 Mary Gannon
Mary Gannon Pulin Che
Pulin Che Yunjia Chen
Yunjia Chen Kai Jiao2
Kai Jiao2 Erik D. Roberson
Erik D. Roberson Qin Wang
Qin Wang- 1Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL, USA
- 2Department of Genetics, University of Alabama at Birmingham, Birmingham, AL, USA
- 3Department of Neurology, University of Alabama at Birmingham, Birmingham, AL, USA
The brain noradrenergic system supplies the neurotransmitter norepinephrine throughout the brain via widespread efferent projections, and plays a pivotal role in modulating cognitive activities in the cortex. Profound noradrenergic degeneration in Alzheimer's disease (AD) patients has been observed for decades, with recent research suggesting that the locus coeruleus (where noradrenergic neurons are mainly located) is a predominant site where AD-related pathology begins. Mounting evidence indicates that the loss of noradrenergic innervation greatly exacerbates AD pathogenesis and progression, although the precise roles of noradrenergic components in AD pathogenesis remain unclear. The aim of this review is to summarize current findings on noradrenergic dysfunction in AD, as well as to point out deficiencies in our knowledge where more research is needed.
Introduction
Noradrenergic neurons are located primarily in the locus coeruleus. Via wide spread efferent projections, these neurons provide norepinephrine throughout the brain (Swanson and Hartman, 1975). Along with dopamine and epinephrine, norepinephrine is a catecholamine that is synthesized through a series of enzymatic steps that begins with the amino acid tyrosine. Tyrosine is first converted to levodopa (L-DOPA) by the enzyme tyrosine hydroxylase, which is the rate-limiting step. Pyridoxal phosphate and DOPA decarboxylase then convert L-DOPA to dopamine, which is subsequently converted to norepinephrine by dopamine beta hydroxylase (DβH).
After synthesis, norepinephrine is released by presynaptic noradrenergic terminals, and can bind to receptors, be degraded by enzymes such as catechol-O-methyltransferases (COMT) or monoamine oxidases (MAO) (Golan et al., 2011), or undergo reuptake into the presynaptic neuron. Norepinephrine elicits its effects through three classes of adrenergic receptors, the α1, the α2, and the β adrenergic receptors, each of which consists of three subtypes (Hein, 2006). While all of the receptors are located at postsynaptic neurons, the α2 receptors are uniquely also located presynaptically where they function as autoreceptors to inhibit the further release of norepinephrine (Knaus et al., 2007). The amount of norepinephrine available to bind to receptors is largely dependent on either degradation or reuptake of norepinephrine to the presynaptic terminals. Norepinephrine reuptake is mediated by the norepinephrine transporter, a Na/K pump that returns norepinephrine to the presynaptic terminal.
Changes in the noradrenergic system have long been observed in Alzheimer's disease (AD). Pathological changes of noradrenergic neurons in the locus coeruleus, including the accumulation of tau protein (Braak and Del Tredici, 2012), begin early in the progression of AD. Later in the disease, profound loss of the noradrenergic neurons is observed, along with compensatory changes such as modifications of neuronal anatomy, neurotransmitter, and noradrenergic receptors. Given the well-established functions of the noradrenergic system in cognition, neuroinflammation, and metabolism, it could be predicted that noradrenergic dysfunction in AD would promote disease progression. Indeed, evidence from both human patients and animal models indicates that loss of noradrenergic innervation greatly exacerbates AD pathogenesis and progression (Marien et al., 2004; Heneka et al., 2006; Grudzien et al., 2007; Kalinin et al., 2007; Pugh et al., 2007). There is currently no effective treatment or cure for AD. Understanding the changes that occur in the noradrenergic system in AD patients could present a unique opportunity to inspire new avenues of treatment. The purpose of this review is to describe what is currently known about noradrenergic dysfunction in AD and its potential contribution to AD pathology and progression, as well as to point out deficiencies in our knowledge where more research is needed.
The Role of the Noradrenergic System in Cognition
The role of the noradrenergic system on cognition has been reviewed extensively elsewhere (Chamberlain and Robbins, 2013). We provide a brief overview of the literature here as an introduction to the role of the noradrenergic system in AD, but we are aware that we have not mentioned all relevant papers to this topic.
Importance of Norepinephrine
Examining the effects of norepinephrine depletion has revealed the importance of norepinephrine in cognitive tasks. Depletion of norepinephrine in animal and cellular models has been achieved in three different ways. First, norepinephrine depletion has been accomplished through degeneration of noradrenergic neurons with DSP-4, a neurotoxin selective for noradrenergic neurons. While this most completely depletes norepinephrine, it also causes structural changes in the brain and therefore the changes may not necessarily be completely attributed to the neurotransmitter loss. Another method of norepinephrine depletion is the use of reserpine to block the vesicular monoamine transporter (VMAT), which transports monoamines into vesicles in the presynaptic terminal for synaptic release. It is important to note that reserpine is not specific to norepinephrine and works on all monoamines, including dopamine and serotonin. A final method of norepinephrine depletion is through the knockout of DβH (the enzyme converting dopamine to norepinephrine) to look exclusively at the effects of norepinephrine. Despite of the potential limitation associated with each method, studies with these independent methods have demonstrated that loss of norepinephrine leads to cognitive impairment in various aspects of cognition.
Depletion of norepinephrine in rats by DSP-4 leads to deficits in working memory (Sontag et al., 2008). This deficit has also been shown to be exacerbated when in combination with the depletion of other neurotransmitters, either dopamine (Pérez et al., 2009) or acetylcholine (Ohno et al., 1997). Treating monkeys with reserpine also induces a working memory deficit (Cai et al., 1993). Deficits in spatial memory, as measured by the Morris water maze, have also been reported following norepinephrine depletion with DSP-4, and this effect is especially significant in stressed and aged rats (Sirviö et al., 1991; Lapiz et al., 2001). In addition, DSP treatment impairs visual memory (Rajabi et al., 2012), olfactory memory (Guan et al., 1993) and avoidance learning, and administration of epinephrine rescues the avoidance learning defect caused by norepinephrine depletion (Bennett et al., 1990). DβH deficient mice also show impaired active-avoidance learning (Thomas and Palmiter, 1997) and social memory (Marino et al., 2005). Together, these studies strongly support a critical role of norepinephrine in cognitive functions.
An alternate method of studying the effect of norepinephrine on cognition is by increasing the availability of this neurotransmitter. The selective norepinephrine reuptake inhibitor atomoxetine increases the amount of norepinephrine available in the synapse by preventing its reuptake by the presynaptic terminals. Increasing norepinephrine improves spatial learning and memory of rats in both the novel object task and the radial arm maze (Tzavara et al., 2006). Additionally, improvements in working memory (Gamo et al., 2010) and attention (Tzavara et al., 2006; Jentsch et al., 2009) have been seen after atomoxetine treatment. These results are consistent with the findings from the depletion studies; while a decrease in norepinephrine induces cognitive deficits, increasing norepinephrine can improve cognitive function.
Differential Roles of the Adrenergic Receptors
All nine adrenergic receptors are members of the G-protein coupled receptor family. The α1 receptors all signal through the Gq signaling pathway and cause increases in both IP3 and calcium upon activation (Chen and Minneman, 2005; Perez, 2007). The β receptors all signal through the Gs signaling pathway and activation of these receptors leads to increased cAMP. The β2 receptor is also able to signal through the Gi signaling pathway to modulate cAMP and lead to ERK activation (Hall, 2004; Xiang, 2011). The α2 receptors signal through the Gi/o signaling pathway to inhibit both calcium and cAMP (Knaus et al., 2007). The α2 receptors can serve as autoreceptors on presynaptic noradrenergic terminals, inhibiting the further release of norepinephrine. The known functions of different adrenergic receptors in cognition are summarized in Table 1.
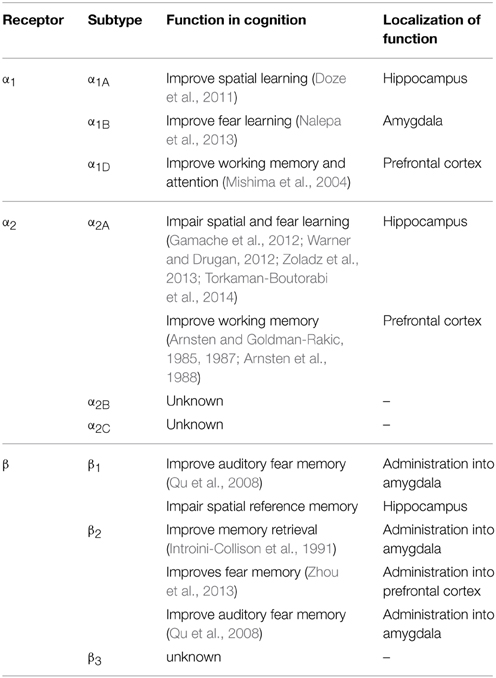
Table 1. The known functions of adrenergic receptors in cognition.
The α1 receptor has been investigated in both fear conditioning and spatial learning. Overall, inhibition of the α1 receptor impairs learning while activation of the receptor improves it. In fear learning, antagonism of the α1 receptor leads to poorer performance in these tasks. Prazosin, an α1antagonist, has been shown to cause both a diminished fear response in an olfactory fear conditioning paradigm (Do Monte et al., 2013) and in an olfactory recall task (Veyrac et al., 2007). In spatial learning, prazosin induces impairment (Torkaman-Boutorabi et al., 2014) and also exacerbates a deficit induced by scopolamine, a muscarinic cholinergic antagonist (Puumala et al., 1998). Consistent with the antagonist data, treatment with α1agonists has been shown to improve spatial memory in the Morris water maze (Puumala et al., 1996, 1998). It is worth noting that prazosin may have an inhibitory effect on overall locomotion in the Morris water maze (Levcik et al., 2013), which would confound the spatial memory test.
Genetic studies have revealed subtype specific roles for the α1 receptors with the α1A knockout mice showing the most dramatic cognitive deficits. The α1A knockout mice display poor spatial learning and memory in both the Barnes maze and Morris water maze (Doze et al., 2011). α1B knockout mice show increased short-term-latency and a decline in long-term-latency in passive avoidance tests, suggesting that the α1B receptor plays a role in fear-motivated exploratory behavior (Nalepa et al., 2013). While α1D knockout mice show no difference in spatial or emotional learning when compared to WT mice (Sadalge et al., 2003), these mice have a deficit in working memory and attention (Mishima et al., 2004).
The α2 receptors are involved in several different paradigms of learning. Antagonism of α2 receptors has been shown to benefit memory and cognition in a number of settings. Yohimbine, an α2 antagonist, increases fear conditioning (Gazarini et al., 2013) and improves spatial cognitive performance in the Morris water maze (Torkaman-Boutorabi et al., 2014). Additionally, yohimbine effectively improves both accuracy and response latencies in the habituated animal (Brown et al., 2012). Another α2 antagonist, dexefaroxan, improves odor learning (Veyrac et al., 2007), spatial and visual memory, and passive avoidance (Chopin et al., 2002). The α2 antagonist atipamezole enhances spatial learning in aged rats (Haapalinna et al., 2000). α2 receptor blockade may also potentiate cholinergic activity in the formation of a long-term memory trace; the α2 antagonists yohimbine, idazoxan, and P86 7480 are all able to enhance passive avoidance learning when co-administered with heptylphysostigmine, a cholinesterase inhibitor (Camacho et al., 1996).
Pharmacological activation of the α2 receptors also leads to cognitive changes. In a model of the effect of stress on spatial learning, clonidine exacerbates the learning deficits produced from the stress of cold water in the Morris water maze (Warner and Drugan, 2012). Clonidine also weakens reconsolidation of fear memories, and has therefore recently been looked at as a potential treatment for PTSD (Gamache et al., 2012; Zoladz et al., 2013). While activation of the α2A receptors induces deficits in spatial and fear learning, it actually has been shown to enhance working memory. Clonidine and guanfacine can improve working memory in aged monkeys with documented memory impairment (Arnsten and Goldman-Rakic, 1985, 1987; Arnsten et al., 1988). It is known that working memory is mainly localized to the prefrontal cortex (Wang et al., 2013c) whereas spatial and fear memory involve the hippocampus (Bird and Burgess, 2008). The difference in clonidine's effects on these different cognitive paradigms is likely due to diverse functions of α2 receptors in regulating neurotransmission in different brain regions. α2 receptor stimulation enhances firing of prefrontal cortical neurons through inhibition of HCN channels (Wang et al., 2007). On the other hand, α2 receptor activation blocks hippocampal long-term plasticity (McMahon and Wang, unpublished findings).
There is limited information on the role of the α2 receptors in cognition from genetically modified mice. α2A(the most prevalent subtype in the CNS) knockout mice show impairment in working memory (Franowicz et al., 2002). Changes in spatial or fear learning in these mice remain uninvestigated.
Much of our knowledge of the role of the β adrenergic receptors in cognition comes from pharmacological studies. Pharmacological activation of β receptors using the agonist isoproterenol has been found to enhance long term potentiation and memory consolidation in the hippocampus (Gelinas et al., 2008). On the other hand, the β antagonist propranolol inhibits memory retrieval in extinction tasks (Ouyang and Thomas, 2005) and in a fear conditioning paradigm (Taherian et al., 2014). Propranolol also inhibits taste memory consolidation (Ruetti et al., 2014) and impairs scent recall (Veyrac et al., 2007). Additionally, administration of propranolol with scopolamine, a muscarinic cholinergic antagonist, or with p-chlorophenylalanine, a serotonergic antagonist, synergistically impairs rat spatial learning in the Morris water maze (Decker et al., 1990; Saber and Cain, 2003; Kenton et al., 2008). Together, these studies indicate a positive role of β adrenergic receptors in memory consolidation in general.
Effects of β1 and β2 subtype-selective ligands on cognition have also been investigated. Infusion of the β2 agonist clenbuterol into the amygdala facilitates memory retention in an inhibitory avoidance task (Introini-Collison et al., 1991), and infusion of clenbuterol into the prefrontal cortex enhances trace fear memory (Zhou et al., 2013). On the other hand, infusion of β1 or β2 antagonists into the amygdala causes a deficit in auditory fear memory (Qu et al., 2008). However, xamoterol, a partial β1 receptor agonist, impairs the retrieval of hippocampus-dependent spatial reference memory in rats (Schutsky et al., 2011), suggesting that different subtypes of β receptors may play different roles in regulating cognition in different brain regions.
Other Functions of the Noradrenergic System Potentially Related to AD
It is well-established that the brains of AD patients have significant neuroinflammation (Hensley, 2010). Both astrocytes and microglia have adrenergic receptors present (Salm and Mccarthy, 1989; Shao and Sutin, 1992; Sutin and Shao, 1992; Tanaka et al., 2002), and norepinephrine regulates inflammatory processes through these receptors. Loss of neurons in the locus coeruleus and the subsequent loss of norepinephrine result in an increase in inflammation in animal models (Heneka et al., 2006, 2010; Jardanhazi-Kurutz et al., 2011). Furthermore, stimulating microglia with norepinephrine led to increased phagocytosis of amyloid-β (Aβ) by microglia (Heneka et al., 2010). Taken together, this indicates that alterations of the noradrenergic system at least partially contribute to the neuroinflammation seen in AD and the pathogenic consequences of that inflammation.
Another symptom of AD is aberrant glucose metabolism (Schubert, 2005), which can be seen in the increased levels of oxidative stress in AD patients that occurs before the appearance of Aβ plaques (Perry et al., 2002). α1 receptors on astrocytes regulate glucose uptake. Additionally, activation of the α2 receptors on astrocytes leads to an increase of glycogenesis, whereas activation of the β-receptors promotes glycogenolysis (O'donnell et al., 2012). This indicates that the adrenergic receptors play a large role in the balance of glycogen in the brain and, therefore, could potentially be targeted to improve the aberrant glucose metabolism seen in AD.
Noradrenergic Changes in AD
There is considerable evidence showing that the normal anatomy and functions of the noradrenergic system are altered in AD. We will consider these data at the level of neurons, neurotransmitters, and receptors.
Changes in Locus Coeruleus Neurons
At an anatomical level, it has been well-established that AD patients show a loss of noradrenergic neurons in the locus coeruleus (Mann et al., 1980; Iversen et al., 1983; Forstl et al., 1994; Matthews et al., 2002; Zarow et al., 2003). Though there is an overall loss in neurons, there is actually an increase in both dendritic and axonal sprouting from the remaining noradrenergic neurons in the locus coeruleus. In patients with dementia, the dendritic sprouting from the locus coeruleus to the prefrontal cortex after neuronal loss keeps the levels of connections stable or even slightly increased, as measured by presynaptic α2 and postsynaptic α1 receptor density (Szot et al., 2007). Axonal sprouting of the remaining locus coeruleus neurons to the hippocampus also occurs in patients with noradrenergic neurodegeneration (Szot et al., 2006). Recently, technological advances have allowed for the detection of locus coeruleus neuron loss in living patients. Using high-resolution fast spin-echo T1-weighted imaging, Takahashi et al. (Takahashi et al., 2015) were able to detect a decrease in locus coeruleus density in AD patients when compared to controls. This advance is exciting as it could allow for the early detection of locus coeruleus neuron loss, which occurs at early stages of AD.
While the noradrenergic changes are commonly seen in AD patients, few commonly used AD mouse models actually show the locus coeruleus degeneration. A Down syndrome mouse model that includes a triplication of the APP gene shows degeneration of the locus coeruleus neurons with aging, which has been linked to the overexpression of APP (Salehi et al., 2009; Lockrow et al., 2011). Tg2576 mice also display some degree of noradrenergic changes, including neurodegeneration in the locus coeruleus (Liu et al., 2008; Guerin et al., 2009; Eimer and Vassar, 2013). However, many other AD rodent models do not have neurodegeneration, which is a limitation of these models.
Changes in Norepinephrine Levels
Despite the loss of the noradrenergic neurons, there have been conflicting reports on levels of norepinephrine in the brain, with some studies showing a decrease in norepinephrine while others showing that norepinephrine levels in AD patients remain constant, or even elevated. Using either HPLC (Martignoni et al., 1992; Nazarali and Reynolds, 1992; Matthews et al., 2002) or a fluorophore based method (Reinikainen et al., 1988), several groups have found a decrease in norepinephrine concentration in various brain regions, with the loss proportional to the level of cognitive deficit (Matthews et al., 2002). Norepinephrine turnover is increased in the remaining locus coeruleus neurons. The concentration of 3-methoxy-4-hydroxyphenylglycol (MHPG), a norepinephrine metabolite, has been used to indicate the norepinephrine turnover rate. AD patients have a higher ratio of the norepinephrine metabolite to norepinephrine (Palmer et al., 1987; Hoogendijk et al., 1999). This increased metabolism of norepinephrine is a possible cause of the decreased norepinephrine levels. On the other hand, a number of studies have shown no change in norepinephrine levels in different brain regions or cerebrospinal fluid (CSF) in both sporadic and familial AD cases (Sparks et al., 1988; Herregodts et al., 1989; Tohgi et al., 1992). In addition, increased norepinephrine levels were reported, which correlated with decreased cognitive function (Tohgi et al., 1992) and aging (Elrod et al., 1997) in AD patients.
There is evidence of compensatory changes that can account for the loss in noradrenergic neurons, yet stable or increased norepinephrine levels. One finding has been that norepinephrine transporter sites are decreased in AD patients. In autoradiograph studies (Gulyas et al., 2010) and radioligand binding studies of post-mortem tissues (Tejani-Butt et al., 1993), the norepinephrine transporter sites were significantly reduced in AD brains compared to normal control samples. As norepinephrine transporter is responsible for norepinephrine reuptake to the presynaptic neurons, a decrease in norepinephrine transporter sites would increase the amount of norepinephrine in the synapse. Another possible mechanism for norepinephrine increase is an increase in the enzymes responsible for the production of norepinephrine. Tyrosine hydroxylase, which catalyzes the rate-limiting step in the production of norepinephrine, is increased in the brain of AD patients (Iversen et al., 1983; Szot et al., 2006, 2007). The enzyme DβH is also increased in serum, CSF, and peripheral blood lymphocytes in AD cases (Miyata et al., 1984; Giubilei et al., 2004).
With researchers reporting both increases and decreases in norepinephrine levels in AD patients, it is necessary to look deeper into the discrepancy. The differences are likely due to the stage of the disease and the brain's ability to compensate for the neuronal loss. One possible theory is that in the early stages of locus coeruleus neuron loss, the remaining neurons undergo compensatory mechanisms to maintain the norepinephrine level. However, as the disease progresses and more neurons are lost, it may become impossible for the remaining neurons to totally compensate. It is also possible that the brain over-compensates for the neuronal loss, which can account for the increased norepinephrine levels reported in the later stages of the disease. Eventually, however, if the disease progresses enough, the loss of locus coeruleus neurons will be too great to overcome and norepinephrine levels will decline. This hypothesis could be tested by using the new high-resolution fast spin-echo T1-weighted imaging to measure locus coeruleus density and correlating it to the norepinephrine levels in the CSF of AD patients (Takahashi et al., 2015).
Changes in Noradrenergic Receptors
In addition to the anatomical and neurotransmitter changes, there are also changes to the adrenergic receptors of AD patients. Changes in the receptors in postmortem brain tissues have been studied using both binding assays and mRNA quantification. These studies indicate several changes in the AD brain, which would lead to altered functional outcome in response to norepinephrine.
α1 receptor density has been examined using non-subtype selective radioligands. Decreases in α1 receptor density have been reported in the prefrontal cortex (Kalaria, 1989), hippocampus, and cerebellum (Shimohama et al., 1986). However, when examining subregions, Szot et. al. found stable to slightly increased α1 binding sites in layers I/II of the prefrontal cortex (Szot et al., 2007) and in the molecular layer of the dentate gyrus of the hippocampus (Szot et al., 2006). Additionally, changes in specific α1 receptor subtypes have been examined, with a reduction of α1C mRNA in layers II/III of the prefrontal cortex and a significant decrease in α1D mRNA in the hippocampus (Szot et al., 2007).
The density of α2 receptors has also been studied by the use of binding assays. Increased α2 receptor density has been found in cortical membranes (Ruiz et al., 1993) and the dentate gyrus granule cell layer (Szot et al., 2006) of post-mortem AD patient brains and in brains of patients with the AD related disease, dementia with Lewy bodies (Leverenz et al., 2001; Szot et al., 2006). The levels of α2 receptors in brain microvessels, which are innervated by locus coeruleus noradrenergic neurons (Kalaria et al., 1989b), are also increased in AD patients (Kalaria et al., 1989a). However, there are also reports of no significant change (Shimohama et al., 1986; Szot et al., 2007) in α2 receptor density in the cortex or a decrease in α2 density in the hippocampus (Pascual et al., 1992) or nucleus basalis of Meynert (Shimohama et al., 1986) of AD patients. One thing that could be contributing to the discrepancy of the above reports is that studying α2 receptor density is a more complicated endeavor due to their unique presence on the presynaptic neuron. This is especially true considering the neuronal loss that occurs in the locus coeruleus of AD patients, and the fact that α2 receptors are located on those neurons. Therefore, even if there is actually an increased density of α2 receptors on the remaining locus coeruleus neurons or at the projection sites, locus coeruleus neuron loss could mask that increase due to the lost receptors on the degenerated neurons. Indeed, Szot et al. (2007) found that the loss of locus coeruleus neurons was greater than the α2 receptor loss in the cortex area with locus coeruleus neuron projections. This probably indicates an increased number of receptors per neuron in AD cases.
Changes in α2 receptor subtypes have also been examined in AD patients. By measuring the mRNA at different projection sites to isolate just the postsynaptic receptors, Szot et al. (2007) found decreased levels of α2A mRNA in the prefrontal cortex, though the reduction was limited to layer II. In the hippocampus, levels of α2A mRNA were unchanged, but there was a significant decrease in α2C mRNA. Szot et al. (2006) also looked at presynaptic receptors in post mortem AD brains by measuring receptor mRNA from the locus coeruleus and found an overall reduction in α2AmRNA in AD patients without change in the number of receptors per cell. While examination of mRNA levels is useful to determine changes in different receptor subtypes, it may not reflect actual changes in receptor density. Therefore, these data should be interpreted with the results from the binding assays.
Finally, β receptor density in AD patient brains has been studied, though there is considerably less information on the β receptor changes. In both AD patients and controls, the β1 receptor density is higher in the cortex than the hippocampus, but there is no difference between the two groups (Lemmer et al., 1993). However, a decrease in β2 receptor density occurs in the microvessels of AD patients (Kalaria et al., 1989a).
Pathological changes of the noradrenergic system in AD are dynamic as the disease progresses. Most current studies look at one snapshot of the pathological process, which likely give discrepant results. Systematic examinations of various noradrenergic components at different disease stages and correlations of these changes with other AD-related pathological and cognitive deficits are necessary in order to fully comprehend noradrenergic dysfunction in AD.
Potential Involvement of Noradrenergic Dysregulation in AD Pathogenesis and Progression
The Role of Norepinephrine
The first evidence for a functional role of noradrenergic dysregulation in AD comes from animal models in which the norepinephrine levels are manipulated. Treatment of AD transgenic mice with DSP-4, a neurotoxin which selectively ablates noradrenergic neurons, increases Aβ deposition (Heneka et al., 2006; Kalinin et al., 2007; Jardanhazi-Kurutz et al., 2010), impairs spatial memory (Jardanhazi-Kurutz et al., 2010) and alters α1, α2, and β1 receptor binding sites and mRNA expression (Jardanhazi-Kurutz et al., 2011). DSP-4 treatment also increases the levels of accumulated hyperphosphorylated tau in cortices of female APP-SL mice (Oikawa et al., 2010). APP/PS1 mice crossed with DβH−/− mice, which are unable to synthesize norepinephrine, have compromised LTP and maze performance, which is worse than in either of the single mutants alone (APP/PS1 or DβH−/−) (Hammerschmidt et al., 2013). APP/PS1 mice crossed with Ear2−/− mice, which show marked locus coeruleus neuron loss, have exacerbated LTP and memory deficits, but do not have plaque deposition different from APP/PS1 mice (Kummer et al., 2014). This indicates that the norepinephrine loss contributes to the cognitive dysfunction of AD, even in the absence of the hallmark pathogenic Aβ plaques. Taken together, this evidence suggests that loss of noradrenergic input to the cerebral cortex exacerbates AD-related pathological and behavioral deficits.
In vivo tests also suggest that norepinephrine supplementation could be beneficial in AD. Norepinephrine levels and microglial function can be rescued by peripherally administering the norepinephrine precursor, L-DOPS, in DSP-4 lesioned AD mice expressing mutant APP (APP/PS1 or APPV717I). Additionally, these same AD model mice have improved Aβ clearance, decreased Aβ plaques, and improved spatial memory after L-DOPS administration (Heneka et al., 2010; Hammerschmidt et al., 2013). Co-administering L-DOPS along with atomoxetine, a norepinephrine reuptake inhibitor, in 5xFAD transgenic mice improves learning in the Morris water maze test (Kalinin et al., 2012).
In addition to animal studies, human association studies support the role of norepinephrine loss in AD. In AD patients, the extent of noradrenergic degeneration correlates with both the degree of pathological changes (including amyloid plaques and neurofibrillary tangles) and the severity of cognitive deficits (Bondareff et al., 1987; Zarow et al., 2003). Additionally, there is a link between a low-activity polymorphism in the DβH gene and AD in a Caucasian population (Combarros et al., 2010). As DβH is required for the production of norepinephrine, lower activity of this enzyme leads to decreased norepinephrine. However, this polymorphism may be population-specific, as a study by Komatsu et al. (2014) looked at that same polymorphism along with another DβH polymorphism and did not find any link between either of the two different DβH polymorphisms and AD in a Japanese population.
Potential Involvement of the Noradrenergic Receptors
The potential role of α1 receptors in modulating AD-related pathological and behavioral changes has been studied using prazosin, an α1 receptor antagonist. Prazosin reduces the generation of Aβ in N2a cells and ameliorates memory loss in APP23 transgenic mice (Katsouri et al., 2013). However, since prazosin is not a subtype-selective antagonist, this study only suggests the involvement of the α1 receptor as a whole, and not the roles of the individual receptor subtypes. Further studies are needed to determine the specific subtypes of α1 receptors involved in the protective effect by prazosin.
Our recent studies have revealed the involvement of the endogenous α2A subtype receptor in AD pathogenesis (Chen et al., 2014). Stimulation of the α2A receptor significantly enhances, while genetic or pharmacological blockade of this receptor reduces, Aβ generation and Aβ-related neuropathology. Activation of α2A receptor signaling disrupts endogenous APP interaction with sorting-related receptor with A repeat (SorLA), and consequently promotes amyloidogenic processing of APP in endosomes. We therefore provided the first evidence that SorLA-dependent APP sorting can be targeted by extracellular signaling to modulate amyloidogenesis. This is particularly important because it suggests a method of reducing Aβ production without targeting the activity of the secretases responsible for its generation. Significantly, there have been reports of decreased SorLA levels in late-onset AD patients (Scherzer et al., 2004) and polymorphisms in the SorLA gene are linked to both early and late-onset AD patients (Rogaeva et al., 2007; Grear et al., 2009; Caglayan et al., 2012; Pottier et al., 2012). These studies suggest that increasing the association between SorLA and APP by inhibiting the α2A receptor could be beneficial for many AD patients. In APP/PS1 mice, a clinically used α2 receptor antagonist, idazoxan, ameliorates AD-related cognitive deficits in both novel object recognition and Morris water maze tests (Chen et al., 2014). Consistently, fluparoxan, another α2 receptor antagonist, improves the spatial working memory in a contextual fear conditioning task in these mice (Scullion et al., 2011). In humans, increased α2A receptor density and/or activity have been associated with type 2 diabetes mellitus and depression (Cottingham et al., 2011; Cottingham and Wang, 2012), both of which are risk factors for AD. Under these disease conditions, the increase in the α2A receptor may lead to α2A receptor-promoted Aβ generation, which may act as a key contributor driving AD-related pathophysiology. Elevated α2 receptor density and response have also been reported in living AD patients. One study has found an increase in the α2 receptor density in platelets (Adunsky et al., 1989), and a few have found that both AD patients and aged individuals show increased norepinephrine levels in the CSF following stimulation or inhibition of the α2 receptors (Peskind et al., 1995; Raskind et al., 1999). Consistent with a role of the α2A receptor promoting AD, our biostatistical analysis of the National Alzheimer's Coordinating Center database indicates that chronic activation of the α2A receptor exacerbates disease progression in AD patients. Therefore, the α2A receptor represents a previously unappreciated therapeutic target for AD.
A role of β receptors in AD pathogenesis and progression has also been suggested by pharmacological studies. The β1 blocker nevibolol reduces Aβ production in TG2576 mice with established impairment, even though it does not improve cognition function (Wang et al., 2013b). The β2 antagonist ICI 118,551 decreases Aβ load, while the β2 agonists isoproterenol and clenbuterol increase it, in APP/PS1 mice (Ni et al., 2006). Activation of the β2 receptor leads to increased trafficking of γ secretase to the late endosomes and lysosomes due to the association between β2 receptor and presenilin-1. The production of Aβ in these organelles is subsequently increased. Blockade of β2 receptors with ICI 118,551 also reduces Aβ generation associated with stress in non-transgenic C57 mice (Yu et al., 2010). However, in a 3xTg model of AD, ICI 118,551 increases Aβ levels and Aβ plaque burden as well as exacerbates cognitive deficits (Branca et al., 2014). The molecular mechanisms underlying the discrepancy of ICI 118,551 effects in different animal models remains to be investigated. Another β blocker, propranolol, which blocks β1 and β2 receptors with equal affinity (Summers, 2006), has been shown to lower BACE1 expression, decrease Aβ42 levels, decrease tau hyperphosphorylation, and improve cognitive impairments in both a mouse model of age-related cognitive decline (SAMP8) or corticosterone-treated mice (Dobarro et al., 2013a,b).
The β2 receptor can also mediate the effect of Aβ on tau phosphorylation (Wang et al., 2013a). In APP/PS1 mice, Aβ binds to and activates the β2 receptors, which leads to an increase in the activities of PKA and JNK. The β2-PKA-JNK pathway results in hyperphosphorylation of tau at specific loci. While the previous studies demonstrate that β agonists promote several aspects of AD pathology, there is also evidence that β agonists reduce, rather than promote, Aβ-related toxicity on neural plasticity and cognition. A β2 agonist, terbutaline, prevents the Aβ evoked inhibition of LTP (Wang et al., 2009b), and a β3 agonist, CL 316243, rescues Aβ induced memory loss in chicks (Gibbs et al., 2010). Overall, β receptors appear to execute diverse functions in multiple aspects related to AD pathology ranging from Aβ metabolism to toxicity, and further studies are needed to determine the role that each receptor subtype plays in the mechanism of AD pathogenesis.
Human genetic studies have indicated association of the β receptors with AD. A polymorphism in the β2 receptor contributes to AD onset in a Chinese group (Yu et al., 2008). Also, a combination of two single-nucleotide polymorphisms, a T allele in the G-protein β3 subunit (GNB3) and a C allele in the β2 adrenergic receptor (ADRB1), is associated with AD susceptibility. This is likely through enhanced cAMP/PKA signaling, which leads to an increase in APP expression (Bullido et al., 2004). Furthermore, hypertension patients who were taking beta blockers showed lower dementia incidence and cognitive decline rate (Khachaturian et al., 2006; Rosenberg et al., 2008), which suggests that blocking β receptors has a beneficial effect on AD.
Summary and Perspectives
The role of the noradrenergic system in cognition has long been known. Cognitive deficits occur after depletion of norepinephrine and improvements in cognitive function occur after increasing the availability of norepinephrine. Norepinephrine elicits its effects through the nine adrenergic receptors to which it binds. While the subtype-specific role of each adrenergic receptor in cognition remains to be elucidated, in general, α1 and β receptors are considered stimulatory, enhancing neurotransmission and plasticity. On the other hand, α2 receptors are inhibitory, reducing NE release and neuronal excitability. In addition to the noradrenergic system being important in cognitive function in normal circumstances, many changes occur in the noradrenergic system in AD patients. The degeneration of noradrenergic neurons in the locus coeruleus of AD patients was an observation made over 30 years ago, and more recent research has found changes that occur within the remaining neurons, likely as compensatory mechanisms. This neuronal loss and the resultant compensatory mechanisms lead to changes in the level of norepinephrine available in the brain, which consequently affect cognitive functions. Evidence from both animal and human studies suggest that loss of noradrenergic input significantly exacerbates AD-related cognitive deficits.
Familial AD with mutations in genes encoding APP or PS1 accounts for less than 10% of AD. In contrast, late-onset sporadic AD likely involves multiple genetic and environmental risk factors that lead to disruption of amyloid homeostasis. The pivotal role that the noradrenergic system plays in supporting interactions with and responses to environmental stimuli, as well as alterations of this system in the early stage of AD, suggest a potential contribution of noradrenergic dysfunction to AD pathogenesis. Supporting this notion, activation of α2A and β2 receptors directly regulates Aβ generation. More studies are needed to address the potential role of each adrenergic receptor subtype in amyloid metabolism and tau pathogenesis. The available receptor knockout mice for each subtype (Philipp and Hein, 2004) provide useful tools to address this. Genetic studies could also be done to look for any polymorphisms within the adrenergic receptor genes that are associated with late-onset AD.
Despite the fact that a clear picture of the roles of various noradrenergic components in AD pathogenesis is still lacking, current evidence points to the α2A receptors as a novel target for AD. This receptor subtype is the key regulator of the noradrenergic activity, inhibiting both noradrenergic input to the cerebral cortex and the resulting response in this brain region (Hein, 2006). In addition, our recent studies have demonstrated that activation of the α2 receptor promotes amyloidogenic processing of the endogenous APP and amyloid-related pathology (Chen et al., 2014). Therefore, a pharmacological blockade of α2A receptors can both increase NE release, which enhances NE-dependent cognitive and neuroprotective effects, and decrease Aβ generation, which reduces Aβ-dependent toxicity. Increased α2A receptor density and/or activity have been observed in living AD patients and in patients at high risk for AD, such as those with type 2 diabetes mellitus and depression. Blockade of α2A receptors would be particularly effective in patients with these receptors upregulated.
While a novel target for AD, adrenergic receptors are an established target for other disorders including cardiovascular, behavioral, and mood disorders. Because of this, extensive drug discovery research has already been done and a number of noradrenergic receptor drugs, including α2 receptor blockers, already exist and are FDA approved. Repurposing these drugs could greatly expedite the timeframe for getting a novel AD drug on the market. In fact, some noradrenergic drugs have been tested in AD patients to alleviate some of the mood symptoms associated with AD. For example, tricyclic antidepressants, which block norepinephrine transporters, have been used to treat depressed AD patients (Sallee and Pollock, 1990; Teri et al., 1991), and both α1 and β receptor antagonists can improve agitation in AD (Pauszek, 1991; Shankle et al., 1995; Peskind et al., 2005; Wang et al., 2009a). While these noradrenergic drugs are effective in the treatment of the behavioral and mood disorders associated with AD, further research is needed to elucidate their effect on cognition. Once this is known, it will be possible to quickly repurpose these drugs in order to treat the cognitive aspects of AD.
Author Contributions
MG, PC, YC, KJ, and QW planned the structure of the manuscript. MG, PC, and YC prepared the manuscript. MG, KJ, ER, and QW edited and restructured the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Chelsea McCoy and Tana Birky for helpful discussion and editing of the manuscript. This study is supported by NIH grants, MH081917 and AG042716.
References
Adunsky, A., Hershkowitz, M., and Rabinowitz, M. (1989). Alzheimer's dementia and binding to alpha 2 adrenoreceptors in platelets. J. Am. Geriatr. Soc. 37, 741–744. doi: 10.1111/j.1532-5415.1989.tb02236.x
Arnsten, A. F., Cai, J. X., and Goldman-Rakic, P. S. (1988). The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for alpha-2 receptor subtypes. J. Neurosci. 8, 4287–4298.
Arnsten, A. F., and Goldman-Rakic, P. S. (1985). Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science 230, 1273–1276. doi: 10.1126/science.2999977
Arnsten, A. F., and Goldman-Rakic, P. S. (1987). Noradrenergic mechanisms in age-related cognitive decline. J. Neural Transm. Suppl. 24, 317–324.
Bennett, M. C., Kaleta-Michaels, S., Arnold, M., and Mcgaugh, J. L. (1990). Impairment of active avoidance by the noradrenergic neurotoxin, DSP4: attenuation by post-training epinephrine. Psychopharmacology 101, 505–510. doi: 10.1007/BF02244229
Bird, C. M., and Burgess, N. (2008). The hippocampus and memory: insights from spatial processing. Nat. Rev. Neurosci. 9, 182–194. doi: 10.1038/nrn2335
Bondareff, W., Mountjoy, C. Q., Roth, M., Rossor, M. N., Iversen, L. L., Reynolds, G. P., et al. (1987). Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1, 256–262. doi: 10.1097/00002093-198701040-00005
Braak, H., and Del Tredici, K. (2012). Where, when, and in what form does sporadic Alzheimer's disease begin? Curr. Opin. Neurol. 25, 708–714. doi: 10.1097/WCO.0b013e32835a3432
Branca, C., Wisely, E. V., Hartman, L. K., Caccamo, A., and Oddo, S. (2014). Administration of a selective beta2 adrenergic receptor antagonist exacerbates neuropathology and cognitive deficits in a mouse model of Alzheimer's disease. Neurobiol. Aging 35, 2726–2735. doi: 10.1016/j.neurobiolaging.2014.06.011
Brown, D. C. II, Co, M. S., Wolff, R. C., and Atzori, M. (2012). Alpha-Adrenergic receptors in auditory cue detection: alpha2 receptor blockade suppresses false alarm responding in the rat. Neuropharmacology 62, 2178–2183. doi: 10.1016/j.neuropharm.2011.12.024
Bullido, M. J., Ramos, M. C., Ruiz-Gomez, A., Tutor, A. S., Sastre, I., Frank, A., et al. (2004). Polymorphism in genes involved in adrenergic signaling associated with Alzheimer's. Neurobiol. Aging 25, 853–859. doi: 10.1016/j.neurobiolaging.2003.10.006
Caglayan, S., Bauerfeind, A., Schmidt, V., Carlo, A. S., Prabakaran, T., Hubner, N., et al. (2012). Identification of Alzheimer disease risk genotype that predicts efficiency of SORL1 expression in the brain. Arch. Neurol. 69, 373–379. doi: 10.1001/archneurol.2011.788
Cai, J. X., Ma, Y. Y., Xu, L., and Hu, X. T. (1993). Reserpine impairs spatial working memory performance in monkeys: reversal by the alpha 2-adrenergic agonist clonidine. Brain Res. 614, 191–196. doi: 10.1016/0006-8993(93)91034-P
Camacho, F., Smith, C. P., Vargas, H. M., and Winslow, J. T. (1996). Alpha 2-adrenoceptor antagonists potentiate acetylcholinesterase inhibitor effects on passive avoidance learning in the rat. Psychopharmacology 124, 347–354. doi: 10.1007/BF02247440
Chamberlain, S. R., and Robbins, T. W. (2013). Noradrenergic modulation of cognition: therapeutic implications. J. Psychopharmacol. 27, 694–718. doi: 10.1177/0269881113480988
Chen, Y., Peng, Y., Che, P., Gannon, M., Liu, Y., Li, L., et al. (2014). alpha(2A) adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proc. Natl. Acad. Sci. U.S.A. 111, 17296–17301. doi: 10.1073/pnas.1409513111
Chen, Z. J., and Minneman, K. P. (2005). Recent progress in alpha1-adrenergic receptor research. Acta Pharmacol. Sin. 26, 1281–1287. doi: 10.1111/j.1745-7254.2005.00224.x
Chopin, P., Colpaert, F. C., and Marien, M. (2002). Effects of acute and subchronic administration of dexefaroxan, an alpha(2)-adrenoceptor antagonist, on memory performance in young adult and aged rodents. J. Pharmacol. Exp. Ther. 301, 187–196. doi: 10.1124/jpet.301.1.187
Combarros, O., Warden, D. R., Hammond, N., Cortina-Borja, M., Belbin, O., Lehmann, M. G., et al. (2010). The dopamine beta-hydroxylase-1021C/T polymorphism is associated with the risk of Alzheimer's disease in the Epistasis Project. BMC Med. Genet. 11, 162. doi: 10.1186/1471-2350-11-162
Cottingham, C., Chen, Y., Jiao, K., and Wang, Q. (2011). The antidepressant desipramine is an arrestin-biased ligand at the alpha(2A)-adrenergic receptor driving receptor down-regulation in vitro and in vivo. J. Biol. Chem. 286, 36063–36075. doi: 10.1074/jbc.M111.261578
Cottingham, C., and Wang, Q. (2012). alpha2 adrenergic receptor dysregulation in depressive disorders: implications for the neurobiology of depression and antidepressant therapy. Neurosci. Biobehav. Rev. 36, 2214–2225. doi: 10.1016/j.neubiorev.2012.07.011
Decker, M. W., Gill, T. M., and Mcgaugh, J. L. (1990). Concurrent muscarinic and beta-adrenergic blockade in rats impairs place-learning in a water maze and retention of inhibitory avoidance. Brain Res. 513, 81–85. doi: 10.1016/0006-8993(90)91091-T
Dobarro, M., Orejana, L., Aguirre, N., and Ramirez, M. J. (2013a). Propranolol reduces cognitive deficits, amyloid beta levels, tau phosphorylation and insulin resistance in response to chronic corticosterone administration. Int. J. Neuropsychopharmacol. 16, 1351–1360. doi: 10.1017/S1461145712001393
Dobarro, M., Orejana, L., Aguirre, N., and Ramirez, M. J. (2013b). Propranolol restores cognitive deficits and improves amyloid and Tau pathologies in a senescence-accelerated mouse model. Neuropharmacology 64, 137–144. doi: 10.1016/j.neuropharm.2012.06.047
Do Monte, F. H., Souza, R. R., Wong, T. T., and Carobrez, A. D. P. (2013). Systemic or intra-prelimbic cortex infusion of prazosin impairs fear memory reconsolidation. Behav. Brain Res. 244, 137–141. doi: 10.1016/j.bbr.2013.01.031
Doze, V. A., Papay, R. S., Goldenstein, B. L., Gupta, M. K., Collette, K. M., Nelson, B. W., et al. (2011). Long-term α1A-adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol. Pharmacol. 80, 747–758. doi: 10.1124/mol.111.073734
Eimer, W. A., and Vassar, R. (2013). Neuron loss in the 5XFAD mouse model of Alzheimer;s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 8:2. doi: 10.1186/1750-1326-8-2
Elrod, R., Peskind, E. R., Digiacomo, L., Brodkin, K. I., Veith, R. C., and Raskind, M. A. (1997). Effects of Alzheimer's disease severity on cerebrospinal fluid norepinephrine concentration. Am. J. Psychiatry 154, 25–30. doi: 10.1176/ajp.154.1.25
Forstl, H., Levy, R., Burns, A., Luthert, P., and Cairns, N. (1994). Disproportionate loss of noradrenergic and cholinergic neurons as cause of depression in Alzheimer's disease - a hypothesis. Pharmacopsychiatry 27, 11–15. doi: 10.1055/s-2007-1014267
Franowicz, J. S., Kessler, L. E., Borja, C. M., Kobilka, B. K., Limbird, L. E., and Arnsten, A. F. (2002). Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J. Neurosci. 22, 8771–8777.
Gamache, K., Pitman, R. K., and Nader, K. (2012). Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology 37, 2789–2796. doi: 10.1038/npp.2012.145
Gamo, N. J., Wang, M., and Arnsten, A. F. (2010). Methylphenidate and atomoxetine enhance prefrontal function through alpha2-adrenergic and dopamine D1 receptors. J. Am. Acad. Child Adolesc. Psychiatry 49, 1011–1023. doi: 10.1016/j.jaac.2010.06.015
Gazarini, L., Stern, C. A., Carobrez, A. P., and Bertoglio, L. J. (2013). Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1-and β-adrenergic receptors. Learn. Mem. 20, 210–219. doi: 10.1101/lm.030007.112
Gelinas, J. N., Tenorio, G., Lemon, N., Abel, T., and Nguyen, P. V. (2008). β-Adrenergic receptor activation during distinct patterns of stimulation critically modulates the PKA-dependence of LTP in the mouse hippocampus. Learn. Mem. 15, 281–289. doi: 10.1101/lm.829208
Gibbs, M. E., Maksel, D., Gibbs, Z., Hou, X., Summers, R. J., and Small, D. H. (2010). Memory loss caused by beta-amyloid protein is rescued by a beta(3)-adrenoceptor agonist. Neurobiol. Aging 31, 614–624. doi: 10.1016/j.neurobiolaging.2008.05.018
Giubilei, F., Calderaro, C., Antonini, G., Sepe-Monti, M., Tisei, P., Brunetti, E., et al. (2004). Increased lymphocyte dopamine beta-hydroxylase immunoreactivity in Alzheimer's disease: compensatory response to cholinergic deficit? Dement. Geriatr. Cogn. Disord. 18, 338–341. doi: 10.1159/000080128
Golan, D. E., Tashjian, A. H., and Armstrong, E. J. (2011). Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Philadelphia, PA: Lippincott Williams & Wilkins.
Grear, K. E., Ling, I. F., Simpson, J. F., Furman, J. L., Simmons, C. R., Peterson, S. L., et al. (2009). Expression of SORL1 and a novel SORL1 splice variant in normal and Alzheimers disease brain. Mol. Neurodegener. 4, 46. doi: 10.1186/1750-1326-4-46
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D. C., and Mesulam, M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol. Aging 28, 327–335. doi: 10.1016/j.neurobiolaging.2006.02.007
Guan, X., Blank, J. L., and Dluzen, D. E. (1993). Role of olfactory bulb norepinephrine in the identification and recognition of chemical cues. Physiol. Behav. 53, 437–441. doi: 10.1016/0031-9384(93)90136-4
Guerin, D., Sacquet, J., Mandairon, N., Jourdan, F., and Didier, A. (2009). Early locus coeruleus degeneration and olfactory dysfunctions in Tg2576 mice. Neurobiol. Aging 30, 272–283. doi: 10.1016/j.neurobiolaging.2007.05.020
Gulyas, B., Brockschnieder, D., Nag, S., Pavlova, E., Kasa, P., Beliczai, Z., et al. (2010). The norepinephrine transporter (NET) radioligand (S,S)-[18F]FMeNER-D2 shows significant decreases in NET density in the human brain in Alzheimer's disease: a post-mortem autoradiographic study. Neurochem. Int. 56, 789–798. doi: 10.1016/j.neuint.2010.03.001
Haapalinna, A., Sirvio, J., Macdonald, E., Virtanen, R., and Heinonen, E. (2000). The effects of a specific alpha(2)-adrenoceptor antagonist, atipamezole, on cognitive performance and brain neurochemistry in aged Fisher 344 rats. Eur. J. Pharmacol. 387, 141–150. doi: 10.1016/S0014-2999(99)00819-5
Hall, R. A. (2004). Beta-adrenergic receptors and their interacting proteins. Semin. Cell Dev. Biol. 15, 281–288. doi: 10.1016/j.semcdb.2003.12.017
Hammerschmidt, T., Kummer, M. P., Terwel, D., Martinez, A., Gorji, A., Pape, H. C., et al. (2013). Selective loss of noradrenaline exacerbates early cognitive dysfunction and synaptic deficits in APP/PS1 mice. Biol. Psychiatry 73, 454–463. doi: 10.1016/j.biopsych.2012.06.013
Hein, L. (2006). Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 326, 541–551. doi: 10.1007/s00441-006-0285-2
Heneka, M. T., Nadrigny, F., Regen, T., Martinez-Hernandez, A., Dumitrescu-Ozimek, L., Terwel, D., et al. (2010). Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U.S.A. 107, 6058–6063. doi: 10.1073/pnas.0909586107
Heneka, M. T., Ramanathan, M., Jacobs, A. H., Dumitrescu-Ozimek, L., Bilkei-Gorzo, A., Debeir, T., et al. (2006). Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 26, 1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006
Hensley, K. (2010). Neuroinflammation in Alzheimer's disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimers Dis. 21, 1–14. doi: 10.3233/JAD-2010-1414
Herregodts, P., Bruyland, M., De Keyser, J., Solheid, C., Michotte, Y., and Ebinger, G. (1989). Monoaminergic neurotransmitters in Alzheimer's disease. An HPLC study comparing presenile familial and sporadic senile cases. J. Neurol. Sci. 92, 101–116. doi: 10.1016/0022-510X(89)90179-2
Hoogendijk, W. J., Feenstra, M. G., Botterblom, M. H., Gilhuis, J., Sommer, I. E., Kamphorst, W., et al. (1999). Increased activity of surviving locus ceruleus neurons in Alzheimer's disease. Ann. Neurol. 45, 82–91.
Introini-Collison, I. B., Miyazaki, B., and Mcgaugh, J. L. (1991). Involvement of the amygdala in the memory-enhancing effects of clenbuterol. Psychopharmacology 104, 541–544. doi: 10.1007/BF02245663
Iversen, L. L., Rossor, M. N., Reynolds, G. P., Hills, R., Roth, M., Mountjoy, C. Q., et al. (1983). Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci. Lett. 39, 95–100. doi: 10.1016/0304-3940(83)90171-4
Jardanhazi-Kurutz, D., Kummer, M. P., Terwel, D., Vogel, K., Dyrks, T., Thiele, A., et al. (2010). Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem. Int. 57, 375–382. doi: 10.1016/j.neuint.2010.02.001
Jardanhazi-Kurutz, D., Kummer, M. P., Terwel, D., Vogel, K., Thiele, A., and Heneka, M. T. (2011). Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience 176, 396–407. doi: 10.1016/j.neuroscience.2010.11.052
Jentsch, J. D., Aarde, S. M., and Seu, E. (2009). Effects of atomoxetine and methylphenidate on performance of a lateralized reaction time task in rats. Psychopharmacology 202, 497–504. doi: 10.1007/s00213-008-1181-0
Kalaria, R. N. (1989). Characterization of [125I]HEAT binding to alpha 1-receptors in human brain: assessment in aging and Alzheimer's disease. Brain Res. 501, 287–294. doi: 10.1016/0006-8993(89)90645-8
Kalaria, R. N., Andorn, A. C., Tabaton, M., Whitehouse, P. J., Harik, S. I., and Unnerstall, J. R. (1989a). Adrenergic receptors in aging and Alzheimer's disease: increased beta 2-receptors in prefrontal cortex and hippocampus. J. Neurochem. 53, 1772–1781. doi: 10.1111/j.1471-4159.1989.tb09242.x
Kalaria, R. N., Stockmeier, C. A., and Harik, S. I. (1989b). Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci. Lett. 97, 203–208. doi: 10.1016/0304-3940(89)90164-X
Kalinin, S., Gavrilyuk, V., Polak, P. E., Vasser, R., Zhao, J., Heneka, M. T., et al. (2007). Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol. Aging 28, 1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003
Kalinin, S., Polak, P. E., Lin, S. X., Sakharkar, A. J., Pandey, S. C., and Feinstein, D. L. (2012). The noradrenaline precursor L-DOPS reduces pathology in a mouse model of Alzheimer's disease. Neurobiol. Aging 33, 1651–1663. doi: 10.1016/j.neurobiolaging.2011.04.012
Katsouri, L., Vizcaychipi, M. P., Mcarthur, S., Harrison, I., Suarez-Calvet, M., Lleo, A., et al. (2013). Prazosin, an alpha(1)-adrenoceptor antagonist, prevents memory deterioration in the APP23 transgenic mouse model of Alzheimer's disease. Neurobiol. Aging 34, 1105–1115. doi: 10.1016/j.neurobiolaging.2012.09.010
Kenton, L., Boon, F., and Cain, D. P. (2008). Combined but not individual administration of beta-adrenergic and serotonergic antagonists impairs water maze acquisition in the rat. Neuropsychopharmacology 33, 1298–1311. doi: 10.1038/sj.npp.1301518
Khachaturian, A. S., Zandi, P. P., Lyketsos, C. G., Hayden, K. M., Skoog, I., Norton, M. C., et al. (2006). Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch. Neurol. 63, 686–692. doi: 10.1001/archneur.63.5.noc60013
Knaus, A. E., Muthig, V., Schickinger, S., Moura, E., Beetz, N., Gilsbach, R., et al. (2007). Alpha2-adrenoceptor subtypes–unexpected functions for receptors and ligands derived from gene-targeted mouse models. Neurochem. Int. 51, 277–281. doi: 10.1016/j.neuint.2007.06.036
Komatsu, M., Shibata, N., Ohnuma, T., Kuerban, B., Tomson, K., Toda, A., et al. (2014). Polymorphisms in the aldehyde dehydrogenase 2 and dopamine beta hydroxylase genes are not associated with Alzheimer's disease. J. Neural. Transm. 121, 427–432. doi: 10.1007/s00702-013-1112-z
Kummer, M. P., Hammerschmidt, T., Martinez, A., Terwel, D., Eichele, G., Witten, A., et al. (2014). Ear2 deletion causes early memory and learning deficits in APP/PS1 mice. J. Neurosci. 34, 8845–8854. doi: 10.1523/JNEUROSCI.4027-13.2014
Lapiz, M. S., Mateo, Y., Durkin, S., Parker, T., and Marsden, C. A. (2001). Effects of central noradrenaline depletion by the selective neurotoxin DSP-4 on the behaviour of the isolated rat in the elevated plus maze and water maze. Psychopharmacology 155, 251–259. doi: 10.1007/s002130100702
Lemmer, B., Langer, L., Ohm, T., and Bohl, J. (1993). Beta-adrenoceptor density and subtype distribution in cerebellum and hippocampus from patients with Alzheimer's disease. Naunyn Schmiedebergs. Arch. Pharmacol. 347, 214–219. doi: 10.1007/BF00169270
Levcik, D., Stuchlik, A., and Klement, D. (2013). Effect of block of α1-adrenoceptors on overall motor activity but not on spatial cognition in the object-position recognition task. Physiol. Res. 62, 561–567.
Leverenz, J. B., Miller, M. A., Dobie, D. J., Peskind, E. R., and Raskind, M. A. (2001). Increased alpha 2-adrenergic receptor binding in locus coeruleus projection areas in dementia with Lewy bodies. Neurobiol. Aging 22, 555–561. doi: 10.1016/S0197-4580(01)00221-4
Liu, Y., Yoo, M. J., Savonenko, A., Stirling, W., Price, D. L., Borchelt, D. R., et al. (2008). Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 28, 13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008
Lockrow, J., Boger, H., Gerhardt, G., Aston-Jones, G., Bachman, D., and Granholm, A.-C. (2011). A noradrenergic lesion exacerbates neurodegeneration in a down syndrome mouse model. J. Alzheimers Dis. 23, 471–489. doi: 10.3233/JAD-2010-101218
Mann, D. M. A., Lincoln, J., Yates, P. O., Stamp, J. E., and Toper, S. (1980). Changes in the monoamine containing neurones of the human CNS in senile dementia. Br. J. Psychiatry 136, 533–541. doi: 10.1192/bjp.136.6.533
Marien, M. R., Colpaert, F. C., and Rosenquist, A. C. (2004). Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Rev. 45, 38–78. doi: 10.1016/j.brainresrev.2004.02.002
Marino, M. D., Bourdelat-Parks, B. N., Cameron Liles, L., and Weinshenker, D. (2005). Genetic reduction of noradrenergic function alters social memory and reduces aggression in mice. Behav. Brain Res. 161, 197–203. doi: 10.1016/j.bbr.2005.02.005
Martignoni, E., Blandini, F., Petraglia, F., Pacchetti, C., Bono, G., and Nappi, G. (1992). Cerebrospinal fluid norepinephrine, 3-methoxy-4-hydroxyphenylglycol and neuropeptide Y levels in Parkinson's disease, multiple system atrophy and dementia of the Alzheimer type. J. Neural Transm. Park. Dis. Dement. Sect. 4, 191–205. doi: 10.1007/BF02260903
Matthews, K. L., Chen, C. P., Esiri, M. M., Keene, J., Minger, S. L., and Francis, P. T. (2002). Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol. Psychiatry 51, 407–416. doi: 10.1016/S0006-3223(01)01235-5
Mishima, K., Tanoue, A., Tsuda, M., Hasebe, N., Fukue, Y., Egashira, N., et al. (2004). Characteristics of behavioral abnormalities in alpha1d-adrenoceptors deficient mice. Behav. Brain Res. 152, 365–373. doi: 10.1016/j.bbr.2003.10.038
Miyata, S., Nagata, H., Yamao, S., Nakamura, S., and Kameyama, M. (1984). Dopamine-beta-hydroxylase activities in serum and cerebrospinal fluid of aged and demented patients. J. Neurol. Sci. 63, 403–409. doi: 10.1016/0022-510X(84)90163-1
Nalepa, I., Kreiner, G., Bielawski, A., Rafa-Zablocka, K., and Roman, A. (2013). alpha1-Adrenergic receptor subtypes in the central nervous system: insights from genetically engineered mouse models. Pharmacol. Rep. 65, 1489–1497. doi: 10.1016/S1734-1140(13)71509-3
Nazarali, A. J., and Reynolds, G. P. (1992). Monoamine neurotransmitters and their metabolites in brain regions in Alzheimer's disease: a postmortem study. Cell. Mol. Neurobiol. 12, 581–587. doi: 10.1007/BF00711237
Ni, Y., Zhao, X., Bao, G., Zou, L., Teng, L., Wang, Z., et al. (2006). Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med. 12, 1390–1396. doi: 10.1038/nm1485
O'donnell, J., Zeppenfeld, D., Mcconnell, E., Pena, S., and Nedergaard, M. (2012). Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 37, 2496–2512. doi: 10.1007/s11064-012-0818-x
Ohno, M., Yoshimatsu, A., Kobayashi, M., and Watanabe, S. (1997). Noradrenergic DSP-4 lesions aggravate impairment of working memory produced by hippocampal muscarinic blockade in rats. Pharmacol.Biochem. Behav. 57, 257–261. doi: 10.1016/S0091-3057(96)00353-X
Oikawa, N., Ogino, K., Masumoto, T., Yamaguchi, H., and Yanagisawa, K. (2010). Gender effect on the accumulation of hyperphosphorylated tau in the brain of locus-ceruleus-injured APP-transgenic mouse. Neurosci. Lett. 468, 243–247. doi: 10.1016/j.neulet.2009.11.005
Ouyang, M., and Thomas, S. A. (2005). A requirement for memory retrieval during and after long-term extinction learning. Proc. Natl. Acad. Sci. U.S.A. 102, 9347–9352. doi: 10.1073/pnas.0502315102
Palmer, A. M., Francis, P. T., Bowen, D. M., Benton, J. S., Neary, D., Mann, D. M., et al. (1987). Catecholaminergic neurones assessed ante-mortem in Alzheimer's disease. Brain Res. 414, 365–375. doi: 10.1016/0006-8993(87)90018-7
Pascual, J., Grijalba, B., Garcia-Sevilla, J. A., Zarranz, J. J., and Pazos, A. (1992). Loss of high-affinity alpha 2-adrenoceptors in Alzheimer's disease: an autoradiographic study in frontal cortex and hippocampus. Neurosci. Lett. 142, 36–40. doi: 10.1016/0304-3940(92)90614-D
Pauszek, M. E. (1991). Propranolol for treatment of agitation in senile dementia. Indiana Med. 84, 16–17.
Perez, D. M. (2007). Structure-function of alpha1-adrenergic receptors. Biochem. Pharmacol. 73, 1051–1062. doi: 10.1016/j.bcp.2006.09.010
Pérez, V., Marin, C., Rubio, A., Aguilar, E., Barbanoj, M., and Kulisevsky, J. (2009). Effect of the additional noradrenergic neurodegeneration to 6-OHDA-lesioned rats in levodopa-induced dyskinesias and in cognitive disturbances. J. Neural. Transm. 116, 1257–1266. doi: 10.1007/s00702-009-0291-0
Perry, G., Cash, A. D., and Smith, M. A. (2002). Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2, 120–123. doi: 10.1155/S1110724302203010
Peskind, E. R., Tsuang, D. W., Bonner, L. T., Pascualy, M., Riekse, R. G., Snowden, M. B., et al. (2005). Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis. Assoc. Disord. 19, 23–28. doi: 10.1097/01.wad.0000155067.16313.5e
Peskind, E. R., Wingerson, D., Murray, S., Pascualy, M., Dobie, D. J., Le Corre, P., et al. (1995). Effects of Alzheimer's disease and normal aging on cerebrospinal fluid norepinephrine responses to yohimbine and clonidine. Arch. Gen. Psychiatry 52, 774–782. doi: 10.1001/archpsyc.1995.03950210068012
Philipp, M., and Hein, L. (2004). Adrenergic receptor knockout mice: distinct functions of 9 receptor subtypes. Pharmacol. Ther. 101, 65–74. doi: 10.1016/j.pharmthera.2003.10.004
Pottier, C., Hannequin, D., Coutant, S., Rovelet-Lecrux, A., Wallon, D., Rousseau, S., et al. (2012). High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 17, 875–879. doi: 10.1038/mp.2012.15
Pugh, P. L., Vidgeon-Hart, M. P., Ashmeade, T., Culbert, A. A., Seymour, Z., Perren, M. J., et al. (2007). Repeated administration of the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) modulates neuroinflammation and amyloid plaque load in mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. J. Neuroinflammation 4:8. doi: 10.1186/1742-2094-4-8
Puumala, T., Greijus, S., Narinen, K., Haapalinna, A., Riekkinen, P. Sr., and Sirvio, J. (1998). Stimulation of alpha-1 adrenergic receptors facilitates spatial learning in rats. Eur. Neuropsychopharmacol. 8, 17–26. doi: 10.1016/S0924-977X(97)00040-0
Puumala, T., Sirvio, J., Ruotsalainen, S., and Riekkinen, P. Sr. (1996). Effects of St-587 and prazosin on water maze and passive avoidance performance of scopolamine-treated rats. Pharmacol. Biochem. Behav. 55, 107–115. doi: 10.1016/0091-3057(95)02231-7
Qu, L. L., Guo, N. N., and Li, B. M. (2008). Beta1- and beta2-adrenoceptors in basolateral nucleus of amygdala and their roles in consolidation of fear memory in rats. Hippocampus 18, 1131–1139. doi: 10.1002/hipo.20478
Rajabi, S., Shamsizadeh, A., Amini, H., Shirazi, M., Allahtavakoli, M., Abbasnejad, M., et al. (2012). Effect of DSP-4 induced central noradrenergic depletion on tactile learning in rat. Neurol. Res. 34, 80–84. doi: 10.1179/1743132811Y.0000000050
Raskind, M. A., Peskind, E. R., Holmes, C., and Goldstein, D. S. (1999). Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer's disease. Biol. Psychiatry 46, 756–765. doi: 10.1016/S0006-3223(99)00008-6
Reinikainen, K. J., Paljarvi, L., Huuskonen, M., Soininen, H., Laakso, M., and Riekkinen, P. J. (1988). A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer's disease. J. Neurol. Sci. 84, 101–116. doi: 10.1016/0022-510X(88)90179-7
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. doi: 10.1038/ng1943
Rosenberg, P. B., Mielke, M. M., Tschanz, J., Cook, L., Corcoran, C., Hayden, K. M., et al. (2008). Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am. J. Geriatr. Psychiatry 16, 883–892. doi: 10.1097/JGP.0b013e318181276a
Ruetti, E., Justel, N., Mustaca, A., and Boccia, M. (2014). Corticosterone and propranolol's role on taste recognition memory. Pharmacol. Biochem. Behav. 127, 37–41. doi: 10.1016/j.pbb.2014.09.013
Ruiz, J., Martin, I., Callado, L. F., Meana, J. J., Barturen, F., and Garcia-Sevilla, J. A. (1993). Non-adrenoceptor [3H]idazoxan binding sites (I2-imidazoline sites) are increased in postmortem brain from patients with Alzheimer's disease. Neurosci. Lett. 160, 109–112. doi: 10.1016/0304-3940(93)90925-B
Saber, A. J., and Cain, D. P. (2003). Combined beta-adrenergic and cholinergic antagonism produces behavioral and cognitive impairments in the water maze: implications for Alzheimer disease and pharmacotherapy with beta-adrenergic antagonists. Neuropsychopharmacology 28, 1247–1256. doi: 10.1038/sj.npp.1300163
Sadalge, A., Coughlin, L., Fu, H., Wang, B., Valladares, O., Valentino, R., et al. (2003). alpha 1d Adrenoceptor signaling is required for stimulus induced locomotor activity. Mol. Psychiatry 8, 664–672. doi: 10.1038/sj.mp.4001351
Salehi, A., Faizi, M., Colas, D., Valletta, J., Laguna, J., Takimoto-Kimura, R., et al. (2009). Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Sci. Transl. Med. 1:7ra17. doi: 10.1126/scitranslmed.3000258
Sallee, F. R., and Pollock, B. G. (1990). Clinical pharmacokinetics of imipramine and desipramine. Clin. Pharmacokinet. 18, 346–364. doi: 10.2165/00003088-199018050-00002
Salm, A. K., and Mccarthy, K. D. (1989). Expression of beta-adrenergic receptors by astrocytes isolated from adult rat cortex. Glia 2, 346–352. doi: 10.1002/glia.440020507
Scherzer, C. R., Offe, K., Gearing, M., Rees, H. D., Fang, G., Heilman, C. J., et al. (2004). Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch. Neurol. 61, 1200–1205. doi: 10.1001/archneur.61.8.1200
Schubert, D. (2005). Glucose metabolism and Alzheimer's disease. Ageing Res. Rev. 4, 240–257. doi: 10.1016/j.arr.2005.02.003
Schutsky, K., Ouyang, M., and Thomas, S. A. (2011). Xamoterol impairs hippocampus-dependent emotional memory retrieval via Gi/o-coupled β2-adrenergic signaling. Learn. Mem. 18, 598–604. doi: 10.1101/lm.2302811
Scullion, G. A., Kendall, D. A., Marsden, C. A., Sunter, D., and Pardon, M. C. (2011). Chronic treatment with the alpha2-adrenoceptor antagonist fluparoxan prevents age-related deficits in spatial working memory in APPxPS1 transgenic mice without altering beta-amyloid plaque load or astrocytosis. Neuropharmacology 60, 223–234. doi: 10.1016/j.neuropharm.2010.09.002
Shankle, W. R., Nielson, K. A., and Cotman, C. W. (1995). Low-dose propranolol reduces aggression and agitation resembling that associated with orbitofrontal dysfunction in elderly demented patients. Alzheimer Dis. Assoc. Disord. 9, 233–237. doi: 10.1097/00002093-199509040-00010
Shao, Y., and Sutin, J. (1992). Expression of adrenergic receptors in individual astrocytes and motor neurons isolated from the adult rat brain. Glia 6, 108–117. doi: 10.1002/glia.440060205
Shimohama, S., Taniguchi, T., Fujiwara, M., and Kameyama, M. (1986). Biochemical characterization of alpha-adrenergic receptors in human brain and changes in Alzheimer-type dementia. J. Neurochem. 47, 1295–1301.
Sirviö, J., Riekkinen, P. Jr., Valjakka, A., Jolkkonen, J., and Riekkinen, P. J. (1991). The effects of noradrenergic neurotoxin, DSP-4, on the performance of young and aged rats in spatial navigation task. Brain Res. 563, 297–302. doi: 10.1016/0006-8993(91)91550-K
Sontag, T. A., Hauser, J., Kaunzinger, I., Gerlach, M., Tucha, O., and Lange, K. W. (2008). Effects of the noradrenergic neurotoxin DSP4 on spatial memory in the rat. J. Neural. Transm. 115, 299–303. doi: 10.1007/s00702-007-0830-5
Sparks, D. L., Dekosky, S. T., and Markesbery, W. R. (1988). Alzheimer's disease: aminergic-cholinergic alterations in hypothalamus. Arch. Neurol. (Chicago) 45, 994–999. doi: 10.1001/archneur.1988.00520330084014
Summers, W. K. (2006). The management of agitation in demented patients with propranolol. J. Alzheimers Dis. 9, 69–75.
Sutin, J., and Shao, Y. (1992). Resting and reactive astrocytes express adrenergic receptors in the adult rat brain. Brain Res. Bull. 29, 277–284. doi: 10.1016/0361-9230(92)90057-5
Swanson, L. W., and Hartman, B. K. (1975). The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J.Comp. Neurol. 163, 467–505. doi: 10.1002/cne.901630406
Szot, P., White, S. S., Greenup, J. L., Leverenz, J. B., Peskind, E. R., and Raskind, M. A. (2006). Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J. Neurosci. 26, 467–478. doi: 10.1523/JNEUROSCI.4265-05.2006
Szot, P., White, S. S., Greenup, J. L., Leverenz, J. B., Peskind, E. R., and Raskind, M. A. (2007). Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience 146, 471–480. doi: 10.1016/j.neuroscience.2007.01.031
Taherian, F., Vafaei, A. A., Vaezi, G. H., Eskandarian, S., Kashef, A., and Rashidy-Pour, A. (2014). Propranolol-induced impairment of contextual fear memory reconsolidation in rats: a similar effect on weak and strong recent and remote memories. Basic Clin. Neurosci. 5, 231–239.
Takahashi, J., Shibata, T., Sasaki, M., Kudo, M., Yanezawa, H., Obara, S., et al. (2015). Detection of changes in the locus coeruleus in patients with mild cognitive impairment and Alzheimer's disease: high-resolution fast spin-echo T1-weighted imaging. Geriatr. Gerontol. Int. 15, 334–340. doi: 10.1111/ggi.12280
Tanaka, K. F., Kashima, H., Suzuki, H., Ono, K., and Sawada, M. (2002). Existence of functional beta1- and beta2-adrenergic receptors on microglia. J. Neurosci. Res. 70, 232–237. doi: 10.1002/jnr.10399
Tejani-Butt, S. M., Yang, J., and Zaffar, H. (1993). Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer's disease. Brain Res. 631, 147–150. doi: 10.1016/0006-8993(93)91201-3
Teri, L., Reifler, B. V., Veith, R. C., Barnes, R., White, E., Mclean, P., et al. (1991). Imipramine in the treatment of depressed Alzheimer's patients: impact on cognition. J. Gerontol. 46, P372–P377. doi: 10.1093/geronj/46.6.P372
Thomas, S. A., and Palmiter, R. D. (1997). Disruption of the dopamine beta-hydroxylase gene in mice suggests roles for norepinephrine in motor function, learning, and memory. Behav. Neurosci. 111, 579–589. doi: 10.1037/0735-7044.111.3.579
Tohgi, H., Ueno, M., Abe, T., Takahashi, S., and Nozaki, Y. (1992). Concentration of monoamines and their metabolites in the cerebrospinal fluid from patients with senile dementia of the Alzheimer type and vascular dementia of the Binswanger type. J. Neural Transm. Park. Dis. Dement. Sect. 4, 69–77. doi: 10.1007/BF02257623
Torkaman-Boutorabi, A., Danyali, F., Oryan, S., Ebrahimi-Ghiri, M., and Zarrindast, M.-R. (2014). Hippocampal α-adrenoceptors involve in the effect of histamine on spatial learning. Physiol. Behav. 129, 17–24. doi: 10.1016/j.physbeh.2014.02.009
Tzavara, E. T., Bymaster, F. P., Overshiner, C. D., Davis, R. J., Perry, K. W., Wolff, M., et al. (2006). Procholinergic and memory enhancing properties of the selective norepinephrine uptake inhibitor atomoxetine. Mol. Psychiatry 11, 187–195. doi: 10.1038/sj.mp.4001763
Veyrac, A., Nguyen, V., Marien, M., Didier, A., and Jourdan, F. (2007). Noradrenergic control of odor recognition in a nonassociative olfactory learning task in the mouse. Learn. Mem. 14, 847–854. doi: 10.1101/lm.708807
Wang, D., Fu, Q., Zhou, Y., Xu, B., Shi, Q., Igwe, B., et al. (2013a). β2 Adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J. Biol. Chem. 288, 10298–10307. doi: 10.1074/jbc.M112.415141
Wang, J., Wright, H. M., Vempati, P., Li, H., Wangsa, J., Dzhuan, A., et al. (2013b). Investigation of nebivolol as a novel therapeutic agent for the treatment of Alzheimer's disease. J. Alzheimers Dis. 33, 1147–1156. doi: 10.3233/JAD-2012-120904
Wang, L., Bodner, M., and Zhou, Y. D. (2013c). Distributed neural networks of tactile working memory. J. Physiol. Paris 107, 452–458. doi: 10.1016/j.jphysparis.2013.06.001
Wang, L. Y., Shofer, J. B., Rohde, K., Hart, K. L., Hoff, D. J., Mcfall, Y. H., et al. (2009a). Prazosin for the treatment of behavioral symptoms in patients with Alzheimer disease with agitation and aggression. Am. J. Geriatr. Psychiatry 17, 744–751. doi: 10.1097/JGP.0b013e3181ab8c61
Wang, M., Ramos, B. P., Paspalas, C. D., Shu, Y., Simen, A., Duque, A., et al. (2007). Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 129, 397–410. doi: 10.1016/j.cell.2007.03.015
Wang, Q. W., Rowan, M. J., and Anwyl, R. (2009b). Inhibition of LTP by beta-amyloid is prevented by activation of beta2 adrenoceptors and stimulation of the cAMP/PKA signalling pathway. Neurobiol. Aging 30, 1608–1613. doi: 10.1016/j.neurobiolaging.2007.12.004
Warner, T. A., and Drugan, R. C. (2012). Morris water maze performance deficit produced by intermittent swim stress is partially mediated by norepinephrine. Pharmacol. Biochem. Behav. 101, 24–34. doi: 10.1016/j.pbb.2011.11.009
Xiang, Y. K. (2011). Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ. Res. 109, 231–244. doi: 10.1161/CIRCRESAHA.110.231340
Yu, J. T., Tan, L., Ou, J. R., Zhu, J. X., Liu, K., Song, J. H., et al. (2008). Polymorphisms at the beta2-adrenergic receptor gene influence Alzheimer's disease susceptibility. Brain Res. 1210, 216–222. doi: 10.1016/j.brainres.2008.03.019
Yu, N. N., Wang, X. X., Yu, J. T., Wang, N. D., Lu, R. C., Miao, D., et al. (2010). Blocking beta2-adrenergic receptor attenuates acute stress-induced amyloid beta peptides production. Brain Res. 1317, 305–310. doi: 10.1016/j.brainres.2009.12.087
Zarow, C., Lyness, S. A., Mortimer, J. A., and Chui, H. C. (2003). Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 60, 337–341. doi: 10.1001/archneur.60.3.337
Zhou, H. C., Sun, Y. Y., Cai, W., He, X. T., Yi, F., Li, B. M., et al. (2013). Activation of beta2-adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP-PKA signaling in the medial prefrontal cortex of rats. Learn. Mem. 20, 274–284. doi: 10.1101/lm.030411.113
Zoladz, P. R., Fleshner, M., and Diamond, D. M. (2013). Differential effectiveness of tianeptine, clonidine and amitriptyline in blocking traumatic memory expression, anxiety and hypertension in an animal model of PTSD. Prog. Neuropsychopharmacol. Biol. Psychiatry 44, 1–16. doi: 10.1016/j.pnpbp.2013.01.001
Keywords: noradrenergic system, norepinephrine, adrenergic receptors, degeneration, dysfunction
Citation: Gannon M, Che P, Chen Y, Jiao K, Roberson ED and Wang Q (2015) Noradrenergic dysfunction in Alzheimer's disease. Front. Neurosci. 9:220. doi: 10.3389/fnins.2015.00220
Received: 24 March 2015; Accepted: 02 June 2015;
Published: 17 June 2015.
Edited by:
Ritchie Williamson, University of Dundee, UKReviewed by:
Joaquin Del Rio, Center for Applied Medical Research, SpainEfthimios M. C. Skoulakis, Biomedical Sciences Research Centre “Alexander Fleming”, Greece
Copyright © 2015 Gannon, Che, Chen, Jiao, Roberson and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Wang, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham,986 MCLM, 1918 University Blvd., Birmingham, AL 35294, USA,cWlud2FuZ0B1YWIuZWR1