 Maria G. Morgese1†
Maria G. Morgese1† Marilena Colaianna1,2†
Marilena Colaianna1,2† Emanuela Mhillaj1,3
Emanuela Mhillaj1,3 Margherita Zotti1
Margherita Zotti1 Stefania Schiavone1
Stefania Schiavone1 Palma D'Antonio1
Palma D'Antonio1 Andrew Harkin4
Andrew Harkin4 Valentina Gigliucci4
Valentina Gigliucci4 Patrizia Campolongo3
Patrizia Campolongo3 Viviana Trezza5
Viviana Trezza5 Angelo De Stradis6
Angelo De Stradis6 Paolo Tucci1
Paolo Tucci1 Vincenzo Cuomo3
Vincenzo Cuomo3 Luigia Trabace1*
Luigia Trabace1*- 1Department of Clinical and Experimental Medicine, University of Foggia, Foggia, Italy
- 2Department of Pathology and Immunology, University of Geneva, Geneva, Switzerland
- 3Department of Physiology and Pharmacology, La Sapienza, University of Rome, Rome, Italy
- 4Neuropsychopharmacology Research Group, School of Pharmacy and Pharmaceutical Sciences and Trinity College Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland
- 5Section of Biomedical Sciences and Technologies, Department of Science, University “Roma Tre,” Rome, Italy
- 6Department of Bio Agro-Food Sciences, The Institute of Sustainable Plant Protection, National Research Council, Bari, Italy
Strong evidence showed neurotoxic properties of beta amyloid (Aβ) and its pivotal role in the Alzheimer's disease (AD) pathogenesis. Beside, experimental data suggest that Aβ may have physiological roles considering that such soluble peptide is produced and secreted during normal cellular activity. There is now suggestive evidence that neurodegenerative conditions, like AD, involve nitric oxide (NO) in their pathogenesis. Nitric oxide also possess potent neuromodulatory actions in brain regions, such as prefrontal cortex (PFC), hippocampus (HIPP), and nucleus accumbens (NAC). In the present study, we evaluated the effect of acute Aβ injection on norepinephrine (NE) content before and after pharmacological manipulations of nitrergic system in above mentioned areas. Moreover, effects of the peptide on NOS activity were evaluated. Our data showed that 2 h after i.c.v. soluble Aβ administration, NE concentrations were significantly increased in the considered areas along with increased iNOS activity. Pre-treatment with NOS inhibitors, 7-Nitroindazole (7-NI), and N6-(1-iminoethyl)-L-lysine-dihydrochloride (L-NIL), reversed Aβ-induced changes. Ultimately, pharmacological block of interleukin1 (IL-1) receptors prevented NE increase in all brain regions. Taken together our findings suggest that NO and IL-1 are critically involved in regional noradrenergic alterations induced by soluble Aβ injection.
Introduction
Amyloid beta peptide (Aβ) is the main component of the amyloid plaques, one of the neuropathological hallmarks of Alzheimer's disease (AD; Di Carlo et al., 2012). It has been hypothesized that the peptide accumulation initiated by a pathogenic cascade ultimately leads to AD (Hardy and Selkoe, 2002). However, mounting evidence suggest that Aβ, besides its well accepted neurotoxic activity, may have physiological roles (Mura et al., 2010a). The peptide, in its soluble form, is produced and secreted during normal cellular activity (Kar et al., 2004) and is able to modulate synaptic activity in the absence of neurotoxicity (Grilli et al., 2010; Mura et al., 2010b). Physiologically, the neuromodulatory role of Aβ would be important for the right functioning of neurotransmitter systems; however, in pathological conditions, Aβ-modulating synaptic activity could trigger functional alterations on neurotransmission (Mura et al., 2012).
On the other hand, several hints point to the involvement of the free radical gas, nitric oxide (NO), in the pathogenesis of neurodegenerative conditions, such as AD (Guix et al., 2005; Fernandez et al., 2010). In the brain, NO is synthesized from L-Arginine (L-Arg) either by inducible nitric oxide synthase (iNOS) in microglia and astrocytes, or by constitutive NOS in neurons and endothelial cells (nNOS and eNOS, respectively). It has been suggested that NO produced by constitutive NOS is accountable for neuroprotection from Aβ-induced cell death, while NO release, following iNOS activation, plays a neurotoxic role (Puzzo et al., 2006). The three NOS isoforms have been proposed to mediate Aβ effects, producing elevated levels of NO, which could contribute to worsen the outcomes of the disease (Fernandez et al., 2010). Although NO behaves as a neurotoxic effector when produced in excessive amount, increasing evidence focus on its neuroprotective properties in mediating cellular transduction mechanisms, regulating neuronal plasticity (Palumbo et al., 2007) and suppressing neuronal apoptotic cell death (Contestabile and Ciani, 2004). By playing a potent neuromodulatory role in several brain regions, such as prefrontal cortex (PFC), hippocampus (HIPP), nucleus accumbens (NAC), and striatum (Trabace et al., 2004, 2007; Saulskaya et al., 2010; Bechade et al., 2011), NO has been presented either as neuroprotective or restorative in neurodegenerative conditions such as AD (Puzzo et al., 2005). Recent evidence linked Aβ effects to the nitrergic pathway; indeed, as suggested by experiments performed in mouse and human cells, Aβ caused a decrease in the activity of the soluble guanylate cyclase sinking NO signaling by binding to the cell surface receptor CD36 (Miller et al., 2010). Therefore, the enhancement of NO levels can be proposed as protective toward neurons from their degeneration and death.
Interestingly, there is several in vivo and in vitro evidence supporting the NO pathway's role in the mechanism of norepinephrine (NE)-induced neuroprotection (Lonart et al., 1992; Chen et al., 2006; Chen and Russo-Neustadt, 2007). Indeed, in primary hippocampal neuronal cell culture, NE stimulation leads to an increased activation of several pathways, whose function has been proposed to be dependent on NO availability. Furthermore, such activation seems to be decreased in presence of NOS inhibition (Chen and Russo-Neustadt, 2007). In addition, it has been shown that NE promotes cell survival both in vivo (Chen and Russo-Neustadt, 2005) and in the primary hippocampal neuronal cell culture (Chen and Russo-Neustadt, 2007). Previous reports also indicate that NE is an important player in innate immunosuppressive maintenance within the central nervous system (Feinstein et al., 2002; Heneka et al., 2002); it has been reported that NE holds anti-inflammatory properties by modulating microglial functions following the degeneration of aminergic locus coeruleus (LC) neurons induced in APP-transgenic mice by using the neurotoxin DSP4 (Heneka et al., 2010). In particular, it has been shown that NE, through the activation of β2 adrenoceptors present on microglial cells, is able to promote endocytosis and degradation of Aβ42 (Kong et al., 2010). Furthermore, NE can significantly reduce glial iNOS via activation of β2 adrenoceptors and cAMP increase (Kalinin et al., 2006).
Based on the above reported background, we were firstly interested in evaluating the interplay among Aβ peptide, noradrenergic, and nitrergic transmission. To this end, we evaluated effects of an acute soluble Aβ injection on noradrenergic neurotransmission; moreover, by using NOS inhibitors, we tested if such pharmacological manipulations of the nitrergic system were able to modulate the neurochemical effects induced by Aβ peptide in brain areas involved in AD, such as PFC, HIPP, and NAC.
In addition, we have previously demonstrated that intrahippocampal injection of soluble Aβ potently increases interleukin-1β levels (Sanz et al., 2009). Such cytokine has been reported to act at distance with NO mediating central NE effect (Hsieh et al., 2010). Thus, we further investigated the effect of the block of IL-1 receptors on NE content of above cited areas in soluble Aβ-treated animals.
Materials and Methods
Animals
A total of 78 young adult male (250–300 g) Wistar rats (Harlan, S. Pietro al Natisone, Udine) were used in this study. They were housed at constant room temperature (22 ± 1°C) and relative humidity (55 ± 5%) under a 12 h light/dark cycle with ad libitum access to standard food and water. Procedures involving animals and their care were conducted in conformity with the institutional guidelines of the Italian Ministry of Health (D.L. 26/2014), the Guide for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council, 2004), the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. All procedures involving animals were conducted in accordance to ARRIVE guidelines. Animal welfare was daily monitored through the entire period of experimental procedures. No signs of distress were evidenced; anyway, all efforts were made to minimize the number of animals used and their suffering.
Cannula Implantation and Aβ Administration
Cannula implantation was performed as previously described (Trabace et al., 2007). Briefly, unilateral 23-gauge stainless steel guide cannulae (Cooper's Needles, Birmingham, UK) were implanted using the following coordinates relative to bregma: AP = −0.5, L = +1.2, H = −3.0 with the incisor bar set at −3.3 mm, according to a stereotaxic atlas (Paxinos and Watson, 1998). On the day of the experiment, Aβ(1-42) fresh solution, at a concentration previously tested (4 μM, in distilled water; Colaianna et al., 2010), was delivered via a 30-gauge needle lowered 2 mm below the cannula tip. The solution was then infused at a flow rate of 2 μL/min for 2 min 30 s through a Hamilton syringe connected to a microdialysis pump (CMA microdialysis, Sweden).
Atomic Force Microscopy (AFM) and Transmission Electron Microscopy (TEM)
Samples for AFM and TEM measurements were obtained by preparing a 4 μM solution of Aβ, both for the “initial state” and the “oligomers” phase. Freshly prepared 4 μM solution of Aβ was used immediately (“initial state solution”). To obtain Aβ oligomers, a different 4 μM solution of Aβ was prepared in 50 mM phosphate buffer and 150 mM NaCl (pH 7.4), and incubated for 24 h at 4°C (“oligomers” solution; Lambert et al., 1998).
For AFM analyses, some drops of the solution were deposited on a freshly cleaved mica substrate. After an incubation of 2 min, samples were washed with ultrapure water and dried for 30 min in a vacuum desiccator.
A Perception Atomic Force Microscope (Assing S.p.A., Italy) was used to record AFM images. Measurements were performed in air, working in the weak repulsive regime of contact mode. Gold coated Si3N4 cantilevers (model MSNL from Bruker Company, Massachusetts, USA) with a spring constant of 0.01 N/m and Silicon tips with a nominal apical radius of 2–12 nm were used for the topographic AFM images. Constant force images were acquired with a scan rate of about 8 s/row, at a resolution of 512 × 512 points.
For TEM analyses, a little drop (20 μL) of each incubated sample solution was applied to carbon coated copper/rhodium grid (400 mesh; TAAB Laboratories Equipment Ltd, Aldermaston, Berks, GB). The coated grid was floated for 2 min on the sample drop and rinsed with 200 mL of double distilled water. Negative staining was performed with 200 μL of 2% w/v uranyl acetate solution (TAAB Laboratories Equipment Ltd). After draining off the excess of staining solution by means of a filter paper, the specimen was transferred for examination in a Philips Morgagni 282D transmission electron microscope, operating at 60 kV. Electron micrographs of negatively stained samples were photographed on Kodak electron microscope film 4489 (Kodak Company, New York, USA).
Pharmacological Manipulation
Thirty minute before Aβ administration, NOS inhibitors 7-Nitroindazole (7-NI) (50 mg kg−1, Vinci Biochem, Florence, Italy), and N6-(1-iminoethyl)-L-lysine-dihydrochloride (L-NIL) (5 mg kg−1, Vinci Biochem, Florence, Italy) were dissolved in saline (1 ml kg−1, veh) and administered intraperitoneally (i.p.) (Trabace et al., 2007) according to the experimental assignment. Interleukin-1 receptor antagonist (IL-1ra, Sigma-Aldrich, Milan, Italy) was applied i.c.v. at a concentration of 100 ng 5 μl−1 per rat (Taepavarapruk and Song, 2010). Drugs were administered 30 min (NOS inhibitors) or 10 min (IL-1ra) before Aβ injection based upon our previous experience and following a survey of the relevant literature (Trabace et al., 2007; Choi et al., 2009).
Post-mortem Tissue Analysis
Rats were euthanized and brains were immediately removed 2 h after Aβ administration. Thereafter, PFC, NAC, and HIPP were collected and stored frozen at −80°C until analyses. Samples were treated and NE and serotonin (5-HT) tissue concentrations were determined by using high performance liquid chromatography (HPLC) coupled with electrochemical detection as previously described (Trabace et al., 2011).
HPLC Analysis
NE and 5-HT concentrations were determined by HPLC coupled with an electrochemical detector (INTRO, Antec Leyden, The Netherlands). Separation was performed by a LC18 reverse phase column (hypersil, 150 × 3 mm, ODS 5 μm; Thermoscientific, Milan, Italy). The detection was accomplished by a Unijet cell (BASi, Kenilworth, U.K.) with a 6 mm diameter glassy carbon electrode at a working potential of 0.65 V vs. Ag/AgCl. The mobile phase used was 85 mM CH3COONa, 0.8 mM octane sulfonic acid, 0.3 mM EDTA, 15 mM NaCl, methanol 6%, in distilled water, buffered at pH 4.85. The flow rate was maintained by an isocratic pump (Shimadzu LC-10 AD, Kyoto, Japan) at 1 ml/min. Data were acquired and integrated using Chromeleon software (version 6.60, Dionex, San Donato Milanese, Italy).
Quantification of NOS Gene Expression
RNA Extraction
Total RNA was extracted from PFC, NAC, and HIPP by using Nucleospin RNA II kits (Macherey-Nagel, Dublin, Ireland) according to the manufacturer's instructions. Total RNA concentrations were measured with a NanoDrop® (Thermo Scientific, Dublin, Ireland) micro-volume spectrophotometer and all RNA samples were equalized with RNase-free water to the lowest detected concentration.
cDNA Synthesis
cDNA was synthesized using the ABI High Capacity cDNA kit (Applied Biosystems, life technologies, Paisley, U.K.) as provided by the protocol. A 2X master-mix solution containing reverse transcription buffer, dNTPs, random primers, and MultiScribeTM reverse transcriptase was made up in RNase-free water and stored on ice. The reverse transcription reaction was performed in a thermocycler (PTC-200, MJ Research) with a three-step program: 10 min at 25°C followed by 120 min at 37°C and a final 5 min step at 85°C. cDNA samples were used immediately for real time PCR or stored at −20°C until needed.
Real-time PCR
Real-Time PCR was performed as multiplex using TaqMan® Gene Expression Assays in accordance with the manufacturer's instructions (Applied Biosystems, life technologies, Paisley, U.K.). The target genes nNOS (Rn00583793), iNOS (Rn00561646), and eNOS (Rn02132634) were identified by FAM-labeled probes, while the housekeeping gene of reference, β–actin (4352340E), was identified by a VIC-labeled probe. The mixture was prepared by adding a 1:5 dilution of the cDNA sample obtained from the previous reaction into a 96-well optical reaction plate (Applied Biosystems, life technologies, Paisley, U.K.), followed by 2X TaqMan® Universal PCR Master Mix (No AmpErase® UNG, Applied Biosystems, life technologies, Paisley, U.K.) and 20X primers for both the target gene and β-actin (to reach a final 1X concentration). Amplification reaction was achieved with a three-stage protocol: 2 min at 50°C followed by 10 min at 95°C, followed again by 40 repetitions of a two-step cycle composed by 15 s at 95°C (denaturation) and 1 min at 60°C (annealing and extension) (ABI Prism 7300 instrument, Applied Biosystems, life technologies, Paisley, U.K.). Data analysis was performed with the 7300 System Software (Applied Biosystems, life technologies, Paisley, U.K.) and RQ values (2-ΔΔCT, where CT is the threshold cycle) of the target genes relative to their own endogenous control were obtained. The RQ values were then converted into fold change values relative to control group. β-actin was used as the reference housekeeping gene in this study.
Nitrite/Nitrate (NOx) Analysis
NOx levels were estimated using an ion chromatographic with conductometric detection method. The brain areas were suspended (1:20, w/v) in ultrapure water and placed at 70°C for 5 min. After cooling, the mixture was centrifuged for 5 min at 1500 × g at room temperature. Chromatographic determinations were performed on a Dionex DX-500 system (Dionex Corporation, Sunnyvale, CA, USA) injecting 25 μL of the supernatant. Chromatographic separations were accomplished using an IonPac AS9-HC column (250 × 4 mm) associated with a pre-column AG9-HC (4 mm, Dionex Corporation) eluted in isocratic mode at a flow rate of 1.0 mL/min. The mobile phase consisted of 9 mM sodium carbonate (total run time, 20 min) (Iammarino et al., 2012).
Statistical Analysis
Results were expressed as percentage of control (SHAM-veh) and mean ± S.E.M. Statistical analyses were performed using Graph Pad 5.0 (GraphPad Software, San Diego, CA) and Systat13 (Systat Software, Inc. San Jose, CA) for Windows. Neurotransmitter concentrations were analyzed using Two-way analysis of variance (ANOVA) followed by Bonferroni multiple comparisons test. NOx concentrations and NOS mRNA content were analyzed by using Student's t-test for independent comparison and Two-way ANOVA followed by Bonferroni multiple comparisons test, as required. Differences were considered statistically significant when P-value was less than 0.05.
Results
Evaluation of Aβ State through AFM and TEM Measurements
Figure 1 shows AFM images of “initial state” and “oligomers” solution of Aβ deposited on a freshly cleaved mica disk. It is evident that in both cases Aβ particles with different sizes are distributed on the mica substrate. The size of such particles was estimated by sphericity assumption, considering height values, because the lateral size is influenced by tip-particle convolution. In fact, Figure 1A showed that the “initial state” solution contained many small particles and very few larger particles. As indicated in Figure 1B, the distribution of height values of the small particles (height less than about 2.5 nm), peaked in the 0.9–1.2 nm range with a mean value of 1.2 ± 0.5 nm. The height of the few larger particles (height more than about 2.5 nm) was more uniformly distributed and was characterized by a mean value of 3.9 ± 0.6 nm. After 24 h incubation at 4°C and pH 7.4, particles assembled in larger aggregates, as shown in the AFM image of “oligomers” solution (Figure 1C). In this case, the aggregate sizes were characterized by a bimodal distribution (Figure 1D), where the two modal values were related to aggregates with a height size value smaller or larger than 9 nm, respectively. The smaller aggregates presented a mean height size of 4.4 ± 1.7 nm, whereas the mean height size of larger aggregates was 22.7 ± 6.7 nm. TEM analyses confirmed results obtained from AFM; indeed, as shown in Figure 1E, in the “initial state solution” the higher percentage of molecules were monomers, while in the “oligomer state solution” oligomers and fibrils were the most representative species (Figure 1F).
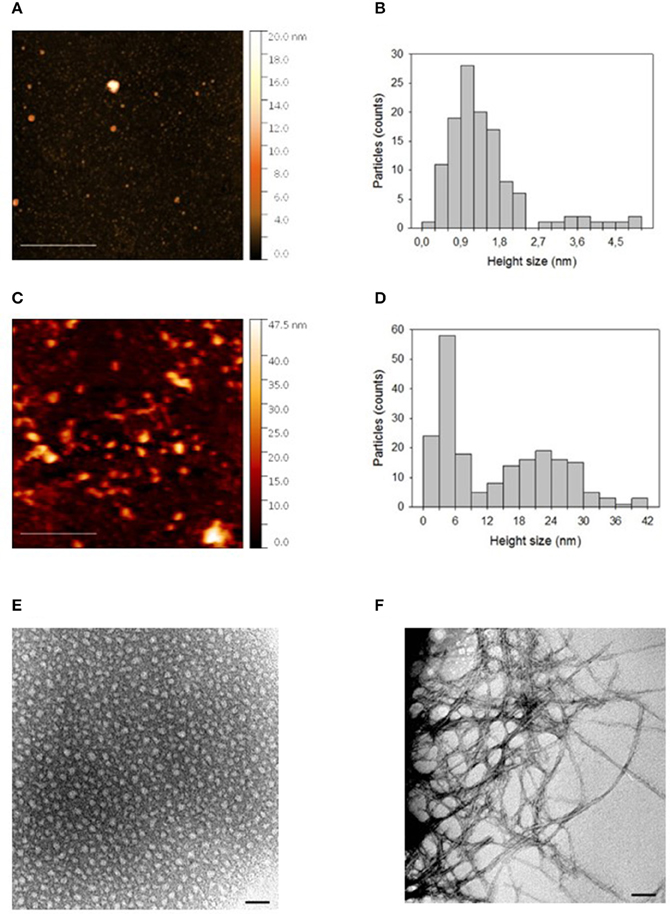
Figure 1. (A) AFM picture measured in contact mode by using Silicon tips having a nominal apical radius of 2–12 nm and (B) histogram representing the distribution of height size values of Aβ “initial state” solution deposited on a freshly cleaved mica disk. In (C,D) are reported the corresponding AFM picture and distribution of height size values for an “oligomer” solution of Aβ deposited on a freshly cleaved mica disk. The color scale reported on the right hand side of each AFM picture represents the height values of the measured structures. In (E,F), TEM pictures of “initial state” and “oligomer state” solutions, respectively, are reported. The horizontal scale bar was 500 nm for AFM and 50 nm for TEM picture.
Effects of Nitrergic Manipulation on Regional NE and 5-HT Concentrations in Aβ-injected Rats
As shown in Figure 2, NE concentrations were higher in Aβ-treated when compared to SHAM rats in all of the three areas investigated (Two-way ANOVA with Bonferroni correction, PFC:P = 0.038; HIPP P = 0.044; and NAC P = 0.023). The administration of NOS inhibitors, L-NIL, and 7-NI, 30 min prior to administration of the soluble peptide, prevented this effect in all brain areas (Figure 2, Two-way ANOVA with Bonferroni correction, n.s.). No differences were found between Aβ-treated and SHAM rats in 5-HT content in all three areas investigated (data not shown), thus no pharmacological manipulation of nitrergic system was further carried out.
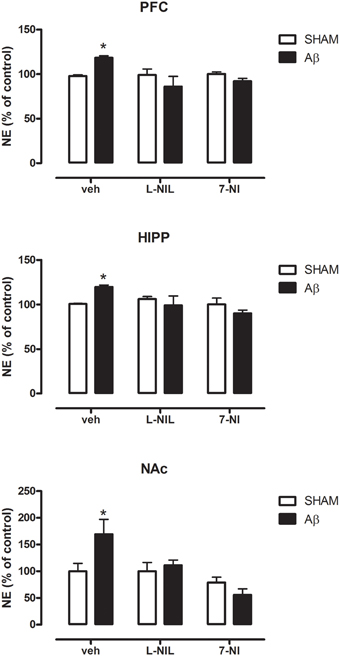
Figure 2. NE levels in PFC, HIPP, and NAC of male Wistar rats 2 h after injection of water (5 μL i.c.v.; SHAM) or Aβ (4 μM, 5 μL i.c.v.; Aβ). Animals were pre-treated with vehicle (saline, veh, 1 ml kg−1), L-NIL (5 mg kg−1, i.p.), or 7-NI (50 mg kg−1, i.p.), 30 min before i.c.v. administration. Data are expressed as percentage of control (n = 6–7 per group; Two-way ANOVA with Bonferroni correction, *P < 0.05).
Effects of Aβ Administration and Pharmacological Manipulation on NOS Expression
To investigate if Aβ administration was able to affect NOS expression, mRNA content of NOS enzymes (neuronal, endothelial, and inducible forms) was evaluated. A significant increase of iNOS was detected in the PFC (Student's t-test, P = 0.028), NAC (Student t-test, P = 0.041) and HIPP (Student's t-test, P = 0.031) of Aβ-treated rats, while nNOS was not affected in these brain areas (Figure 3). Moreover, results showed that eNOS mRNA was decreased only in HIPP (Student's t-test, P = 0.016), while no difference was evident in PFC and NAC (Figure 3). To evaluate iNOS activity, NOx levels were measured. Statistical analysis showed that the soluble peptide induced a strong increase in NOx levels in PFC (Student's t-test, P = 0.046), HIPP (Student's t-test, P = 0.003), and NAC (Student's t-test, P = 0.042; Figure 4).
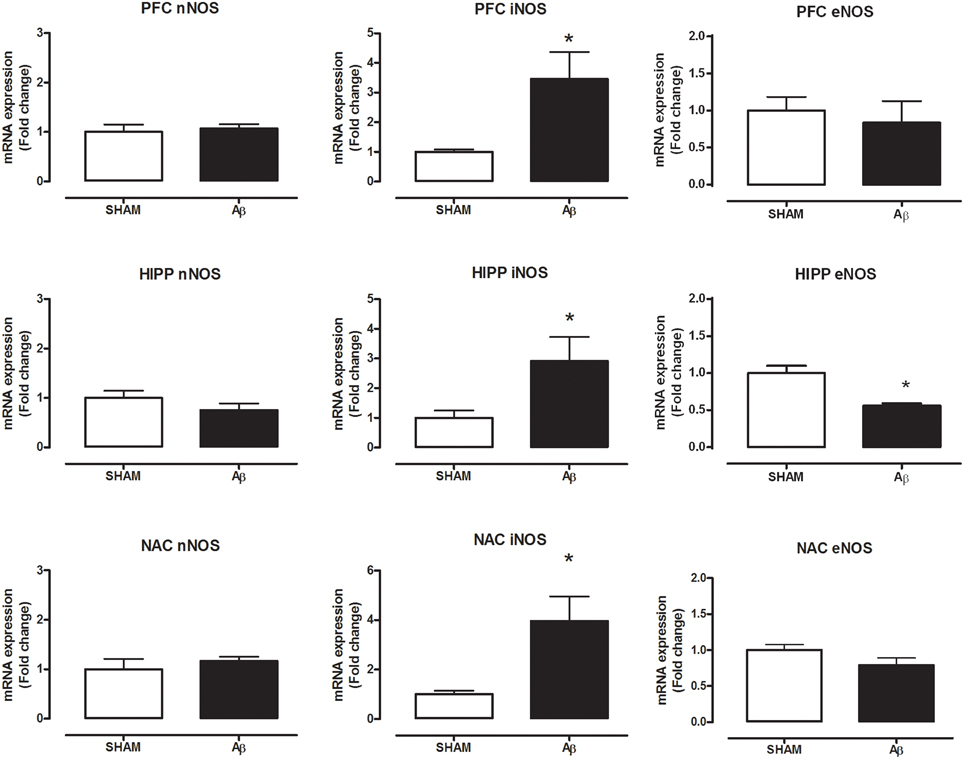
Figure 3. Neuronal NOS, iNOS, and eNOS mRNA content in PFC, HIPP, and NAC of male Wistar rats 2 h after injection of water (5 μL i.c.v.; SHAM) or Aβ (4 μM, 5 μL i.c.v.; Aβ). Data are expressed as mean ± SEM (n = 6 per group; Student's t-test, *P < 0.05).
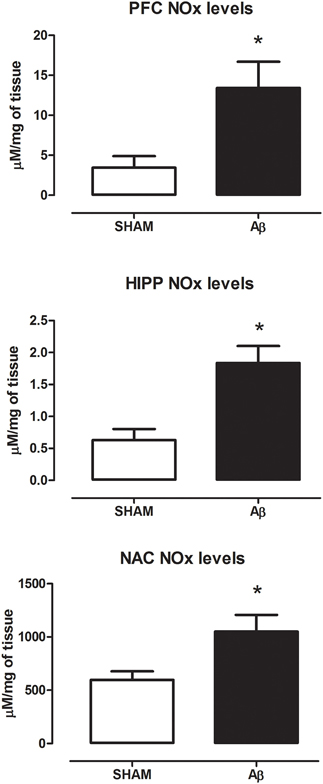
Figure 4. NOx levels in PFC, HIPP, and NAC of male Wistar rats 2 h after injection of water (5 μL i.c.v.; SHAM) or Aβ (4 μM, 5 μL i.c.v.; Aβ). Data are expressed as mean ± SEM (n = 6 per group; Student's t-test, *P < 0.05 for PFC and NAC and *P < 0.01 for HIPP).
Effects of Block of IL-1β Receptors on Aβ-induced NE Increase
In order to possibly establish a putative mechanism for the increase in NE Aβ-induced, we pre-treated the animals with the antagonist of IL-1 receptors, IL1ra. Results showed that in all investigated areas such treatment was able to prevent NE increase in Aβ-treated rats (Figure 5, Two-way ANOVA followed by Bonferroni post hoc test, n.s.).
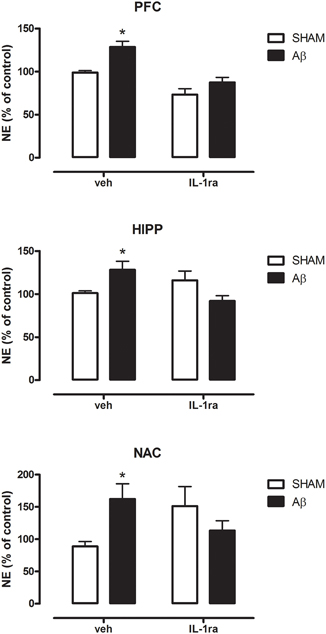
Figure 5. NE levels in PFC, HIPP, and NAC of male Wistar rats 2 h after injection of water (5 μL i.c.v.; SHAM) or Aβ (4 μM, 5 μL i.c.v.; Aβ). Animals received 10 min before an i.c.v. injection of vehicle (saline, veh, 5 μl) or interleukin-1 receptor antagonist (IL-1ra, 100 ng 5 μl−1). Data are expressed as percentage of control (n = 5–6 per group; Two-way ANOVA with Bonferroni correction, *P < 0.05).
Discussion
In the present study, we found that a single i.c.v. injection of freshly prepared Aβ solution induced a significant increase of NE concentrations in the PFC, NAC, and HIPP when measured 2 h after administration. Moreover, treatment with nitrergic modulators and with the antagonist of IL-1 receptor prevented NE increase in Aβ-treated rats. We also found that the increase was accompanied with an increase in iNOS mRNA levels along with NOx concentrations in all brain areas investigated.
Although alterations in neurotransmitter secretion have been extensively reported in the case of other monoaminergic transmitters (Trabace et al., 2007; Preda et al., 2008; Morgese et al., 2014), the exact mechanism through which Aβ may alter NE concentrations in these areas is not completely understood yet. However, we hypothesized that the altered Aβ-induced NE content may be caused by messengers/neurotransmitters such as NO. The free-radical gas NO is produced by NOS and it modulates vital physiological functions (Moncada et al., 1991; Murad, 2003) acting as an intercellular messenger, which possess also neuromodulatory actions (Garthwaite, 1991; Snyder and Bredt, 1991). Patel and coworkers have demonstrated in vitro that, at hippocampal level, the activation of the noradrenergic system is neuroprotective in stressful condition and that, at least partially, these protective actions are mediated by NO signaling (Patel et al., 2010). Additionally, NE seems to control both enzymatic activity and expression of nNOS and iNOS in rat magnocellular neurons (Grange-Messent et al., 2004). In line with these observations, in the present study, we demonstrated that the increase in NE concentrations was accompanied with higher iNOS mRNA, along with increased NOx concentrations, suggesting that the effects of Aβ could be associated with NO-related actions on the noradrenergic system. Indeed, we have found that pharmacological inhibition of the nitrergic system, 30 min before Aβ injection, prevented the increase in NE concentrations. Our hypothesis that noradrenergic system activation could be mediated by NO release after NOS induction was supported by the observation that the increase in NE concentrations was prevented in Aβ-treated rats with the administration of different NOS inhibitors, 7-NI, and L-NIL. Indeed, the administration of both compounds completely prevented the increase in NE concentrations in all areas investigated, indicating that activation of NOS is necessary for this action to occur.
Furthermore, the altered release of NE may be explained through the release of other neuromodulators, such as cytokines. In this regard, Kamikawa and colleagues have reported that increased NE levels occur in mPFC via indirect glutamate release. In particular, the activation of IL-1β receptors at glutamatergic terminals leads to the release of glutamate acting at non-NMDA receptors in nerve terminals that induce NOS activation and then NO production (Kamikawa et al., 1998). We have previously demonstrated that an intra-hippocampal administration of Aβ evoked an increased release of IL-1β through a purinergic receptor (P2X7)-mediated mechanism (Sanz et al., 2009). Thus, we hypothesized that Aβ-dependent release of IL-1β may be able to generate such a cascade. Our hypothesis was pharmacologically confirmed, considering that blocking the IL-1 receptors inhibited the Aβ-induced increase of NE. Furthermore, a direct action of IL-1β on its receptors localized on glutamatergic terminals in the mPFC seems to be responsible for an initial release of NE after IL-1β injection (Kamikawa et al., 1998). This observation appears very interesting for our research, since we have recently demonstrated that, in a microdialysis study conducted in this same experimental model, Aβ was able to elicit an increase in the release of glutamate in PFC 2h after a single i.c.v. injection (Tucci et al., 2014).
On the other hand, many reports have indicated that Aβ directly interacts with the noradrenergic system and in particular, that Aβ directly binds to adrenergic receptors (Igbavboa et al., 2006; Wang et al., 2011). Wang et al. (2011) demonstrated, in vitro that Aβ may cause desensitization and subsequently internalization of β2 adrenergic receptors in prefrontal cortical neurons (Wang et al., 2011).
This increase in noradrenergic tone could also reflect a neuroprotective phenomenon. In this regard, it has been reported that NA can regulate glial activation and a valid approach for neurodegeneration could rely on pharmacological strategies leading to increase NA (Braun et al., 2014). Furthermore, in vitro studies have evidenced a protective effect of NA toward toxicity Aβ-induced by increasing neurotrophic factor expression via activation of β adrenergic receptor signaling cascade (Counts and Mufson, 2010; Liu et al., 2015). Reduced NA concentrations in LC projecting areas facilitates the inflammatory reaction of microglial cells after Aβ exposure, thus impairing microglial migration and phagocytosis, thereby decreasing Aβ clearance (Heneka et al., 2010). Interestingly, it has been reported that NA inhibits iNOS induction after inflammatory stimuli in astrocytes (Feinstein et al., 1993) and microglia (Dello Russo et al., 2004).
On the other hand, in vitro studies indicate that Aβ can alter neurotransmitter release by interacting with presynaptic proteins (Russell et al., 2012). In particular, the peptide was reported to be able to be internalized at presynaptic level where it can disrupt the complex synaptophysin/vesicle associate membrane protein 2 (VAMP2) leading to increased vesicle priming and exocytosis (Russell et al., 2012). NO can also interfere with neurotransmission considering that cGMP signaling and S-nitrosylation of proteins can cause enhanced presynaptic binding of syntaxin with VAMP and synaptosomal-associated protein-25 (SNAP 25) (Meffert et al., 1996). Therefore, we can hypothesize that the increased NA levels found in our model can be the result of a presynaptic interaction of Aβ and NO signaling molecules. Accordingly, it has been shown that NO recruitment may represent a compensatory mechanism to enhance synaptic transmission and plasticity in a transgenic model of early AD (Chakroborty et al., 2015).
Here we also found that Aβ was able to decrease eNOS mRNA only in HIPP. Such result is in agreement with literature data showing that Aβ reduces eNOS activity (Gentile et al., 2004). However, in our experience only HIPP seems to be affected. A possible explanation may rely on anatomical structure since HIPP is a close region to injection area. Moreover, it has been reported that eNOS deficient mice have higher Aβ levels in HIPP, indicating a putative interconnection in this area considering that NO supplementation reduced Aβ production (Austin et al., 2010, 2013). In this light, several reports indicated that the effect of Aβ may be region specific (Kar et al., 2004; Mura et al., 2012).
Ultimately, in our experimental conditions, we might possibly exclude a widespread toxic property of soluble Aβ on brain neurotransmitters. Indeed, 5-HT concentrations were not altered by Aβ administration in all investigated areas at this time-point, while we have previously found that in PFC the peptide reduced such neurotransmitter content 7 days after i.c.v. infusion, indicating that 5-HT impairment needs a longer time to occur (Colaianna et al., 2010). Furthermore, we have previously shown that i.c.v. injection or retrodialysis of Aβ leads to a significant reduction in DA concentrations in the PFC (Trabace et al., 2007). Thus, the effects of Aβ seem to be neurotransmitter-specific. Moreover, we have previously shown that the used concentrations of Aβ are not associated, at least acutely, with gross neurotoxicity as shown by Hoechst nuclear staining (Trabace et al., 2007). However, we cannot exclude, after Aβ treatment, the presence of more subtle signs of toxicity. In this regard, evaluation of sign of toxicity/apoptotic pathway activation could represent an interesting future field of research.
In conclusion, we have demonstrated that the noradrenergic system seems to be activated possibly as a compensatory mechanism following soluble Aβ increased levels. In particular, the modulation of nitrergic system and involvement of IL-1 receptors seem to play a crucial role in this process. Our results contribute to shed light on the physiological interplay among soluble Aβ, nitrergic and noradrenergic transmissions in the brain.
Author Contributions
MM and MC, wrote the paper, and performed experiments (animal surgery, post mortem NE quantification). EM, MZ, and SS performed experiments (PCR analyses and Nox quantification). PD, AH, VG, AD performed experiments (PCR analyses, AFM quantifications and TEM analyses). PC, VT, and PT analyzed the data. VC and LT revised the paper and designed the research study. All the authors critically revised the work and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. A. Di Taranto, from the “Istituto Zooprofilattico Sperimentale della Puglia e della Basilicata, Via Manfredonia 20, 71121 Foggia, Italy” for her expert technical competence and assistance in NOx analyses. This study was supported by PRIN 2012 (to VC) from MIUR, by PRIN 2011 (to PT) from MIUR, by G. Montel award (to MM) from University of Foggia and by G. Montel award (to MC) from University of Foggia.
Abbreviations
AD, Alzheimer's disease; Aβ, soluble beta amyloid; 7-NI, 7-Nitroindazole; L-NIL, N6-(1-iminoethyl)-L-lysine-dihydrochloride; PFC, prefrontal cortex; HIPP, hippcampus; NAC, nucleus accumbens; NO, nitric oxide; iNOS, inducible Nitric oxide synthase; eNOS, endotelial Nitric oxide synthase; nNOS, neuronal Nitric oxide synthase; NOx, nitrates and nitrites; NE, norepinephrine; IL-1, interleukin 1; IL-1ra, interleukin 1 receptor antagonist.
References
Austin, S. A., d'Uscio, L. V., and Katusic, Z. S. (2013). Supplementation of nitric oxide attenuates AbetaPP and BACE1 protein in cerebral microcirculation of eNOS-deficient mice. J. Alzheimers Dis. 33, 29–33. doi: 10.3233/JAD-2012-121351
Austin, S. A., Santhanam, A. V., and Katusic, Z. S. (2010). Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ. Res. 107, 1498–1502. doi: 10.1161/CIRCRESAHA.110.233080
Béchade, C., Pascual, O., Triller, A., and Bessis, A. (2011). Nitric oxide regulates astrocyte maturation in the hippocampus: involvement of NOS2. Mol. Cell. Neurosci. 46, 762–769. doi: 10.1016/j.mcn.2011.02.009
Braun, D., Madrigal, J. L., and Feinstein, D. L. (2014). Noradrenergic regulation of glial activation: molecular mechanisms and therapeutic implications. Curr. Neuropharmacol. 12, 342–352. doi: 10.2174/1570159X12666140828220938
Chakroborty, S., Kim, J., Schneider, C., West, A. R., and Stutzmann, G. E. (2015). Nitric oxide signaling is recruited as a compensatory mechanism for sustaining synaptic plasticity in Alzheimer's disease mice. J. Neurosci. 35, 6893–6902. doi: 10.1523/JNEUROSCI.4002-14.2015
Chen, M. J., Ivy, A. S., and Russo-Neustadt, A. A. (2006). Nitric oxide synthesis is required for exercise-induced increases in hippocampal BDNF and phosphatidylinositol 3′ kinase expression. Brain Res. Bull. 68, 257–268. doi: 10.1016/j.brainresbull.2005.08.013
Chen, M. J., and Russo-Neustadt, A. A. (2005). Exercise activates the phosphatidylinositol 3-kinase pathway. Brain Res. Mol. Brain Res. 135, 181–193. doi: 10.1016/j.molbrainres.2004.12.001
Chen, M. J., and Russo-Neustadt, A. A. (2007). Nitric oxide signaling participates in norepinephrine-induced activity of neuronal intracellular survival pathways. Life Sci. 81, 1280–1290. doi: 10.1016/j.lfs.2007.09.003
Choi, D. Y., Liu, M., Hunter, R. L., Cass, W. A., Pandya, J. D., Sullivan, P. G., et al. (2009). Striatal neuroinflammation promotes Parkinsonism in rats. PLoS ONE 4:e5482. doi: 10.1371/journal.pone.0005482
Colaianna, M., Tucci, P., Zotti, M., Morgese, M. G., Schiavone, S., Govoni, S., et al. (2010). Soluble beta amyloid(1-42): a critical player in producing behavioural and biochemical changes evoking depressive-related state? Br. J. Pharmacol. 159, 1704–1715. doi: 10.1111/j.1476-5381.2010.00669.x
Contestabile, A., and Ciani, E. (2004). Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation. Neurochem. Int. 45, 903–914. doi: 10.1016/j.neuint.2004.03.021
Counts, S. E., and Mufson, E. J. (2010). Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J. Neurochem. 113, 649–660. doi: 10.1111/j.1471-4159.2010.06622.x
Dello Russo, C., Boullerne, A. I., Gavrilyuk, V., and Feinstein, D. L. (2004). Inhibition of microglial inflammatory responses by norepinephrine: effects on nitric oxide and interleukin-1beta production. J. Neuroinflammation 1:9. doi: 10.1186/1742-2094-1-9
Di Carlo, M., Giacomazza, D., and San Biagio, P. L. (2012). Alzheimer's disease: biological aspects, therapeutic perspectives and diagnostic tools. J. Phys. Condens. Matter 24:244102. doi: 10.1088/0953-8984/24/24/244102
Feinstein, D. L., Galea, E., and Reis, D. J. (1993). Norepinephrine suppresses inducible nitric oxide synthase activity in rat astroglial cultures. J. Neurochem. 60, 1945–1948. doi: 10.1111/j.1471-4159.1993.tb13425.x
Feinstein, D. L., Heneka, M. T., Gavrilyuk, V., Dello Russo, C., Weinberg, G., and Galea, E. (2002). Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 41, 357–365. doi: 10.1016/S0197-0186(02)00049-9
Fernandez, A. P., Pozo-Rodrigalvarez, A., Serrano, J., and Martinez-Murillo, R. (2010). Nitric oxide: target for therapeutic strategies in Alzheimer's disease. Curr. Pharm. Des. 16, 2837–2850. doi: 10.2174/138161210793176590
Garthwaite, J. (1991). Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 14, 60–67. doi: 10.1016/0166-2236(91)90022-M
Gentile, M. T., Vecchione, C., Maffei, A., Aretini, A., Marino, G., Poulet, R., et al. (2004). Mechanisms of soluble beta-amyloid impairment of endothelial function. J. Biol. Chem. 279, 48135–48142. doi: 10.1074/jbc.M407358200
Grange-Messent, V., Raison, D., Dugas, B., and Calas, A. (2004). Noradrenaline up-regulates the neuronal and the inducible nitric oxide synthase isoforms in magnocellular neurons of rat brain slices. J. Neurosci. Res. 78, 683–690. doi: 10.1002/jnr.20331
Grilli, M., Lagomarsino, F., Zappettini, S., Preda, S., Mura, E., Govoni, S., et al. (2010). Specific inhibitory effect of amyloid-beta on presynaptic muscarinic receptor subtypes modulating neurotransmitter release in the rat nucleus accumbens. Neuroscience 167, 482–489. doi: 10.1016/j.neuroscience.2010.01.058
Guix, F. X., Uribesalgo, I., Coma, M., and Muñoz, F. J. (2005). The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 76, 126–152. doi: 10.1016/j.pneurobio.2005.06.001
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Heneka, M. T., Galea, E., Gavriluyk, V., Dumitrescu-Ozimek, L., Daeschner, J., O'Banion, M. K., et al. (2002). Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer's disease. J. Neurosci. 22, 2434–2442. doi: 10.1136/jnnp.2005.075358
Heneka, M. T., Nadrigny, F., Regen, T., Martinez-Hernandez, A., Dumitrescu-Ozimek, L., Terwel, D., et al. (2010). Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U.S.A. 107, 6058–6063. doi: 10.1073/pnas.0909586107
Hsieh, C. H., Li, H. Y., and Chen, J. C. (2010). Nitric oxide and interleukin-1beta mediate noradrenergic induced corticotrophin-releasing hormone release in organotypic cultures of rat paraventricular nucleus. Neuroscience 165, 1191–1202. doi: 10.1016/j.neuroscience.2009.12.003
Iammarino, M., Di Taranto, A., and Muscarella, M. (2012). Investigation on the presence of sulphites in fresh meat preparations: estimation of an allowable maximum limit. Meat Sci. 90, 304–308. doi: 10.1016/j.meatsci.2011.07.015
Igbavboa, U., Johnson-Anuna, L. N., Rossello, X., Butterick, T. A., Sun, G. Y., and Wood, W. G. (2006). Amyloid beta-protein1-42 increases cAMP and apolipoprotein E levels which are inhibited by beta1 and beta2-adrenergic receptor antagonists in mouse primary astrocytes. Neuroscience 142, 655–660. doi: 10.1016/j.neuroscience.2006.06.056
Kalinin, S., Polak, P. E., Madrigal, J. L., Gavrilyuk, V., Sharp, A., Chauhan, N., et al. (2006). Beta-amyloid-dependent expression of NOS2 in neurons: prevention by an alpha2-adrenergic antagonist. Antioxid. Redox Signal. 8, 873–883. doi: 10.1089/ars.2006.8.873
Kamikawa, H., Hori, T., Nakane, H., Aou, S., and Tashiro, N. (1998). IL-1beta increases norepinephrine level in rat frontal cortex: involvement of prostanoids, NO, and glutamate. Am. J. Physiol. 275, R803–R810.
Kar, S., Slowikowski, S. P., Westaway, D., and Mount, H. T. (2004). Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer's disease. J. Psychiatry Neurosci. 29, 427–441.
Kong, Y., Ruan, L., Qian, L., Liu, X., and Le, Y. (2010). Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 30, 11848–11857. doi: 10.1523/JNEUROSCI.2985-10.2010
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Liu, X., Ye, K., and Weinshenker, D. (2015). Norepinephrine protects against amyloid-beta toxicity via TrkB. J. Alzheimers. Dis. 44, 251–260. doi: 10.3233/JAD-141062
Lonart, G., Wang, J., and Johnson, K. M. (1992). Nitric oxide induces neurotransmitter release from hippocampal slices. Eur. J. Pharmacol. 220, 271–272. doi: 10.1016/0014-2999(92)90759-W
Meffert, M. K., Calakos, N. C., Scheller, R. H., and Schulman, H. (1996). Nitric oxide modulates synaptic vesicle docking fusion reactions. Neuron 16, 1229–1236. doi: 10.1016/S0896-6273(00)80149-X
Miller, T. W., Isenberg, J. S., Shih, H. B., Wang, Y., and Roberts, D. D. (2010). Amyloid-beta inhibits No-cGMP signaling in a CD36- and CD47-dependent manner. PLoS ONE 5:e15686. doi: 10.1371/journal.pone.0015686
Moncada, S., Palmer, R. M., and Higgs, E. A. (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43, 109–142.
Morgese, M. G., Tucci, P., Colaianna, M., Zotti, M., Cuomo, V., Schiavone, S., et al. (2014). Modulatory activity of soluble beta amyloid on HPA axis function in rats. Curr. Pharm. Des. 20, 2539–2546. doi: 10.2174/13816128113199990500
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sarà, M., Racchi, M., et al. (2010a). Beta-amyloid: a disease target or a synaptic regulator affecting age-related neurotransmitter changes? Curr. Pharm. Des. 16, 672–683. doi: 10.2174/138161210790883723
Mura, E., Preda, S., Govoni, S., Lanni, C., Trabace, L., Grilli, M., et al. (2010b). Specific neuromodulatory actions of amyloid-beta on dopamine release in rat nucleus accumbens and caudate putamen. J. Alzheimers. Dis. 19, 1041–1053. doi: 10.3233/JAD-2010-1299
Mura, E., Zappettini, S., Preda, S., Biundo, F., Lanni, C., Grilli, M., et al. (2012). Dual effect of beta-amyloid on alpha7 and alpha4beta2 nicotinic receptors controlling the release of glutamate, aspartate and GABA in rat hippocampus. PLoS ONE 7:e29661. doi: 10.1371/journal.pone.0029661
Murad, F. (2003). The excitement and rewards of research with our discovery of some of the biological effects of nitric oxide. Circ. Res. 92, 339–341. doi: 10.1161/01.RES.0000061772.73917.99
Palumbo, M. L., Fosser, N. S., Rios, H., Zorrilla Zubilete, M. A., Guelman, L. R., Cremaschi, G. A., et al. (2007). Loss of hippocampal neuronal nitric oxide synthase contributes to the stress-related deficit in learning and memory. J. Neurochem. 102, 261–274. doi: 10.1111/j.1471-4159.2007.04528.x
Patel, N. J., Chen, M. J., and Russo-Neustadt, A. A. (2010). Norepinephrine and nitric oxide promote cell survival signaling in hippocampal neurons. Eur. J. Pharmacol. 633, 1–9. doi: 10.1016/j.ejphar.2010.01.012
Paxinos, G., and Watson, C. (1998). The Rat Brain in Stereotaxic Coordinates. New York, NY: Elsevier Academic Press.
Preda, S., Govoni, S., Lanni, C., Racchi, M., Mura, E., Grilli, M., et al. (2008). Acute beta-amyloid administration disrupts the cholinergic control of dopamine release in the nucleus accumbens. Neuropsychopharmacology 33, 1062–1070. doi: 10.1038/sj.npp.1301485
Puzzo, D., Palmeri, A., and Arancio, O. (2006). Involvement of the nitric oxide pathway in synaptic dysfunction following amyloid elevation in Alzheimer's disease. Rev. Neurosci. 17, 497–523. doi: 10.1515/REVNEURO.2006.17.5.497
Puzzo, D., Vitolo, O., Trinchese, F., Jacob, J. P., Palmeri, A., and Arancio, O. (2005). Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J. Neurosci. 25, 6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005
Russell, C. L., Semerdjieva, S., Empson, R. M., Austen, B. M., Beesley, P. W., and Alifragis, P. (2012). Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS ONE 7:e43201. doi: 10.1371/journal.pone.0043201
Sanz, J. M., Chiozzi, P., Ferrari, D., Colaianna, M., Idzko, M., Falzoni, S., et al. (2009). Activation of microglia by amyloid β requires P2X7 receptor expression. J. Immunol. 182, 4378–4385. doi: 10.4049/jimmunol.0803612
Saulskaya, N. B., Fofonova, N. V., and Sudorgina, P. V. (2010). Activation of the NO-ergic system of the nucleus accumbens on presentation of contextual danger signals. Neurosci. Behav. Physiol. 40, 907–912. doi: 10.1007/s11055-010-9344-y
Snyder, S. H., and Bredt, D. S. (1991). Nitric oxide as a neuronal messenger. Trends Pharmacol. Sci. 12, 125–128. doi: 10.1016/0165-6147(91)90526-X
Taepavarapruk, P., and Song, C. (2010). Reductions of acetylcholine release and nerve growth factor expression are correlated with memory impairment induced by interleukin-1beta administrations: effects of omega-3 fatty acid EPA treatment. J. Neurochem. 112, 1054–1064. doi: 10.1111/j.1471-4159.2009.06524.x
Trabace, L., Cassano, T., Tucci, P., Steardo, L., Kendrick, K. M., and Cuomo, V. (2004). The effects of nitric oxide on striatal serotoninergic transmission involve multiple targets: an in vivo microdialysis study in the awake rat. Brain Res. 1008, 293–298. doi: 10.1016/j.brainres.2004.01.090
Trabace, L., Kendrick, K. M., Castrignanò, S., Colaianna, M., De Giorgi, A., Schiavone, S., et al. (2007). Soluble amyloid beta1-42 reduces dopamine levels in rat prefrontal cortex: relationship to nitric oxide. Neuroscience 147, 652–663. doi: 10.1016/j.neuroscience.2007.04.056
Trabace, L., Zotti, M., Morgese, M. G., Tucci, P., Colaianna, M., Schiavone, S., et al. (2011). Estrous cycle affects the neurochemical and neurobehavioral profile of carvacrol-treated female rats. Toxicol. Appl. Pharmacol. 255, 169–175. doi: 10.1016/j.taap.2011.06.011
Tucci, P., Mhillaj, E., Morgese, M. G., Colaianna, M., Zotti, M., Schiavone, S., et al. (2014). Memantine prevents memory consolidation failure induced by soluble beta amyloid in rats. Front. Behav. Neurosci. 8:332. doi: 10.3389/fnbeh.2014.00332
Keywords: soluble beta amyloid, norepinephrine, nitric oxide, interleukin-1, prefrontal cortex, hippocampus, nucleus accumbens
Citation: Morgese MG, Colaianna M, Mhillaj E, Zotti M, Schiavone S, D'Antonio P, Harkin A, Gigliucci V, Campolongo P, Trezza V, De Stradis A, Tucci P, Cuomo V and Trabace L (2015) Soluble beta amyloid evokes alteration in brain norepinephrine levels: role of nitric oxide and interleukin-1. Front. Neurosci. 9:428. doi: 10.3389/fnins.2015.00428
Received: 03 September 2015; Accepted: 21 October 2015;
Published: 05 November 2015.
Edited by:
Andrew C. McCreary, Brains On-Line, NetherlandsReviewed by:
Habibeh Khoshbouei, Univeristy of Florida, USAFrancesco Rossi, Second University of Naples, Italy
Copyright © 2015 Morgese, Colaianna, Mhillaj, Zotti, Schiavone, D'Antonio, Harkin, Gigliucci, Campolongo, Trezza, De Stradis, Tucci, Cuomo and Trabace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigia Trabace, bHVpZ2lhLnRyYWJhY2VAdW5pZmcuaXQ=
†These authors have contributed equally to this work.