 Judith M. van der Harg1,2
Judith M. van der Harg1,2 Leslie Eggels3
Leslie Eggels3 Silvie R. Ruigrok2
Silvie R. Ruigrok2 Jeroen J. M. Hoozemans4
Jeroen J. M. Hoozemans4 Susanne E. la Fleur3
Susanne E. la Fleur3 Wiep Scheper1,2,5*
Wiep Scheper1,2,5*- 1Department of Genome Analysis, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Departments of Functional Genomics and Molecular and Cellular Neuroscience, Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, VU University Amsterdam, Amsterdam, Netherlands
- 3Department of Endocrinology and Metabolism, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 4Department of Pathology, Neuroscience Campus Amsterdam, VU University Medical Center, Amsterdam, Netherlands
- 5Department of Clinical Genetics and Alzheimer Center, VU University Medical Center, Amsterdam, Netherlands
Abnormal phosphorylation and aggregation of tau is a key hallmark of Alzheimer's disease (AD). AD is a multifactorial neurodegenerative disorder for which Diabetes Mellitus (DM) is a risk factor. In animal models for DM, the phosphorylation and aggregation of tau is induced or exacerbated, however the underlying mechanism is unknown. In addition to the metabolic dysfunction, DM is characterized by chronic low-grade inflammation. This was reported to be associated with a neuroinflammatory response in the hypothalamus of DM animal models. Neuroinflammation is also implicated in the development and progression of AD. It is unknown whether DM also induces neuroinflammation in brain areas affected in AD, the cortex and hippocampus. Here we investigated whether neuroinflammation could be the mechanistic trigger to induce tau phosphorylation in the brain of DM animals. Two distinct diabetic animal models were used; rats on free-choice high-fat high-sugar (fcHFHS) diet that are insulin resistant and streptozotocin-treated rats that are insulin deficient. The streptozotocin-treated animals demonstrated increased tau phosphorylation in the brain as expected, whereas the fcHFHS diet fed animals did not. Remarkably, neither of the diabetic animal models showed reactive microglia or increased GFAP and COX-2 levels in the cortex or hippocampus. From this, we conclude: 1. DM does not induce neuroinflammation in brain regions affected in AD, and 2. Neuroinflammation is not a prerequisite for tau phosphorylation. Neuroinflammation is therefore not the mechanism that explains the close connection between DM and AD.
Introduction
Alzheimer's disease (AD) is a multifactorial neurodegenerative disorder characterized by aggregation of Aβ and abnormal phosphorylation and aggregation of tau (Grundke-Iqbal et al., 1986; Selkoe, 1991). Neuroinflammation is associated with the development and progression of AD. Different genome-wide association studies identified genes involved in the innate immune system as risk factors for AD; CR1, CD33, and TREM2, which are all expressed in microglia (Bertram et al., 2008; Harold et al., 2009; Lambert et al., 2009; Jonsson et al., 2013). Moreover, microglia are activated early in AD pathology (Hoozemans et al., 2005). Microglial activation is already observed in patients with mild cognitive impairment (MCI, Okello et al., 2009). Interestingly, the density of activated microglial correlates inversely with cognitive performance of AD patients (Combs, 2009). These data strongly indicate the involvement of neuroinflammation in AD pathogenesis.
However, the exact underlying pathomechanism of neuroinflammation in AD pathology is complex. Neuroinflammation influences both the clearance and production of toxic tau and Aβ species. Proinflammatory cytokines reduce the clearance capacity of glial cells thereby resulting in a disbalance of production and clearance of accumulated proteins (Lee and Landreth, 2010). In addition, BACE1 activity is increased by inflammation in vitro and is reduced after using nonsteroidal anti-inflammatory drugs in AD mice (Sastre et al., 2003, 2006; Lee et al., 2008). Tau phosphorylation is increased by a shift in the balance of tau kinase and phosphatase activity (Arnaud et al., 2006). The activity of the tau kinases GSK3β, Cdk5, and p38-MAPK is increased upon inflammation. In addition, a different pathway of inducing tau pathology by neuroinflammation was described by Arnaud et al. (2009) showing that inflammation leads to tau cleavage into an aggregation-prone form known to seed tau aggregation.
Epidemiological studies show that Diabetes Mellitus (DM) is a risk factor for AD and that the incidence of AD is higher in people with DM (Biessels et al., 2006; Kopf and Frölich, 2009). Moreover, DM is associated with higher risk for MCI (Luchsinger et al., 2007). DM is characterized by marked high levels of blood glucose and occurs in two forms: type 1 DM (T1DM), which results from insulin deficiency, and type 2 DM (T2DM) which starts with overproduction of insulin due to insulin resistance and over time results like T1DM in extreme hyperglycemia. In transgenic AD models, both insulin deficiency and insulin resistance exacerbate tau pathology (Ke et al., 2009; Park, 2011). Interestingly, various studies show induction of endogenous tau phosphorylation in the brains of T1DM animal models (reviewed by Park, 2011; El Khoury et al., 2014). An increased level of endogenous tau phosphorylation is also reported in some animals on high-caloric diet that develops insulin resistance. However, this is not consistently observed (Table 1).
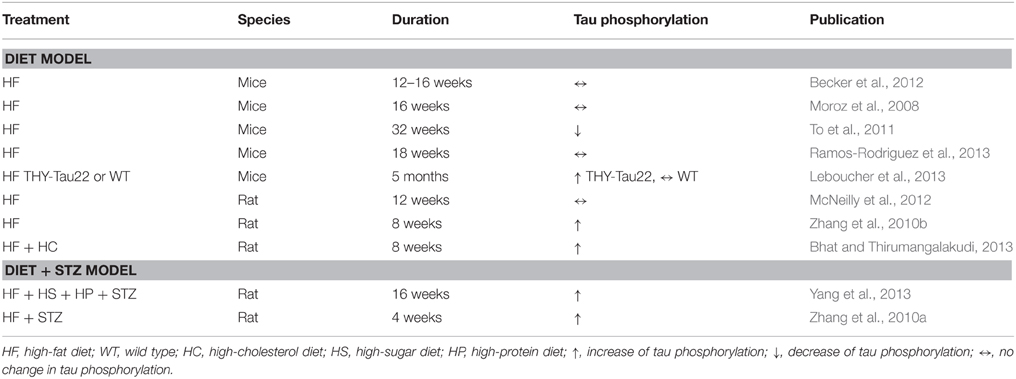
Table 1. Overview of tau phosphorylation in diet-induced diabetic models.
Interestingly, DM is characterized by low-grade systemic inflammation. Inflammation has been implicated in the progression and peripheral complications of both T1DM and T2DM (King, 2008; Gustafson, 2010; Vykoukal and Davies, 2011). This peripheral inflammation can be accompanied by neuroinflammation in specific regions of the central nervous system. Reactive glial cells and activation of different cytokines are reported in the hypothalamus of insulin deficient (Luo et al., 2002) as well as insulin resistant animals and in obese humans (Thaler et al., 2012). However, the adverse effects of insulin deficiency or insulin resistance on regions of the brain involved in cognition (cortex and hippocampus) are hardly investigated. Therefore, we investigated whether neuroinflammation could be the mechanistic trigger to induce tau pathology in the brain of DM animals. Two distinct diabetic animal models were used to study neuroinflammation in the cortex and the hippocampus, brain areas primarily affected in AD. The first model mimics T1DM by destroying the pancreatic β cells with streptozotocin (STZ) resulting in insulin deficiency and extreme hyperglycemia (Qu et al., 2011). In the second model, rats are fed a free-choice high-fat high-sugar (fcHFHS) diet for 10 weeks to model obesity-induced insulin resistance. Previously we showed that rats have increased body weight, slight hyperglycemia, hyperinsulinemia, glucose intolerance and a diminished insulin response to a glucose load after a 4-week fcHFHS diet (la Fleur et al., 2011). In this study, we investigated whether inflammation, a common dominator in both insulin deficient and insulin resistant animals, can lead to tau phosphorylation using these two animal models.
Materials and Methods
Animals
This study was performed with male Wistar rats (250–350 g; Charles River, Sulzfeld, Germany). Rats were individually housed under a 12:12 h light/dark cycle (lights on 07.00 h) at 21–23°C with ad libitum access to standard chow (Special Diets Services, Essex, United Kingdom) and tap water. Two distinct diabetic animal models were used: an insulin resistance diet model and insulin deficient model. Rats on fcHFHS diet received in addition to the control groups a dish of saturated fat [beef tallow (Ossewit/Blanc de Boeuf, Vandermoortele, Belgium)] and a bottle of 30% sugar water (1 M sucrose mixed from commercial grade sugar and water) in the cage for 1 week (n = 6), 4 weeks (n = 6), or 10 weeks (n = 9). For the second diabetic model, T1DM, rats received under anesthesia of isoflurane subcutaneous an injection of STZ (experimental group) (Sigma Aldrich, St. Louis, MO, USA; 65 mg/kg in 0.3 ml citrate buffer pH 4.2) or vehicle (citrate buffer). Animals were sacrificed 20 days after injection (n > 5 animals per group). For western blot analysis, animals were decapitated under CO2/O2 and brains were quickly removed and collected on dry ice before frozen in −80°C. Subsequently, hippocampus and cortex were dissected for protein lysates. For immunohistological analysis, animals were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde under Nembutal anesthetics (120 mg/kg). Brains were embedded in paraffin. During the experiment body weight, food and water intake was measured 3 times a week. Glucose and insulin levels were not determined since the therefore required clamp studies or fasting of the animals both have a profound effect on the brain and could interfere with our results (Faggioni et al., 2000; Rummel et al., 2010; Lavin et al., 2011; Fuente-Martin et al., 2012; Bowe et al., 2014; Routh et al., 2014; Vasconcelos et al., 2014). However, previously fasting hyperinsulinemia and slight increases in glucose levels have been shown in the fcHFHS model used in this study (la Fleur et al., 2010, 2011; Harris and Apolzan, 2012). In STZ model trunk blood was collected to measure plasma glucose concentration with the Biosen (EKF diagnostics, Cardiff, UK) following the assay protocol. A significant increase in plasma glucose levels without fasting of STZ-treated animals compared to control rats was detectable due to the extreme hyperglycemia levels of STZ-treated rats (6.28 ± 0.44 mmol/L basal glucose levels in control rats vs. 20.9 ± 4.95 mmol/L basal glucose levels in STZ-treated rats). The experiment was approved by the Committee for Animal Experimentation of the Academic Medical Center of the University of Amsterdam, the Netherlands.
SDS-PAGE and Western Blotting
Brain lysates were homogenized with a seirin needle and incubated for 30 min on ice in RIPA buffer (50 mM TrisHCl, 150 mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, 0.1% SDS, 2 mM EDTA) supplemented with protease and phosphatase inhibitors. Subsequently, brain lysates were centrifuged for 15 min at 20.000 × g at 4°C. Supernatant protein content was determined by BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded on 10% polyacrylamide gels and blotted onto nitrocellulose membrane (Millipore, Billerica, MA, USA). Blots were pre-incubated with 5% bovine serum albumin (BSA; Boehringer, Mannheim, Germany) in TBS-T [0.05% Tween-20 in Tris buffered saline (TBS)] for 60 min at room temperature and subsequently incubated at 4°C overnight with primary antibodies. Membranes were washed 3 × 10 min in TBS-T and subsequently incubated with species-specific secondary antibodies conjugated to horseradish peroxidase (dilution 1:2000, Dako, Glostrup, Denmark). Reactive protein bands were visualized using LumiLightPLUS Western blotting substrate (Roche Applied Science). Results were analyzed using Advanced Image Data Analyzer software (Raytest, Straubenhardt, Germany) version 3.44.035 and using Image Studio Version 2.0 software (Li-cor, Lincoln, NE, USA). Brain samples from the experimental and the control group were all loaded on the same SDS-PAGE gel. The primary antibodies and their dilution factors are listed in Table 2.
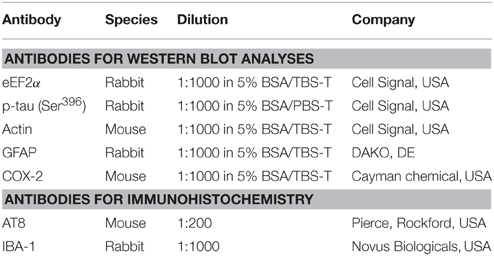
Table 2. Primary antibodies.
Immunohistochemistry
Brains were cut into 5 μm sagittal sections. The sections were immersed in 0.3% H2O2 in TBS for 30 min to quench endogenous peroxidase activity. Sections were treated with 10 mmol/l, pH 6.0, sodium citrate buffer (AT8) or with 10 mM Tris and 1 mM EDTA buffer, pH 9.0 (IBA-1) for 10 min at 99°C for antigen retrieval and subsequently incubated with primary antibodies at 4°C overnight. Antibodies (Table 2) were diluted in TBS containing 0.5% triton-X-100. Negative controls for all immunostainings were generated by omission of primary antibodies. Sections were washed with TBS and subsequently incubated for 120 min with undiluted EnVision/HRP anti-rabbit/mouse (Dako, Hamburg, Germany). Color was developed using 3, 3′-diaminobenzidine (EnVision detection system/HRP 1:50, DakoCytomation, Glostrup, Denmark) as chromogen. Sections were counterstained with haematoxylin and mounted using Depex (BDH Laboratories Supplies, East Grinstead, UK).
Statistical Analysis
GraphPad Prism software was used for graphs and statistical analysis. All data are compared to the control group. Two-sided unpaired Student's t-test was used for single statistical comparison. No significance difference was defined as P > 0.05.
Results
To investigate whether there is neuroinflammation in AD-affected brain areas as a complication of diabetes we used a STZ model and fcHFHS diet model. In both diabetic models body weight gain was measured at the end of the experiment to ensure effectiveness of the treatment. As expected, STZ treatment reduced body weight (Figure 1A). In line with a 4-week fcHFHS diet, body weight gain was significantly increased after a 10-week fcHFHS diet compared to standard chow diet (Figure 1B). We first investigated endogenous levels of phosphorylated tau in the brain of both diabetic models. Immunohistochemistry for phosphorylated tau using the AT8 antibody on brain tissue of rats after 10-weeks fcHFHS diet showed no positive reactivity (Figure 2A). In contrast, the STZ model showed increased tau phosphorylation in the cortex, as previously reported in several studies (Park, 2011; El Khoury et al., 2014). To ascertain that tau phosphorylation did not occur as an adaptive response just after the start of the fcHFHS diet that disappeared after adjustment to the diet, tau phosphorylation was studied at earlier time points. Western blot analyses of hippocampal protein lysates of rats fed a fcHFHS diet for 4-weeks (Figure 2B) and 1-week (Figure 2C) were performed. There was no increased tau phosphorylation at Ser396 observed after 4-week or 1-week diet. As a positive control, western blot analyses of tau phosphorylation at Ser396 were performed in STZ-treated rats showing increased levels of phosphorylated tau in the STZ model using this method as well (Supplementary Figure 1). These results demonstrate that -in contrast to the STZ model- the fcHFHS diet model does not induce tau phosphorylation.
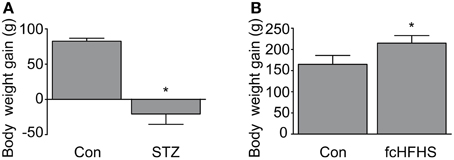
Figure 1. Body weight gain in diabetic rats. Body weight gain 20 days after citrate buffer (con) or STZ injection is shown (A). Data is presented as mean ± SD of n = 6 animals per group. STZ treatment results in loss of approximately 20 grams (g) of body weight from the start of the experiment. Body weight gain after 10-weeks standard chow diet (con) or fcHFHS diet (B) are shown as mean ± SD of n = 9 animals per group. Rats on fcHFHS diet gain on average 50 g more weight than rats on standard chow diet. (*p < 0.01).
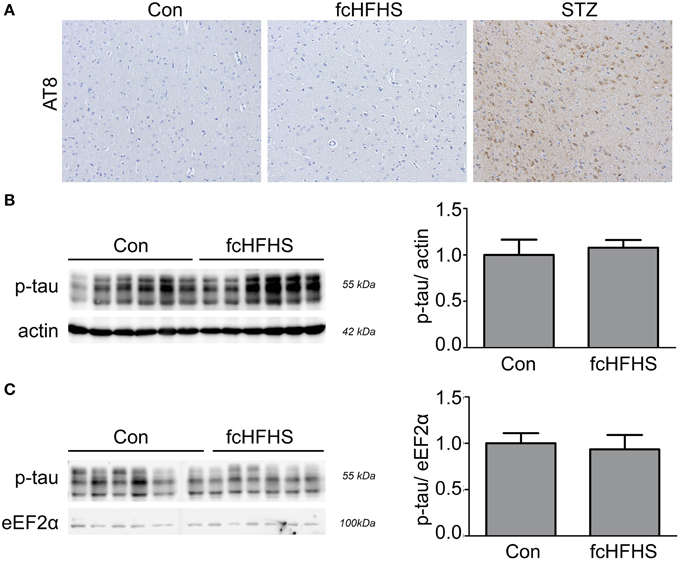
Figure 2. Tau phosphorylation in diabetic rats. Sagittal brain sections of control rats (con), rats on 10-weeks fcHFHS diet and STZ-treated rats were stained for tau using the AT8 antibody. Representative immunohistochemical images of the cortex are shown (A). Tau phosphorylation was observed in STZ-treated rats, but no positive reactivity was found in the animals on the fcHFHS diet. Western blot analyses of hippocampus protein lysates of animals on fcHFHS diet for 4 weeks (B) or 1 week (C) did not show an increase in tau Ser396 phosphorylation (p-tau) compared to control animals on standard chow diet (con). Quantification of western blot is presented as mean ± SEM of n = 6 animals per group.
The differential effects of the STZ treatment and the fcHFHS diet on tau phosphorylation provides the opportunity to study whether neuroinflammation is inextricably linked to increased tau phosphorylation. Inflammation has been associated with the progression and peripheral complications of both T1DM and T2DM (King, 2008; Gustafson, 2010; Vykoukal and Davies, 2011). It is not known whether this is associated with an inflammatory response in the brain as well, therefore, we investigated neuroinflammation in the cortex and the hippocampus in both models. IBA-1 staining was performed to study the morphology of microglia. Reactive microglia are clustered and characterized by shorter and thicker processes. We did not observe a difference in microglial morphology in the brains 20 days after STZ injection (Figure 3A) nor did we observe a difference in microglial morphology in animals after a 10-week fcHFHS diet compared to standard chow diet (Figure 3B). Therefore, the cortex and hippocampus did not show differences in reactive microglia. To exclude that a moderate neuroinflammatory response is induced cyclooxygenase-2 (COX-2) was studied. COX-2 is upregulated in activated microglia and dynamically regulated by pro-inflammatory signals (Minghetti et al., 1999). Western blot analyses of COX-2 in STZ-treated animals (Figures 4A–C) and in animals after 4-weeks fcHFHS diet (Figures 4E–G) did not show upregulation of COX-2 in the cortex and the hippocampus. Finally, glial fibrillary acidic protein (GFAP) was studied as marker for reactive astrocytes. Also GFAP levels in STZ-treated animals (Figures 4A,B,D) and in animals after 4-weeks fcHFHS diet (Figures 4E,F,H) were not increased in the cortex and the hippocampus. A 4-week fcHFHS diet even resulted in a significant decrease of GFAP levels. These data demonstrate that neuroinflammation is not a complication of diabetes in brain areas affected in AD. Moreover, increased tau phosphorylation can occur independently of neuroinflammation in insulin deficient and insulin resistant animal models.
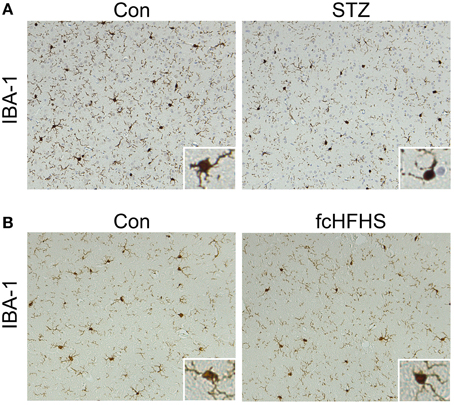
Figure 3. No reactive microglia in diabetic rat cortex. IBA-1 immunohistochemistry was performed on sagittal brain sections of rats 20 days after injection of citrate buffer (con) or STZ (A) and of rats on a 10-week standard chow diet (con) or fcHFHS diet (B). Representative images of the cortex are showed of n = 9 animals per diet group and n = 6 animals per injected group. No difference in microglial morphology is observed compared to control group either in STZ-treated rats or in rats on fcHFHS diet.
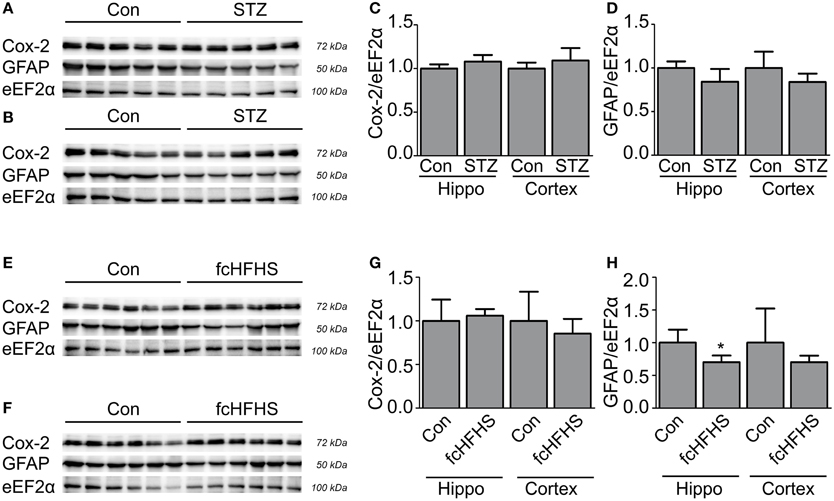
Figure 4. No increased COX-2 and GFAP levels in diabetic rat cortex or hippocampus. Western blot analyses of protein lysates of STZ-treated animals (A–D) and animals on 4-week fcHFHS diet (E–H) of the hippocampus (Hippo) (A,E) and cortex (B,F) were performed. Quantification of COX-2 levels in STZ model (C) and diet model (G) did not show a difference compared to the control. Quantification of GFAP levels in STZ-treated animals did not show a change (D). GFAP levels after 4-week diet even showed a decrease (H). Quantification of western blot is presented as mean ± SD of n = 5 or 6 animals per group. (*p < 0.01).
Discussion
The present study demonstrates absence of reactive microglia and increased levels of GFAP and COX-2 in the hippocampus and the cortex of two distinct diabetic animal models. This indicates that DM does not directly lead to neuroinflammation in AD-affected brain areas. An increased level of endogenous tau phosphorylation was observed in STZ-treated rats demonstrating that neuroinflammation is not a prerequisite for diabetes-induced tau phosphorylation. The absence of increased tau phosphorylation in our fcHFHS diet model is in accordance with the literature (Table 1). Other studies using a high-fat diet also did not observe an increased level of endogenous tau phosphorylation (Moroz et al., 2008; To et al., 2011; Becker et al., 2012; McNeilly et al., 2012; Leboucher et al., 2013; Ramos-Rodriguez et al., 2013). The studies that did observe diet-induced tau phosphorylation, used a tau transgenic mouse or combined the diet with a STZ treatment at the end of the experiment to mimic the hyperglycemic state of T2DM (Zhang et al., 2010a; Leboucher et al., 2013; Yang et al., 2013). Only one study using a high-fat diet with hyperglycemia levels to the extent observed in STZ-induced animals and one study with a high-fat and high-cholesterol diet for 2 months reported an increased level of endogenous phosphorylated tau (Zhang et al., 2010b; Bhat and Thirumangalakudi, 2013). Overall, this suggests that insulin resistance due to a high-caloric diet is not sufficient to induce tau phosphorylation. Nevertheless, the observation that both diabetic models regardless of tau phosphorylation showed no neuroinflammation indicates that neuroinflammation is not the mechanistic trigger to induce tau phosphorylation in the diabetic brain.
Inflammation was reported in the hypothalamus of STZ-treated animals and animals on a high-caloric diet (Luo et al., 2002; Thaler et al., 2012). Microglial activation in the hypothalamus, particularly in the arcuate nucleus, is already observed after 1 day. The permeable blood brain barrier of this area may explain the neuroinflammatory response whereas other brain areas are less accessible for influences from the periphery (Davidson et al., 2012). Indeed, bypassing of the blood brain barrier by direct intracerebroventricular injection of STZ results in glial activation and neuroinflammation in the hippocampus and the cortex (Prickaerts et al., 1999; Chen et al., 2013). This suggests that the adverse effects of STZ resulting in neuroinflammation do not reach the AD-affected areas after intraperitoneal STZ injection. Finally, some studies show increased glial activation in the hippocampus or the cortex after prolonged treatment. One study showed only astrocytic activation in the cortex after 21-weeks of 41%-fat diet and extensive neuroinflammation after 21-weeks of 60%-fat diet (Pistell et al., 2010). In addition, increased astrocytic reactivity was found in the hippocampus of mice 4-weeks after a single intraperitoneal STZ injection (Saravia et al., 2002). This indicates that neuroinflammation in AD-affected brain areas upon diet and STZ treatment occurs only upon prolonged and extensive treatment. Since tau phosphorylation is already observed 20 days after STZ treatment, neuroinflammation is also unrelated to increased phosphorylated tau levels in these studies.
Although we demonstrate that neuroinflammation is not a prerequisite for increased tau phosphorylation, our data do not exclude that peripheral inflammation could have an effect on the brain resulting in increased tau phosphorylation independent of neuroinflammation. In addition, DM may alter the neuroinflammatory response or even trigger neuroinflammation after a priming stimulus and thus facilitate AD pathology. As mentioned above there is a body of evidence to suggest that neuroinflammation plays a role in the progression of AD (Hoozemans and O'Banion, 2005; Bertram et al., 2008; Combs, 2009; Harold et al., 2009; Lambert et al., 2009; Okello et al., 2009; Jonsson et al., 2013). Interestingly, in an AD animal model for Aβ pathology neuroinflammation occurred after the increase of Aβ levels, but before the onset of plaque formation (Hanzel et al., 2014). Indeed, neuropathological and experimental studies demonstrate that Aβ can activate the immune system including the pro-inflammatory cytokine cascade (Eikelenboom et al., 2012; Heneka et al., 2014). In addition, in transgenic animals modeling tau pathology neuroinflammation was observed after initiation of tau pathology and in turn, tau phosphorylation was exacerbated by lipopolysacharide injections into the brain (Zilka et al., 2009; Lee et al., 2010). This indicates that neuroinflammation is a secondary response upon increased Aβ and phosphorylated tau. Subsequently, neuroinflammation can increase Aβ production and tau phosphorylation resulting in a vicious cycle that accelerates AD pathology. This is in accordance with our observations in the T1DM model, which mimics the early phase of tau pathology and shows no neuroinflammation.
As neuroinflammation is not the trigger for increased tau phosphorylation the question remains which mechanism induces endogenous tau phosphorylation in the diabetic brain. Interestingly, dysfunction in insulin signaling has been reported in post-mortem AD brain material and in animal models of AD (Steen et al., 2005; Lester-Coll et al., 2006; de la Monte, 2009; Moloney et al., 2010; Talbot et al., 2012). Since increased tau phosphorylation was not observed in both the STZ model and fcHFHS diet model, insulin deficiency and insulin resistance may have a differential effect on tau phosphorylation. In addition, it should be mentioned that the STZ model has extreme hyperglycemia to the extent of T1DM that may contribute to tau phosphorylation whereas high-caloric diet models show slight hyperglycemia not comparable with the final state of T2DM. Finally, we previously reported increased tau phosphorylation upon metabolic stress in a physiological hypometabolic model in vivo indicating that also metabolic stress in DM could be the trigger to induce tau phosphorylation (van der Harg et al., 2014). Further research is needed to better understand the differences of insulin deficiency and insulin resistance on tau phosphorylation and to find the underlying mechanism for diabetic-induced tau phosphorylation.
In conclusion, our data demonstrate that increased tau phosphorylation occurs independently of neuroinflammation. Eliminating neuroinflammation as causal factor facilitates further elucidation of the complex connection between DM and AD and helps to put the multiple underlying mechanisms contributing to AD in an integrated framework (van Dijk et al., 2015). Since the incidence of both DM and AD is increasing due to the aging population, better understanding of the connection between these diseases is crucial. Further research is necessary to discover the mechanistic trigger for increased tau phosphorylation in the diabetic brain.
Author Contributions
All authors substantial contributed to the conception and design of the work and the critically drafting of the manuscript. All authors approved the manuscript to be published and agreed to be accountable for the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Internationale Stichting Alzheimer Onderzoek Nederland (ISAO #10502) to WS, Hersenstichting Nederland 2013(1)-90 to SlF and an AMC PhD fellowship to JvdH. We thank Frank Baas for stimulating discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2015.00432
References
Arnaud, L., Robakis, N. K., and Figueiredo-Pereira, M. E. (2006). It may take inflammation, phosphorylation and ubiquitination to ‘tangle’ in Alzheimer's disease. Neurodegener. Dis. 3, 313–319. doi: 10.1159/000095638
Arnaud, L. T., Myeku, N., and Figueiredo-Pereira, M. E. (2009). Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J. Neurochem. 110, 328–342. doi: 10.1111/j.1471-4159.2009.06142.x
Becker, K., Freude, S., Zemva, J., Stöhr, O., Krone, W., and Schubert, M. (2012). Chronic peripheral hyperinsulinemia has no substantial influence on tau phosphorylation in vivo. Neurosci. Lett. 516, 306–310. doi: 10.1016/j.neulet.2012.04.022
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Bhat, N. R., and Thirumangalakudi, L. (2013). Increased tau phosphorylation and impaired brain insulin/IGF signaling in mice fed a high fat/high cholesterol diet. J. Alzheimers Dis. 36, 781–789. doi: 10.3233/JAD-2012-121030
Biessels, G. J., Staekenborg, S., Brunner, E., Brayne, C., and Scheltens, P. (2006). Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 5, 64–74. doi: 10.1016/S1474-4422(05)70284-2
Bowe, J. E., Franklin, Z. J., Hauge-Evans, A. C., King, A. J., Persaud, S. J., and Jones, P. M. (2014). Metabolic phenotyping guidelines: assessing glucose homeostasis in rodent models. J. Endocrinol. 222, G13–G25. doi: 10.1530/joe-14-0182
Chen, Y., Liang, Z., Blanchard, J., Dai, C. L., Sun, S., Lee, M. H., et al. (2013). A non-transgenic mouse model (icv-STZ mouse) of Alzheimer's disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 47, 711–725. doi: 10.1007/s12035-012-8375-5
Combs, C. K. (2009). Inflammation and microglia actions in Alzheimer's disease. J. Neuroimmune Pharmacol. 4, 380–388. doi: 10.1007/s11481-009-9165-3
Davidson, T. L., Monnot, A., Neal, A. U., Martin, A. A., Horton, J. J., and Zheng, W. (2012). The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood-brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiol. Behav. 107, 26–33. doi: 10.1016/j.physbeh.2012.05.015
de la Monte, S. M. (2009). Insulin resistance and Alzheimer's disease. BMB Rep. 42, 475–481. doi: 10.5483/BMBRep.2009.42.8.475
Eikelenboom, P., Hoozemans, J. J., Veerhuis, R., van Exel, E., Rozemuller, A. J., and van Gool, W. A. (2012). Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimers Res. Ther. 4, 15. doi: 10.1186/alzrt118
El Khoury, N. B., Gratuze, M., Papon, M. A., Bretteville, A., and Planel, E. (2014). Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 8:22. doi: 10.3389/fncel.2014.00022
Faggioni, R., Moser, A., Feingold, K. R., and Grunfeld, C. (2000). Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am. J. Pathol. 156, 1781–1787. doi: 10.1016/S0002-9440(10)65049-3
Fuente-Martin, E., García-Cáceres, C., Granado, M., de Ceballos, M. L., Sánchez-Garrido, M. A., Sarman, B., et al. (2012). Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Invest. 122, 3900–3913. doi: 10.1172/JCI64102
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Gustafson, B. (2010). Adipose tissue, inflammation and atherosclerosis. J. Atheroscler. Thromb. 17, 332–341. doi: 10.5551/jat.3939
Hanzel, C. E., Pichet-Binette, A., Pimentel, L. S., Iulita, M. F., Allard, S., Ducatenzeiler, A., et al. (2014). Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer's disease. Neurobiol. Aging 35, 2249–2262. doi: 10.1016/j.neurobiolaging.2014.03.026
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Harris, R. B., and Apolzan, J. W. (2012). Changes in glucose tolerance and leptin responsiveness of rats offered a choice of lard, sucrose, and chow. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R1327–R1339. doi: 10.1152/ajpregu.00477.2011
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Hoozemans, J. J., and O'Banion, M. K. (2005). The role of COX-1 and COX-2 in Alzheimer's disease pathology and the therapeutic potentials of non-steroidal anti-inflammatory drugs. Curr. Drug Targets CNS Neurol. Disord. 4, 307–315. doi: 10.2174/1568007054038201
Hoozemans, J. J., van Haastert, E. S., Veerhuis, R., Arendt, T., Scheper, W., Eikelenboom, P., et al. (2005). Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer's disease. J. Neuroinflammation 2:27. doi: 10.1186/1742-2094-2-27
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Ke, Y. D., Delerue, F., Gladbach, A., Götz, J., and Ittner, L. M. (2009). Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer's disease. PLoS ONE 4:e7917. doi: 10.1371/journal.pone.0007917
King, G. L. (2008). The role of inflammatory cytokines in diabetes and its complications. J. Periodontol. 79, 1527–1534. doi: 10.1902/jop.2008.080246
Kopf, D., and Frölich, L. (2009). Risk of incident Alzheimer's disease in diabetic patients: a systematic review of prospective trials. J. Alzheimers Dis. 16, 677–685. doi: 10.3233/JAD-2009-1011
la Fleur, S. E., Luijendijk, M. C., van Rozen, A. J., Kalsbeek, A., and Adan, R. A. (2011). A free-choice high-fat high-sugar diet induces glucose intolerance and insulin unresponsiveness to a glucose load not explained by obesity. Int. J. Obes. (Lond). 35, 595–604. doi: 10.1038/ijo.2010.164
la Fleur, S. E., van Rozen, A. J., Luijendijk, M. C., Groeneweg, F., and Adan, R. A. (2010). A free-choice high-fat high-sugar diet induces changes in arcuate neuropeptide expression that support hyperphagia. Int. J. Obes. (Lond). 34, 537–546. doi: 10.1038/ijo.2009.257
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lavin, D. N., Joesting, J. J., Chiu, G. S., Moon, M. L., Meng, J., Dilger, R. N., et al. (2011). Fasting induces an anti-inflammatory effect on the neuroimmune system which a high-fat diet prevents. Obesity (Silver Spring) 19, 1586–1594. doi: 10.1038/oby.2011.73
Leboucher, A., Laurent, C., Fernandez-Gomez, F. J., Burnouf, S., Troquier, L., Eddarkaoui, S., et al. (2013). Detrimental effects of diet-induced obesity on tau pathology are independent of insulin resistance in tau transgenic mice. Diabetes 62, 1681–1688. doi: 10.2337/db12-0866
Lee, C. Y., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960. doi: 10.1007/s00702-010-0433-4
Lee, D. C., Rizer, J., Selenica, M. L., Reid, P., Kraft, C., Johnson, A., et al. (2010). LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation 7:56. doi: 10.1186/1742-2094-7-56
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
Lester-Coll, N., Rivera, E. J., Soscia, S. J., Doiron, K., Wands, J. R., and de la Monte, S. M. (2006). Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J. Alzheimers Dis. 9, 13–33.
Luchsinger, J. A., Reitz, C., Patel, B., Tang, M. X., Manly, J. J., and Mayeux, R. (2007). Relation of diabetes to mild cognitive impairment. Arch. Neurol. 64, 570–575. doi: 10.1001/archneur.64.4.570
Luo, Y., Kaur, C., and Ling, E. A. (2002). Neuronal and glial response in the rat hypothalamus-neurohypophysis complex with streptozotocin-induced diabetes. Brain Res. 925, 42–54. doi: 10.1016/S0006-8993(01)03258-9
McNeilly, A. D., Williamson, R., Balfour, D. J., Stewart, C. A., and Sutherland, C. (2012). A high-fat-diet-induced cognitive deficit in rats that is not prevented by improving insulin sensitivity with metformin. Diabetologia 55, 3061–3070. doi: 10.1007/s00125-012-2686-y
Minghetti, L., Walsh, D. T., Levi, G., and Perry, V. H. (1999). In vivo expression of cyclooxygenase-2 in rat brain following intraparenchymal injection of bacterial endotoxin and inflammatory cytokines. J. Neuropathol. Exp. Neurol. 58, 1184–1191. doi: 10.1097/00005072-199911000-00008
Moloney, A. M., Griffin, R. J., Timmons, S., O'Connor, R., Ravid, R., and O'Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243. doi: 10.1016/j.neurobiolaging.2008.04.002
Moroz, N., Tong, M., Longato, L., Xu, H., and de la Monte, S. M. (2008). Limited Alzheimer-type neurodegeneration in experimental obesity and type 2 diabetes mellitus. J. Alzheimers Dis. 15, 29–44.
Okello, A., Edison, P., Archer, H. A., Turkheimer, F. E., Kennedy, J., Bullock, R., et al. (2009). Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72, 56–62. doi: 10.1212/01.wnl.0000338622.27876.0d
Park, S. A. (2011). A common pathogenic mechanism linking type-2 diabetes and Alzheimer's disease: evidence from animal models. J. Clin. Neurol. 7, 10–18. doi: 10.3988/jcn.2011.7.1.10
Pistell, P. J., Morrison, C. D., Gupta, S., Knight, A. G., Keller, J. N., Ingram, D. K., et al. (2010). Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 219, 25–32. doi: 10.1016/j.jneuroim.2009.11.010
Prickaerts, J., Fahrig, T., and Blokland, A. (1999). Cognitive performance and biochemical markers in septum, hippocampus and striatum of rats after an i.c.v. injection of streptozotocin: a correlation analysis. Behav. Brain Res. 102, 73–88. doi: 10.1016/S0166-4328(98)00158-2
Qu, Z., Jiao, Z., Sun, X., Zhao, Y., Ren, J., and Xu, G. (2011). Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res. 1383, 300–306. doi: 10.1016/j.brainres.2011.01.084
Ramos-Rodriguez, J. J., Ortiz, O., Jimenez-Palomares, M., Kay, K. R., Berrocoso, E., Murillo-Carretero, M. I., et al. (2013). Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 38, 2462–2475. doi: 10.1016/j.psyneuen.2013.05.010
Routh, V. H., Hao, L., Santiago, A. M., Sheng, Z., and Zhou, C. (2014). Hypothalamic glucose sensing: making ends meet. Front. Syst. Neurosci. 8:236. doi: 10.3389/fnsys.2014.00236
Rummel, C., Inoue, W., Poole, S., and Luheshi, G. N. (2010). Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol. Psychiatry 15, 523–534. doi: 10.1038/mp.2009.98
Saravia, F. E., Revsin, Y., Gonzalez Deniselle, M. C., Gonzalez, S. L., Roig, P., Lima, A., et al. (2002). Increased astrocyte reactivity in the hippocampus of murine models of type 1 diabetes: the nonobese diabetic (NOD) and streptozotocin-treated mice. Brain Res. 957, 345–353. doi: 10.1016/S0006-8993(02)03675-2
Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., and van Leuven, F. (2003). Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 23, 9796–9804.
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., et al. (2006). Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448. doi: 10.1073/pnas.0503839103
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6, 487–498. doi: 10.1016/0896-6273(91)90052-2
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80.
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Thaler, J. P., Yi, C. X., Schur, E. A., Guyenet, S. J., Hwang, B. H., Dietrich, M. O., et al. (2012). Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122, 153–162. doi: 10.1172/JCI59660
To, A. W., Ribe, E. M., Chuang, T. T., Schroeder, J. E., and Lovestone, S. (2011). The epsilon3 and epsilon4 alleles of human APOE differentially affect tau phosphorylation in hyperinsulinemic and pioglitazone treated mice. PLoS ONE 6:e16991. doi: 10.1371/journal.pone.0016991
van der Harg, J. M., Nölle, A., Zwart, R., Boerema, A. S., van Haastert, E. S., Strijkstra, A. M., et al. (2014). The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis. 5, e1393. doi: 10.1038/cddis.2014.354
van Dijk, G., van Heijningen, S., Reijne, A. C., Nyakas, C., van der Zee, E. A., and Eisel, U. L. (2015). Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front. Neurosci. 9:173. doi: 10.3389/fnins.2015.00173
Vasconcelos, A. R., Yshii, L. M., Viel, T. A., Buck, H. S., Mattson, M. P., Scavone, C., et al. (2014). Intermittent fasting attenuates lipopolysaccharide-induced neuroinflammation and memory impairment. J. Neuroinflammation 11:85. doi: 10.1186/1742-2094-11-85
Vykoukal, D., and Davies, M. G. (2011). Vascular biology of metabolic syndrome. J. Vasc. Surg. 54, 819–831. doi: 10.1016/j.jvs.2011.01.003
Yang, Y., Ma, D., Wang, Y., Jiang, T., Hu, S., Zhang, M., et al. (2013). Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J. Alzheimers Dis. 33, 329–338. doi: 10.3233/JAD-2012-121294
Zhang, L., Yang, J., Chen, X. Q., Zan, K., Wen, X. D., Chen, H., et al. (2010a). Antidiabetic and antioxidant effects of extracts from Potentilla discolor Bunge on diabetic rats induced by high fat diet and streptozotocin. J. Ethnopharmacol. 132, 518–524. doi: 10.1016/j.jep.2010.08.053
Zhang, T., Pan, B. S., Sun, G. C., Sun, X., and Sun, F. Y. (2010b). Diabetes synergistically exacerbates poststroke dementia and tau abnormality in brain. Neurochem. Int. 56, 955–961. doi: 10.1016/j.neuint.2010.04.003
Keywords: neuroinflammation, Alzheimer's disease, Diabetes Mellitus, phosphorylated tau, cortex, hippocampus
Citation: van der Harg JM, Eggels L, Ruigrok SR, Hoozemans JJM, la Fleur SE and Scheper W (2015) Neuroinflammation is not a Prerequisite for Diabetes-induced Tau Phosphorylation. Front. Neurosci. 9:432. doi: 10.3389/fnins.2015.00432
Received: 05 September 2015; Accepted: 23 October 2015;
Published: 09 November 2015.
Edited by:
Marc Lee Goalstone, University of Colorado Denver, USAReviewed by:
Brian M. Shewchuk, Brody School of Medicine at East Carolina University, USASachin Laldas Badole, University of South Florida, USA
Copyright © 2015 van der Harg, Eggels, Ruigrok, Hoozemans, la Fleur and Scheper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wiep Scheper, dy5zY2hlcGVyQHZ1bWMubmw=