 Gaurav Bedse
Gaurav Bedse Fabio Di Domenico
Fabio Di Domenico Gaetano Serviddio
Gaetano Serviddio Tommaso Cassano
Tommaso Cassano- 1Department of Physiology and Pharmacology “V. Erspamer,” Sapienza University of Rome, Rome, Italy
- 2Department of Biochemical Sciences, Sapienza University of Rome, Rome, Italy
- 3Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy
- 4Department of Clinical and Experimental Medicine, University of Foggia, Foggia, Italy
Alzheimer's disease (AD) is the most common form of dementia affecting elderly people. AD is a multifaceted pathology characterized by accumulation of extracellular neuritic plaques, intracellular neurofibrillary tangles (NFTs) and neuronal loss mainly in the cortex and hippocampus. AD etiology appears to be linked to a multitude of mechanisms that have not been yet completely elucidated. For long time, it was considered that insulin signaling has only peripheral actions but now it is widely accepted that insulin has neuromodulatory actions in the brain. Insulin signaling is involved in numerous brain functions including cognition and memory that are impaired in AD. Recent studies suggest that AD may be linked to brain insulin resistance and patients with diabetes have an increased risk of developing AD compared to healthy individuals. Indeed insulin resistance, increased inflammation and impaired metabolism are key pathological features of both AD and diabetes. However, the precise mechanisms involved in the development of AD in patients with diabetes are not yet fully understood. In this review we will discuss the role played by aberrant brain insulin signaling in AD. In detail, we will focus on the role of insulin signaling in the deposition of neuritic plaques and intracellular NFTs. Considering that insulin mitigates beta-amyloid deposition and phosphorylation of tau, pharmacological strategies restoring brain insulin signaling, such as intranasal delivery of insulin, could have significant therapeutic potential in AD treatment.
Introduction
Alzheimer's disease (AD) is the most common cause of dementia. About 35.6 million people worldwide are now suffering from AD, and disease prevalence is expected to affect 115 million by 2050 (Wortmann, 2012). AD is a progressive, degenerative, and irreversible neurological disorder that causes deterioration of memory, judgment, and reasoning in the elderly (Querfurth and Laferla, 2010). Patients suffering from AD exhibit cognitive impairment, memory loss, and behavioral changes (Querfurth and Laferla, 2010). The neurodegeneration in AD is characterized by neuronal loss and synaptic injury (Dekosky and Scheff, 1990). The main pathological hallmarks of AD are extracellular insoluble beta amyloid (Aβ) plaques (Selkoe, 1989) and intracellular neurofibrillary tangles (NFTs). AD may be classified in two types based on genetic endowment. The first type is inherited via an autosomal dominant pattern, i.e., familial AD, and the second type is sporadic AD. Familial AD displays early disease onset, whereas sporadic AD cases mostly develop the disorder at an older age (Cruts and Van Broeckhoven, 1998). Although AD was discovered a century ago, the etiology of sporadic AD is not well understood. Over decades, it was hypothesized that neurodegeneration in AD is mainly caused by Aβ accumulation, phosphorylated tau aggregation, and/or neuroinflammation. However, recent human and preclinical studies have provided convincing evidence that AD is a degenerative metabolic disease, which is mediated by impairments in brain insulin responsiveness, glucose utilization, and energy metabolism leading to increased oxidative stress, inflammation, and worsening of insulin resistance (Hoyer, 2002, 2004; Schubert et al., 2004; Rivera et al., 2005; Steen et al., 2005; Watson and Craft, 2006; Craft, 2007; Neumann et al., 2008; Krikorian et al., 2010; Luchsinger, 2010; Baker et al., 2011; Talbot et al., 2012; Butterfield et al., 2014a,b; de La Monte, 2014).
The accumulating evidence that reduced glucose utilization and deficient energy metabolism occur early in the course of disease, suggests a role for impaired insulin signaling in the pathogenesis of neurodegenerative diseases. In brain, the insulin/insulin-like growth factor (IGF) signaling is important for neuronal growth, synaptic maintenance and neuroprotection (Stockhorst et al., 2004; Van Dam and Aleman, 2004). It is now proposed that impairments in brain insulin/IGF signaling is associated with increased accumulation of Aβ, phosphorylated tau, reactive oxygen/nitrogen species, pro-inflammatory and pro-apoptosis molecules (de La Monte et al., 2009; de La Monte, 2012a,b, 2014). Both restoration of insulin responsiveness and use of insulin therapy can lead to improved cognitive performance. This review focuses on the recent progress in our understanding of the neuronal insulin signaling in the pathogenesis and progression of AD. The review will also discuss therapeutic augmentation of brain insulin signaling by intranasal insulin delivery as a promising treatment for AD.
Insulin Signaling in the Brain
The role of insulin in the brain has been little studied compared with its role in peripheral tissue. However, there is evidence that insulin has important functions in the brain including metabolic, neurotrophic, neuromodulatory and neuroendocrine actions. Recent studies have demonstrated that insulin and insulin receptor (IR) are ubiquitously expressed in the brain as in peripheral tissues (Schulingkamp et al., 2000; Van Der Heide et al., 2006). Brain insulin levels can reach 10- to 100- fold greater than in plasma, especially in hippocampus, hypothalamus, cortex, olfactory bulb, substantia nigra and pituitary (Frolich et al., 1998; Van Der Heide et al., 2006). IRs are abundantly expressed in neurons and less abundantly in glia (Frolich et al., 1998). Insulin is a hormone that has wide range of functions. Insulin is important for cell growth and survival. In the brain, insulin, IGFs and their receptors regulate dendritic sprouting, neuronal stem cell activation, cell growth, repair, synaptic maintenance and neuroprotection (Craft and Watson, 2004; Hoyer, 2004; Stockhorst et al., 2004; Van Dam and Aleman, 2004; Kleinridders et al., 2014). Insulin not only regulates glucose and lipid metabolism in the brain, but also plays an important role in learning and memory (Zhao et al., 2004b). Insulin is actively transported across blood–brain-barrier (BBB) and it is also produced locally in the brain (Banks et al., 2012). Similar to insulin, IGF-1 is present also in rodent and human brain and can cross BBB (Duarte et al., 2012).
IR and IGF receptor type-1 (IGF-1R) are tetrameric glycoproteins that belong to the receptor tyrosine (Tyr) kinase superfamily, composed of two α and two β subunits (Schlessinger, 2000; Wada et al., 2005; Moloney et al., 2010; Duarte et al., 2012). Two different types of IR have been reported: neuron specific type (extensively expressed in neurons) and peripheral-like type (with lower density in glia cells) (Moreira et al., 2009). Due to structural and functional similarity, insulin and IGF-1 can activate both IR and IGF-1R, however they exhibit higher affinity to own receptors (<1 nM) (Conejo and Lorenzo, 2001). Once bound to their respective receptors, insulin or IGF-1 promotes autophosphorylation of tyrosin residue, triggering its intrinsic tyrosin activity and phosphorylating insulin receptor substrate (IRS) docking protein at tyrosine residue (Duarte et al., 2012). IRS-1 and IRS-2 are ubiquitously expressed and are the primary mediators of insulin-dependent mitogenesis and regulation of glucose metabolism in most cell types. IRS-1 was the first substrate identified and represents the prototype of the IRS family proteins, while IRS-2 was initially identified as an alternative substrate for the IR in animals with targeted disruption of the IRS-1 gene (Sesti et al., 2001). IRS is a critical switch in the insulin-signaling pathway, and also interacts with other receptor tyrosine kinases including IGF1/2, tropomyosin-related kinase receptor B (TrkB) and ErbB. The phosphorylation of IRS1 on tyrosine residues leads to the downstream activation of Akt, the mammalian target of rapamycin (mTOR) and glycogen synthase kinase 3 (GSK3), among other pathways; moreover, the phosphorylation of IRS1 on multiple serine (Ser) residues can inhibit IRS1 activity, leading to insulin resistance (Yarchoan et al., 2014). Several reports suggested that insulin signaling is altered in aging as well as in AD. Table 1 summarizes alteration of insulin signaling components in aging as well as in the AD. Figure 1 illustrates how aberrant insulin signaling is linked to AD pathology and how it forms a vicious cycle, which results in deterioration of learning and memory and neuronal loss. In the following section we will discuss the evidence of relation of insulin signaling deficiency with AD pathology and disease progression.
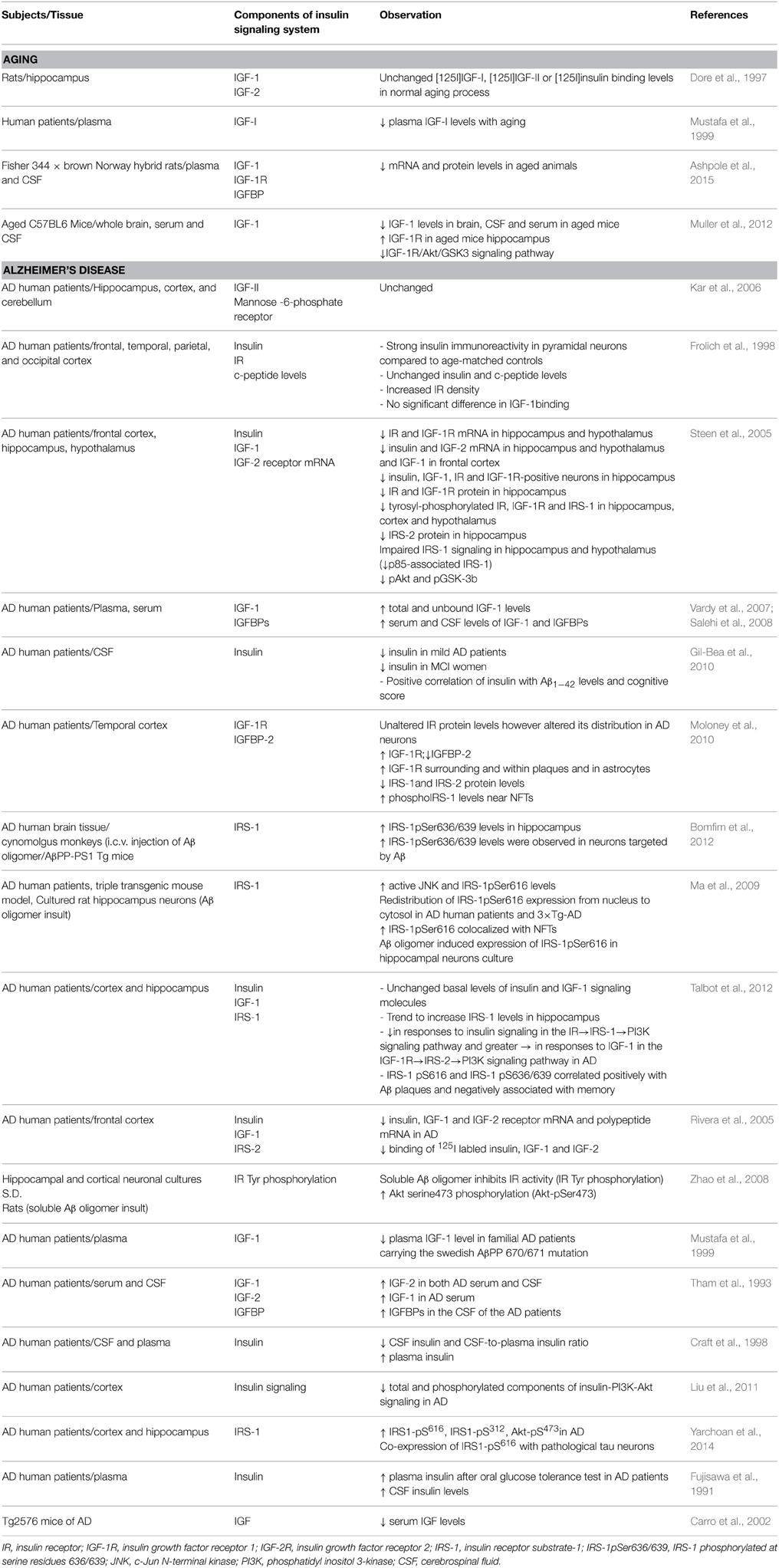
Table 1. Altered insulin signaling in normal aging and Alzheimer's diseases.
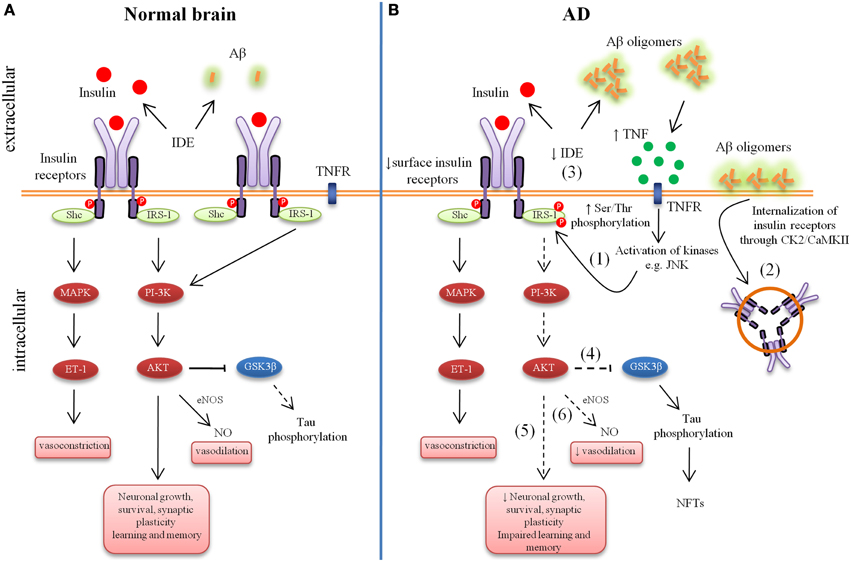
Figure 1. Aberrant brain insulin signaling in Alzheimer's Disease (AD). Schematic outline of neuronal insulin signaling in the normal brain (A) and AD brain (B). Under physiological conditions, insulin binding to its receptor triggers phosphorylation of insulin receptor substrate-1 (IRS-1). This results in phosphoinositide 3-kinase (PI3K) activation and downstream cellular responses that facilitate neuronal growth, neuronal survival, synaptic plasticity, learning and memory, etc. Activation of the IR can result in both vasodilatation and vasoconstriction and under physiological conditions there is a balance of both processes to regulate the immediate metabolic requirements of various tissues. In AD, accumulation of amyloid-β (Aβ) oligomers leads to increased tumor necrosis factor-alpha (TNF-α) levels and activation of stress kinases such as c-Jun N-terminal kinase (JNK) resulting in inhibitory serine phosphorylation of IRS-1 (1). Aβ oligomers cause removal of IRs from the cell surface mediated by Casein Kinase 2 (CK2) and Ca2+/Calmodulin-Dependent Kinase II (CaMKII) and redistribute them to cell bodies (2). Insulin resistance lowers the expression of Aβ-degrading insulin degrading enzyme (IDE) (3). Lowered IDE expression further decreases the availability of IDE for Aβ degradation. The reduction in brain insulin signaling increases GSK-3β activity (4), which increases abnormal tau phosphorylation. Deficient insulin signaling leads to impairment in nerve growth, synaptic plasticity, learning and memory, etc. (5). Aberrant phosphorylation of IRS causes an imbalance in homeostatic regulation of vascular function (6). This decreased production of NO may results in decreased cerebral blood flow and increased pro-inflammatory cytokines and reactive oxygen species production.
Insulin Signaling and AD Pathology
Ample number of evidence suggested that alterations in brain insulin metabolism could be one pathological factor for neurodegenerative diseases including AD. In support of this hypothesis, AD patients have shown reduced brain insulin receptor sensitivity, hyperphosphorylation of insulin receptor and downstream second messenger such as insulin receptor substrate-1 (IRS-1) and attenuated insulin and insulin-like growth factor receptor expression (Watson and Craft, 2003; Rivera et al., 2005; Steen et al., 2005; Holscher and Li, 2010; Talbot et al., 2012; de La Monte, 2012c; Freiherr et al., 2013). Moreover, reduced CSF insulin levels have been observed in AD. However, few studies have demonstrated either normal or increased CSF insulin levels in AD patients (Freiherr et al., 2013). However, CNS has hyper-insulinemia in AD is still controversial.
Neuritic (Amyloid Beta) Plaques
The amyloid plaques found in the brains of patients with AD are mainly composed of Aβ, a peptide derived from a larger molecule that is known as the amyloid precursor protein (AβPP). AβPP is normally cleaved within its extracellular domain by a protease known as “α-secretase,” and the processing releases a large, soluble APP fragment (sAPPα) into the extracellular space (Querfurth and Laferla, 2010). Because cleavage occurs within the Aβ domain, α-secretase processing does not generate Aβ. Aggregation prone and damaging Aβ originate from proteolysis of the AβPP by the sequential enzymatic actions of beta-site amyloid precursor protein–cleaving enzyme 1 (BACE-1), a β-secretase, and γ-secretase (Querfurth and Laferla, 2010). Aβ peptides are natural products of metabolism consisting of 36–43 amino acids. Monomers of Aβ40 are much more prevalent than the aggregation-prone and damaging Aβ42 species. An imbalance between production and clearance, and aggregation of peptides, causes Aβ to accumulate, and this excess may be the initiating factor in AD (Querfurth and Laferla, 2010).
Several lines of evidence suggested a link between energy metabolism defects to functional alterations associated with pathogenesis of AD. Inhibition of energy metabolism can alter AβPP processing and induce amyloidogenic products (Gabuzda et al., 1994). The relationship between insulin and Aβ metabolism is receiving increasing attention. Recently, a direct link between the Aβ metabolism and insulin pathways has been described in neuronal cell lines (Solano et al., 2000; Gasparini et al., 2001). Insulin treatment elevated sAPPα secretion in a concentration- and tyrosin kinase-dependent manner in the neuronal cells and also reduced the Aβ accumulation in neuronal cells (Solano et al., 2000; Gasparini et al., 2001). It has been demonstrated that insulin and IGFs can protect neurons from Aβ-induced neurotoxicity in neuronal cultures (Mattson, 1997). Therefore, sAPPα derived from insulin-mediated metabolism of AβPP may function in modulation of neuronal excitability, synaptic plasticity, neurite outgrowth, synaptogenesis, and cell survival (Mattson, 1997). It has been recently shown that IGF-1 enhances clearance of brain Aβ by modulating transport and/or production Aβ carriers at the blood–brain interface in the choroid plexus (Ashpole et al., 2015). IGF-1 seems to increase Aβ clearance by enhancing transport of Aβ carrier proteins such as albumin and transthyretin into the brain (Carro et al., 2002). Under physiological conditions insulin degrading enzyme (IDE) is secreted at high levels from the microglial cells, and degrades Aβ extracellularly (Qiu et al., 1998). In addition to insulin degradation, IDE has been found to degrade Aβ in neuronal and microglial cell cultures, and to eliminate neurotoxic effects of Aβ (Qiu et al., 1998; Vekrellis et al., 2000; Sudoh et al., 2002). Insulin can modulate the extracellular degradation of Aβ through insulin degrading enzyme (IDE), which is involved in insulin catabolism (Kurochkin and Goto, 1994; Authier et al., 1996; Qiu et al., 1998; Farris et al., 2003). The expression of IDE is under control of insulin levels. Insulin via phosphatidylinositol-3-kinase (PI3K) pathway increases IDE protein levels (Zhao et al., 2004a). In line with this, IDE knockout mice exhibited elevated levels of cerebral Aβ (Farris et al., 2003). In contrary, overexpression of IDE in the AβPP double transgenic mice decreased their brain Aβ levels, and completely prevented Aβ plaque formation (Leissring et al., 2003). Further support for this link was provided by the findings that deficient insulin signaling (decreased PI3K subunit P85) was correlated with reduced IDE in AD brains and in Tg2576 Swedish AβPP transgenic mice (Zhao et al., 2004a). Aβ degrading activity of IDE was shown to be lower in AD brains compared to controls (Perez et al., 2000). On the other hand, Aβ oligomers cause rapid and significant disruption of signaling by brain cell IRs.
It has been demonstrated that, acute treatment with Aβ oligomer to hippocampus caused a rapid and substantial loss of neuronal surface IRs specifically on dendrites bound (Zhao et al., 2008; de Felice et al., 2009). Aβ oligomer-treated neurons showed elevated levels of IRs in their cell bodies, suggesting redistribution of IRs (Figure 1). The rapid redistribution of surface receptors rather than net receptor loss in short term Aβ oligomer treatment was further supported by Western blots, which showed no Aβ oligomer-induced changes in total IR levels (Zhao et al., 2008). This leads to decreased responsiveness to insulin, revealed by impaired insulin-induced receptor protein tyrosine kinase activity in cultured neurons exposed to oligomers (Zhao et al., 2008; de Felice et al., 2009). The Aβ oligomer-induced redistribution of IRs is consistent with the reports in which other synaptic proteins, such as NMDA receptors subunits and EphB2 receptor tyrosin kinase, also showed surface loss by soluble Aβ oligomers (Snyder et al., 2005; Lacor et al., 2007). Although Aβ oligomer-induced removal of NMDA receptors is mediated by α-7 nicotinic receptors, the Aβ oligomers-induced loss of dendritic IRs dose not follow similar mechanism (Snyder et al., 2005). Aβ oligomers are known to cause internalization of NMDA receptors through Casein Kinase 2 (CK2) and Ca2+/Calmodulin-Dependent Kinase II (CaMKII) (Chung et al., 2004). Therefore it was hypothesis that Aβ oligomer-induce internalization of NMDA receptors and IRs might share common mechanisms involving CK2 and CaMKII. Consistent with this hypothesis De Felice et al showed that Aβ oligomer caused major downregulation of membrane surface IRs via mechanism sensitive to CK2 and CaMKII (de Felice et al., 2009).
IRs play key roles in the important neurological processes including learning and memory and tau phosphorylation. Thus, Aβ oligomer-induced loss of membrane IRs might represent an important early mechanism underlying memory impairment and other pathological features of AD. Therefore, Aβ oligomers-induced loss of IR receptors can lead to learning and memory impairments and over tau phosphorylation. Further research in this field revealed that Aβ oligomers-induced neuronal oxidative stress. IR down-regulation and synaptic loss are markedly decreased by insulin treatment (de Felice et al., 2009). Neuroprotection by insulin requires IR tyrosin kinase activity, which suggested that insulin through IR might downregulate Aβ oligomer binding sites in the synapse instead of a simple competition between Aβ oligomers and insulin for a common binding site on the neuronal surface.
Inhibition of brain insulin signaling by streptozotocin, a compound known to induce diabetes, treatment to rat astrocytoma cells induced amyloidogenic protein expression as evidenced by the increase in AβPP, BACE-1, and Aβ42 expression (Rajasekar et al., 2014). Furthermore, it has been demonstrated that Aβ oligomer-induces elevation in proinflammatory tumor necrosis factor-alpha (TNF-α) levels and it triggers aberrant activation of c-Jun N-terminal kinase (JNK) in neurons, which ultimately leads to serine phosphorylation of IRS-1 (Bomfim et al., 2012). IRS-1 serine phosphorylation (IRS-1pSer) blocks the downstream insulin signaling, which triggers, in turn, peripheral insulin resistance (de Felice, 2013). Aberrant insulin signaling accelerates plaque production in the brain by enhancing the amyloidogenic processing of the AβPP (Wang et al., 2010; Son et al., 2012), and also increases Aβ aggregation through monosialotetrahexosylganglioside (GM1) clustering and membrane signaling (Yamamoto et al., 2012). Thus, Aβ oligomer-induced insulin resistance may create a vicious cycle in which oligomers upregulate their own production and aggregation by disrupting insulin physiological actions. Such a mechanism could account in part for Aβ oligomer build up in AD brains.
Neurofibrillary Tangles
Tau is a neuronal microtubule-associated protein found in axons. Tau plays an important role in assembly and stability of microtubules as well as in vesicle transport in neurons. Tau, in its hyperphosphorylated form, is the major component of paired helical filaments (PHFs), the building block of neurofibrillary lesions in AD brain. Hyperphosphorylated tau is insoluble, does not have affinity for microtubules, and self-associates into paired helical filament structure (Querfurth and Laferla, 2010). Abnormal hyperphosphorylation of tau prompts an accumulation of NFTs in axons of neurons, can impair normal axon transport, disrupt synaptic plasticity and finally induce cell loss (Querfurth and Laferla, 2010; Bedse et al., 2015). Evidence suggests that abnormal activation of kinases like GSK-3β as well as caspases may be responsible for hyperphosphorylation of tau (Churcher, 2006; Rohn, 2010). IR activation, through phosphorylated IRS proteins, results in activation of multiple signaling pathways including PI3K and extracellular signal-regulated kinase (ERK) that directly regulated various physiological processes (Figure 1) (Saltiel, 2001). Activation of PI3K→Akt cascade promotes neuronal growth and survival (Rodgers and Theibert, 2002). Akt inactivates GSK-3β, which inhibits tau phosphorylation (Zhao et al., 2009; Perluigi et al., 2014). There are number of evidence showing that insulin regulated tau phosphorylation and increased rate of NFT development (Hong and Lee, 1997; Lesort and Johnson, 2000; Schubert et al., 2003; Cheng et al., 2005; Freude et al., 2005). It has been shown that insulin and IGF-1 regulate tau phosphorylation through the inhibition of GSK-3β in cultural neurons. These effects of insulin and IGF-1 are mediated through the inhibition of GSK-3β via the PI3K-protein kinase B (PI3K-PKB) signaling pathway (Hong and Lee, 1997). Moreover, peripheral hyperinsulinemia promotes tau phosphorylation in vivo (Freude et al., 2005). When IGF-1 and IRS-2 gene were deleted, tau phosphorylation was dramatically increased in IGF-1 and IRS-2 knockout mice (Schubert et al., 2003; Cheng et al., 2005). IGF-1 genetic deletion specifically increased tau phosphorylation at two specific residues, Ser-293 and Ser-202, both GSK-3β targeted sites (Cheng et al., 2005). Inhibition of brain insulin signaling by intra cerebroventricular administration of streptozotocin also induces hyperphosphorylation of tau at multiple sites (Deng et al., 2009; Chen et al., 2013). This treatment dramatically increased total tau and hyperphosphorylated tau in the hippocampus of triple transgenic AD (3×Tg-AD) mice (Chen et al., 2013, 2014a). These results suggested that insulin and IGF-1 signaling normally prevents tau hyperphosphorylation in the brain. As mentioned earlier in the review, diabetes is characterized by insulin resistance, hyperinsulinemia and impaired insulin signaling. In type 2 diabetes increased GSK-3β activity might lead to an elevation of Aβ production (Phiel et al., 2003) and increased tau phosphorylation (Freude et al., 2005; Sims-Robinson et al., 2010). On the other hand, Aβ oligomer induced JNK activation leading to phosphorylation and degradation of the adaptor protein IRS-1 (Ma et al., 2009). IRS deficiency contributes to insulin resistance in diabetes. Significantly reduced IRS-1 and IRS-2 levels occur in AD brain, accompanied by elevated cytosolic phosphor-IRS1 (Ser 312 and 316) (Ma et al., 2009; Bomfim et al., 2012; Talbot et al., 2012; Yarchoan et al., 2014). Phosphorylation of IRS-1 (Ser 312 and 316) inhibits the regulation of insulin on GSK-3β activity, which leads to further increase in hyperphosphorylation of tau (Ma et al., 2009). The association of IRS1 abnormalities and tau was further supported by double immunofluorescence experiments demonstrating frequent co-expression of IRS1-pS616 with tau lesions in neurons and dystrophic neuritis (Yarchoan et al., 2014). The activity of GSK3 can be down regulated in response to insulin or IGF-1 through the activation of the PI3K pathway.
Reduced Cerebral Blood Flow
Neurons depend on blood vessels for their oxygen and nutrient supplies, and for the removal of carbon dioxide and other potentially toxic metabolites from the brain's interstitial fluid. Recent evidence suggests that vascular dysfunction leads to neuronal dysfunction and neurodegeneration, and that it might contribute to the development of proteinaceous brain and cerebrovascular “storage” disorders (Zlokovic, 2011). AD is characterized by a decreased regional cerebral blood flow that could result in decrease brain supply of oxygen, glucose, and nutrients. Insulin signaling regulates vasodilation and vasoconstriction (Aulston et al., 2013). IR activation mediates vasodilation through PI3K→Akt pathway. It stimulates endothelial nitric oxide synthase (eNOS) resulting in the production of nitric oxide (NO) and vascular relaxation (Figure 1A) (Bolotina et al., 1994; Kahn et al., 1997). IR activation can also mediate vasoconstriction. Activation of IR can also lead to phosphorylation of the Src homology containing (Shc) protein, which in turn binds the Growth factor receptor-bound protein 2 (Grb-2) resulting in the activation of Son of sevemless (Sos) protein. This complex then activates the Rat sarcoma (Ras) protein leading to phosphorylation of Rapidly accelerated fibrosarcoma (Raf) protein kinase that results in activation of Mitogen-activated protein kinase (MAPK). Activation of MAPK stimulates release of endothelin-1 (ET-1), a vasoconstrictor (Figure 1A) (Potenza et al., 2005, 2006; Formoso et al., 2006). By mediating vascular properties, insulin signaling plays an important role in glucose and oxygen availability to the brain (Aulston et al., 2013). In insulin resistant state, there is a specific impairment in the vasodilatory PI3K pathway, whereas the Ras/MAPK-dependent pathway is unaffected (Jiang et al., 1999; Cusi et al., 2000). This results in decreased NO production and increased production of ET-1 in humans leading to vasoconstriction (Figure 1B) (Piatti et al., 1996). NO further plays important role in protecting blood vessels from endogenous injury by preventing platelet and leukocyte interactions (Kubes et al., 1991; Sarkar et al., 1996). Decreased production of NO allows for increased expression of proinflammatory transcription of the nuclear factor-κB (NFκB), and production of chemokines and cytokines (Zeiher et al., 1995). The resultant decrease in nutrient availability to the brain due to decreased NO production results in an increase of oxidative stress and reactive oxygen species (ROS) production and an increased inflammatory response. Released pro-inflammatory cytokines and macrophage recruitment instigates the onset of atherosclerosis, ultimately leading to macrovascular complications (Aulston et al., 2013).
Moreover, insulin can improve dendritic spine density and rescue spine loss caused by apolipoprotein E 4 (APOE4). APOE4, a major genetic risk factor for AD, exerts neuropathological effects through multiple pathways, including impairment of dendritic spine structure and mitochondrial function. It has been shown that insulin sensitizers, such as rosiglitazone [peroxisome proliferator-activated receptor gamma (PPAR-γ)], significantly increased dendritic spine density and rescued detrimental effects of APOE4 on dendritic spine (Brodbeck et al., 2008). Thus, it is suggested that rosiglitazone might improve cognition in AD patients by increasing dendritic spine density. There is also evidence that insulin sensitizers modulate mitochondrial function (Wang et al., 2002). Fuenzalida and co-workers reported that rosiglitazone treatment in neuronal cells up-regulates Bcl-2 thereby stabilizing mitochondrial potential and protecting against apoptosis (Fuenzalida et al., 2007).
Intranasal Insulin Delivery to the Brain
Thus, impaired insulin sensitivity of IR and increased insulin resistance at brain may contribute to number of pathological processes that lead to acceleration of AD pathology. Therefore, restoring insulin to normal levels in the brain by insulin treatment, as showed in laboratory animals, may provide therapeutic benefits in AD subjects. Unlike, insulin cannot be administered orally, since it is degraded in the gastrointestinal track by various enzymes. Increasing brain insulin levels in AD patients by intravenous administration has been shown to acutely improve performance on hippocampus-dependent memory task (Craft et al., 1996). However, high systemic doses would be needed to achieve functionally effective insulin concentrations in the brain, causing strong peripheral side effects such as hypoglycemia and induction and/or exacerbation of peripheral insulin resistance (Benedict et al., 2011). Therefore oral and intravenous route of administration is not viable in clinical setting. In contrast, intranasal administration is a promising approach to selectively enhance brain insulin levels while avoiding adverse side effects (Born et al., 2002). This route offers a relatively high bioavailability, avoidance of the first-pass effect and invasive administration. Intranasal administration of insulin provides rapid delivery of insulin to the central nervous system via bulk flow via along olfactory and trigeminal perivascular channels, and slower delivery via olfactory bulb axonal transports (Thorne and Frey, 2001; Craft et al., 2012). Clinical studies have shown that intranasally administered insulin reaches to cerebrospinal fluid (CSF) within 30 min without substantial uptake into the bloodstream, bypassing the bloodstream (Born et al., 2002). Moreover, insulin exerts rapid changes on oscillatory electroencephalogram (EEG) parameters (Hallschmid et al., 2004). However, such comparable effects on EEG were not observed to those induced by intravenous bolus injection of insulin, suggesting that following intranasal administration, a significant amount of administered insulin dose reaches to the brain in a functional active state (Hallschmid et al., 2004). Recently, National Institute of Health (NIH) has selected intranasal insulin administration as one of the two therapeutic strategies receiving substantial funding as part of the National Alzheimer's Plan in the US (Wadman, 2012). This plan is a part of the initiative to find a therapeutic treatment to cure AD by 2025.
Intranasal Insulin Improves Memory in the Healthy Humans as Well as in the AD Patients
Enhancing central nervous insulin action has been shown to improve memory functions in animals as well as in humans (see Table 2) (Benedict et al., 2011). In a human study, the effects of 8 weeks of intranasal insulin (4 × 40 IU/d) and placebo were assessed on hippocampus-dependent declarative memory in 38 health subjects (Benedict et al., 2004). At the beginning and end of treatment, in total 30 words were orally presented to subjects. The subjects were asked to write down all words they still remembered after 3 min (immediate recall session) and one week later (delayed recall session) after presentation of words. The subjects treated with the insulin remembered significantly more words in delayed recall session, but not in immediate recall session, compared to placebo group. In this study the improving effect of subchronic intranasal insulin administration seemed to be specific for hippocampus dependent declarative memory, without any peripheral side effects. As far as the increased delayed recall authors pointed out that the improvement in the insulin-treated group occurred on a background of a generally decreasing performance. In addition, authors suggested that it might reflects an influence on encoding of the words at learning or a direct influence on retrieval, rather than an effect on the proper consolidation of memory. The same research group further studied effects of rapid-acting insulin analog insulin aspart, regular human insulin and placebo under similar experimental conditions (Benedict et al., 2007). Authors hypothesized that rapidly absorbed insulin aspart would show stronger effects on memory functions than regular human insulin due to the higher efficiency to reach brain. After 8 weeks of treatment subjects treated with both regular human insulin and insulin aspart remembered significantly more words than placebo group. Insulin aspart-treated subjects performed even better that those treated with regular human insulin (Benedict et al., 2007).
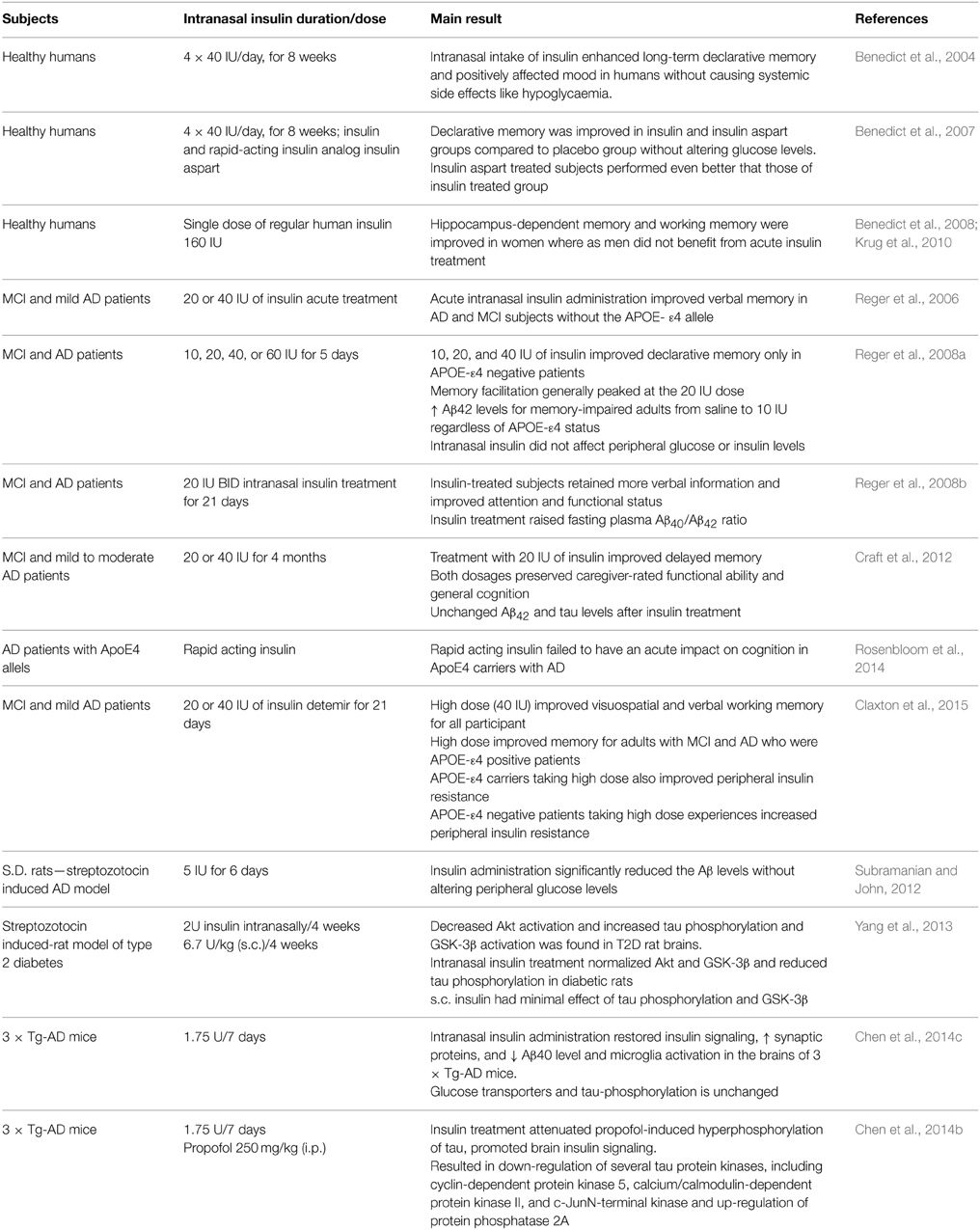
Table 2. Intranasal insulin improves memory function.
In a study, gender differences regarding acute effects of central nervous insulin 160 IU were studied in 32 young healthy subjects (18 women and 14 men) in hippocampus-dependent two-dimensional-object location task and working memory task (digit span) (Benedict et al., 2008). Acute insulin treatment significantly improved hippocampus-dependent memory and working memory in women but not in men. The same group of researchers further showed that these effects were not restricted only to young women but also for postmenopausal women (Krug et al., 2010). Based on the encouraging effects of intranasal insulin that demonstrated improvement of memory without any side effects in healthy humans, few clinical trial have been carried out in patients with mild cognitive impairments (MCI) and AD patients. In a first study, 26 memory-impaired subjects (13 with early AD and 13 with mild MCI) and 35 normal controls received intranasal insulin (20 or 40 IU) or saline (placebo) treatments (Reger et al., 2006). Cognition was tested 15 min post-treatment, and blood was acquired at baseline and 45 min after treatment. Intranasal insulin treatment did not change plasma insulin or glucose levels. Insulin treatment facilitated recall on two measures of verbal memory in memory-impaired APOE negative adults. These effects were stronger for memory-impaired APOE negative subjects than for memory-impaired APOE positive subjects and normal adults. Unexpectedly, memory-impaired APOE positive subjects showed poorer recall following insulin administration on one test of memory. This study provided further evidence for APOE-related differences in insulin metabolism. Another study supported the same observation that intranasal administration improves cognition in APOE negative patients (Reger et al., 2008a). Recently, it has been demonstrated that high dose of insulin (40 IU) can improve visuospatial and verbal working memory in APOE positive MCI and AD patients (Claxton et al., 2015). High dose of insulin also improved peripheral insulin resistance in APOE positive patients. Conversely, high dose of insulin experienced increased peripheral insulin resistance in APOE negative patients (Claxton et al., 2015). These findings suggest that groups with different genetic risks for AD may show differential dose-response curves following intranasal insulin administration (Reger et al., 2008a; Rosenbloom et al., 2014). Further, it is reported that insulin treated subjects retained more verbal information, improved attention and raised fasting plasma Aβ40/Aβ42 ratio (Reger et al., 2008b). Another study supported the notion that the intranasal administration of insulin improves cognition in patients affected by mild cognitive impairment or AD (Craft et al., 2012). However, authors did not observe any changes in the Aβ42 and tau levels in the CSF after treatment.
Conclusions
The advances in AD research in the last decade have revealed that AD is linked to insulin signaling deficiency in the brain. This hypothesis was further strengthen when intracerebroventricular administration of streptozotocin resulted in AD-like cognitive impairments, neurodegeneration and insulin resistance (Lester-Coll et al., 2006; de La Monte, 2014). Insulin/IGF signaling promotes the trafficking of AβPP-Aβ (Watson et al., 2003) and also enhances clearance of Aβ by modulating Aβ transporters and carriers at the BBB (Carro et al., 2002; Ashpole et al., 2015). Moreover, insulin/IGF negatively controls Aβ intracellular deposition, tau phosphorylation and degradation by IDE (Gasparini et al., 2001, 2002). Deregulation of insulin/IGF signaling increases Aβ deposition, tau phosphorylation, reactive species and decreases cerebral blood flow (Figure 1) (de La Monte, 2014). Accumulation of Aβ oligomers further worsen insulin deficiency by decreasing insulin's binding affinity to its receptors, reducing and desensitizing cell surface IRs and phosphorylating IRS-1 (Figure 1) (Lacor et al., 2007; Zhao et al., 2008; Ma et al., 2009; Fernandez and Torres-Aleman, 2012; Talbot et al., 2012). This review proposed intra nasal administration as potential therapeutics, which has been shown to improve cognition (Table 2) without any side effects in clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ashpole, N. M., Sanders, J. E., Hodges, E. L., Yan, H., and Sonntag, W. E. (2015). Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 68, 76–81. doi: 10.1016/j.exger.2014.10.002
Aulston, B. D., Odero, G. L., Aboud, Z., and Glazner, G. W. (2013). “Alzheimer's Disease and Diabetes,” in Understanding Alzheimer's Disease, ed I. Zerr (InTech), Available online at: http://www.intechopen.com/books/understanding-alzheimer-s-disease/alzheimer-s-disease-and-diabetes
Authier, F., Posner, B. I., and Bergeron, J. J. (1996). Insulin-degrading enzyme. Clin. Invest. Med. 19, 149–160.
Baker, L. D., Cross, D. J., Minoshima, S., Belongia, D., Watson, G. S., and Craft, S. (2011). Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 68, 51–57. doi: 10.1001/archneurol.2010.225
Banks, W. A., Owen, J. B., and Erickson, M. A. (2012). Insulin in the brain: there and back again. Pharmacol. Ther. 136, 82–93. doi: 10.1016/j.pharmthera.2012.07.006
Bedse, G., Romano, A., Lavecchia, A. M., Cassano, T., and Gaetani, S. (2015). The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in Alzheimer's disease. J. Alzheimers Dis. 43, 1115–1136. doi: 10.3233/jad-141635
Benedict, C., Frey, W. H. II, Schioth, H. B., Schultes, B., Born, J., and Hallschmid, M. (2011). Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 46, 112–115. doi: 10.1016/j.exger.2010.08.026
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. doi: 10.1016/j.psyneuen.2004.04.003
Benedict, C., Hallschmid, M., Schmitz, K., Schultes, B., Ratter, F., Fehm, H. L., et al. (2007). Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243. doi: 10.1038/sj.npp.1301193
Benedict, C., Kern, W., Schultes, B., Born, J., and Hallschmid, M. (2008). Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 93, 1339–1344. doi: 10.1210/jc.2007-2606
Bolotina, V. M., Najibi, S., Palacino, J. J., Pagano, P. J., and Cohen, R. A. (1994). Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 368, 850–853. doi: 10.1038/368850a0
Bomfim, T. R., Forny-Germano, L., Sathler, L. B., Brito-Moreira, J., Houzel, J. C., Decker, H., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease- associated Abeta oligomers. J. Clin. Invest. 122, 1339–1353. doi: 10.1172/JCI57256
Born, J., Lange, T., Kern, W., Mcgregor, G. P., Bickel, U., and Fehm, H. L. (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat. Neurosci. 5, 514–516. doi: 10.1038/nn0602-849
Brodbeck, J., Balestra, M. E., Saunders, A. M., Roses, A. D., Mahley, R. W., and Huang, Y. (2008). Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 1343–1346. doi: 10.1073/pnas.0709906104
Butterfield, D. A., Di Domenico, F., and Barone, E. (2014a). Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim. Biophys. Acta 1842, 1693–1706. doi: 10.1016/j.bbadis.2014.06.010
Butterfield, D. A., Gu, L., Di Domenico, F., and Robinson, R. A. (2014b). Mass spectrometry and redox proteomics: applications in disease. Mass Spectrom. Rev. 33, 277–301. doi: 10.1002/mas.21374
Carro, E., Trejo, J. L., Gomez-Isla, T., Leroith, D., and Torres-Aleman, I. (2002). Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 8, 1390–1397. doi: 10.1038/nm1202-793
Chen, Y., Deng, Y., Zhang, B., and Gong, C. X. (2014a). Deregulation of brain insulin signaling in Alzheimer's disease. Neurosci. Bull. 30, 282–294. doi: 10.1007/s12264-013-1408-x
Chen, Y., Liang, Z., Blanchard, J., Dai, C. L., Sun, S., Lee, M. H., et al. (2013). A non-transgenic mouse model (icv-STZ mouse) of Alzheimer's disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 47, 711–725. doi: 10.1007/s12035-012-8375-5
Chen, Y., Run, X., Liang, Z., Zhao, Y., Dai, C. L., Iqbal, K., et al. (2014b). Intranasal insulin prevents anesthesia-induced hyperphosphorylation of tau in 3xTg-AD mice. Front. Aging Neurosci. 6:100. doi: 10.3389/fnagi.2014.00100
Chen, Y., Zhao, Y., Dai, C. L., Liang, Z., Run, X., Iqbal, K., et al. (2014c). Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Abeta level and microglia activation in the brains of 3xTg-AD mice. Exp. Neurol. 261, 610–619. doi: 10.1016/j.expneurol.2014.06.004
Cheng, C. M., Tseng, V., Wang, J., Wang, D., Matyakhina, L., and Bondy, C. A. (2005). Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology 146, 5086–5091. doi: 10.1210/en.2005-0063
Chung, H. J., Huang, Y. H., Lau, L. F., and Huganir, R. L. (2004). Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J. Neurosci. 24, 10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004
Churcher, I. (2006). Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr. Top. Med. Chem. 6, 579–595. doi: 10.2174/156802606776743057
Claxton, A., Baker, L. D., Hanson, A., Trittschuh, E. H., Cholerton, B., Morgan, A., et al. (2015). Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer's Disease dementia. J. Alzheimers Dis. 44, 897-906. doi: 10.3233/jad-141791
Conejo, R., and Lorenzo, M. (2001). Insulin signaling leading to proliferation, survival, and membrane ruffling in C2C12 myoblasts. J. Cell Physiol. 187, 96–108. doi: 10.1002/1097-4652(2001)9999:9999<::aid-jcp1058>3.0.co;2-v
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. doi: 10.1001/archneurol.2011.233
Craft, S., Newcomer, J., Kanne, S., Dagogo-Jack, S., Cryer, P., Sheline, Y., et al. (1996). Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol. Aging 17, 123–130. doi: 10.1016/0197-4580(95)02002-0
Craft, S., Peskind, E., Schwartz, M. W., Schellenberg, G. D., Raskind, M., and Porte, D. Jr. (1998). Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 50, 164–168. doi: 10.1212/WNL.50.1.164
Craft, S., and Watson, G. S. (2004). Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 3, 169–178. doi: 10.1016/S1474-4422(04)00681-7
Craft, S. (2007). Insulin resistance and Alzheimer's disease pathogenesis: potential mechanisms and implications for treatment. Curr. Alzheimer Res. 4, 147–152. doi: 10.2174/156720507780362137
Cruts, M., and Van Broeckhoven, C. (1998). Molecular genetics of Alzheimer's disease. Ann. Med. 30, 560–565. doi: 10.3109/07853899809002605
Cusi, K., Maezono, K., Osman, A., Pendergrass, M., Patti, M. E., Pratipanawatr, T., et al. (2000). Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Invest. 105, 311–320. doi: 10.1172/JCI7535
de Felice, F. G., Vieira, M. N., Bomfim, T. R., Decker, H., Velasco, P. T., Lambert, M. P., et al. (2009). Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 1971–1976. doi: 10.1073/pnas.0809158106
de Felice, F. G. (2013). Alzheimer's disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest. 123, 531–539. doi: 10.1172/JCI64595
de La Monte, S. M., Longato, L., Tong, M., and Wands, J. R. (2009). Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs 10, 1049–1060.
de La Monte, S. M. (2012c). Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr. Alzheimer Res. 9, 35–66. doi: 10.2174/156720512799015037
de La Monte, S. M. (2012a). Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer's disease. Drugs 72, 49–66. doi: 10.2165/11597760-000000000-00000
de La Monte, S. M. (2012b). Triangulated mal-signaling in Alzheimer's disease: roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J. Alzheimers Dis. 30(Suppl. 2), S231–S249. doi: 10.3233/jad-2012-111727
de La Monte, S. M. (2014). Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur. Neuropsychopharmacol. 24, 1954–1960. doi: 10.1016/j.euroneuro.2014.06.008
Dekosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Deng, Y., Li, B., Liu, Y., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2009). Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: implication for Alzheimer's disease. Am. J. Pathol. 175, 2089–2098. doi: 10.2353/ajpath.2009.090157
Dore, S., Kar, S., Rowe, W., and Quirion, R. (1997). Distribution and levels of [125I]IGF-I, [125I]IGF-II and [125I]insulin receptor binding sites in the hippocampus of aged memory-unimpaired and -impaired rats. Neuroscience 80, 1033–1040. doi: 10.1016/S0306-4522(97)00154-1
Duarte, A. I., Moreira, P. I., and Oliveira, C. R. (2012). Insulin in central nervous system: more than just a peripheral hormone. J. Aging Res. 2012:384017. doi: 10.1155/2012/384017
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Fernandez, A. M., and Torres-Aleman, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Formoso, G., Chen, H., Kim, J. A., Montagnani, M., Consoli, A., and Quon, M. J. (2006). Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium. Mol. Endocrinol. 20, 1153–1163. doi: 10.1210/me.2005-0266
Freiherr, J., Hallschmid, M., Frey, W. H., Brünner, Y. F., Chapman, C. D., Hölscher, C., et al. (2013). Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs 27, 505–514. doi: 10.1007/s40263-013-0076-8
Freude, S., Plum, L., Schnitker, J., Leeser, U., Udelhoven, M., Krone, W., et al. (2005). Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes 54, 3343–3348. doi: 10.2337/diabetes.54.12.3343
Frolich, L., Blum-Degen, D., Bernstein, H. G., Engelsberger, S., Humrich, J., Laufer, S., et al. (1998). Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J. Neural Transm. 105, 423–438. doi: 10.1007/s007020050068
Fuenzalida, K., Quintanilla, R., Ramos, P., Piderit, D., Fuentealba, R. A., Martinez, G., et al. (2007). Peroxisome Proliferator-activated Receptor γ Up-regulates the Bcl-2 Anti- apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol.Chem. 282, 37006–37015. doi: 10.1074/jbc.M700447200
Fujisawa, Y., Sasaki, K., and Akiyama, K. (1991). Increased insulin levels after OGTT load in peripheral blood and cerebrospinal fluid of patients with dementia of Alzheimer type. Biol. Psychiatry 30, 1219–1228. doi: 10.1016/0006-3223(91)90158-I
Gabuzda, D., Busciglio, J., Chen, L. B., Matsudaira, P., and Yankner, B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 269, 13623–13628.
Gasparini, L., Gouras, G. K., Wang, R., Gross, R. S., Beal, M. F., Greengard, P., et al. (2001). Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 21, 2561–2570.
Gasparini, L., Netzer, W. J., Greengard, P., and Xu, H. (2002). Does insulin dysfunction play a role in Alzheimer's disease? Trends Pharmacol. Sci. 23, 288–293. doi: 10.1016/S0165-6147(02)02037-0
Gil-Bea, F. J., Solas, M., Solomon, A., Mugueta, C., Winblad, B., Kivipelto, M., et al. (2010). Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer's disease. J. Alzheimers Dis. 22, 405–413. doi: 10.3233/JAD-2010-100795
Hallschmid, M., Schultes, B., Marshall, L., Molle, M., Kern, W., Bredthauer, J., et al. (2004). Transcortical direct current potential shift reflects immediate signaling of systemic insulin to the human brain. Diabetes 53, 2202–2208. doi: 10.2337/diabetes.53.9.2202
Holscher, C., and Li, L. (2010). New roles for insulin-like hormones in neu- ronal signalling and protection: new hopes for novel treatments of Alzheimer's disease? Neurobiol. Aging, 31, 1495–1502. doi: 10.1016/j.neurobiolaging.2008.08.023
Hong, M., and Lee, V. M. (1997). Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 272, 19547–19553. doi: 10.1074/jbc.272.31.19547
Hoyer, S. (2002). The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J. Neural Transm. 109, 341–360. doi: 10.1007/s007020200028
Hoyer, S. (2004). Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 490, 115–125. doi: 10.1016/j.ejphar.2004.02.049
Jiang, Z. Y., Lin, Y. W., Clemont, A., Feener, E. P., Hein, K. D., Igarashi, M., et al. (1999). Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Invest. 104, 447–457. doi: 10.1172/JCI5971
Kahn, A. M., Husid, A., Allen, J. C., Seidel, C. L., and Song, T. (1997). Insulin acutely inhibits cultured vascular smooth muscle cell contraction by a nitric oxide synthase-dependent pathway. Hypertension 30, 928–933. doi: 10.1161/01.HYP.30.4.928
Kar, S., Poirier, J., Guevara, J., Dea, D., Hawkes, C., Robitaille, Y., et al. (2006). Cellular distribution of insulin-like growth factor-II/mannose-6-phosphate receptor in normal human brain and its alteration in Alzheimer's disease pathology. Neurobiol. Aging 27, 199–210. doi: 10.1016/j.neurobiolaging.2005.03.005
Kleinridders, A., Ferris, H. A., Cai, W., and Kahn, C. R. (2014). Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63, 2232–2243. doi: 10.2337/db14-0568
Krikorian, R., Eliassen, J. C., Boespflug, E. L., Nash, T. A., and Shidler, M. D. (2010). Improved cognitive-cerebral function in older adults with chromium supplementation. Nutr. Neurosci. 13, 116–122. doi: 10.1179/147683010X12611460764084
Krug, R., Benedict, C., Born, J., and Hallschmid, M. (2010). Comparable sensitivity of postmenopausal and young women to the effects of intranasal insulin on food intake and working memory. J. Clin. Endocrinol. Metab. 95, E468–E472. doi: 10.1210/jc.2010-0744
Kubes, P., Suzuki, M., and Granger, D. N. (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655. doi: 10.1073/pnas.88.11.4651
Kurochkin, I. V., and Goto, S. (1994). Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 345, 33–37. doi: 10.1016/0014-5793(94)00387-4
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J. Neurosci. 27, 796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/S0896-6273(03)00787-6
Lesort, M., and Johnson, G. V. (2000). Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience 99, 305–316. doi: 10.1016/S0306-4522(00)00200-1
Lester-Coll, N., Rivera, E. J., Soscia, S. J., Doiron, K., Wands, J. R., and de La Monte, S. M. (2006). Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J. Alzheimers Dis. 9, 13–33.
Liu, Y., Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2011). Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J. Pathol. 225, 54–62. doi: 10.1002/path.2912
Luchsinger, J. A. (2010). Type 2 diabetes, related conditions, in relation and dementia: an opportunity for prevention? J. Alzheimers Dis. 20, 723–736. doi: 10.3233/jad-2010-091687
Ma, Q. L., Yang, F., Rosario, E. R., Ubeda, O. J., Beech, W., Gant, D. J., et al. (2009). Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29, 9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009
Mattson, M. P. (1997). Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 77, 1081–1132.
Moloney, A. M., Griffin, R. J., Timmons, S., O'connor, R., Ravid, R., and O'Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243. doi: 10.1016/j.neurobiolaging.2008.04.002
Moreira, P. I., Duarte, A. I., Santos, M. S., Rego, A. C., and Oliveira, C. R. (2009). An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer's disease. J. Alzheimers Dis. 16, 741–761. doi: 10.3233/jad-2009-0972
Muller, A. P., Fernandez, A. M., Haas, C., Zimmer, E., Portela, L. V., and Torres-Aleman, I. (2012). Reduced brain insulin-like growth factor I function during aging. Mol. Cell Neurosci. 49, 9–12. doi: 10.1016/j.mcn.2011.07.008
Mustafa, A., Lannfelt, L., Lilius, L., Islam, A., Winblad, B., and Adem, A. (1999). Decreased plasma insulin-like growth factor-I level in familial Alzheimer's disease patients carrying the Swedish APP 670/671 mutation. Dement. Geriatr. Cogn. Disord. 10, 446–451. doi: 10.1159/000017188
Neumann, K. F., Rojo, L., Navarrete, L. P., Farias, G., Reyes, P., and Maccioni, R. B. (2008). Insulin resistance and Alzheimer's disease: molecular links & clinical implications. Curr. Alzheimer Res. 5, 438–447. doi: 10.2174/156720508785908919
Perez, A., Morelli, L., Cresto, J. C., and Castano, E. M. (2000). Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 25, 247–255. doi: 10.1023/A:1007527721160
Perluigi, M., Pupo, G., Tramutola, A., Cini, C., Coccia, R., Barone, E., et al. (2014). Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta. 1842, 1144–1153. doi: 10.1016/j.bbadis.2014.04.007
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423, 435–439. doi: 10.1038/nature01640
Piatti, P. M., Monti, L. D., Conti, M., Baruffaldi, L., Galli, L., Phan, C. V., et al. (1996). Hypertriglyceridemia and hyperinsulinemia are potent inducers of endothelin-1 release in humans. Diabetes 45, 316–321. doi: 10.2337/diab.45.3.316
Potenza, M. A., Marasciulo, F. L., Chieppa, D. M., Brigiani, G. S., Formoso, G., Quon, M. J., et al. (2005). Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am. J. Physiol. Heart Circ. Physiol. 289, H813–H822. doi: 10.1152/ajpheart.00092.2005
Potenza, M. A., Marasciulo, F. L., Tarquinio, M., Quon, M. J., and Montagnani, M. (2006). Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes 55, 3594–3603. doi: 10.2337/db06-0667
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
Querfurth, H. W., and Laferla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rajasekar, N., Dwivedi, S., Nath, C., Hanif, K., and Shukla, R. (2014). Protection of streptozotocin induced insulin receptor dysfunction, neuroinflammation and amyloidogenesis in astrocytes by insulin. Neuropharmacology 86, 337–352. doi: 10.1016/j.neuropharm.2014.08.013
Reger, M. A., Watson, G. S., Frey, W. H. II, Baker, L. D., Cholerton, B., Keeling, M. L., et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., et al. (2008a). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 13, 323–331.
Reger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., Baker, L. D., Cholerton, B., et al. (2008b). Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70, 440–448. doi: 10.1212/01.WNL.0000265401.62434.36
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., and De La Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J. Alzheimers Dis. 8, 247–268.
Rodgers, E. E., and Theibert, A. B. (2002). Functions of PI 3-kinase in development of the nervous system. Int. J. Dev. Neurosci. 20, 187–197. doi: 10.1016/S0736-5748(02)00047-3
Rohn, T. T. (2010). The role of caspases in Alzheimer's disease; potential novel therapeutic opportunities. Apoptosis 15, 1403–1409. doi: 10.1007/s10495-010-0463-2
Rosenbloom, M. H., Barclay, T. R., Pyle, M., Owens, B. L., Cagan, A. B., Anderson, C. P., et al. (2014). A single-dose pilot trial of intranasal rapid-acting insulin in apolipoprotein e4 carriers with mild-moderate Alzheimer's disease. CNS Drugs 28, 1185–1189. doi: 10.1007/s40263-014-0214-y
Salehi, Z., Mashayekhi, F., and Naji, M. (2008). Insulin like growth factor-1 and insulin like growth factor binding proteins in the cerebrospinal fluid and serum from patients with Alzheimer's disease. Biofactors 33, 99–106. doi: 10.1002/biof.5520330202
Saltiel, A. R. (2001). New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104, 517–529. doi: 10.1016/S0092-8674(01)00239-2
Sarkar, R., Meinberg, E. G., Stanley, J. C., Gordon, D., and Webb, R. C. (1996). Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ. Res. 78, 225–230. doi: 10.1161/01.RES.78.2.225
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell 103, 211–225. doi: 10.1016/S0092-8674(00)00114-8
Schubert, M., Brazil, D. P., Burks, D. J., Kushner, J. A., Ye, J., Flint, C. L., et al. (2003). Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 23, 7084–7092.
Schubert, M., Gautam, D., Surjo, D., Ueki, K., Baudler, S., Schubert, D., et al. (2004). Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 101, 3100–3105. doi: 10.1073/pnas.0308724101
Schulingkamp, R. J., Pagano, T. C., Hung, D., and Raffa, R. B. (2000). Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci. Biobehav. Rev. 24, 855–872. doi: 10.1016/S0149-7634(00)00040-3
Selkoe, D. J. (1989). Amyloid beta protein precursor and the pathogenesis of Alzheimer's disease. Cell 58, 611–612. doi: 10.1016/0092-8674(89)90093-7
Sesti, G., Federici, M., Hribal, M. L., Lauro, D., Sbraccia, P., and Lauro, R. (2001). Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 15, 2099–2111. doi: 10.1096/fj.01-0009rev
Sims-Robinson, C., Kim, B., Rosko, A., and Feldman, E. L. (2010). How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 6, 551–559. doi: 10.1038/nrneurol.2010.130
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–8105. doi: 10.1038/nn1503
Solano, D. C., Sironi, M., Bonfini, C., Solerte, S. B., Govoni, S., and Racchi, M. (2000). Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 14, 1015–1022.
Son, S. M., Song, H., Byun, J., Park, K. S., Jang, H. C., Park, Y. J., et al. (2012). Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes 61, 3126–3138. doi: 10.2337/db11-1735
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80.
Stockhorst, U., De Fries, D., Steingrueber, H. J., and Scherbaum, W. A. (2004). Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol. Behav. 83, 47–54. doi: 10.1016/j.physbeh.2004.07.022
Subramanian, S., and John, M. (2012). Intranasal administration of insulin lowers amyloid-beta levels in rat model of diabetes. Indian J. Exp. Biol. 50, 41–44.
Sudoh, S., Frosch, M. P., and Wolf, B. A. (2002). Differential effects of proteases involved in intracellular degradation of amyloid beta-protein between detergent-soluble and -insoluble pools in CHO-695 cells. Biochemistry 41, 1091–1099. doi: 10.1021/bi011193l
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Tham, A., Nordberg, A., Grissom, F. E., Carlsson-Skwirut, C., Viitanen, M., and Sara, V. R. (1993). Insulin-like growth factors and insulin-like growth factor binding proteins in cerebrospinal fluid and serum of patients with dementia of the Alzheimer type. J. Neural Transm. Park. Dis. Dement. Sect. 5, 165–176. doi: 10.1007/BF02257671
Thorne, R. G., and Frey, W. H. II. (2001). Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin. Pharmacokinet. 40, 907–946. doi: 10.2165/00003088-200140120-00003
Van Dam, P. S., and Aleman, A. (2004). Insulin-like growth factor-I, cognition and brain aging. Eur. J. Pharmacol. 490, 87–95. doi: 10.1016/j.ejphar.2004.02.047
Van Der Heide, L. P., Ramakers, G. M., and Smidt, M. P. (2006). Insulin signaling in the central nervous system: learning to survive. Prog. Neurobiol. 79, 205–221. doi: 10.1016/j.pneurobio.2006.06.003
Vardy, E. R., Rice, P. J., Bowie, P. C., Holmes, J. D., Grant, P. J., and Hooper, N. M. (2007). Increased circulating insulin-like growth factor-1 in late-onset Alzheimer's disease. J. Alzheimers Dis. 12, 285–290.
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., et al. (2000). Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665.
Wada, A., Yokoo, H., Yanagita, T., and Kobayashi, H. (2005). New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J. Pharmacol. Sci. 99, 128–143. doi: 10.1254/jphs.CRJ05006X
Wadman, M. (2012). US government sets out Alzheimer's plan. Nature 485, 426–427. doi: 10.1038/485426a
Wang, X., Zheng, W., Xie, J. W., Wang, T., Wang, S. L., Teng, W. P., et al. (2010). Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol. Neurodegener. 5:46. doi: 10.1186/1750-1326-5-46
Wang, Y. L., Frauwirth, K. A., Rangwala, S. M., Lazar, M. A., and Thompson, C. B. (2002). Thiazolidinedione activation of peroxisome proliferator-activated receptor γ can enhance mitochondrial potential and promote cell survival. J. Biol. Chem. 277, 31781–31788. doi: 10.1074/jbc.M204279200
Watson, G. S., and Craft, S. (2003). The role of insulin resistance in the pathogenesis of Alzheimer's disease: implications for treatment. CNS Drugs 17, 27–45.
Watson, G. S., and Craft, S. (2006). Insulin resistance, inflammation, and cognition in Alzheimer's Disease: lessons for multiple sclerosis. J. Neurol. Sci. 245, 21–33. doi: 10.1016/j.jns.2005.08.017
Watson, G. S., Peskind, E. R., Asthana, S., Purganan, K., Wait, C., Chapman, D., et al. (2003). Insulin increases CSF Abeta42 levels in normal older adults. Neurology 60, 1899–1903. doi: 10.1212/01.WNL.0000065916.25128.25
Wortmann, M. (2012). Dementia: a global health priority - highlights from an ADI and World Health Organization report. Alzheimers Res. Ther. 4:40. doi: 10.1186/alzrt143
Yamamoto, N., Matsubara, T., Sobue, K., Tanida, M., Kasahara, R., Naruse, K., et al. (2012). Brain insulin resistance accelerates Abeta fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 121, 619–628. doi: 10.1111/j.1471-4159.2012.07668.x
Yang, Y., Ma, D., Wang, Y., Jiang, T., Hu, S., Zhang, M., et al. (2013). Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J. Alzheimers Dis. 33, 329–338. doi: 10.3233/JAD-2012-121294
Yarchoan, M., Toledo, J. B., Lee, E. B., Arvanitakis, Z., Kazi, H., Han, L. Y., et al. (2014). Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer's disease and tauopathies. Acta Neuropathol. 128, 679–689. doi: 10.1007/s00401-014-1328-5
Zeiher, A. M., Fisslthaler, B., Schray-Utz, B., and Busse, R. (1995). Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ. Res. 76, 980–986. doi: 10.1161/01.RES.76.6.980
Zhao, L., Teter, B., Morihara, T., Lim, G. P., Ambegaokar, S. S., Ubeda, O. J., et al. (2004a). Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J. Neurosci. 24, 11120–11126. doi: 10.1523/JNEUROSCI.2860-04.2004
Zhao, W. Q., Chen, H., Quon, M. J., and Alkon, D. L. (2004b). Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 490, 71–81. doi: 10.1016/j.ejphar.2004.02.045
Zhao, W. Q., De Felice, F. G., Fernandez, S., Chen, H., Lambert, M. P., Quon, M. J., et al. (2008). Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 22, 246–260. doi: 10.1096/fj.06-7703com
Zhao, W. Q., Lacor, P. N., Chen, H., Lambert, M. P., Quon, M. J., Krafft, G. A., et al. (2009). Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}. J. Biol. Chem. 284, 18742–18753. doi: 10.1074/jbc.M109.011015
Keywords: insulin signaling, insulin-like growth factor, Alzheimer's disease, beta amyloid, phosphorylated tau
Citation: Bedse G, Di Domenico F, Serviddio G and Cassano T (2015) Aberrant insulin signaling in Alzheimer's disease: current knowledge. Front. Neurosci. 9:204. doi: 10.3389/fnins.2015.00204
Received: 11 March 2015; Accepted: 22 May 2015;
Published: 16 June 2015.
Edited by:
Ashok Kumar, University of Florida, USAReviewed by:
Karthik Bodhinathan, Sanford Burnham Medical Research Institute, USAParamita Chakrabarty, University of Florida, USA
Copyright © 2015 Bedse, Di Domenico, Serviddio and Cassano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tommaso Cassano, Department of Clinical and Experimental Medicine, University of Foggia, Viale Luigi Pinto 1, 71100 Foggia, Italy,dG9tbWFzby5jYXNzYW5vQHVuaWZnLml0