 Ornella Cuomo1
Ornella Cuomo1 Antonio Vinciguerra1
Antonio Vinciguerra1 Pierpaolo Cerullo1
Pierpaolo Cerullo1 Serenella Anzilotti2Paola Brancaccio1
Serenella Anzilotti2Paola Brancaccio1 Leonilda Bilo3
Leonilda Bilo3 Antonella Scorziello1
Antonella Scorziello1 Pasquale Molinaro1
Pasquale Molinaro1 Gianfranco Di Renzo1
Gianfranco Di Renzo1 Giuseppe Pignataro1*
Giuseppe Pignataro1*- 1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, Federico II University of Naples, Naples, Italy
- 2SDN Istituto di Ricerca Diagnostica e Nucleare, Naples, Italy
- 3Division of Neurology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, Federico II University of Naples, Naples, Italy
Most of the current focus on developing neuroprotective therapies is aimed at preventing neuronal death. However, these approaches have not been successful despite many years of clinical trials mainly because the numerous side effects observed in humans and absent in animals used at preclinical level. Recently, the research in this field aims to overcome this problem by developing strategies which induce, mimic, or boost endogenous protective responses and thus do not interfere with physiological neurotransmission. Preconditioning is a protective strategy in which a subliminal stimulus is applied before a subsequent harmful stimulus, thus inducing a state of tolerance in which the injury inflicted by the challenge is mitigated. Tolerance may be observed in ischemia, seizure, and infection. Since it requires protein synthesis, it confers delayed and temporary neuroprotection, taking hours to develop, with a pick at 1–3 days. A new promising approach for neuroprotection derives from post-conditioning, in which neuroprotection is achieved by a modified reperfusion subsequent to a prolonged ischemic episode. Many pathways have been proposed as plausible mechanisms to explain the neuroprotection offered by preconditioning and post-conditioning. Although the mechanisms through which these two endogenous protective strategies exert their effects are not yet fully understood, recent evidence highlights that the maintenance of ionic homeostasis plays a key role in propagating these neuroprotective phenomena. The present article will review the role of protein transporters and ionic channels involved in the control of ionic homeostasis in the neuroprotective effect of ischemic preconditioning and post-conditioning in adult brain, with particular regards to the Na+/Ca2+ exchangers (NCX), the plasma membrane Ca2+-ATPase (PMCA), the Na+/H+ exchange (NHE), the Na+/K+/2Cl− cotransport (NKCC) and the acid-sensing cation channels (ASIC). Ischemic stroke is the third leading cause of death and disability. Up until now, all clinical trials testing potential stroke neuroprotectants failed. For this reason attention of researchers has been focusing on the identification of brain endogenous neuroprotective mechanisms activated after cerebral ischemia. In this context, ischemic preconditioning and ischemic post-conditioning represent two neuroprotecive strategies to investigate in order to identify new molecular target to reduce the ischemic damage.
Ischemic Preconditioning
The concept of ischemic preconditioning was introduced first in the heart (Murry et al., 1986) and later on in the brain (Murry et al., 1986; Schurr et al., 1986). Ischemic preconditioning is neuroprotective strategy in which a subliminal stimulus is applied before a subsequent harmful ischemia (Kirino, 2002; Dirnagl et al., 2003; Gidday, 2006). The state of tolerance mediated by this strategy is composed of two phases of neuroprotection. The first phase, named rapid or acute preconditioning (Perez-Pinzon et al., 1997), starts 3–5 min after the preconditioning stimulus and ends 1 h later and is due to rapid post-translational protein modifications (Barone et al., 1998; Meller et al., 2006). The second phase, named delayed preconditioning (Perez-Pinzon et al., 1997), starts 2–3 days after preconditioning and ends 1 week later and mainly involves de novo protein synthesis (Barone et al., 1998; Dirnagl et al., 2003; Meller et al., 2005).
Mechanisms Involved in Ischemic Preconditioning Neuroprotection
The molecular mechanisms underlying ischemic tolerance are not yet fully understood because of its extreme complexity, involving many signaling pathways and alterations in gene expression. Several studies have investigated the signaling cascades and key molecules both in in vitro and in vivo models of ischemic preconditioning. Most of the stressors induce both rapid and delayed tolerance. As recently reviewed (Obrenovitch, 2008), the features of ischemic tolerance partially are similar to naturally occurring adaptive mechanisms, including at cellular levels:
1. Alterations in cellular energy metabolism: preconditioning preserves mitochondrial function (Dave et al., 2001; Racay et al., 2007) and mediates an increase in expression of genes playing a role in energy metabolism.
2. Preservation of mitochondrial membrane potential: preconditioning preserves respiratory complexes, mitochondrial oxidative phosphorylation and antioxidant enzymes such as superoxide dismutases (SODs), catalase, glutathione peroxidase, and thioredoxin system (Danielisová et al., 2006; Racay et al., 2007).
3. Modulation of neuronal excitotoxicity: preconditioning mediates a shift to inhibitory neurotransmission, suppressing glutamate release, downregulating AMPA and NMDA receptors, and glial glutamate transporters, increasing GABA release and GABA-A receptors expression (Dave et al., 2005).
4. Suppression of cell death and apoptotic mechanisms: preconditioning reduces the release of cytochrome c, inhibits caspases, and proapoptotic genes, and activates survival pathways such as serine/threonine activated kinases (Akt) and extracellular signal-regulated kinases (ERK) and trophic factors (Lehotský et al., 2009).
5. Activation of the mechanisms mediating DNA repair and plasticity by increasing the activity nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) (Gidday, 2006; Lehotský et al., 2009).
6. Reduction of inflammatory response: preconditioning activates Toll-like receptor, suppresses induced proinflammatory cytokines (TNFs) and activates transcription nuclear factor κB (NFκB) (Gidday, 2006).
7. Cerebrovascular adaptation by vascular remodeling: preconditioning activates vascular endothelial growth factors (VEGF), erythropoietin and heme oxygenase 1 as targets of hypoxia inducing factor 1α (HIF-1α). The blood–brain barrier was preserved thanks to the reduced activity of matrix metalloproteinases and cell adhesion molecules (Gidday, 2006).
8. Improved capacity to preserve cellular ionic and pH homeostasis: preconditioning regulates expression and activity of ion transport systems (Ca2+-ATPase, acid-sensing ion channels, Na+/H+ exchange, Na+/K+/2Cl− cotransport and Na+/Ca2+ exchanger) and transporters for metabolites.
Ischemic Post-conditioning
Unlike ischemic preconditioning, ischemic post-conditioning is a relatively novel neuroprotective strategy (Zhao et al., 2006; Pignataro et al., 2008). Rapid revascularization of the occluded vessels represent one of the most effective approaches currently used for the treatment of acute ischemic stroke. However, during the early reperfusion phase an increase in reactive oxygen species and intracellular free Ca2+ may occur, thus leading to additional injury (Kuroda and Siesjö, 1997). Ischemic post-conditioning is realized by applying brief interruptions of reperfusion after ischemia. That a protocol of cycles of brief reperfusion followed by re-occlusion was able to reduce the ischemic damage was first demonstrated during cardiac ischemia (Zhao et al., 2003). More recently, it has been demonstrated that ischemic post-conditioning induces neuroprotection also in a rat hippocampal slice model of cerebral ischemia (Scartabelli et al., 2008), in rodent models of spinal cord injury (Jiang et al., 2006), and in models of focal (Zhao et al., 2006; Pignataro et al., 2008, 2013b; Xing et al., 2008), and global ischemia (Pateliya et al., 2008; Wang et al., 2008).
Mechanisms Involved in Ischemic Post-conditioning Neuroprotection
At present a few information are available about the mechanisms involved in the neuroprotection mediated by ischemic post-conditioning. Since post-conditioning is applied after the ischemic injury, the changes observed in the brain have to be ascribed to the combined effects of post-conditioning and stroke, rather than the effect of the post-conditioning only. Thus, it is difficult to sunder the protective mechanisms of post-conditioning from the consequences of its protection. However, up until now, many mechanisms have been suggested (Zhao, 2009), such as:
1. Reduction of hyperemia consequent to the reperfusion, that may be accompanied other toxic events, such as the production of free radicals, loss of blood–brain barrier integrity, and activation of inflammation cascade (Schaller and Graf, 2004).
2. Blocking of apoptosis. Ischemic post-conditioning reduced ROS production (Zhao et al., 2006), attenuates lipid peroxidate levels (Xing et al., 2008) and increased the activities of antioxidant enzymes, such as superoxide dismutase and catalase (Danielisová et al., 2006). Furthermore, rapid post-conditioning reduced the release of cytochrome c from the mitochondria to the cytosol, a critical cascade for the induction of apoptosis, thus suggesting that post-conditioning may reduce ischemic injury by blocking apoptosis (Zhao et al., 2012).
3. Inhibition of inflammation. Rapid post-conditioning inhibits inflammation after stroke. Indeed, rapid post-conditioning inhibits myeloperoxidase activity, an indicator of leukocyte accumulation (Xing et al., 2008), attenuates the expressions of IL-1β, TNF-α, and the ICAM-1 protein expression (Xing et al., 2008).
4. Activation of Akt Pathway. In agreement with previous studies demonstrating that the Akt pathway promotes neuronal survival after stroke (Zhao et al., 2006), rapid post-conditioning increases Akt phosphorylation and Akt activity (Gao et al., 2008; Pignataro et al., 2008). Furthermore, the protection mediated by rapid post-conditioning was prevented by Akt inhibition (Gao et al., 2008; Pignataro et al., 2008) and abolished the protective effect post-conditioning in hippocampal slice cultures (Scartabelli et al., 2008), overall, ischemic post-conditioning exerts its neuroprotective effectiveness also through Akt activation.
5. Cellular ionic homeostasis and energy metabolism. Post-conditioning-mediated neuroprotection is correlated to the maintainance of ionic homeostasis occurring by modulating expression and activity of several proteins involved in ionic homeostasis, some of which will be treated below (Figure 1, Table 1).
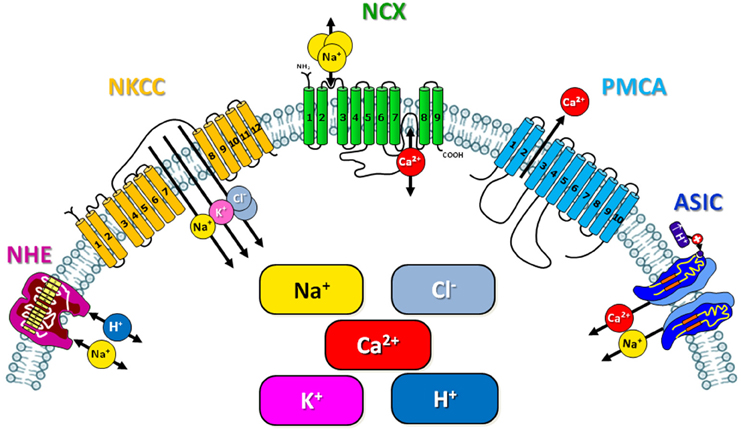
Figure 1. Schematic representation of the ionic exchangers/transporters involved in the neuroprotection mediated by ischemic preconditioning and post-conditioning.
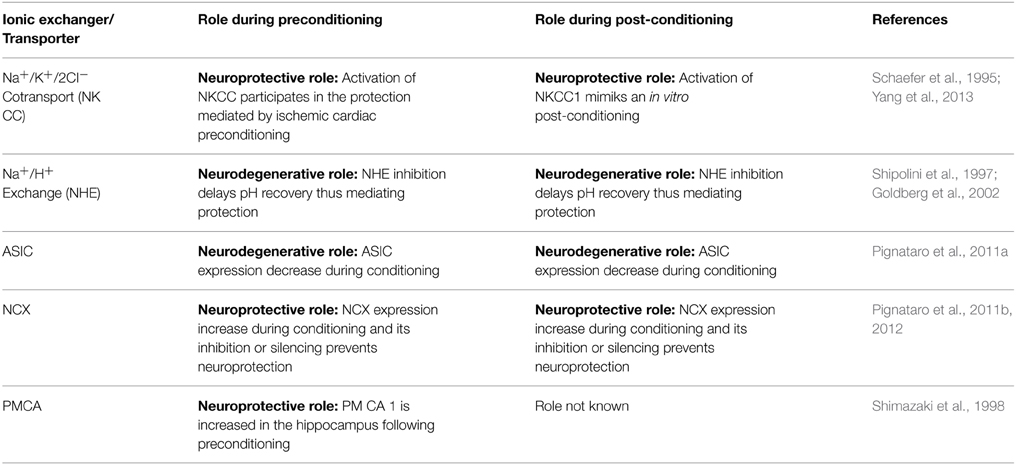
Table 1. Role of ionic exchangers/transporters during pre-and post-conditioning.
Brain Ionic Transporters during Ischemic Conditioning
Ischemic brain conditioning activates intracellular biological responses to a potential lethal insult; therefore it is predictable that organs reinforce their tolerance when exposed to a sublethal insult prior or after a harmful episode of ischemia by increasing energy metabolism or delaying anoxic depolarization after the onset of the subsequent ischemic insult (Stenzel-Poore et al., 2003; Pignataro et al., 2008, 2009). Indeed, an impairment in the activity of voltage-gated potassium channels has been observed in cortical neurons exposed to a brief non-injurious oxygen and glucose deprivation (OGD). Moreover, it has been demonstrated that ischemic conditioning prevented the inhibition of Na+/K+-ATPase activity after brain ischemia in hippocampal and cortical neurons of rats subjected to global forebrain ischemia. Instead, in vivo experiments in gerbils showed that both Ca2+-ATPase activity and mitochondrial calcium internalization increased in CA1 hippocampal neurons exposed to preconditioning (Ohta et al., 1996). Accordingly, in hippocampal neurons obtained from preconditioned gerbils [Ca2+]i increased after anoxic and aglycemic episodes whereas this increase was inhibited in ischemic-tolerant animals (Shimazaki et al., 1998). The molecular mechanisms of this phenomenon are still under investigation. An explanation may be the increase in the expression of the plasma membrane calcium ATPase 1 (PMCA-1) as recently demonstrated (Kato et al., 2005). Moreover, many evidence support the idea that other transporters are involved in the protection mediated by brain conditioning. In this regard, a key role is exerted by the sodium calcium exchanger (NCX), in fact its expression was reduced during cerebral ischemia (Pignataro et al., 2004; Boscia et al., 2006). More important, NCX downregulation was prevented when rats were subjected either to ischemic preconditioning or post-conditioning (Pignataro et al., 2013a). In addition Na+/H+ exchanger and Na+/K+/2Cl− cotransporter are critical ion transporters in the context of cerebral ischemia, as they contribute to the regulation of intracellular pH and cell volume (Pedersen, 2006). Finally, acid-sensing cation channels (ASIC) have also been studied during ischemic conditions since they represent key targets in many different aspects of acidosis, a condition that occurs during ischemic stroke. Brain preconditioning as an adaptive response to stroke requires in particular new protein transcription and translation; giving that HIF-1–dependent transcription is being clearly common in activating pathways of several exchangers/transporters/ionic channels involved in stroke, scientific community has assigned a key role to HIF-1α, as a master transcriptional regulator of cellular and developmental response to hypoxia. In effect stable HIF-1 can bind to its heterodimeric partner HIF-1β, and together these proteins, once stabilized during hypoxic-ischemic conditions, can act in the nucleus to transactivate genes involved in adaptation to hypoxic-ischemic stress. HIF represents the “hyphen” that definitely binds to each other all the different mechanisms converging on the same signaling cascade, responsible for the homeostatic regulation in protected brain (Ratan et al., 2004). In the light of these premises, the present article will review the main role of protein transporters and exchangers controlling ionic homeostasis in the neuroprotective effect of ischemic preconditioning and post-conditioning, with particular regards to the Na+/Ca2+ exchangers (NCX), the plasma membrane Ca2+-ATPase (PMCA), the Na+/H+ exchange (NHE), the Na+/K+/2Cl− cotransport (NKCC) and the acid-sensing cation channels (ASIC).
Role of Na+/H+ Exchange (NHE) and Na+/K+/2Cl− Cotransport (NKCC) In Brain Conditioning
The ubiquitous plasma membrane Na+/H+ exchanger is protein highly conserved across vertebrate species and characterized as a major membrane transporter involved in the regulation of cellular pH and volume in the nephron of the kidney. Specifically it is present in the intercalary cells of the collecting duct (Pedersen, 2006). The Na+/K+/2Cl− cotransporter (NKCC) is a protein that aids in the active transport of sodium, potassium, and chloride into and out of cells; it is widely distributed throughout the body and it has important functions in organs that secrete fluids. NKCC2 is found specifically in the kidney, where it serves to extract sodium, potassium, and chloride from the urine so that they can be reabsorbed into the blood (Haas, 1994).
Na+/H+ exchanger and Na+/K+/2Cl− cotransporter are critical ion transporters in the context of cerebral ischemia, as they contribute to the regulation of intracellular pH and cell volume (Pedersen, 2006). In particular, while the mammalian NHE is an integral membrane transport protein that mediates an electroneutral 1:1 exchange of intracellular H+ for extracellular Na+ (Xia et al., 2003), the electroneutral NKCC contransporter mediates the influx of Na+, K+, and Cl− with a stoichiometry of 1Na+:1K+:2Cl−.
Due to this dual role it is difficult to predict whether a change in the expression and in the activity of these transporters may ameliorate or impair the susceptibility of cells to ischemia; for example, enhanced Na+/H+ exchange would help to reduce the intracellular pH but, on the other hand, it would promote cell swelling and further Ca2+ influx through Na+/Ca2+ exchange. In particular, in cortical astrocyte cultures obtained from Na+/H+ exchanger isoform 1-deficient NHE1(-/-) mice the lack of NHE1 attenuated the damage induced by conditions mimicking cerebral ischemia in vitro, and this effect was confirmed also in vivo (Kintner et al., 2004). Indeed, the pharmacological blockade of NHE-1 reduces infarct volume in animal models of brain ischemia. In addition, ischemia/reperfusion causes an increase in NHE-1 expression/activity (Ramasamy et al., 1995).
Although NHE activation is essential for the restoration of physiological pHi, hyperactivation of NHE1 in neurons, in response to the metabolic acidification associated with an ischemic–hypoxic insult (Vornov et al., 1996; Luo et al., 2005; Hwang et al., 2008; Kersh et al., 2009), disrupts the intracellular ion balance, causing intracellular Na+ and Ca2+ overload (Matsumoto et al., 2004), which eventually leads to cell death. Consequently the in vitro and in vivo approach of several studies have demonstrated neuroprotection with NHE inhibitors.
Similar data were obtained with genetic manipulations of NKCC1 in mice and by the inhibition of NKCC1 using bumetanide (Chen and Sun, 2005). NKCC1 is expressed in neurons throughout the brain where it contributes to the maintenance of [Cl−]i. Thus, it may affect neuronal excitability by regulating [Cl−]i. Expression of NKCC1 protein has also been found in astrocytes and oligodendrocytes (Lenart et al., 2004). The pharmacological inhibition or transgenic ablation of NKCC1 significantly attenuates infarction and swelling after brain ischemia induced by transient middle cerebral artery occlusion (tMCAO) (Chen and Sun, 2005). Therefore, it may be hypothesized that NKCC1 represents an attractive target for conditioning strategies against stroke.
NHE and NKCC Role during Ischemic Preconditioning
NHE and NKCC role during ischemic preconditioning is still controversial and very little is known about their role in the processes triggered by brain preconditioning, since all the studies were conducted in the heart. According to a first theory, activation of both transporters participates in the protection mediated by ischemic cardiac preconditioning. Indeed, the reduced acidification occurring during ischemia (Schaefer et al., 1995) along with the increase in intracellular sodium following preconditioning is consistent with a modulation of these proteins by preconditioning (Ramasamy et al., 1995). These mechanisms may include the activation of both NKCC and NHE, potentially resulting in reduced intracellular acidosis during ischemia. NKCC regulation is multifactorial, with evidence that the cotransporter is regulated by changes in cell volume (O'Donnell, 1993) and cAMP-dependent (Pewitt et al., 1990; Lytle et al., 1992; Klein, 1993) and non cAMP-dependent protein phosphorylation as well as ionic concentration gradients. Increased inward cotransporter flux during ischemia after preconditioning could be beneficial in protecting the heart through its functional coupling with the Cl−/HCO3− exchanger (Anderson et al., 1990). In fact, chloride transported into the cell by the Na+-K+-2Cl− cotransporter may exit via Cl−/HCO−3 exchange, thus increasing intracellular HCO3− and limiting acidosis.
The primary factors regulating NHE include the intracellular proton concentration and the phosphorylation state. Under baseline conditions, the exchanger contributes to the extracellular/intracellular proton gradient with net outward transport of protons (Piwnica-Worms et al., 1986). Under conditions of ischemia in which protons are generated and intracellular pH decreases, the exchanger is stimulated to increase extrusion of protons, thus increasing the inward transport of sodium ions. However, this hypothesis has been more recently disproved. In fact, it has been highlighted that the above discussed study was carried out in hearts perfused with a medium containing HCO3− and the method used to induce acute intracellular acidosis resulted in greater acidosis in preconditioned than in control hearts. For these reasons, the faster initial recovery from acidosis in preconditioned hearts may be a consequence of the lower starting pHi. Indeed, de Albuquerque et al. (1995) recently showed that, in a similar condition, the rate of pHi recovery is similar in control and preconditioned rat hearts. More recently, it has been demonstrated that NHE activity did not contribute to the cardioprotection mediated by ischemic preconditioning. Indeed, inhibition of Na+/H+ exchanger isoform-1 (NHE1) is protective in adult myocardium in a model of ischemia followed by reperfusion; however, the effect is unclear in immature myocardium (Cun et al., 2007). More important, the administration of HOE, a NHE inhibitor, did not prevent the protection induced by preconditioning, thus suggesting that NHE inhibition during the prolonged ischemic period may enhance the protection afforded by preconditioning (Shipolini et al., 1997). Accordingly, another group demonstrated that both NHE inhibition and ischemic preconditioning eliminated the increase in intramyocyte Na+ content that otherwise occurred with cardioplegic arrest and reperfusion in a porcine model. Because their mechanisms are distinct, the authors proposed that an additive beneficial effect against ischemia-reperfusion injury can be achieved by using NHE inhibition together with a preconditioning stimulus as prereperfusion therapy (Goldberg et al., 2002).
NHE and NKCC Role during Ischemic Post-conditioning
Up until now, there is few information about the role of NHE1 exchanger in brain post-conditioning since many studies have demonstrated its neurodegenerative role in brain ischemia upon activation. According to these data, the ischemic neuroprotection induced by post-conditioning may be associated with the downregulation of NHE1 expression or activity. These hypothesis are supported by few experimental evidences. In this regard, some information were obtained in the heart where it has been demonstrated the role of NHE in a delayed recovery of pHi after heart post-conditioning. Indeed, it has been suggested that prolongation of acidosis during reperfusion is important for the post-conditioning-mediated neuroprotection. Inserte et al. demonstrated that such post-conditioning protocols that significantly prolong pHi recovery during initial reperfusion are protective for the heart. Accordingly, there is a narrow inverse correlation between the level of the delay and the extent of cell death, demonstrating the key role of the delay of intracellular acidosis in the protection mediated by post-conditioning (Inserte et al., 2009). The normalization of pHi during reperfusion is mediated by the combined action of different transport systems, including Na+/H+-exchanger, Na+/HCO3− symport, and H+-coupled lactate efflux (Vandenberg et al., 1993). These systems appear to be mainly redundant, since inhibition of one of them does not result in significant delay in pHi recovery. For instance, inhibition of Na+/H+-exchanger by selective agents administered at the onset of reperfusion has a very small effect on pHi recovery (Docherty et al., 1997; Ten Hove and Van Echteld, 2004). By contrast, simultaneous inhibition of Na+/H+-exchanger and Na+/HCO3− symport delay normalization of pHi (Docherty et al., 1997; Schäfer et al., 2000; Inserte et al., 2008).
The concept that pHi correction was correlated to the washout of lactate, H+ and CO2 was supported by the strong correlation between the delay in pHi recovery and the levels of lactate measured in the coronary effluent (blood outflow through coronaries) during the first 2 min of reperfusion (Vandenberg et al., 1993; Inserte et al., 2008). Thus, reduced catabolite washout caused an attenuation of the transmembrane H+ gradient and a decreased activity of Na+/H+-exchanger and Na+/HCO3− symport.
The protective effect of prolongation of intracellular acidosis during reperfusion has been solidly demonstrated in different models, including isolated cardiomyocytes (Schäfer et al., 2000) and intact hearts (Kitakaze et al., 1988; Ohashi et al., 1996; Preckel et al., 1998; Inserte et al., 2008). Intriguingly, there is a correlation between delayed acidosis and other cardioprotective pathways during post-conditioning such as Reperfusion Injury Salvage Kinases (RISK) (Fujita et al., 2007). According to this theory, alkalotic buffer reduced Akt and ERK phosporylation in post-conditioned myocardium.
Regarding the role of Na+-dependent chloride transporter (NKCC) during brain post-conditioning few information are available. In a recent work it has been demonstrated that in hippocampal neurons exposed to hypoxia conditioning mimiking an in vitro post-conditioning, NKCC1 is strongly activated through binding with STE20/SPS1-related proline/alanine-rich kinase (SPAK) (Yang et al., 2013). This finding could be considered as starting point for future studies.
Role of Acid-Sensing Cation Channels (ASIC) in Brain Conditioning
Acid-sensing Ion Channels (ASICs) are neuronal voltage-insensitive cationic channels activated by extracellular protons. ASIC proteins are a subfamily of the ENaC/Deg superfamily of ion channels. ASICs are potential drug targets for treating a wide variety of conditions linked to both the CNS and PNS. Acid-sensing cation channels (ASIC) have been extensively studied both in physiological and in pathological conditions for their wide distribution in the nervous system and since they represent key targets in many different aspects of acidosis, a condition that occurs during ischemic stroke, as previously analyzed (Waldmann et al., 1997; Wemmie et al., 2006). Until now, six ASIC subunit proteins, encoded by four genes, have been identified: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, and ASIC4; among these, the subtypes 1a, 2a, and 2b are expressed in neurons of the CNS. All ASICs belong to the degenerin/epithelial Na+ channel (DEG/ENaC) superfamily, which are Na+- selective cation channels sensitive to amiloride (Crowell and Kaufmann, 1961; Sutherland et al., 2000). Blockade of ASIC1a and deletion of the ASIC1 gene rescued neurons from ischemic cell death. In CNS neurons, ASIC1a responds to extracellular pH reduction ranging from 6.9 to 5.0 to generate rapid depolarizing currents (Waldmann et al., 1997), and its activation enhances the probability of action potential initiation (Vukicevic and Kellenberger, 2004). ASIC1a channels are activated at the pH values associated with CNS diseases (ischemia, ~pH 6.5–6.0; seizure, ~pH 6.8; AD, ~pH 6.6). In the case of homomeric ASIC1a channels, acid activation also induces Ca2+ entry directly through these channels (Crowell and Kaufmann, 1961; Nedergaard et al., 1991; Dietrich and Morad, 2010). In addition, the ASIC-mediated membrane depolarization may facilitate the activation of voltage gated Ca2+ channels and NMDA receptor-gated channels (Gao et al., 2005), further promoting neuronal excitation and [Ca2+]i accumulation.
Acidosis usually occurs during many central nervous system (CNS) diseases. Indeed, ischemic brain pH falls to 6.0 due to the accumulation of lactic acid and to the protons produced by ATP hydrolysis (Nedergaard et al., 1991; Gao et al., 2005). So the reasonable strategy to contrast pH lowering, sustained by ASICs activation, is to block its activity, since acidosis is a common feature of acute neurological conditions such as ischemic stroke, brain trauma, and epileptic seizures (Rehncrona, 1985). It is rational to suggest that restoration of acid–base balance and blockade of the down-stream pathways of acidosis may represent two promising strategies to avoid the consequences of acidosis.
In this direction ASIC involvement in neuroprotection elicited in the brain by pre- and post-conditioning has been investigated (Pignataro et al., 2007).
Indeed, Pignataro and colleagues demonstrated that ASIC1a is downregulated during both preconditioning and post-conditioning and that this downregulation was dependent on p-Akt. Particularly, p-Akt is up-regulated during ischemic pre- and post-conditioning. In fact, during harmful ischemia, Akt is transiently phosphorylated, and then activated for a short time interval after reperfusion (Beg et al., 2008), whereas after brain conditioning the phosphorylation of Akt persists longer, even 24 h later, the same time interval at which an ASIC1a downregulation occurs. Interestingly, selective p-Akt inhibition prevented ASIC1a downregulation thus reverting the conditioning-induced neuroprotection.
That p-Akt is the mediator of the downregulation of ASIC1a during pre- and post-conditioning are supported by previous papers showing that Reperfusion Injury Salvage Kinasesì (RISKs), including Akt (Nedergaard et al., 1991; Xu and Xiong, 2007; Beg et al., 2008; Pignataro et al., 2011a) are activated during conditioning. In conclusion, since reduction of ASIC1a is a key factor in the neuroprotective effect of pre- and post-conditioning, the downregulation of its expression and activity might be a potential strategy to ameliorate the ischemic damage. Besides ASIC1a, ASIC2a has also been linked to ischemic preconditioning neuroprotection. In fact, the upregulation of ASIC2a is associated to the neuroprotective effect of ischemic preconditioning on global brain ischemia (Miao et al., 2010).
Role of Sodium/Calcium Exchanger (NCX) during Brain Conditioning
The sodium-calcium exchanger (Na+/Ca2+ exchanger, NCX, or exchange protein) is an antiporter membrane protein that removes calcium from cells. It consists of nine transmembrane segments that can mediate Ca2+ and Na+ fluxes across the plasma membrane. Depending on the intracellular concentrations of Ca2+, [Ca2+]i, and Na+, [Na+]i, NCX can operate either in the forward mode, coupling the uphill extrusion of Ca2+ to the influx of Na+ ions, or in the reverse mode, mediating the extrusion of Na+ and the influx of the Ca2+ ions (Pignataro et al., 2004). The NCX is considered one of the most important cellular mechanisms for removing Ca2+. The maintenance of Na+ and Ca2+ homeostasis, mediated by this exchanger, is a crucial physiological phenomenon in neuronal and non-neuronal cells. NCX countertransports four or three Na+ ions in change of one Ca2+ ion (Reeves and Hale, 1984; Fujioka et al., 2000; Kang and Hilgemann, 2004); it is a high-capacity and low-affinity ionic transporter. When intracellular Ca2+ concentrations ([Ca2+]i) increase this exchanger mediates the entrance of sodium ions and the extrusion of calcium ions in tight dependence of their electrochemical gradient. The way in which the antiporter works (forward or reverse) depends on several factors such as membrane potential, Na+ and Ca2+ gradient. Therefore, NCX represents an important actor in the regulation of pathophysiological and physiological responses to increases of [Ca2+]i and [Na+]i (Haddad and Jiang, 1997; Dirnagl et al., 1999; Annunziato et al., 2004) In particular, three distinct NCX genes, ncx1-3, and several splicing variants have been so far described (Nicoll et al., 1990, 1996). The CNS is the only organ that expresses all three NCX gene products. NCX is particularly expressed in neurons at the level of synapses (Canitano et al., 2002) where, in the course of an action potential, Ca2+ crosses the plasma membrane. Calcium entry allows the fusion of synaptic vesicles with the plasmamembrane and permits the neurotransmitter release. After that, plasma membrane Ca2+ ATPase and NCX mediate the extrusion of the residual intracellular Ca2+. In presence of [Ca2+]i higher than 500 nM, NCX become the prevalent Ca2+ extrusion mechanism (Pignataro et al., 2014). At the same time, the deregulation of [Ca2+]i and [Na+]i homeostasis may mediate the damage of neuronal and glial cells occurring after stroke and other neurodegenerative disorders.
Na+/Ca2+ Exchanger in Brain Preconditioning
It is well-known that NCX may play a role in the maintenance of ionic homeostasis during ischemic conditioning (Pignataro et al., 2004, 2012; Molinaro et al., 2013). In this respect, NCX is downregulated during brain ischemia and this downregulation occurs in a manner dependent on the exchanger isoform and on the brain region interested by the damage (Pignataro et al., 2004; Boscia et al., 2006). It has been recently demonstrated that NCX1 and NCX3 isoforms represent two new effectors of the preconditioning-mediated neuroprotection (Pignataro et al., 2012). In fact, while the absence of NCX1 and NCX3 is able to partially prevent the neuroprotection mediated by ischemic preconditioning, the pro-survival factor p-Akt mediated preconditioning-induced brain protection by inducing an upregulation of NCX1 and NCX3. In this field, these results have been obtained in an experimental model of ischemic preconditioning in which preconditioning was mediated in rats by 30 min occlusion of the middle cerebral artery (MCA), followed by a subsequent harmful stimulus of 100 min of MCA occlusion, applied 72 h after the preconditioning stimulus (Pignataro et al., 2012). The correlation between NCX and p-Akt was demonstrated also by confocal fluorescence experiments, that showed a colocalization among NCX1, NCX3, and p-Akt in the temporoparietal cortex of preconditioned rats (Formisano et al., 2008; Pignataro et al., 2012). Moreover, the PI3-K inhibitor, LY294002, reverts the protective outcome elicited by ischemic preconditioning, thus giving further support to the role of p-Akt in this phenomenon (Endo et al., 2006; Pignataro et al., 2008). The overexpression p-Akt-dependent of NCX1 and NCX3 elicited by brain preconditioning may be considered a response of cells to overcome the deregulation of ionic homeostasis occurring in the brain under anoxic conditions. On the other hand, p-Akt is not the only transducer able to activate NCX1 and NCX3. Indeed, Valsecchi and colleagues underlineed that after ischemic preconditioning, hypoxia-inducible factor, HIF-1α, is strongly increased and mediated the increased expression of NCX1 expression, thus contributing to brain protection induced by ischemic preconditioning (Valsecchi et al., 2011) and suggesting that ncx1 gene represents a new HIF-1 target (Valsecchi et al., 2011). Summarizing, NCX1 and NCX3 overexpression may be considered as a mechanism that preconditioning activates in neurons and glial cellsin order to overcome the loss of ionic homeostasis caused by harmful ischemia. Interestingly, the activation of these mechanisms are long lasting, since NCX1 and NCX3 upregulation is still present 72 h after preconditioning induction, thus suggesting that these two isoforms can be included in the list of effectors able to mediate the so-called delayed preconditioning (Pignataro et al., 2013a).
Na+/Ca2+ Exchanger in Brain Post-conditioning
Regarding NCX role during brain post-conditioning, NCX3 has been confirmed as a molecular effector involved in the neuroprotection (Pignataro et al., 2011b). In particular, interesting results have been obtained in an experimental model of ischemic post-conditioning, performed in the laboratory of Pignataro and colleagues by subjecting adult male rats to 10 min of MCAO 10 min after 100 min of MCAO. Results showed that p-Akt expression after post-conditioning increased and timely mirrors the increase in NCX3 expression. On the other hand, NCX3 silencing reverted the post-conditioning-mediated neuroprotection and the selective inhibition of p-Akt prevented NCX3 upregulation and the neuroprotection (Pignataro et al., 2011b). NCX3 overexpression during post-conditioning may contribute to counteract the ionic homeostasis dysregulation. By contrast NCX1 silencing did not revert the post-conditioning-induced neuroprotection, thus showing that NCX1 does not play a relevant role during post-conditioning. This result can be explained by the observation that unlike NCX1 and NCX2, the isoform NCX3 may be still operative during ATP depletion (Linck et al., 1998; Secondo et al., 2007). In addition, the differences in NCX1 and NCX3 promoters render NCX3 a better target for the prosurvival kinase cAMP response element-binding protein (CREB), an Akt downstream player (Gabellini et al., 2003). After post-conditioning, the phosphorylation of Akt is present longer (Pignataro et al., 2008) and timely parallels the intervals of NCX3 upregulation. Furthermore, confocal experiments in the temporoparietal confirmed that the increased expression of these two proteins after post-conditioning occurs in the same cells. These considerations, beside showing that p-Akt palys a key role in neuroprotection, more important suggest NCX3 as one of the additional p-Akt effectors involved in the neuroprotective effect of ischemic post-conditioning.
Role of Plasma Membrane Ca2+ ATPase (PMCA) in Brain Conditioning
The plasma membrane Ca2+ ATPase (PMCA) is a transport protein in the plasma membrane of cells that functions removing calcium (Ca2+) from the cell; so it is also an important regulator of the calcium concentration in the extracellular space. This protein is the only plasmamembrane high affinity Ca2+ transporting system. It removes Ca2+ from the cytosol in all eukaryotic cells, and for most of them it is the only Ca2+ exporting system together with Na+/Ca2+ exchanger which represent the principal Ca2+ exchanger in heart cells and neurons (Jensen et al., 1993). The high Ca2+ affinity of the pump, however renders it the only system capable of fine-tuning its exchanges with the external ambient (Brini et al., 2013).
Conversely to other plasma membrane buffering systems, such as NCX and ATPases, plasma membrane Ca2+ -ATPase may export intracellular Ca2+ even with relatively lower transport capacity during prolonged membrane hyperpolarization and during conditions of increased intracellular Na+ concentrations, as occurred under ischemic conditions. Ohta et al. (1996) suggested that ischemia-tolerant CA1 neurons, in a model of bilateral common carotid arteries occlusion in gerbils, export more efficiently a larger amount of Ca2+ (Kato et al., 2005).
Moreover, in gerbils, ischemic preconditioning by pre-exposure to 2-min ischemia is extremely effective in protecting against ischemic cell death since there is a modification of the electrophysiological properties of CA1 neurons (Kawai et al., 1998; Shimazaki et al., 2000). In this experimental conditions, expression of PMCA1 is increased in the hippocampus following 2-min ischemia (Kato et al., 2005). The same authors evaluated PMCA1 expression also in a model of tolerance induced by a low dose treatment with 3-Nitro Propionic Acid (3-NPA). They observed an increase in the expression of Bcl-2 in the hippocampus in both the models of preconditioning. However, PMCA1 increased just in the 2-min ischemia model and not in the 3-NPA model, thus suggesting that it plays an important but not essential role in the enhancement of ischemic tolerance (Kato et al., 2005).
Conclusions
The identification of the players involved in the control of ionic homeostasis during pre and post-conditioning should provide more direct opportunities for translational neuroprotection trials. Indeed, the present review, summarizing the specific effect of the plasmamembrane proteins involved in the maintenance of cellular ionic concentration, would provide information of which transporters needed to be activated or inhibited in order to protect the brain by the ischemic injury.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, S. E., Murphy, E., Steenbergen, C., London, R. E., and Cala, P. M. (1990). Na-H exchange in myocardium: effects of hypoxia and acidification on Na and Ca. Am. J. Physiol. 259(6 Pt 1), C940–C948.
Annunziato, L., Pignataro, G., and Di Renzo, G. F. (2004). Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol. Rev. 56, 633–654. doi: 10.1124/pr.56.4.5
Barone, F. C., White, R. F., Spera, P. A., Ellison, J., Currie, R. W., Wang, X., et al. (1998). Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 29, 1937–1950, discussion 1950–1931.
Beg, A. A., Ernstrom, G. G., Nix, P., Davis, M. W., and Jorgensen, E. M. (2008). Protons act as a transmitter for muscle contraction in C. elegans. Cell 132, 149–160. doi: 10.1016/j.cell.2007.10.058
Boscia, F., Gala, R., Pignataro, G., de Bartolomeis, A., Cicale, M., Ambesi-Impiombato, A., et al. (2006). Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J. Cereb. Blood Flow Metab. 26, 502–517. doi: 10.1038/sj.jcbfm.9600207
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2013). The plasma membrane calcium pump in health and disease. FEBS J. 280, 5385–5397. doi: 10.1111/febs.12193
Canitano, A., Papa, M., Boscia, F., Castaldo, P., Sellitti, S., Taglialatela, M., et al. (2002). Brain distribution of the Na+/Ca2+ exchanger-encoding genes NCX1, NCX2, and NCX3 and their related proteins in the central nervous system. Ann. N.Y. Acad. Sci. 976, 394–404. doi: 10.1111/j.1749-6632.2002.tb04766.x
Chen, H., and Sun, D. (2005). The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol. Res. 27, 280–286. doi: 10.1179/016164105X25243
Crowell, J. W., and Kaufmann, B. N. (1961). Changes in tissue pH after circulatory arrest. Am. J. Physiol. 200, 743–745.
Cun, L., Ronghua, Z., Bin, L., Jin, L., and Shuyi, L. (2007). Preconditioning with Na+/H+ exchange inhibitor HOE642 reduces calcium overload and exhibits marked protection on immature rabbit hearts. ASAIO J. 53, 762–765. doi: 10.1097/MAT.0b013e31815766e3
Danielisová, V., Némethová, M., Gottlieb, M., and Burda, J. (2006). The changes in endogenous antioxidant enzyme activity after postconditioning. Cell. Mol. Neurobiol. 26, 1181–1191. doi: 10.1007/s10571-006-9034-z
Dave, K. R., Lange-Asschenfeldt, C., Raval, A. P., Prado, R., Busto, R., Saul, I., et al. (2005). Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J. Neurosci. Res. 82, 665–673. doi: 10.1002/jnr.20674
Dave, K. R., Saul, I., Busto, R., Ginsberg, M. D., Sick, T. J., and Pérez-Pinzón, M. A. (2001). Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. J. Cereb. Blood Flow Metab. 21, 1401–1410. doi: 10.1097/00004647-200112000-00004
de Albuquerque, C. P., Gerstenblith, G., and Weiss, R. G. (1995). Myocardial buffering capacity in ischemia preconditioned rat hearts. J. Mol. Cell. Cardiol. 27, 777–781. doi: 10.1016/0022-2828(95)90084-5
Dietrich, C. J., and Morad, M. (2010). Synaptic acidification enhances GABAA signaling. J. Neurosci. 30, 16044–16052. doi: 10.1523/JNEUROSCI.6364-09.2010
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397. doi: 10.1016/S0166-2236(99)01401-0
Dirnagl, U., Simon, R. P., and Hallenbeck, J. M. (2003). Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 26, 248–254. doi: 10.1016/S0166-2236(03)00071-7
Docherty, J. C., Yang, L., Pierce, G. N., and Deslauriers, R. (1997). Na(+)-H+ exchange inhibition at reperfusion is cardioprotective during myocardial ischemia-reperfusion; 31P NMR studies. Mol. Cell. Biochem. 176, 257–264. doi: 10.1023/A:1006861414737
Endo, H., Nito, C., Kamada, H., Nishi, T., and Chan, P. H. (2006). Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 26, 1479–1489. doi: 10.1038/sj.jcbfm.9600303
Formisano, L., Saggese, M., Secondo, A., Sirabella, R., Vito, P., Valsecchi, V., et al. (2008). The two isoforms of the Na+/Ca2+ exchanger, NCX1 and NCX3, constitute novel additional targets for the prosurvival action of Akt/protein kinase B pathway. Mol. Pharmacol. 73, 727–737. doi: 10.1124/mol.107.042549
Fujioka, S., Kitaura, Y., Ukimura, A., Deguchi, H., Kawamura, K., Isomura, T., et al. (2000). Evaluation of viral infection in the myocardium of patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 36, 1920–1926. doi: 10.1016/S0735-1097(00)00955-4
Fujita, M., Asanuma, H., Hirata, A., Wakeno, M., Takahama, H., Sasaki, H., et al. (2007). Prolonged transient acidosis during early reperfusion contributes to the cardioprotective effects of postconditioning. Am. J. Physiol. Heart Circ. Physiol. 292, H2004–H2008. doi: 10.1152/ajpheart.01051.2006
Gabellini, N., Bortoluzzi, S., Danieli, G. A., and Carafoli, E. (2003). Control of the Na+/Ca2+ exchanger 3 promoter by cyclic adenosine monophosphate and Ca2+ in differentiating neurons. J. Neurochem. 84, 282–293. doi: 10.1046/j.1471-4159.2003.01511.x
Gao, J., Duan, B., Wang, D. G., Deng, X. H., Zhang, G. Y., Xu, L., et al. (2005). Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron 48, 635–646. doi: 10.1016/j.neuron.2005.10.011
Gao, X., Zhang, H., Takahashi, T., Hsieh, J., Liao, J., Steinberg, G. K., et al. (2008). The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 105, 943–955. doi: 10.1111/j.1471-4159.2008.05218.x
Gidday, J. M. (2006). Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448. doi: 10.1038/nrn1927
Goldberg, S. P., Digerness, S. B., Skinner, J. L., Killingsworth, C. R., Katholi, C. R., and Holman, W. L. (2002). Ischemic preconditioning and Na+/H+ exchange inhibition improve reperfusion ion homeostasis. Ann. Thorac. Surg. 73, 569–574. doi: 10.1016/S0003-4975(01)03309-4
Haddad, G. G., and Jiang, C. (1997). O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annu. Rev. Physiol. 59, 23–42. doi: 10.1146/annurev.physiol.59.1.23
Hwang, I. K., Yoo, K. Y., An, S. J., Li, H., Lee, C. H., Choi, J. H., et al. (2008). Late expression of Na+/H+ exchanger 1 (NHE1) and neuroprotective effects of NHE inhibitor in the gerbil hippocampal CA1 region induced by transient ischemia. Exp. Neurol. 212, 314–323. doi: 10.1016/j.expneurol.2008.04.007
Inserte, J., Barba, I., Hernando, V., Abellán, A., Ruiz-Meana, M., Rodríguez-Sinovas, A., et al. (2008). Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc. Res. 77, 782–790. doi: 10.1093/cvr/cvm082
Inserte, J., Barba, I., Hernando, V., and Garcia-Dorado, D. (2009). Delayed recovery of intracellular acidosis during reperfusion prevents calpain activation and determines protection in postconditioned myocardium. Cardiovasc. Res. 81, 116–122. doi: 10.1093/cvr/cvn260
Jensen, P. E., Mulvany, M. J., Aalkjaer, C., Nilsson, H., and Yamaguchi, H. (1993). Free cytosolic Ca2+ measured with Ca(2+)-selective electrodes and fura 2 in rat mesenteric resistance arteries. Am. J. Physiol. 265(2 Pt 2), H741–H746.
Jiang, X., Shi, E., Nakajima, Y., and Sato, S. (2006). Postconditioning, a series of brief interruptions of early reperfusion, prevents neurologic injury after spinal cord ischemia. Ann. Surg. 244, 148–153. doi: 10.1097/01.sla.0000217608.08582.35
Kang, T. M., and Hilgemann, D. W. (2004). Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature 427, 544–548. doi: 10.1038/nature02271
Kato, K., Shimazaki, K., Kamiya, T., Amemiya, S., Inaba, T., Oguro, K., et al. (2005). Differential effects of sublethal ischemia and chemical preconditioning with 3-nitropropionic acid on protein expression in gerbil hippocampus. Life Sci. 77, 2867–2878. doi: 10.1016/j.lfs.2005.01.037
Kawai, K., Nakagomi, T., Kirino, T., Tamura, A., and Kawai, N. (1998). Preconditioning in vivo ischemia inhibits anoxic long-term potentiation and functionally protects CA1 neurons in the gerbil. J. Cereb. Blood Flow Metab. 18, 288–296. doi: 10.1097/00004647-199803000-00007
Kersh, A. E., Hartzler, L. K., Havlin, K., Hubbell, B. B., Nanagas, V., Kalra, A., et al. (2009). pH regulating transporters in neurons from various chemosensitive brainstem regions in neonatal rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1409–R1420. doi: 10.1152/ajpregu.91038.2008
Kintner, D. B., Su, G., Lenart, B., Ballard, A. J., Meyer, J. W., Ng, L. L., et al. (2004). Increased tolerance to oxygen and glucose deprivation in astrocytes from Na(+)/H(+) exchanger isoform 1 null mice. Am. J. Physiol. Cell Physiol. 287, C12–C21. doi: 10.1152/ajpcell.00560.2003
Kirino, T. (2002). Ischemic tolerance. J. Cereb. Blood Flow Metab. 22, 1283–1296. doi: 10.1097/00004647-200211000-00001
Kitakaze, M., Weisfeldt, M. L., and Marban, E. (1988). Acidosis during early reperfusion prevents myocardial stunning in perfused ferret hearts. J. Clin. Invest. 82, 920–927. doi: 10.1172/JCI113699
Klein, M. (1993). Differential cyclic AMP dependence of facilitation at Aplysia sensorimotor synapses as a function of prior stimulation: augmentation versus restoration of transmitter release. J. Neurosci. 13, 3793–3801.
Kuroda, S., and Siesjö, B. K. (1997). Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clin. Neurosci. 4, 199–212.
Lehotský, J., Burda, J., Danielisová, V., Gottlieb, M., Kaplán, P., and Saniová, B. (2009). Ischemic tolerance: the mechanisms of neuroprotective strategy. Anat. Rec. (Hoboken) 292, 2002–2012. doi: 10.1002/ar.20970
Lenart, B., Kintner, D. B., Shull, G. E., and Sun, D. (2004). Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J. Neurosci. 24, 9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004
Linck, B., Qiu, Z., He, Z., Tong, Q., Hilgemann, D. W., and Philipson, K. D. (1998). Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3). Am. J. Physiol. 274(2 Pt 1), C415–C423.
Luo, J., Chen, H., Kintner, D. B., Shull, G. E., and Sun, D. (2005). Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J. Neurosci. 25, 11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005
Lytle, C., and Forbush, B. III. (1992). The Na-K-Cl cotransport protein of shark rectal gland. II. Regulation by direct phosphorylation. J. Biol. Chem. 267, 25438–25443.
Matsumoto, Y., Yamamoto, S., Suzuki, Y., Tsuboi, T., Terakawa, S., Ohashi, N., et al. (2004). Na+/H+ exchanger inhibitor, SM-20220, is protective against excitotoxicity in cultured cortical neurons. Stroke 35, 185–190. doi: 10.1161/01.STR.0000106910.42815.C2
Meller, R., Cameron, J. A., Torrey, D. J., Clayton, C. E., Ordonez, A. N., Henshall, D. C., et al. (2006). Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J. Biol. Chem. 281, 7429–7436. doi: 10.1074/jbc.M512138200
Meller, R., Minami, M., Cameron, J. A., Impey, S., Chen, D., Lan, J. Q., et al. (2005). CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J. Cereb. Blood Flow Metab. 25, 234–246. doi: 10.1038/sj.jcbfm.9600024
Miao, Y., Zhang, W., Lin, Y., Lu, X., and Qiu, Y. (2010). Neuroprotective effects of ischemic preconditioning on global brain ischemia through up-regulation of acid-sensing ion channel 2a. Int. J. Mol. Sci. 11, 140–153. doi: 10.3390/ijms11010140
Molinaro, P., Cantile, M., Cuomo, O., Secondo, A., Pannaccione, A., Ambrosino, P., et al. (2013). Neurounina-1, a novel compound that increases Na+/Ca2+ exchanger activity, effectively protects against stroke damage. Mol. Pharmacol. 83, 142–156. doi: 10.1124/mol.112.080986
Murry, C. E., Jennings, R. B., and Reimer, K. A. (1986). Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. doi: 10.1161/01.CIR.74.5.1124
Nedergaard, M., Kraig, R. P., Tanabe, J., and Pulsinelli, W. A. (1991). Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 260(3 Pt 2), R581–R588.
Nicoll, D. A., Longoni, S., and Philipson, K. D. (1990). Molecular cloning and functional expression of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. Science 250, 562–565. doi: 10.1126/science.1700476
Nicoll, D. A., Quednau, B. D., Qui, Z., Xia, Y. R., Lusis, A. J., and Philipson, K. D. (1996). Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J. Biol. Chem. 271, 24914–24921. doi: 10.1074/jbc.271.40.24914
Obrenovitch, T. P. (2008). Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol. Rev. 88, 211–247. doi: 10.1152/physrev.00039.2006
O'Donnell, M. E. (1993). Role of Na-K-Cl cotransport in vascular endothelial cell volume regulation. Am. J. Physiol. 264(5 Pt 1), C1316–C1326.
Ohashi, T., Yamamoto, F., Yamamoto, H., Ichikawa, H., Shibata, T., and Kawashima, Y. (1996). Transient reperfusion with acidic solution affects postischemic functional recovery: studies in the isolated working rat heart. J. Thorac. Cardiovasc. Surg. 111, 613–620. doi: 10.1016/S0022-5223(96)70313-6
Ohta, S., Furuta, S., Matsubara, I., Kohno, K., Kumon, Y., and Sakaki, S. (1996). Calcium movement in ischemia-tolerant hippocampal CA1 neurons after transient forebrain ischemia in gerbils. J. Cereb. Blood Flow Metab. 16, 915–922. doi: 10.1097/00004647-199609000-00015
Pateliya, B. B., Singh, N., and Jaggi, A. S. (2008). Possible role of opioids and KATP channels in neuroprotective effect of postconditioning in mice. Biol. Pharm. Bull. 31, 1755–1760. doi: 10.1248/bpb.31.1755
Pedersen, S. F. (2006). The Na+/H+ exchanger NHE1 in stress-induced signal transduction: implications for cell proliferation and cell death. Pflugers Arch. 452, 249–259. doi: 10.1007/s00424-006-0044-y
Perez-Pinzon, M. A., Xu, G. P., Dietrich, W. D., Rosenthal, M., and Sick, T. J. (1997). Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J. Cereb. Blood Flow Metab. 17, 175–182. doi: 10.1097/00004647-199702000-00007
Pewitt, E. B., Hegde, R. S., Haas, M., and Palfrey, H. C. (1990). The regulation of Na/K/2Cl cotransport and bumetanide binding in avian erythrocytes by protein phosphorylation and dephosphorylation. Effects of kinase inhibitors and okadaic acid. J. Biol. Chem. 265, 20747–20756.
Pignataro, G., Boscia, F., Esposito, E., Sirabella, R., Cuomo, O., Vinciguerra, A., et al. (2012). NCX1 and NCX3: two new effectors of delayed preconditioning in brain ischemia. Neurobiol. Dis. 45, 616–623. doi: 10.1016/j.nbd.2011.10.007
Pignataro, G., Cuomo, O., Esposito, E., Sirabella, R., Di Renzo, G., and Annunziato, L. (2011a). ASIC1a contributes to neuroprotection elicited by ischemic preconditioning and postconditioning. Int. J. Physiol. Pathophysiol. Pharmacol. 3, 1–8. Available online at: http://www.ijppp.org/files/IJPPP1008003.pdf
Pignataro, G., Cuomo, O., Vinciguerra, A., Sirabella, R., Esposito, E., Boscia, F., et al. (2013a). NCX as a key player in the neuroprotection exerted by ischemic preconditioning and postconditioning. Adv. Exp. Med. Biol. 961, 223–240. doi: 10.1007/978-1-4614-4756-6_19
Pignataro, G., Esposito, E., Cuomo, O., Sirabella, R., Boscia, F., Guida, N., et al. (2011b). The NCX3 isoform of the Na+/Ca2+ exchanger contributes to neuroprotection elicited by ischemic postconditioning. J. Cereb. Blood Flow Metab. 31, 362–370. doi: 10.1038/jcbfm.2010.100
Pignataro, G., Esposito, E., Sirabella, R., Vinciguerra, A., Cuomo, O., Di Renzo, G., et al. (2013b). nNOS and p-ERK involvement in the neuroprotection exerted by remote postconditioning in rats subjected to transient middle cerebral artery occlusion. Neurobiol. Dis. 54, 105–114. doi: 10.1016/j.nbd.2013.02.008
Pignataro, G., Gala, R., Cuomo, O., Tortiglione, A., Giaccio, L., Castaldo, P., et al. (2004). Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke 35, 2566–2570. doi: 10.1161/01.STR.0000143730.29964.93
Pignataro, G., Meller, R., Inoue, K., Ordonez, A. N., Ashley, M. D., Xiong, Z., et al. (2008). In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J. Cereb. Blood Flow Metab. 28, 232–241. doi: 10.1038/sj.jcbfm.9600559
Pignataro, G., Scorziello, A., Di Renzo, G., and Annunziato, L. (2009). Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J. 276, 46–57. doi: 10.1111/j.1742-4658.2008.06769.x
Pignataro, G., Simon, R. P., and Xiong, Z. G. (2007). Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 130(Pt 1), 151–158. doi: 10.1093/brain/awl325
Pignataro, G., Sirabella, R., Anzilotti, S., Di Renzo, G., and Annunziato, L. (2014). Does Na(+)/Ca(2)(+) exchanger, NCX, represent a new druggable target in stroke intervention? Transl. Stroke Res. 5, 145–155. doi: 10.1007/s12975-013-0308-8
Piwnica-Worms, D., Jacob, R., Shigeto, N., Horres, C. R., and Lieberman, M. (1986). Na/H exchange in cultured chick heart cells: secondary stimulation of electrogenic transport during recovery from intracellular acidosis. J. Mol. Cell. Cardiol. 18, 1109–1116. doi: 10.1016/S0022-2828(86)80036-0
Preckel, B., Schlack, W., Obal, D., Barthel, H., Ebel, D., Grunert, S., et al. (1998). Effect of acidotic blood reperfusion on reperfusion injury after coronary artery occlusion in the dog heart. J. Cardiovasc. Pharmacol. 31, 179–186. doi: 10.1097/00005344-199802000-00002
Racay, P., Tatarkova, Z., Drgova, A., Kaplan, P., and Dobrota, D. (2007). Effect of ischemic preconditioning on mitochondrial dysfunction and mitochondrial p53 translocation after transient global cerebral ischemia in rats. Neurochem. Res. 32, 1823–1832. doi: 10.1007/s11064-007-9437-3
Ramasamy, R., Liu, H., Anderson, S., Lundmark, J., and Schaefer, S. (1995). Ischemic preconditioning stimulates sodium and proton transport in isolated rat hearts. J. Clin. Invest. 96, 1464–1472. doi: 10.1172/JCI118183
Ratan, R. R., Siddiq, A., Aminova, L., Lange, P. S., Langley, B., Ayoub, I., et al. (2004). Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke 35(11 Suppl. 1), 2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e
Reeves, J. P., and Hale, C. C. (1984). The stoichiometry of the cardiac sodium-calcium exchange system. J. Biol. Chem. 259, 7733–7739.
Rehncrona, S. (1985). Brain acidosis. Ann. Emerg. Med. 14, 770–776. doi: 10.1016/S0196-0644(85)80055-X
Scartabelli, T., Gerace, E., Landucci, E., Moroni, F., and Pellegrini-Giampietro, D. E. (2008). Neuroprotection by group I mGlu receptors in a rat hippocampal slice model of cerebral ischemia is associated with the PI3K-Akt signaling pathway: a novel postconditioning strategy? Neuropharmacology 55, 509–516. doi: 10.1016/j.neuropharm.2008.06.019
Schaefer, S., Carr, L. J., Prussel, E., and Ramasamy, R. (1995). Effects of glycogen depletion on ischemic injury in isolated rat hearts: insights into preconditioning. Am. J. Physiol. 268(3 Pt 2), H935–H944.
Schäfer, C., Ladilov, Y. V., Siegmund, B., and Piper, H. M. (2000). Importance of bicarbonate transport for protection of cardiomyocytes against reoxygenation injury. Am. J. Physiol. Heart Circ. Physiol. 278, H1457–H1463.
Schaller, B., and Graf, R. (2004). Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J. Cereb. Blood Flow Metab. 24, 351–371. doi: 10.1097/00004647-200404000-00001
Schurr, A., Reid, K. H., Tseng, M. T., West, C., and Rigor, B. M. (1986). Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res. 374, 244–248. doi: 10.1016/0006-8993(86)90418-X
Secondo, A., Staiano, R. I., Scorziello, A., Sirabella, R., Boscia, F., Adornetto, A., et al. (2007). BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: possible relationship with mitochondrial membrane potential. Cell Calcium 42, 521–535. doi: 10.1016/j.ceca.2007.01.006
Shimazaki, K., Nakamura, T., Nakamura, K., Oguro, K., Masuzawa, T., Kudo, Y., et al. (1998). Reduced calcium elevation in hippocampal CA1 neurons of ischemia-tolerant gerbils. Neuroreport 9, 1875–1878. doi: 10.1097/00001756-199806010-00038
Shimazaki, K., Urabe, M., Monahan, J., Ozawa, K., and Kawai, N. (2000). Adeno-associated virus vector-mediated bcl-2 gene transfer into post-ischemic gerbil brain in vivo: prospects for gene therapy of ischemia-induced neuronal death. Gene Ther. 7, 1244–1249. doi: 10.1038/sj.gt.3301211
Shipolini, A. R., Yokoyama, H., Galinanes, M., Edmondson, S. J., Hearse, D. J., and Avkiran, M. (1997). Na+/H+ exchanger activity does not contribute to protection by ischemic preconditioning in the isolated rat heart. Circulation 96, 3617–3625. doi: 10.1161/01.CIR.96.10.3617
Stenzel-Poore, M. P., Stevens, S. L., Xiong, Z., Lessov, N. S., Harrington, C. A., Mori, M., et al. (2003). Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362, 1028–1037. doi: 10.1016/S0140-6736(03)14412-1
Sutherland, S. P., Cook, S. P., and McCleskey, E. W. (2000). Chemical mediators of pain due to tissue damage and ischemia. Prog. Brain Res. 129, 21–38. doi: 10.1016/S0079-6123(00)29003-1
Ten Hove, M., and Van Echteld, C. J. (2004). Limited effects of post-ischemic NHE blockade on [Na+]i and pHi in rat hearts explain its lack of cardioprotection. Cardiovasc. Res. 61, 522–529. doi: 10.1016/j.cardiores.2003.07.005
Valsecchi, V., Pignataro, G., Del Prete, A., Sirabella, R., Matrone, C., Boscia, F., et al. (2011). NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke 42, 754–763. doi: 10.1161/STROKEAHA.110.597583
Vandenberg, J. I., Metcalfe, J. C., and Grace, A. A. (1993). Mechanisms of pHi recovery after global ischemia in the perfused heart. Circ. Res. 72, 993–1003. doi: 10.1161/01.RES.72.5.993
Vornov, J. J., Thomas, A. G., and Jo, D. (1996). Protective effects of extracellular acidosis and blockade of sodium/hydrogen ion exchange during recovery from metabolic inhibition in neuronal tissue culture. J. Neurochem. 67, 2379–2389. doi: 10.1046/j.1471-4159.1996.67062379.x
Vukicevic, M., and Kellenberger, S. (2004). Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am. J. Physiol. Cell Physiol. 287, C682–C690. doi: 10.1152/ajpcell.00127.2004
Waldmann, R., Champigny, G., Bassilana, F., Heurteaux, C., and Lazdunski, M. (1997). A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177. doi: 10.1038/386173a0
Wang, J. Y., Shen, J., Gao, Q., Ye, Z. G., Yang, S. Y., Liang, H. W., et al. (2008). Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke 39, 983–990. doi: 10.1161/STROKEAHA.107.499079
Wemmie, J. A., Price, M. P., and Welsh, M. J. (2006). Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 29, 578–586. doi: 10.1016/j.tins.2006.06.014
Xia, Y., Zhao, P., Xue, J., Gu, X. Q., Sun, X., Yao, H., et al. (2003). Na+ channel expression and neuronal function in the Na+/H+ exchanger 1 null mutant mouse. J. Neurophysiol. 89, 229–236. doi: 10.1152/jn.00488.2002
Xing, B., Chen, H., Zhang, M., Zhao, D., Jiang, R., Liu, X., et al. (2008). Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke 39, 2362–2369. doi: 10.1161/STROKEAHA.107.507939
Xu, T. L., and Xiong, Z. G. (2007). Dynamic regulation of acid-sensing ion channels by extracellular and intracellular modulators. Curr. Med. Chem. 14, 1753–1763. doi: 10.2174/092986707781058977
Yang, L., Cai, X., Zhou, J., Chen, S., Chen, Y., Chen, Z., et al. (2013). STE20/SPS1-related proline/alanine-rich kinase is involved in plasticity of GABA signaling function in a mouse model of acquired epilepsy. PLoS ONE 8:e74614. doi: 10.1371/journal.pone.0074614
Zhao, H. (2009). Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J. Cereb. Blood Flow Metab. 29, 873–885. doi: 10.1038/jcbfm.2009.13
Zhao, H., Ren, C., Chen, X., and Shen, J. (2012). From rapid to delayed and remote postconditioning: the evolving concept of ischemic postconditioning in brain ischemia. Curr. Drug Targets 13, 173–187. doi: 10.2174/138945012799201621
Zhao, H., Sapolsky, R. M., and Steinberg, G. K. (2006). Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J. Cereb. Blood Flow Metab. 26, 1114–1121. doi: 10.1038/sj.jcbfm.9600348
Zhao, Z. Q., Corvera, J. S., Halkos, M. E., Kerendi, F., Wang, N. P., Guyton, R. A., et al. (2003). Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 285, H579–H588. doi: 10.1152/ajpheart.01064.2002
Keywords: transporters, ionic exchangers, stroke, preconditioning/post-conditioning, brain ischemia
Citation: Cuomo O, Vinciguerra A, Cerullo P, Anzilotti S, Brancaccio P, Bilo L, Scorziello A, Molinaro P, Di Renzo G and Pignataro G (2015) Ionic homeostasis in brain conditioning. Front. Neurosci. 9:277. doi: 10.3389/fnins.2015.00277
Received: 26 March 2015; Accepted: 23 July 2015;
Published: 10 August 2015.
Edited by:
Mark P. Burns, Georgetown University Medical Center, USAReviewed by:
Zhihui Yang, University of Florida, USAPatrick A. Forcelli, Georgetown University, USA
Copyright © 2015 Cuomo, Vinciguerra, Cerullo, Anzilotti, Brancaccio, Bilo, Scorziello, Molinaro, Di Renzo and Pignataro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Pignataro, Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, “Federico II” University of Naples, Via Pansini 5, 80131 Naples, Italy,Z3BpZ25hdGFAdW5pbmEuaXQ=