 Gunjan Joshi
Gunjan Joshi Michael E. Bekier II
Michael E. Bekier II Yanzhuang Wang
Yanzhuang Wang- 1Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI, USA
- 2Department of Neurology, University of Michigan School of Medicine, Ann Arbor, MI, USA
The Golgi apparatus is an essential cellular organelle for post-translational modifications, sorting, and trafficking of membrane and secretory proteins. Proper functionality of the Golgi requires the formation of its unique cisternal-stacking morphology. The Golgi structure is disrupted in a variety of neurodegenerative diseases, suggesting a common mechanism and contribution of Golgi defects in neurodegenerative disorders. A recent study on Alzheimer's disease (AD) revealed that phosphorylation of the Golgi stacking protein GRASP65 disrupts its function in Golgi structure formation, resulting in Golgi fragmentation. Inhibiting GRASP65 phosphorylation restores the Golgi morphology from Aβ-induced fragmentation and reduces Aβ production. Perturbing Golgi structure and function in neurons may directly impact trafficking, processing, and sorting of a variety of proteins essential for synaptic and dendritic integrity. Therefore, Golgi defects may ultimately promote the development of AD. In the current review, we focus on the cellular impact of impaired Golgi morphology and its potential relationship to AD disease development.
Introduction
The Golgi apparatus is a membranous cellular organelle that mediates proper trafficking, post-translational processing, and sorting of membrane and secretory proteins. The Golgi is often localized to the perinuclear region of the cell, which depends on an intact microtubule network and a motor protein dynein. Golgi membranes form multilayer stacks that are laterally linked into a ribbon. Formation of stable multilayer stacks and ribbons appears to be essential for proper functioning of the Golgi (Rambourg et al., 1987; Ladinsky et al., 1999; Klumperman, 2011; Klute et al., 2011). The Golgi structure is controlled by Golgi matrix proteins, a group of peripheral and membrane proteins localized on the cytoplasmic surface of Golgi membranes, including the coiled-coil domain-containing golgins and the stacking proteins GRASP55/65 (Ramirez and Lowe, 2009; Xiang and Wang, 2011; Wong and Munro, 2014). Depletion of Golgi matrix proteins such as GRASP65 (Xiang et al., 2013; Veenendaal et al., 2014), GRASP55 (Feinstein and Linstedt, 2008; Xiang and Wang, 2010), GM130 (Puthenveedu et al., 2006), golgin-84 (Diao et al., 2003), and golgin-160 (Williams et al., 2006), or treatment of cells with pharmacological drugs such as Brefeldin A (BFA) (Klausner et al., 1992), results in an abnormal, fragmented Golgi morphology. Disruption of normal Golgi morphology directly impacts protein trafficking and processing. For example, breaking down the Golgi ribbon of mammalian cells by nocodazole treatment strongly inhibits intra-Golgi transport of large cargoes without altering the rate of transport of smaller cargoes (Lavieu et al., 2014), while inhibition of stack formation by knocking down GRASP55/65 by RNA interference (RNAi) accelerates protein trafficking, impairs proper glycosylation, and leads to missorting and delivery of lysosomal proteins to the extracellular space (Xiang et al., 2013). Thus, the structure of the Golgi is closely linked to vital cellular processes.
Golgi fragmentation has been observed in neurodegenerative diseases, including Alzheimer's (AD) (Stieber et al., 1996; Huse et al., 2002), Parkinson's (PD) (Mizuno et al., 2001), and Huntington's (HD) (Hilditch-Maguire et al., 2000) diseases and amyotrophic lateral sclerosis (ALS) (Mourelatos et al., 1996; Gonatas et al., 1998; Fujita and Okamoto, 2005). However, the molecular basis of Golgi fragmentation and its role in disease progression remain largely unexplored. Golgi defects may impact the trafficking and processing of many proteins essential for neuronal functions. Thus, morphological and subsequent functional impairments of the Golgi may contribute to the neurotoxicity associated with neurodegenerative diseases. Understanding the mechanisms that cause Golgi fragmentation and its downstream effects on neuronal functions are likely important for understanding the molecular basis of the diseases. Furthermore, targeting Golgi structural defects may represent a novel approach to treating or preventing related diseases with Golgi defects. In this review, we summarize the current literature on the cause and effect of Golgi fragmentation in AD, the leading cause of dementia in adults (Alzheimer's, 2015).
Golgi and Amyloid Plaque Formation
One neuropathological hallmark of AD is the formation of extracellular amyloid plaques by secreted amyloid beta (Aβ) peptides (Nelson et al., 2009), which is highly related to Golgi structure and function (Figure 1). Aβ is derived from the amyloid precursor protein (APP), a type I membrane protein that travels through the exocytic and endocytic pathways and undergoes sequential proteolysis by the action of β- and γ-secretases (Vassar et al., 1999). In neurons, APP is transported from the Golgi to many sub-cellular compartments (Haass et al., 1992), including the soma, dendrites and axons, through the exocytic and endocytic pathways. Despite the abundant literature demonstrating the critical role of endosomes in APP processing (for review see Suh and Checler, 2002; Small and Gandy, 2006), it has been indicated that the Golgi (in particular the trans-Golgi network) may be a site where Aβ is generated in the cell (Greenfield et al., 1999; Burgos et al., 2010; Choy et al., 2012). In addition, proper functioning of the Golgi is required for trafficking and maturation of both APP and its processing enzymes. For instance, the activity of the γ-secretase depends on the trafficking and maturation of nicastrin (Chung and Struhl, 2001) and other components of the γ-secretase complex through the Golgi (Herreman et al., 2003). Nicastrin is not catalytically active, but is important for the maturation and proper trafficking of the γ-secretase complex (Zhang et al., 2005). Nicastrin functions to stabilize presenilins (PSs), the catalytic subunit of the γ-secretase complex, and mediates PS trafficking to the cell surface by an unknown mechanism (Edbauer et al., 2002; Hu et al., 2002). Nicastrin also binds to the N-terminal domain of APP, and facilitates APP trafficking and cleavage (Yu et al., 2000; Kimberly et al., 2002). APP travels from the endoplasmic reticulum (ER) through the Golgi to the plasma membrane. The majority of APP localizes to the Golgi where it undergoes post-translational modifications and only a small fraction resides in the ER and the plasma membrane (Thinakaran and Koo, 2008). Finally, the α-secretatse ADAM10 is transported from dendritic Golgi outposts to synaptic membranes, a reaction modulated by the synapse-associated protein-97 (SAP97) (Saraceno et al., 2014). Thus, APP processing and Aβ production are intrinsically linked to the proper morphology and functionality of the Golgi.
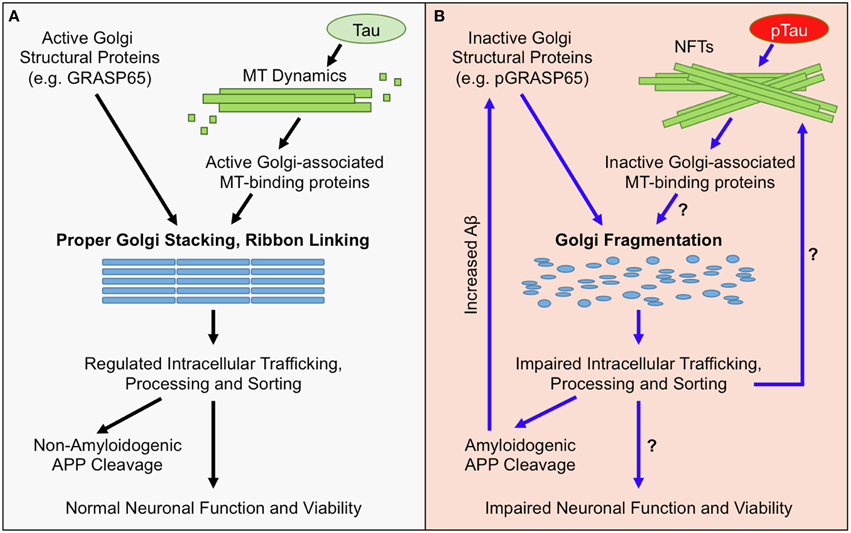
Figure 1. Golgi Morphological and Functional Defects in AD. (A) Under normal conditions, the structure of the Golgi is maintained by active Golgi structural proteins such as GRASP65 (non-phosphorylated) and an intact microtubule (MT) network. Maintaining the Golgi structure is essential for proper trafficking and processing of APP and its processing enzymes. The majority of APP undergoes non-amyloidogenic processing, and cell-surface proteins, lipids, and polysaccharides, which are essential for neuronal function, are properly sorted and transported. Together, these factors maintain neuronal functionality and viability. (B) In AD, the Golgi is fragmented due to inactivation of Golgi structural proteins, such as degradation or phosphorylation of GRASP65 (pGRASP65), or tau hyper phosphorylation (pTau) and NFT formation that disrupt MT dynamics and protein trafficking. Golgi fragmentation impairs trafficking, processing, and sorting of APP and APP-processing enzymes, which stimulates amyloidogenic APP cleavage and further inactivates GRASP65. Additionally, Golgi fragmentation is predicted to alter trafficking, processing, and sorting of proteins, lipids, and polysaccharides that are essential for neuronal function; which could ultimately promote neuronal dysfunction and/or cell death.
Golgi and Tau Pathology in AD
Another pathological distinction in AD is the formation of neurofibrillary tangles (NFTs) caused by precipitation of hyperphosphorylated tau protein, which is also related to Golgi structure and function (Figure 1) (Morishima-Kawashima and Ihara, 2002). Tau is an abundant protein that binds and stabilizes microtubules in neurons (Gong and Alonso Adel, 2013). Neurons have long protrusions such as axons; transport of membrane organelles and proteins into and from these protrusions highly depends on an intact microtubule network (Baas, 2002). Long microtubules emanate out of the centrosomes and function as the track for rapid transport of membranes by kinesin and dynein motors (Hancock, 2014). Membranes and proteins for synaptic vesicle formation and synaptic function are essentially all transported in this way (Gallant, 2000; Goldstein and Yang, 2000).
In AD, hyperphosphorylation of tau results in NFT formation, which impairs microtubule integrity and blocks membrane transport (Patrick et al., 1999; Avila, 2006; Baloyannis, 2014). Interestingly, the Golgi, whose formation relies on an intact microtubule network (Rios and Bornens, 2003), may also function as an important microtubule organization center (Lewis and Polleux, 2012; Ori-McKenney et al., 2012; Zhu and Kaverina, 2013). Multiple microtubule-binding proteins, such as γ-tubulin (Radulescu et al., 2011), CLASP (Liu et al., 2007; Miller et al., 2009), GMAP-210 (Infante et al., 1999; Roboti et al., 2015), and GM130 (Rivero et al., 2009), are localized on the Golgi membranes and modulate microtubule network formation and membrane trafficking in neurons (Rios, 2014; Tang et al., 2015) and astrocytes (Yoshiyama et al., 2003). It has been reported that tau interacts with Golgi membranes and mediates their association with microtubules (Farah et al., 2006). Overexpression of wild-type and mutant human tau proteins causes Golgi fragmentation in primary hippocampal neurons (Liazoghli et al., 2005), implying that overabundance of tau, and potentially NFT formation, precedes Golgi morphological defects. Additionally, Golgi fragmentation may affect tau phosphorylation (Jiang et al., 2014) and promote NFTs formation in AD (Grundke-Iqbal et al., 1986). Therefore, there is a close connection between Golgi morphology and function, microtubule organization, and tau pathology in AD.
Golgi Morphological Defects in AD
Golgi morphological alterations have been observed in neurons of AD patients (Dal Canto, 1996; Stieber et al., 1996; Gonatas et al., 1998; Huse et al., 2002), which occur even before the formation of NFTs and neuritic plaques (Baloyannis, 2014). Golgi fragmentation has been recently confirmed in AD tissue culture and mouse models (Joshi et al., 2014). Expression of the Swedish mutant APP (APPswe) and the exon 9-deletion mutant PS1 (PS1ΔE9) in cells and mouse brain, or treatment of neurons with synthetic Aβ peptides, resulted in Golgi fragmentation. Under fluorescence microscopy, Golgi elements are unlinked and dispersed into the cytoplasm. Under electron microscopy (EM), the disconnected Golgi stacks exhibit reduced number of cisternae per stack, shorter cisternae, more vesicles surrounding each stack and a dilated Golgi structure compared with wild type cells and untreated neurons (Joshi et al., 2014). In this study, the direct cause of Golgi fragmentation is Aβ accumulation, as Aβ treatment causes Golgi fragmentation in cultured neurons and other cell types; this effect is reversible upon removal of Aβ from the tissue culture medium (Joshi et al., 2014). Golgi fragmentation has also been observed under physiological conditions, such as during migration, upon growth factor treatment, and in neurons with increased neuronal activity (Bisel et al., 2008; Thayer et al., 2013), suggesting that Golgi fragmentation may not be an immediate pathological response, but rather a compensatory reaction to allow fast transport of proteins to their final destinations when the cells are under stress. Golgi fragmentation, however, impacts protein glycosylation and sorting (Wang et al., 2008; Xiang et al., 2013), as we discuss below.
Mechanisms of Golgi Morphological Defects in AD
The formation and maintenance of the perinuclear localization of the Golgi ribbon rely on an intact microtubule cytoskeleton emanating from the perinuclear centrosomes (Glick and Nakano, 2009; Yadav and Linstedt, 2011; Rios, 2014). The minus end-directed motor dynein associates with the Golgi and moves the membranes along the microtubules toward the centrosomes, leading to the concentration of the Golgi stacks in the pericentriolar region and formation of the Golgi ribbon (Rios and Bornens, 2003; Miller et al., 2009). The basic structural and functional unit of the Golgi is a stack of flattened cisternae. The exact mechanism for Golgi stack formation is not fully understood, but is believed to rely on Golgi structural proteins (Wang and Seemann, 2011; Xiang and Wang, 2011). Among the well characterized Golgi structural proteins, GRASP65 (Wang et al., 2003) and GRASP55 (Xiang and Wang, 2010) play essential roles in Golgi structure formation, whereas others, including GM130 (Lowe et al., 1998), golgin-160 (Hicks and Machamer, 2002), and p115 (Chiu et al., 2002), are more important for trafficking across the Golgi stack (Xiang and Wang, 2011). For example, GRASP65 forms oligomers that tether the cisternae into stacks (Wang et al., 2003; Tang et al., 2010; Lavieu et al., 2013) and ribbons (Puthenveedu et al., 2006). Recently a GRASP65 knockout mouse has been reported, with defects in cis-Golgi ribbon-linking and no apparent neurological phenotype (Veenendaal et al., 2014). One potential concern regarding this mouse strain is that some mRNA encoding exon 1–3 is still present (~20% of the mRNA level in wild-type mice). This truncated mRNA encodes a 115 aa N-terminal fragment of GRASP65. If translated (although not detected by available antibodies raised against the full length protein), this fragment would be sufficient for Golgi stacking, according to biochemical and structural studies (Tang et al., 2010; Truschel et al., 2011). However, a more recent structure (Feng et al., 2013) indicates that a longer fragment is needed for GRASP65 function in Golgi stack formation (Wang et al., 2003, 2005). The lack of a more prominent Golgi stacking and neurological phenotype may also be due to the complementation by GRASP55, the homolog of GRASP65 that shares some redundancy in stacking (Shorter et al., 1999; Lee et al., 2014).
The mechanism of Golgi fragmentation in AD has not been well studied, but likely involves multiple mechanisms (Figure 1). One possibility is through the disruption of the microtubule network by tau precipitation and NFT formation (Liazoghli et al., 2005). Microtubule defects may affect both the central localization of the Golgi in the cell and ER-Golgi-plasma membrane trafficking that indirectly impacts the size and morphology of the Golgi (Fokin et al., 2014). Tau could also affect vesicle trafficking by inhibiting the binding of motor proteins such as kinesins to microtubules (Seitz et al., 2002). Another possibility is through modulation of Golgi structural proteins (Figure 2). Both mitotic phosphorylation and apoptotic cleavage of Golgi proteins results in Golgi fragmentation (Wang and Seemann, 2011). For instance, GRASP65 is phosphorylated by mitotic kinases Cdk1 and Polo-like kinase (Plk1) during mitosis (Wang et al., 2003) and cleaved by caspase-3 in apoptosis (Lane et al., 2002), both of which cause Golgi fragmentation (Tang et al., 2008; Wang and Seemann, 2011). In tissue culture and mouse models of AD, GRASP65 phosphorylation was implicated as a major cause of Golgi fragmentation (Joshi et al., 2014; Joshi and Wang, 2015). At the molecular level, Aβ accumulation triggers Ca2+ influx (Zempel et al., 2010), which activates Calpain, a protease known to increase the cleavage of p35 to p25 (Lee et al., 2000), p25 then activates Cdk5. It has been previously reported that p35 and Cdk5 are associated with Golgi membranes and regulate membrane traffic (Paglini et al., 2001). Subsequently, Cdk5 (also known to phosphorylate tau in AD) phosphorylates GRASP65, which negatively regulates GRASP65, leading to Golgi fragmentation. Consequently, Golgi fragmentation enhances APP trafficking and increases Aβ production (Joshi et al., 2014). Fragmentation of the Golgi was rapidly reversible by the use of Cdk5-specific inhibitors, or by expression of non-phosphorylatable GRASP proteins, both of which significantly reduced APP trafficking and Aβ production (Figure 2). In the same study, degradation of Golgi structural proteins was not detected (Joshi et al., 2014). These results suggest that Golgi fragmentation in AD, at least in the early stage, is caused by phosphorylation of Golgi structural proteins, an event that occurs in parallel with tau hyper-phosphorylation during the development of the disease. Overall, the causes of Golgi structural defects in AD are expected to be manifold and require further investigation to determine the precise mechanisms.
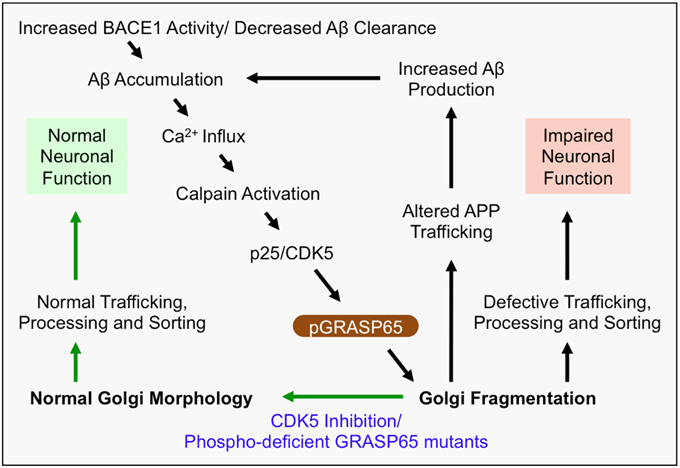
Figure 2. Mechanism of Golgi defects in AD. Increased BACE1 activity and/or decreased Aβ clearance from the extracellular space leads to the accumulation of Aβ. In turn, Aβ induces Ca2+ influx, which activates Calpain and induces cleavage of p35 to p25. Consequently, p25 activates CDK5, which in turn phosphorylates and inactivates the Golgi structural protein GRASP65 (i.e., pGRASP65). Inactivation of GRASP65 causes Golgi fragmentation, which alters trafficking of APP, and potentially the secretases, leading to increased Aβ production. This deleterious feedback loop (indicated by black arrows) would impair the integrity of the secretory pathway for sorting, trafficking and modifications of many essential proteins, which may compromise neuronal function, activate inflammatory responses, or cause neuronal cell death. Inhibition of CDK5 or expression of non-phosphorylatable GRASP65 mutants restores the normal Golgi morphology and reduces APP trafficking and Aβ production (indicated by green arrows). Therefore, rescue of the defective Golgi may delay AD development.
Functional Consequences of Golgi Fragmentation in AD
Proper Golgi structure formation controls the sequence and speed of protein transport through the Golgi membranes for proper trafficking, maturation, sorting, and processing of not only APP, but also many neuronal proteins (Dries and Yu, 2008; Joshi and Wang, 2015). When BACE1 activity is increased or Aβ clearance is decreased, Aβ accumulation induces Golgi fragmentation through modification of GRASP65, and other Golgi structural proteins (Joshi et al., 2014; Joshi and Wang, 2015). Fragmentation of the Golgi, as one possible outcome, enhances vesicle budding from the Golgi membranes, accelerates protein trafficking, and impairs accurate glycosylation (Wang et al., 2008; Xiang et al., 2013), and thus increases Aβ production by enhancing amyloidogenic cleavage (Figure 2). APP and the β-secretase BACE1 are sorted by the Golgi into different compartments and the loss of the sorting function of the Golgi by fragmentation in AD results in defective trafficking of APP and BACE1, which promotes Aβ production (Tan and Evin, 2012; Das et al., 2013; Joshi et al., 2014). This deleterious feedback loop may impair the integrity of the secretory pathway for sorting, trafficking and modifications of many essential neuronal proteins. For example, many AD-related proteins that are essential for neuronal function, such as NMDA and AMPA receptors (Pérez-Otaño and Ehlers, 2005; Greger and Esteban, 2007; Shepherd and Huganir, 2007), and synaptic integrity proteins such as neurexins (Fairless et al., 2008; Reissner et al., 2013), are processed and trafficked through the Golgi. Thus, Golgi fragmentation might impair trafficking of membrane receptors and ion channels that regulate neuronal function and/or viability (Figure 1).
Long term Golgi defects might also result in a significant change in the composition of proteins, lipids, and polysaccharides at the cell surface (Figure 1). A change in the composition of cell-surface molecules could not only directly impair neuronal activity and synaptic integrity, but also potentially trigger an immune response against neurons with Golgi defects that leads to cell death. In addition, Golgi defects may impact microtubule organization, tau function, and cell polarity. The Golgi also functions as a storage reservoir for Ca2+ (Pinton et al., 1998; Griesbeck et al., 2001), a small molecule essential for neurotransmission. Ca2+ is a key regulator of membrane fusion (Pang and Sudhof, 2010), synaptic plasticity (Neher and Sakaba, 2008), and neurite growth (Tojima et al., 2011). Defects in calcium signaling has been observed in neurodegeneration (Wojda et al., 2008), and prolonged neuronal hyperexcitability and neuronal activity lead to Golgi fragmentation (Thayer et al., 2013). Taken together, Golgi defects may compromise neuron activity and survival through multiple pathways and serve as an important mechanism of pathogenesis in AD and other neurodegenerative diseases.
Conclusion
The Golgi apparatus plays an essential role in trafficking and sorting of proteins that are vital for neuronal functions. The Golgi is fragmented in many neurodegenerative diseases, suggesting that Golgi defects may contribute to neurodegeneration. Golgi fragmentation likely involves multiple mechanisms. In a recent study on AD, Golgi fragmentation is caused by phosphorylation of the Golgi structural protein GRASP65 and Golgi fragmentation results in enhanced APP trafficking and Aβ production. Golgi fragmentation is predicted to impair intracellular trafficking of many proteins that are essential for neuronal function. Restoring Golgi morphological defects might be an attractive approach to treating, or even preventing, AD. Understanding the impact of Golgi pathology and determining the molecular mechanisms that cause Golgi fragmentation is expected to shed insight into novel therapeutic approaches for treating AD and potentially other neurodegenerative disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Henry Paulson, Raymond Scott Turner, Geoffrey Murphy, and Wang lab members for insightful discussions. This work was supported in part by the National Institutes of Health (Grants GM087364, GM105920, and GM112786), the American Cancer Society (Grant RGS-09-278-01-CSM), the National Institutes of Health-funded Michigan Alzheimer's Disease Research Center (Grant P50 AG08761), MCubed and the Fastforward Protein Folding Disease Initiative of the University of Michigan, and an anonymous donation to YW.
References
Alzheimer's, A. (2015). 2015 Alzheimer's disease facts and figures. Alzheimers. Dement. 11, 332–384. doi: 10.1016/j.jalz.2015.02.003
Avila, J. (2006). Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 580, 2922–2927. doi: 10.1016/j.febslet.2006.02.067
Baas, P. W. (2002). Microtubule transport in the axon. Int. Rev. Cytol. 212, 41–62. doi: 10.1016/S0074-7696(01)12003-6
Baloyannis, S. J. (2014). Golgi apparatus and protein trafficking in Alzheimer's disease. J. Alzheimers. Dis. 42(Suppl. 3), S153–S162. doi: 10.3233/JAD-132660
Bisel, B., Wang, Y., Wei, J. H., Xiang, Y., Tang, D., Miron-Mendoza, M., et al. (2008). ERK regulates Golgi and centrosome orientation towards the leading edge through GRASP65. J. Cell Biol. 182, 837–843. doi: 10.1083/jcb.200805045
Burgos, P. V., Mardones, G. A., Rojas, A. L., Dasilva, L. L., Prabhu, Y., Hurley, J. H., et al. (2010). Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18, 425–436. doi: 10.1016/j.devcel.2010.01.015
Chiu, R., Novikov, L., Mukherjee, S., and Shields, D. (2002). A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J. Cell Biol. 159, 637–648. doi: 10.1083/jcb.200208013
Choy, R. W., Cheng, Z., and Schekman, R. (2012). Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 109, E2077–E2082. doi: 10.1073/pnas.1208635109
Chung, H. M., and Struhl, G. (2001). Nicastrin is required for Presenilin-mediated transmembrane cleavage in Drosophila. Nat. Cell Biol. 3, 1129–1132. doi: 10.1038/ncb1201-1129
Dal Canto, M. C. (1996). The Golgi apparatus and the pathogenesis of Alzheimer's disease. Am. J. Pathol. 148, 355–360.
Das, U., Scott, D. A., Ganguly, A., Koo, E. H., Tang, Y., and Roy, S. (2013). Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79, 447–460. doi: 10.1016/j.neuron.2013.05.035
Diao, A., Rahman, D., Pappin, D. J., Lucocq, J., and Lowe, M. (2003). The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J. Cell Biol. 160, 201–212. doi: 10.1083/jcb.200207045
Dries, D. R., and Yu, G. (2008). Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer's disease. Curr. Alzheimer Res. 5, 132–146. doi: 10.2174/156720508783954695
Edbauer, D., Winkler, E., Haass, C., and Steiner, H. (2002). Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc. Natl. Acad. Sci. U.S.A. 99, 8666–8671. doi: 10.1073/pnas.132277899
Fairless, R., Masius, H., Rohlmann, A., Heupel, K., Ahmad, M., Reissner, C., et al. (2008). Polarized targeting of neurexins to synapses is regulated by their C-terminal sequences. J. Neurosci. 28, 12969–12981. doi: 10.1523/JNEUROSCI.5294-07.2008
Farah, C. A., Perreault, S., Liazoghli, D., Desjardins, M., Anton, A., Lauzon, M., et al. (2006). Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil. Cytoskeleton 63, 710–724. doi: 10.1002/cm.20157
Feinstein, T. N., and Linstedt, A. D. (2008). GRASP55 regulates Golgi ribbon formation. Mol. Biol. Cell 19, 2696–2707. doi: 10.1091/mbc.E07-11-1200
Feng, Y., Yu, W., Li, X., Lin, S., Zhou, Y., Hu, J., et al. (2013). Structural insight into Golgi membrane stacking by GRASP65 and GRASP55 proteins. J. Biol. Chem. 288, 28418–28427. doi: 10.1074/jbc.M113.478024
Fokin, A. I., Brodsky, I. B., Burakov, A. V., and Nadezhdina, E. S. (2014). Interaction of early secretory pathway and Golgi membranes with microtubules and microtubule motors. Biochemistry (Mosc) 79, 879–893. doi: 10.1134/S0006297914090053
Fujita, Y., and Okamoto, K. (2005). Golgi apparatus of the motor neurons in patients with amyotrophic lateral sclerosis and in mice models of amyotrophic lateral sclerosis. Neuropathology 25, 388–394. doi: 10.1111/j.1440-1789.2005.00616.x
Gallant, P. E. (2000). Axonal protein synthesis and transport. J. Neurocytol. 29, 779–782. doi: 10.1023/A:1010961022455
Glick, B. S., and Nakano, A. (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25, 113–132. doi: 10.1146/annurev.cellbio.24.110707.175421
Goldstein, L. S., and Yang, Z. (2000). Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu. Rev. Neurosci. 23, 39–71. doi: 10.1146/annurev.neuro.23.1.39
Gonatas, N. K., Gonatas, J. O., and Stieber, A. (1998). The involvement of the Golgi apparatus in the pathogenesis of amyotrophic lateral sclerosis, Alzheimer's disease, and ricin intoxication. Histochem. Cell Biol. 109, 591–600. doi: 10.1007/s004180050257
Gong, C. X., and Alonso Adel, C. (2013). Tau and beyond for Alzheimer's disease. J. Alzheimers. Dis. 37, 465–466. doi: 10.3233/JAD-130490
Greenfield, J. P., Tsai, J., Gouras, G. K., Hai, B., Thinakaran, G., Checler, F., et al. (1999). Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 96, 742–747. doi: 10.1073/pnas.96.2.742
Greger, I. H., and Esteban, J. A. (2007). AMPA receptor biogenesis and trafficking. Curr. Opin. Neurobiol. 17, 289–297. doi: 10.1016/j.conb.2007.04.007
Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., and Tsien, R. Y. (2001). Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276, 29188–29194. doi: 10.1074/jbc.M102815200
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Haass, C., Koo, E. H., Mellon, A., Hung, A. Y., and Selkoe, D. J. (1992). Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357, 500–503. doi: 10.1038/357500a0
Hancock, W. O. (2014). Bidirectional cargo transport: moving beyond tug of war. Nat. Rev. Mol. Cell Biol. 15, 615–628. doi: 10.1038/nrm3853
Herreman, A., Van Gassen, G., Bentahir, M., Nyabi, O., Craessaerts, K., Mueller, U., et al. (2003). gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 116, 1127–1136. doi: 10.1242/jcs.00292
Hicks, S. W., and Machamer, C. E. (2002). The NH2-terminal domain of Golgin-160 contains both Golgi and nuclear targeting information. J. Biol. Chem. 277, 35833–35839. doi: 10.1074/jbc.M206280200
Hilditch-Maguire, P., Trettel, F., Passani, L. A., Auerbach, A., Persichetti, F., and Macdonald, M. E. (2000). Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 9, 2789–2797. doi: 10.1093/hmg/9.19.2789
Hu, Y., Ye, Y., and Fortini, M. E. (2002). Nicastrin is required for gamma-secretase cleavage of the Drosophila Notch receptor. Dev. Cell 2, 69–78. doi: 10.1016/S1534-5807(01)00105-8
Huse, J. T., Liu, K., Pijak, D. S., Carlin, D., Lee, V. M., and Doms, R. W. (2002). Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J. Biol. Chem. 277, 16278–16284. doi: 10.1074/jbc.M111141200
Infante, C., Ramos-Morales, F., Fedriani, C., Bornens, M., and Rios, R. M. (1999). GMAP-210, A cis-Golgi network-associated protein, is a minus end microtubule-binding protein. J. Cell Biol. 145, 83–98. doi: 10.1083/jcb.145.1.83
Jiang, Q., Wang, L., Guan, Y., Xu, H., Niu, Y., Han, L., et al. (2014). Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol. Aging 35, 1352–1363. doi: 10.1016/j.neurobiolaging.2013.11.022
Joshi, G., Chi, Y., Huang, Z., and Wang, Y. (2014). Abeta-induced Golgi fragmentation in Alzheimer's disease enhances Abeta production. Proc. Natl. Acad. Sci. U.S.A. 111, E1230–E1239. doi: 10.1073/pnas.1320192111
Joshi, G., and Wang, Y. (2015). Golgi defects enhance APP amyloidogenic processing in Alzheimer's disease. Bioessays 37, 240–247. doi: 10.1002/bies.201400116
Kimberly, W. T., Lavoie, M. J., Ostaszewski, B. L., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2002). Complex N-linked glycosylated nicastrin associates with active gamma-secretase and undergoes tight cellular regulation. J. Biol. Chem. 277, 35113–35117. doi: 10.1074/jbc.M204446200
Klausner, R. D., Donaldson, J. G., and Lippincott-Schwartz, J. (1992). Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080. doi: 10.1083/jcb.116.5.1071
Klumperman, J. (2011). Architecture of the mammalian Golgi. Cold Spring Harb. Perspect. Biol. 3:a005181. doi: 10.1101/cshperspect.a005181
Klute, M. J., Melançon, P., and Dacks, J. B. (2011). Evolution and diversity of the Golgi. Cold Spring Harb. Perspect. Biol. 3:a007849. doi: 10.1101/cshperspect.a007849
Ladinsky, M. S., Mastronarde, D. N., McIntosh, J. R., Howell, K. E., and Staehelin, L. A. (1999). Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J. Cell Biol. 144, 1135–1149. doi: 10.1083/jcb.144.6.1135
Lane, J. D., Lucocq, J., Pryde, J., Barr, F. A., Woodman, P. G., Allan, V. J., et al. (2002). Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 156, 495–509. doi: 10.1083/jcb.200110007
Lavieu, G., Dunlop, M. H., Lerich, A., Zheng, H., Bottanelli, F., and Rothman, J. E. (2014). The Golgi ribbon structure facilitates anterograde transport of large cargoes. Mol. Biol. Cell 25, 3028–3036. doi: 10.1091/mbc.E14-04-0931
Lavieu, G., Zheng, H., and Rothman, J. E. (2013). Stapled Golgi cisternae remain in place as cargo passes through the stack. Elife 2:e00558. doi: 10.7554/eLife.00558
Lee, I., Tiwari, N., Dunlop, M. H., Graham, M., Liu, X., and Rothman, J. E. (2014). Membrane adhesion dictates Golgi stacking and cisternal morphology. Proc. Natl. Acad. Sci. U.S.A. 111, 1849–1854. doi: 10.1073/pnas.1323895111
Lee, M. S., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L. H. (2000). Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364. doi: 10.1038/35012636
Lewis, T. L. Jr., and Polleux, F. (2012). Neuronal morphogenesis: Golgi outposts, acentrosomal microtubule nucleation, and dendritic branching. Neuron 76, 862–864. doi: 10.1016/j.neuron.2012.11.019
Liazoghli, D., Perreault, S., Micheva, K. D., Desjardins, M., and Leclerc, N. (2005). Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am. J. Pathol. 166, 1499–1514. doi: 10.1016/S0002-9440(10)62366-8
Liu, Z., Vong, Q. P., and Zheng, Y. (2007). CLASPing Microtubules at the trans-Golgi Network. Dev. Cell 12, 839–840. doi: 10.1016/j.devcel.2007.05.007
Lowe, M., Rabouille, C., Nakamura, N., Watson, R., Jackman, M., Jamsa, E., et al. (1998). Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94, 783–793. doi: 10.1016/S0092-8674(00)81737-7
Miller, P. M., Folkmann, A. W., Maia, A. R., Efimova, N., Efimov, A., and Kaverina, I. (2009). Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 11, 1069–1080. doi: 10.1038/ncb1920
Mizuno, Y., Hattori, N., Kitada, T., Matsumine, H., Mori, H., Shimura, H., et al. (2001). Familial Parkinson's disease. Alpha-synuclein and parkin. Adv. Neurol. 86, 13–21.
Morishima-Kawashima, M., and Ihara, Y. (2002). Alzheimer's disease: beta-Amyloid protein and tau. J. Neurosci. Res. 70, 392–401. doi: 10.1002/jnr.10355
Mourelatos, Z., Gonatas, N. K., Stieber, A., Gurney, M. E., and Dal Canto, M. C. (1996). The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. U.S.A. 93, 5472–5477. doi: 10.1073/pnas.93.11.5472
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872. doi: 10.1016/j.neuron.2008.08.019
Nelson, P. T., Braak, H., and Markesbery, W. R. (2009). Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J. Neuropathol. Exp. Neurol. 68, 1–14. doi: 10.1097/NEN.0b013e3181919a48
Ori-McKenney, K. M., Jan, L. Y., and Jan, Y. N. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76, 921–930. doi: 10.1016/j.neuron.2012.10.008
Paglini, G., Peris, L., Diez-Guerra, J., Quiroga, S., and Caceres, A. (2001). The Cdk5-p35 kinase associates with the Golgi apparatus and regulates membrane traffic. EMBO Rep. 2, 1139–1144. doi: 10.1093/embo-reports/kve250
Pang, Z. P., and Südhof, T. C. (2010). Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 22, 496–505. doi: 10.1016/j.ceb.2010.05.001
Patrick, G. N., Zukerberg, L., Nikolic, M., De La Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Pérez-Otaño, I., and Ehlers, M. D. (2005). Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 28, 229–238. doi: 10.1016/j.tins.2005.03.004
Pinton, P., Pozzan, T., and Rizzuto, R. (1998). The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 17, 5298–5308. doi: 10.1093/emboj/17.18.5298
Puthenveedu, M. A., Bachert, C., Puri, S., Lanni, F., and Linstedt, A. D. (2006). GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 8, 238–248. doi: 10.1038/ncb1366
Radulescu, A. E., Mukherjee, S., and Shields, D. (2011). The Golgi protein p115 associates with gamma-tubulin and plays a role in Golgi structure and mitosis progression. J. Biol. Chem. 286, 21915–21926. doi: 10.1074/jbc.M110.209460
Rambourg, A., Clermont, Y., Hermo, L., and Segretain, D. (1987). Tridimensional structure of the Golgi apparatus of nonciliated epithelial cells of the ductuli efferentes in rat: an electron microscope stereoscopic study. Biol. Cell 60, 103–115. doi: 10.1111/j.1768-322X.1987.tb00550.x
Ramirez, I. B., and Lowe, M. (2009). Golgins and GRASPs: holding the Golgi together. Semin. Cell Dev. Biol. 20, 770–779. doi: 10.1016/j.semcdb.2009.03.011
Reissner, C., Runkel, F., and Missler, M. (2013). Neurexins. Genome Biol. 14:213. doi: 10.1186/gb-2013-14-9-213
Rios, R. M. (2014). The centrosome-Golgi apparatus nexus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130462. doi: 10.1098/rstb.2013.0462
Rios, R. M., and Bornens, M. (2003). The Golgi apparatus at the cell centre. Curr. Opin. Cell Biol. 15, 60–66. doi: 10.1016/S0955-0674(02)00013-3
Rivero, S., Cardenas, J., Bornens, M., and Rios, R. M. (2009). Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 28, 1016–1028. doi: 10.1038/emboj.2009.47
Roboti, P., Sato, K., and Lowe, M. (2015). The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 128, 1595–1606. doi: 10.1242/jcs.166710
Saraceno, C., Marcello, E., Di Marino, D., Borroni, B., Claeysen, S., Perroy, J., et al. (2014). SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 5:e1547. doi: 10.1038/cddis.2014.492
Seitz, A., Kojima, H., Oiwa, K., Mandelkow, E. M., Song, Y. H., and Mandelkow, E. (2002). Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 21, 4896–4905. doi: 10.1093/emboj/cdf503
Shepherd, J. D., and Huganir, R. L. (2007). The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643. doi: 10.1146/annurev.cellbio.23.090506.123516
Shorter, J., Watson, R., Giannakou, M. E., Clarke, M., Warren, G., and Barr, F. A. (1999). GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 18, 4949–4960. doi: 10.1093/emboj/18.18.4949
Small, S. A., and Gandy, S. (2006). Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31. doi: 10.1016/j.neuron.2006.09.001
Stieber, A., Mourelatos, Z., and Gonatas, N. K. (1996). In Alzheimer's disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 148, 415–426.
Suh, Y. H., and Checler, F. (2002). Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol. Rev. 54, 469–525. doi: 10.1124/pr.54.3.469
Tan, J., and Evin, G. (2012). Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer's disease pathogenesis. J. Neurochem. 120, 869–880. doi: 10.1111/j.1471-4159.2011.07623.x
Tang, D., Mar, K., Warren, G., and Wang, Y. (2008). Molecular mechanism of mitotic Golgi disassembly and reassembly revealed by a defined reconstitution assay. J. Biol. Chem. 283, 6085–6094. doi: 10.1074/jbc.M707715200
Tang, D., Yuan, H., and Wang, Y. (2010). The role of GRASP65 in Golgi cisternal stacking and cell cycle progression. Traffic 11, 827–842. doi: 10.1111/j.1600-0854.2010.01055.x
Tang, Z., Ioja, E., Bereczki, E., Hultenby, K., Li, C., Guan, Z., et al. (2015). mTor mediates tau localization and secretion: implication for Alzheimer's disease. Biochim. Biophys. Acta 1853, 1646–1657. doi: 10.1016/j.bbamcr.2015.03.003
Thayer, D. A., Jan, Y. N., and Jan, L. Y. (2013). Increased neuronal activity fragments the Golgi complex. Proc. Natl. Acad. Sci. U.S.A. 110, 1482–1487. doi: 10.1073/pnas.1220978110
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.R800019200
Tojima, T., Hines, J. H., Henley, J. R., and Kamiguchi, H. (2011). Second messengers and membrane trafficking direct and organize growth cone steering. Nat. Rev. Neurosci. 12, 191–203. doi: 10.1038/nrn2996
Truschel, S. T., Sengupta, D., Foote, A., Heroux, A., MacBeth, M. R., and Linstedt, A. D. (2011). Structure of the membrane-tethering GRASP domain reveals a unique PDZ ligand interaction that mediates Golgi biogenesis. J. Biol. Chem. 286, 20125–20129. doi: 10.1074/jbc.C111.245324
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Veenendaal, T., Jarvela, T., Grieve, A. G., Van Es, J. H., Linstedt, A. D., and Rabouille, C. (2014). GRASP65 controls the cis Golgi integrity in vivo. Biol. Open 3, 431–443. doi: 10.1242/bio.20147757
Wang, Y., Satoh, A., and Warren, G. (2005). Mapping the functional domains of the Golgi stacking factor GRASP65. J. Biol. Chem. 280, 4921–4928. doi: 10.1074/jbc.M412407200
Wang, Y., and Seemann, J. (2011). Golgi biogenesis. Cold Spring Harb. Perspect. Biol. 3:a005330. doi: 10.1101/cshperspect.a005330
Wang, Y., Seemann, J., Pypaert, M., Shorter, J., and Warren, G. (2003). A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 22, 3279–3290. doi: 10.1093/emboj/cdg317
Wang, Y., Wei, J. H., Bisel, B., Tang, D., and Seemann, J. (2008). Golgi cisternal unstacking stimulates COPI vesicle budding and protein transport. PLoS ONE 3:e1647. doi: 10.1371/journal.pone.0001647
Williams, D., Hicks, S. W., MacHamer, C. E., and Pessin, J. E. (2006). Golgin-160 is required for the Golgi membrane sorting of the insulin-responsive glucose transporter GLUT4 in adipocytes. Mol. Biol. Cell 17, 5346–5355. doi: 10.1091/mbc.E06-05-0386
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590. doi: 10.1002/iub.91
Wong, M., and Munro, S. (2014). Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 346:1256898. doi: 10.1126/science.1256898
Xiang, Y., and Wang, Y. (2010). GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 188, 237–251. doi: 10.1083/jcb.200907132
Xiang, Y., and Wang, Y. (2011). New components of the Golgi matrix. Cell Tissue Res. 344, 365–379. doi: 10.1007/s00441-011-1166-x
Xiang, Y., Zhang, X., Nix, D. B., Katoh, T., Aoki, K., Tiemeyer, M., et al. (2013). Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 4:1659. doi: 10.1038/ncomms2669
Yadav, S., and Linstedt, A. D. (2011). Golgi positioning. Cold Spring Harb. Perspect. Biol. 3:a005322. doi: 10.1101/cshperspect.a005322
Yoshiyama, Y., Zhang, B., Bruce, J., Trojanowski, J. Q., and Lee, V. M. (2003). Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J. Neurosci. 23, 10662–10671.
Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., et al. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54. doi: 10.1038/35024009
Zempel, H., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2010). Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
Zhang, Y. W., Luo, W. J., Wang, H., Lin, P., Vetrivel, K. S., Liao, F., et al. (2005). Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. J. Biol. Chem. 280, 17020–17026. doi: 10.1074/jbc.M409467200
Keywords: Alzheimer's disease, amyloid precursor protein, amyloid beta, Golgi, GRASP65, phosphorylation
Citation: Joshi G, Bekier ME II and Wang Y (2015) Golgi fragmentation in Alzheimer's disease. Front. Neurosci. 9:340. doi: 10.3389/fnins.2015.00340
Received: 23 July 2015; Accepted: 08 September 2015;
Published: 24 September 2015.
Edited by:
Catherine Rabouille, Hubrecht Institute of the KNAW, NetherlandsReviewed by:
Nigel Hooper, University of Manchester, UKPeng Lei, The University of Melbourne, Australia
Martin Lowe, University of Manchester, UK
Copyright © 2015 Joshi, Bekier and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanzhuang Wang, Department of Molecular, Cellular and Developmental Biology, University of Michigan, 830 North University Avenue, Ann Arbor, MI 48109-1048, USA,eXp3YW5nQHVtaWNoLmVkdQ==
†These authors have contributed equally to this work.