 Mitsuhiro Kato
Mitsuhiro Kato- Department of Pediatrics, Yamagata University Faculty of Medicine, Yamagata, Japan
Neuronal migration disorders are human (or animal) diseases that result from a disruption in the normal movement of neurons from their original birth site to their final destination during early development. As a consequence, the neurons remain somewhere along their migratory route, their location depending on the pathological mechanism and its severity. The neurons form characteristic abnormalities, which are morphologically classified into several types, such as lissencephaly, heterotopia, and cobblestone dysplasia. Polymicrogyria is classified as a group of malformations that appear secondary to post-migration development; however, recent findings of the underlying molecular mechanisms reveal overlapping processes in the neuronal migration and post-migration development stages. Mutations of many genes are involved in neuronal migration disorders, such as LIS1 and DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. ARX is of particular interest from basic and clinical perspectives because it is critically involved in tangential migration of GABAergic interneurons in the forebrain and its mutations cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. The recent advances in gene and genome analysis technologies will enable the genetic basis of neuronal migration disorders to be unraveled, which, in turn, will facilitate genotype-phenotype correlations to be determined.
Introduction
The characteristic six-layered neocortex in the human brain is formed by two types of neuron, projection neurons and interneurons, which migrate from their birth places, such as the ventricular zone and ganglionic eminence, respectively. Neuronal migration disorders are human (or animal) diseases that result from the disruption of normal movement of neurons from their original birth site to their final destination during early development. As a consequence, the neurons remain somewhere along their migratory route, their location depending on the pathological mechanism and its severity. Many genes have been found to be responsible for neuronal migration disorders, such as LIS1 and DCX in classical lissencephaly spectrum, TUBA1A in lissencephaly with cerebellar hypoplasia, ARX in X-linked lissencephaly with abnormal genitalia (XLAG), FLNA and ARGEF2 in periventricular heterotopia, FCMD and glycosylation-related genes, such as POMT1, POMT2, POMGNT1, POMGNT2, FKRP, LARGE, TMEM5, POMK, ISPD, GMPPB, B3GNT1, and B3GALNT2 in cobblestone dysplasias, GPR56, SRPX2, and some tubulin-related genes, e.g., TUBA8, TUBB2B, and TUBB3, in polymicrogyria (Kato and Dobyns, 2003; Vuillaumier-Barrot et al., 2012; Buysse et al., 2013; Stevens et al., 2013; Fry et al., 2014). Recently, we found that mutations in COL4A1, which encodes type IV collagen alpha 1 subunit, cause schizencephaly accompanied by polymicrogyria in the adjacent cortex of the transmantle cleft as well as focal cortical dysplasia (Yoneda et al., 2013). Historically, brain malformations including neuronal migration disorders have been classified based on a postmortem examination. The advancement and spread of neuroimaging techniques, particularly magnetic resonance imaging (MRI), make it easier to find out many types of brain malformations, but make it more complicated to classify them. Moreover, the unveiling of responsible genes for brain malformations has changed the classification scheme and causes most neuroscientists and even physicians trouble to follow it. Here, I review the clinical manifestation of neuronal migration disorders, focusing mainly on lissencephaly, in terms of genotype-phenotype correlations.
Lissencephaly Spectrum: Classical Lissencephaly to Subcortical Band Heterotopia
Lissencephaly is classified as a spectrum of disorders caused by widespread abnormal transmantle migration, ranging from classical lissencephaly (agyria or pachygyria) to subcortical band heterotopia or double-cortex syndrome (Barkovich et al., 2012). Classical lissencephaly is characterized by a smooth (lissos in Greek) brain surface with a decreased number of sulci and wide gyri. Mutations in LIS1, located on chromosome 17p13.3, or DCX on Xq23 are the main cause for classical lissencephaly (Table 1) (Kato and Dobyns, 2003). Mutations in DCX are causative for classical lissencephaly in male individuals and subcortical band heterotopia in female individuals. A combination of a severity grading scale [the most severe form, Grade 1 (total agyria) to the mildest form, Grade 6 (subcortical band heterotopia) via the intermediate forms comprised of a combination of agyria, pachygyria, and subcortical band heterotopia] and an anterior or posterior gradient scale is useful to predict the causative gene for lissencephaly spectrum (Kato and Dobyns, 2003). For instance, mutations of LIS1, ARX, or TUBA1A result in a posterior more severe than anterior gradient, while mutations of DCX or RELN lead to an anterior more severe than posterior gradient. LIS1 participates in cytoplasmic dynein-mediated nucleokinesis, somal translocation, and cell motility (Smith et al., 2000) as well as mitosis or neurogenesis and chromosomal segregation (Faulkner et al., 2000). DCX is a microtubule-associated protein and is involved in microtubule polymerization and stabilization (Gleeson et al., 1999). Missense mutations in DCX responsible for lissencephaly spectrum are mainly located in two tandem repeats (N-terminal or C-terminal doublecortin domains), which bind to microtubules or free tubulin and other components (Friocourt et al., 2005), respectively.
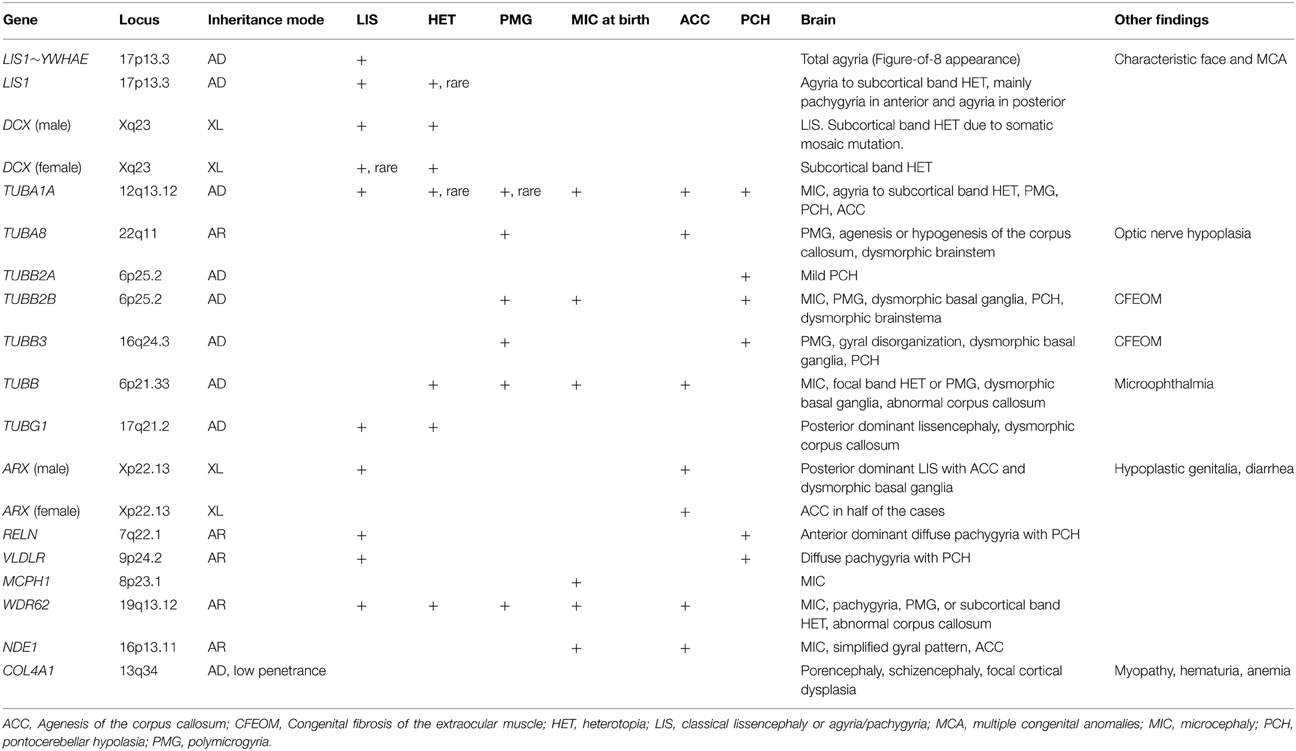
Table 1. Clinical features of gene mutations causing cortical disruptions.
MRI of the brain is useful to discriminate agyria, pachygyria, and subcortical band heterotopia. Agyria is generally characterized by the disappearance of deep sulci in more than one lobe and the thickness of the cortex is 10–20 mm (Figure 1). The gyri in pachygyria are wider than in the normal cortex and the thickness of the cortex is 4–9 mm (Figure 2). Brain MRI of subcortical band heterotopia shows bilateral continuous symmetric bands of gray matter underlying an almost normal cortical mantle with relatively shallow sulci (Figure 3). More than 90% of patients with subcortical band heterotopia are female and the cause is usually heterozygous DCX mutation. Subcortical band heterotopia in male patients is caused by somatic mosaic DCX mutations or LIS1 mutations (Gleeson et al., 2000; Kato et al., 2001; D'agostino et al., 2002; Poolos et al., 2002). Coexistence of agyria and pachygyria or pachygyria and subcortical band heterotopia can be seen in the same patient, suggesting common mechanisms for these phenotypes. Microscopically, agyria and pachygyria present a four-layered cortex with an outer molecular layer, superficial layer, cell sparse layer, and deep cellular layer. In the marginal zone between pachygyria and subcortical band heterotopia, the outer molecular layer corresponds to layer I of the normal six-layered cortex, the superficial layer corresponds to layers II–VI, the cell sparse layer corresponds to subcortical white matter, and the deep cellular layer corresponds to band heterotopia with a mass of unlayered ectopic neurons (Figure 4). The primary pathology of lissencephaly due to the DCX mutations shows only minor differences compared with that caused by LIS1 mutations, for example, inferior olivary ectopia is present in LIS1 mutation brains but is absent in DCX mutation brain (Berg et al., 1998); however, Viot et al. report a different cortical architecture for DCX lissencephaly (Viot et al., 2004).
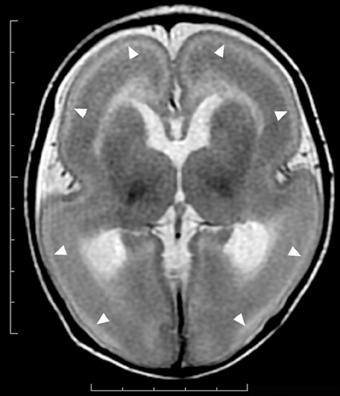
Figure 1. Complete agyria in a DCX mutation patient (Grade 1 on the severity scale). T2-weighted axial MRI image. Wide shallow sylvian fissures create a figure-of-eight appearance. The thickness of the cortex is over 10 mm. A high-intensity (white) line (arrow heads) beneath the cerebral surface is consistent with a cell sparse layer of the four-layered cortex.
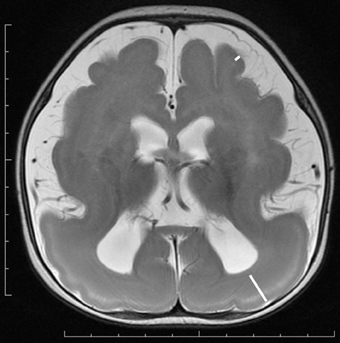
Figure 2. Anterior pachygyria and posterior agyria in a LIS1 mutation patient (Grade 3 on the severity scale). T2-weighted axial MRI image. Note the difference in the width of gyri, the depth of sulci and the thickness of the cortex (bars) between anterior and posterior regions.
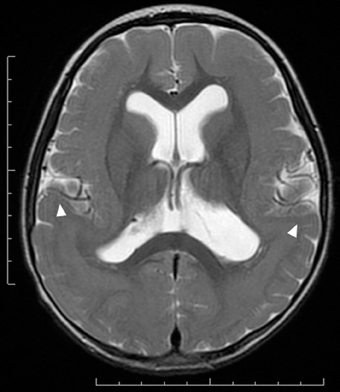
Figure 3. Subcortical band heterotopia or double cortex syndrome in a DCX mutation patient (Grade 5 on the severity scale). T2-weighted axial MRI image. Subcortical heterotopic gray matter in the posterior region fuses into the pachygyric cortex in the anterior region (arrowheads).
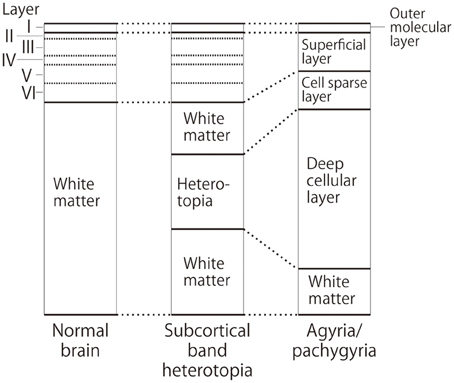
Figure 4. Schematic diagram of cortical layers in the lissencephaly spectrum compared to the normal brain. Deep cellular layer of the pachygyric or agyric cortex fuses with laminar or band heterotopia in the subcortical white matter, but not with normal six-layered cortex.
The severity of the clinical manifestations of lissencephaly spectrum is correlated with the degree of brain malformation. Patients with agyria show severe muscle hypotonia from infancy (known as floppy infant) and achieve neither head control nor are they able to say meaningful words. A specific form of epileptic seizure, epileptic spasms, occurs in 80% of patients with agyria or pachygyria, although electroencephalography (EEG) may not present with typical hypsarrhythmia, which is characteristically seen in infantile spasms or West syndrome (Guerrini, 2005). However, the main clinical features of subcortical band heterotopia are intellectual disability and epileptic seizures, both of which are milder than those of agyria or pachygyria. Intellectual disability ranges from normal to severe retardation and correlates with the thickness of the band and the degree of pachygyria (Barkovich et al., 1994; Bahi-Buisson et al., 2013). Genetic counseling is particularly important for parents that have a boy with classical lissencephaly or a girl with subcortical band heterotopia because the mother may be a heterozygous carrier of the DCX mutation.
Miller-Dieker syndrome is a contiguous gene syndrome caused by a microdeletion in 17p13.3, a region that contains LIS1 and YWHAE (which encodes 14-3-3 protein epsilon). Phenotypes of Miller-Dieker syndrome are more severe than that of classical lissencephaly because of an isolated LIS1 mutation. They are characterized by complete agyria and facial abnormalities including prominent forehead, bitemporal hollowing, short nose with upturned nares, prominent upper lip with downturned vermilion border and small jaw, and sometimes other congenital defects involving the heart, kidneys, intestine, or fingers (Kato and Dobyns, 2003). Neurological findings of Miller–Dieker syndrome are similar to those of patients with agyria, such as severe developmental delay with weak muscle tone and profound intellectual disability, intractable seizures, dysphagia, and poor prognosis with recurrent infection of the respiratory system.
Tubulin-Related Disorders, Tubulinopathies
Microtubules provide the main structural framework for the shafts of axons and dendrites, and with actin serve as tracks for intracellular trafficking and to provide the driving force underlying neurite extension and intracellular movement of organelles during mitosis (Flynn et al., 2013). Recently, genes involved in microtubule function have been identified to be causative for various human diseases, such as lissencephaly (Keays et al., 2007; Poirier et al., 2007), polymicrogyria (Abdollahi et al., 2009; Jaglin et al., 2009; Jansen et al., 2011), simplified gyral patter in which the cortical thickness is normal (Cushion et al., 2014), complex brain malformations (Poirier et al., 2010, 2013; Breuss et al., 2012), abnormal eye movement (Tischfield et al., 2010), torsion dystonia (Hersheson et al., 2013), and hypomyelinating leukodystrophy (Simons et al., 2013). All the above are classified as tubulinopathies (Cushion et al., 2013; Bahi-Buisson et al., 2014). Microtubules are assembled from soluble tubulin heterodimers consisting of alpha- and beta-tubulin. Multiple isoforms of both tubulins are encoded by different genes. Mutations of TUBA1A, which encodes alpha tubulin, cause lissencephaly spectrum, particularly diffuse agyria or perisylvian pachygyria, with microcephaly, agenesis of the corpus callosum, and cerebellar hypoplasia (Figure 5) (Bahi-Buisson et al., 2008). TUBA1A mutations account for only 1% of isolated classical lissencephaly; however, they account for approximately 30% of patients with lissencephaly associated with cerebellar hypoplasia (Kumar et al., 2010). Dysgenesis of the anterior limb of the internal capsule and disorganization of the hippocampus are other neuroimaging features for TUBA1A mutation (Poirier et al., 2007). Mutations of TUBA1A cause polymicrogyria as well. Interestingly, mutations of TUBB2B cause polymicrogyria with or without congenital fibrosis of the external ocular muscles as well as bilateral perisylvian pachygyria(Cederquist et al., 2012; Romaniello et al., 2014). Polymicrogyria is classified as a group of malformations that appear secondary to post-migration development; however, recent findings of the underlying molecular mechanisms reveal overlapping process in neuronal migration and post-migration development stages.
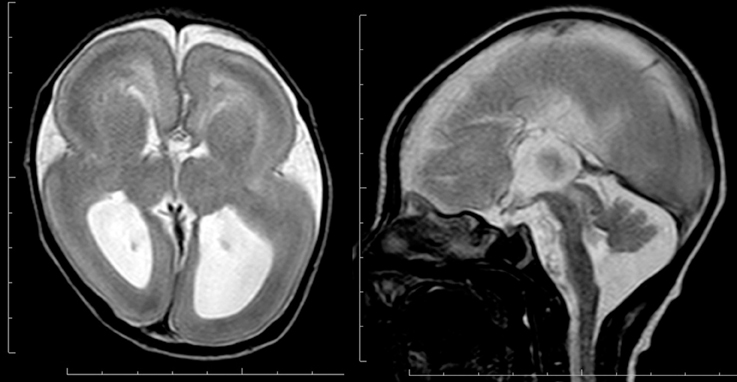
Figure 5. Complete agyria in a TUBA1A mutation patient (Grade 1 on the severity scale). T2-weighted axial MRI image (left) and midsagittal image (right). The boundary of the caudate nucleus and lentiform nucleus is obscure. Complete agenesis of the corpus callosum and pontocerebellar hypoplasia are also seen.
Mutations of TUBA8 cause polymicrogyria with optic nerve hypoplasia and display autosomal recessive inheritance (Abdollahi et al., 2009). Mutations of TUBB2A, which encodes beta-tubulin, cause infantile-onset epilepsy with simplified gyral patterning (Cederquist et al., 2012; Cushion et al., 2014; Romaniello et al., 2014). Mutations of TUBB3 cause two distinct forms. One is congenital fibrosis of the external ocular muscles or oculomotor nerve hypoplasia and later-onset peripheral axon degeneration with dysgenesis of the corpus callosum, anterior commissure, and internal capsule, but with no cortical dysplasia suggesting migrational defects (Tischfield et al., 2010). Another is cortical dysgenesis including polymicrogyria, pontocerebellar hypoplasia, and abnormal basal ganglia, but with no ocular motility defects (Poirier et al., 2010). The main mechanisms underlying the phenotypes caused byTUBB3 mutations are impaired axon guidance owing to disrupted microtubule dynamics and kinesin interaction (Tischfield et al., 2010). Tubulinopathies caused by the mutations of the genes encoding alpha- or beta-tubulin demonstrate more extensive phenotypes compared to other gene mutations, such as LIS1, DCX, or RELN. Mutations of TUBA1A, which encodes alpha-tubulin 1A, is the most frequently found in patients with brain malformations, while more genes encoding beta-tubulin, such as TUBB2A, TUBB2B, TUBB3, TUBB4A, and TUBB, are identified in a wide spectrum of disorders besides brain malformations. Pathological mechanisms and discrepancy between alpha- and beta-tubulinopathies should be elucidated.
ARX-Related Disorders, Interneuronopathies
The embryonic cerebral cortex at the stage of neuronal migration contains neuronal cells with two modes of migration; radial migration from the ventricular zone toward the pia and tangential migration from ganglionic eminence along a tangential trajectory into the developing cortex. Radially migrating neurons in the cortex are mainly excitatory projection neurons expressing glutamate as a neurotransmitter. Tangentially migrating neurons are inhibitory interneurons expressing the neurotransmitter GABA. XLAG is caused by mutation of ARX, which is expressed in the embryonic ganglionic eminence, neocortex, and hippocampus and plays important roles in neuronal proliferation, interneuronal migration, and differentiation in the embryonic forebrain, as well as a secondary role in differentiation of the testes (Kitamura et al., 2002). Patients with XLAG present occipital-predominant classical lissencephaly, particularly anterior pachygyria and posterior agyria, or a simplified gyral pattern, agenesis of the corpus callosum, and abnormal basal ganglia (Kato et al., 2004). In the most severe form of XLAG, patients show hydranencephaly with a large occipital cavity. Female carriers of ARX mutations causing XLAG have a risk of agenesis of the corpus callosum with no cortical defects. Abnormalities of external genitalia range from hypoplastic penis or undescended testes to complete female appearance, while the karyotype is 46,XY. Neuropathological studies show a complete loss or a decreased number of cortical interneurons in human XLAG and in Arx-null mice (Bonneau et al., 2002; Kitamura et al., 2002) and a three-layered cortex in human XLAG (Forman et al., 2005). Patients with XLAG show intractable seizures soon after birth, suggesting a great disparity between excitatory projection neurons and inhibitory interneurons. ARX mutations in patients with XLAG are null mutations or non-conservative missense mutations at critical amino acids in the homeodomain, while other missense mutations or expansion mutations in the polyalanine tract result in X-linked intellectual disability with or without dystonia, West syndrome, Ohtahara syndrome, or early infantile epileptic encephalopathy with suppression burst on EEG but with no brain malformation (Bienvenu et al., 2002; Stromme et al., 2002; Guerrini et al., 2007; Kato et al., 2007, 2010). Interestingly, longer polyalanine expansion is correlated with more severe and earlier onset phenotypes. A wide spectrum of ARX-related disorders forms a group of interneuronopathies based on the role of ARX during neurogenesis, as seen in patients and in the Arx-null mouse model (Kato and Dobyns, 2005; Marsh et al., 2009).
Classical Lissencephalies Associated with Other Forms of Brain Malformation
Classical lissencephaly caused by LIS1 or DCX mutations usually exist in isolated forms and only show cortical dysplasia on brain MRI. Rare variant forms of lissencephaly are associated with congenital microcephaly, cerebellar hypoplasia, or agenesis of the corpus callosum. Each form demonstrates characteristic radiological findings and some of the causative genes have been identified.
A lissencephaly group with cerebellar hypoplasia can be classified into several types according to brain imaging, additional clinical features, and causative genes (Ross et al., 2001). Among them, frontal predominant mild lissencephaly (diffuse pachygyria) with severe hippocampal and cerebellar hypoplasia or Reelin-type lissencephaly is caused by mutation of either RELN or VLDLR and shows autosomal recessive inheritance (Hong et al., 2000; Boycott et al., 2005). Dysequilibrium syndrome is an allelic disorder of the VLDLR locus (Moheb et al., 2008). Reelin-type lissencephaly has an inverted or no clear pattern of cortical lamination attributable to abnormal migration of the neurons in an outside-in birth order (Cooper, 2008; Dekimoto et al., 2010).
Lissencephaly can be associated with congenital microcephaly, though the head circumference of lissencephaly caused by the LIS1 or DCX mutations is usually within the normal range. Lissencephaly with a head circumference of less than −3 SD at birth is classified as microlissencephaly (Barkovich et al., 2005) or microcephaly with lissencephaly (Barkovich et al., 2012). Although many genes identified to be responsible for primary microcephaly, such as MCPH1, ASPM, CENPJ, CDK5RAP2, and PNKP, are involved with the cell-cycle phase of mitosis affecting neurogenesis (Barbelanne and Tsang, 2014), the causative genes for microlissencephaly remain unknown in many cases. Mutations of WDR62, which encodes a protein localized to centrosomes throughout mitosis and nucleoli during interphase, cause microcephaly with pachygyria or polymicrogyria (Bilguvar et al., 2010). Mutations of NDE1, which encodes a protein that binds dynein and functions in centrosome duplication, as well as the TUBA1A mutations mentioned above, cause microcephaly with a simplified gyral pattern, agenesis of the corpus callosum, and cerebellar hypoplasia (Alkuraya et al., 2011; Bakircioglu et al., 2011).
Conclusion
Neuronal migration disorders are classified based on causative genes as well as on brain MRI and neuropathological findings. There are strong relationships between clinical manifestations and mutation of a particular gene, in accordance with the expression and functions of that gene. Recent advances in gene and genome analysis technology will enable the genetic basis of neuronal migration disorders to be readily determined, facilitating the elucidation of genotype-phenotype correlations.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is grateful to the patients and their families for their kind cooperation. This study was partially funded by the Ministry of Health, Labour and Welfare of Japan (26310301), and an Intramural Research Grant (24-7, 27-5) for Neurological and Psychiatric Disorders from the National Center of Neurology & Psychiatry. This study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdollahi, M. R., Morrison, E., Sirey, T., Molnar, Z., Hayward, B. E., Carr, I. M., et al. (2009). Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet. 85, 737–744. doi: 10.1016/j.ajhg.2009.10.007
Alkuraya, F. S., Cai, X., Emery, C., Mochida, G. H., Al-Dosari, M. S., Felie, J. M., et al. (2011). Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am. J. Hum. Genet. 88, 536–547. doi: 10.1016/j.ajhg.2011.04.003
Bahi-Buisson, N., Poirier, K., Boddaert, N., Saillour, Y., Castelnau, L., Philip, N., et al. (2008). Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J. Med. Genet. 45, 647–653. doi: 10.1136/jmg.2008.058073
Bahi-Buisson, N., Poirier, K., Fourniol, F., Saillour, Y., Valence, S., Lebrun, N., et al. (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137, 1676–1700. doi: 10.1093/brain/awu082
Bahi-Buisson, N., Souville, I., Fourniol, F. J., Toussaint, A., Moores, C. A., Houdusse, A., et al. (2013). New insights into genotype-phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 136, 223–244. doi: 10.1093/brain/aws323
Bakircioglu, M., Carvalho, O. P., Khurshid, M., Cox, J. J., Tuysuz, B., Barak, T., et al. (2011). The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am. J. Hum. Genet. 88, 523–535. doi: 10.1016/j.ajhg.2011.03.019
Barbelanne, M., and Tsang, W. Y. (2014). Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res. Int. 2014:547986. doi: 10.1155/2014/547986
Barkovich, A. J., Guerrini, R., Battaglia, G., Kalifa, G., N'guyen, T., Parmeggiani, A., et al. (1994). Band heterotopia: correlation of outcome with magnetic resonance imaging parameters. Ann. Neurol. 36, 609–617. doi: 10.1002/ana.410360409
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., and Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135, 1348–1369. doi: 10.1093/brain/aws019
Barkovich, A. J., Kuzniecky, R. I., Jackson, G. D., Guerrini, R., and Dobyns, W. B. (2005). A developmental and genetic classification for malformations of cortical development. Neurology 65, 1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d
Berg, M. J., Schifitto, G., Powers, J. M., Martinez-Capolino, C., Fong, C. T., Myers, G. J., et al. (1998). X-linked female band heterotopia-male lissencephaly syndrome. Neurology 50, 1143–1146. doi: 10.1212/WNL.50.4.1143
Bienvenu, T., Poirier, K., Friocourt, G., Bahi, N., Beaumont, D., Fauchereau, F., et al. (2002). ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum. Mol. Genet. 11, 981–991. doi: 10.1093/hmg/11.8.981
Bilguvar, K., Ozturk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., et al. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. doi: 10.1038/nature09327
Bonneau, D., Toutain, A., Laquerriere, A., Marret, S., Saugier-Veber, P., Barthez, M. A., et al. (2002). X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann. Neurol. 51, 340–349. doi: 10.1002/ana.10119
Boycott, K. M., Flavelle, S., Bureau, A., Glass, H. C., Fujiwara, T. M., Wirrell, E., et al. (2005). Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am. J. Hum. Genet. 77, 477–483. doi: 10.1086/444400
Breuss, M., Heng, J. I., Poirier, K., Tian, G., Jaglin, X. H., Qu, Z., et al. (2012). Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2, 1554–1562. doi: 10.1016/j.celrep.2012.11.017
Buysse, K., Riemersma, M., Powell, G., van Reeuwijk, J., Chitayat, D., Roscioli, T., et al. (2013). Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet. 22, 1746–1754. doi: 10.1093/hmg/ddt021
Cederquist, G. Y., Luchniak, A., Tischfield, M. A., Peeva, M., Song, Y., Menezes, M. P., et al. (2012). An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum. Mol. Genet. 21, 5484–5499. doi: 10.1093/hmg/dds393
Cooper, J. A. (2008). A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 31, 113–119. doi: 10.1016/j.tins.2007.12.003
Cushion, T. D., Dobyns, W. B., Mullins, J. G., Stoodley, N., Chung, S. K., Fry, A. E., et al. (2013). Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 136, 536–548. doi: 10.1093/brain/aws338
Cushion, T. D., Paciorkowski, A. R., Pilz, D. T., Mullins, J. G., Seltzer, L. E., Marion, R. W., et al. (2014). De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am. J. Hum. Genet. 94, 634–641. doi: 10.1016/j.ajhg.2014.03.009
D'agostino, M. D., Bernasconi, A., Das, S., Bastos, A., Valerio, R. M., Palmini, A., et al. (2002). Subcortical band heterotopia (SBH) in males: clinical, imaging and genetic findings in comparison with females. Brain 125, 2507–2522. doi: 10.1093/brain/awf248
Dekimoto, H., Terashima, T., and Katsuyama, Y. (2010). Dispersion of the neurons expressing layer specific markers in the reeler brain. Dev. Growth Differ. 52, 181–193. doi: 10.1111/j.1440-169X.2009.01153.x
Faulkner, N. E., Dujardin, D. L., Tai, C. Y., Vaughan, K. T., O'connell, C. B., Wang, Y., et al. (2000). A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2, 784–791. doi: 10.1038/35041020
Flynn, K. C., Stiess, M., and Bradke, F. (2013). “Role of the cytoskeleton and membrane trafficking in axon-dendrite morphogenesis,” in Cellular Migration and Formation of Neuronal Connections: Comprehensive Developmental Neuroscience, eds J. L. Rubenstein and P. Rakic (San Diego,CA: Academic Press), 82–116.
Forman, M. S., Squier, W., Dobyns, W. B., and Golden, J. A. (2005). Genotypically defined lissencephalies show distinct pathologies. J. Neuropathol. Exp. Neurol. 64, 847–857. doi: 10.1097/01.jnen.0000182978.56612.41
Friocourt, G., Kappeler, C., Saillour, Y., Fauchereau, F., Rodriguez, M. S., Bahi, N., et al. (2005). Doublecortin interacts with the ubiquitin protease DFFRX, which associates with microtubules in neuronal processes. Mol. Cell. Neurosci. 28, 153–164. doi: 10.1016/j.mcn.2004.09.005
Fry, A. E., Cushion, T. D., and Pilz, D. T. (2014). The genetics of lissencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 198–210. doi: 10.1002/ajmg.c.31402
Gleeson, J. G., Lin, P. T., Flanagan, L. A., and Walsh, C. A. (1999). Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23, 257–271. doi: 10.1016/S0896-6273(00)80778-3
Gleeson, J. G., Minnerath, S., Kuzniecky, R. I., Dobyns, W. B., Young, I. D., Ross, M. E., et al. (2000). Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am. J. Hum. Genet. 67, 574–581. doi: 10.1086/303043
Guerrini, R. (2005). Genetic malformations of the cerebral cortex and epilepsy. Epilepsia 46(Suppl. 1), 32–37. doi: 10.1111/j.0013-9580.2005.461010.x
Guerrini, R., Moro, F., Kato, M., Barkovich, A. J., Shiihara, T., McShane, M. A., et al. (2007). Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 69, 427–433. doi: 10.1212/01.wnl.0000266594.16202.c1
Hersheson, J., Mencacci, N. E., Davis, M., MacDonald, N., Trabzuni, D., Ryten, M., et al. (2013). Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann. Neurol. 73, 546–553. doi: 10.1002/ana.23832
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O., et al. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96. doi: 10.1038/79246
Jaglin, X. H., Poirier, K., Saillour, Y., Buhler, E., Tian, G., Bahi-Buisson, N., et al. (2009). Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 41, 746–752. doi: 10.1038/ng.380
Jansen, A. C., Oostra, A., Desprechins, B., De Vlaeminck, Y., Verhelst, H., Regal, L., et al. (2011). TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology 76, 988–992. doi: 10.1212/WNL.0b013e31821043f5
Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K., Abuelo, D. N., et al. (2004). Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 23, 147–159. doi: 10.1002/humu.10310
Kato, M., and Dobyns, W. B. (2003). Lissencephaly and the molecular basis of neuronal migration. Hum. Mol. Genet. 12 Spec No 1, R89–R96. doi: 10.1093/hmg/ddg086
Kato, M., and Dobyns, W. B. (2005). X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy.” J. Child Neurol. 20, 392–397. doi: 10.1177/08830738050200042001
Kato, M., Kanai, M., Soma, O., Takusa, Y., Kimura, T., Numakura, C., et al. (2001). Mutation of the doublecortin gene in male patients with double cortex syndrome: somatic mosaicism detected by hair root analysis. Ann. Neurol. 50, 547–551. doi: 10.1002/ana.1231
Kato, M., Koyama, N., Ohta, M., Miura, K., and Hayasaka, K. (2010). Frameshift mutations of the ARX gene in familial Ohtahara syndrome. Epilepsia 51, 1679–1684. doi: 10.1111/j.1528-1167.2010.02559.x
Kato, M., Saitoh, S., Kamei, A., Shiraishi, H., Ueda, Y., Akasaka, M., et al. (2007). A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 81, 361–366. doi: 10.1086/518903
Keays, D. A., Tian, G., Poirier, K., Huang, G. J., Siebold, C., Cleak, J., et al. (2007). Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57. doi: 10.1016/j.cell.2006.12.017
Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-Kogo, A., Kusaka, M., et al. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369. doi: 10.1038/ng1009
Kumar, R. A., Pilz, D. T., Babatz, T. D., Cushion, T. D., Harvey, K., Topf, M., et al. (2010). TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 19, 2817–2827. doi: 10.1093/hmg/ddq182
Marsh, E., Fulp, C., Gomez, E., Nasrallah, I., Minarcik, J., Sudi, J., et al. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132, 1563–1576. doi: 10.1093/brain/awp107
Moheb, L. A., Tzschach, A., Garshasbi, M., Kahrizi, K., Darvish, H., Heshmati, Y., et al. (2008). Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome. Eur. J. Hum. Genet. 16, 270–273. doi: 10.1038/sj.ejhg.5201967
Poirier, K., Keays, D. A., Francis, F., Saillour, Y., Bahi, N., Manouvrier, S., et al. (2007). Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 28, 1055–1064. doi: 10.1002/humu.20572
Poirier, K., Lebrun, N., Broix, L., Tian, G., Saillour, Y., Boscheron, C., et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639–647. doi: 10.1038/ng.2613
Poirier, K., Saillour, Y., Bahi-Buisson, N., Jaglin, X. H., Fallet-Bianco, C., Nabbout, R., et al. (2010). Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 19, 4462–4473. doi: 10.1093/hmg/ddq377
Poolos, N. P., Das, S., Clark, G. D., Lardizabal, D., Noebels, J. L., Wyllie, E., et al. (2002). Males with epilepsy, complete subcortical band heterotopia, and somatic mosaicism for DCX. Neurology 58, 1559–1562. doi: 10.1212/WNL.58.10.1559
Romaniello, R., Arrigoni, F., Cavallini, A., Tenderini, E., Baschirotto, C., Triulzi, F., et al. (2014). Brain malformations and mutations in alpha- and beta-tubulin genes: a review of the literature and description of two new cases. Dev. Med. Child Neurol. 56, 354–360. doi: 10.1111/dmcn.12370
Ross, M. E., Swanson, K., and Dobyns, W. B. (2001). Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics 32, 256–263. doi: 10.1055/s-2001-19120
Simons, C., Wolf, N. I., McNeil, N., Caldovic, L., Devaney, J. M., Takanohashi, A., et al. (2013). A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 92, 767–773. doi: 10.1016/j.ajhg.2013.03.018
Smith, D. S., Niethammer, M., Ayala, R., Zhou, Y., Gambello, M. J., Wynshaw-Boris, A., et al. (2000). Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2, 767–775. doi: 10.1038/35041000
Stevens, E., Carss, K. J., Cirak, S., Foley, A. R., Torelli, S., Willer, T., et al. (2013). Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 92, 354–365. doi: 10.1016/j.ajhg.2013.01.016
Stromme, P., Mangelsdorf, M. E., Shaw, M. A., Lower, K. M., Lewis, S. M., Bruyere, H., et al. (2002). Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 30, 441–445. doi: 10.1038/ng862
Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87. doi: 10.1016/j.cell.2009.12.011
Viot, G., Sonigo, P., Simon, I., Simon-Bouy, B., Chadeyron, F., Beldjord, C., et al. (2004). Neocortical neuronal arrangement in LIS1 and DCX lissencephaly may be different. Am. J. Med. Genet. A 126, 123–128. doi: 10.1002/ajmg.a.20569
Vuillaumier-Barrot, S., Bouchet-Seraphin, C., Chelbi, M., Devisme, L., Quentin, S., Gazal, S., et al. (2012). Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 91, 1135–1143. doi: 10.1016/j.ajhg.2012.10.009
Keywords: lissencephaly, heterotopia, polymicrogyria, tubulinopathy, interneuronopathy, LIS1, DCX, ARX
Citation: Kato M (2015) Genotype-phenotype correlation in neuronal migration disorders and cortical dysplasias. Front. Neurosci. 9:181. doi: 10.3389/fnins.2015.00181
Received: 01 December 2014; Accepted: 06 May 2015;
Published: 21 May 2015.
Edited by:
Chiaki Ohtaka-Maruyama, Tokyo Metropolitan Institute of Medical Science, JapanReviewed by:
Brian Key, Asdasdasd, AustriaMasaharu Hayashi, Tokyo Metropolitan Institute of Medical Science, Japan
Copyright © 2015 Kato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitsuhiro Kato, Department of Pediatrics, Yamagata University Faculty of Medicine, 2-2-2 Iida-nishi, Yamagata 990-9585, Japan,a3RtdC1ocm9AdW1pbi5hYy5qcA==
†Present Address: Mitsuhiro Kato, Department of Pediatrics, Showa University School of Medicine, Tokyo, Japan