014). Neuron-specific overexpression of the truncated DISC1 also resulted in drastic phenotypes: enlarged lateral ventricle, reduced number of PV-positive interneurons and schizophrenia-like behavioral abnormalities (Hikida et al., 2007; Pletnikov et al., 2008; Shen et al., 2008; Ayhan et al., 2011) (Table 2). Furthermore, a technique of inducible transgene expression enabled a specific expression of truncated DISC1 during only prenatal period, only postnatal period or both periods (Ayhan et al., 2011). Prenatal expression only led to decreased brain volume and decreased number of PV-positive interneurons. Enlarged lateral ventricle seems to be affected by postnatal expression of truncated DISC1. In contrast, enhanced responsiveness to psychostimulant required prenatal and postnatal continuous expression (Ayhan et al., 2011), which suggests that both neurodevelopmental abnormality and neuronal functional impairment caused by truncated DISC1 might be essential for pathogenesis of schizophrenia.
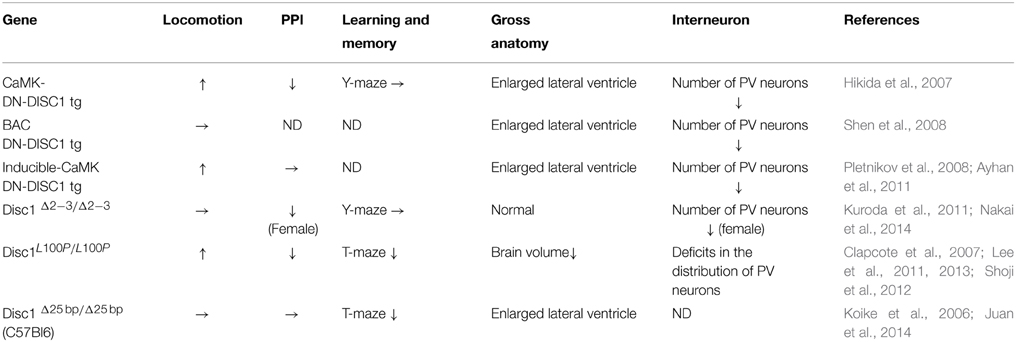
Table 2. Summary of mutant Disc1 mouse models.
22q11 Deletion Syndrome
22q11.2 deletion syndrome (22q11DS) is the most frequent known genetic cause of schizophrenia (Pulver et al., 1994). 22q11DS accounts for about 1% of schizophrenia cases (Karayiorgou et al., 1995; Manolio et al., 2009). Prior brain imaging studies of human 22q11 DS have indicated reduced cerebral volume, ventricular enlargement and reduced hippocampal volume (Eliez et al., 2000, 2001; Chow et al., 2002; Simon et al., 2005). All of these brain anomalies have also been reported in schizophrenia (Shenton et al., 2001). All of the genes except for one gene in human 22q11.2 locus exist on mouse chromosome 16 (Puech et al., 1997). This has facilitated the generation of mouse models of 22q11 DS, which carry a hemizygous deletion of 22q11-related region of mouse chromosome16 (Lindsay et al., 1999; Paylor and Lindsay, 2006; Stark et al., 2008). These animal models show schizophrenia-related behavioral abnormalities such as working memory deficits, sensory information-processing deficits, and enhanced responsiveness to psychostimulants (Paylor et al., 2001; Stark et al., 2008; Earls et al., 2011; Kimoto et al., 2012), which are recognized as major deficits of schizophrenia (Elvevag and Goldberg, 2000; Green et al., 2000; Swerdlow et al., 2001). Animal models of 22q11DS showed reduced density of layer II–IV projection neurons in a medial PFC (Meechan et al., 2009), reduced volume of a perinatal HP dentate gyrus (Toritsuka et al., 2013), delayed migration of hippocampal dentate neuronal progenitors and cortical interneurons, and altered distribution of PV-positive interneurons (Meechan et al., 2009, 2012; Toritsuka et al., 2013), although it remains to be elucidated these deficits in neurogenesis lead to excitatory/inhibitory imbalance or not. Perinatal hippocampal DG and interneuronal migration abnormalities are caused by Cxcl12/Cxcr4 signaling deficits (Toritsuka et al., 2013). Cxcl12/Cxcr4 signaling might play pivotal roles in the pathogenesis of schizophrenia. Previous studies also suggest a possible involvement of Cxcl12/Cxcr4 signaling in the neurodevelopmental disorders of GD17 MAM-treated animal model of schizophrenia (Paredes et al., 2006). The expression of CXCL12 is decreased in olfactory neurons from sporadic cases with schizophrenia compared with normal controls (Toritsuka et al., 2013).
Among of genes deleted in 22q11DS, Dgcr8 is a promising candidate gene for schizophrenia-related phenotypes. Dgcr8 forms the microprocessor complex of microRNA (miRNA) with Drosha, which is essential for miRNA production. Overexpression of Dgcr8 rescued interneuronal migration deficits of 22q11DS model mice, and the migration of hippocampal DG and interneuronal progenitors were also affected inDgcr8+/− mice (Toritsuka et al., 2013). These observations demonstrated the important roles of Dgcr8 in the pathogenesis of 22q11DS. miRNA–mediated regulation network fine tunes the balance of signaling and confers robustness to the system (Herranz and Cohen, 2010). miRNA-mediated regulation can buffer increases or reductions in gene dosage (Staton et al., 2011). Haplodeletion of Dgcr8 causes 20–70% reduction of a specific subsets of mature miRNAs both in Dgcr8 heterozygous and 22q11DS model mice (Stark et al., 2008). Dgcr8 heterozygousity might uncover the effects of 22q11 microdeletion through the disruption of miRNA-mediated buffering effects. In mice, heterozygous deletion of Dgcr8 alone showed working memory deficits, sensory information-processing deficits and some of neurodevelopmental abnormalities such as reduced cortical neuronal densities (Stark et al., 2008; Fenelon et al., 2011). The behavioral abnormalities and neurodevelopmental disorders of Dgcr8+/− mice are similar but some of them are milder than those of 22q11DS model mice,(Stark et al., 2008; Meechan et al., 2009; Fenelon et al., 2011), which might suggest that additional haplodeletion of other genes in 22q11-related regions might be required for the complete reconstitution of phenotypes of 22q11DS model mice. It remains to be elucidated whether behavioral abnormalities of 22q11DS model mice are directly caused by neuronal migration deficit and Cxcr4 signaling defects.
Concluding Remarks
Elucidating the relationship between neurodevelopmental abnormalities and the pathogenesis of schizophrenia would be exceptionally difficult. In order to dissect the complex causal relations, more sophisticated genetic manipulation would be required. Combination of various techniques such as conditional knockout, inducible transgene expression and virus-mediated gene delivery will enable cell type-specific and developmental stage-specific knockout or rescue experiments. In the future comprehensive profile of neurodevelopmental deficits-behavioral abnormalities will provide significant insights into mental disease pathogenesis of all these neurodevelopmental genes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agim, Z. S., Esendal, M., Briollais, L., Uyan, O., Meschian, M., Martinez, L. A., et al. (2013). Discovery, validation and characterization of Erbb4 and Nrg1 haplotypes using data from three genome-wide association studies of schizophrenia. PLoS ONE 8:e53042. doi: 10.1371/journal.pone.0053042
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akbarian, S., Bunney, W. E. Jr. Potkin, S. G., Wigal, S. B., Hagman, J. O., Sandman, C. A., et al. (1993a). Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch. Gen. Psychiatry 50, 169–177. doi: 10.1001/archpsyc.1993.01820150007001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E. Jr., et al. (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266. doi: 10.1001/archpsyc.1995.03950160008002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akbarian, S., Kim, J. J., Potkin, S. G., Hetrick, W. P., Bunney, W. E. Jr., and Jones, E. G. (1996). Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients. Arch. Gen. Psychiatry 53, 425–436. doi: 10.1001/archpsyc.1996.01830050061010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akbarian, S., Vinuela, A., Kim, J. J., Potkin, S. G., Bunney, W. E. Jr., and Jones, E. G. (1993b). Distorted distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase neurons in temporal lobe of schizophrenics implies anomalous cortical development. Arch. Gen. Psychiatry 50, 178–187. doi: 10.1001/archpsyc.1993.01820150016002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akil, M., and Lewis, D. A. (1997). Cytoarchitecture of the entorhinal cortex in schizophrenia. Am. J. Psychiatry 154, 1010–1012. doi: 10.1176/ajp.154.7.1010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anton, E. S., Marchionni, M. A., Lee, K. F., Rakic, P., Rio, C., Rieff, H. I., et al. (1997). Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex Neuregulin and erbB receptors play a critical role in neuronal migration. Development 124, 3501–3510.
Arnold, S. E., Hyman, B. T., Van Hoesen, G. W., and Damasio, A. R. (1991). Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch. Gen. Psychiatry 48, 625–632. doi: 10.1001/archpsyc.1991.01810310043008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arnold, S. E., Ruscheinsky, D. D., and Han, L. Y. (1997). Further evidence of abnormal cytoarchitecture of the entorhinal cortex in schizophrenia using spatial point pattern analyses. Biol. Psychiatry 42, 639–647. doi: 10.1016/S0006-3223(97)00142-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ayhan, Y., Abazyan, B., Nomura, J., Kim, R., Ladenheim, B., Krasnova, I. N., et al. (2011). Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol. Psychiatry 16, 293–306. doi: 10.1038/mp.2009.144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Badner, J. A., and Gershon, E. S. (2002). Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol. Psychiatry 7, 405–411. doi: 10.1038/sj.mp.4001012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beasley, C. L., Cotter, D. R., and Everall, I. P. (2002). Density and distribution of white matter neurons in schizophrenia, bipolar disorder and major depressive disorder: no evidence for abnormalities of neuronal migration. Mol. Psychiatry 7, 564–570. doi: 10.1038/sj.mp.4001038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beasley, C. L., Honavar, M., Everall, I. P., and Cotter, D. (2009). Two-dimensional assessment of cytoarchitecture in the superior temporal white matter in schizophrenia, major depressive disorder and bipolar disorder. Schizophr. Res. 115, 156–162. doi: 10.1016/j.schres.2009.09.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benzel, I., Bansal, A., Browning, B. L., Galwey, N. W., Maycox, P. R., McGinnis, R., et al. (2007). Interactions among genes in the ErbB-Neuregulin signalling network are associated with increased susceptibility to schizophrenia. Behav. Brain Funct. 3:31. doi: 10.1186/1744-9081-3-31
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernstein, H. G., Krell, D., Baumann, B., Danos, P., Falkai, P., Diekmann, S., et al. (1998). Morphometric studies of the entorhinal cortex in neuropsychiatric patients and controls: clusters of heterotopically displaced lamina II neurons are not indicative of schizophrenia. Schizophr. Res. 33, 125–132. doi: 10.1016/S0920-9964(98)00071-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boucher, A. A., Arnold, J. C., Duffy, L., Schofield, P. R., Micheau, J., and Karl, T. (2007). Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Delta9-tetrahydrocannabinol. Psychopharmacology (Berl). 192, 325–336. doi: 10.1007/s00213-007-0721-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braff, D. L., and Geyer, M. A. (1990). Sensorimotor gating and schizophrenia. Human and animal model studies. Arch. Gen. Psychiatry 47, 181–188. doi: 10.1001/archpsyc.1990.01810140081011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., et al. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J. Neurosci. 28, 6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y. J., Zhang, M., Yin, D. M., Wen, L., Ting, A., Wang, P., et al. (2010). ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 107, 21818–21823. doi: 10.1073/pnas.1010669107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cho, R. Y., Konecky, R. O., and Carter, C. S. (2006). Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 103, 19878–19883. doi: 10.1073/pnas.0609440103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chow, E. W., Zipursky, R. B., Mikulis, D. J., and Bassett, A. S. (2002). Structural brain abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biol. Psychiatry 51, 208–215. doi: 10.1016/S0006-3223(01)01246-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., et al. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402. doi: 10.1016/j.neuron.2007.04.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corbin, J. G., Nery, S., and Fishell, G. (2001). Telencephalic cells take a tangent: non-radial migration in the mammalian forebrain. Nat. Neurosci. 4, 1177–1182. doi: 10.1038/nn749
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deakin, I. H., Law, A. J., Oliver, P. L., Schwab, M. H., Nave, K. A., Harrison, P. J., et al. (2009). Behavioural characterization of neuregulin 1 type I overexpressing transgenic mice. Neuroreport 20, 1523–1528. doi: 10.1097/WNR.0b013e328330f6e7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deakin, I. H., Nissen, W., Law, A. J., Lane, T., Kanso, R., Schwab, M. H., et al. (2012). Transgenic overexpression of the type I isoform of neuregulin 1 affects working memory and hippocampal oscillations but not long-term potentiation Behavioural characterization of neuregulin 1 type I overexpressing transgenic mice. Cereb. Cortex 22, 1520–1529. doi: 10.1093/cercor/bhr223
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Del Pino, I., Garcia-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martinez de Lagran, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. doi: 10.1016/j.neuron.2013.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duffy, L., Cappas, E., Scimone, A., Schofield, P. R., and Karl, T. (2008). Behavioral profile of a heterozygous mutant mouse model for EGF-like domain neuregulin 1. Behav. Neurosci. 122, 748–759. doi: 10.1037/0735-7044.122.4.748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Earls, L. R., Bayazitov, I. T., Fricke, R. G., Berry, R. B., Illingworth, E., Mittleman, G., et al. (2011). Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J. Neurosci. 30, 15843–15855. doi: 10.1523/JNEUROSCI.1425-10.2010
Ehrlichman, R. S., Luminais, S. N., White, S. L., Rudnick, N. D., Ma, N., Dow, H. C., et al. (2009). Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 1294, 116–127. doi: 10.1016/j.brainres.2009.07.065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ekelund, J., Hennah, W., Hiekkalinna, T., Parker, A., Meyer, J., Lonnqvist, J., et al. (2004). Replication of 1q42 linkage in Finnish schizophrenia pedigrees Chromosome 1 loci in Finnish schizophrenia families. Mol. Psychiatry 9, 1037–1041. doi: 10.1038/sj.mp.4001536
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ekelund, J., Hovatta, I., Parker, A., Paunio, T., Varilo, T., Martin, R., et al. (2001). Chromosome 1 loci in Finnish schizophrenia families. Hum. Mol. Genet. 10, 1611–1617. doi: 10.1093/hmg/10.15.1611
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eliez, S., Blasey, C. M., Schmitt, E. J., White, C. D., Hu, D., and Reiss, A. L. (2001). Velocardiofacial syndrome: are structural changes in the temporal and mesial temporal regions related to schizophrenia? Am. J. Psychiatry 158, 447–453. doi: 10.1176/appi.ajp.158.3.447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eliez, S., Schmitt, J. E., White, C. D., and Reiss, A. L. (2000). Children and adolescents with velocardiofacial syndrome: a volumetric MRI study. Am. J. Psychiatry 157, 409–415. doi: 10.1176/appi.ajp.157.3.409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elvevag, B., and Goldberg, T. E. (2000). Cognitive impairment in schizophrenia is the core of the disorder. Crit. Rev. Neurobiol. 14, 1–21. doi: 10.1615/CritRevNeurobiol.v14.i1.10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Falkai, P., Schneider-Axmann, T., and Honer, W. G. (2000). Entorhinal cortex pre-alpha cell clusters in schizophrenia: quantitative evidence of a developmental abnormality. Biol. Psychiatry 47, 937–943. doi: 10.1016/S0006-3223(99)00250-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fazzari, P., Paternain, A. V., Valiente, M., Pla, R., Lujan, R., Lloyd, K., et al. (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380. doi: 10.1038/nature08928
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feldman, M. L., and Peters, A. (1978). The forms of non-pyramidal neurons in the visual cortex of the rat. J. Comp. Neurol. 179, 761–793. doi: 10.1002/cne.901790406
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fenelon, K., Mukai, J., Xu, B., Hsu, P. K., Drew, L. J., Karayiorgou, M., et al. (2011). Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 4447–4452 doi: 10.1073/pnas.1101219108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fisahn, A., Neddens, J., Yan, L., and Buonanno, A. (2009). Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia. Cereb. Cortex 19, 612–618. doi: 10.1093/cercor/bhn107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flagstad, P., Glenthoj, B. Y., and Didriksen, M. (2005). Cognitive deficits caused by late gestational disruption of neurogenesis in rats: a preclinical model of schizophrenia. Neuropsychopharmacology 30, 250–260. doi: 10.1038/sj.npp.1300625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flagstad, P., Mork, A., Glenthoj, B. Y., van Beek, J., Michael-Titus, A. T., and Didriksen, M. (2004). Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology 29, 2052–2064. doi: 10.1038/sj.npp.1300516
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flames, N., Long, J. E., Garratt, A. N., Fischer, T. M., Gassmann, M., Birchmeier, C., et al. (2004). Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron 44, 251–261. doi: 10.1016/j.neuron.2004.09.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fung, S. J., Webster, M. J., Sivagnanasundaram, S., Duncan, C., Elashoff, M., and Weickert, C. S. (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 167, 1479–1488. doi: 10.1176/appi.ajp.2010.09060784
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394. doi: 10.1038/378390a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gill, K. M., Lodge, D. J., Cook, J. M., Aras, S., and Grace, A. A. (2011). A novel alpha5GABA(A)R-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology 36, 1903–1911. doi: 10.1038/npp.2011.76
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glover, J. C., Rakic, P., Ayoub, A. E., Breunig, J. J., Dominguez, M. H., Gal, J. S., et al. (2009). “The developmental and functional logic of neuronal circuits”: commentary on the Kavli Prize in Neuroscience. Neuroscience 163, 977–984. doi: 10.1016/j.neuroscience.2009.07.047
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldman-Rakic, P. S. (1994). Working memory dysfunction in schizophrenia. J. Neuropsychiatry Clin. Neurosci. 6, 348–357. doi: 10.1176/jnp.6.4.348
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Golub, M. S., Germann, S. L., and Lloyd, K. C. (2004). Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav. Brain Res. 153, 159–170. doi: 10.1016/j.bbr.2003.11.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gourevitch, R., Rocher, C., Le Pen, G., Krebs, M. O., and Jay, T. M. (2004). Working memory deficits in adult rats after prenatal disruption of neurogenesis. Behav. Pharmacol. 15, 287–292. doi: 10.1097/01.fbp.0000135703.48799.71
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grace, A., and Moore, H. (1998). “Regulation of information flow in the nucleus accumbens: amodel for the pathophysiology of schizophrenia,” in Origins and Development of Schizophrenia: Advances in Experimental Psychopathology, eds M. F. Lenzenweger and R. H. Dworkin (Washington, DC: American Psychological Association Press), 123–157.
Green, M. F., Kern, R. S., Braff, D. L., and Mintz, J. (2000). Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr. Bull. 26, 119–136. doi: 10.1093/oxfordjournals.schbul.a033430
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamshere, M. L., Bennett, P., Williams, N., Segurado, R., Cardno, A., Norton, N., et al. (2005). Genomewide linkage scan in schizoaffective disorder: significant evidence for linkage at 1q42 close to DISC1, and suggestive evidence at 22q11 and 19p13. Arch. Gen. Psychiatry 62, 1081–1088. doi: 10.1001/archpsyc.62.10.1081
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hansen, D. V., Lui, J. H., Flandin, P., Yoshikawa, K., Rubenstein, J. L., Alvarez-Buylla, A., et al. (2013). Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat. Neurosci. 16, 1576–1587. doi: 10.1038/nn.3541
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hashimoto, R., Numakawa, T., Ohnishi, T., Kumamaru, E., Yagasaki, Y., Ishimoto, T., et al. (2006). Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum. Mol. Genet. 15, 3024–3033. doi: 10.1093/hmg/ddl244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., et al. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326. doi: 10.1084/jem.20051342
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heckers, S. (2004). The hippocampus in schizophrenia. Am. J. Psychiatry 161, 2138–2139. doi: 10.1176/appi.ajp.161.11.2138-a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herranz, H., and Cohen, S. M. (2010). MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 24, 1339–1344. doi: 10.1101/gad.1937010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., et al. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506. doi: 10.1073/pnas.0704774104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honea, R., Crow, T. J., Passingham, D., and Mackay, C. E. (2005). Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am. J. Psychiatry 162, 2233–2245. doi: 10.1176/appi.ajp.162.12.2233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Howard, M. W., Rizzuto, D. S., Caplan, J. B., Madsen, J. R., Lisman, J., Aschenbrenner-Scheibe, R., et al. (2003). Gamma oscillations correlate with working memory load in humans. Cereb. Cortex 13, 1369–1374. doi: 10.1093/cercor/bhg084
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishizuka, K., Kamiya, A., Oh, E. C., Kanki, H., Seshadri, S., Robinson, J. F., et al. (2011). DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473, 92–96. doi: 10.1038/nature09859
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jakob, H., and Beckmann, H. (1986). Prenatal developmental disturbances in the limbic allocortex in schizophrenics. J. Neural Transm. 65, 303–326. doi: 10.1007/BF01249090
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Joshi, D., Fullerton, J. M., and Weickert, C. S. (2014). Elevated ErbB4 mRNA is related to interneuron deficit in prefrontal cortex in schizophrenia. J. Psychiatr. Res. 53, 125–132. doi: 10.1016/j.jpsychires.2014.02.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Juan, L. W., Liao, C. C., Lai, W. S., Chang, C. Y., Pei, J. C., Wong, W. R., et al. (2014). Phenotypic characterization of C57BL/6J mice carrying the Disc1 gene from the 129S6/SvEv strain. Brain Struct. Funct. 219, 1417–1431. doi: 10.1007/s00429-013-0577-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., et al. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178. doi: 10.1038/ncb1328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karayiorgou, M., Morris, M. A., Morrow, B., Shprintzen, R. J., Goldberg, R., Borrow, J., et al. (1995). Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl. Acad. Sci. U.S.A. 92, 7612–7616. doi: 10.1073/pnas.92.17.7612
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kimoto, S., Muraki, K., Toritsuka, M., Mugikura, S., Kajiwara, K., Kishimoto, T., et al. (2012). Selective overexpression of Comt in prefrontal cortex rescues schizophrenia-like phenotypes in a mouse model of 22q11 deletion syndrome. Transl. Psychiatry 2:e146. doi: 10.1038/tp.2012.70
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koike, H., Arguello, P. A., Kvajo, M., Karayiorgou, M., and Gogos, J. A. (2006). Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3693–3697. doi: 10.1073/pnas.0511189103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kovelman, J. A., and Scheibel, A. B. (1984). A neurohistological correlate of schizophrenia. Biol. Psychiatry 19, 1601–1621.
Kramer, R., Bucay, N., Kane, D. J., Martin, L. E., Tarpley, J. E., and Theill, L. E. (1996). Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc. Natl. Acad. Sci. U.S.A. 93, 4833–4838. doi: 10.1073/pnas.93.10.4833
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krimer, L. S., Herman, M. M., Saunders, R. C., Boyd, J. C., Hyde, T. M., Carter, J. M., et al. (1997). A qualitative and quantitative analysis of the entorhinal cortex in schizophrenia. Cereb. Cortex 7, 732–739. doi: 10.1093/cercor/7.8.732
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kubo, K., Tomita, K., Uto, A., Kuroda, K., Seshadri, S., Cohen, J., et al. (2010). Migration defects by DISC1 knockdown in C57BL/6, 129X1/SvJ, and ICR strains via in utero gene transfer and virus-mediated RNAi. Biochem. Biophys. Res. Commun. 400, 631–637. doi: 10.1016/j.bbrc.2010.08.117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuroda, K., Yamada, S., Tanaka, M., Iizuka, M., Yano, H., Mori, D., et al. (2011). Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum. Mol. Genet. 20, 4666–4683. doi: 10.1093/hmg/ddr400
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Law, A. J., Kleinman, J. E., Weinberger, D. R., and Weickert, C. S. (2007). Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum. Mol. Genet. 16, 129–141. doi: 10.1093/hmg/ddl449
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Law, A. J., Lipska, B. K., Weickert, C. S., Hyde, T. M., Straub, R. E., Hashimoto, R., et al. (2006). Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc. Natl. Acad. Sci. U.S.A. 103, 6747–6752. doi: 10.1073/pnas.0602002103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, F. H., Fadel, M. P., Preston-Maher, K., Cordes, S. P., Clapcote, S. J., Price, D. J., et al. (2011). Disc1 point mutations in mice affect development of the cerebral cortex. J. Neurosci. 31, 3197–3206. doi: 10.1523/JNEUROSCI.4219-10.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, F. H., Zai, C. C., Cordes, S. P., Roder, J. C., and Wong, A. H. (2013). Abnormal interneuron development in disrupted-in-schizophrenia-1 L100P mutant mice. Mol. Brain. 6:20. doi: 10.1186/1756-6606-6-20
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Le Pen, G., Gourevitch, R., Hazane, F., Hoareau, C., Jay, T. M., and Krebs, M. O. (2006). Peri-pubertal maturation after developmental disturbance: a model for psychosis onset in the rat. Neuroscience 143, 395–405. doi: 10.1016/j.neuroscience.2006.08.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levinson, D. F., Duan, J., Oh, S., Wang, K., Sanders, A. R., Shi, J., et al. (2011). Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168, 302–316. doi: 10.1176/appi.ajp.2010.10060876
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewis, C. M., Levinson, D. F., Wise, L. H., DeLisi, L. E., Straub, R. E., Hovatta, I., et al. (2003). Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am. J. Hum. Genet. 73, 34–48. doi: 10.1086/376549
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewis, D. A., Curley, A. A., Glausier, J. R., and Volk, D. W. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35, 57–67. doi: 10.1016/j.tins.2011.10.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, D., Collier, D. A., and He, L. (2006). Meta-analysis shows strong positive association of the neuregulin 1 (NRG1) gene with schizophrenia. Hum. Mol. Genet. 15, 1995–2002. doi: 10.1093/hmg/ddl122
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, H., Chou, S. J., Hamasaki, T., Perez-Garcia, C. G., and O'Leary, D. D. (2012). Neuregulin repellent signaling via ErbB4 restricts GABAergic interneurons to migratory paths from ganglionic eminence to cortical destinations. Neural Dev. 7:10. doi: 10.1186/1749-8104-7-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liddle, P. F., and Morris, D. L. (1991). Schizophrenic syndromes and frontal lobe performance. Br. J. Psychiatry 158, 340–345. doi: 10.1192/bjp.158.3.340
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lindsay, E. A., Botta, A., Jurecic, V., Carattini-Rivera, S., Cheah, Y. C., Rosenblatt, H. M., et al. (1999). Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401, 379–383. doi: 10.1038/43900
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y. L., Fann, C. S., Liu, C. M., Chen, W. J., Wu, J. Y., Hung, S. I., et al. (2006). A single nucleotide polymorphism fine mapping study of chromosome 1q42.1 reveals the vulnerability genes for schizophrenia, GNPAT and DISC1: association with impairment of sustained attention. Biol. Psychiatry 60, 554–562. doi: 10.1016/j.biopsych.2006.04.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lodge, D. J., Behrens, M. M., and Grace, A. A. (2009). A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 29, 2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lodge, D. J., and Grace, A. A. (2007). Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 27, 11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, X., He, W., Hu, X., and Yan, R. (2014). Reversible overexpression of bace1-cleaved neuregulin-1 N-terminal fragment induces schizophrenia-like phenotypes in mice. Biol. Psychiatry 76, 120–127. doi: 10.1016/j.biopsych.2013.09.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, T., Wang, C., Wang, L., Zhou, X., Tian, M., Zhang, Q., et al. (2013). Subcortical origins of human and monkey neocortical interneurons. Nat. Neurosci. 16, 1588–1597. doi: 10.1038/nn.3536
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Macgregor, S., Visscher, P. M., Knott, S. A., Thomson, P., Porteous, D. J., Millar, J. K., et al. (2004). A genome scan and follow-up study identify a bipolar disorder susceptibility locus on chromosome 1q42. Mol. Psychiatry 9, 1083–1090. doi: 10.1038/sj.mp.4001544
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031. doi: 10.1016/j.cell.2008.12.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marin, O., and Rubenstein, J. L. (2003). Cell migration in the forebrain. Annu. Rev. Neurosci. 26, 441–483. doi: 10.1146/annurev.neuro.26.041002.131058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Medoff, D. R., Holcomb, H. H., Lahti, A. C., and Tamminga, C. A. (2001). Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus 11, 543–550. doi: 10.1002/hipo.1070
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meechan, D. W., Tucker, E. S., Maynard, T. M., and LaMantia, A. S. (2009). Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 16434–16445. doi: 10.1073/pnas.0905696106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meechan, D. W., Tucker, E. S., Maynard, T. M., and LaMantia, A. S. (2012). Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc. Natl. Acad. Sci. U.S.A. 109, 18601–18606. doi: 10.1073/pnas.1211507109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., et al. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191. doi: 10.1126/science.1112915
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Minzenberg, M. J., Firl, A. J., Yoon, J. H., Gomes, G. C., Reinking, C., and Carter, C. S. (2010). Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 35, 2590–2599. doi: 10.1038/npp.2010.150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miyoshi, G., Hjerling-Leffler, J., Karayannis, T., Sousa, V. H., Butt, S. J., Battiste, J., et al. (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci. 30, 1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moore, H., Jentsch, J. D., Ghajarnia, M., Geyer, M. A., and Grace, A. A. (2006). A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol. Psychiatry 60, 253–264. doi: 10.1016/j.biopsych.2006.01.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munafo, M. R., Thiselton, D. L., Clark, T. G., and Flint, J. (2006). Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol. Psychiatry 11, 539–546. doi: 10.1038/sj.mp.4001817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakai, T., Nagai, T., Wang, R., Yamada, S., Kuroda, K., Kaibuchi, K., et al. (2014). Alterations of GABAergic and dopaminergic systems in mutant mice with disruption of exons 2 and 3 of the Disc1 gene. Neurochem. Int. 74, 74–83. doi: 10.1016/j.neuint.2014.06.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neddens, J., and Buonanno, A. (2010). Selective populations of hippocampal interneurons express ErbB4 and their number and distribution is altered in ErbB4 knockout mice. Hippocampus 20, 724–744. doi: 10.1002/hipo.20675
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nuechterlein, K. H., Barch, D. M., Gold, J. M., Goldberg, T. E., Green, M. F., and Heaton, R. K. (2004). Identification of separable cognitive factors in schizophrenia. Schizophr. Res. 72, 29–39. doi: 10.1016/j.schres.2004.09.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O'Connell, P., Woodruff, P. W., Wright, I., Jones, P., and Murray, R. M. (1997). Developmental insanity or dementia praecox: was the wrong concept adopted? Schizophr. Res. 23, 97–106. doi: 10.1016/S0920-9964(96)00110-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O'Tuathaigh, C. M., Babovic, D., O'Sullivan, G. J., Clifford, J. J., Tighe, O., Croke, D. T., et al. (2007). Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience 147, 18–27. doi: 10.1016/j.neuroscience.2007.03.051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O'Tuathaigh, C. M., Harte, M., O'Leary, C., O'Sullivan, G. J., Blau, C., Lai, D., et al. (2010). Schizophrenia-related endophenotypes in heterozygous neuregulin-1 ‘knockout’ mice. Eur. J. Neurosci. 31, 349–358. doi: 10.1111/j.1460-9568.2009.07069.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ozeki, Y., Tomoda, T., Kleiderlein, J., Kamiya, A., Bord, L., Fujii, K., et al. (2003). Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc. Natl. Acad. Sci. U.S.A. 100, 289–294. doi: 10.1073/pnas.0136913100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paredes, M. F., Li, G., Berger, O., Baraban, S. C., and Pleasure, S. J. (2006). Stromal-derived factor-1 (CXCL12) regulates laminar position of Cajal-Retzius cells in normal and dysplastic brains. J. Neurosci. 26, 9404–9412. doi: 10.1523/JNEUROSCI.2575-06.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paylor, R., and Lindsay, E. (2006). Mouse models of 22q11 deletion syndrome. Biol. Psychiatry 59, 1172–1179. doi: 10.1016/j.biopsych.2006.01.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paylor, R., McIlwain, K. L., McAninch, R., Nellis, A., Yuva-Paylor, L. A., Baldini, A., et al. (2001). Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum. Mol. Genet. 10, 2645–2650. doi: 10.1093/hmg/10.23.2645
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pei, J. C., Liu, C. M., and Lai, W. S. (2014). Distinct phenotypes of new transmembrane-domain neuregulin 1 mutant mice and the rescue effects of valproate on the observed schizophrenia-related cognitive deficits. Front. Behav. Neurosci. 8:126. doi: 10.3389/fnbeh.2014.00126
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pletnikov, M. V., Ayhan, Y., Xu, Y., Nikolskaia, O., Ovanesov, M., Huang, H., et al. (2008). Enlargement of the lateral ventricles in mutant DISC1 transgenic mice. Mol. Psychiatry 13:115. doi: 10.1038/sj.mp.4002144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Puech, A., Saint-Jore, B., Funke, B., Gilbert, D. J., Sirotkin, H., Copeland, N. G., et al. (1997). Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization. Proc. Natl. Acad. Sci. U.S.A. 94, 14608–14613. doi: 10.1073/pnas.94.26.14608
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pulver, A. E., Nestadt, G., Goldberg, R., Shprintzen, R. J., Lamacz, M., Wolyniec, P. S., et al. (1994). Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 182, 476–478. doi: 10.1097/00005053-199408000-00010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rakic, P. (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. doi: 10.1038/nrn2719
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rees, E., Walters, J. T., Georgieva, L., Isles, A. R., Chambert, K. D., Richards, A. L., et al. (2014). Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 204, 108–114. doi: 10.1192/bjp.bp.113.131052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rio, C., Rieff, H. I., Qi, P., Khurana, T. S., and Corfas, G. (1997). Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron 19, 39–50. doi: 10.1016/S0896-6273(00)80346-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ripke, S., O'Dushlaine, C., Chambert, K., Moran, J. L., Kahler, A. K., Akterin, S., et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159. doi: 10.1038/ng.2742
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sasaki, S., Shionoya, A., Ishida, M., Gambello, M. J., Yingling, J., Wynshaw-Boris, A., et al. (2000). A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28, 681–696. doi: 10.1016/S0896-6273(00)00146-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schizophrenia Working Group of the Psychiztric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schobel, S. A., Lewandowski, N. M., Corcoran, C. M., Moore, H., Brown, T., Malaspina, D., et al. (2009). Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch. Gen. Psychiatry 66, 938–946. doi: 10.1001/archgenpsychiatry.2009.115
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shamir, A., Kwon, O. B., Karavanova, I., Vullhorst, D., Leiva-Salcedo, E., Janssen, M. J., et al. (2012). The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J. Neurosci. 32, 2988–2997 doi: 10.1523/JNEUROSCI.1899-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, S., Lang, B., Nakamoto, C., Zhang, F., Pu, J., Kuan, S. L., et al. (2008). Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J. Neurosci. 28, 10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shenton, M. E., Dickey, C. C., Frumin, M., and McCarley, R. W. (2001). A review of MRI findings in schizophrenia. Schizophr. Res. 49, 1–52. doi: 10.1016/S0920-9964(01)00163-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shi, J., Levinson, D. F., Duan, J., Sanders, A. R., Zheng, Y., Pe'er, I., et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757. doi: 10.1038/nature08192
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shoji, H., Toyama, K., Takamiya, Y., Wakana, S., Gondo, Y., and Miyakawa, T. (2012). Comprehensive behavioral analysis of ENU-induced Disc1-Q31L and -L100P mutant mice. BMC Res. Notes 5:108. doi: 10.1186/1756-0500-5-108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Silberberg, G., Darvasi, A., Pinkas-Kramarski, R., and Navon, R. (2006). The involvement of ErbB4 with schizophrenia: association and expression studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 142–148. doi: 10.1002/ajmg.b.30275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Simon, T. J., Ding, L., Bish, J. P., McDonald-McGinn, D. M., Zackai, E. H., and Gee, J. (2005). Volumetric, connective, and morphologic changes in the brains of children with chromosome 22q11.2 deletion syndrome: an integrative study. Neuroimage 25, 169–180. doi: 10.1016/j.neuroimage.2004.11.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singh, K. K., De Rienzo, G., Drane, L., Mao, Y., Flood, Z., Madison, J., et al. (2011). Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 72, 545–558. doi: 10.1016/j.neuron.2011.09.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singh, K. K., Ge, X., Mao, Y., Drane, L., Meletis, K., Samuels, B. A., et al. (2010). Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron 67, 33–48. doi: 10.1016/j.neuron.2010.06.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. doi: 10.1038/ng.138
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Staton, A. A., Knaut, H., and Giraldez, A. J. (2011). miRNA regulation of Sdf1 chemokine signaling provides genetic robustness to germ cell migration. Nat. Genet. 43, 204–211. doi: 10.1038/ng.758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
St Clair, D., Blackwood, D., Muir, W., Carothers, A., Walker, M., Spowart, G., et al. (1990). Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16. doi: 10.1016/0140-6736(90)91520-K
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892. doi: 10.1086/342734
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinecke, A., Gampe, C., Nitzsche, F., and Bolz, J. (2014). DISC1 knockdown impairs the tangential migration of cortical interneurons by affecting the actin cytoskeleton. Front. Cell. Neurosci. 8:190. doi: 10.3389/fncel.2014.00190
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinecke, A., Gampe, C., Valkova, C., Kaether, C., and Bolz, J. (2012). Disrupted-in-Schizophrenia 1 (DISC1) is necessary for the correct migration of cortical interneurons. J. Neurosci. 32, 738–745. doi: 10.1523/JNEUROSCI.5036-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sullivan, P. F., Daly, M. J., and O'Donovan, M. (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551. doi: 10.1038/nrg3240
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swerdlow, N. R., Braff, D. L., Taaid, N., and Geyer, M. A. (1994). Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch. Gen. Psychiatry 51, 139–154. doi: 10.1001/archpsyc.1994.03950020063007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swerdlow, N. R., Geyer, M. A., and Braff, D. L. (2001). Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl). 156, 194–215. doi: 10.1007/s002130100799
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taya, S., Shinoda, T., Tsuboi, D., Asaki, J., Nagai, K., Hikita, T., et al. (2007). DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 27, 15–26. doi: 10.1523/JNEUROSCI.3826-06.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thomson, P. A., Parla, J. S., McRae, A. F., Kramer, M., Ramakrishnan, K., Yao, J., et al. (2014). 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol. Psychiatry 19, 668–675. doi: 10.1038/mp.2013.68
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Toritsuka, M., Kimoto, S., Muraki, K., Landek-Salgado, M. A., Yoshida, A., Yamamoto, N., et al. (2013). Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc. Natl. Acad. Sci. U.S.A. 110, 17552–17557. doi: 10.1073/pnas.1312661110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Volk, D. W., Austin, M. C., Pierri, J. N., Sampson, A. R., and Lewis, D. A. (2000). Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch. Gen. Psychiatry 57, 237–245. doi: 10.1001/archpsyc.57.3.237
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weickert, C. S., Tiwari, Y., Schofield, P. R., Mowry, B. J., and Fullerton, J. M. (2012). Schizophrenia-associated HapICE haplotype is associated with increased NRG1 type III expression and high nucleotide diversity. Transl. Psychiatry 2:e104. doi: 10.1038/tp.2012.25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669. doi: 10.1001/archpsyc.1987.01800190080012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yau, H. J., Wang, H. F., Lai, C., and Liu, F. C. (2003). Neural development of the neuregulin receptor ErbB4 in the cerebral cortex and the hippocampus: preferential expression by interneurons tangentially migrating from the ganglionic eminences. Cereb. Cortex 13, 252–264. doi: 10.1093/cercor/13.3.252
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, D. M., Chen, Y. J., Lu, Y. S., Bean, J. C., Sathyamurthy, A., Shen, C., et al. (2013). Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron 78, 644–657. doi: 10.1016/j.neuron.2013.03.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: schizophrenia, GABAergic interneuron, mouse models, Neuregulin1, DISC1, 22q11 deletion syndrome, Dgcr8, Cxcr4
Citation: Muraki K and Tanigaki K (2015) Neuronal migration abnormalities and its possible implications for schizophrenia. Front. Neurosci. 9:74. doi: 10.3389/fnins.2015.00074
Received: 30 November 2014; Paper pending published: 27 December 2014;
Accepted: 20 February 2015; Published: 10 March 2015.
Edited by:
Chiaki Ohtaka-Maruyama, Tokyo Metropolitan Institute of Medical Science, JapanReviewed by:
Luca Bonfanti, University of Turin, ItalyEmanuel DiCicco-Bloom, University of Medicine and Dentistry of New Jersey/Robert Wood Johnson Medical School, USA
Copyright © 2015 Muraki and Tanigaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenji Tanigaki, Shiga Medical Center, Research Institute, Moriyama 5-4-30, Shiga 524-8524, JapandGFuaWdha2lAcmVzLm1lZC5zaGlnYS1wcmVmLmpw