 Tzyy-Nan Huang
Tzyy-Nan Huang Yi-Ping Hsueh
Yi-Ping Hsueh- Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan
T-brain-1 (TBR1) is a brain-specific T-box transcription factor. In 1995, Tbr1 was first identified from a subtractive hybridization that compared mouse embryonic and adult telencephalons. Previous studies of Tbr1−∕− mice have indicated critical roles for TBR1 in the development of the cerebral cortex, amygdala, and olfactory bulb. Neuronal migration and axonal projection are two important developmental features controlled by TBR1. Recently, recurrent de novo disruptive mutations in the TBR1 gene have been found in patients with autism spectrum disorders (ASDs). Human genetic studies have identified TBR1 as a high-confidence risk factor for ASDs. Because only one allele of the TBR1 gene is mutated in these patients, Tbr1+∕− mice serve as a good genetic mouse model to explore the mechanism by which de novo TBR1 mutation leads to ASDs. Although neuronal migration and axonal projection defects of cerebral cortex are the most prominent phenotypes in Tbr1−∕− mice, these features are not found in Tbr1+∕− mice. Instead, inter- and intra-amygdalar axonal projections and NMDAR expression and activity in amygdala are particularly susceptible to Tbr1 haploinsufficiency. The studies indicated that both abnormal brain wiring (abnormal amygdalar connections) and excitation/inhibition imbalance (NMDAR hypoactivity), two prominent models for ASD etiology, are present in Tbr1+∕− mice. Moreover, calcium/calmodulin-dependent serine protein kinase (CASK) was found to interact with TBR1. The CASK–TBR1 complex had been shown to directly bind the promoter of the Grin2b gene, which is also known as Nmdar2b, and upregulate Grin2b expression. This molecular function of TBR1 provides an explanation for NMDAR hypoactivity in Tbr1+∕− mice. In addition to Grin2b, cell adhesion molecules—including Ntng1, Cdh8, and Cntn2—are also regulated by TBR1 to control axonal projections of amygdala. Taken together, the studies of Tbr1 provide an integrated picture of ASD etiology at the cellular and circuit levels.
Introduction
Autism spectrum disorders (ASDs) are heterogeneous and highly heritable neuropsychiatric disorders. Hundreds of genes with de novo copy-number variations or de novo point mutations have been identified in thousands of patients with ASDs (Gilman et al., 2011; Neale et al., 2012; O'roak et al., 2012a,b; De Rubeis et al., 2014; Iossifov et al., 2014). Although this variety of ASD-associated genes reflects the high heterogeneity of ASDs, ~26 high-confidence risk genes for ASDs have been summarized from large scale whole-exome sequencing (O'roak et al., 2012a; De Rubeis et al., 2014; Table 1). Among these high-confidence risk genes, 11 encode either transcription factors or chromatin remodeling factors, indicating that the dysregulation of gene expression is a common pathogenic mechanism for ASDs (Table 1). To date, T-BRAIN-1 (TBR-1) is the best studied transcription regulator among the high-confidence risk genes for ASDs. In this review, we summarize the physiological functions of TBR1 and the currently understood mechanisms by which TBR1 mutations cause ASDs. Based on the data accumulated from the mouse model, we suggest that abnormal brain wiring and reduced neuronal activity in the amygdala are the primary causes for TBR1-dependent ASDs.

Table 1. High-confidence risk factors for ASDs.
Identification of TBR1 in the Regulation of Brain Development
TBR1 contains a T-box DNA binding domain (Figure 1) and belongs to the T-box transcription factor family (Papaioannou, 2014). Twenty years ago, Dr. John Rubenstein's laboratory first identified Tbr1 from a subtractive hybridization screen using cDNA libraries made from mouse embryonic day 14.5 (E14.5) and adult telencephalons (Bulfone et al., 1995). Tbr1 mRNA levels were approximately 10-fold higher in E14.5 telencephalons than in adult telencephalons (Bulfone et al., 1995), suggesting a role for TBR1 in brain development. In situ hybridization and immunofluorescence staining indicate that Tbr1 is expressed in the postmitotic neurons of the cerebral cortex, hippocampus, olfactory bulb and amygdala at the embryonic stages (Bulfone et al., 1995, 1998; Remedios et al., 2007; Huang et al., 2014). Using markers of projection neurons, including glutamate and CaMKII, TBR1 has been found to be further restricted to the projection neurons of the cerebral cortex, amygdala and olfactory bulb (Bulfone et al., 1998; Hevner et al., 2001; Huang et al., 2014). In the cerebral cortex, layer 6 neurons express the highest levels of TBR1. Projection neurons in the remaining layers also express TBR1, though the expression levels are lower (Hevner et al., 2001). In the amygdala, TBR1 is only expressed in the projection neurons of the lateral and basal amygdala (Huang et al., 2014). These studies clearly show that TBR1 is a projection neuron-specific T-box factor highly enriched in embryonic telencephalons.
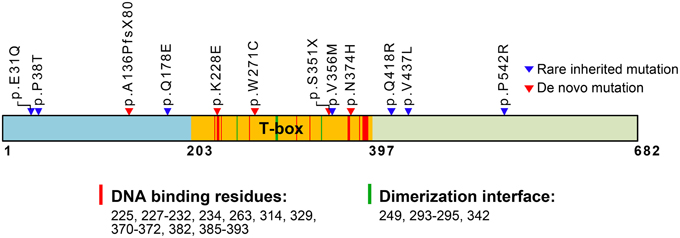
Figure 1. Schematic domain structure of TBR1 and identified mutations in patients with ASDs. The T-box DNA binding domain extends from amino acid (aa) residue 203–397. The predicted aa residues for DNA binding and dimerization based on the T-box structure of Brachyury (T protein) are also indicated and labeled with red and green strips in the T-box. The positions of de novo mutations are labeled with red triangles; the positions of rare inherited mutations are labeled with blue triangles. The functions of the residues in the T-box are predicted based on comparison with the Brachury T-box (pfam00907: T-box, http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=250216).
Dr. John Rubenstein and colleagues generated Tbr1−∕− mice to investigate the physiological function of Tbr1 (Bulfone et al., 1998; Hevner et al., 2001). A homozygous deficiency of Tbr1 results in neonatal lethality within 1–2 days after birth, indicating that Tbr1 is essential for survival. Most projection neurons in the olfactory bulb, including mitral and tufted cells, and axonal output to the lateral olfactory tract are lost in Tbr1−∕− mice (Bulfone et al., 1998). In the cerebral cortex, the inside-out pattern of neuronal migration is completely disrupted, as the six-layer laminar structure of the cortex is disorganized. Moreover, the contralateral axonal projections of the cerebral cortex and both corticothalamic and thalamocortical axonal projections are also defective, since they end mid-way to their final destinations in Tbr1−∕− mice (Hevner et al., 2001). TBR1 is also required for neuronal migration in the amygdala. A portion of the dorsal pallium that migrates from the caudal telencephalon pole toward the rostral telencephalon forms the basal and lateral amygdala. In Tbr1−∕− mice, this caudal-to-rostral migration is disrupted and thus impairs amygdala development (Remedios et al., 2007). Although TBR1 is also expressed in the hippocampus, its importance in hippocampus development and function remains unclear.
Based on the studies using Tbr1−∕− mice, it is clear that TBR1 is critical for development of projection neurons in the cerebral cortex, olfactory bulb and amygdala.
TBR1 Downstream Target Genes
Target genes of the TBR1 transcription factor were first identified by searching a database using the target sequence of the T-box DNA binding domain (Hsueh et al., 2000; Wang et al., 2004a,c). Because members of the T-box protein family share a DNA binding sequence, this method cannot ensure that the target genes are specific for TBR1. Because TBR1 is neuron-specific, neuronal expression is the first criterion to further screen the TBR1 target genes identified from sequence analysis. Results of an electrophoretic mobility shift assay, chromatin immunoprecipitation and a luciferase reporter assay have shown that TBR1 directly binds to the promoters and regulates the promoter activity of Grin2b (Glutamate receptor, ionotropic, N-methyl-aspartate 2b, also known as Nmdar2b) and Reln (Reelin) (Hsueh et al., 2000; Wang et al., 2004a,c). Changes in RELN and NMDAR2B protein levels have also been confirmed in Tbr1−∕− mice (Hevner et al., 2001; Wang et al., 2004c). Because Reln encodes an extracellular protein that is critical for neuronal migration (Martinez-Cerdeno and Noctor, 2014; Ohshima, 2014; Sekine et al., 2014), regulation of Reln expression by TBR1 could explain the migration phenotype in Tbr1−∕− mice. Regulation of Grin2b expression by TBR1 is critical for neuronal activation, which we discuss further in a later section.
Both our and Dr. Robert Hevner's laboratories independently applied microarray analyses to identify TBR1 downstream genes. Using E14.5 and P0.5 mouse brains, Dr. Hevner's laboratory focused on the arealization and lamination of the cerebral cortex (Bedogni et al., 2010). Tbr1 exhibits a high rostral and low caudal expression pattern in the cortex (Bulfone et al., 1995). At both E14.5 and P0.5, a Tbr1 deletion noticeably alters the expression of regional markers. In general, rostral genes are downregulated in Tbr1−∕− brains, while caudal genes are upregulated. For cortical layer markers, most markers of layer 6, subplate and Cajal–Retzius cells exhibit noticeably reduced expression levels in Tbr1−∕− brains. The majority of layer 2–5 markers are upregulated (Bedogni et al., 2010). The markers of lamination and arealization whose expression levels are altered in Tbr1−∕− brains are listed in Table 2. These studies indicate that TBR1 is critical for controlling the neuronal specification of the cerebral cortex.
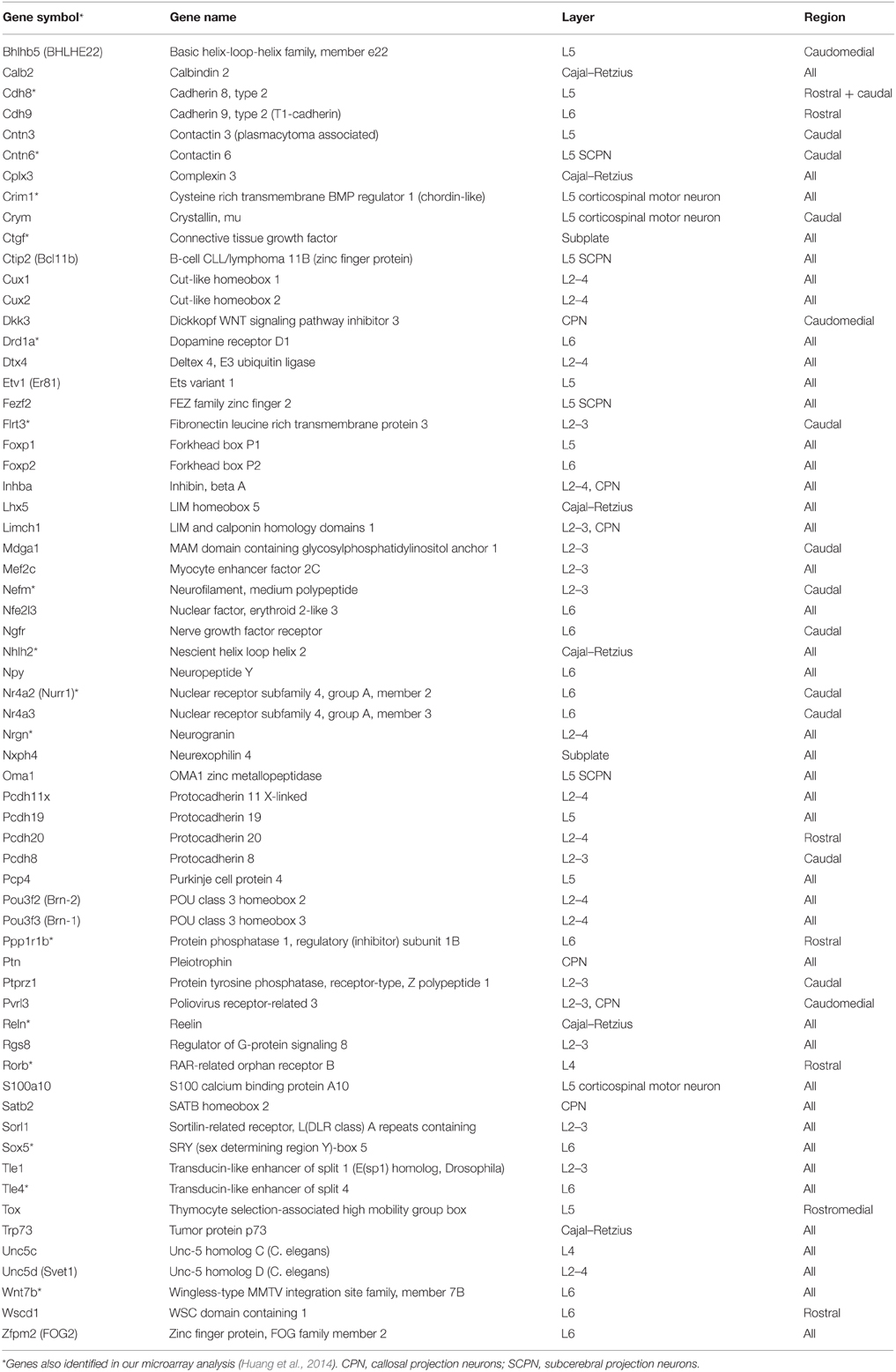
Table 2. Tbr1 deletion alters expression of genes with layer- or region-specific distribution in the cerebral cortex.
In our laboratory, we have identified more than 124 genes that are regulated by TBR1 at E16.5 (Huang et al., 2014; Chuang et al., 2015). The 16 region- or layer-specific genes presented in work by Bedogni et al. (2010) from Dr. Hevner's laboratory are also included in our gene list (Table 2). Moreover, based on a literature search and the database of the Simons Foundation Autism Research Initiative (https://gene.sfari.org/autdb/Welcome.do), 23 ASD-associated genes and a dyslexia causative gene, Kiaa0319, have also been found to be regulated by TBR1. Changes in the expression of these ASD- and dyslexia-associated genes (Table 3) provide support for the influence of TBR1 in ASDs. Tbr1 might act as master gene controlling the expression of a panel of ASD-associated genes and thus influence neural development and function (Chuang et al., 2015).
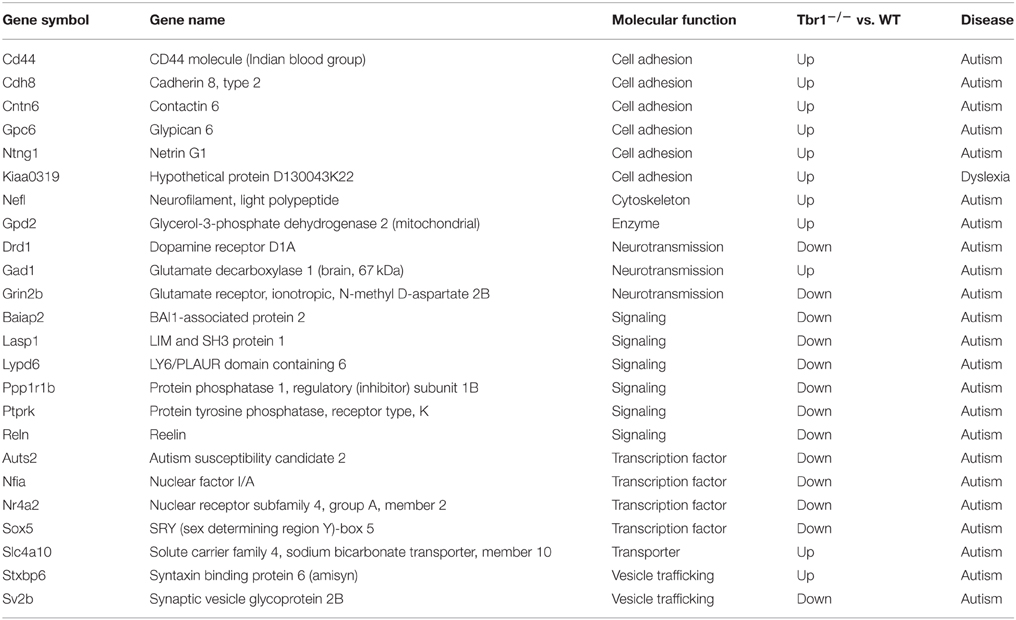
Table 3. TBR1 influences the expression (upregulation or downregulation) of genes associated with autism or dyslexia.
Moreover, the expression levels of 15 transcriptional regulators are reduced, while those of three transcription factors are upregulated in Tbr1−∕− brains compared with wild-type littermates (Table 4). These changes suggest that, in addition to directly regulating gene expression, TBR1 also controls transcriptional networks to influence neuronal development. Indeed, evidence has indicated that TBR1 directly binds to the locus of Fezf2, a layer 5-specific transcription factor, and represses Fezf2 expression in layer 6 to specify the corticothalamal projections of layer 6 neurons (Han et al., 2011; McKenna et al., 2011). The second transcriptional regulator directly controlled by TBR1 is autistic susceptibility gene2 (Auts2). TBR1 binds to the region around the Auts2 transcriptional start site and activates expression of the Auts2 gene (Bedogni et al., 2010). AUTS2 is part of polycomb repressive complex I (PRCI) that catalyzes the monoubiquitination of histone H2A and epigenetically represses gene expression, particularly during the developmental stage (de Napoles et al., 2004; Wang et al., 2004b). In contrast to the canonical role of PRCI in gene repression, the PRCI–AUST2 complex activates neuronal gene expression by recruiting casein kinase 2 and p300 to chromatin (Gao et al., 2014). The activation of Auts2 expression by TBR1 supports the influence of TBR1 on global gene expression in neurons.
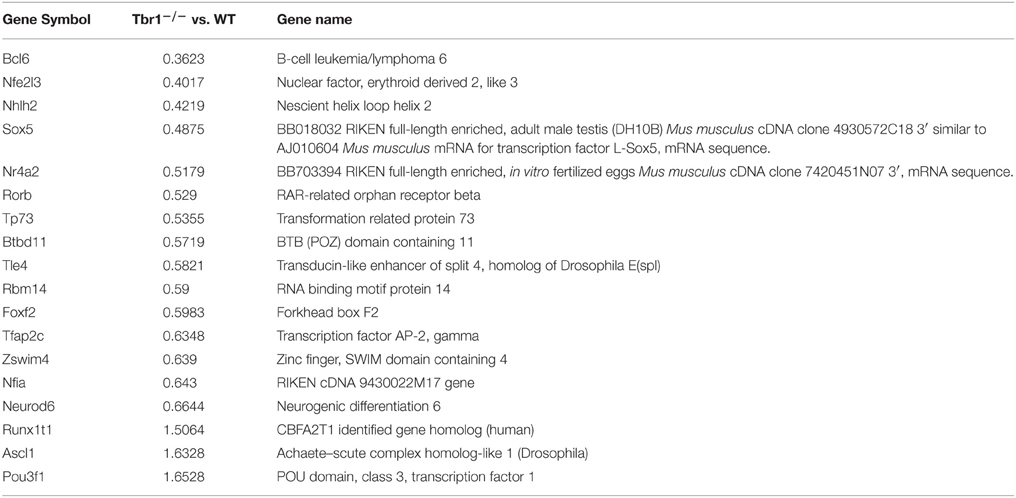
Table 4. Deletion of Tbr1 influences the expression of a panel of transcription factors in neurons.
In addition to using a transcriptional cascade to indirectly control gene expression, TBR1 may also alter the relative number of projection neurons and interneurons in the brain and influence the total expression levels of certain genes, such as Gad1, which encodes glutamate decarboxylase 1 (GAD67)—an essential gene of GABAergic neurons. In Tbr1 deletion mice, the expression of Gad1 is noticeably upregulated (Chuang et al., 2015). Because Tbr1 is specifically expressed in glutamatergic projection neurons, it is possible to speculate that increased Gad1 expression is indirectly linked to a reduction in the population of glutamatergic neurons.
Consistent with the function of TBR1 in the regulation of axonal projection, TBR1 also regulates eight membrane proteins (CNTN2, CDH8, GPC6, CD44, FLRT3, CNTN6, NTNG1, and KIAA0319) that are involved in cell adhesion; although it is still unclear whether these genes are directly or indirectly regulated by TBR1 (Chuang et al., 2015). Interestingly, seven of these eight membrane proteins are upregulated in Tbr1−∕− brains (Table 5). Because these genes control cell adhesion and axonal growth, the impairment of axonal projection in Tbr1 deficient neurons is likely due to imbalanced cell–cell and cell–matrix interactions. Alteration of the strength of these interactions may preclude neurite growth and extension (Chuang et al., 2015).
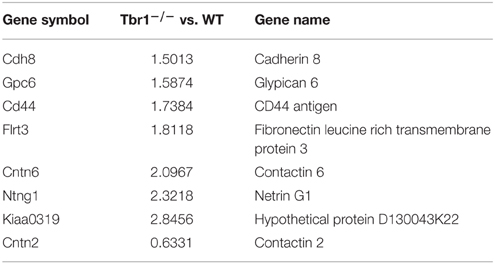
Table 5. Tbr1 deletion mainly upregulates expression of cell adhesion molecules in neurons.
In conclusion, TBR1 controls the expression of a series of genes that regulate cell-cell adhesion, axonal growth, neurotransmission and gene expression.
TBR1 Interacting Proteins
To date, only two proteins, CASK and FOXP2, have been identified as interacting partners with TBR1. Both CASK and FOXP2 are associated with ASDs (Samuels et al., 2007; O'roak et al., 2011). TBR1 was identified as a binding partner for CASK from a yeast two-hybrid screen using the guanylate kinase domain of CASK as bait (Hsueh et al., 2000). The C-terminal region of TBR1 is required for the interaction with CASK (Hsueh et al., 2000). CASK—a multidomain adaptor protein—is widely distributed in various subcellular compartments and interacts with more than two dozen cellular proteins (Hsueh, 2006). The interaction with CASK increases the transcriptional activity of TBR1 (Hsueh et al., 2000) by recruiting a nucleosome assembly protein CINAP (CASK interacting nucleosome assembly protein, also known as testis specific protein Y-encoded like 2, TSPYL2) to the promoter region containing the T-box DNA binding motif (Wang et al., 2004a). CINAP also interacts with the guanylate kinase domain of CASK. However, it does not compete with TBR1 for CASK binding. Instead, TBR1, CASK and CINAP form a tripartite complex to regulate Grin2b expression (Wang et al., 2004a,c). CASK is well-known as a causative gene in X-linked mental retardation (Najm et al., 2008). The interaction of CASK and TBR1 and the consequent effect on the regulation of Grin2b expression and neural development has been suggested to contribute to the phenotype of patients with CASK mutations (Hsueh, 2009). It can also be speculated that Grin2b expression, as controlled by the TBR1–CASK complex, might also be involved in ASDs due to TBR1 or CASK mutations.
FOXP2 is a critical transcription factor that controls speech (Lai et al., 2001; Enard et al., 2002) and is also associated with ASDs (Gong et al., 2004; Li et al., 2005). In contrast to the interaction between CASK and TBR1, the interaction between TBR1 and FOXP2 is less clear. Research suggests that both the T-box and C-terminal regions of TBR1 are involved in the interaction with FOXP2 (Deriziotis et al., 2014). For FOXP2, both its N- and C-terminal regions contribute to the interaction between FOXP2 and TBR1 (Deriziotis et al., 2014). Although it has been speculated that the interaction of FOXP2 and TBR1 is likely relevant to the verbal deficits in ASD patients, the molecular function of the TBR1-FOXP2 interaction is unclear. Furthermore, FOXP2 and TBR1 are only coexpressed in layer 6 of the cerebral cortex and not in other layers of the cerebral cortex and amygdala. Thus, the interaction with FOXP2 can only partly account for the function of TBR1.
TBR1, its binding partners CASK and FOXP2 and its direct downstream target GRIN2B, are all associated with ASDs, reinforcing the role of TBR1 in ASDs.
TBR1 Mutations Associate with Neurological Disorders
Genetic analyses of patients have identified TBR1 as a high-confidence risk factor for ASDs (https://gene.sfari.org/autdb/GSGeneList.do?c=1). Identified mutations in TBR1 genes are summarized in Figure 1. Both de novo and inherited mutations in TBR1 have been found in patients with ASDs (Figure 1). Two of the mutations, p.A136PfsX80 and p.S351X, result in early termination and generate truncated proteins that lack a functional DNA-binding T-box domain (O'roak et al., 2012a,b; De Rubeis et al., 2014). These two truncated mutants can no longer function in transcription or in interactions with CASK and FOXP2. The remaining three de novo mutations are p.K228E, p.W271C, and p.N374H (Figure 1). Based on simulations with the T-box DNA binding domain of Brachury (http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=250216), the K228 residue is predicted to directly contribute to DNA binding (Figure 1). Thus, the p.K228E mutation is expected to disrupt the DNA binding ability of TBR1. The residues W271 and N374 are adjacent to the DNA binding residues (Figure 1). Thus, the p.W271C and p.N374H mutations could alter protein conformation and indirectly influence DNA binding. The p.V356M inherited mutation is localized in the T-box, but it is relatively far from the DNA binding and dimerization motifs. The remaining inherited mutations, including p.E31Q, p.P38T, p.Q178E, p.Q418R, p.V437L, and p.P542R, are localized to the N- and C-terminal regions and are not known to influence DNA binding. The impact of these inherited mutations is unclear.
To date, only two studies have analyzed the effects of these ASD mutations on TBR1 function. We contributed to the first functional study, which examined axonal growth in the amygdalar neurons of the TBR1 N374H mutant (Huang et al., 2014). An experiment comparing Tbr1+∕− and wild-type amygdalar neurons showed that the deletion of one Tbr1 gene results in multiple and shorter axons in amygdalar neurons (Huang et al., 2014). The reintroduction of wild-type Tbr1 into Tbr1+∕− amygdalar neurons effectively promotes axon growth and reduces the percentage of neurons carrying multiple axons to the levels seen in wild-type neurons. However, the N374H mutant fails to rescue the axonal defects of Tbr1+∕− amygdalar neurons, suggesting that the p.N374H mutation identified in patients with ASD results in a loss of function (Huang et al., 2014).
The second study focused on the effect of ASD mutations on the subcellular distribution, transcriptional activity, dimerization and protein-protein interaction of TBR1 using heterologous HEK293 cells as a model (Deriziotis et al., 2014). De novo mutations, including p.K228E and p.N374H, change the subcellular distribution of TBR1 in HEK293 cells. The mutant proteins tend to form large aggregates in the nuclei. The impact of these two mutations on the transcriptional activity of TBR1 is unclear because the luciferase reporter assay did not show a difference between the wild-type TBR1 and K228E and N374H mutants. However, similar to the truncated mutants, the K228E and N374H mutants no longer interact with FOXP2, which is consistent with the observation that the T-box domain is also involved in FOXP2 interactions, as described above. The mechanisms by which rare inherited mutations impair the function of TBR1 remain largely unclear, except for the p.Q418R mutation, which is known to reduce the interaction between TBR1 and FOXP2. Because TBR1 is a projection neuron-specific transcription factor, the relevance of the interaction between TBR1 and FOXP2 must be investigated in neurons instead of HEK293 cells.
In addition to ASDs, TBR1 is also associated with intellectual disability. The TBR1 locus is at chromosome 2q24.2. Both a microdeletion of the chromosome region that contains 2q24.2 and de novo mutations of the TBR1 gene have been found in patients with intellectual disabilities (Traylor et al., 2012; Burrage et al., 2013; Hamdan et al., 2014; Palumbo et al., 2014). Moreover, the expression levels of TBR1 are increased in patients that suffer from schizophrenia (Molnar et al., 2003). Taken together, TBR1 is closely associated with ASDs, schizophrenia and intellectual disability.
Tbr1 Haploinsufficiency Results in Neuronal Defects
In ASD patients, only one of the two TBR1 alleles is mutated (Neale et al., 2012; O'roak et al., 2012a,b; De Rubeis et al., 2014; Deriziotis et al., 2014). Several possibilities may explain the effect of TBR1 heterozygosity on brain function: haploinsufficiency or a dominant negative or gain-of-function effect of the mutated allele. Because two of the de novo mutations of the TBR1 gene, p.A136PfsX80, and p.S351X, result in early termination and generate truncated proteins that lack a full length T-box DNA binding domain and dimerization domain (Figure 1), the mutants are not expected to exert a dominant negative effect on the activity of TBR1 based on the known molecular function of TBR1. Instead, the defects are likely caused by haploinsufficiency. Tbr1+∕− neurons are characterized by shorter and multiple axons (Huang et al., 2014), indicating that loss of a copy of the Tbr1 gene results in abnormal neuronal differentiation. Thus, TBR1 deficits in patients are likely due to haploinsufficiency.
Tbr1+∕− Mice Serve as a Mouse Model for ASDs
Because only one of two TBR1 alleles is mutated in patients with ASDs and Tbr1 heterozygosity does not influence survival and the general health of mice, Tbr1+∕− mice serve as a good animal model to elucidate the role of Tbr1 in ASDs. The core symptoms of patients with ASDs are both verbal and non-verbal communication defects, impaired social interaction and cognitive inflexibility. ASDs are also frequently associated with learning disability. A series of behavior paradigms have been applied to characterize the behavioral defects of Tbr1 (Table 6). Compared with wild-type littermates, the locomotor and exploratory activities, the level of anxiety and the hippocampus-dependent memory of Tbr1+∕− mice are normal (Huang et al., 2014). However, the amygdala-dependent behaviors of Tbr1+∕− mice are noticeably affected. Conditioned taste aversion and auditory fear conditioning—two amygdala-dependent learning and memory paradigms—are both impaired in Tbr1+∕− mice. Cognitive flexibility, as examined by appetitive-motivated T-maze and two-choice digging tests, is also noticeably reduced in Tbr1+∕− mice. The three-chamber test, reciprocal social interactions and social transmission of food preferences have also been applied to characterize the social interactions of Tbr1+∕− mice. These paradigms all indicate that the social interactions of Tbr1+∕− mice are impaired. Moreover, the frequency of ultrasonic vocalization is significantly lower in isolated Tbr1+∕− pups. Thus, these behavioral analyses strongly support that Tbr1+∕− mice exhibit autism-like behaviors (Huang et al., 2014).
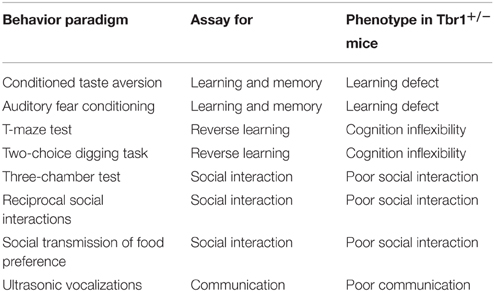
Table 6. Tbr1 haploinsufficiency results in autism-like behaviors in mice.
Defects in Amygdalar Circuits and Activation are Critical for ASDs Caused by Tbr1 Deficiency
When Tbr1 is completely deleted from mice, the most prominent phenotypes are observed in the neuronal migration and axonal projection of the embryonic cerebral cortex (Bulfone et al., 1998; Hevner et al., 2001). However, none of these defects are found in the Tbr1+∕− brain (Huang et al., 2014). Cortical lamination, contralateral cortical projection, corticothalamal projection and the size of the cerebral cortex of Tbr1+∕− brains are comparable to those of wild-type brains (Huang et al., 2014). Unexpectedly, the posterior part of the anterior commissure is either missing or dramatically reduced in Tbr1+∕− mice (Huang et al., 2014). This defect is 100% penetrant in all Tbr1+∕− mice. Thus, the posterior part of the anterior commissure is the structure most sensitive to Tbr1 haploinsufficiency. Consequently, defects of the posterior part of the anterior commissure are more relevant to the pathogenesis of TBR1-dependent ASDs.
The posterior part of the anterior commissure serves to connect the contralateral amygdalae (interamygdalar projections). The amygdala contains three major nuclei, namely the lateral, basal and central amygdala (Figure 2). Interamygdalar projections emerge from the lateral and basal amygdala. These two nuclei also project to the ipsilateral central amygdala (intraamygdalar projections). The lateral and basal amygdalae are the nuclei that receive inputs from the cortex, thalamus and hippocampus. To induce a freezing response, the lateral and basal amygdalae deliver the signals to the central amygdala, and the central amygdala further projects to the brainstem and hypothalamus. In addition to the central amygdala, the lateral and basal amygdalae also project back to the cortex, hippocampus, and thalamus, which are believed to regulate memory and social behavior (Lee et al., 2013; Janak and Tye, 2015). Because the amygdala is the pivotal brain structure for social intelligence, the amygdala is an obvious target for the etiology of TBR1-dependent ASDs.
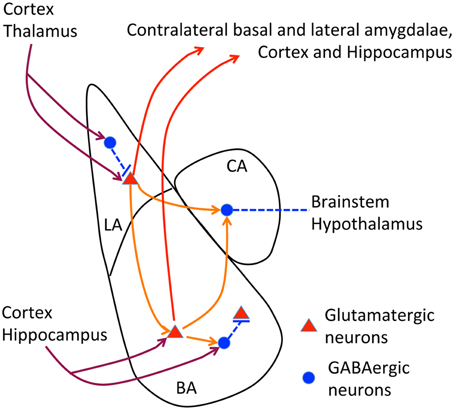
Figure 2. Neural circuits of amygdala. The relative position of lateral, basal and central amygdalae and the known axonal connections of amygdala are summarized. In lateral and basal amygdalae, glutamatergic neurons comprise the majority of cells. In contrast, GABAergic neurons are the major population in the central amygdala. Dark red lines, afferent pathways from cortex, thalamus and hippocampus; red lines, efferent pathways to contralateral amygdala, cortex and hippocampus; orange lines, intraamygdalar pathways. The central amygdala also delivers signals to the brainstem and hypothalamus to regulate freezing and emotion responses.
The results of DiI tracing and retrograde red bead labeling show that both inter- and intra-amygdalar axonal projections are noticeably impaired in Tbr1+∕− mice (Figure 3; Huang et al., 2014). The lateral and basal amygdala neurons are the major target of Tbr1 haploinsufficiency. Consistent with the axonal projection defects in the brain, Tbr1+∕− amygdala neurons possess shorter and multiple axons, which suggests that Tbr1 haploinsufficiency results in a cell-autonomous effect that restricts axonal extension and differentiation (Huang et al., 2014). To investigate how TBR1 controls axonal growth, we examined the TBR1 downstream target genes in a gene list from our microarray data. Specifically, we examined Cntn2, Cdh8, and Ntng1 because these three genes encode adhesion proteins that regulate neurite outgrowth and fasciculation (Furley et al., 1990; Stoeckli and Landmesser, 1995; Kunz et al., 1998; Nakashiba et al., 2002; Bekirov et al., 2008). Restoring the expression levels of Cntn2, Cdh8, and Ntng1 in Tbr1+∕− amygdalar neurons effectively ameliorates axonal growth and differentiation in cultures and promotes axonal projection to form the posterior part of the anterior commissure in vivo. Thus, TBR1 controls the expression of a panel of genes that regulate amygdalar axonal projections. Note that although the axonal projections of lateral and basal amygdalae are significantly impaired in Tbr1+∕− brains, the size and cell density of the lateral and basal amygdalae do not differ between Tbr1+∕− mice and wild-type littermates. It is unclear whether Tbr1+∕− amygdalar neurons mistarget to other brain regions. More investigations are needed to characterize this regulation in detail.
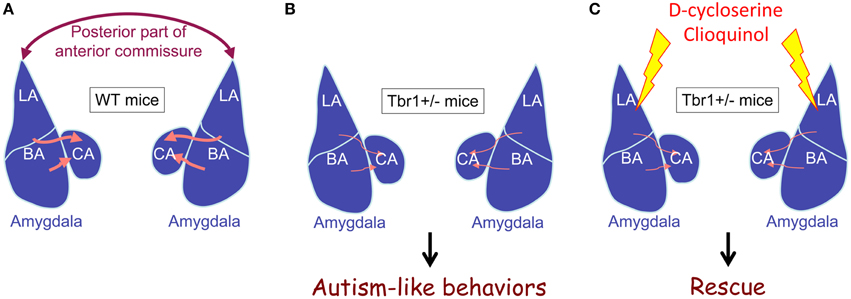
Figure 3. Tbr1 haploinsufficiency results in defects of intra- and inter-amygdalar axonal projections. (A) Wild-type mice. (B) Tbr1+∕− mice. (C) Tbr1+∕− mice with D-cycloserine or clioquinol injection. The interamygdalar projection via the posterior part of the anterior commissure is significantly impaired in Tbr1+∕− mice. Intraamygdalar connections between basolateral and central amygdalae are also noticeably reduced in Tbr1+∕− mice. Tbr1+∕− mice are characterized by autism-like behaviors. Increased neuronal activity in the amygdala using either D-cycloserine or clioquinol restores behavioral defects to normal levels, although axonal projection defects are not rescued.
Although the significance of interamygdalar connections is unclear, a reduction of intraamygdalar axonal projections implies that the amygdala is functionally impaired in Tbr1+∕− mice. Indeed, neuronal activation of the amygdala is impaired in Tbr1+∕− mice (Huang et al., 2014). Two amygdala-dependent learning/memory paradigms—conditioned taste aversion and auditory fear conditioning—have been used to investigate amygdalar responses. Both c-FOS and NMDAR2B protein levels were used to monitor neuronal activation in amygdalae. These experiments showed that the induction of both c-FOS and NMDAR2B are either much lower or completely absent in the lateral and basal amygdalae of Tbr1+∕− mice after conditioned taste aversion training and auditory fear conditioning (Huang et al., 2014; Chuang et al., 2015). Thus, both axonal projection and neuronal activation are defective in Tbr1+∕− amygdalae.
Possibly due to an impairment of NMDAR induction in the Tbr1+∕− amygdala, electrophysiological recording data showed that the NMDA/AMPA ratio is noticeably lower in the thalamic-lateral amygdala synapses of Tbr1+∕− brains compared with those of wild-type brains (Lee et al., 2015b). Consistent with amygdala-specific defects in Tbr1+∕− mice, the hippocampal Schaffer collateral-CA1 pyramidal synapses do not exhibit this abnormal NMDA/AMPA ratio (Lee et al., 2015b). These electrophysiological studies clearly demonstrate a functional NMDAR deficiency in Tbr1+∕− amygdalae.
If impairment of amygdala activation, and particularly reduced NMDAR activity, is critical for autism-like behaviors in Tbr1+∕− mice, the activation of amygdalar neurons should ameliorate the behavioral defects of Tbr1+∕− mice. Indeed, a bilateral local infusion of D-cycloserine (a coagonist of NMDAR) into the amygdala clearly ameliorates the reciprocal social interaction and conditioned taste aversion defects seen in Tbr1+∕− mice (Huang et al., 2014). D-cycloserine applied 30 min before the behavioral assay does not influence the expression of TBR1 target genes, suggesting the behavioral effects are mediated by an acute enhancement of NMDAR transmission. Because administration of D-cycloserine to the amygdala is sufficient to ameliorate the behavioral defects of Tbr1+∕− mice, the etiology of autism-like behaviors in Tbr1+∕− mice very likely involves amygdala defects (Huang et al., 2014).
In addition to local infusion, systemic administration of D-cycloserine via an intraperitoneal injection also effectively restores neuronal activation of the Tbr1+∕− amygdala and improves social interaction, cognitive inflexibility and associative memory of Tbr1+∕− mice (Huang et al., 2014). These results indicate a potential therapeutic avenue for ASD patients possessing TBR1 gene mutations. Tbr1+∕− mice are not the only mouse model to have been used to demonstrate the beneficial effect of D-cycloserine. The behavioral deficits of Shank2−∕−, Nlgn1−∕−, and Grid1−∕− mice can also be ameliorated by systemic administration of D-cycloserine. Specifically, D-cycloserine improves social interactions in both Shank2−∕− and Grid1−∕− mice in a three-chamber test (Won et al., 2012; Yadav et al., 2012). Furthermore, D-cycloserine reduces the repetitive grooming behavior of Nlgn1−∕− mice (Blundell et al., 2010). These three mutant mice all show NMDAR defects, which is consistent with the idea that NMDAR deficits are critical to the etiology of ASDs (Lee et al., 2015a). Improving NMDAR activity can ameliorate the behavioral defects of these mutant mice.
To further support the NMDAR deficit hypothesis in ASDs, we recently showed that by improving NMDAR activity via the administration of clioquinol, the social defects of Tbr1+∕− mice are rescued (Lee et al., 2015b). Clioquinol is a zinc chelator and ionophore that promotes the mobilization of zinc from presynaptic vesicles to the postsynaptic site. The postsynaptic elevation of zinc activates the protein tyrosine kinase SRC and consequently enhances NMDAR activity. Systemic administration of clioquinol noticeably improves the sociability of the mutant mice in the three-chamber test. Consistent with the behavioral rescue, the defective electrophysiological responses of mutant brains are also ameliorated by clioquinol treatment. In Tbr1+∕− brains, clioquinol can restore the reduced NMDA/AMPA ratio of the thalamic-lateral amygdala synapses. Clioquinol treatment also shows a beneficial effect on Shank2−∕− mice. It enhances the NMDAR activity of hippocampal Schaffer collateral-CA1 pyramidal synapses in Shank2−∕− mice (Lee et al., 2015b). Even though the molecular mechanisms responsible for the NMDAR deficit differ between Tbr1+∕− and Shank2−∕− mice, increasing NMDAR activity via D-cycloserine or clioquinol efficiently ameliorates the behavioral defects of these two mutant mice. These data support that an NMDAR deficit is likely to be a common pathogenic mechanism of ASDs. Moreover, studies using D-cycloserine and clioquinol suggest that activation of amygdalar neurons using suitable pharmacological treatments can ameliorate the behavioral defects caused by Tbr1 haploinsufficiency, even though the axonal projection defects of the Tbr1+∕− amygdala cannot be rescued in adult animals.
Tbr1 Serves as Immediate Early Gene to Control Neuronal Activation in Mature Neurons
Although the expression levels of Tbr1 gradually decline after birth, the protein levels of TBR1 remain detectable in adult mouse brains (Hsueh et al., 2000; Hong and Hsueh, 2007). Based on the following scenario, TBR1 may also play a role in the adult brain. TBR1 regulates Grin2b expression (Wang et al., 2004a), and CASK phosphorylation by protein kinase A (PKA) enhances this regulation. CASK phosphorylation increases the interaction between TBR1 and CASK and thus upregulates Grin2b promoter activity (Huang et al., 2010). Therefore, PKA phosphorylation may increase the ability of TBR1 to influence Grin2b expression, even though the expression levels of Tbr1 are lower in adult brains.
A study of in vitro cultured neurons unexpectedly showed that glutamate and bicuculline treatments noticeably upregulates Tbr1 expression (Chuang et al., 2014). Two to six hours after glutamate or bicuculline treatment, both the RNA and protein levels of Tbr1 are obviously increased. This induction is transient. The RNA levels of Tbr1 are decreased to basal levels 12 h after stimulation. This feature is shared among the cortical, hippocampal, and amygdalar neurons, although the induction of Tbr1 expression is much less pronounced in hippocampal neurons (Chuang et al., 2014). In addition to cultured neurons, behavioral stimulation also changes Tbr1 expression levels in mouse brains. When conditioned taste aversion is applied to stimulate neuronal activation, similar to c-Fos induction, Tbr1 RNA levels in the lateral amygdala, the insular cortex and the ventral hippocampus are also transiently increased 2 h after training (Chuang et al., 2014). Neuronal activation also induces Grin2b expression in vitro and in vivo, but this induction occurs several hours after that of Tbr1. Moreover, deletion of Tbr1 completely blocks Grin2b induction in culture (Chuang et al., 2014). Both NMDAR and CaMKII are required to induce Tbr1 expression (Figure 4). Thus, in addition to regulating axonal differentiation and neuronal migration during the early developmental stage, Tbr1 also acts as an immediate early gene in response to synaptic stimulation in mature neurons, which might contribute to the etiology of TBR1-related ASDs. In particular, the cerebral cortex of Tbr1+∕− mice likely exhibits defective electrophysiological responses and thus influences behaviors, even though anatomic defects of the cerebral cortex have not been identified in Tbr1+∕− mice. More investigations need to be conducted to address this possibility.
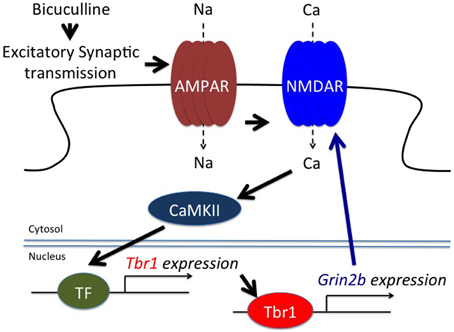
Figure 4. TBR1 acts as an immediate early gene to induce Grin2b expression during neuronal activation. Both bicuculline, an antagonist of GABAA receptors, and glutamate treatment induce Tbr1 expression. Although the transcription factor (TF) required for Tbr1 expression is unknown, evidence indicates that CaMKII is involved in Tbr1 upregulation. TBR1 can then induce Grin2b expression to enhance the synaptic responses (adapted from Chuang et al., 2014).
Abnormal Brain Wiring and Excitation/Inhibition Imbalance—two Prominent Models for the Pathogenic Mechanism of ASDs
Although the etiology of ASDs is heterogeneous, the two most prominent models for autism pathogenesis in the literature are abnormal brain wiring and an imbalance of neuronal activity (excitatory/inhibitory imbalance; Rubenstein and Merzenich, 2003; Walsh et al., 2008; Bernardinelli et al., 2014; Cellot and Cherubini, 2014; Deidda et al., 2014). These two defects lead to abnormal information processing and result in autism-like behaviors. These two models are interconnected to a certain extent. During neurodevelopment, neurons must extend their axons and form synapses with their target neurons, which allows the activity of the downstream target neurons to be regulated. In the absence of correct excitatory or inhibitory inputs, the activity of target neurons will be either too low or too high. These inappropriate levels of neuronal activity result in abnormal information processing, which leads to aberrant behaviors. Moreover, the imbalanced activity of neurons also influences (either strengthens or attenuates) their connections to other neurons. When the connection is too weak, it may be eliminated, which may alter brain wiring. In the Tbr1+∕− mouse model, amygdalar axonal projections are defective. Both inter- and intra-amygdalar connections are noticeably impaired. Moreover, the NMDAR activity of amygdalar neurons is also much lower in Tbr1+∕− brains. Thus, Tbr1+∕− brains are characterized by both abnormal brain wiring and defective neuronal activation. Further investigation is required to see whether one deficit contributes more substantially to ASD pathology.
Concluding Remarks
Tbr1+∕− mice constitute the first genetic mouse model to show that defects in amygdalar circuits and activity result in autism-like behaviors. TBR1 controls the expression of a panel of genes that is associated with ASDs. TBR1 regulates axonal growth and the neuronal activation of amygdalar neurons by regulating downstream genes. Enhancing NMDAR activation with D-cycloserine and clioquinol to increase neuronal activity can ameliorate the behavioral defects of Tbr1+∕− mice, although the anatomic defects caused by Tbr1 haploinsufficiency are not rescued. Tbr1+∕− mice thereby serve as a model to elucidate how mutation of an autism causative gene influences brain wiring and impairs neuronal activity and consequently results in autism-like behaviors. Nevertheless, several issues remain unresolved. First, why is the amygdala the brain structure most sensitive to Tbr1 haploinsufficiency? Second, since Tbr1+∕− amygdalar neurons do not correctly form inter- and intra-amygdalar connections, a study of the mistargeting of Tbr1+∕− amygdalar axons might further illustrate the neural circuit defects caused by Tbr1 haploinsufficiency. Third, although the anatomical features of the cerebral cortex of Tbr1+∕− mice are normal, the electrophysiological responses of the Tbr1+∕− cerebral cortex remain to be measured. Fourth, the TBR1 downstream genes have not been annotated in detail. Only four TBR1 direct target genes have so far been identified. Further work is necessary to understand the individual actions of other TBR1 downstream target genes and how their dysfunction could generate autism-like behaviors. Finally, it is unclear how TBR1 functions as a transcriptional activator in some cases but acts as a repressor in others. This phenomenon also deserves further investigation. Addressing these questions will further elucidate the roles of TBR1 in brains and potentially impact on autism research.
Author Contributions
Both YH and TH wrote the manuscript and prepared the tables and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Academia Sinica (AS-103-TP-B05) and the Ministry of Science and Technology (MOST 103-2321-B-001-002, 103-2321-B-001-018, and 104-2321-B-001-050) to YH. TH is supported by a Postdoctoral Fellowship from Academia Sinica.
References
Bedogni, F., Hodge, R. D., Elsen, G. E., Nelson, B. R., Daza, R. A., Beyer, R. P., et al. (2010). Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. U.S.A. 107, 13129–13134. doi: 10.1073/pnas.1002285107
Bekirov, I. H., Nagy, V., Svoronos, A., Huntley, G. W., and Benson, D. L. (2008). Cadherin-8 and N-cadherin differentially regulate pre- and postsynaptic development of the hippocampal mossy fiber pathway. Hippocampus 18, 349–363. doi: 10.1002/hipo.20395
Bernardinelli, Y., Nikonenko, I., and Muller, D. (2014). Structural plasticity: mechanisms and contribution to developmental psychiatric disorders. Front. Neuroanat. 8:123. doi: 10.3389/fnana.2014.00123
Blundell, J., Blaiss, C. A., Etherton, M. R., Espinosa, F., Tabuchi, K., Walz, C., et al. (2010). Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010
Bulfone, A., Smiga, S. M., Shimamura, K., Peterson, A., Puelles, L., and Rubenstein, J. L. (1995). T-brain-1: a homolog of Brachyury whose expression defines molecularly distinct domains within the cerebral cortex. Neuron 15, 63–78. doi: 10.1016/0896-6273(95)90065-9
Bulfone, A., Wang, F., Hevner, R., Anderson, S., Cutforth, T., Chen, S., et al. (1998). An olfactory sensory map develops in the absence of normal projection neurons or GABAergic interneurons. Neuron 21, 1273–1282. doi: 10.1016/S0896-6273(00)80647-9
Burrage, L. C., Eble, T. N., Hixson, P. M., Roney, E. K., Cheung, S. W., and Franco, L. M. (2013). A mosaic 2q24.2 deletion narrows the critical region to a 0.4 Mb interval that includes TBR1, TANK, and PSMD14. Am. J. Med. Genet. A 161A, 841–844. doi: 10.1002/ajmg.a.35751
Cellot, G., and Cherubini, E. (2014). GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr 2:70. doi: 10.3389/fped.2014.00070
Chuang, H. C., Huang, T. N., and Hsueh, Y. P. (2014). Neuronal excitation upregulates Tbr1, a high-confidence risk gene of autism, mediating Grin2b expression in the adult brain. Front. Cell. Neurosci. 8:280. doi: 10.3389/fncel.2014.00280
Chuang, H. C., Huang, T. N., and Hsueh, Y. P. (2015). T-Brain-1 - A Potential Master Regulator in Autism Spectrum Disorders. Autism Res. 8:412. doi: 10.1002/aur.1456
Deidda, G., Bozarth, I. F., and Cancedda, L. (2014). Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci. 8:119. doi: 10.3389/fncel.2014.00119
de Napoles, M., Mermoud, J. E., Wakao, R., Tang, Y. A., Endoh, M., Appanah, R., et al. (2004). Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676. doi: 10.1016/j.devcel.2004.10.005
Deriziotis, P., O'roak, B. J., Graham, S. A., Estruch, S. B., Dimitropoulou, D., Bernier, R. A., et al. (2014). De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat. Commun. 5, 4954. doi: 10.1038/ncomms5954
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Enard, W., Przeworski, M., Fisher, S. E., Lai, C. S., Wiebe, V., Kitano, T., et al. (2002). Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418, 869–872. doi: 10.1038/nature01025
Furley, A. J., Morton, S. B., Manalo, D., Karagogeos, D., Dodd, J., and Jessell, T. M. (1990). The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 61, 157–170. doi: 10.1016/0092-8674(90)90223-2
Gao, Z., Lee, P., Stafford, J. M., von Schimmelmann, M., Schaefer, A., and Reinberg, D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. doi: 10.1038/nature13921
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi: 10.1016/j.neuron.2011.05.021
Gong, X., Jia, M., Ruan, Y., Shuang, M., Liu, J., Wu, S., et al. (2004). Association between the FOXP2 gene and autistic disorder in Chinese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 127B, 113–116. doi: 10.1002/ajmg.b.20162
Hamdan, F. F., Srour, M., Capo-Chichi, J. M., Daoud, H., Nassif, C., Patry, L., et al. (2014). De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10:e1004772. doi: 10.1371/journal.pgen.1004772
Han, W., Kwan, K. Y., Shim, S., Lam, M. M., Shin, Y., Xu, X., et al. (2011). TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc. Natl. Acad. Sci. U.S.A. 108, 3041–3046. doi: 10.1073/pnas.1016723108
Hevner, R. F., Shi, L., Justice, N., Hsueh, Y., Sheng, M., Smiga, S., et al. (2001). Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29, 353–366. doi: 10.1016/S0896-6273(01)00211-2
Hong, C. J., and Hsueh, Y. P. (2007). Cytoplasmic distribution of T-box transcription factor Tbr-1 in adult rodent brain. J. Chem. Neuroanat. 33, 124–130. doi: 10.1016/j.jchemneu.2007.01.005
Hsueh, Y. P. (2006). The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 13, 1915–1927. doi: 10.2174/092986706777585040
Hsueh, Y. P. (2009). Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann. Neurol. 66, 438–443. doi: 10.1002/ana.21755
Hsueh, Y. P., Wang, T. F., Yang, F. C., and Sheng, M. (2000). Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404, 298–302. doi: 10.1038/35005118
Huang, T. N., Chang, H. P., and Hsueh, Y. P. (2010). CASK phosphorylation by PKA regulates the protein-protein interactions of CASK and expression of the NMDAR2b gene. J. Neurochem. 112, 1562–1573. doi: 10.1111/j.1471-4159.2010.06569.x
Huang, T. N., Chuang, H. C., Chou, W. H., Chen, C. Y., Wang, H. F., Chou, S. J., et al. (2014). Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci. 17, 240–247. doi: 10.1038/nn.3626
Iossifov, I., O'roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Janak, P. H., and Tye, K. M. (2015). From circuits to behaviour in the amygdala. Nature 517, 284–292. doi: 10.1038/nature14188
Kunz, S., Spirig, M., Ginsburg, C., Buchstaller, A., Berger, P., Lanz, R., et al. (1998). Neurite fasciculation mediated by complexes of axonin-1 and Ng cell adhesion molecule. J. Cell Biol. 143, 1673–1690. doi: 10.1083/jcb.143.6.1673
Lai, C. S., Fisher, S. E., Hurst, J. A., Vargha-Khadem, F., and Monaco, A. P. (2001). A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413, 519–523. doi: 10.1038/35097076
Lee, E. J., Choi, S. Y., and Kim, E. (2015a). NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol. 20, 8–13. doi: 10.1016/j.coph.2014.10.007
Lee, E. J., Lee, H., Huang, T. N., Chung, C., Shin, W., Kim, K., et al. (2015b). Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat. Commun. 6, 7168. doi: 10.1038/ncomms8168
Lee, S., Kim, S. J., Kwon, O. B., Lee, J. H., and Kim, J. H. (2013). Inhibitory networks of the amygdala for emotional memory. Front. Neural Circuits 7:129. doi: 10.3389/fncir.2013.00129
Li, H., Yamagata, T., Mori, M., and Momoi, M. Y. (2005). Absence of causative mutations and presence of autism-related allele in FOXP2 in Japanese autistic patients. Brain Dev. 27, 207–210. doi: 10.1016/j.braindev.2004.06.002
Martinez-Cerdeno, V., and Noctor, S. C. (2014). Cajal, Retzius, and Cajal-Retzius cells. Front. Neuroanat. 8:48. doi: 10.3389/fnana.2014.00048
McKenna, W. L., Betancourt, J., Larkin, K. A., Abrams, B., Guo, C., Rubenstein, J. L., et al. (2011). Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J. Neurosci. 31, 549–564. doi: 10.1523/JNEUROSCI.4131-10.2011
Molnar, M., Potkin, S. G., Bunney, W. E., and Jones, E. G. (2003). MRNA expression patterns and distribution of white matter neurons in dorsolateral prefrontal cortex of depressed patients differ from those in schizophrenia patients. Biol. Psychiatry 53, 39–47. doi: 10.1016/S0006-3223(02)01456-7
Najm, J., Horn, D., Wimplinger, I., Golden, J. A., Chizhikov, V. V., Sudi, J., et al. (2008). Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 40, 1065–1067. doi: 10.1038/ng.194
Nakashiba, T., Nishimura, S., Ikeda, T., and Itohara, S. (2002). Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mech. Dev. 111, 47–60. doi: 10.1016/S0925-4773(01)00600-1
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Ohshima, T. (2014). Neuronal migration and protein kinases. Front. Neurosci. 8:458. doi: 10.3389/fnins.2014.00458
O'roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
O'roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O'roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Palumbo, O., Fichera, M., Palumbo, P., Rizzo, R., Mazzolla, E., Cocuzza, D. M., et al. (2014). TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med. Genet. A 164A, 828–833. doi: 10.1002/ajmg.a.36363
Papaioannou, V. E. (2014). The T-box gene family: emerging roles in development, stem cells and cancer. Development 141, 3819–3833. doi: 10.1242/dev.104471
Remedios, R., Huilgol, D., Saha, B., Hari, P., Bhatnagar, L., Kowalczyk, T., et al. (2007). A stream of cells migrating from the caudal telencephalon reveals a link between the amygdala and neocortex. Nat. Neurosci. 10, 1141–1150. doi: 10.1038/nn1955
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183X.2003.00037.x
Samuels, B. A., Hsueh, Y. P., Shu, T., Liang, H., Tseng, H. C., Hong, C. J., et al. (2007). Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron 56, 823–837. doi: 10.1016/j.neuron.2007.09.035
Sekine, K., Kubo, K., and Nakajima, K. (2014). How does Reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci. Res. 86, 50–58. doi: 10.1016/j.neures.2014.06.004
Stoeckli, E. T., and Landmesser, L. T. (1995). Axonin-1, Nr-CAM, and Ng-CAM play different roles in the in vivo guidance of chick commissural neurons. Neuron 14, 1165–1179. doi: 10.1016/0896-6273(95)90264-3
Traylor, R. N., Dobyns, W. B., Rosenfeld, J. A., Wheeler, P., Spence, J. E., Bandholz, A. M., et al. (2012). Investigation of TBR1 hemizygosity: four individuals with 2q24 microdeletions. Mol. Syndromol. 3, 102–112. doi: 10.1159/000342008
Walsh, C. A., Morrow, E. M., and Rubenstein, J. L. (2008). Autism and brain development. Cell 135, 396–400. doi: 10.1016/j.cell.2008.10.015
Wang, G. S., Hong, C. J., Yen, T. Y., Huang, H. Y., Ou, Y., Huang, T. N., et al. (2004a). Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 42, 113–128. doi: 10.1016/S0896-6273(04)00139-4
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004b). Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878. doi: 10.1038/nature02985
Wang, T. F., Ding, C. N., Wang, G. S., Luo, S. C., Lin, Y. L., Ruan, Y., et al. (2004c). Identification of Tbr-1/CASK complex target genes in neurons. J. Neurochem. 91, 1483–1492. doi: 10.1111/j.1471-4159.2004.02845.x
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Keywords: amygdala, axonal projection, autism, cerebral cortex, intellectual disability, neural circuit, neurodevelopmental disorders, TBR1
Citation: Huang T-N and Hsueh Y-P (2015) Brain-specific transcriptional regulator T-brain-1 controls brain wiring and neuronal activity in autism spectrum disorders. Front. Neurosci. 9:406. doi: 10.3389/fnins.2015.00406
Received: 29 April 2015; Accepted: 12 October 2015;
Published: 03 November 2015.
Edited by:
Gul Dolen, Johns Hopkins University, USAReviewed by:
Marc Fuccillo, University of Pennsylvania, USAMollie Meffert, Johns Hopkins School of Medicine, USA
Copyright © 2015 Huang and Hsueh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Ping Hsueh, eXBoQGdhdGUuc2luaWNhLmVkdS50dw==