 Diana Amantea1*
Diana Amantea1* Giuseppe Micieli2
Giuseppe Micieli2 Cristina Tassorelli2,3
Cristina Tassorelli2,3 María I. Cuartero4
María I. Cuartero4 Iván Ballesteros4
Iván Ballesteros4 Michelangelo Certo1
Michelangelo Certo1 María A. Moro4
María A. Moro4 Ignacio Lizasoain4
Ignacio Lizasoain4 Giacinto Bagetta1,5
Giacinto Bagetta1,5- 1Section of Preclinical and Translational Pharmacology, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Rende, Italy
- 2C. Mondino National Neurological Institute, Pavia, Italy
- 3Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy
- 4Unidad de Investigación Neurovascular, Departamento de Farmacología, Facultad de Medicina, Universidad Complutense de Madrid and Instituto de Investigación Hospital 12 de Octubre, Madrid, Spain
- 5Section of Neuropharmacology of Normal and Pathological Neuronal Plasticity, University Consortium for Adaptive Disorders and Head Pain, University of Calabria, Rende, Italy
The innate immune system plays a dualistic role in the evolution of ischemic brain damage and has also been implicated in ischemic tolerance produced by different conditioning stimuli. Early after ischemia, perivascular astrocytes release cytokines and activate metalloproteases (MMPs) that contribute to blood–brain barrier (BBB) disruption and vasogenic oedema; whereas at later stages, they provide extracellular glutamate uptake, BBB regeneration and neurotrophic factors release. Similarly, early activation of microglia contributes to ischemic brain injury via the production of inflammatory cytokines, including tumor necrosis factor (TNF) and interleukin (IL)-1, reactive oxygen and nitrogen species and proteases. Nevertheless, microglia also contributes to the resolution of inflammation, by releasing IL-10 and tumor growth factor (TGF)-β, and to the late reparative processes by phagocytic activity and growth factors production. Indeed, after ischemia, microglia/macrophages differentiate toward several phenotypes: the M1 pro-inflammatory phenotype is classically activated via toll-like receptors or interferon-γ, whereas M2 phenotypes are alternatively activated by regulatory mediators, such as ILs 4, 10, 13, or TGF-β. Thus, immune cells exert a dualistic role on the evolution of ischemic brain damage, since the classic phenotypes promote injury, whereas alternatively activated M2 macrophages or N2 neutrophils prompt tissue remodeling and repair. Moreover, a subdued activation of the immune system has been involved in ischemic tolerance, since different preconditioning stimuli act via modulation of inflammatory mediators, including toll-like receptors and cytokine signaling pathways. This further underscores that the immuno-modulatory approach for the treatment of ischemic stroke should be aimed at blocking the detrimental effects, while promoting the beneficial responses of the immune reaction.
Introduction
As highlighted by recent expression profiling studies, the majority of the genes acutely modulated in the blood of stroke patients are implicated in the regulation of the innate immune system (Tang et al., 2006; Barr et al., 2010; Oh et al., 2012; Brooks et al., 2014). Moreover, serum levels of markers of acute inflammation correlate with the severity of brain damage and neurological deficit (Fassbender et al., 1994; Smith et al., 2004; Basic Kes et al., 2008; Whiteley et al., 2009; Chang et al., 2010).
Indeed, the innate immune system plays a pivotal role in the evolution of ischemic cerebral injury, as soluble mediators (i.e., cytokines and chemokines) and specialized cells, activated in the brain or recruited from the periphery, actively participate to the detrimental processes implicated in tissue damage, as well as to the repair and regeneration phases (Kamel and Iadecola, 2012; Amantea et al., 2014a). The dualistic role exerted by several mediators of the immune reaction may explain why most anti-inflammatory approaches, conceived disregarding the potential beneficial function of the target, have failed to reach the clinical setting.
In addition, a subdued activation of the immune system has been involved in ischemic tolerance, since different preconditioning stimuli act by reprogramming the immune response, through the modulation of inflammatory mediators, including toll-like receptors (TLRs) and cytokine signaling pathways (Garcia-Bonilla et al., 2014a). This further underscores that promoting the endogenous neuroprotective reactions of the innate immune system represents an attractive opportunity to develop novel effective stroke therapeutics.
This review has been conceived to describe the role played by the diverse mediators of the innate immune system in ischemic brain damage, also highlighting their beneficial role with the ambition to stimulate more extensive research aimed at selectively targeting these processes.
Cellular Mediators of the Immune Response
Resident Immune Cells
All the cellular components of the neurovascular unit participate to the inflammatory reaction involved in ischemic stroke injury (Figure 1). On the intravascular side, platelets and the complement system are rapidly activated after vessel occlusion, thus providing the first trigger for the inflammatory response (Atkinson et al., 2006; Nieswandt et al., 2011). Elevated endothelial expression of the adhesion molecules P-selectin and intercellular adhesion molecule (ICAM)-1 promotes polymorphonuclear leukocytes (PMN) recruitment that exacerbates microvessel obstruction, also due to the reduced bioavailability of nitric oxide (NO) (Granger et al., 1989; Mori et al., 1992; Wong and Crack, 2008). Recently, Sreeramkumar et al. (2014) have demonstrated that the interaction between PMN and platelets within the microvasculature of infarcted brains is inhibited by blocking P-selectin glycoprotein ligand-1 (PSGL-1), and this correlates with a significant decrease in the infarct volume after permanent occlusion of the middle cerebral artery. Moreover, the ischemia-induced release of pro-inflammatory cytokines [e.g., interleukin (IL)-1 and tumor necrosis factor (TNF)] further promotes adhesion molecules expression and, together with the activation of proteases [i.e., matrix metalloproteases (MMPs)], prompts blood-brain barrier (BBB) breakdown leading to leukocytes extravasation in the injured brain (Ishikawa et al., 2004; Amantea et al., 2007; Yemisci et al., 2009; Yilmaz and Granger, 2010).
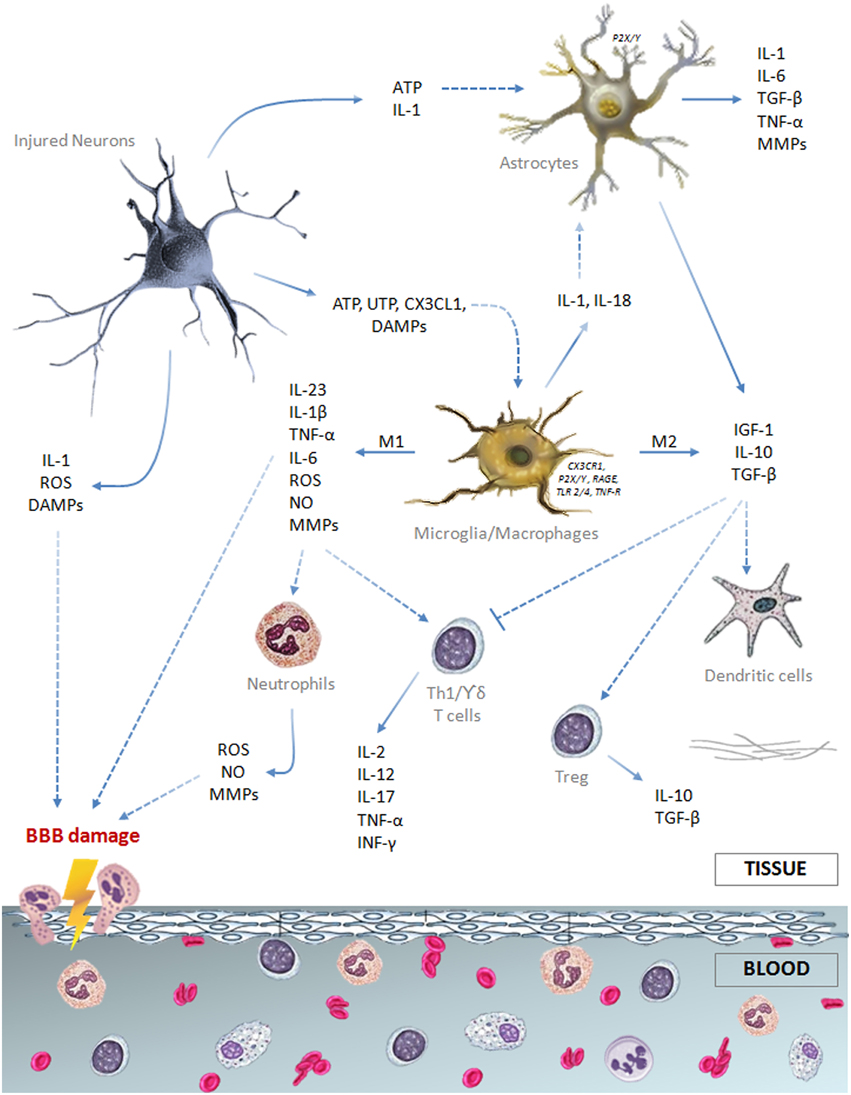
Figure 1. Schematic drawing summarizing the major cellular and soluble mediators of the immune response elicited by an ischemic insult.
Astrocytes are the most abundant glial cells of the human brain and form part of the BBB. Ultrastructural studies have shown that pericapillary astrocyte end-feet are the first cellular elements to swell during cerebral ischemia (Dodson et al., 1977). Cytokines and reactive oxygen species (ROS), released by neurons and glial cells few minutes after the ischemic insult, alter molecular expression patterns in astrocytes and induce cellular hypertrophy, proliferation and scar formation (Sofroniew, 2009). Moreover, stimulation of P2Y(1) receptors by adenosine 5′-triphosphate (ATP), released or leaked from injured cells, prompts production of pro-inflammatory cytokines and chemokines by astrocytes via activation of a phosphorylated-p65 subunit (RelA)-mediated NF-κB pathway (Kuboyama et al., 2011). IL-1β and MMPs produced by perivascular astrocytes participate to BBB disruption and vasogenic edema (Rosenberg et al., 1998; del Zoppo and Hallenbeck, 2000; Amantea et al., 2010). Nevertheless, astrocytes exposed to an ischemic insult may also participate to neuroprotective and reparative responses (Table 1) by extracellular glutamate uptake (Stanimirovic et al., 1997; Rossi et al., 2007), BBB rebuilding (Kinoshita et al., 1990; del Zoppo, 2009) and neurotrophic factors release (Shen et al., 2010; Barreto et al., 2011). Accordingly, impairment of astrocyte function amplifies ischemic neuronal death (Nakase et al., 2003; Ouyang et al., 2007) and ablation of their reactivity and proliferation delays neurovascular remodeling and disrupts scar formation, negatively affecting functional recovery after focal cerebral ischemia in rodents (Nawashiro et al., 2000; Li et al., 2008; Hayakawa et al., 2010). Alternatively, proliferation of astrocytes induced by environmental enrichment or by pharmacological induction of signal transducer and activator of transcription factor (STAT)-3 phosphorylation ameliorates histological and functional outcomes in stroke models (Keiner et al., 2008; Amantea et al., 2011). Thus, reactive astrogliosis may exert a dualistic role on the propagation of ischemic brain damage, depending on the polarization of astrocytes toward specific phenotypes (Zamanian et al., 2012; Rusnakova et al., 2013) and on their interaction with surrounding neurons and microglia (Bezzi et al., 2001; Kang et al., 2012).
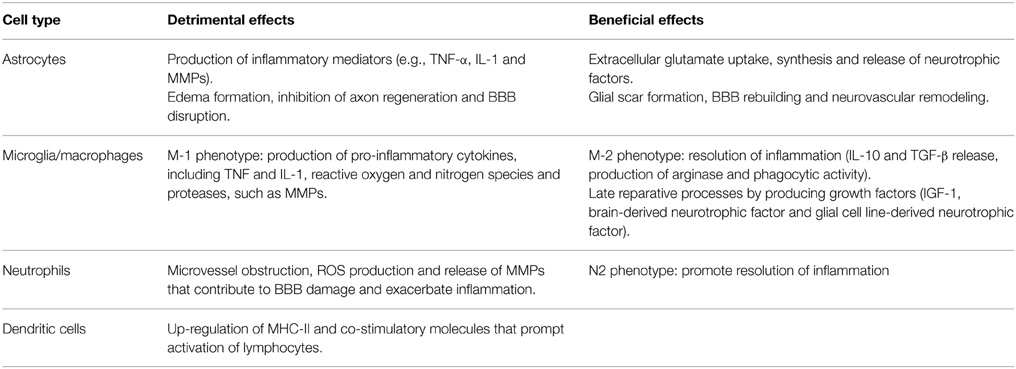
Table 1. Dualistic effects of innate immune cells activated after ischemic brain injury.
After an ischemic insult, microglia, the resident immune cells of the central nervous system, is rapidly activated by ATP released by damaged neurons and other glial cells, acting on P2X7 receptors to prompt production and release of pro-inflammatory mediators (Melani et al., 2006; Dénes et al., 2007). Stimulation of microglia also relies on TLR4 stimulation, fractalkine receptor (CX3CR1) modulation and/or reduced CD200 receptor stimulation evoked by ischemia-induced disturbance of neuron-microglia cross-talk (Lehnardt et al., 2003; Dénes et al., 2008; Dentesano et al., 2012). Moreover, the increased release of specific neurotransmitters, such as glutamate and γ-aminobutyric acid, may elicit an inflammatory or a neuroprotective phenotype in microglia by signaling through Nox (Pocock and Kettenmann, 2007; Mead et al., 2012).
Microglial activation is accompanied by a substantial morphological transformation characterized in the early stages by retraction of cellular processes and enlargement of cell bodies, to ultimately acquire an amoeboid macrophage-like phenotype (Davis et al., 1994; Zhang et al., 1997; Schilling et al., 2003; Jung and Schwartz, 2012). As reported both in ischemia animal models (Zhang et al., 1997; Stoll et al., 1998; Dénes et al., 2007) and in stroke patients (Gerhard et al., 2005; Price et al., 2006), the reactivity and proliferation of microglia reach a peak few days after the insult and may persist for several weeks.
Reactive microglia may enhance inflammation and ischemic tissue injury via increased production and release of cytokines, such as IL-1 and TNF (Barone et al., 1997; Rothwell et al., 1997; Lambertsen et al., 2005; Amantea et al., 2010), reactive oxygen and nitrogen species (Green et al., 2001) and proteases (del Zoppo et al., 2007). These detrimental effects are underscored by the evidence that pharmacological- or microRNA-induced suppression of microglial activity limits ischemic cerebral injury (Hailer, 2008; Fagan et al., 2011; Zhang et al., 2012). However, other studies have demonstrated beneficial effects exerted by microglia in experimental stroke settings (Kitamura et al., 2004; Imai et al., 2007; Lalancette-Hébert et al., 2007). Indeed, by producing IL-10, transforming growth factor (TGF)-β and insulin-like growth factor (IGF)-1, microglia promotes resolution of the inflammatory reaction and reparative mechanisms involved in late tissue recovery (O'Donnell et al., 2002; Lalancette-Hébert et al., 2007; Neumann et al., 2008; Ransohoff and Cardona, 2010).
The reason for these apparently discrepant results can be found in the aptitude of microglia/macrophages to differentiate toward diverse phenotypes, depending on the dynamic evolution of the ischemic damage (Clausen et al., 2008; Perego et al., 2011). Indeed, the classic pro-inflammatory M1 phenotype is activated by interferon (INF)-γ or through TLRs modulation, whereas the alternatively activated M2 phenotype is induced by regulatory factors, including interleukins 4, 10, 13, or TGF-β (Italiani and Boraschi, 2014) (Table 1). Early after an ischemic insult, local microglia assumes the M2 “beneficial” phenotype, to then develop into a pro-inflammatory M1 phenotype prompted by the ischemic neurons (Hu et al., 2012). Thus, in addition to the classical approach aimed at suppressing detrimental M1 functions (i.e., production of TNF-α, IL-1β, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, and IL-6), preservation of the alternatively activated M2 phenotype may represent an innovative strategy for stroke neuroprotection, as demonstrated in mice lacking the class-A scavenger receptor (Xu et al., 2012) or the myeloid-specific mineralcorticoid receptor (Frieler et al., 2011). Given the similarities between local microglia and macrophages, as well as the ability of microglia to develop active phenotypes indistinguishable from circulating macrophages, further insights into their polarization will be given in the following section.
Blood-Borne Cells
Genomic profile studies indicate a critical role of innate immunity in regulating stroke response and recovery. The genes identified in these profiles are involved in immune signaling at different levels, including the cerebral microenvironment, the vasculature and, most notably, the peripheral circulation (Brooks et al., 2014). In the first analysis performed in stroke patients, Moore et al. (2005) detected the expression of genes associated with the regulation of the cerebral microenvironment in peripheral blood mononuclear cells (PMBCs), thus supporting the important finding that peripheral blood is reflective of changes in the brain.
Interestingly, the majority of the genes identified in all the stroke-specific profiles published to date are related to the immune system (Moore et al., 2005; Tang et al., 2006; Barr et al., 2010; Oh et al., 2012). Among these studies, Brooks et al. (2014) recently identified a panel of overlapping genes, significantly expressed within 3 h from stroke onset, including arginase1 (ARG1), carbonic anhydrase 4 (CA4), lymphocyte antigen 96 (LY96), matrix metalloproteinase 9 (MMP9), and S100 calcium binding protein A12 (S100A12). Specifically, four of these genes (ARG1, LY96, MMP9, S100A12) are implicated in the innate immune response, the fifth (CA4) being highly expressed in the BBB (Brooks et al., 2014).
These findings strongly support the theory that the immune response to the ischemic insult engages specialized circulating cells, such as neutrophils, macrophages, dendritic cells and T lymphocytes, that are recruited and migrate to the brain, upon activation triggered by soluble factors released by injured and dying cells (Price et al., 2004; Buck et al., 2008; Felger et al., 2010; Yilmaz and Granger, 2010; Kamel and Iadecola, 2012). Infiltrating leukocytes affect the evolution of tissue damage by releasing a series of mediators including purines, ROS and danger associated molecular patterns (DAMPs), such as high mobility group box (HMGB)-1 protein, heat shock protein 60, β-amyloid, DNA or RNA immune complexes (Amantea et al., 2014a).
Interestingly, the majority of the genes acutely regulated in the blood of stroke patients are expressed in neutrophils and, to a lesser extent, in macrophages (Tang et al., 2006). Accordingly, these are the first cells to infiltrate the ischemic brain, reaching a peak within 24–72 h after the insult (Clark et al., 1994; Garcia et al., 1994; Gelderblom et al., 2009). In patients, higher peripheral leukocyte and neutrophil counts, but not lymphocyte counts, are associated with larger infarct volumes (Buck et al., 2008), and brain accumulation of neutrophils correlates with poor neurological outcome and brain damage severity both in humans (Akopov et al., 1996) and in rodents (Matsuo et al., 1994a,b; Connolly et al., 1996; Atochin et al., 2000). In fact, neutrophils prompt microvessel obstruction/thrombosis (del Zoppo et al., 1991; Ritter et al., 2005), production of ROS and release of MMPs (Justicia et al., 2003; Gidday et al., 2005; Bao Dang et al., 2013); thus, their modulation may represent a useful strategy to ameliorate stroke outcome. This may be achieved by pharmacological approaches, as well as by remote ischemic preconditioning, as documented by the evidence that forearm transient ischemia reduces neutrophil function, including adhesion, exocytosis, phagocytosis and cytokine secretion (Shimizu et al., 2010).
Interestingly, recent findings have highlighted the ability of neutrophils to polarize toward beneficial N2 phenotypes. In the setting of stroke, neutrophil reprogramming can be induced by activation of the nuclear peroxisome proliferator-activated receptor (PPAR)-γ (Cuartero et al., 2013). Thus, despite promoting early neutrophil infiltration to the ischemic core, the PPAR-γ agonist rosiglitazone provides neuroprotection and resolution of inflammation after experimental stroke induced by permanent MCAo by promoting N2-polarization and increased neutrophil clearance (Cuartero et al., 2013).
Likewise neutrophils, hematogenous macrophages infiltrating the ischemic brain (Schilling et al., 2003; Jander et al., 2007), exert a dualistic role on the evolution of tissue damage (Frieler et al., 2011; Hu et al., 2012; Xu et al., 2012). In fact, they possess the ability to switch between the classically activated M1 phenotype and alternatively activated M2 phenotypes (Ballesteros et al., 2014a) (Table 1). The M1 cells initiate and sustain inflammation by releasing neurotoxic factors and ROS that underlie macrophage/microglia-mediated neurotoxicity after stroke; whereas, M2-polarized cells are involved in beneficial responses by clearing debris and by promoting angiogenesis, tissue remodeling and repair (Gliem et al., 2012; Shechter and Schwartz, 2013). Up-regulation of M2 markers observed in the ischemic brain (Frieler et al., 2011; Perego et al., 2011; Hu et al., 2012; Zarruk et al., 2012; Ballesteros et al., 2014a) is due to an increased cerebral infiltration of alternatively activated blood-borne monocytes (Perego et al., 2011), as well as to the ability of local microglia/macrophages to assume an M2 phenotype (Hu et al., 2012). Indeed, local microglia and newly recruited macrophages assume the M2 phenotype at early stages of ischemic stroke but, upon priming by ischemic neurons, gradually transform into the M1 phenotype (Hu et al., 2012). The exact mechanisms that control macrophage polarization in the setting of stroke have not been fully elucidated, as well as it is not clear whether the acquisition of a specific phenotype involves recruitment of circulating precursors or in situ cell re-instruction. Endogenous production of the M2-polarizing cytokine IL-4, triggered by MCAo in mice, has been shown to promote Th2 polarization and, thus, beneficial effects on stroke outcome (Xiong et al., 2011). Further studies have shown that a subpopulation of bone marrow-derived monocytes/macrophages, recruited via CCR2 and acting through TGF-β1, maintains the integrity of the neurovascular unit in murine stroke models (Gliem et al., 2012).
To date, only few studies have assessed the therapeutic benefits of reducing the M1/M2 ratio in stroke setting. Frieler et al. (2011) showed that deficiency of the mineralcorticoid receptor (MR) decreases the expression of M1 markers, while preserving the ischemia-induced expression of M2 markers. The resulting elevation of M2 polarized myeloid cells in the ischemic brain was correlated with a better stroke outcome in MR−/− mice (Frieler et al., 2011). Similarly, deficiency of the fractalkine receptor CX3CR1 has been associated with a protective inflammatory milieu, characterized by the promotion of M2 polarization markers (Fumagalli et al., 2013). PPAR-γ-mediated CD36 up-regulation has been involved in the modulation of microglia phenotype, promoting phagocytosis of apoptotic neutrophils, and thus contributing to the resolution of inflammation after stroke (Ballesteros et al., 2014b). By contrast, elevation of M1/M2 ratio promoted by the class A scavenger receptor expressed in microglia/macrophages has been associated with exacerbation of ischemic brain injury (Xu et al., 2012). At variance with the latter, the selective cannabionoid receptor 2 agonist, JWH-133, provides neuroprotection in the acute phase of ischemic stroke by reducing microglia activation, without affecting M2 polarization (Zarruk et al., 2012).
Although preclinical findings strongly suggest the therapeutic usefulness of M2-polarizing agents, the exact mechanisms modulating M1/M2 ratio need further investigation and their relevance for stroke outcome in human stroke has to be validated.
At later stages after the ischemic insult, a significant elevation of dendritic cells occurs in the injured brain hemisphere, reaching a peak 72 h after the insult (Kostulas et al., 2002; Reichmann et al., 2002; Gelderblom et al., 2009). Both peripheral and brain resident dendritic cells are pivotally involved in bridging innate and adaptive immunity by up-regulating major histocompatibility complex (MHC)-II and co-stimulatory molecules that contribute to the activation of lymphocytes (Gelderblom et al., 2009; Felger et al., 2010). Moreover, resident dendritic cells participate in orchestrating the early local immune response and in the late recruitment of lymphocytes (Felger et al., 2010) following activation by INF-γ (Gottfried-Blackmore et al., 2009). Brain infiltration of T lymphocytes occurs relatively late (i.e., 3–7 days) after ischemia (Jander et al., 1995; Schwab et al., 2001; Gelderblom et al., 2009); nevertheless, these cells contribute to the progression of brain damage (Yilmaz et al., 2006; Hurn et al., 2007; Jin et al., 2010), exerting distinct effects depending on the specific cell subset recruited (Amantea et al., 2014a). Recent evidence demonstrates that the functional sphingosine-1-phosphate receptor agonist FTY720 (fingolimod), minimizes brain damage and functional deficits in experimental stroke (Shichita et al., 2009; Hasegawa et al., 2010; Wei et al., 2011). Interestingly, the putative elevation of the incidence of bacterial pneumonia caused by the inhibition of the adaptive immunity by fingolimod does not seem to be actually relevant for the neuroprotective effects of the drug (Pfeilschifter et al., 2011).
Thus, the ischemic insult is associated to a relevant activation of the innate immune system, involving both local and blood-borne specialized cells that, upon recruitment, modulate the cerebral inflammatory response to stroke and set the stage for the activation of adaptive immunity (Figure 1).
Molecular Mediators of the Immune Response
Receptors
As stated above, activation of P2X7 receptors on microglia prompts the processing and release of pro-inflammatory cytokines (Brough et al., 2002; Melani et al., 2006). Moreover, overactivation of P2X7 receptors is involved in excitotoxic neuronal death (Arbeloa et al., 2012) and participates to ischemia-induced damage to olygodendrocytes and myelin (Domercq et al., 2010).
Activation of TLRs by HMGB1, peroxiredoxin (Prx) proteins and other DAMPS, plays an important role in ischemic brain injury (Fossati and Chiarugi, 2007; Shichita et al., 2012a,b; Pradillo et al., 2014). In particular, TLR2 and TLR4 crucially contribute to the induction of the inflammatory response and to the evolution of brain damage, as documented by the evidence that TLR2- or TLR4-deficiency is associated to reduced ischemic brain damage and to suppression of ischemia-induced expression and release of inflammatory cytokines (Tang et al., 2007; Hyakkoku et al., 2010). Indeed, TLR4 deficient mice display significant suppression of IκB phosphorylation, NFκB activity, pro-inflammatory mediators, including TNF-α and IL-6 (Cao et al., 2007; Hyakkoku et al., 2010) and the enzymes inducible nitric oxide synthase (NOS) and cyclooxygenase (COX)-2 (Caso et al., 2007, 2008).
The relevance of TLR2 and TLR4 has also been demonstrated in ischemic stroke patients, since up-regulation of these receptors is associated with greater inflammatory responses and with poor functional outcome (Brea et al., 2011). The stimulation of macrophages and T cells by TLRs-associated pathways induces strong inflammatory responses (Shichita et al., 2012a). Following cerebral ischemia, the activation of TLR4 by HMGB-1 induces MMP-9 up-regulation in neurons and astrocytes (Qiu et al., 2010) and promotes detrimental effects by macrophages infiltrating the injured brain (Yang et al., 2011). In fact, cerebral microinjection of HMGB-1 increases the transcript levels of pro-inflammatory mediators and sensitizes the tissue to ischemic injury (Faraco et al., 2007). In addition, deficiency of TLR4 in young animals subjected to focal cerebral ischemia, promotes subventricular zone cell proliferation, increasing the number of the transit-amplifying cells (type C cells; prominin-1+/EGFR+/nestin- cells) at 24 and 48 h, of proliferating immature (BrdU+) cells at 7d and of neuroblast cells (type A cells; doublecortin+ cells) at 14d (Moraga et al., 2014). Despite a negative effect on SVZ cell proliferation, TLR4 plays an important role in stroke-induced neurogenesis by promoting neuroblasts migration and increasing the number of new cortical neurons after stroke (Moraga et al., 2014).
Although the exact mechanisms by which TLRs modulate the evolution of ischemic brain injury have not been fully elucidated, pharmacological inhibition of TLR2 and TLR4 and/or blockade of some of their endogenous ligands (i.e., cellular fibronectin or heat shock protein 60), represent promising therapeutic options, effective in reducing the inflammatory response to stroke injury (Brea et al., 2011).
Paradoxically, by reprogramming TLRs signaling, stimulation of TLRs before ischemia leads to suppression of pro-inflammatory responses and to enhanced expression of numerous anti-inflammatory mediators that collectively contribute to neuroprotection (Pradillo et al., 2009; Vartanian et al., 2011). Activation of TLR4 by low doses of LPS reduces synthesis and release of some pro-inflammatory cytokines, and inhibits microglial activation and neutrophil infiltration, thus reducing ischemic brain injury (Rosenzweig et al., 2004; Pradillo et al., 2009). Tolerance to brain ischemia induced by low doses of the major TLR4 ligand, LPS, administered 1–3 days before the insult, has been demonstrated in several experimental stroke models (Tasaki et al., 1997; Hickey et al., 2007; Yu et al., 2010). Recent work has also demonstrated that ischemic preconditioning reduces brain damage from permanent middle cerebral artery occlusion in mice by increasing expression of TLR3 in cortical astrocytes (Pan et al., 2014). Therefore, induction of ischemic tolerance by subdued TLRs activation represents an interesting opportunity to exploit these receptors for stroke therapy.
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin family of cell surface receptors and has been implicated in the development and progression of stroke. The full-length, membrane-bound RAGE isoform (fl-RAGE) is mainly expressed in neurons and in microglia/macrophages. Up-regulation of this receptor has been documented after both permanent and transient focal cerebral ischemia in rodents (Qiu et al., 2008; Zhai et al., 2008; Hassid et al., 2009) and in the ischemic hemisphere of stroke patients (Hassid et al., 2009). More specifically, ischemia-induced modifications of the expression of RAGE and its isoforms in the brain strongly depends on the intensity and on the propagation of the insult; showing a distinct modulation in the core and penumbra regions (Greco et al., 2012, 2014). By activating fl-RAGE on microglia/macrophages, HMGB-1 protein released from dying neurons, contributes to the development of ischemic brain damage (Muhammad et al., 2008; Qiu et al., 2008). Conversely, blockade of fl-RAGE signaling promotes cell survival and reduces stroke infarct volume in animal models (Kim et al., 2006; Liu et al., 2007; Muhammad et al., 2008).
A reduced plasma level of soluble RAGE isoforms (sRAGE) has been reported in rats subjected to either transient or permanent focal brain ischemia (Greco et al., 2012, 2014). This is of significance since sRAGE, generated either by alternative splicing or by proteolysis of the full-length form, effectively bind AGEs, thereby competing with the cell surface fl-RAGE, thus providing a “decoy” function that may counteract the detrimental effects of receptor signaling in neurons (Koyama et al., 2007; Muhammad et al., 2008; Tang et al., 2013). Interestingly, the reduction of sRAGE levels induced by transient MCAo in rats, is minimized by pre-treatment with a neuroprotective dose of the poly(ADP-ribose) polymerase (PARP) inhibitor PJ34 (Greco et al., 2014), suggesting that sRAGE may represent a useful biomarker of stroke severity and of effective neuroprotective treatment. In patients, some studies have reported an association between circulating sRAGE levels and brain infarct volume, stroke severity (Park et al., 2004; Yokota et al., 2009) and inflammatory status (Cui et al., 2013), others have suggested that sRAGE levels at onset may predict cognitive impairment after cerebral ischemia (Qian et al., 2012).
Inflammatory Cytokines
The inflammatory response to ischemic brain injury is orchestrated by a variety of cytokines released by resident brain cells, such as neurons and glia, and by blood-borne immune cells. TNF, IL-1 and IL-6 strongly affect the development of ischemic brain damage in animal models, and their levels are increased in the blood and in the cerebrospinal fluid of stroke patients (Lambertsen et al., 2012), thus attracting considerable interest as putative markers of stroke severity and neurologic outcome (Emsley et al., 2007; Jickling and Sharp, 2011). By contrast, other cytokines, including IL-10 and TGF-β exert immunoregulatory and anti-inflammatory effects, thus promoting reparative processes. In addition, mild systemic inflammation induced by remote preconditioning is neuroprotective in stroke models (Petcu et al., 2008) and several soluble mediators of the innate immune system have been involved in ischemic tolerance produced by diverse preconditioning stimuli (Garcia-Bonilla et al., 2014a).
A significant and rapid up-regulation of TNF-α occurs following focal cerebral ischemia both in animal models and in stroke patients. In fact, expression of this cytokine is elevated in neurons during the first hours after the insult; whereas, at later stages, it is increased in microglia/macrophages and in blood-borne immune cells (Gregersen et al., 2000; Dziewulska and Mossakowski, 2003). Accordingly, recent flow cytometry experiments have demonstrated that the major source of this cytokine in the stroke-lesioned mouse brain is microglia and macrophages (Clausen et al., 2008; Lambertsen et al., 2012).
TNF-α is believed to play a detrimental role in ischemic injury, since its neutralization with specific monoclonal antibodies or binding proteins provides neuroprotection in experimental stroke models (Barone et al., 1997; Nawashiro et al., 1997; Lavine et al., 1998; Lambertsen et al., 2012). Nevertheless, studies from transgenic animals demonstrate that the cytokine may also exert beneficial effects through the activation of p55 TNF receptor (TNF-RI) (Bruce et al., 1996; Gary et al., 1998; Taoufik et al., 2007; Lambertsen et al., 2009). In rodents, expression of TNF-RI is elevated in neurons and in non-neuronal cells few hours after MCAo, whereas up-regulation of TNF-RII occurs from 24 h after injury in resident microglia and infiltrating macrophages (Botchkina et al., 1997; Dziewulska and Mossakowski, 2003; Yin et al., 2004; Pradillo et al., 2005; Lambertsen et al., 2007). Despite its predominant inflammatory role, TNF-RII has also been implicated in neuroprotection (Marchetti et al., 2004), and this further complicates the interpretation of the pleiotropic effects of TNF in ischemic neuronal damage (Hallenbeck, 2002; McCoy and Tansey, 2008).
Moreover, the TNF pathway has been involved in ischemic, hypoxic, endotoxic and exercise-induced preconditioning (Garcia-Bonilla et al., 2014a). In fact, LPS-preconditioned TNF-α null mice are not protected from ischemic brain injury (Rosenzweig et al., 2007) and administration of a neutralizing TNF binding protein nullifies the beneficial effect of LPS pre-treatment in spontaneously hypertensive rats subjected to permanent MCAo (Tasaki et al., 1997). Paradoxically, TNF-α has a dualistic effect in stroke, since it's up-regulation has been shown to underlie LPS-induced tolerance in mice subjected to focal cerebral ischemia, whereas suppression of TNF-α signaling during ischemia confers neuroprotection after LPS preconditioning (Rosenzweig et al., 2007). Up-regulation of TNF-α converting enzyme (TACE) and increased serum levels of TNF-α have also been involved in preconditioning induced by prolonged and intermittent normobaric hyperoxia in rats subjected to MCAo (Bigdeli and Khoshbaten, 2008; Bigdeli et al., 2008). In rats, the increased expression of TNF-α lasting for 2–3 weeks of physical activity underlies amelioration of downstream inflammatory events, reduced BBB disruption and the consequent ischemic neuroprotection induced by exercise preconditioning (Ding et al., 2005, 2006; Guo et al., 2008b).
IL-1 is a proinflammatory cytokine that plays a pivotal role in the neurodegenerative processes triggered by the ischemic insult (Olsson et al., 2012; Dénes et al., 2013). Cerebral levels of both IL-1α and IL-1β are elevated within hours of reperfusion after focal ischemia (Hara et al., 1997; Amantea et al., 2007, 2010; Luheshi et al., 2011). IL-1α is mainly induced in microglia, whereas IL-1β can be released by all the elements of the neurovascular unit, including neurons, astrocytes, microglia/macrophages and endothelial cells, being its cellular source strongly dependent on the spatio-temporal evolution of the damage (Amantea et al., 2010; Luheshi et al., 2011; Giles et al., 2015). In astrocytes and microglia, ischemia-induced production of IL-1β involves activation of TLR4 (Simi et al., 2007) and p38 mitogen-activated protein kinase (MAPK) (Walton et al., 1998; Irving et al., 2000). Interestingly, under ischemic conditions, caspase-1-independent pathways seem to contribute to IL-1β maturation. Accordingly, it has been recently demonstrated that early elevation of IL-1β in the ischemic cortex of rats subjected to transient MCAo is not associated with caspase-1 activation, whereas production of the mature cytokine is strongly dependent on gelatinases, i.e., MMP2 and MMP-9 activity (Amantea et al., 2007, 2014b).
It is intriguing to observe that IL-1 is not directly toxic to healthy neurons, whereas it may become harmful via the modulation of other elements of the neurovascular unit, such as astrocytes and the endothelium. IL-1 stimulates astrogliosis (Herx and Yong, 2001) and prompts the release of cytokines, chemokines (Andre et al., 2005) and the activation of MMP-9 in astrocytes (Thornton et al., 2008). Furthermore, this cytokine induces endothelial expression of adhesion molecules, including ICAM-1 and vascular cell adhesion molecule (VCAM)-1, that together with the local stimulation of the release of chemokines, promotes neutrophil adhesion and infiltration in the injured hemisphere (Thornton et al., 2011; Allen et al., 2012).
Despite their established detrimental roles, both IL-1α and IL-1β have been shown to underlie tolerance to global ischemia induced by common carotid artery occlusion in gerbil (Ohtsuki et al., 1996). Moreover, ischemic preconditioning produced by bilateral common carotid artery occlusion protects mice against subsequent MCAo injury by differentially regulating cortical IL-1β and IL-1 receptor antagonist (IL-1ra) expression in order to promote a shift toward an anti-inflammatory state that contributes to neuroprotection (Shin et al., 2009). The regulation of the expression and of the effects of IL-1 has also been implicated in LPS preconditioning via the TLR4 signaling pathway (Gong et al., 2014).
Other cytokines involved in the evolution of brain infarction and contributing to aggravate neurological functions are IL-17 and IL-23 (Shichita et al., 2009; Konoeda et al., 2010; Ma et al., 2013; Swardfager et al., 2013). IL-17A secreted by γδ T cells promotes neutrophil recruitment and its blockade with specific antibodies exerts neuroprotection (Gelderblom et al., 2012). Despite the presence of IL-17A-positive lymphocytes in autoptic brain of stroke patients, IL-17 blood level does not seem to be a good predictor of stroke outcome as compared to other cytokines such as IL-6 (Zeng et al., 2013). In fact, plasma and cerebrospinal fluid levels of IL-6 correlate with stroke severity and poor clinical outcome in patients (Smith et al., 2004; Waje-Andreassen et al., 2005; Whiteley et al., 2009) and reduced blood concentrations of this cytokine have been correlated to the improved outcome induced by treatment with IL-1ra (Emsley et al., 2005). By contrast, in stroke animal models, IL-6 appears to play a neuroprotective role (Matsuda et al., 1996; Loddick et al., 1998; Herrmann et al., 2003), suggesting that its rapid and persistent elevation in the ischemic brain (Suzuki et al., 1999; Ali et al., 2000) may represent a compensatory mechanism to counteract the damaging effects of the insult. In fact, by activating its receptor and the downstream phosphorylation of STAT-3, IL-6 may induce neuroprotection (Yamashita et al., 2005; Jung et al., 2011). Moreover, IL-6 enhances the effectiveness of cell transplantation therapy in ischemic stroke, by reprogramming neural stem cells to tolerate oxidative stress and to induce angiogenesis through STAT-3 activation (Perini et al., 2001).
Anti-Inflammatory Cytokines
While plasma concentrations of detrimental cytokines are elevated, levels of IL-10, associated with better outcome, are decreased in patients (Perini et al., 2001; Vila et al., 2003; Basic Kes et al., 2008). Results from animal models demonstrate that IL-10 represents a major downregulator of the detrimental effects of proinflammatory mediators during stroke and modulates neuronal vulnerability to excitotoxic ischemic damage (Spera et al., 1998; Grilli et al., 2000; Frenkel et al., 2005).
Another cytokine playing beneficial effects in cerebral ischemia is TGF-β (Gliem et al., 2012). Levels of this cytokine are elevated in the blood of patients 1 day after ischemic stroke (Yan et al., 2012) and in activated astrocytes and microglia/macrophages of the ischemic brain for at least 1 week in animal models (Lehrmann et al., 1998; Yamashita et al., 1999; Doyle et al., 2010). In rats, administration of a TGF-β antagonist aggravates brain damage caused by focal cerebral ischemia (Ruocco et al., 1999); whereas, in mice, intranasal delivery of this cytokine after stroke reduces infarct volume and increases neurogenesis in the subventricular zone (Ma et al., 2008). The beneficial effects of TGF-β involve both anti-inflammatory effects, including inhibition of brain elevation of MCP-1 and MIP-1α (Pang et al., 2001), but also induction of glial scar formation (Logan et al., 1994) and anti-apoptotic effects (Zhu et al., 2002).
In mice, preconditioning with low dose LPS is associated with upregulation of anti-inflammatory cytokines, including TGF-β in brain and IL-10 in blood (Vartanian et al., 2011).
Chemokines
In patients with acute ischemic stroke, serum levels of stromal cell-derived factor (SDF)-1α have been correlated with favorable long-term outcome (Kim et al., 2012) and this chemokine has been suggested to be a predictor of future stroke (Schutt et al., 2012). SDF-1α promotes bone marrow-derived cell targeting to the ischemic brain and improves local cerebral blood flow (Cui et al., 2007; Shyu et al., 2008), thus representing a promising target to implement stem cell therapy in patients. In fact, in the injured hemisphere, it activates CXCR4 on neural progenitor cells guiding their specific migration to the site of damage (Robin et al., 2006). Moreover, SDF-1α participates to the regulation of post-ischemic inflammation and it is involved in neurovascular repair (Wang et al., 2012). Despite these documented beneficial effects, some detrimental roles have also been suggested to be mediated by this chemokine, since pharmacological blockade of CXCR4 is neuroprotective in stroke by reducing BBB damage and inflammatory processes (Huang et al., 2013). Soluble fractalkine (CX3CL1) released by neurons upon an ischemic insult controls leukocyte trafficking and participates to the activation and chemoattraction of microglia to the injured brain via the activation of CX3CR1 receptors (Tarozzo et al., 2002; Dénes et al., 2008; Zhu et al., 2009). The detrimental effects of this chemokine are mediated by IL-1β and TNF-α in stroke mouse models (Soriano et al., 2002; Dénes et al., 2008). By contrast, in ischemic stroke patients, higher plasma concentrations of fractalkine are associated with better outcome and with low levels of systemic markers of inflammation (Donohue et al., 2012). In fact, by inhibiting caspase-3 and by activating adenosine receptors, exogenous administration of fractalkine is neuroprotective in wild-type rodents undergone permanent ischemia (Cipriani et al., 2011; Rosito et al., 2014). The apparent discrepancies between this latter finding and those produced in transgenic animals may be explained by the altered microglia responsiveness to fractalkine in the absence of constitutive fractalkine-CX3CR1 signaling (Cipriani et al., 2011).
Among the chemokines implicated in cerebral ischemia, MCP-1 (also known as CCL2) and MIP-1α have been shown to play an important role in promoting tissue damage via recruitment of inflammatory cells (Wang et al., 1995; Che et al., 2001; Takami et al., 2001; Minami and Satoh, 2003). The mRNA levels of both chemokines are elevated in the ischemic brain of rodents and MCP-1 levels are also increased in the cerebrospinal fluid of stroke patients (Losy and Zaremba, 2001). Mice lacking MCP-1 or its receptor, CCR2, display reduced infarct volume along with impaired leukocyte recruitment and reduced expression of inflammatory mediators in the injured brain (Hughes et al., 2002; Dimitrijevic et al., 2007; Strecker et al., 2011; Schuette-Nuetgen et al., 2012). Conversely, overexpression of MCP-1 prompts exacerbation of brain injury and increased cerebral recruitment of inflammatory cells (Chen et al., 2003). Moreover, MCP-1, as well as SDF-1α, promotes migration of newly formed neuroblasts from neurogenic regions to ischemic damaged areas (Robin et al., 2006; Yan et al., 2007).
Up-regulation of MCP-1 has been shown to underlie hypoxic preconditioning-induced stroke tolerance in mice (Stowe et al., 2012; Wacker et al., 2012); whereas, activation of CCR2 is involved in the neuroprotective effects exerted by both ischemic preconditioning and post-conditioning in mice subjected to global cerebral ischemia (Rehni and Singh, 2012).
Monocyte chemotactic protein-induced protein 1 (MCPIP1) deficiency exacerbates ischemic brain damage by upregulation of proinflammatory cytokines and this Zn finger-containing immunoregulatory protein also participates in LPS- and electroacupuncture-induced ischemic stroke tolerance (Liang et al., 2001; Jin et al., 2013).
Enzymes: MMPs, COX, and NOS
MMPs play a crucial role in the evolution of the inflammatory response to ischemic brain injury (Cunningham et al., 2005). Stroke-induced damage to the BBB and hemorrhagic transformation are both induced by the activation of the gelatinases, MMP-2 and MMP-9, as demonstrated in animal models (Romanic et al., 1998; Rosenberg et al., 1998; Asahi et al., 2000) and in patients (Horstmann et al., 2003; Rosell et al., 2006, 2008). RNA-expression levels of MMP-9 in circulating monocytes have been correlated with the brain infarct lesion in stroke patients (Ulrich et al., 2013); while, serum levels of this enzyme have been associated with clinical diffusion mismatch (Rodríguez-Yáñez et al., 2011).
Cerebral expression and activity of gelatinases increase very early after an ischemic insult, with a specific cellular expression pattern dependent on the spatio-temporal evolution of the damage (Rosenberg et al., 1998; Planas et al., 2001; Yang et al., 2007; Amantea et al., 2008). Pharmacological inhibition of gelatinases, as well as gene deletion of MMP-9, reduces infarct volume caused by focal cerebral ischemia in rodents (Romanic et al., 1998; Asahi et al., 2000; Gasche et al., 2001; Amantea et al., 2007, 2014b). The mechanisms by which gelatinases contribute to ischemic brain damage include disruption of BBB integrity, hemorrhagic transformation and white matter myelin degradation (Cunningham et al., 2005). Moreover, MMPs and their endogenous inhibitors (TIMPs) regulate neuronal cell death through modulation of excitotoxicity (Jourquin et al., 2003), DNA repairing enzymes (Hill et al., 2012), anoikis (Gu et al., 2002), calpain activity (Copin et al., 2005) and production of neurotoxic products (Gu et al., 2002; Zhang et al., 2003), including pro-inflammatory cytokines (Amantea et al., 2007, 2014b). Moreover, the degradation of tight junction proteins claudin-5 and occludin by MMPs prompts hemorrhagic transformation, suggesting that these enzymes may represent a promising target for reducing the hemorrhagic complications associated with thrombolytic therapy (Yang and Rosenberg, 2011; Liu et al., 2012).
The up-regulation of the extracellular MMP inducer (EMMPRIN) occurring in peri-ischemic regions 2–7 days after focal ischemia in mice is coincident with the delayed increase of MMP-9, suggesting its involvement in neurovascular remodeling (Zhu et al., 2008). In fact, the late activation of MMP-9 promotes vascular endothelial growth factor (VEGF) signaling, contributing to neuronal survival and new vessels formation (Zhao et al., 2006). Thus, MMPs play a dual role in stroke injury, including early detrimental effects and beneficial roles at later stages after the insult. Moreover, MMPs participate to the protective response evoked by several preconditioning stimuli. In fact, tolerance produced by ischemic preconditioning or pre-ischemic exercise has been shown to be associated with downregulation of MMP-9 and subsequent amelioration of brain oedema and BBB disruption in rats undergone focal cerebral ischemia (Zhang et al., 2006; Davis et al., 2007; Guo et al., 2008a). Similarly, the neuroprotective action of TNF-α induced by pre-ischemic physical exercise has been demonstrated to occur via reduced MMP-9 activity and amelioration of BBB dysfunction in rats subjected to transient MCAo, through the involvement of extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation (Guo et al., 2008b; Chaudhry et al., 2010). Reduced activity of gelatinases was also associated with the neuroprotective effects exerted by hyperbaric oxygen preconditioning in hyperglycemic rats subjected to MCAo (Soejima et al., 2013).
After an ischemic insult, the expression of COX-2 is elevated in neurons, vascular cells and neutrophils, as demonstrated both in stroke patients and in animal models (Nogawa et al., 1997; Iadecola et al., 1999, 2001; Chakraborti et al., 2010). This enzyme contributes to post-ischemic inflammation through the production of toxic prostanoids and superoxide, and its deficiency or pharmacological inhibition leads to reduce BBB damage and to lower cerebral infiltration of leukocytes in rodent models of ischemic stroke (Iadecola et al., 2001; Candelario-Jalil et al., 2007). COX-2-derived prostaglandin E2 may also contribute to ischemic cell damage by disrupting Ca2+ homeostasis in neurons through activation of EP1 receptors (Kawano et al., 2006). Interestingly, an association between functional outcome and specific COX-2 variants has recently been demonstrated in ischemic stroke patients (Maguire et al., 2011).
COX-2 has been implicated in hyperbaric oxygen preconditioning, since pharmacological inhibition of this enzyme abolishes the beneficial effects of the conditioning stimulus in a rat model of transient global cerebral ischemia (Cheng et al., 2011). Similarly, COX-2 induction participates to ischemic tolerance induced by cortical spreading depression (Horiguchi et al., 2006) or by a brief ischemic episode in rats (Choi et al., 2006; Pradillo et al., 2009), likely via the stimulation of the PGE2/PI3K/Akt pathway (Park et al., 2008). The elevated expression of COX-2 induced by ischemic preconditioning has been suggested to occur via a cascade involving epsilon protein kinase C and ERK1/2 activation, as well as NFkB nuclear translocation, as demonstrated in vitro (Kim et al., 2007, 2010).
While elevated expression of inducible NOS is associated with ischemic (Cho et al., 2005), TLR4-mediated (Pradillo et al., 2009) and anesthetic preconditioning (Kapinya et al., 2002), induction of this enzyme has also been implicated in the release of toxic amounts of NO by infiltrating neutrophils, microglia/macrophages and endothelial cells (Nakashima et al., 1995; Iadecola et al., 1996; Forster et al., 1999; Garcia-Bonilla et al., 2014b). NO released by endothelial cells early after the ischemic insult plays a beneficial role by inducing vasodilatation; whereas, at later stages, overactivation of neuronal NOS and, more importantly, de novo expression of inducible NOS contribute to ischemic tissue damage (Iadecola et al., 1996, 1997; Moro et al., 2004; Murphy and Gibson, 2007). In fact, excessive production of NO by inducible NOS is cytotoxic by promoting NFκB activation, by inhibiting ATP-producing enzymes, by producing peroxynitrite and by stimulating other pro-inflammatory enzymes such as COX-2 (Nogawa et al., 1998; Greco et al., 2011). Moreover, NO contributes to ischemic cell death via S-nitrosylation and, thereby, activation of glutamate receptor (GluR)-6 signaling (Yu et al., 2008) and MMP-9 (Gu et al., 2002).
NO plays a crucial role in cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats (Horiguchi et al., 2005). Moreover, recent findings have demonstrated the involvement of endothelial NOS in remote ischemic preconditioning (Peng et al., 2012), while neuronal NOS has been involved in the neuroprotection exerted by remote post-conditioning (Pignataro et al., 2013).
Concluding Remarks
Although ischemic stroke is a major cause of mortality and long-term disability worldwide (Go et al., 2014), current therapeutic approaches for its acute treatment only rely on blood flow restoration by thrombus lysis or removal (Mangiafico and Consoli, 2014; Berkhemer et al., 2015; Hacke, 2015). The therapeutic window, intended as the temporal range during which the endovascular treatment may reach the target of a useful recanalization, is conventionally set at a threshold of 6 h; therefore, only less that 10% of patients may actually benefit from these procedures. Thus, the identification of novel targets that allow widening the time-window for pharmacological intervention, as well as the possibility of limiting the development of ischemic brain damage by promoting innate beneficial responses, is a urgent challenge. To date, the clinical translation of immunomodulatory drugs has been hampered by the fact that most strategies tested in humans were purely based on anti-inflammatory approaches (Table 2), disregarding the beneficial roles of some elements of the immune reaction to stroke injury (Amantea et al., 2014a). In this context, targeting immune responses that evolve during hours or days after the ischemic insult, by selectively promoting their beneficial components, represents a promising avenue for the development of more effective and safe stroke therapeutics.
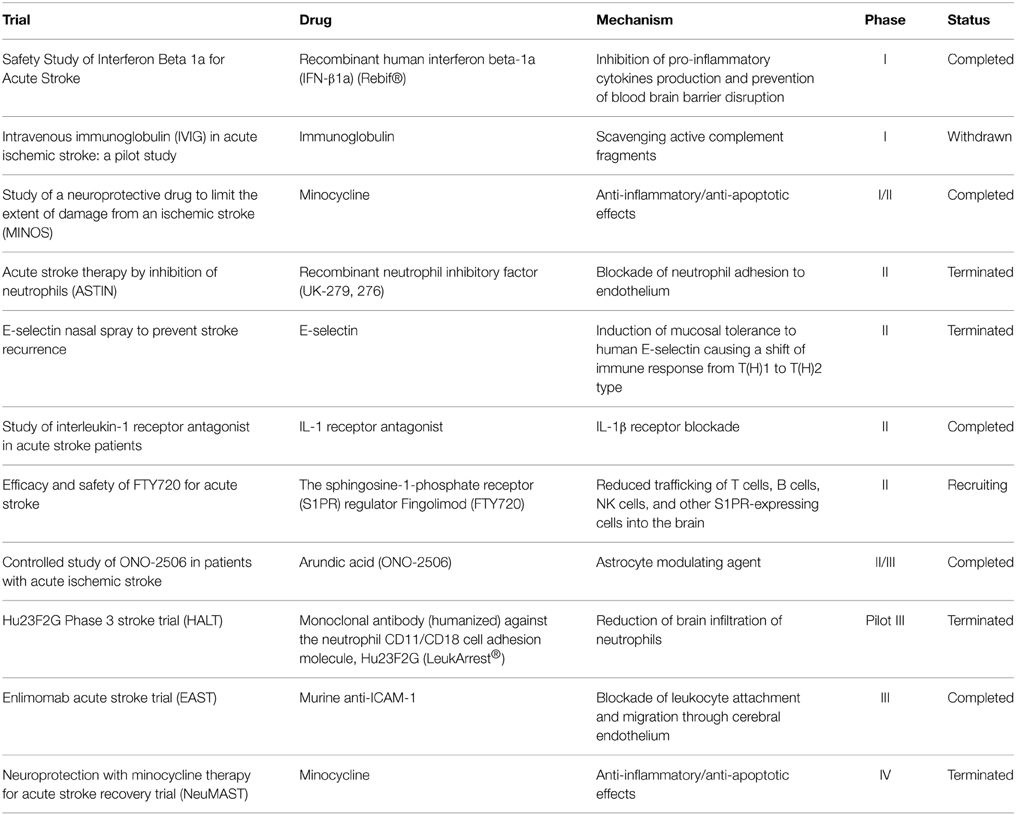
Table 2. Acute ischemic stroke trials for the clinical validation of anti-inflammatory and immunomodulatory drugs.
Author Contributions
All the authors participated in the collection, review, and analysis of the relevant literature, as well as to drafting and revising of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partially funded by the Italian Ministry of University and Research (PRIN prot. 20089 BARSR_004 to GB), the University of Calabria (ex quota 60%) and the Italian Ministry of Health (Ricerca Corrente 2013 to GM and CT). This work was also partially supported by grants from Spanish Ministry of Economy And Competitiveness SAF2012-33216 (MM), CSD2010-00045 (MM) and SAF2014-52225 (IL), from Fondo Europeo de Desarrollo Regional (FEDER) RETICS RD12/0014/0003 (IL), and from Regional Madrid Government S2010/BMD-2336 (MM) and S2010/BMD-2349 (IL).
References
Akopov, S. E., Simonian, N. A., and Grigorian, G. S. (1996). Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke 27, 1739–1743. doi: 10.1161/01.STR.27.10.1739
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ali, C., Nicole, O., Docagne, F., Lesne, S., MacKenzie, E. T., Nouvelot, A., et al. (2000). Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J. Cereb. Blood Flow Metab. 20, 956–966. doi: 10.1097/00004647-200006000-00008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Allen, C., Thornton, P., Denes, A., McColl, B. W., Pierozynski, A., Monestier, M., et al. (2012). Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 189, 381–392. doi: 10.4049/jimmunol.1200409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amantea, D., Tassorelli, C., Russo, R., Petrelli, F., Morrone, L. A., Bagetta, G., et al. (2011). Neuroprotection by leptin in a rat model of permanent cerebral ischemia: effects on STAT3 phosphorylation in discrete cells of the brain. Cell Death Dis. 8, 2:e238. doi: 10.1038/cddis.2011.125
Amantea, D., Tassorelli, C., Petrelli, F., Certo, M., Bezzi, P., Micieli, G., et al. (2014a). Understanding the multifaceted role of inflammatory mediators in ischemic stroke. Curr. Med. Chem. 21, 2098–2117. doi: 10.2174/0929867321666131227162634
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amantea, D., Certo, M., Russo, R., Bagetta, G., Corasaniti, M. T., and Tassorelli, C. (2014b). Early reperfusion injury is associated to MMP2 and IL-1β elevation in cortical neurons of rats subjected to middle cerebral artery occlusion. Neuroscience 277, 755–763. doi: 10.1016/j.neuroscience.2014.07.064
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amantea, D., Corasaniti, M. T., Mercuri, N. B., Bernardi, G., and Bagetta, G. (2008). Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience 152, 8–17. doi: 10.1016/j.neuroscience.2007.12.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amantea, D., Bagetta, G., Tassorelli, C., Mercuri, N. B., and Corasaniti, M. T. (2010). Identification of distinct cellular pools of interleukin-1β during the evolution of the neuroinflammatory response induced by transient middle cerebral artery occlusion in the brain of rat. Brain Res. 1313, 259–269 doi: 10.1016/j.brainres.2009.12.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amantea, D., Russo, R., Gliozzi, M., Fratto, V., Berliocchi, L., Bagetta, G., et al. (2007). Early upregulation of matrix metalloproteinases following reperfusion triggers neuroinflammatory mediators in brain ischemia in rat. Int. Rev. Neurobiol. 82, 149–169. doi: 10.1016/S0074-7742(07)82008-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andre, R., Pinteaux, E., Kimber, I., and Rothwell, N. J. (2005). Differential actions of IL-1 alpha and IL-1 beta in glial cells share common IL-1 signalling pathways. Neuroreport 16, 153–157. doi: 10.1097/00001756-200502080-00017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arbeloa, J., Pérez-Samartín, A., Gottlieb, M., and Matute, C. (2012). P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol. Dis. 45, 954–961. doi: 10.1016/j.nbd.2011.12.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Asahi, M., Asahi, K., Jung, J. C., del Zoppo, G. J., Fini, M. E., and Lo, E. H. (2000). Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockdown and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 20, 1681–1689. doi: 10.1097/00004647-200012000-00007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atkinson, C., Zhu, H., Qiao, F., Varela, J. C., Yu, J., Song, H., et al. (2006). Complement-dependent P-selectin expression and injury following ischemic stroke. J. Immunol. 177, 7266–7274. doi: 10.4049/jimmunol.177.10.7266
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atochin, D. N., Fisher, D., Demchenko, I. T., and Thom, S. R. (2000). Neutrophil sequestration and the effect of hyperbaric oxygen in a rat model of temporary middle cerebral artery occlusion. Undersea Hyperb. Med. 27, 185–190.
Ballesteros, I., Cuartero, M. I., de la Parra, J., Pérez-Ruiz, A., Hurtado, O., Lizasoain, I., et al. (2014a). “Polarization of macrophages/microglia toward an M2 phenotype as a therapeutic strategy for stroke treatment,” in Rational Basis for Clinical Translation in Stroke Therapy, eds G. Micieli and D. Amantea (Boca Raton, FL: CRC press), 381–392.
Ballesteros, I., Cuartero, M. I., Pradillo, J. M., de la Parra, J., Pérez-Ruiz, A., Corbí, A., et al. (2014b). Rosiglitazone-induced CD36 up-regulation resolves inflammation by PPARgamma and 5-LO-dependent pathways. J. Leukoc. Biol. 95, 587–598 doi: 10.1189/jlb.0613326
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bao Dang, Q., Lapergue, B., Tran-Dinh, A., Diallo, D., Moreno, J. A., Mazighi, M., et al. (2013). High-density lipoproteins limit neutrophil-induced damage to the blood-brain barrier in vitro. J. Cereb. Blood Flow Metab. 33, 575–582. doi: 10.1038/jcbfm.2012.206
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barone, F. C., Arvin, B., White, R. F., Miller, A., Webb, C. L., Willette, R. N., et al. (1997). Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 28, 1233–1244. doi: 10.1161/01.STR.28.6.1233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barr, T. L., Conley, Y., Ding, J., Dillman, A., Warach, S., Singleton, A., et al. (2010). Genomic biomarkers and cellular pathways of ischemic stroke by RNA gene expression profiling. Neurology 75, 1009–1014. doi: 10.1212/WNL.0b013e3181f2b37f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barreto, G., Whilte, R. E., Ouyang, Y., Xu, L., and Giffard, R. G. (2011). Astrocytes: targets for neuroprotection in stroke. Cent. Nerv. Syst. Agents Med. Chem. 11, 164–173. doi: 10.2174/187152411796011303
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basic Kes, V., Simundic, A. M., Nikolac, N., Topic, E., and Demarin, V. (2008). Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clin. Biochem. 41, 1330–1334. doi: 10.1016/j.clinbiochem.2008.08.080
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berkhemer, O. A., Fransen, P. S., Beumer, D., van den Berg, L. A., Lingsma, H. F., Yoo, A. J., et al. (2015). A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med. 372, 11–20. doi: 10.1056/NEJMoa1411587
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., et al. (2001). CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710. doi: 10.1038/89490
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bigdeli, M. R., Hajizadeh, S., Froozandeh, M., Heidarianpour, A., Rasoulian, B., Asgari, A. R., et al. (2008). Normobaric hyperoxia induces ischemic tolerance and upregulation of glutamate transporters in the rat brain and serum TNF-alpha level. Exp. Neurol. 212, 298–306. doi: 10.1016/j.expneurol.2008.03.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bigdeli, M. R., and Khoshbaten, A. (2008). In vivo preconditioning with normobaric hyperoxia induces ischemic tolerance partly by triggering tumor necrosis factor-alpha converting enzyme/tumor necrosis factor-alpha/nuclear factor-kappaB. Neuroscience 153, 671–678. doi: 10.1016/j.neuroscience.2008.02.064
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Botchkina, G. I., Meistrell, M. E. 3rd. Botchkina, I. L., and Tracey, K. J. (1997). Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol. Med. 3, 765–781.
Brea, D., Blanco, M., Ramos-Cabrer, P., Moldes, O., Arias, S., Pérez-Mato, M., et al. (2011). Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J. Cereb. Blood Flow Metab. 31, 1424–1431. doi: 10.1038/jcbfm.2010.231
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brooks, S. D., Van Gilder, R., Frisbee, J. C., and Barr, T. L. (2014). “Genomics for the advancement of clinical translation in stroke,” in Rational Basis for Clinical Translation in Stroke Therapy, eds G. Micieli and D. Amantea (Boca Raton, FL: CRC press), 123–136.
Brough, D., Le Feuvre, R. A., Iwakura, Y., and Rothwell, N. J. (2002). Purinergic (P2X7) receptor activation of microglia induces cell death via an interleukin-1-independent mechanism. Mol. Cell. Neurosci. 19, 272–280. doi: 10.1006/mcne.2001.1054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bruce, A. J., Boling, W., Kindy, M. S., Peschon, J., Kraemer, P. J., Carpenter, M. K., et al. (1996). Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 2, 788–794. doi: 10.1038/nm0796-788
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buck, B. H., Liebeskind, D. S., Saver, J. L., Bang, O. Y., Yun, S. W., Starkman, S., et al. (2008). Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. doi: 10.1161/STROKEAHA.107.490128
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Candelario-Jalil, E., González-Falcón, A., García-Cabrera, M., León, O. S., and Fiebich, B. L. (2007). Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J. Neurochem. 100, 1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cao, C. X., Yang, Q. W., Lv, F. L., Cui, J., Fu, H. B., and Wang, J. Z. (2007). Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem. Biophys. Res. Comm. 353, 509–514. doi: 10.1016/j.bbrc.2006.12.057
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caso, J. R., Pradillo, J. M., Hurtado, O., Lorenzo, P., Moro, M. A., and Lizasoain, I. (2007). Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 115, 1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caso, J. R., Pradillo, J. M., Hurtado, O., Leza, J. C., Moro, M. A., and Lizasoain, I. (2008). Toll-like receptor4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke 39, 1314–1320. doi: 10.1161/STROKEAHA.107.498212
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chakraborti, A. K., Garg, S. K., Kumar, R., Motiwala, H. F., and Jadhavar, P. S. (2010). Progress in COX-2 inhibitors: a journey so far. Curr. Med. Chem. 17, 1563–1593. doi: 10.2174/092986710790979980
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, L. T., Yuen, C. M., Liou, C. W., Lu, C. H., Chang, W. N., Youssef, A. A., et al. (2010). Link between interleukin-10 level and outcome after ischemic stroke. Neuroimmunomodulation 17, 223–228. doi: 10.1159/000290038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chaudhry, K., Rogers, R., Guo, M., Lai, Q., Goel, G., Liebelt, B., et al. (2010). Matrix metalloproteinase-9 (MMP-9) expression and extracellular signal-regulated kinase 1 and 2 (ERK1/2) activation in exercise-reduced neuronal apoptosis after stroke. Neurosci. Lett. 474, 109–114. doi: 10.1016/j.neulet.2010.03.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Che, X., Ye, W., Panga, L., Wu, D. C., and Yang, G. Y. (2001). Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 902, 171–177. doi: 10.1016/S0006-8993(01)02328-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Hallenbeck, J. M., Ruetzler, C., Bol, D., Thomas, K., Berman, N. E., et al. (2003). Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J. Cereb. Blood Flow Metab. 23, 748–755. doi: 10.1097/01.WCB.0000071885.63724.20
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cheng, O., Ostrowski, R. P., Wu, B., Liu, W., Chen, C., and Zhang, J. H. (2011). Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral ischemia. Stroke 42, 484–490. doi: 10.1161/STROKEAHA.110.604421
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cho, S., Park, E. M., Zhou, P., Frys, K., Ross, M. E., and Iadecola, C. (2005). Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J. Cereb. Blood FlowMetab. 25, 493–501. doi: 10.1038/sj.jcbfm.9600058
Choi, J. S., Kim, H. Y., Chun, M. H., Chung, J. W., and Lee, M. Y. (2006). Differential regulation of cyclooxygenase-2 in the rat hippocampus after cerebral ischemia and ischemic tolerance. Neurosci. Lett. 393, 231–236. doi: 10.1016/j.neulet.2005.09.074
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E., et al. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clark, R. K., Lee, E. V., White, R. F., Jonak, Z. L., Feuerstein, G. Z., and Barone, F. C. (1994). Reperfusion following focal stroke hastens inflammation and resolution of ischemic injured tissue. Brain Res. Bull. 35, 387–392. doi: 10.1016/0361-9230(94)90119-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clausen, B. H., Lambertsen, K. L., Babcock, A. A., Holm, T. H., Dagnaes-Hansen, F., and Finsen, B. (2008). Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke mice. J. Neuroinflamm. 5:46. doi: 10.1186/1742-2094-5-46
Connolly, E. S. Jr. Winfree, C. J., Springer, T. A., Naka, Y., Liao, H., Yan, S. D., et al. (1996). Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Invest. 97, 209–216. doi: 10.1172/JCI118392
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Copin, J. C., Goodyear, M. C., Gidday, J. M., Shah, A. R., Gascon, E., Dayer, A., et al. (2005). Role of matrix metalloproteinases in apoptosis after transient focal cerebral ischemia in rats and mice. Eur. J. Neurosci. 22, 1597–1608. doi: 10.1111/j.1460-9568.2005.04367.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cuartero, M. I., Ballesteros, I., Moraga, A., Nombela, F., Vivancos, J., Hamilton, J. A., et al. (2013). N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke 44, 3498–3508. doi: 10.1161/STROKEAHA.113.002470
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cui, X. P., Chen, H. J., Hou, X., Wang, S. S., Jayaram, S., and Zheng, Z. C. (2013). Polymorphism of the RAGE affects the serum inflammatory levels and risk of ischemic stroke in a Chinese population. Cell Physiol. Biochem. 32, 986–996. doi: 10.1159/000354494
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cui, X., Chen, J., Zacharek, A., Li, Y., Roberts, C., Kapke, A., et al. (2007). Nitric oxide donor upregulation of stromal cell-derived factor-1/chemokine (CXC motif) receptor 4 enhances bone marrow stromal cell migration into ischemic brain after stroke. Stem Cells 25, 2777–2785. doi: 10.1634/stemcells.2007-0169
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cunningham, L. A., Wetzel, M., and Rosenberg, G. A. (2005). Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 50, 329–339. doi: 10.1002/glia.20169
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, E. J., Foster, T. D., and Thomas, W. E. (1994). Cellular forms and functions of brain microglia. Brain Res. Bull. 34, 73–78. doi: 10.1016/0361-9230(94)90189-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, W., Mahale, S., Carranza, A., Cox, B., Hayes, K., Jimenez, D., et al. (2007). Exercise pre-conditioning ameliorates blood-brain barrier dysfunction in stroke by enhancing basal lamina. Neurol. Res. 29, 382–387. doi: 10.1179/016164107X204701
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
del Zoppo, G. J., Schmid-Schonbein, G. W., Mori, E., Copeland, B. R., and Chang, C. M. (1991). Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke 22, 1276–1283. doi: 10.1161/01.STR.22.10.1276
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
del Zoppo, G. J., and Hallenbeck, J. M. (2000). Advances in the vascular pathophysiology of ischemic stroke. Thromb. Res. 98, 73–81. doi: 10.1016/S0049-3848(00)00218-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
del Zoppo, G. J. (2009). Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 158, 972–982. doi: 10.1016/j.neuroscience.2008.08.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
del Zoppo, G. J., Milner, R., Mabuchi, T., Hung, S., Wang, X., Berg, G. I., et al. (2007). Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 38, 646–651. doi: 10.1161/01.STR.0000254477.34231.cb
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dénes, A., Ferenczi, S., Halász, J., Környei, Z., and Kovács, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dénes, A., Vidyasagar, R., Feng, J., Narvainen, J., McColl, B. W., Kauppinen, R. A., et al. (2007). Proliferating resident microglia after focal cerebral ischaemia in mice. J. Cereb. Blood Flow Metab. 27, 1941–1953. doi: 10.1038/sj.jcbfm.9600495
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dénes, A., Wilkinson, F., Bigger, B., Chu, M., Rothwell, N. J., and Allan, S. M. (2013). Central and haematopoietic interleukin-1 both contribute to ischaemic brain injury in mice. Dis. Model Mech. 6, 1043–1048. doi: 10.1242/dmm.011601
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dentesano, G., Straccia, M., Ejarque-Ortiz, A., Tusell, J. M., Serratosa, J., Saura, J., et al. (2012). Inhibition of CD200R1 expression by C/EBP beta in reactive microglial cells. J. Neuroinflamm. 9:165. doi: 10.1186/1742-2094-9-165
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F., and Andjelkovic, A. V. (2007). Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ding, Y. H., Young, C. N., Luan, X., Li, J., Rafols, J. A., Clark, J. C., et al. (2005). Exercise preconditioning ameliorates inflammatory injury in ischemic rats during reperfusion. Acta Neuropathol. 109, 237–246. doi: 10.1007/s00401-004-0943-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ding, Y. H., Mrizek, M., Lai, Q., Wu, Y., Reyes, R. Jr Li, J., et al. (2006). Exercise preconditioning reduces brain damage and inhibits TNF-alpha receptor expression after hypoxia/reoxygenation: an in vivo and in vitro study. Curr. Neurovasc. Res. 3, 263–271. doi: 10.2174/156720206778792911
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dodson, R. F., Chu, L. W., Welch, K. M., and Achar, V. S. (1977). Acute tissue response to cerebral ischemia in the gerbil: an ultrastructural study. J. Neurol. Sci. 33, 161–170. doi: 10.1016/0022-510X(77)90190-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Domercq, M., Perez-Samartin, A., Aparicio, D., Alberdi, E., Pampliega, O., and Matute, C. (2010). P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 58, 730–740. doi: 10.1002/glia.20958
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donohue, M. M., Cain, K., Zierath, D., Shibata, D., Tanzi, P. M., and Becker, K. J. (2012). Higher plasma fractalkine is associated with better 6-month outcome from ischemic stroke. Stroke 43, 2300–2306. doi: 10.1161/STROKEAHA.112.657411
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doyle, K. P., Cekanaviciute, E., Mamer, L. E., and Buckwalter, M. S. (2010). TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflamm. 7:62. doi: 10.1186/1742-2094-7-62
Dziewulska, D., and Mossakowski, M. J. (2003). Cellular expression of tumor necrosis factor alpha and its receptors in human ischemic stroke. Clin. Neuropathol. 22, 35–40.
Emsley, H. C., Smith, C. J., Gavin, C. M., Georgiou, R. F., Vail, A., Barberan, E. M., et al. (2007). Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 7:5. doi: 10.1186/1471-2377-7-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Emsley, H. C., Smith, C. J., Georgiou, R. F., Vail, A., Hopkins, S. J., Rothwell, N. J., et al. (2005). A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 76, 1366–1372. doi: 10.1136/jnnp.2004.054882
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fagan, S. C., Cronic, L. E., and Hess, D. C. (2011). Minocycline development for acute ischemic stroke. Transl. Stroke Res. 2, 202–208. doi: 10.1007/s12975-011-0072-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Faraco, G., Fossati, S., Bianchi, M. E., Patrone, M., Pedrazzi, M., Sparatore, B., et al. (2007). High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 103, 590–603. doi: 10.1111/j.1471-4159.2007.04788.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fassbender, K., Rossol, S., Kammer, T., Daffertshofer, M., Wirth, S., Dollman, M., et al. (1994). Proinflammatory cytokines in serum of patients with acute cerebral damage and outcome of disease. J. Neurol. Sci. 122, 135–139. doi: 10.1016/0022-510X(94)90289-5
Felger, J., Abe, T., Kaunzner, U., Gottfried-Blackmore, A., Gal-Toth, J., McEwen, B., et al. (2010). Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav. Immun. 24, 724–737. doi: 10.1016/j.bbi.2009.11.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Forster, C., Clark, H. B., Ross, M. E., and Iadecola, C. (1999). Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol. 97, 215–220. doi: 10.1007/s004010050977
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fossati, S., and Chiarugi, A. (2007). Relevance of high-mobility group protein box 1 to neurodegeneration. Int. Rev. Neurobiol. 82, 137–148. doi: 10.1016/S0074-7742(07)82007-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frenkel, D., Huang, Z., Maron, R., Koldzic, D. N., Moskowitz, M. A., and Weiner, H. L. (2005). Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J. Neurol. Sci. 233, 125–132. doi: 10.1016/j.jns.2005.03.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frieler, R. A., Meng, H., Duan, S. Z., Berger, S., Schütz, G., He, Y., et al. (2011). Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke 42, 179–185. doi: 10.1161/STROKEAHA.110.598441
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fumagalli, S., Perego, C., Ortolano, F., and De Simoni, M. G. (2013). CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia 61, 827–842. doi: 10.1002/glia.22474
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia, J. H., Liu, K. F., Yoshida, Y., Lian, J., Chen, S., and del Zoppo, G. J. (1994). Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am. J. Pathol. 144, 188–199.
Garcia-Bonilla, L., Benakis, C., Moore, J., Iadecola, C., and Anrather, J. (2014a). Immune mechanisms in cerebral ischemic tolerance. Front. Neurosci. 8:44. doi: 10.3389/fnins.2014.00044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia-Bonilla, L., Moore, J. M., Racchumi, G., Zhou, P., Butler, J. M., Iadecola, C., et al. (2014b). Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J. Immunol. 193, 2531–2537. doi: 10.4049/jimmunol.1400918
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gary, D. S., Bruce-Keller, A. J., Kindy, M. S., and Mattson, M. P. (1998). Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 18, 1283–1287. doi: 10.1097/00004647-199812000-00001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gasche, Y., Copin, J. C., Sugawara, T., Fujimura, M., and Chan, P. H. (2001). Matrix metalloproteinase inhibition prevents oxidative stress associated blood-brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 21, 1393–1400. doi: 10.1097/00004647-200112000-00003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gelderblom, M., Leypoldt, F., Steinbach, K., Behrens, D., Choe, C. U., Siler, D. A., et al. (2009). Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/STROKEAHA.108.534503
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gelderblom, M., Weymar, A., Bernreuther, C., Velden, J., Arunachalam, P., Steinbach, K., et al. (2012). Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 120, 3793–3802. doi: 10.1182/blood-2012-02-412726
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gerhard, A., Schwarz, J., Myers, R., Wise, R., and Banati, R. B. (2005). Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. Neuroimage 24, 591–595. doi: 10.1016/j.neuroimage.2004.09.034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gidday, J. M., Gasche, Y. G., Copin, J. C., Shah, A. R., Perez, R. S., Shapiro, S. D., et al. (2005). Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. 289, H558–H568. doi: 10.1152/ajpheart.01275.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giles, J. A., Greenhalgh, A. D., Davies, C. L., Denes, A., Shaw, T., et al. (2015). Requirement for interleukin-1 to drive brain inflammation reveals tissue-specific mechanisms of innate immunity. Eur. J. Immunol. 45, 525–530. doi: 10.1002/eji.201444748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gliem, M., Mausberg, A. K., Lee, J. I., Simiantonakis, I., Rooijen, N., Hartung, H. P., et al. (2012). Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752. doi: 10.1002/ana.23529
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Go, A. S., Mozaffarian, D., Roger, V. L., Benjamin, E. J., Berry, J. D., Blaha, M. J., et al. (2014). American heart association statistics committee and stroke statistics subcommittee. Executive summary: heart disease and stroke statistics-2014 update: a report from the american heart association. Circulation 129, 399–410. doi: 10.1161/01.cir.0000442015.53336.12
Gong, G., Bai, S., Wu, W., Hu, L., Liu, Y., Niu, J., et al. (2014). Lrg participates in lipopolysaccharide preconditioning-induced brain ischemia injury via TLR4 signaling pathway. J. Mol. Neurosci. 54, 20–26. doi: 10.1007/s12031-014-0240-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gottfried-Blackmore, A., Kaunzner, U., Idoyaga, J., Felger, J., McEwen, B., and Bulloch, K. (2009). Acute in vivo exposure to interferon-gamma enables resident brain dendritic cells to become effective antigen presenting cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20918–20923. doi: 10.1073/pnas.0911509106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Granger, D. N., Benoit, J. N., Suzuki, M., and Grisham, M. B. (1989). Leukocyte adherence to venular endothelium during ischemia-reperfusion. Am. J. Physiol. 257, G683–G688.
Greco, R., Amantea, D., Mangione, A. S., Petrelli, F., Gentile, R., Nappi, G., et al. (2012). Modulation of RAGE isoforms expression in the brain and plasma of rats exposed to transient focal cerebral ischemia. Neurochem. Res. 37, 1508–1516. doi: 10.1007/s11064-012-0778-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greco, R., Mangione, A. S., Amantea, D., Bagetta, G., Nappi, G., and Tassorelli, C. (2011). IkappaB-alpha expression following transient focal cerebral ischemia is modulated by nitric oxide. Brain Res. 1372, 145–151. doi: 10.1016/j.brainres.2010.11.071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greco, R., Tassorelli, C., Mangione, S., Levandis, G., Certo, M., Nappi, G., et al. (2014). Neuroprotection by the PARP inhibitor PJ34 modulates cerebral and circulating RAGE levels in rats exposed to focal brain ischemia. Eur. J. Pharmacol. 744, 91–97. doi: 10.1016/j.ejphar.2014.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Green, S. P., Cairns, B., Rae, J., Erret-Baroncini, C., Hongo, J. A., Erickson, R. W., et al. (2001). Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 21, 374–384. doi: 10.1097/00004647-200104000-00006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gregersen, R., Lambertsen, K., and Finsen, B. (2000). Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 20, 53–65. doi: 10.1097/00004647-200001000-00009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grilli, M., Barbieri, I., Basudev, H., Brusa, R., Casati, C., Lozza, G., et al. (2000). Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur. J. Neurosci. 12, 2265–2272. doi: 10.1046/j.1460-9568.2000.00090.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gu, Z., Kaul, M., Yan, B., Kriedel, S. J., Cul, J., Strongin, A., et al. (2002). S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297, 1186–1190. doi: 10.1126/science.1073634
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, M., Cox, B., Mahale, S., Davis, W., Carranza, A., Hayes, K., et al. (2008a). Pre-ischemic exercise reduces matrix metalloproteinase-9 expression and ameliorates blood-brain barrier dysfunction in stroke. Neuroscience 151, 340–351. doi: 10.1016/j.neuroscience.2007.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, M., Lin, V., Davis, W., Huang, T., Carranza, A., Sprague, S., et al. (2008b). Preischemic induction of TNF-alpha by physical exercise reduces blood-brain barrier dysfunction in stroke. J. Cereb. Blood Flow Metab. 28, 1422–1430. doi: 10.1038/jcbfm.2008.29
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hacke, W. (2015). Interventional Thrombectomy for major stroke - A step in the right direction. N. Engl. J. Med. 372, 76–77. doi: 10.1056/NEJMe1413346
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hailer, N. P. (2008). Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 84, 211–233. doi: 10.1016/j.pneurobio.2007.12.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hallenbeck, J. M. (2002). The many faces of tumor necrosis factor in stroke. Nat. Med. 8, 1363–1368. doi: 10.1038/nm1202-1363
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hara, H., Friedlander, R. M., Gagliardini, V., Ayata, C., Fink, K., Huang, Z., et al. (1997). Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. U.S.A. 94, 2007–2012. doi: 10.1073/pnas.94.5.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hasegawa, Y., Suzuki, H., Sozen, T., Rolland, W., and Zhang, J. H. (2010). Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41, 368–374. doi: 10.1161/STROKEAHA.109.568899
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hassid, B. G., Nair, M. N., Ducruet, A. F., Otten, M. L., Komotar, R. J., Pinsky, D. J., et al. (2009). Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J. Clin. Neurosci. 16, 302–306. doi: 10.1016/j.jocn.2007.12.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayakawa, K., Nakano, T., Irie, K., Higuchi, S., Fujioka, M., Orito, K., et al. (2010). Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 30, 871–882. doi: 10.1038/jcbfm.2009.257
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herrmann, O., Tarabin, V., Suzuki, S., Attigah, N., Coserea, I., Schneider, A., et al. (2003). Regulation of body temperature and neuroprotection by endogenous interleukin-6 in cerebral ischemia. J. Cereb. Blood Flow Metab. 23, 406–415. doi: 10.1097/00004647-200304000-00004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herx, L. M., and Yong, V. W. (2001). Interleukin-1 beta is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 60, 961–971.
Hickey, E. J., You, X., Kaimaktchiev, V., Stenzel-Poore, M., and Ungerleider, R. M. (2007). Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J. Thorac. Cardiovasc. Surg. 133, 1588–1596. doi: 10.1016/j.jtcvs.2006.12.056
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hill, J. W., Poddar, R., Thompson, J. F., Rosenberg, G. A., and Yang, Y. (2012). Intranuclear matrix metalloproteinases promote DNA damage and apoptosis induced by oxygen-glucose deprivation in neurons. Neuroscience 220, 277–290. doi: 10.1016/j.neuroscience.2012.06.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Horiguchi, T., Snipes, J. A., Kis, B., Shimizu, K., and Busija, D. W. (2005). The role of nitric oxide in the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Brain Res. 1039, 84–89. doi: 10.1016/j.brainres.2005.01.047
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Horiguchi, T., Snipes, J. A., Kis, B., Shimizu, K., and Busija, D. W. (2006). Cyclooxygenase-2 mediates the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Neuroscience 140, 723–730. doi: 10.1016/j.neuroscience.2006.02.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Horstmann, S., Kalb, P., Koziol, J., Gardner, H., and Wagner, S. (2003). Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke 34, 2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S., et al. (2012). Microglia/Macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/STROKEAHA.112.659656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, J., Li, Y., Tang, Y., Tang, G., Yang, G. Y., and Wang, Y. (2013). CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 44, 190–197. doi: 10.1161/STROKEAHA.112.670299
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hughes, P. M., Allegrini, P. R., Rudin, M., Perry, V. H., Mir, A. K., and Wiessner, C. (2002). Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J. Cereb. Blood Flow Metab. 22, 308–317. doi: 10.1097/00004647-200203000-00008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hurn, P. D., Subramanian, S., Parker, S. M., Afentoulis, M. E., Kaler, L. J., Vandenbark, A. A., et al. (2007). T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J. Cereb. Blood Flow Metab. 27, 1798–1805. doi: 10.1038/sj.jcbfm.9600482
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hyakkoku, K., Hamanaka, J., Tsuruma, K., Shimazawa, M., Tanaka, H., Uematsu, S., et al. (2010). Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171, 258–267. doi: 10.1016/j.neuroscience.2010.08.054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., Forster, C., Nogawa, S., Clark, H. B., and Ross, M. E. (1999). Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol. 98, 9–14. doi: 10.1007/s004010051045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., Niwa, K., Nogawa, S., Zhao, X., Nagayama, M., Araki, E., et al. (2001). Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1294–1299. doi: 10.1073/pnas.98.3.1294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., Zhang, F., Casey, R., Clark, H. B., and Ross, M. E. (1996). Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke 27, 1373–1380. doi: 10.1161/01.STR.27.8.1373
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., Zhang, F., Casey, R., Nagayama, M., and Ross, M. E. (1997). Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 17, 9157–9164.
Imai, F., Suzuki, H., Oda, J., Ninomiya, T., Ono, K., Sano, H., et al. (2007). Neuroprotective effect of exogenous microglia in global brain ischemia. J. Cereb. Blood Flow Metab. 27, 488–500. doi: 10.1038/sj.jcbfm.9600362
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Irving, E. A., Barone, F. C., Reith, A. D., Hadingham, S. J., and Parsons, A. A. (2000). Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res. 77, 65–75. doi: 10.1016/S0169-328X(00)00043-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishikawa, M., Zhang, J. H., Nanda, A., and Granger, D. N. (2004). Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front. Biosci. 9, 1339–1347. doi: 10.2741/1330
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Italiani, P., and Boraschi, D. (2014). From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front. Immunol. 5:514. doi: 10.3389/fimmu.2014.00514
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jander, S., Kraemer, M., Schroeter, M., Witte, O., and Stoll, G. (1995). Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J. Cereb. Blood Flow Metab. 15, 42–51. doi: 10.1038/jcbfm.1995.5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jander, S., Schroeter, M., and Saleh, A. (2007). Imaging inflammation in acute brain ischemia. Stroke 38, 642–645. doi: 10.1161/01.STR.0000250048.42916.ad
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jickling, G. C., and Sharp, F. R. (2011). Blood biomarkers of ischemic stroke. Neurotherapeutics 8, 349–360. doi: 10.1007/s13311-011-0050-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jin, Z., Liang, J., Wang, J., and Kolattukudy, P. E. (2013). Delayed brain ischemia tolerance induced by electroacupuncture pretreatment is mediated via MCP-induced protein 1. J. Neuroinflamm. 10:63. doi: 10.1186/1742-2094-10-63
Jin, R., Yang, G., and Li, G. (2010). Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 87, 779–789. doi: 10.1189/jlb.1109766
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jourquin, J., Tremblay, E., Decanis, N., Charton, G., Hanessian, S., Chollet, A. M., et al. (2003). Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur. J. Neurosci. 18, 1507–1517. doi: 10.1046/j.1460-9568.2003.02876.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jung, J. E., Kim, G. S., and Chan, P. H. (2011). Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke 42, 3574–3579. doi: 10.1161/STROKEAHA.111.626648
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jung, S., and Schwartz, M. (2012). Non-identical twins - microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation. Front. Immunol. 3:89. doi: 10.3389/fimmu.2012.00089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Justicia, C., Panés, J., Solé, S., Cervera, A., Deulofeu, R., Chamorro, A., et al. (2003). Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J. Cereb. Blood Flow Metab. 23, 1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lehrmann, E., Kiefer, R., Christensen, T., Toyka, K. V., Zimmer, J., Diemer, N. H., et al. (1998). Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia 24, 437–448.
Kamel, H., and Iadecola, C. (2012). Brain-immune interactions and ischemic stroke. Clinical implications. Arch. Neurol. 69, 576–581. doi: 10.1001/archneurol.2011.3590
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, S. S., Keasey, M. P., Cai, J., and Hagg, T. (2012). Loss of neuron-astroglial interaction rapidly induces protective CNTF expression after stroke in mice. J. Neurosci. 32, 9277–9287. doi: 10.1523/JNEUROSCI.1746-12.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kapinya, K. J., Löwl, D., Fütterer, C., Maurer, M., Waschke, K. F., Isaev, N. K., et al. (2002). Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 33, 1889–1898. doi: 10.1161/01.STR.0000020092.41820.58
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawano, T., Anrather, J., Zhou, P., Park, L., Wang, G., Frys, K. A., et al. (2006). Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat. Med. 12, 225–229. doi: 10.1038/nm1362
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keiner, S., Wurm, F., Kunze, A., Witte, O. W., and Redecker, C. (2008). Rehabilitative therapies differentially alter proliferation and survival of glial cell populations in the perilesional zone of cortical infarcts. Glia 56, 516–527. doi: 10.1002/glia.20632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, Y. S., Baek, W., Kim, M. K., Kim, H. Y., Lee, K. Y., Lee, Y. J., et al. (2012). Association between serum stromal cell-derived factor-1α and long-term outcome of acute ischemic stroke. Eur. Neurol. 67, 363–369. doi: 10.1159/000335351
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. B., Sig Choi, J., Yu, Y. M., Nam, K., Piao, C. S., Kim, S. W., et al. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, E., Raval, A. P., Defazio, R. A., and Perez-Pinzon, M. A. (2007). Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience 145, 931–941. doi: 10.1016/j.neuroscience.2006.12.063
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, E. J., Raval, A. P., Hirsch, N., and Perez-Pinzon, M. A. (2010). Ischemic preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa B activation in mixed cortical neuronal cultures. Transl. Stroke Res. 1, 40–47. doi: 10.1007/s12975-009-0006-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kinoshita, A., Yamada, K., Kohmura, E., and Hayakawa, T. (1990). Effect of astrocyte-derived factors on ischemic brain edema induced by rat MCA occlusion. APMIS 98, 851–857. doi: 10.1111/j.1699-0463.1990.tb05006.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kitamura, Y., Takata, K., Inden, M., Tsuchiya, D., Yanagisawa, D., Nakata, J., et al. (2004). Intracerebroventricular injection of microglia protects against focal brain ischemia. J. Pharmacol. Sci. 94, 203–206. doi: 10.1254/jphs.94.203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Konoeda, F., Shichita, T., Yoshida, H., Sugiyama, Y., Muto, G., Hasegawa, E., et al. (2010). Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem. Biophys. Res. Commun. 402, 500–506. doi: 10.1016/j.bbrc.2010.10.058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kostulas, N., Li, H. L., Xiao, B. G., Huang, Y. M., Kostulas, V., and Link, H. (2002). Dendritic cells are present in ischemic brain after permanent middle cerebral artery occlusion in the rat. Stroke 33, 1129–1134. doi: 10.1161/hs0402.105379
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koyama, H., Yamamoto, H., and Nishizawa, Y. (2007). RAGE and soluble RAGE: potential therapeutic targets for cardiovascular diseases. Mol. Med. 13, 625–635. doi: 10.2119/2007-00087.Koyama
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuboyama, K., Harada, H., Tozaki-Saitoh, H., Tsuda, M., Ushijima, K., and Inoue, K. (2011). Astrocytic P2Y(1) receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J. Cereb. Blood Flow Metab. 31, 1930–1941. doi: 10.1038/jcbfm.2011.49
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglia cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambertsen, K. L., Biber, K., and Finsen, B. (2012). Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 32, 1677–1698. doi: 10.1038/jcbfm.2012.88
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambertsen, K. L., Clausen, B. H., Babcock, A. A., Gregersen, R., Fenger, C., Nielsen, H. H., et al. (2009). Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 29, 1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambertsen, K. L., Clausen, B. H., Fenger, C., Wulf, H., Owens, T., Dagnaes-Hansen, F., et al. (2007). Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience 144, 934–949. doi: 10.1016/j.neuroscience.2006.10.046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambertsen, K. L., Meldgaard, M., Ladeby, R., and Finsen, B. (2005). A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 25, 119–135. doi: 10.1038/sj.jcbfm.9600014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lavine, S. D., Hofman, F. M., and Zlokovic, B. V. (1998). Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J. Cereb. Blood Flow Metab. 18, 52–58. doi: 10.1097/00004647-199801000-00005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lehnardt, S., Massillon, L., Follett, P., Jensen, F. E., Ratan, R., Rosenberg, P. A., et al. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 8514–8519. doi: 10.1073/pnas.1432609100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, L., Lundkvist, A., Andersson, D., Wilhelmsson, U., Nagai, N., Pardo, A. C., et al. (2008). Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 28, 468–481. doi: 10.1038/sj.jcbfm.9600546
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liang, J., Wang, J., Saad, Y., Warble, L., Becerra, E., and Kolattukudy, P. E. (2001). Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J. Neuroinflamm. 8:182. doi: 10.1186/1742-2094-8-182
Liu, F., Yao, Y. M., Dong, N., Xu, S., and Sheng, Z. Y. (2007). The receptor mechanism of high mobility group box-1 protein induced apoptosis in peritoneal macrophages in mice. Zhonghua Shao Shang Za Zhi 23, 432–435.
Liu, J., Jin, X., Liu, K. J., and Liu, W. (2012). Matrix metalloproteinase-2-mediated occluding degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci. 32, 3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loddick, S. A., Turnbull, A. V., and Rothwell, N. J. (1998). Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18, 176–179. doi: 10.1097/00004647-199802000-00008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Logan, A., Berry, M., Gonzalez, A. M., Frautschy, S. A., Sporn, M. B., and Baird, A. (1994). Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur. J. Neurosci. 6, 355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Losy, J., and Zaremba, J. (2001). Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke 32, 2695–2696. doi: 10.1161/hs1101.097380
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luheshi, N. M., Kovács, K. J., Lopez-Castejon, G., Brough, D., and Denes, A. (2011). Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflamm. 8:186. doi: 10.1186/1742-2094-8-186
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, M., Ma, Y., Yi, X., Guo, R., Zhu, W., Fan, X., et al. (2008). Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 9:117. doi: 10.1186/1471-2202-9-117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, S., Zhong, D., Chen, H., Zheng, Y., Sun, Y., Luo, J., et al. (2013). The immunomodulatory effect of bone marrow stromal cells (BMSCs) on interleukin (IL)-23/IL-17-mediated ischemic stroke in mice. J. Neuroimmunol. 257, 28–35. doi: 10.1016/j.jneuroim.2013.01.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maguire, J., Thakkinstian, A., Levi, C., Lincz, L., Bisset, L., Sturm, J., et al. (2011). Impact of COX-2 rs5275 and rs20417 and GPIIIa rs5918 polymorphisms on 90-day ischemic stroke functional outcome: a novel finding. J. Stroke Cerebrovasc. Dis. 20, 134–144. doi: 10.1016/j.jstrokecerebrovasdis.2009.10.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangiafico, S., and Consoli, A. (2014). “Overview of endovascular treatment and revascularization strategies in acute ischemic stroke,” in Rational Basis for Clinical Translation in Stroke Therapy, eds G. Micieli and D. Amantea (Boca Raton, FL: CRC press), 24–36.
Marchetti, L., Klein, M., Schlett, K., Pfizenmaier, K., and Eisel, U. L. (2004). Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 279, 32869–32881. doi: 10.1074/jbc.M311766200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsuda, S., Wen, T. C., Morita, F., Otsuka, H., Igase, K., Yoshimura, H., et al. (1996). Interleukin-6 prevents ischemia-induced learning disability and neuronal and synaptic loss in gerbils. Neurosci. Lett. 204, 109–112. doi: 10.1016/0304-3940(96)12340-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsuo, Y., Onodera, H., Shiga, Y., Nakamura, M., Ninomiya, M., Kihara, T., et al. (1994a). Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke 25, 1469–1475. doi: 10.1161/01.STR.25.7.1469
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsuo, Y., Onodera, H., Shiga, Y., Shozuhara, H., Ninomiya, M., Kihara, T., et al. (1994b). Role of cell adhesion molecules in brain injury after transient middle cerebral artery occlusion in the rat. Brain Res. 656, 344–352. doi: 10.1016/0006-8993(94)91478-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McCoy, M. K., and Tansey, M. G. (2008). TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 5:45. doi: 10.1186/1742-2094-5-45
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mead, E. L., Mosley, A., Eaton, S., Dobson, L., Heales, S. J., and Pocock, J. M. (2012). Microglial neurotransmitter receptors trigger superoxide production in microglia, consequences for microglial-neuronal interactions. J. Neurochem. 121, 287–301. doi: 10.1111/j.1471-4159.2012.07659.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Melani, A., Amadio, S., Gianfriddo, M., Vannucchi, M. G., Volontè, C., Bernardi, G., et al. (2006). P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 26, 974–982. doi: 10.1038/sj.jcbfm.9600250
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Minami, M., and Satoh, M. (2003). Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Sci. 74, 321–327. doi: 10.1016/j.lfs.2003.09.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moore, D. F., Li, H., Jeffries, N., Wright, V., Cooper, R. A., Elkahloun, A., et al. (2005). Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischemic stroke: a pilot investigation. Circulation 111, 212–221. doi: 10.1161/01.CIR.0000152105.79665.C6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moraga, A., Pradillo, J. M., Cuartero, M. I., Hernández-Jiménez, M., Oses, M., Moro, M. A., et al. (2014). Toll-like receptor 4 modulates cell migration and cortical neurogenesis after focal cerebral ischemia. FASEB J. 28, 4710–4718. doi: 10.1096/fj.14-252452
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mori, E., del Zoppo, G. J., Chambers, J. D., Copeland, B. R., and Arfors, K. E. (1992). Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 23, 712–718. doi: 10.1161/01.STR.23.5.712
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moro, M. A., Cárdenas, A., Hurtado, O., Leza, J. C., and Lizasoain, I. (2004). Role of nitric oxide after brain ischaemia. Cell Calcium 36, 265–275. doi: 10.1016/j.ceca.2004.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muhammad, S., Barakat, W., Stoyanov, S., Murikinati, S., Yang, H., Tracey, K. J., et al. (2008). The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 28, 12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murphy, S., and Gibson, C. L. (2007). Nitric oxide, ischaemia and brain inflammation. Biochem. Soc. Trans. 35, 1133–1137. doi: 10.1042/BST0351133
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakase, T., Fushiki, S., and Naus, C. C. (2003). Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke 34, 1987–1993. doi: 10.1161/01.STR.0000079814.72027.34
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakashima, M. N., Yamashita, K., Kataoka, Y., Yamashita, Y. S., and Niwa, M. (1995). Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell. Mol. Neurobiol. 15, 341–349. doi: 10.1007/BF02089944
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nawashiro, H., Brenner, M., Fukui, S., Shima, K., and Hallenbeck, J. M. (2000). High susceptibility to cerebral ischemia in GFAP-null mice. J. Cereb. Blood Flow Metab. 20, 1040–1044. doi: 10.1097/00004647-200007000-00003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nawashiro, H., Martin, D., and Hallenbeck, J. M. (1997). Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J. Cereb. Blood Flow Metab. 17, 229–232. doi: 10.1097/00004647-199702000-00013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neumann, J., Sauerzweig, S., Rönicke, R., Gunzer, F., Dinkel, K., Ullrich, O., et al. (2008). Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J. Neurosci. 28, 5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nieswandt, B., Pleines, I., and Bender, M. (2011). Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J. Thromb. Haemost. 1, 92–104. doi: 10.1111/j.1538-7836.2011.04361.x
Nogawa, S., Forster, C., Zhang, F., Nagayama, M., Ross, M. E., and Iadecola, C. (1998). Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 95, 10966–10971. doi: 10.1073/pnas.95.18.10966
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nogawa, S., Zhang, F., Ross, M. E., and Iadecola, C. (1997). Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J. Neurosci. 17, 2746–2755.
O'Donnell, S. L., Frederick, T. J., Krady, J. K., Vannucci, S. J., and Wood, T. L. (2002). IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39, 85–97. doi: 10.1002/glia.10081
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oh, S. H., Kim, O. J., Shin, D. A., Song, J., Yoo, H., Kim, Y. K., et al. (2012). Alteration of immunologic responses on peripheral blood in the acute phase of ischemic stroke: blood genomic profiling study. J. Neuroimmunol. 249, 60–65. doi: 10.1016/j.jneuroim.2012.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohtsuki, T., Ruetzler, C. A., Tasaki, K., and Hallenbeck, J. M. (1996). Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J. Cereb. Blood Flow Metab. 16, 1137–1142. doi: 10.1097/00004647-199611000-00007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Olsson, S., Holmegaard, L., Jood, K., Sjögren, M., Engström, G., Lövkvist, H., et al. (2012). Genetic variation within the interleukin-1 gene cluster and ischemic stroke. Stroke 43, 2278–2282. doi: 10.1161/STROKEAHA.111.647446
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ouyang, Y. B., Voloboueva, L. A., Xu, L. J., and Giffard, R. G. (2007). Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 27, 4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pan, L. N., Zhu, W., Li, Y., Xu, X. L., Guo, L. J., Lu, Q., et al. (2014). Astrocytic Toll-like receptor 3 is associated with ischemic preconditioning-induced protection against brain ischemia in rodents. PLoS ONE 9:e99526. doi: 10.1371/journal.pone.0099526
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pang, L., Ye, W., Che, X. M., Roessler, B. J., Betz, A. L., and Yang, G. Y. (2001). Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-β1 expression. Stroke 32, 544–552. doi: 10.1161/01.STR.32.2.544
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, M. K., Kang, Y. J., Lee, H. S., Kim, H. J., Seo, H. G., Lee, J. H., et al. (2008). The obligatory role of COX-2 expression for induction of HO-1 in ischemic preconditioned rat brain. Biochem. Biophys. Res. Commun. 377, 1191–1194. doi: 10.1016/j.bbrc.2008.10.149
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, I. H., Yeon, S. I., Youn, J. H., Choi, J. E., Sasaki, N., Choi, I. H., et al. (2004). Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 40, 1203–1211. doi: 10.1016/j.molimm.2003.11.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peng, B., Guo, Q. L., He, Z. J., Ye, Z., Yuan, Y. J., Wang, N., et al. (2012). Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K/Akt pathway. Brain Res. 1445, 92–102. doi: 10.1016/j.brainres.2012.01.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Perego, C., Fumagalli, S., and De Simoni, M. G. (2011). Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 8:174. doi: 10.1186/1742-2094-8-174
Perini, F., Morra, M., Alecci, M., Galloni, E., Marchi, M., and Toso, V. (2001). Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci. 22, 289–296. doi: 10.1007/s10072-001-8170-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petcu, E. B., Kocher, T., Kuhr, A., Buga, A. M., Klöting, I., Herndon, J. G., et al. (2008). Mild systemic inflammation has a neuroprotective effect after stroke in rats. Curr. Neurovasc. Res. 5, 214–223. doi: 10.2174/156720208786413424
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pfeilschifter, W., Czech-Zechmeister, B., Sujak, M., Foerch, C., Wichelhaus, T. A., and Pfeilschifter, J. (2011). Treatment with the immunomodulator FTY720 does not promote spontaneous bacterial infections after experimental stroke in mice. Exp. Transl. Stroke Med. 9, 3–2. doi: 10.1186/2040-7378-3-2
Pignataro, G., Esposito, E., Sirabella, R., Vinciguerra, A., Cuomo, O., Di Renzo, G., et al. (2013). nNOS and p-ERK involvement in the neuroprotection exerted by remote postconditioning in rats subjected to transient middle cerebral artery occlusion. Neurobiol. Dis. 54, 105–114. doi: 10.1016/j.nbd.2013.02.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Planas, A. M., Sole, S., and Justicia, C. (2001). Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol. Dis 8, 834–846. doi: 10.1006/nbdi.2001.0435
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pocock, J. M., and Kettenmann, H. (2007). Neurotransmitter receptors on microglia. Trends Neurosci. 30, 527–535. doi: 10.1016/j.tins.2007.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pradillo, J. M., Fernández-López, D., García-Yébenes, I., Sobrado, M., Hurtado, O., Moro, M. A., et al. (2009). Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J. Neurochem. 109, 287–294. doi: 10.1111/j.1471-4159.2009.05972.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pradillo, J. M., Moraga, A., Garcia-Culebras, A., Palma-Tortosa, S., Oses, M., Amantea, D., et al. (2014). “Immne response in cerebral ischemia: role of TLRs,” in Rational Basis for Clinical Translation in Stroke Therapy, eds G. Micieli and D. Amantea (Boca Raton, FL: CRC press), 367–379.
Pradillo, J. M., Romera, C., Hurtado, O., Cárdenas, A., Moro, M. A., Leza, J. C., et al. (2005). TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J. Cereb. Blood Flow Metab. 25, 193–203. doi: 10.1038/sj.jcbfm.9600019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Price, C. J., Wang, D., Menon, D. K., Guadagno, J. V., Cleij, M., Fryer, T., et al. (2006). Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke 37, 1749–1753. doi: 10.1161/01.STR.0000226980.95389.0b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Price, C. J., Menon, D. K., Peters, A. M., Ballinger, J. R., Barber, R. W., Balan, K. K., et al. (2004). Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke 35, 1659–1664. doi: 10.1161/01.STR.0000130592.71028.92
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qian, L., Ding, L., Cheng, L., Zhu, X., Zhao, H., Jin, J., et al. (2012). Early biomarkers for post-stroke cognitive impairment. J. Neurol. 259, 2111–2118. doi: 10.1007/s00415-012-6465-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, J., Nishimura, M., Wang, Y., Sims, J. R., Qiu, S., Savitz, S. I., et al. (2008). Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 28, 927–938. doi: 10.1038/sj.jcbfm.9600582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, J., Xu, J., Zheng, Y., Wei, Y., Zhu, X., Lo, E. H., et al. (2010). High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 41, 2077–2082. doi: 10.1161/STROKEAHA.110.590463
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ransohoff, R. M., and Cardona, A. E. (2010). The myeloid cells of the central nervous system parenchyma. Nature 468, 253–262. doi: 10.1038/nature09615
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rehni, A. K., and Singh, T. G. (2012). Involvement of CCR-2 chemokine receptor activation in ischemic preconditioning and postconditioning of brain in mice. Cytokine 60, 83–89. doi: 10.1016/j.cyto.2012.05.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reichmann, G., Schroeter, M., Jander, S., and Fischer, H. G. (2002). Dendritic cells and dendritic-like microglia in focal cortical ischemia of the mouse brain. J. Neuroimmunol. 129, 125–132. doi: 10.1016/S0165-5728(02)00184-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ritter, L. S., Stempel, K. M., Coull, B. M., and McDonagh, P. F. (2005). Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol. Res. Nurs. 6, 281–288. doi: 10.1177/1099800405274579
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Robin, A. M., Zhang, Z. G., Wang, L., Zhang, R. L., Katakowski, M., Zhang, L., et al. (2006). Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 26, 125–134. doi: 10.1038/sj.jcbfm.9600172
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rodríguez-Yáñez, M., Sobrino, T., Arias, S., Vázquez-Herrero, F., Brea, D., Blanco, M., et al. (2011). Early biomarkers of clinical-diffusion mismatch in acute ischemic stroke. Stroke 42, 2813–2818. doi: 10.1161/STROKEAHA.111.614503
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Romanic, A. M., White, R. F., Arleth, A. J., Ohlstein, E. H., and Barone, F. C. (1998). Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29, 1020–1030. doi: 10.1161/01.STR.29.5.1020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosell, A., Ortega-Aznar, A., Alvarez-Sabin, J., Fernandez-Cadenas, I., Ribo, M., Molina, C. A., et al. (2006). Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke 37, 1399–1406. doi: 10.1161/01.STR.0000223001.06264.af
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosell, A., Cuadrado, E., Ortega-Aznar, A., Hernández-Guillamon, M., Lo, E. H., and Montaner, J. (2008). MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39, 1121–1126. doi: 10.1161/STROKEAHA.107.500868
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenberg, G. A., Estrada, E. Y., and Dencoff, J. E. (1998). Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke 29, 2189–2195. doi: 10.1161/01.STR.29.10.2189
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenzweig, H. L., Lessov, N. S., Henshall, D. C., Minami, M., Simon, R. P., and Stenzel-Poore, M. P. (2004). Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 35, 2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenzweig, H. L., Minami, M., Lessov, N. S., Coste, S. C., Stevens, S. L., Henshall, D. C., et al. (2007). Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J. Cereb. Blood Flow Metab. 27, 1663–1674. doi: 10.1038/sj.jcbfm.9600464
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosito, M., Lauro, C., Chece, G., Porzia, A., Monaco, L., Mainiero, F., et al. (2014). Trasmembrane chemokines CX3CL1 and CXCL16 drive interplay between neurons, microglia and astrocytes to counteract pMCAO and excitotoxic neuronal death. Front. Cell. Neurosci. 8:193. doi: 10.3389/fncel.2014.00193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rossi, D. J., Brady, J. D., and Mohr, C. (2007). Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 10, 1377–1386. doi: 10.1038/nn2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rothwell, N., Allan, S., and Toulmond, S. (1997). The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J. Clin. Invest. 100, 2648–2652. doi: 10.1172/JCI119808
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruocco, A., Nicole, O., Docagne, F., Ali, C., Chazalviel, L., Komesli, S., et al. (1999). A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J. Cereb. Blood Flow Metab. 19, 1345–1353. doi: 10.1097/00004647-199912000-00008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rusnakova, V., Honsa, P., Dzamba, D., Ståhlberg, A., Kubista, M., and Anderova, M. (2013). Heterogeneity of astrocytes: from development to injury - single cell gene expression. PLoS ONE 8:e69734. doi: 10.1371/journal.pone.0069734
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schilling, M., Besselmann, M., Leonhard, C., Mueller, M., Ringelstein, E. B., and Kiefer, R. (2003). Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 183, 25–33. doi: 10.1016/S0014-4886(03)00082-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schuette-Nuetgen, K., Strecker, J. K., Minnerup, J., Ringelstein, E. B., and Schilling, M. (2012). MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp. Neurol. 233, 849–858. doi: 10.1016/j.expneurol.2011.12.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schutt, R. C., Burdick, M. D., Strieter, R. M., Mehrad, B., and Keeley, E. C. (2012). Plasma CXCL12 levels as a predictor of future stroke. Stroke 43, 3382–3386. doi: 10.1161/STROKEAHA.112.660878
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwab, J. M., Nguyen, T. D., Meyermann, R., and Schluesener, H. J. (2001). Human focal cerebral infarctions induce differential lesional interleukin-16 (IL-16) expression confined to infiltrating granulocytes, CD8+ T-lymphocytes and activated microglia/macrophages. J. Neuroimmunol. 114, 232–241. doi: 10.1016/S0165-5728(00)00433-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shechter, R., and Schwartz, M. (2013). Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer ‘if’ but ‘how’. J. Pathol. 229, 332–346. doi: 10.1002/path.4106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, L. H., Li, Y., and Chopp, M. (2010). Astrocytic endogenous glial cell derived neurotrophic factor production is enhanced by bone marrow stromal cell transplantation in the ischemic boundary zone after stroke in adult rats. Glia 58, 1074–1081 doi: 10.1002/glia.20988
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shichita, T., Ago, T., Kamouchi, M., Kitazono, T., Yoshimura, A., and Ooboshi, H. (2012a). Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J. Neurochem. 123, 29–38. doi: 10.1111/j.1471-4159.2012.07941.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shichita, T., Hasegawa, E., Kimura, A., Morita, R., Sakaguchi, R., Takada, I., et al. (2012b). Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917. doi: 10.1038/nm.2749
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shichita, T., Sugiyama, Y., Ooboshi, H., Sugimori, H., Nakagawa, R., Takada, I., et al. (2009). Pivotal role of cerebral interleukin-17-producing Tcells in the delayed phase of ischemic brain injury. Nat. Med. 15, 946–950. doi: 10.1038/nm.1999
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shimizu, M., Saxena, P., Konstantinov, I. E., Cherepanov, V., Cheung, M. M. H., Wearden, P., et al. (2010). Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J. Surg. Res. 158, 155–161. doi: 10.1016/j.jss.2008.08.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, J. A., Park, E. M., Choi, J. S., Seo, S. M., Kang, J. L., Lee, K. E., et al. (2009). Ischemic preconditioning-induced neuroprotection is associated with differential expression of IL-1beta and IL-1 receptor antagonist in the ischemic cortex. J. Neuroimmunol. 217, 14–19. doi: 10.1016/j.jneuroim.2009.06.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shyu, W. C., Lin, S. Z., Yen, P. S., Su, C. Y., Chen, D. C., Wang, H. J., et al. (2008). Stromal cell-derived factor-1 alpha promotes neuroprotection, angiogenesis, and mobilization/homing of bone marrow-derived cells in stroke rats. J. Pharmacol. Exp. Ther. 324, 834–849. doi: 10.1124/jpet.107.127746
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Simi, A., Lerouet, D., Pinteaux, E., and Brough, D. (2007). Mechanisms of regulation for interleukin-1β in neurodegenerative disease. Neuropharmacology 52, 1563–1569. doi: 10.1016/j.neuropharm.2007.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, C. J., Emsley, H. C. A., Gavin, C. M., Georgiou, R. F., Vail, A., Barberan, E. M., et al. (2004). Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 4:2. doi: 10.1186/1471-2377-4-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Soejima, Y., Hu, Q., Krafft, P. R., Fujii, M., Tang, J., and Zhang, J. H. (2013). Hyperbaric oxygen preconditioning attenuates hyperglycemia-enhanced hemorrhagic transformation by inhibiting matrix metalloproteinases in focal cerebral ischemia in rats. Exp. Neurol. 247, 737–743. doi: 10.1016/j.expneurol.2013.03.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R., et al. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65. doi: 10.1016/S0165-5728(02)00033-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spera, P., Ellison, J., Feuerstein, G., and Barone, F. (1998). IL-10 reduces rat brain injury following focal stroke. Neurosci. Lett. 251, 189–192. doi: 10.1016/S0304-3940(98)00537-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sreeramkumar, V., Adrover, J. M., Ballesteros, I., Cuartero, M. I., Rossaint, J., Bilbao, I., et al. (2014). Neutrophils scan for activated platelets to initiate inflammation. Science 346, 1234–1238. doi: 10.1126/science.1256478
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stanimirovic, D. B., Ball, R., and Durkin, J. P. (1997). Stimulation of glutamate uptake and Na,K-ATPase activity in rat astrocytes exposed to ischemia-like insults. Glia 19, 123–134.
Stoll, G., Jander, S., and Schroeter, M. (1998). Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 56, 149–171. doi: 10.1016/S0301-0082(98)00034-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stowe, A. M., Wacker, B. K., Cravens, P. D., Perfater, J. L., Li, M. K., Hu, R., et al. (2012). CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J. Neuroinflamm. 9:33. doi: 10.1186/1742-2094-9-33
Strecker, J. K., Minnerup, J., Gess, B., Ringelstein, E. B., Schäbitz, W. R., and Schilling, M. (2011). Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1β and G-CSF after transient focal ischemia in mice. PLoS ONE 6:e25863. doi: 10.1371/journal.pone.0025863
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suzuki, S., Tanaka, K., Nogawa, S., Nagata, E., Ito, D., Dembo, T., et al. (1999). Temporal profile and cellular localization of interleukin-6 protein after focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 19, 1256–1262. doi: 10.1097/00004647-199911000-00010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swardfager, W., Winer, D. A., Herrmann, N., Winer, S., and Lanctôt, K. L. (2013). Interleukin-17 in post-stroke neurodegeneration. Neurosci. Biobehav. Rev. 37, 436–447. doi: 10.1016/j.neubiorev.2013.01.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takami, S., Minami, M., Nagata, I., Namura, S., and Satoh, M. (2001). Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 21, 1430–1435. doi: 10.1097/00004647-200112000-00007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, S. C., Arumugam, T. V., Xu, X., Cheng, A., Mughal, M. R., Jo, D. G., et al. (2007). Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. U.S.A. 104, 13798–13803. doi: 10.1073/pnas.0702553104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, S. C., Wang, Y. C., Li, Y. I., Lin, H. C., Manzanero, S., Hsieh, Y. H., et al. (2013). Functional role of soluble receptor for advanced glycation end products in stroke. Arterioscler. Thromb. Vasc. Biol. 33, 585–594. doi: 10.1161/ATVBAHA.112.300523
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, Y., Xu, H., Du, X., Lit, L., Walker, W., Lu, A., et al. (2006). Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J. Cereb. Blood Flow Metab. 26, 1089–1102. doi: 10.1038/sj.jcbfm.9600264
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taoufik, E., Valable, S., Muller, G. J., Roberts, M. L., Divoux, D., Tinel, A., et al. (2007). FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J. Neurosci. 27, 6633–6646. doi: 10.1523/JNEUROSCI.1091-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tarozzo, G., Campanella, M., Ghiani, M., Bulfone, A., and Beltramo, M. (2002). Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur. J. Neurosci. 15, 1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tasaki, K., Ruetzler, C. A., Ohtsuki, T., Martin, D., Nawashiro, H., and Hallenbeck, J. M. (1997). Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 748, 267–270. doi: 10.1016/S0006-8993(96)01383-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thornton, P., McColl, B. W., Cooper, L., Rothwell, N. J., and Allan, S. M. (2011). Interleukin-1 drives cerebrovascular inflammation via map kinase-independent pathways. Curr. Neurovasc. Res. 7, 330–340. doi: 10.2174/156720210793180800
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thornton, P., Pinteaux, E., Allan, S. M., and Rothwell, N. J. (2008). Matrix metalloproteinase-9 and urokinase plasminogen activator mediate interleukin-1-induced neurotoxicity. Mol. Cell Neurosci. 37, 135–142. doi: 10.1016/j.mcn.2007.09.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ulrich, N. H., Dehmel, T., Wittsack, H. J., Kieseier, B. C., and Seitz, R. J. (2013). Peripheral blood levels of matrix metalloproteinase-9 predict lesion volume in acute stroke. Neurol. Sci. 34, 379–382. doi: 10.1007/s10072-012-0999-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vartanian, K. B., Stevens, S. L., Marsh, B. J., Williams-Karnesky, R., Lessov, N. S., and Stenzel-Poore, M. P. (2011). LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflamm. 8:140. doi: 10.1186/1742-2094-8-140
Vila, N., Castillo, J., Davalos, A., Esteve, A., Planas, A. M., and Chamorro, A. (2003). Levels of anti-inflammatory cytokines and neurological worsening in acute ischemic stroke. Stroke 34, 671–675. doi: 10.1161/01.STR.0000057976.53301.69
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wacker, B. K., Perfater, J. L., and Gidday, J. M. (2012). Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem. 123, 954–962. doi: 10.1111/jnc.12047
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waje-Andreassen, U., Kråkenes, J., Ulvestad, E., Thomassen, L., Myhr, K. M., Aarseth, J., et al. (2005). IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol. Scand. 111, 360–365. doi: 10.1111/j.1600-0404.2005.00416.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walton, K. M., DiRocco, R., Bartlett, B. A., Koury, E., Marcy, V. R., Jarvis, B., et al. (1998). Activation of p38MAPK in microglia after ischemia. J. Neurochem. 70, 1764–1767. doi: 10.1046/j.1471-4159.1998.70041764.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Huang, J., Li, Y., and Yang, G. Y. (2012). Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr. Drug Targets 13, 166–172. doi: 10.2174/138945012799201603
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., Yue, T. L., Barone, F. C., and Feuerstein, G. Z. (1995). Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke 26, 661–665. doi: 10.1161/01.STR.26.4.661
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wei, Y., Yemisci, M., Kim, H. H., Yung, L. M., Shin, H. K., Hwang, S. K., et al. (2011). Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 69, 119–129. doi: 10.1002/ana.22186
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Whiteley, W., Jackson, C., Lewis, S., Lowe, G., Rumley, A., Sandercock, P., et al. (2009). Inflammatory markers and poor outcome after stroke: a prospective cohort study and systematic review of interleukin-6. PLoS Med. 6:e1000145. doi: 10.1371/journal.pmed.1000145
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wong, C. H., and Crack, P. J. (2008). Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr. Med. Chem. 15, 1–14. doi: 10.2174/092986708783330665
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xiong, X., Barreto, G. E., Xu, L., Ouyang, Y. B., Xie, X., and Giffard, R. G. (2011). Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42, 2026–2032. doi: 10.1161/STROKEAHA.110.593772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Qian, L., Zong, G., Ma, K., Zhu, X., Zhang, H., et al. (2012). Class A scavenger receptor promotes cerebral ischemic injury by pivoting microglia/macrophage polarization. Neuroscience 218, 35–48. doi: 10.1016/j.neuroscience.2012.05.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamashita, K., Gerken, U., Vogel, P., Hossmann, K., and Wiessner, C. (1999). Biphasic expression of TGF-beta1 mRNA in the rat brain following permanent occlusion of the middle cerebral artery. Brain Res. 836, 139–145. doi: 10.1016/S0006-8993(99)01626-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamashita, T., Sawamoto, K., Suzuki, S., Suzuki, N., Adachi, K., Kawase, T., et al. (2005). Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J. Neurochem. 94, 459–468. doi: 10.1111/j.1471-4159.2005.03227.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, J., Greer, J. M., and McCombe, P. A. (2012). Prolonged elevation of cytokine levels after human acute ischaemic stroke with evidence of individual variability. J. Neuroimmunol. 246, 78–84. doi: 10.1016/j.jneuroim.2012.02.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, Y. P., Sailor, K. A., Lang, B. T., Park, S. W., Vemuganti, R., and Dempsey, R. J. (2007). Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 27, 1213–1224. doi: 10.1038/sj.jcbfm.9600432
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Estrada, E. Y., Thompson, J. F., Liu, W., and Rosenberg, G. A. (2007). Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 27, 697–709. doi: 10.1038/sj.jcbfm.9600375
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Q. W., Lu, F. L., Zhou, Y., Wang, L., Zhong, Q., Lin, S., et al. (2011). HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J. Cereb. Blood Flow Metab. 31, 593–605. doi: 10.1038/jcbfm.2010.129
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Y., and Rosenberg, G. A. (2011). MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol. Biol. 762, 333–345. doi: 10.1007/978-1-61779-185-7_24
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yemisci, M., Gursoy-Ozdemir, Y., Vural, A., Can, A., Topalkara, K., and Dalkara, T. (2009). Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037. doi: 10.1038/nm.2022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yilmaz, G., Arumugam, T. V., Stokes, K. Y., and Granger, D. N. (2006). Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation 113, 2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yilmaz, G., and Granger, D. N. (2010). Leukocyte recruitment and ischemic brain injury. Neuromol. Med. 12, 193–204. doi: 10.1007/s12017-009-8074-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, L., Ohtaki, H., Nakamachi, T., Kudo, Y., Makino, R., and Shioda, S. (2004). Delayed expressed TNFR1 co-localize with ICAM-1 in astrocyte in mice brain after transient focal ischemia. Neurosci. Lett. 370, 30–35. doi: 10.1016/j.neulet.2004.07.083
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yokota, C., Minematsu, K., Tomii, Y., Naganuma, M., Ito, A., Nagasawa, H., et al. (2009). Low levels of plasma soluble receptor for advanced glycation end products are associated with severe leukoaraiosis in acute stroke patients. J. Neurol. Sci. 287, 41–44. doi: 10.1016/j.jns.2009.09.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, J. T., Lee, C. H., Yoo, K.-Y., Choi, J. H., Li, H., Park, O. K., et al. (2010). Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. J. Neurol. Sci. 296, 69–78. doi: 10.1016/j.jns.2010.06.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, H. M., Xu, J., Li, C., Zhou, C., Zhang, F., Han, D., et al. (2008). Coupling between neuronal nitric oxide synthase and glutamate receptor 6-mediated c-Jun N-terminal kinase signaling pathway via S-nitrosylation contributes to ischemia neuronal death. Neuroscience 155, 1120–1132. doi: 10.1016/j.neuroscience.2008.03.061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zarruk, J. G., Fernández-López, D., García-Yébenes, I., García-Gutiérrez, M. S., Vivancos, J., Nombela, F., et al. (2012). Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 43, 211–219. doi: 10.1161/STROKEAHA.111.631044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeng, L., Liu, J., Wang, Y., Wang, L., Wenig, S., Chen, S., et al. (2013). Cocktail blood biomarkers: prediction of clinical outcomes in patients with acute ischemic stroke. Eur. Neurol. 69, 68–75. doi: 10.1159/000342896
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhai, D. X., Kong, Q. F., Xu, W. S., Bai, S. S., Peng, H. S., Zhao, K., et al. (2008). RAGE expression is up-regulated in human cerebral ischemia and pMCAO rats. Neurosci. Lett. 445, 117–121. doi: 10.1016/j.neulet.2008.08.077
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, F. Y., Chen, X. C., Ren, H. M., and Bao, W. M. (2006). Effects of ischemic preconditioning on blood-brain barrier permeability and MMP-9 expression of ischemic brain. Neurol. Res. 28, 21–24. doi: 10.1179/016164106X91825
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Chopp, M., and Powers, C. (1997). Temporal profile of microglial response following transient (2h) middle cerebral artery occlusion. Brain Res. 744, 189–198. doi: 10.1016/S0006-8993(96)01085-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, L., Dong, L. Y., Li, Y. J., Hong, Z., and Wei, W. S. (2012). The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor. J. Neuroinflamm. 9:211. doi: 10.1186/1742-2094-9-211
Zhang, K., McQuibban, G. A., Silva, C., Butler, G. S., Johnston, J. B., Holden, J., et al. (2003). HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 6, 1064–1071. doi: 10.1038/nn1127
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, B. Q., Wang, S., Kim, H. Y., Storrie, H., Rosen, B. R., Mooney, D. J., et al. (2006). Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 12, 441–445. doi: 10.1038/nm1387
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, W., Khachi, S., Hao, Q., Shen, F., Young, W. L., Yang, G. Y., et al. (2008). Upregulation of EMMPRIN after permanent focal cerebral ischemia. Neurochem. Int. 52, 1086–1091. doi: 10.1016/j.neuint.2007.11.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, Y., Yang, G. Y., Ahlemeyer, B., Pang, L., Che, X. M., Culmsee, C., et al. (2002). Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J. Neurosci. 22, 3898–3909.
Zhu, J., Zhou, Z., Liu, Y., and Zheng, J. (2009). Fractalkine and CX3CR1 are involved in the migration of intravenously grafted human bone marrow stromal cells toward ischemic brain lesion in rats. Brain Res. 1287, 173–183. doi: 10.1016/j.brainres.2009.06.068
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: cytokines, immune system, ischemic stroke, ischemic tolerance, macrophages, neutrophils, preconditioning
Citation: Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I and Bagetta G (2015) Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 9:147. doi: 10.3389/fnins.2015.00147
Received: 26 January 2015; Accepted: 09 April 2015;
Published: 29 April 2015.
Edited by:
Giuseppe Pignataro, Federico II University of Naples, ItalyReviewed by:
Samir Kumar-Singh, University of Antwerp, BelgiumJan Mulder, Karolinska Institute, Sweden
Copyright © 2015 Amantea, Micieli, Tassorelli, Cuartero, Ballesteros, Certo, Moro, Lizasoain and Bagetta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Amantea, Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Via P. Bucci, Ed. Polifunzionale, I-87036 Rende, Italy,YW1hbnRlYUB1bmljYWwuaXQ=