 Marina Grimaldi1,2,3,4
Marina Grimaldi1,2,3,4 Abdelhay Boulahtouf1,2,3,4
Abdelhay Boulahtouf1,2,3,4 Vanessa Delfosse5,6
Vanessa Delfosse5,6 Erwan Thouennon1,2,3,4
Erwan Thouennon1,2,3,4 William Bourguet5,6
William Bourguet5,6 Patrick Balaguer1,2,3,4*
Patrick Balaguer1,2,3,4*- 1Institut de Recherche en Cancérologie de Montpellier, Montpellier, France
- 2Institut National de la Santé et de la Recherche Médicale U1194, Montpellier, France
- 3Université Montpellier, Montpellier, France
- 4Institut Reìgional du Cancer de Montpellier, Montpellier, France
- 5Institut National de la Santé et de la Recherche Médicale U1054, Montpellier, France
- 6Centre National de la Recherche Scientifique UMR5048, Centre de Biochimie Structurale, Université Montpellier, Montpellier, France
Zebrafish is increasingly used as an animal model to study the effects of environmental nuclear receptors (NRs) ligands. As most of these compounds have only been tested on human NRs, it is necessary to measure their effects on zebrafish NRs. Estrogen receptors (ER) α and β and peroxysome proliferator activated receptor (PPAR) γ are main targets of environmental disrupting compounds (EDCs). In humans there are two distinct nuclear ERs (hERα and hERβ), whereas the zebrafish genome encodes three ERs, zfERα, zfERβ1, and zfERβ2. Only one isoform of PPARγ is expressed in both humans and zebrafish. In this review, we described reporter cell lines that we established to study the interaction of EDCs with human and zebrafish ERs and PPARγ. Using these cell lines, we observed that zfERs are thermo-sensitive while zfPPARγ is not. We also showed significant differences in the ability of environmental and synthetic ligands to modulate activation of zfERs and zfPPARγ in comparison to hERs and hPPARγ. Some environmental estrogens (bisphenol A, mycoestrogens) which are hER panagonists displayed greater potency for zfERα as compared to zfERβs. hERβ selective agonists (8βVE2, DPN, phytoestrogens) also displayed zfERα selectivity. Among hERα selective synthetic agonists, 16α-LE2 was the most zfERα selective compound. Almost all zfPPARγ environmental ligands (halogenated bisphenol A derivatives, phthalates, perfluorinated compounds) displayed similar affinity for human and zebrafish PPARγ while pharmaceutical hPPARγ agonists like thiazolidones are not recognized by zfPPARγ. Altogether, our studies show that all hERs and hPPARγ ligands do not control in a similar manner the transcriptional activity of zfERs and zfPPARγ and point out that care has to be taken in transposing the results obtained using the zebrafish as a model for human physiopathology.
Introduction
Human nuclear hormone receptors (NHRs) are a family of 48 transcription factors, many of which have been shown to be activated by ligands. NHRs regulate cognate gene networks involved in key physiological functions such as cell growth and differentiation, development, homeostasis, or metabolism (Gronemeyer et al., 2004; Germain et al., 2006). Consequently, inappropriate exposure to environmental pollutants often leads to proliferative, reproductive, and metabolic diseases, including hormonal cancers, infertility, obesity or diabetes. NHRs are modular proteins composed of several domains, most notably an N-terminal domain, which harbors a ligand-independent activation function (AF-1), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) hosting a ligand-dependent transcriptional activation function (AF-2) (Gronemeyer et al., 2004). In the absence of the cognate ligand, some NHRs are located in the nucleus, bind to the DNA response elements of their target genes, and recruit corepressors, while others are located in the cytoplasm in an inactive complex with chaperones.
Ligand binding induces major structural alterations of the receptor LBDs, leading to (1) destabilization of corepressor or chaperone interfaces, (2) exposure of nuclear localization signals to allow nuclear translocation and DNA binding of cytoplasmic receptors, and (3) recruitment of coactivators triggering gene transcription through chromatin remodeling and activation of the general transcription machinery. The crystal structures of many NHR LBDs have been determined, revealing a conserved core of 12 α-helices (H1–H12) and a short two-stranded antiparallel β-sheet (S1 and S2) arranged into a three-layered sandwich fold. This arrangement generates a mostly hydrophobic cavity in the lower half of the domain, which can accommodate the cognate ligand. In all hormone-bound LBD structures, the ligand-binding pocket (LBP) is sealed by helix H12. This conformation is specifically induced by the binding of hormones or synthetic agonists and is referred to as the “active conformation” because it allows the dissociation of corepressors and favors the recruitment of transcriptional coactivators (Bourguet et al., 2000; Renaud and Moras, 2000; Pike, 2006).
In contrast to agonist binding, interaction with antagonists prevents the correct positioning of helix H12, thus avoiding association with the LxxLL motifs of coactivators. The LBD also contributes to the modulation of the N-terminal AF-1 through interdomain crosstalk so that both AF-1 and AF-2 domains can recruit a range of coregulatory proteins and act individually or in a synergistic manner (Benecke et al., 2000; Bommer et al., 2002; Wilson, 2011).
Among nuclear receptors, ERs and PPARγ are main targets of numerous synthetic substances released into the environment by human activities. These substances can act as endocrine-disrupting chemicals (EDCs) causing reproductive, developmental, metabolic, or neurological diseases as well as hormone-related cancers (Diamanti-Kandarakis et al., 2009). Many EDCs are man-made compounds, for example bisphenols, phthalates, parabens, dioxins, pesticides, alkylphenols, organotins, polychlorinated biphenyls, or perfluoroalkyl compounds. Some natural EDCs can also be found in plants and fungi. Standard methods to study interaction of EDCs with these nuclear receptors use stable cell reporter gene assays based on human ERs and PPARγ activation (Balaguer et al., 1999; Legler et al., 1999; Seimandi et al., 2005; Riu et al., 2011a). To address whether chemicals exert an effect at the organismal level, ER activity assays have been developed for zebrafish. In these animals, GFP reporter constructs are designed to act in certain tissues exclusively (such as liver or brain) (Kurauchi et al., 2005; Brion et al., 2012) or in all tissues of embryos and larvae (Gorelick and Halpern, 2011; Lee et al., 2012). Zebrafish has also been used as an in vivo model to study the effect of environmental compounds on PPARγ (Riu et al., 2014). Zebrafish stores neutral lipid triglycerides in visceral, intramuscular, and subcutaneous adipocyte depots (Tingaud-Sequeira et al., 2012). Studies of the zebrafish embryo, which is optically transparent thus facilitating the labeling and detection of lipid depots using lipid staining (Minchin and Rawls, 2011), have shown that white adipose tissue appearance is correlated with size rather than the age of the fish. By using zebrafish as a PPARγ ligand screening model, we have showed that halogenated-BPA analogs are potent inducers of lipid accumulation in vivo through PPARγ signaling (Riu et al., 2014).
In order to evaluate the effects of environmental and pharmaceutical compounds on the transcriptional activity of zfERs and zfPPARγ and to compare the data with their activity on hERs and hPPARγ, we established human and zebrafish ERs and PPARγ reporter cell lines in the same cellular context (Balaguer et al., 1999; Seimandi et al., 2005; Pinto et al., 2014; Riu et al., 2014). In HeLa cells stably expressing an ERE-driven luciferase reporter (HELN cells), we expressed the full-length hERα, hERβ, zfERα, zfERβ1, and zfERβ2, respectively. Similarly, in HeLa cells stably expressing a GAL4RE-driven luciferase reporter (HG5LN cells), we expressed a fusion protein consisting of the hPPARγ or zfPPARγ ligand binding domain (LBD) and the DNA binding domain (DBD) of the yeast transcription factor GAL4 (GAL4-PPARγ).
The resulting HELN-ERs and HG5LN PPARγ cell lines were used to evaluate the effects of environmental compounds on gene transactivation by the five ERs and the two PPARγ, and to compare these effects with results obtained on hER and PPARγ orthologs. Since zebrafish is used as a model for studying the effects of environmental compounds in vivo, determining the transcriptional profiles of these compounds on the zfERs and zfPPARγ is crucial to support the zebrafish model for ER- and PPARγ-related studies and their extrapolation to the mammalian system.
Estrogen Receptors
Estrogen signaling is mainly mediated by the two estrogen receptors ERα (also called NR3A1) and ERβ (also called NR3A2) (Jensen and Jordan, 2003; Dahlman-Wright et al., 2006) which play important roles in the growth and maintenance of various tissues such as the mammary gland, uterus, bones, or the cardiovascular system. Like most NRs, ERs bind as dimers to DNA response elements in the promoter region of target genes and respond to the naturally occurring sex hormone 17β-estradiol (E2). Both hERs are widely distributed throughout the body, displaying distinct but overlapping expression patterns in a variety of tissues (Couse and Korach, 1999). hERα is primarily expressed in the uterus, liver, kidney, and heart, whereas hERβ is preferentially expressed in the ovary, prostate, lung, gastrointestinal tract, bladder, and hematopoietic and central nervous systems (Kuiper et al., 1997). However, hERα and hERβ are coexpressed in a number of tissues including the mammary gland, thyroid, adrenal, bones, and some regions of the brain. Although hERα and hERβ share similar mechanisms of action, several differences in the transcriptional abilities of each receptor and distinct phenotypes between gene-null animals have been identified, suggesting that these receptors may regulate distinct cellular pathways (Curtis et al., 1996; Couse and Korach, 1999). Interestingly, when hERs are coexpressed, hERβ exhibits an inhibitory action on ERα-mediated gene expression (Pettersson et al., 2000; Liu et al., 2002), so that hERβ has been shown to antagonize several hERα-mediated effects including fat reduction and cell proliferation in breast, uterus, or prostate (Ogawa et al., 1998; Weihua et al., 2000; Lindberg et al., 2003). Furthermore, in addition to controlling the normal development and function of the reproductive system and other tissues, estrogens are key regulators of primary breast and prostatic cancer growth (Jensen and Jordan, 2003). Roughly 40% of human cancers require steroid hormones for their growth and the first-line therapy for treatment of hormone-dependent cancers is based on androgen and estrogen antagonists interacting with AR or ERs and shutting down the corresponding hormone-responsive pathway. Interestingly, ERβ has been shown to antagonize ERα-mediated effects on cell proliferation in the breast, uterus, ovary, and prostate (Weihua et al., 2000; Lindberg et al., 2003; Ellem and Risbridger, 2009). In this regard, estrogens with selectivity for either ER subtypes may produce different biological outcomes, particularly on cancer cell proliferation. Given the widespread role of ERs in human physiology, it is not surprising that environmental compounds which bind to ERs, thus substituting for the natural hormone and deregulating the fine-tuned action of E2, can lead to ER-related disorders including breast, endometrial, colorectal, or prostate cancers, as well as neurodegenerative, inflammatory, immune, cardiovascular, and metabolic diseases.
Small fish including zebrafish (Danio rerio) are increasingly being used as model species to study in vivo effects of EDCs (Segner, 2009; Vosges et al., 2010; Brion et al., 2012). In zebrafish, three zfER subtypes (zfERα, zfERβ1, and zfERβ2) are present (Menuet et al., 2002; Hawkins and Thomas, 2004). Zebrafish ERα (esr1) is orthologous to the human ERα, while ERβ1 (esr2b) and ERβ2 (esr2a) are orthologs of the human ERβ (Bardet et al., 2002). The overall amino-acid sequence identity between the zfER subtypes and their corresponding human ER orthologs is approximately 50% (Menuet et al., 2002). ZfERs are differently expressed and regulated in reproductive tissue like gonads, liver, as well as in brain. In adult liver, E2 induces zfERα expression while it has no effect on zfERβ2 and represses zfERβ1 expression (Menuet et al., 2002). Moreover, both zfERα and zfERβ2 upregulate zfERα expression after E2 exposure, whereas zfERβ1 has no effect on this expression (Menuet et al., 2004). These studies suggest that the different forms of zfERs have partially distinct and nonredundant functions. Hence, in the perspective of developing fish in vitro assays, it is essential to take into account all zfER subtypes in the assessment of chemical estrogenicity in zebrafish. Since these three zfERs are thought to mediate different biological effects, there is an increased interest in finding subtype-selective zfER ligands.
Estrogen Receptors Reporter Cell Lines
To understand and to evaluate impact of xenoestrogens on ER-signaling pathway, it is necessary to develop cell-based transcription assay systems that could reflect different cellular contexts and/or different model species. In vitro assays based on reporter gene driven by ERE have been proven to be useful and relevant screening tools to address the large number of chemicals yet needed to be tested for their estrogenic potential. We and other groups have developed stable reporter gene assays based on human ERα and ERβ activation in different cell contexts and successfully used them to characterize estrogenic potency of chemicals (Balaguer et al., 1999; Legler et al., 1999; Wilson et al., 2004; Sotoca et al., 2008; Docquier et al., 2013). In order to take into account the species of origin of studied receptor in hazard assessment of estrogenic chemicals in fish, we have developed in vitro stable reporter gene assays derived from fish species (Molina-Molina et al., 2008; Cosnefroy et al., 2012; Pinto et al., 2014). Among them, HELN-zfERα, -zfERβ1, and -zfERβ2 (Pinto et al., 2014) reporter cell lines were established in a similar way than HELN-hERα and -hERβ cell lines (Pinto et al., 2014). Briefly, HELN-ERs cell lines cells were obtained by transfection of HELN cells (HeLa cells stably transfected with the ERE-β Globin-Luc-SVNeo plasmid) (Balaguer et al., 1999) by the corresponding pSG5-puro plasmids (pSG5-hERα-puro, -hERβ-puro -zfERα-puro, -zfERβ1-puro, and -zfERβ2-puro, respectively).
Selectivity of Chemicals for Human and Zebrafish Estrogen Receptors
Screening of endogenous, environmental and synthetic ligands in the HELN-zfER cell lines showed that known mammalian ER ligands are also able to induce transcriptional activity of zebrafish ER subtypes (Pinto et al., 2014). This screening allowed us to assess differences in the potency of the estrogenic compounds among the three zfER subtypes, and compare their selectivity toward hERs using a similar human cellular context. The HELN-zfERs cells were incubated at 28°C after addition of chemicals to the cells because it is a more physiologically relevant temperature for zebrafish, which increased the potency of estradiol approximately 10-fold compared to incubation at 37°C. Temperature sensitivity of fish ERs has already been reported using reporter gene assays (Matthews et al., 2002; Cosnefroy et al., 2009) and the reason seems to be thermo-dependence of estrogen binding (Tan et al., 1999; Matthews et al., 2002; Sumida et al., 2003).
We have shown that there are clear differences between the selectivity of various (anti)estrogens for zebrafish and human ER isoforms, establishing the fact that a direct translation of (anti)estrogenic effects (activities or potencies) from mammals to zebrafish is not possible. Although none of the tested compounds specifically activated either zebrafish or human ERs, transcriptional activities toward human and zebrafish ERs need to be studied.
Natural (E2) and pharmaceutical (EE2) estrogens display similar affinities for hERs and zfERs. Some environmental estrogens (α-zearalanol, bisphenol-A) with similar affinity for hERs preferentially activated zfERα rather than zfERβs. Other environmental estrogens (nonylphenol mixture, 4-tert-octylphenol) with similar affinity for hERs displayed slightly higher affinity for zfERα and zfERβ2 than for zfERβ1. Benzophenone 2 and phytoestrogens (genistein, liquiritigenin) which have higher affinity for hERβ than for hERα also displayed slightly higher affinity for zfERα and zfERβ2 than for zfERβ1. Finally, hERβ selective synthetic compounds (8β-vE2, DPN) preferentially activated zfERα compared to zfERβ s. On the contrary and similar to hERs, the synthetic compound 16α-E2, which has 1000-fold more selectivity for hERα (Escande et al., 2006), also exhibited higher affinity for zfERα compared to the zfERβ subtypes and is the most selective compound for zfERα nowadays (Figure 1; Table 1).
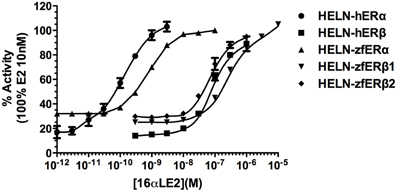
Figure 1. Transcriptional activity of hERα, hERβ, zfERα, zfERβ1, and zfERβ2 in response to the synthetic pharmaceutical compound 16α-LE2. HELN-hERα (●), -hERβ1 (■), -zfERα (○), HELN-zfERβ1 (□), HELN-zfERβ2 (◊) cells were exposed to different concentrations of 16α-LE2. Results are expressed as % of 10 nM E2 treatment and are derived from Escande et al. (2006) and Pinto et al. (2014).
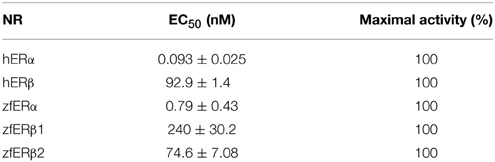
Table 1. 16αLE2 ERs EC50 and maximal activities.
To gain structural insights into the zfERα selectivity of 16α-LE2 in human and zebrafish ERs, we used the web-based server EDMon (Endocrine Disruptor Monitoring; http://atome2.cbs.cnrs.fr/AT2B/SERVER/EDMon.html) (Delfosse et al., 2012) to model zfERs in complex with this ligand. The structural basis of the hERα and hERβ selectivity toward certain ligands has been associated with two amino acid differences in their ligand-binding pockets. Indeed, L384 and M421 of hERα are replaced by M336 and I373 in hERβ, respectively (Figure 2 and Manas et al., 2004). Superimposition of the 16α-LE2-bound zfERα model on the crystal structure of hERα in complex with E2 (PDB code 3UUD) showed that the phenol ring of 16α-LE2 occupies the same position as that of E2 and is engaged in a network of hydrogen bonds with E353 from helix 3 (H3) and R394 from H5 (Figure 2A). On the other side of the ligand-binding pocket (LBP), it appears that the hydrogen bond observed between the 17-hydroxyl group of E2 and H524 (H11) is conserved in 16α-LE2. The difference between the two complexes resides in the lactone ring of 16α-LE2 which points toward M421 (H7) that must undergo a large conformational change to accommodate this additional group. In hERβ, the linear M421 is replaced by the branched residue Ileu 373 characterized by a much smaller intrinsic flexibility (Figure 2B). As a consequence, I373 maintains the synthetic ligand in a position where it interacts unfavorably with M336 (H3). Therefore, 16α-LE2 adopts different positions in hERα and hERβ, the more constrained environment provided by the latter accounting for the weaker affinity of the ligand for this receptor subtype. The affinity values measured with the zebrafish receptors reflect the variations in the space constraints provided by the different combinations of residues in the three receptor subtypes. With H3 and H7 residues identical to those of the human receptor, zfERα interacts with 16α-LE2 with the highest affinity. The slight difference in the binding affinity of 16α-LE2 for hERα and zfERα relies most likely on the replacement of L349 (H3) by a methionine residue (M317) (Figure 2A) and a possible loss of a favorable interaction provided by the branched but not by the linear residue (Figure 2A). With a conserved isoleucine in H7 (I406) and a leucine residue in H3 (L369) (Figure 2B), zfERβ1 displays the most constrained LBP reflecting the weakest binding affinity for 16α-LE2. This receptor combines two large residues with low (isoleucine) and medium (leucine) flexibilities. The replacement of I406 in H7 of zfERβ1 by a leucine residue (L391) (Figure 2B) in zfERβ2 provides a slight gain in LBP plasticity, in agreement with the slightly better affinity of 16α-LE2 for the latter.
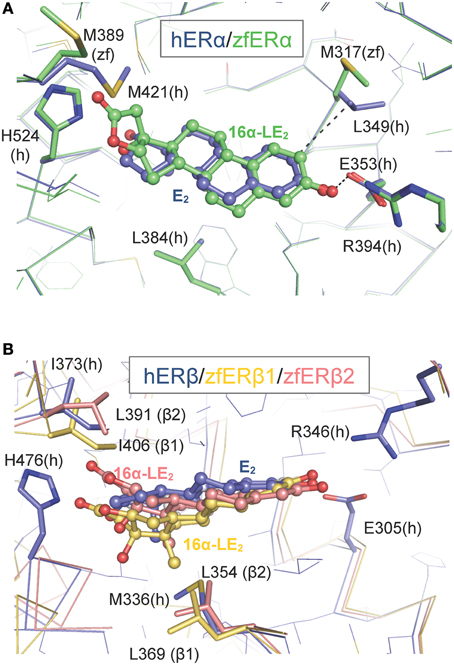
Figure 2. Modeling of the interaction between 16α-LE2 and the human (h) and zebrafish (zf) estrogen receptors. Superposition of the structures of hERα (A) and hERβ (B) LBDs bound to E2 (blue) on to the molecular models of the 16α-LE2-bound zfERα (A) (green) and 16α-LE2-bound hERβ1 (red) and 16α-LE2-bound hERβ2 (yellow) (B) LBDs. In zfERα, the lactone ring of 16α-LE2 points toward M421 (H7) which undergoes a large conformational change (black arrow) to accommodate this additional group. In hERβ, the linear M421 present in ERα (M389 in zfERα) is replaced by branched residues (I373 in hERβ, I406 in zfERβ1 and L391 in zfERβ2), which are characterized by a much smaller intrinsic flexibility that maintains the synthetic ligand in a position where it interacts unfavorably with M336 in hERβ, L369 in zfERβ1 and L354 in zfERβ2. This figure is derived from Pinto et al. (2014).
The inability of hERβ-selective phytoestrogens (genistein and liquiritigenin) and pharmaceuticals (8bv-E2, DPN) to activate preferentially the zfERβ isoforms is explained by the mutation of a critical amino acid involved in genistein binding in hERβ. In all zfERs, the position homologous to hERβ M336 is occupied, as in hERα, by a leucine residue (Figure 2) (Sassi-Messai et al., 2009). This amino acid change most likely accounts for the lack of obvious selectivity of the phytoestrogens toward the zfERβ subtypes.
Peroxysome Proliferator γ
PPARs are involved in the regulation of glucose, lipid, and cholesterol metabolism in response to fatty acids and their derivatives, eicosanoids, and drugs used in the treatment of hyperlipidemia and diabetes. The human PPAR subfamily contains three members known as hPPARα, hPPARβ, and hPPARγ. Each hPPAR subtype shows a distinct tissue distribution and ligand preference. hPPARγ is highly expressed in adipose tissue and is a central regulator of lipid storage and adipocyte gene expression and differentiation (Tontonoz et al., 1995) and is involved in various pathophysiological disorders, including metabolic disease, insulin resistance, and diabetes (Rosen and Spiegelman, 2001). hPPARγ is the target for antidiabetic agents of the thiazolidinedione class, which includes troglitazone, pioglitazone, and rosiglitazone. The LBD of hPPARγ is rather large and the diversity of ligands that can be accommodated within its pocket, mainly represented by lipid derivatives, may contribute to the large array of roles that have been assigned to hPPARγ. Given the physiological role of hPPARγ in adipose tissue development and maintenance, it has been proposed that disruption of regulation pathways under the control of hPPARγ may be involved in the onset of diabetes and obesity (Swedenborg et al., 2009). Indeed, activation of this receptor by certain xenobiotic compounds has been shown to stimulate adipogenesis in vitro and in vivo through induction of the differentiation of preadipocytes of the fibroblastic lineage into mature adipocytes (Grun and Blumberg, 2009; le Maire et al., 2009; Janesick and Blumberg, 2011; Riu et al., 2011a). This has led to the “obesogen hypothesis,” according to which, in addition to disruption of the balance between caloric intake and expenditure characterizing modern life-style, the rapidly growing obesity epidemic could also implicate environmental risk factors including an increased exposure to chemicals that interfere with any aspects of metabolism (Grun and Blumberg, 2009; Janesick and Blumberg, 2011, 2012). Accordingly, compounds that have the potential to disrupt any metabolic signaling pathways and lead to increased fat accumulation and obesity are referred to as “obesogens” (Grun and Blumberg, 2006).
Like for ERs, zebrafish begin to be used as model species to study in vivo effects of EDCs on PPARγ (Lyche et al., 2011; Riu et al., 2014). Similar to mammals, zebrafish store neutral lipid triglycerides in the visceral, intramuscular, and subcutaneous adipocyte depots. The first adipocytes, which can be observed from day 8 to 12, or at a minimal size of about 5 mm (Imrie and Sadler, 2010), appear in the pancreatic region, then in the viscera, and later on, in the subcutaneous and cranial regions (Flynn et al., 2009; Imrie and Sadler, 2010). Lipid staining can be detected before this stage; however, at this time point, the lipids are not stored in adipocytes, but rather in the yolk, hepatocytes, blood vessels, skeletal myocytes, jaw chondrocytes, and neuronal tissue in the brain (Imrie and Sadler, 2010).
Pparγ reporter cell lines
HG5LN-hPPARγ and -zfPPARγ reporter cell lines were established in a similar way (Seimandi et al., 2005; Riu et al., 2014). Briefly, HG5LN-PPARγ cell line was obtained by transfection of HG5LN cells (HeLa cells stably transfected with the GALRE5-β Globin-Luc-SVNeo plasmid) (Seimandi et al., 2005) by the corresponding pSG5-puro plasmids [pSG5-GAL4(DBD)-hPPARγ(LBD)-puro and -zfPPARγ(LBD)-puro, respectively]. Interestingly, the thermodependence observed for zfERs is not shared by zfPPARγ (Riu et al., 2014).
Selectivity of chemicals for human and zebrafish PPARγ
Screening of environmental and pharmaceutical ligands in the HG5LN-zfPPARγ cell lines showed that known hPPARγ ligands are not always able to induce transcriptional activity of zebrafish PPARγ (Riu et al., 2011a). Pharmaceutical hPPARγ ligands like thiazolidones (rosiglitazone, troglitazone) do not or very weakly bind to zfPPARγ. On the contrary, environmental PPARγ compounds including phthalates (MEHP), perfluorinated compounds (PFOA, PFOS) and halogenated derivatives of BPA (TBBPA, TCBPA) are common activators of hPPARγ and zfPPARγ (Figures 3, 4; Table 2). We also provide evidence that activation of ERs and PPARγ depends on the halogenation degree of BPA analogs. The bulkier are brominated BPA analogs, the greater is their capability to activate PPARγ and the weaker is their estrogenic potential (Riu et al., 2011b).
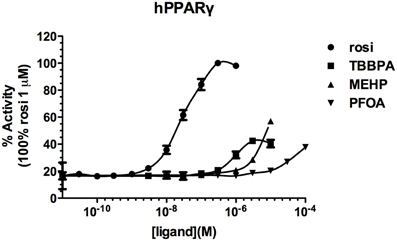
Figure 3. Transcriptional activity of hPPARγ in response to the synthetic pharmaceutical compound rosiglitazone and the environmental compounds TBBPA, MEHP, and PFOA. HG5LN hPPARγ and zfPPARγ cells were exposed to different concentrations of rosiglitazone (●), TBBPA (□), MEHP (○), and PFOA (◊). Results are expressed as % of basal activity and are derived from Riu et al. (2011a).
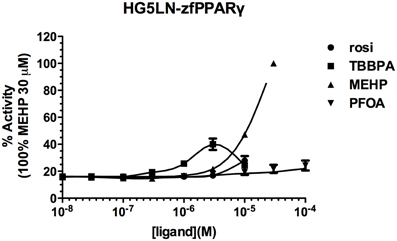
Figure 4. Transcriptional activity of zfPPARγ in response to the synthetic pharmaceutical compound rosiglitazone and the environmental compounds TBBPA, MEHP, and PFOA. HG5LN hPPARγ and zfPPARγ cells were exposed to different concentrations of rosiglitazone (●), TBBPA (□), MEHP (○), and PFOA (◊). Results are expressed as % of basal activity and are derived from Riu et al. (2011a) and Riu et al. (2014).
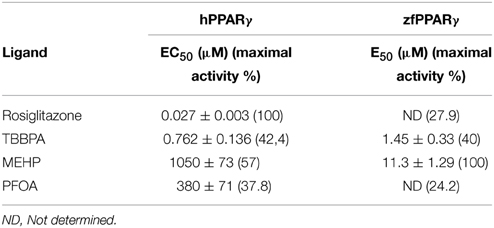
Table 2. EC50 and maximal activities of PPARγ ligands.
Comparison of human and zebrafish PPARγ sequences reveals several residue differences which could explain the differential ligand specificity of the various species (Figure 5A). In particular, the replacement of human PPARγ Gly284 and Cys285 by serine and tyrosine residues in zebrafish PPARγ provides a rationale for the weak binding affinity of rosiglitazone for this receptor as compared to that observed for the human homolog (Figure 4B). In contrast, the different binding mode of halogenated compounds allows both hPPARγ and zfPPARγ to accommodate TBBPA and TCBPA (Figure 5B).
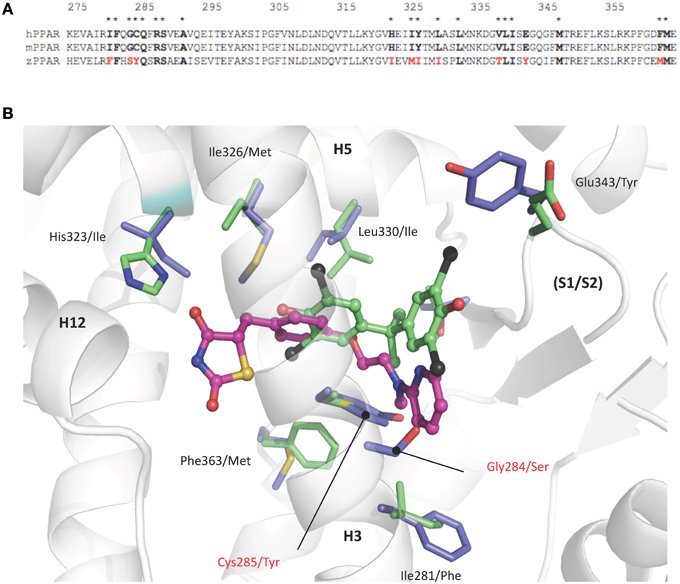
Figure 5. (A) Sequence alignment of human, mouse, and zebrafish PPARγ ligand binding pocket residues. Asterisks denote residues in contact with TBBPA and/or rosiglitazone (PDB code 2PRG). Interacting residues that differ between sequences are highlighted in red. (B) TBBPA (carbon atoms colored in green) and rosiglitazone (magenta, PDB code 2PRG) as they are positioned in the human PPARγ. Residues that differ in the ligand binding pocket of human and zebrafish PPARγ are displayed as green and blue sticks, respectively. The human PPARγ Gly284 and Cys285 which are replaced by serine and tyrosine residues in zebrafish PPARγ are indicated in red. This figure is derived from Riu et al. (2011a).
Structural and biophysical studies revealed that TBT binds to both hRXR and hPPARγ through formation of a covalent bond between the tin atom and the sulfur atom of cysteine residues located in the LBP of both receptors (le Maire et al., 2009; Delfosse et al., 2014). In RXR, this cysteine (Cys432) is located in helix H11 and is conserved in several species. In contrast, the cysteine residue of PPARγ (Cys285) resides in H3 and is not conserved in several species including zebrafish.
Conclusion
We have shown above that there are clear differences between the activity of various EDCs for zebrafish and human ERs and PPARs, demonstrating that a direct translation of effects from mammals to zebrafish is not possible. The differences revealed in this study, in terms of transcriptional activities toward human and zebrafish ERs and PPARs, highlight the need to take into account the species of origin when assessing the potency of chemicals. This is particularly important with regard to EDCs screening for hazard assessment since at the present time established test guidelines are only based on human cell lines expressing human nuclear receptors.
To this end, such in vitro cell lines expressing zebrafish nuclear receptors can serve as useful screening tools to address nuclear receptor potency of chemicals for fish models. Hence, an initial screening should be followed up with an NR-subtype specific analysis using both human and zebrafish NRs to elucidate the full spectrum of NR-mediated EDCs effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the financial support from the Agence Nationale de la Recherche, project PROOFS and TOXSYN.
References
Balaguer, P., François, F., Comunale, F., Fenet, H., Boussioux, A. M., Pons, M., et al. (1999). Reporter cell lines to study the estrogenic effects of xenoestrogens. Sci. Total Environ. 233, 47–56. doi: 10.1016/S0048-9697(99)00178-3
Bardet, P. L., Horard, B., Robinson-Rechavi, M., Laudet, V., and Vanacker, J. M. (2002). Characterization of oestrogen receptors in zebrafish (Danio rerio). J. Mol. Endocrinol. 28, 153–163. doi: 10.1677/jme.0.0280153
Benecke, A., Chambon, P., and Gronemeyer, H. (2000). Synergy between estrogen receptor alpha activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep. 1, 151–157. doi: 10.1093/embo-reports/kvd028
Bommer, M., Benecke, A., Gronemeyer, H., and Rochette-Egly, C. (2002). TIF2 mediates the synergy between RARalpha 1 activation functions AF-1 and AF-2. J. Biol. Chem. 277, 37961–37966. doi: 10.1074/jbc.M206001200
Bourguet, W., Germain, P., and Gronemeyer, H. (2000). Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 21, 381–388. doi: 10.1016/S0165-6147(00)01548-0
Brion, F., Le Page, Y., Piccini, B., Cardoso, O., Tong, S. K., Chung, B. C., et al. (2012). Screening estrogenic activities of chemicals or mixtures in vivo using transgenic (cyp19a1b-GFP) zebrafish embryos. PLoS ONE 7:e36069. doi: 10.1371/journal.pone.0036069
Cosnefroy, A., Brion, F, Guillet, B, Laville, N, Porcher, J. M., Balaguer, P., et al. (2009). A stable fish reporter cell line to study estrogen receptor transactivation by environmental (xeno)estrogens. Toxicol. In Vitro 23, 1450–1454. doi: 10.1016/j.tiv.2009.07.003
Cosnefroy, A., Brion, F., Maillot-Maréchal, E., Porcher, J. M., Pakdel, F., Balaguer, P., et al. (2012). Selective activation of zebrafish estrogen receptor subtypes by chemicals by using stable reporter gene assay developed in a zebrafish liver cell line. Toxicol. Sci. 125, 439–449. doi: 10.1093/toxsci/kfr297
Couse, J. F., and Korach, K. S. (1999). Estrogen receptor null mice: what have we learned and where will they lead us? Endocr. Rev. 20, 358–417. doi: 10.1210/edrv.20.3.0370
Curtis, S. W., Washburn, T., Sewall, C., DiAugustine, R., Lindzey, J., Couse, J. F., et al. (1996). Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. U.S.A. 93, 12626–12630 doi: 10.1073/pnas.93.22.12626
Dahlman-Wright, K., Cavailles, V., Fuqua, S. A., Jordan, V. C., Katzenellenbogen, J. A., Korach, K. S., et al. (2006). International union of pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 58, 773–781. doi: 10.1124/pr.58.4.8
Delfosse, V., Grimaldi, M., Cavaillès, V., Balaguer, P., and Bourguet, W. (2014). Structural and functional profiling of environmental ligands for estrogen receptors. Environ. Health Perspect. 122, 1306–1313. doi: 10.1289/ehp.1408453
Delfosse, V., Grimaldi, M., Pons, J. L., Boulahtouf, A., le Maire, A., Cavailles, V., et al. (2012). Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. U.S.A. 109, 14930–14935. doi: 10.1073/pnas.1203574109
Diamanti-Kandarakis, E., Bourguignon, J. P., Giudice, L. C., Hauser, R., Prins, G. S., Soto, A. M., et al. (2009). Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr. Rev. 30, 293–342. doi: 10.1210/er.2009-0002
Docquier, A., Garcia, A., Savatier, J., Boulahtouf, A., Bonnet, S., Bellet, V., et al. (2013). Negative regulation of estrogen signaling by ERβ and RIP140 in ovarian cancer cells. Mol. Endocrinol. 27, 1429–1441. doi: 10.1210/me.2012-1351
Ellem, S. J., and Risbridger, G. P. (2009). The dual, opposing roles of estrogen in the prostate. Ann. N.Y. Acad. Sci. 1155, 174–186. doi: 10.1111/j.1749-6632.2009.04360.x
Escande, A., Pillon, A., Servant, N., Cravedi, J. P., Larrea, F., Muhn, P., et al. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol. 71, 1459–1469. doi: 10.1016/j.bcp.2006.02.002
Flynn, E. J. III., Trent, C. M., and Rawls, J. F. (2009). Ontogeny and nutritional control of adipogenesis in zebrafish (Danio rerio). J. Lipid. Res. 50, 1641–1652. doi: 10.1194/jlr.M800590-JLR200
Germain, P., Staels, B., Dacquet, C., Spedding, M., and Laudet, V. (2006). Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 58, 685–704. doi: 10.1124/pr.58.4.2
Gorelick, D. A., and Halpern, M. E. (2011). Visualization of estrogen receptor transcriptional activation in zebrafish. Endocrinology 152, 2690–2703. doi: 10.1210/en.2010-1257
Gronemeyer, H., Gustafsson, J. A., and Laudet, V. (2004). Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 3, 950–964. doi: 10.1038/nrd1551
Grun, F., and Blumberg, B. (2006). Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 147, S50–S55 doi: 10.1210/en.2005-1129
Grun, F., and Blumberg, B. (2009). Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 304, 19–29. doi: 10.1016/j.mce.2009.02.018
Hawkins, M. B., and Thomas, P. (2004). 17alpha-ethinylestradiol disrupts the ontogeny of the forebrain GnRH system and the expression of brain aromatase during early development of zebrafish. Endocrinology 145, 2968–2977. doi: 10.1210/en.2003-0806
Imrie, D., and Sadler, K. C. (2010). White adipose tissue development in zebrafish is regulated by both developmental time and fish size. Dev. Dyn. 239, 3013–3023. doi: 10.1002/dvdy.22443
Janesick, A., and Blumberg, B. (2011). Minireview: PPARγ as the target of obesogens. J. Steroid Biochem. Mol. Biol. 127, 4–8. doi: 10.1016/j.jsbmb.2011.01.005
Janesick, A., and Blumberg, B. (2012). stem cells and the developmental programming of obesity. Int. J. Androl. 35, 437–448. doi: 10.1111/j.1365-2605.2012.01247.x
Jensen, E. V., and Jordan, V. C. (2003). The estrogen receptor: a model for molecular medicine. Clin. Cancer Res. 9, 1980–1989.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Häggblad, J., Nilsson, S., et al. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138, 863–870.
Kurauchi, K., Nakaguchi, Y., Tsutsumi, M., Hori, H., Kurihara, R., Hashimoto, S., et al. (2005). In vivo visual reporter system for detection of estrogen-like substances by transgenic medaka. Environ. Sci. Technol. 39, 2762–2768. doi: 10.1021/es0486465
Lee, O., Tyler, C. R., and Kudoh, T. (2012). Development of a transient expression assay for detecting environmental oestrogens in zebrafish and medaka embryos. BMC Biotechnol. 12:32. doi: 10.1186/1472-6750-12-32
Legler, J., van den Brink, C. E., Brouwer, A., Murk, A. J., van der Saag, P. T., Vethaak, A. D., et al. (1999). Development of a stably transfected estrogen receptor-mediated luciferase reporter gene assay in the human T47D breast cancer cell line. Toxicol. Sci. 48, 55–66. doi: 10.1093/toxsci/48.1.55
le Maire, A., Grimaldi, M., Roecklin, D., Dagnino, S., Vivat-Hannah, V., Balaguer, P., et al. (2009). Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 10, 367–373. doi: 10.1038/embor.2009.8
Lindberg, M. K., Moverare, S., Skrtic, S., Gao, H., Dahlman-Wright, K., Gustafsson, J. A., et al. (2003). Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol. Endocrinol. 17, 203–208. doi: 10.1210/me.2002-0206
Liu, M. M., Albanese, C., Anderson, C. M., Hilty, K., Webb, P., Uht, R. M., et al. (2002). Opposing action of estrogen receptors alpha and beta on cyclin D1 gene expression. J. Biol. Chem. 277, 24353–24360. doi: 10.1074/jbc.M201829200
Lyche, J. L., Nourizadeh-Lillabadi, R, Karlsson, C, Stavik, B, Berg, V, Skåre, J. U., et al. (2011). Natural mixtures of POPs affected body weight gain and induced transcription of genes involved in weight regulation and insulin signaling. Aquat. Toxicol. 102, 197–204. doi: 10.1016/j.aquatox.2011.01.017
Manas, E. S., Xu, Z. B., Unwalla, R. J., and Somers, W. S. (2004). Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure 12, 2197–2207. doi: 10.1016/j.str.2004.09.015
Matthews, J. B., Fertuck, K. C., Celius, T., Huang, Y. W., Fong, C. J., and Zacharewski, T. R. (2002). Ability of structurally diverse natural products and synthetic chemicals to induce gene expression mediated by estrogen receptors from various species. J. Steroid Biochem. Mol. Biol. 82, 181–194. doi: 10.1016/S0960-0760(02)00159-0
Menuet, A., Le Page, Y., Torres, O., Kern, L., Kah, O., and Pakdel, F. (2004). Analysis of the estrogen regulation of the zebrafish estrogen receptor (ER) reveals distinct effects of ERalpha, ERbeta1 and ERbeta2. J. Mol. Endocrinol. 32, 975–986. doi: 10.1677/jme.0.0320975
Menuet, A., Pellegrini, E, Anglade, I, Blaise, O, Laudet, V, Kah, O., et al. (2002). Molecular characterization of three estrogen receptor forms in zebrafish: binding characteristics, transactivation properties, and tissue distributions. Biol. Reprod. 66, 1881–1892. doi: 10.1095/biolreprod66.6.1881
Minchin, J. E., and Rawls, J. F. (2011). In vivo analysis of white adipose tissue in zebrafish. Methods Cell Biol. 105, 63–86. doi: 10.1016/b978-0-12-381320-6.00003-5
Molina-Molina, J. M., Escande, A., Pillon, A., Gomez, E., Pakdel, F., Cavaillès, V., et al. (2008). Profiling of benzophenone derivatives using fish and human estrogen receptor-specific in vitro bioassays. Toxicol. Appl. Pharmacol. 232, 384–395. doi: 10.1016/j.taap.2008.07.017
Ogawa, S., Eng, V., Taylor, J., Lubahn, D. B., Korach, K. S., and Pfaff, D. W. (1998). Roles of estrogen receptor alpha gene expression in reproduction-related behaviors in female mice. Endocrinology 139:5070–5081.
Pettersson, K., Delaunay, F., and Gustafsson, J. A. (2000). Estrogen receptor beta acts as a dominant regulator of estrogen signaling Oncogene 19, 4970–4978 doi: 10.1038/sj.onc.1203828
Pike, A. C. (2006). Lessons learnt from structural studies of the oestrogen receptor. Best Pract. Res. Clin. Endocrinol. Metab. 20, 1–14. doi: 10.1016/j.beem.2005.09.002
Pinto, C., Grimaldi, M., Boulahtouf, A., Pakdel, F., Brion, F., Aït-Aïssa, S., et al. (2014). Selectivity of natural, synthetic and environmental estrogens for zebrafish estrogen receptors. Toxicol. Appl. Pharmacol. 280, 60–69. doi: 10.1016/j.taap.2014.07.020
Renaud, J. P., and Moras, D. (2000). Structural studies on nuclear receptors. Cell. Mol. Life Sci. 57, 1748–1769. doi: 10.1007/PL00000656
Riu, A., Grimaldi, M., le Maire, A., Bey, G., Phillips, K., Boulahtouf, A., et al. (2011a). Peroxisome proliferator-activated receptor γ is a target for halogenated analogs of bisphenol A. Environ. Health Perspect. 119, 1227–1232. doi: 10.1289/ehp.1003328
Riu, A., le Maire, A., Grimaldi, M., Audebert, M., Hillenweck, A., Bourguet, W., et al. (2011b). Characterization of novel ligands of ERα, ERβ, and PPARγ: the case of halogenated bisphenol A and their conjugated metabolites. Toxicol. Sci. 122, 372–382. doi: 10.1093/toxsci/kfr132
Riu, A., McCollum, C. W., Pinto, C. L., Grimaldi, M., Hillenweck, A., Perdu, E., et al. (2014). Halogenated bisphenol-A analogs act as obesogens in zebrafish larvae (Danio rerio). Toxicol. Sci. 139, 48–58. doi: 10.1093/toxsci/kfu036
Rosen, E. D., and Spiegelman, B. M. (2001). PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 276, 37731–37734. doi: 10.1074/jbc.R100034200
Sassi-Messai, S., Gibert, Y., Bernard, L., Nishio, S., Ferri Lagneau, K. F., Molina, J., et al. (2009). The phytoestrogen genistein affects zebrafish development through two different pathways. PLoS ONE 4:e4935. doi: 10.1371/journal.pone.0004935
Segner, H. (2009). Zebrafish (Danio rerio) as a model organism for investigating endocrine disruption. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 149, 187–195. doi: 10.1016/j.cbpc.2008.10.099
Seimandi, M., Lemaire, G., Pillon, A., Perrin, A., Carlavan, I., Voegel, J. J., et al. (2005). Differential responses of PPARα, PPARδ, and PPARγ reporter cell lines to selective PPAR synthetic ligands. Anal. Biochem. 344, 8–15. doi: 10.1016/j.ab.2005.06.010
Sotoca, A. M., Ratman, D., van der Saag, P., Ström, A., Gustafsson, J. A., Vervoort, J., et al. (2008). Phytoestrogen-mediated inhibition of proliferation of the human T47D breast cancer cells depends on the ERalpha/ERbeta ratio. J. Steroid Biochem. Mol. Biol. 112, 171–178. doi: 10.1016/j.jsbmb.2008.10.002
Sumida, K., Ooe, N, Saito, K, and Kaneko, H. (2003). Limited species differences in estrogen receptor alpha-medicated reporter gene transactivation by xenoestrogens. J. Steroid. Biochem. Mol. Biol. 84, 33–40. doi: 10.1016/S0960-0760(03)00003-7
Swedenborg, E., Ruegg, J., Makela, S., and Pongratz, I. (2009). Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J. Mol. Endocrinol. 43, 1–10. doi: 10.1677/JME-08-0132
Tan, N. S., Frecer, V., Lam, T. J., and Ding, J. L. (1999). Temperature dependence of estrogen binding: importance of a subzone in the ligand binding domain of a novel piscine estrogen receptor. Biochim. Biophys. Acta 1452, 103–120. doi: 10.1016/S0167-4889(99)00128-7
Tingaud-Sequeira, A., Knoll-Gellida, A., André, M., and Babin, P. J. (2012). Vitellogenin expression in white adipose tissue in female teleost fish. Biol. Reprod. 86, 38. doi: 10.1095/biolreprod.111.093757
Tontonoz, P., Hu, E., and Spiegelman, B. M. (1995). Regulation of adipocyte gene expression and differentiation by peroxisome proliferator activated receptor gamma. Curr. Opin. Genet. Dev. 5, 571–576. doi: 10.1016/0959-437X(95)80025-5
Vosges, M., Le Page, Y., Chung, B. C., Combarnous, Y., Porcher, J. M., Kah, O., et al. (2010). 17alpha-ethinylestradiol disrupts the ontogeny of the forebrain GnRH system and the expression of brain aromatase during early development of zebrafish. Aquat Toxicol. 99, 479–491. doi: 10.1016/j.aquatox.2010.06.009
Weihua, Z., Saji, S., Makinen, S., Cheng, G., Jensen, E. V., Warner, M., et al. (2000). Estrogen receptor (ER) beta, a modulator of ERalpha in the uterus. Proc. Natl. Acad. Sci. U.S.A. 97, 5936–5941. doi: 10.1073/pnas.97.11.5936
Wilson, E. M. (2011). Analysis of interdomain interactions of the androgen receptor. Methods Mol. Biol. 776, 113–129. doi: 10.1007/978-1-61779-243-4_8
Keywords: estrogen receptor, peroxysome proliferator activated receptor γ, environmental disrupting compounds, reporter cell lines, human, zebrafish
Citation: Grimaldi M, Boulahtouf A, Delfosse V, Thouennon E, Bourguet W and Balaguer P (2015) Reporter cell lines to evaluate the selectivity of chemicals for human and zebrafish estrogen and peroxysome proliferator activated γ receptors Front. Neurosci. 9:212. doi: 10.3389/fnins.2015.00212
Received: 11 December 2014; Accepted: 26 May 2015;
Published: 09 June 2015.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Berta Levavi-Sivan, The Hebrew University, IsraelTaisen Iguchi, National Institute for Basic Biology, Japan
Copyright © 2015 Grimaldi, Boulahtouf, Delfosse, Thouennon, Bourguet and Balaguer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Balaguer, Institut de Recherche en Cancérologie de Montpellier, Institut National de la Santé et de la Recherche Médicale 1194, Institut Reìgional du Cancer de Montpellier, Parc Euromédecine, 208 rue des Apothicaires, 34090 Montpellier, France,cGF0cmljay5iYWxhZ3VlckBpbnNlcm0uZnI=