 Shail A. Jagmag
Shail A. Jagmag Naveen Tripathi
Naveen Tripathi Sunil D. Shukla
Sunil D. Shukla Sankar Maiti
Sankar Maiti Sukant Khurana
Sukant Khurana- 1Department of Biology, Indian Institute of Science Education and Research, Kolkata, India
- 2Department of Zoology, Government Meera Girl's College, Udaipur, India
Parkinson's disease is one of the most common neurodegenerative diseases. Animal models have contributed a large part to our understanding and therapeutics developed for treatment of PD. There are several more exhaustive reviews of literature that provide the initiated insights into the specific models; however a novel synthesis of the basic advantages and disadvantages of different models is much needed. Here we compare both neurotoxin based and genetic models while suggesting some novel avenues in PD modeling. We also highlight the problems faced and promises of all the mammalian models with the hope of providing a framework for comparison of various systems.
Parkinson's Disease—Model Utilization for Therapeutics
Parkinson's disease (PD) is a common neurodegenerative disorder, with cardinal features of akinesia, bradykinesia, rigidity, and tremors (Rodriguez-Oroz et al., 2009). The neuropathological hallmarks of PD are the loss of dopaminergic neurons in the SubstantiaNigra pars compacta (SNc) and the formation of intra-neuronal proteinaceous inclusions, called Lewy Bodies (LBs). Loss of neurons from other brain regions has also been observed in the later stages of the disease, such as the cholinergic nucleus basalis of Meynert, many subnuclei in the thalamus and amygdala, and the serotoninergic neurons of the raphe nucleus (Jellinger, 1991; Braak et al., 2000, 2003). In most cases of PD, injury or environmental insult induced changes in the brain connectivity and gene expression, with genetic factors contributing to the predisposition, are suspected to cause the disease but in a smaller fraction of cases, between 10 and 20%, genes are known to be the culprits for causation. Genetic defects in mitochondrial function (Winklhofer and Haass, 2010), dysfunction of the ubiquitin-proteosome pathway (McNaught et al., 2001), and alterations of free radical formation (Palacino et al., 2004) have been shown to play a role in familial PD. Studies have reported increases in the sensitivity of mice with these defects to neurotoxins (Nieto et al., 2006; Haque et al., 2012).
The use of animals to model different aspects of PD phenotype allows us the ability to study both disease progression and explore possible treatments. While none of the currently available models of PD completely phenocopy the disease but they have contributed extensively to our knowledge of PD. So far experimental models have been of two major types: (A) Toxin models and (B) Genetic models. An understanding of different PD models can enhance the ability of PD researcher to employ appropriate models for their experiments.
Toxin Based Models
These are based on neurotoxins which allow for testing of nigrostrial DA neuron degeneration. Figure 1 summarizes major toxin models.
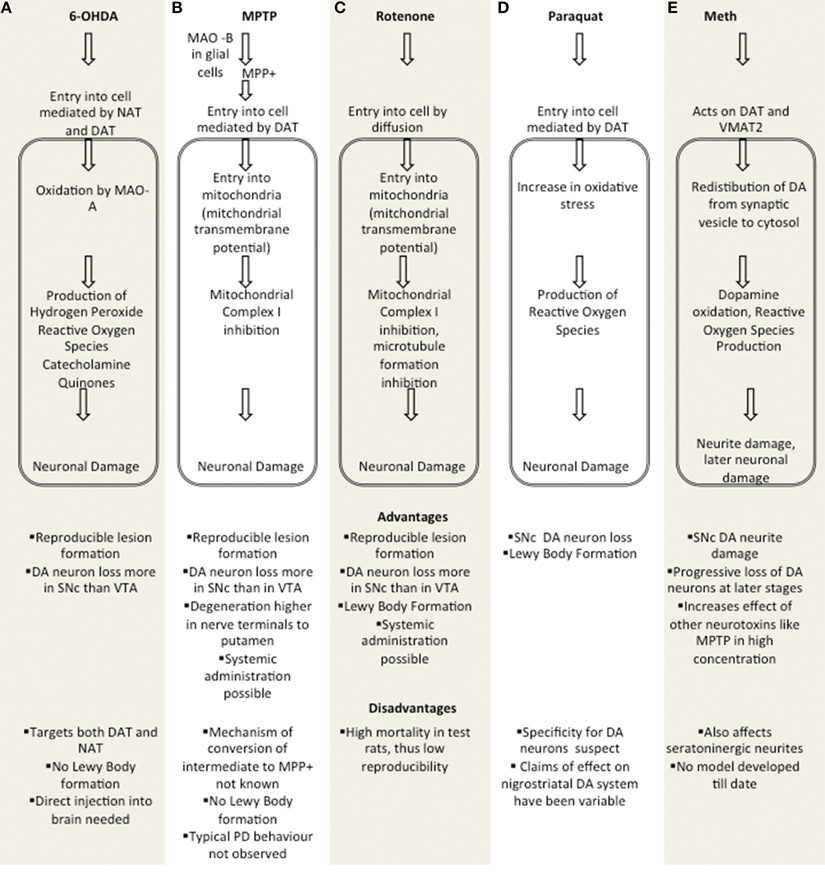
Figure 1. Comparison of major toxin models. (A) 6-OHDA, (B) MPTP, (C) Rotenone, (D) Paraquat, (E) Methamphetamine. MAO A, Monoamine oxidase A; MAO B, Monamine Oxidase B; DAT, Dopaminergic Transporter; NAT, Noradrenergic Transporter; SNc, Substantia Nigra pars Compacta; VTA, Ventral tegmental area; VMAT2, Vesicular Monoamine Transporter 2.
6-OHDA
6-hydroxydopamine (6-OHDA), presented in Figure 1A. is a selective neurotoxin that was first reported to cause lesions in nigrostriatal DA neurons in rats (Ungerstedt, 1968) but has been subsequently shown to work in other animals such as mice (da Conceição et al., 2010; Thiele et al., 2012). 6-OHDA accumulates in the cytosol and promotes formation of hydrogen peroxide, other reactive oxygen species and quinines by auto-oxidation (Cohen, 1984; Simola et al., 2007). Down regulation of dopamine synthesis in the lesioned striatum has also been observed with the non-lesioned striatum compensating for this by increased dopamine production (Del-Bel et al., 2014) 6-OHDA being hydrophilic cannot cross the blood brain barrier and thus administration is carried out by direct injection in the Substantia Nigra pars Compacta (SNc), Medial Forebrain Bundle (MFB) or striatum, depending on the rate at which lesion formation is desired. Injection into the SNc, MFB causes DA neuronal death in less than 24 h (Jeon et al., 1995). Striatal injection causes death of DA neurons over the course of 1–3 weeks. Injection of 6-OHDA causes progressive retrograde neuronal degeneration in the SNc and Ventral Tegmental Area (VTA) (Sauer and Oertel, 1994; Przedborski et al., 1995). In addition to DA transporters, it also targets noradrenergic transporters (Luthman et al., 1989). Thus, in addition to inducing PD symptoms, 6-OHDA also causes damage to other parts of the brain. The other disadvantage of 6-OHDA is that production of LB-like inclusions is not seen (Dauer and Przedborski, 2003). Behavioral wise, unilaterally lesioned rodents show drug induced rotational behavior (Blandini et al., 2008). Motor impairments are also observed, primarily due to impairment of limbs contralateral to the hemisphere in which the 6-OHDA is administered (Simola et al., 2007).
MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), presented in Figure 1B. is a neurotoxin precursor of 1-methyl-4-phenylpyridinium (MPP+), which causes damage to the nigrostriatal DA pathway with a significant loss of DA neurons in the striatum and SNc, more similar to that seen in PD. MPTP susceptibility increases with age (Ovadia et al., 1995). MPTP is converted to an intermediate 1-methyl-4-phenyl-2,3-dihydropyridinium by the action of monoamine oxidase B in glial cells. This intermediate is then oxidized to MPP+ (Jackson-Lewis and Przedborski, 2007). MPP+ has a high affinity for the plasma membrane dopamine transporter with comparatively lower affinities for the norepinephrine and serotonin transporters (Javitch et al., 1985). Once inside dopaminergic neurons, MPP+ can be sequestrated into synaptosomal vesicles or be concentrated within the mitochondria (Ramsay and Singer, 1986) utilizing mitochondrial transmembrane potential. In the mitochondria, MPP+ blocks the electron transport chain by inhibiting Mitochondrial Complex I (Varastet et al., 1994). Due to rapid conversion of MPTP to MPP+, most chronic treatments are actually serial acute insults (Jackson-Lewis and Przedborski, 2007). Thus, a true chronic model would require continuous delivery of MPTP using devices such as osmotic pumps (Fornai et al., 2005). Rats have proven to be resistant to MPTP induced toxicity (Riachi et al., 1990). The reason for this resistance has been speculated to be because of differential MPP+ sequestration (Schmidt and Ferger, 2001). While MPTP produces the best results when used in monkeys including formation of LB like inclusions (Kowall et al., 2000) taking into account practical considerations, the MPTP mouse model is more popular. MPTP causes greater damage to DA neurons in the SNc than in VTA (Blesa et al., 2011, 2012). Recent dopaminergic neuron characterisation has found a specific DA subtype in the SNc is more vulnerable to MPTP (Poulin et al., 2014). Also the degeneration of nerve terminals to the putamen is higher than those to the caudate nucleus (Blesa et al., 2010). This too resembles PD phenotype.
Mice treated with MPTP also do not show behavior typical of PD, however alterations in motor movement are observed, where significant dopaminergic neuron loss is present (Jackson-Lewis and Przedborski, 2007). This model also has a significant weakness of the lack of formation of LBs in mice. Care must be taken to study the interaction of the drug tested with MPP+ before conclusions about its efficacy are drawn as some drugs might reduce the oxidative stress induced by MPTP. Also the conversion of MPTP to MPP+ includes an intermediate, which is oxidized to MPP+, thus an antioxidant treatment protocol might give good results by targeting this step without actually preventing DA neuron loss but this remains to be tested. Use of probenicid to competitively inhibit renal excretion of MPTP has also been shown to increase SNc neuronal loss (Meredith et al., 2002). MPTP has a big advantage because of its lipophylic nature as it can cross the blood brain barrier, thus allowing greater ease in administration, including systemic administration.
Rotenone
Rotenone, presented in Figure 1C. occurs naturally in several plants and has been used as a broad spectrum insecticide, and pesticide. It functions by blocking the mitochondrial electron transport chain through inhibition of complex I, as seen in MPTP. Rotenone also blocks mitosis and inhibits cell proliferation. This is by perturbation of microtubule assembly and decreasing the GTP hydrolysis rate (Srivastava and Panda, 2007). Chronic systemic exposure to rotenone in rats causes many features of PD, including nigrostriatal DA degeneration. This model has been shown to reproduce almost all the features of PD, including the formation of intracellular inclusions that resemble LB (Sherer et al., 2003). Rotenone can be injected intraperitoneally, intravenously or subcutaneously for systemic treatment. It has also been directly injected into the brain stereotaxically (Xiong et al., 2009). However, despite demonstrating the slow and specific loss of DA neurons, this model is difficult to replicate due to the high mortality observed in rats, when treated with rotenone (Fleming et al., 2004). Rotenone is highly lipophilic and easily crosses the blood brain barrier (Talpade et al., 2000).
Paraquat
N,N′-dimethyl-4,4′-bipyridinium dichloride (Paraquat), presented in Figure 1D. is one of the most widely used herbicides. It shares structural similarity to MPP+. Parquat causes oxidative stress in the cell through generation of reactive oxygen species. It has been shown to cause SNc DA neuron degeneration and like rotenone also induces formation of LB in DA neurons in mice and rats (McCormack et al., 2002; Cicchetti et al., 2005). However, large variability has been observed in cell death and specificity for DA neurons including some contradictions, with some researchers claiming that Paraquat does not cause changes in the nigrostriatal DA system (Miller, 2007). Paraquat has been used in conjuction with 2-(dithiocarboxy)aminoethylcarbamodithioato(2-)-kS, kS' manganese also called Maneb, a fungicide, which has been shown to potentiate the effects of both MPTP and Paraquat. Maneb on its own has also been shown to decrease locomotor activity and produce SNc neurons loss (Thrash et al., 2007).
Amphetamine Based Models
Treatment of rodents and primates with high doses of methamphetamine has shown selective DA, serotonergic nerve terminal as well as SNc neuronal loss (Wagner et al., 1979; Thrash et al., 2009). Methamphetamine, presented in Figure 1E. causes this damage by promoting change in the distribution of DA from the synaptic vesicle to the cytosol (Howard et al., 2011). To do so, it interacts with both the dopamine transporter (DAT) and the vesicular monoamine transporter (VMAT2), resulting in promoting the collapse of vesicular proton gradients. This leads to DA oxidation within the neuronal cytosol, which causes oxidative stress within the cell by generation of hydroxyl and superoxide Reactive Oxygen Species (ROS) (Larsen et al., 2002; Cadet et al., 2007). Subtoxic concentrations of methamphetamine has been shown to protect DA neurons cells against 6-OHDA toxicity, whereas higher concentrations of methamphetamine exacerbated it (El Ayadi and Zigmond, 2011). On the other hand, despite affecting mainly the serotonergic system, MDMA can also affect DA neurons, with the repeated administration of MDMA producing degeneration of DA terminals in the striatum, and neuronal loss in the SNc (Granado et al., 2008a,b). Development of an amphetamine model of PD, especially in conjunction with other neurotoxins such as MPTP, and Paraquat might be useful.
Genetic Models
In theory genetic model of a simple disease or a syndrome can be made of a mutant gene involved in the progression of the disease in patients or even a gene that might not be validated to be involved in patients but can recapitulate some key features of the disease in the model system. The goal of making genetic model is 3-fold:
1. Understand the signaling and pathways associated with known causal gene.
2. Understand disease signaling by introducing a perturbation in signaling through a gene not found to be causal in patients but can mimic key disease and equally importantly disease like phenotypes.
3. To enable therapeutic screens.
Five genes are frequently targeted as disease models for PD and they have all been known to have causal connection in familial PD. One of the largest genome wide analysis studies for PD to date has implicated 28 independent variants across 24 loci (Nalls et al., 2014). What is not obvious is whether the genes encoded (Nalls et al., 2014) within these loci function as the main drivers, carriers or helpers of disease progression. In more common forms of PD, several gene functions are likely to be altered, hence monogenic models are expected to be less successful than toxin-induced models. That said genetic models have been of use in modeling familial PD and also have shed some light on more common PD mechanisms. Some of the model organisms have been invertebrates and one might be led to question the utility of invertebrate models that do not have SNc. While invertebrate models mimic more simplistic features such as loss of DA neurons, they provide a good vehicle to understand the genetic network, molecular signaling, and provide for first round of screening that can be followed up with further work in mammalian models. Tables 1–4 describe the various common rodent genetic models for PD, while Table 5 details fruit fly models. Some of the models are described below:
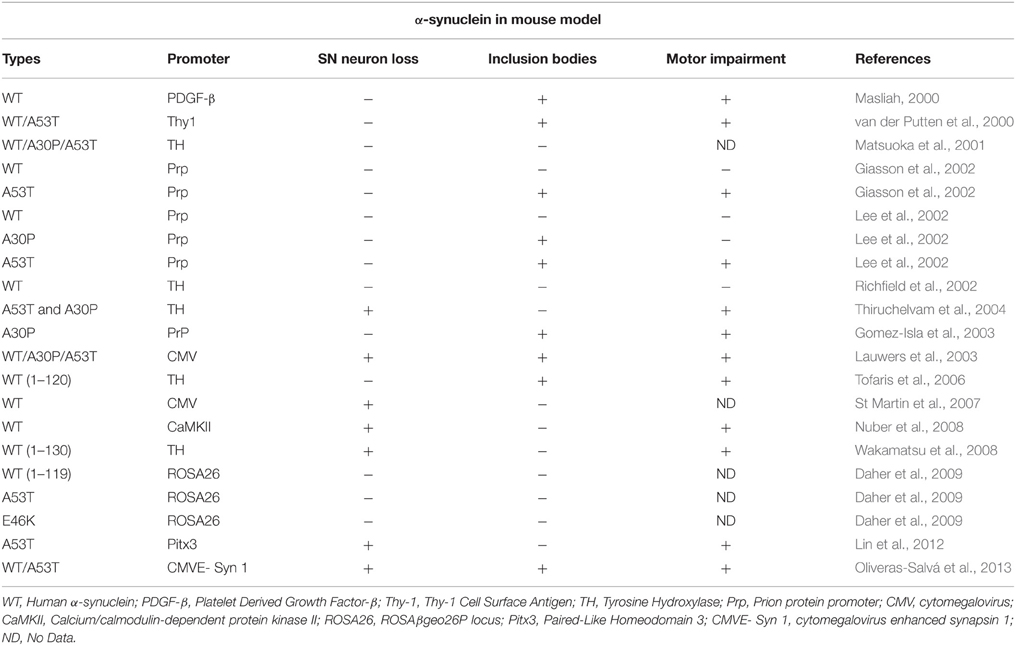
Table 1. α-synuclein models in mice.
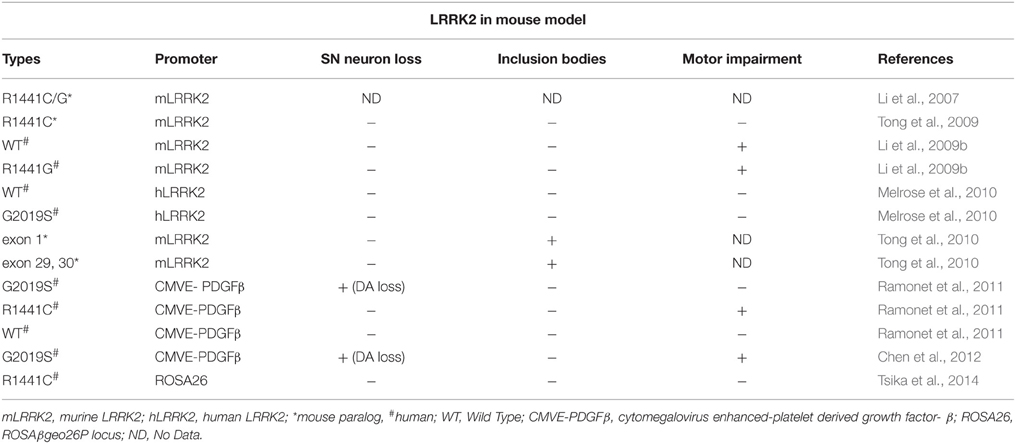
Table 2. LRRK2 models in mice.

Table 3. Parkin, PINK1, and DJ-1 mice models.
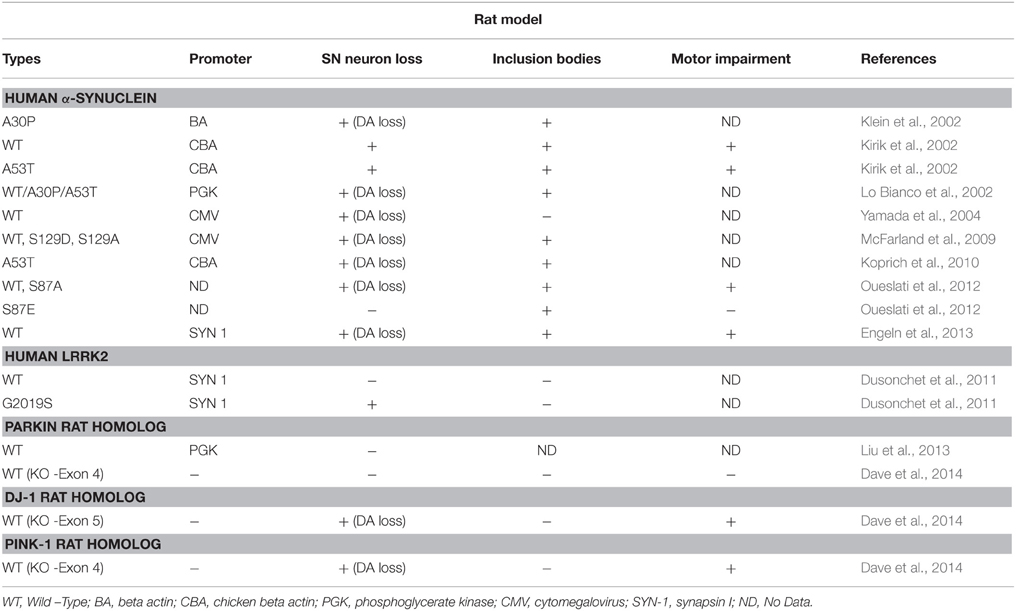
Table 4. Genetic models in rats.
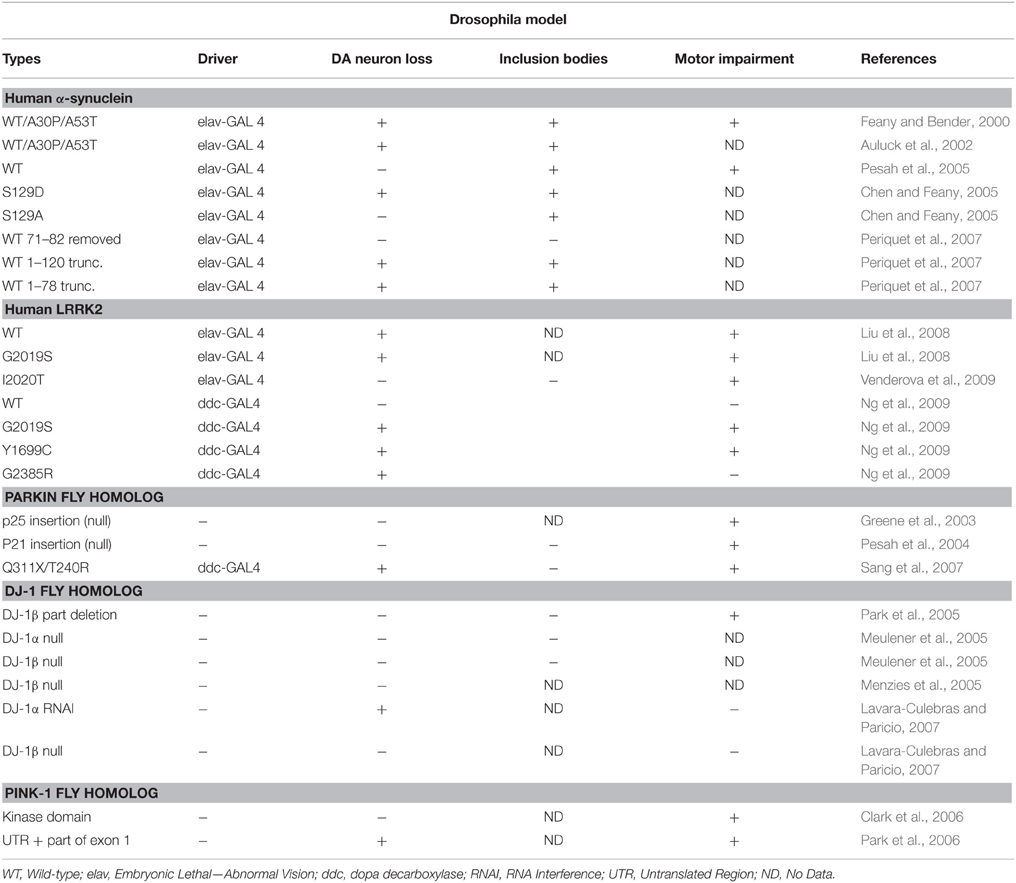
Table 5. Genetic models in fruit flies.
α-Synuclein
This gene is linked to a dominant type of familial PD and the α-synuclein protein is a major part of LBs observed in the brains of PD patients (Iwatsubo, 2003). Table 1 catalogs α-synuclein mice models, while Table 4 has rat genetic models, including α-synuclein. Mutations in five locations have so far been identified in familial PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Singleton et al., 2003; Chartier-Harlin et al., 2004; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Kiely et al., 2013; Proukakis et al., 2013). Injection of wild type or mutant α-synucelin protein has been shown to induce loss of DA neurons, and cause motor impairment in both mice and rats (Oliveras-Salvá et al., 2013). Several mutant lines have been developed in mice that show decreases in striatal DA, exhibit inclusion bodies, and show motor impairmentsbut several fail to show significant degeneration of nigrostrial PD neurons (Masliah, 2000; van der Putten et al., 2000; Giasson et al., 2002; Lee et al., 2002; Richfield et al., 2002; Gomez-Isla et al., 2003; Fernagut and Chesselet, 2004; Thiruchelvam et al., 2004; Tofaris et al., 2006; St Martin et al., 2007; Nuber et al., 2008; Wakamatsu et al., 2008; Daher et al., 2009). Recent use of the Pitx3 promoter shows promise as the line shows progressive SNc DA neuronal loss too along with decrease in DA release and significant motor defects (Li et al., 2009a; Lin et al., 2012). Viral Vectors such as Lentiviruses, Adeno-associated Viruses have been directly injected into the brain at the SNc near the cell bodies of DA neurons in both mice and rats (Lauwers et al., 2003, 2007). Mutant lines with DA loss and inclusion bodies have also been developed in rats (Klein et al., 2002; Lo Bianco et al., 2002; Yamada et al., 2004; McFarland et al., 2009; Koprich et al., 2010; Oueslati et al., 2012; Engeln et al., 2013). While so many models have been developed with α-synuclein, its exact function is not known. Available data suggests that α-synuclein might be a presynaptic regulator of DA release, synthesis or storage, and has been shown to be a regulator of paired-stimulus depression (PSD) (Maries et al., 2003). It also seems to play a role in neuroprotection (Quilty et al., 2006).
LRRK 2
Mutations to this gene are known to cause an autosomal familial form of PD (Funayama et al., 2002; Paisán-Ruíz et al., 2004). Mice LRRK2 lines are compared in Table 2 and rat lines in Table 4. Mitochondrial dysfunction enhances LRRK2 neurodegeneration in some models through unclear mechanisms (Winklhofer and Haass, 2010). LRRK 2 knockout mice have been demonstrated to show abnormal aggregation and accumulation of proteins including α-synuclein, while otherwise not showing any nigrostriatal degeneration (Li et al., 2007, 2009b; Melrose et al., 2010; Tong et al., 2010; Ramonet et al., 2011; Hinkle et al., 2012; Tsika et al., 2014). Virus based models have so far shown some nigrostriatal degeneration however only partial PD phenotypes have so far been developed (Dusonchet et al., 2011; Chen et al., 2012). The LRRK2 gene codes for a 2527 amino acid long protein with multiple domains (Anand and Braithwaite, 2009). Of these domains, two enzymatic domains, the kinase domain and the GTPase domain are of particular interest. In addition multiple protein-protein interaction regions suggest that LRRK may have a role as a major signaling complex (Marín, 2006; Mata et al., 2006). More information on LRRK 2 interactions is needed.
Parkin
Parkin mutations have been seen in cases of familial PD. Table 3 covers Parkin mice strains. Parkin is an integral ligase in the ubiquitin proteosome system (Lücking et al., 2000). Most Parkin transgenic rodents do not exhibit loss of DA neurons in the SNc (Goldberg et al., 2003; Itier et al., 2003; Palacino et al., 2004; Von Coelln et al., 2004; Perez and Palmiter, 2005; Lu et al., 2009; Bian et al., 2012; Liu et al., 2013). Some recent transgenic rodent models have demonstrated modest loss of DA neurons (Kitada et al., 2009; Dave et al., 2014; Van Rompuy et al., 2014). Popularadoption of these models awaits successful reproduction of the results.
DJ-1
DJ-1 is molecular chaperone that under redox reductions plays a role in inhibition of α-synuclein aggregate formation (Shendelman et al., 2004). DJ-1 mutations are linked to autosomal recessive, early onset PD and genetic models using DJ-1 are cataloged in Table 3. Rat model of DJ-1 is presented in Table 4. KO models of DJ-1 show decreased DA release in the striatum but no loss of SNc DA neurons (Goldberg et al., 2005; Andres-Mateos et al., 2007; Manning-Boğ et al., 2007; Yamaguchi and Shen, 2007; Chandran et al., 2008). Hypersensitivity to neurotoxins, such as MPTP, was also observed in DJ-1 deficient mice (Kim et al., 2005). One new model, the DJ1-C57 mouse, shows promise with dramatic unilateral loss of dopaminergic (DA) neurons in the SNc that progresses to bilateral degeneration of the nigrostriatal axis with aging and mild motor behavior deficits (Rousseaux et al., 2012). If reproduced, this model would be highly beneficial to study early onset PD. A transgenic rat model of DJ-1 has also been produced, which exhibits dopaminergic neuron loss and motor abnormalities (Dave et al., 2014).
PINK1
Mutations in the PARK6 locus of PINK1 cause a form of early-onset autosomal PD. Table 3 presents mice model of PINK1 and Table 4 rat models. PINK1 codes for a mitochondrial kinase, which recruits Parkin from the cytosol to the mitochondria, increases the ubiquitination activity of Parkin, and induces Parkin-mediated mitophagy (Lazarou et al., 2013). Since PINK1 and the Parkin function in the same pathway, the phenotypes of PINK1 and Parkin KO mice are very similar. No significant DA neuron abnormalities or LB formation have been observed in PINK1 KO mice however mitochondrial functional defects and increased sensitivity to oxidative stress were observed (Kim et al., 2005; Kitada et al., 2007). Increased levels of α-synuclein through overexpression in PINK1 KO mice results in DA loss but no degeneration in the SNc (Oliveras-Salvá et al., 2013). PINK1 KO rats exhibiting DA loss and motor impairment have been developed recently which more closely mimics PD phenotype (Dave et al., 2014).
Non-Mammalian Genetic Models of PD
Barring α-synuclein, most familial PD genes have at least one drosophila homolog. This includes homologs of PINK1, Parkin, DJ-1, and LRKK2 that have been presented in Table 5. Models with human α-synuclein and LRRK2 have also been developed (Feany and Bender, 2000; Auluck et al., 2002; Chen and Feany, 2005; Pesah et al., 2005; Periquet et al., 2007; Liu et al., 2008; Ng et al., 2009). These transgenic flies show some of the traits of familial PD, with well characterized loss of dopaminergic neurons and motor impairment, except in the case of DJ-1 in which only motor impairment has been observed in DJ-1 β partial deletion (Greene et al., 2003; Pesah et al., 2004; Chen and Feany, 2005; Meulener et al., 2005; Park et al., 2005, 2006; Clark et al., 2006; Lavara-Culebras and Paricio, 2007; Sang et al., 2007) D. melanogaster transgenic models have also helped in elucidating the role of DJ-1, Parkin and PINK1 in mitochondrial physiology (Venderova et al., 2009; Cookson, 2012). Further the study of interactions of human α-synuclein, LRRK2, Parkin, PINK1, and DJ-1 genes has also been possible in the drosophila system (Hirth, 2010).
Like D. melanogaster, D. rerio homologs of most familial PD genes have been discovered. Unlike the rodent and drosophila genetic models of PD, comparatively less characterisation has been carried out in D. rerio. Expression of human α-synuclein and knockouts of Parkin, PINK1, DJ-1 and LRRK2 have been generated, which show some success in mimicking symptoms of familial PD (Park et al., 2006; Bretaud et al., 2007; Anichtchik et al., 2008; Flinn et al., 2009; Fett et al., 2010; Sheng et al., 2010; Milanese et al., 2012; Priyadarshini et al., 2013; O'Donnell et al., 2014). Verification of the results and further behavioral testing is required to establish these models for therapeutic screens.
The advantages of transparency and complete cell lineage information make C. elegans an interesting model for neurodegenerative diseases. Homologs of human PD-related proteins, including Parkin, LRRK2, PINK1 and DJ-1, have been found in C. elegans (Springer et al., 2005; Sakaguchi-Nakashima et al., 2007; Sämann et al., 2009; Kamp et al., 2010; Lee et al., 2013; Chen et al., 2015). Transgenic models developed for these genes have shown increase sensitivity to neurotoxins like MPTP (Ved et al., 2005).
Conclusion
Both toxin and genetic based models have their advantages and disadvantages. However, the use of the two in combination would be quite beneficial. Thus, a multi gene modulated transgenic model in combination with a reliable and effective neurotoxin might allow us to model the PD phenotype better. Addition of a miRNA or siRNA cocktail to the appropriate model systems could potentially allow for the creation of a very robust and accurate PD model showing all the symptoms of PD. Development of primary cell culture models might allow for mimicking slow development of PD cellular damage phenotype too, and be useful for drug discovery. In coming years, we expect to see better models for both basic understanding of PD and also for improved high-throughput drug-discovery.
Author Contributions
SJ conducted overall review of the field and wrote major part of this manuscript. NT co-conducted the review of the field. SS and SM provided expertise on selected topics, wrote and edited select parts of the manuscript. SK envisaged the overall study, guided the work of SJ and NT, and conducted the final editing.
Funding
SJ was funded by CSIR Ph.D. fellowship. SK's lab is funded by intramural IISER-K funding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer Dr Ines Moreno-Gonzalez and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
Anand, V. S., and Braithwaite, S. P. (2009). LRRK2 in Parkinson's disease: biochemical functions. FEBS J. 276, 6428–6435. doi: 10.1111/j.1742-4658.2009.07341.x
Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T. M., Thomas, B., et al. (2007). DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 14807–14812. doi: 10.1073/pnas.0703219104
Anichtchik, O., Diekmann, H., Fleming, A., Roach, A., Goldsmith, P., and Rubinsztein, D. C. (2008). Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J. Neurosci. 28, 8199–8207. doi: 10.1523/JNEUROSCI.0979-08.2008
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Auluck, P. K., Chan, H. Y. E., Trojanowski, J. Q., Lee, V. M. Y., and Bonini, N. M. (2002). Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868. doi: 10.1126/science.1067389
Bian, M., Liu, J., Hong, X., Yu, M., Huang, Y., Sheng, Z., et al. (2012). Overexpression of Parkin Ameliorates Dopaminergic Neurodegeneration Induced by 1- Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in Mice. PLoS ONE 7:e39953. doi: 10.1371/journal.pone.0039953
Blandini, F., Armentero, M.-T., and Martignoni, E. (2008). The 6-hydroxydopamine model: news from the past. Parkinsonism Relat. Disord. 14 (Suppl. 2), S124–S129. doi: 10.1016/j.parkreldis.2008.04.015
Blesa, J., Juri, C., Collantes, M., Peñuelas, I., Prieto, E., Iglesias, E., et al. (2010). Progression of dopaminergic depletion in a model of MPTP-induced Parkinsonism in non-human primates. An 18F-DOPA and 11C-DTBZ PET study. Neurobiol. Dis. 38, 456–463. doi: 10.1016/j.nbd.2010.03.006
Blesa, J., Juri, C., García-Cabezas, M. Á., Adánez, R., Sánchez-González, M. Á., Cavada, C., et al. (2011). Inter-hemispheric asymmetry of nigrostriatal dopaminergic lesion: a possible compensatory mechanism in Parkinson's disease. Front. Syst. Neurosci. 5:92. doi: 10.3389/fnsys.2011.00092
Blesa, J., Pifl, C., Sánchez-González, M. A., Juri, C., García-Cabezas, M. A., Adánez, R., et al. (2012). The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiol. Dis. 48, 79–91. doi: 10.1016/j.nbd.2012.05.018
Braak, H., Rüb, U., Sandmann-Keil, D., Gai, W. P., de Vos, R. A., Jansen Steur, E. N., et al. (2000). Parkinson's disease: affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol. 99, 489–495. doi: 10.1007/s004010051150
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Bretaud, S., Allen, C., Ingham, P. W., and Bandmann, O. (2007). p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson's disease. J. Neurochem. 100, 1626–1635. doi: 10.1111/j.1471-4159.2006.04291.x
Cadet, J. L., Krasnova, I. N., Jayanthi, S., and Lyles, J. (2007). Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox. Res. 11, 183–202. doi: 10.1007/BF03033567
Chandran, J. S., Lin, X., Zapata, A., Höke, A., Shimoji, M., Moore, S. O., et al. (2008). Progressive behavioral deficits in DJ-1-deficient mice are associated with normal nigrostriatal function. Neurobiol. Dis. 29, 505–514. doi: 10.1016/j.nbd.2007.11.011
Chartier-Harlin, M.-C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet (London, England) 364, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Chen, C.-Y., Weng, Y.-H., Chien, K.-Y., Lin, K.-J., Yeh, T.-H., Cheng, Y.-P., et al. (2012). (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 19, 1623–1633. doi: 10.1038/cdd.2012.42
Chen, L., and Feany, M. B. (2005). Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663. doi: 10.1038/nn1443
Chen, P., DeWitt, M. R., Bornhorst, J., Soares, F. A., Mukhopadhyay, S., Bowman, A. B., et al. (2015). Age- and manganese-dependent modulation of dopaminergic phenotypes in a C. elegans DJ-1 genetic model of Parkinson's disease. Metallomics 7, 289–98. doi: 10.1039/C4MT00292J
Cicchetti, F., Lapointe, N., Roberge-Tremblay, A., Saint-Pierre, M., Jimenez, L., Ficke, B. W., et al. (2005). Systemic exposure to paraquat and Maneb models early Parkinson's disease in young adult rats. Neurobiol. Dis. 20, 360–371. doi: 10.1016/j.nbd.2005.03.018
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Cookson, M. R. (2012). Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb. Perspect. Med. 2:a009415. doi: 10.1101/cshperspect.a009415
da Conceição, F. S., Ngo-Abdalla, S., Houzel, J. C., and Rehen, S. K. (2010). Murine model for Parkinson's disease: from 6-OH dopamine lesion to behavioral test. J. Vis. Exp. 35:1376. doi: 10.3791/1376
Daher, J. P. L., Ying, M., Banerjee, R., McDonald, R. S., Hahn, M. D., Yang, L., et al. (2009). Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol. Neurodegener. 4:34. doi: 10.1186/1750-1326-4-34
Dauer, W., and Przedborski, S. (2003). Parkinson's Disease. Neuron 39, 889–909. doi: 10.1016/S0896-6273(03)00568-3
Dave, K. D., De Silva, S., Sheth, N. P., Ramboz, S., Beck, M. J., Quang, C., et al. (2014). Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. Neurobiol. Dis. 70, 190–203. doi: 10.1016/j.nbd.2014.06.009
Del-Bel, E., Padovan-Neto, F. E., Szawka, R. E., da-Silva, C. A., Raisman-Vozari, R., Anselmo-Franci, J., et al. (2014). Counteraction by nitric oxide synthase inhibitor of neurochemical alterations of dopaminergic system in 6-OHDA-lesioned rats under L-DOPA treatment. Neurotox. Res. 25, 33–44. doi: 10.1007/s12640-013-9406-3
Dusonchet, J., Kochubey, O., Stafa, K., Young, S. M., Zufferey, R., Moore, D. J., et al. (2011). A rat model of progressive nigral neurodegeneration induced by the Parkinson's disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 907–912. doi: 10.1523/JNEUROSCI.5092-10.2011
El Ayadi, A., and Zigmond, M. J. (2011). Low concentrations of methamphetamine can protect dopaminergic cells against a larger oxidative stress injury: mechanistic study. PLoS ONE 6:e24722. doi: 10.1371/journal.pone.0024722
Engeln, M., Fasano, S., Ahmed, S. H., Cador, M., Baekelandt, V., Bezard, E., et al. (2013). Levodopa gains psychostimulant-like properties after nigral dopaminergic loss. Ann. Neurol. 74, 140–144. doi: 10.1002/ana.23881
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson's disease. Nature 404, 394–398. doi: 10.1038/35006074
Fernagut, P.-O., and Chesselet, M.-F. (2004). Alpha-synuclein and transgenic mouse models. Neurobiol. Dis. 17, 123–130. doi: 10.1016/j.nbd.2004.07.001
Fett, M. E., Pilsl, A., Paquet, D., van Bebber, F., Haass, C., Tatzelt, J., et al. (2010). Parkin is protective against proteotoxic stress in a transgenic zebrafish model. PLoS ONE 5:e11783. doi: 10.1371/journal.pone.0011783
Fleming, S. M., Zhu, C., Fernagut, P. O., Mehta, A., DiCarlo, C. D., Seaman, R. L., et al. (2004). Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp. Neurol. 187, 418–429. doi: 10.1016/j.expneurol.2004.01.023
Flinn, L., Mortiboys, H., Volkmann, K., Köster, R. W., Ingham, P. W., and Bandmann, O. (2009). Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132, 1613–1623. doi: 10.1093/brain/awp108
Fornai, F., Schlüter, O. M., Lenzi, P., Gesi, M., Ruffoli, R., Ferrucci, M., et al. (2005). Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and -synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 3413–3418. doi: 10.1073/pnas.0409713102
Funayama, M., Hasegawa, K., and Kowa, H. (2002). A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11. 2–q13. 1. Ann. Neurol. 51, 296–301. doi: 10.1002/ana.10113
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., and Lee, V. M.-Y. (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. doi: 10.1016/S0896-6273(02)00682-7
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient Mice Exhibit Nigrostriatal Deficits but not Loss of Dopaminergic Neurons. J. Biol. Chem. 278, 43628–43635. doi: 10.1074/jbc.M308947200
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal Dopaminergic Deficits and Hypokinesia Caused by Inactivation of the Familial Parkinsonism-Linked Gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Gomez-Isla, T., Irizarry, M. C., Mariash, A., Cheung, B., Soto, O., Schrump, S., et al. (2003). Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol. Aging 24, 245–258. doi: 10.1016/S0197-4580(02)00091-X
Granado, N., Escobedo, I., O'shea, E., Colado, M. I., and Moratalla, R. (2008a). Early loss of dopaminergic terminals in striosomes after MDMA administration to mice. Synapse 62, 80–84. doi: 10.1002/syn.20466
Granado, N., O'shea, E., Bove, J., Vila, M., Colado, M. I., and Moratalla, R. (2008b). Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J. Neurochem. 107, 1102–1112. doi: 10.1111/j.1471-4159.2008.05705.x
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083. doi: 10.1073/pnas.0737556100
Haque, M. E., Mount, M. P., Safarpour, F., Abdel-Messih, E., Callaghan, S., Mazerolle, C., et al. (2012). Inactivation of Pink1 gene in vivo Sensitizes dopamine-producing neurons to 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson Disease genes, parkin or DJ-1. J. Biol. Chem. 287, 23162–23170. doi: 10.1074/jbc.M112.346437
Hinkle, K. M., Yue, M., Behrouz, B., Dächsel, J. C., Lincoln, S. J., Bowles, E. E., et al. (2012). LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol. Neurodegener. 7:25. doi: 10.1186/1750-1326-7-25
Hirth, F. (2010). Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol. Disord. Drug Targets 9, 504–523. doi: 10.2174/187152710791556104
Howard, C. D., Keefe, K. A., Garris, P. A., and Daberkow, D. P. (2011). Methamphetamine neurotoxicity decreases phasic, but not tonic, dopaminergic signaling in the rat striatum. J. Neurochem. 118, 668–676. doi: 10.1111/j.1471-4159.2011.07342.x
Itier, J. M., Ibáñez, P., Mena, M. A., Abbas, N., Cohen-Salmon, C., Bohme, G. A., et al. (2003). Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 12, 2277–2291. doi: 10.1093/hmg/ddg239
Iwatsubo, T. (2003). Aggregation of α-synuclein in the pathogenesis of Parkinson's disease. J. Neurol. 250, 1. doi: 10.1007/s00415-003-1303-x
Jackson-Lewis, V., and Przedborski, S. (2007). Protocol for the MPTP mouse model of Parkinson's disease. Nat. Protoc. 2, 141–151. doi: 10.1038/nprot.2006.342
Javitch, J. A., D'Amato, R. J., Strittmatter, S. M., and Snyder, S. H. (1985). Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. U.S.A.82, 2173–2177.
Jellinger, K. A. (1991). Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 14, 153–197. doi: 10.1007/BF03159935
Jeon, B. S., Jackson-Lewis, V., and Burke, R. E. (1995). 6-Hydroxydopamine lesion of the rat substantia nigra: time course and morphology of cell death. Neurodegeneration 4, 131–137. doi: 10.1006/neur.1995.0016
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Kiely, A. P., Asi, Y. T., Kara, E., Limousin, P., Ling, H., Lewis, P., et al. (2013). α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 125, 753–769. doi: 10.1007/s00401-013-1096-7
Kim, R. H., Smith, P. D., Aleyasin, H., Hayley, S., Mount, M. P., Pownall, S., et al. (2005). Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220. doi: 10.1073/pnas.0501282102
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., et al. (2002). Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791.
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446. doi: 10.1073/pnas.0702717104
Kitada, T., Tong, Y., Gautier, C. A., and Shen, J. (2009). Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J. Neurochem. 111, 696–702. doi: 10.1111/j.1471-4159.2009.06350.x
Klein, R. L., King, M. A., Hamby, M. E., and Meyer, E. M. (2002). Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 13, 605–612. doi: 10.1089/10430340252837206
Koprich, J. B., Johnston, T. H., Reyes, M. G., Sun, X., and Brotchie, J. M. (2010). Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pat. Mol. Neurodegener. 5:43. doi: 10.1186/1750-1326-5-43
Kowall, N. W., Hantraye, P., Brouillet, E., Beal, M. F., McKee, A. C., and Ferrante, R. J. (2000). MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 11, 211–213. doi: 10.1097/00001756-200001170-00041
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Larsen, K. E., Fon, E. A., Hastings, T. G., Edwards, R. H., and Sulzer, D. (2002). Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 22, 8951–8960.
Lauwers, E., Bequé, D., Van Laere, K., Nuyts, J., Bormans, G., Mortelmans, L., et al. (2007). Non-invasive imaging of neuropathology in a rat model of α-synuclein overexpression. Neurobiol. Aging 28, 248–257. doi: 10.1016/j.neurobiolaging.2005.12.005
Lauwers, E., Debyser, Z., Van Dorpe, J., De Strooper, B., Nuttin, B., and Baekelandt, V. (2003). Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector-mediated overexpression of alpha-synuclein. Brain Pathol. 13, 364–372. doi: 10.1111/j.1750-3639.2003.tb00035.x
Lavara-Culebras, E., and Paricio, N. (2007). Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene 400, 158–165. doi: 10.1016/j.gene.2007.06.013
Lazarou, M., Narendra, D. P., Jin, S. M., Tekle, E., Banerjee, S., and Youle, R. J. (2013). PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol. 200, 163–172. doi: 10.1083/jcb.201210111
Lee, J.-Y., Kim, C., Kim, J., and Park, C. (2013). DJR-1.2 of Caenorhabditis elegans is induced by DAF-16 in the dauer state. Gene 524, 373–376. doi: 10.1016/j.gene.2013.04.032
Lee, M. K., Stirling, W., Xu, Y., Xu, X., Qui, D., Mandir, A. S., et al. (2002). Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 8968–8973. doi: 10.1073/pnas.132197599
Li, J., Dani, J. A., and Le, W. (2009a). The role of transcription factor Pitx3 in dopamine neuron development and Parkinson's disease. Curr. Top. Med. Chem. 9, 855–859. doi: 10.2174/156802609789378236
Li, X., Tan, Y.-C., Poulose, S., Olanow, C. W., Huang, X.-Y., and Yue, Z. (2007). Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J. Neurochem. 103, 238–247. doi: 10.1111/j.1471-4159.2007.04743.x
Li, Y., Liu, W., Oo, T. F., Wang, L., Tang, Y., Jackson-Lewis, V., et al. (2009b). Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat. Neurosci. 12, 826–828. doi: 10.1038/nn.2349
Lin, X., Parisiadou, L., Sgobio, C., Liu, G., Yu, J., Sun, L., et al. (2012). Conditional expression of Parkinson's disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 32, 9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012
Liu, B., Traini, R., Killinger, B., Schneider, B., and Moszczynska, A. (2013). Overexpression of parkin in the rat nigrostriatal dopamine system protects against methamphetamine neurotoxicity. Exp. Neurol. 247, 359–372. doi: 10.1016/j.expneurol.2013.01.001
Liu, Z., Wang, X., Yu, Y., Li, X., Wang, T., Jiang, H., et al. (2008). A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 105, 2693–2698. doi: 10.1073/pnas.0708452105
Lo Bianco, C., Ridet, J.-L., Schneider, B. L., Deglon, N., and Aebischer, P. (2002). alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10813–10818. doi: 10.1073/pnas.152339799
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Lu, X.-H., Fleming, S. M., Meurers, B., Ackerson, L. C., Mortazavi, F., Lo, V., et al. (2009). Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase k-resistant -synuclein. J. Neurosci. 29, 1962–1976. doi: 10.1523/JNEUROSCI.5351-08.2009
Luthman, J., Fredriksson, A., Sundström, E., Jonsson, G., and Archer, T. (1989). Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: motor behavior and monoamine alterations at adult stage. Behav. Brain Res. 33, 267–277. doi: 10.1016/S0166-4328(89)80121-4
Manning-Boğ, A. B., Caudle, W. M., Perez, X. A., Reaney, S. H., Paletzki, R., Isla, M. Z., et al. (2007). Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol. Dis. 27, 141–150. doi: 10.1016/j.nbd.2007.03.014
Maries, E., Dass, B., Collier, T. J., Kordower, J. H., and Steece-Collier, K. (2003). The role of alpha-synuclein in Parkinson's disease: insights from animal models. Nat. Rev. Neurosci. 4, 727–738. doi: 10.1038/nrn1199
Marín, I. (2006). The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol. Biol. Evol. 23, 2423–2433. doi: 10.1093/molbev/msl114
Masliah, E. (2000). Dopaminergic loss and inclusion body formation in -synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269. doi: 10.1126/science.287.5456.1265
Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P., and Gallo, K. A. (2006). LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286–293. doi: 10.1016/j.tins.2006.03.006
Matsuoka, Y., Vila, M., Lincoln, S., McCormack, A., Picciano, M., LaFrancois, J., et al. (2001). Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol. Dis. 8, 535–539. doi: 10.1006/nbdi.2001.0392
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, J. W., Cory-Slechta, D. A., et al. (2002). Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127. doi: 10.1006/nbdi.2002.0507
McFarland, N. R., Fan, Z., Xu, K., Schwarzschild, M. A., Feany, M. B., Hyman, B. T., et al. (2009). Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524. doi: 10.1097/NEN.0b013e3181a24b53
McNaught, K. S., Olanow, C. W., Halliwell, B., Isacson, O., and Jenner, P. (2001). Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat. Rev. Neurosci. 2, 589–594. doi: 10.1038/35086067
Melrose, H. L., Dächsel, J. C., Behrouz, B., Lincoln, S. J., Yue, M., Hinkle, K. M., et al. (2010). Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517. doi: 10.1016/j.nbd.2010.07.010
Menzies, F. M., Yenisetti, S. C., and Min, K. T. (2005). Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 15, 1578–1582. doi: 10.1016/j.cub.2005.07.036
Meredith, G. E., Totterdell, S., Petroske, E., Santa Cruz, K., Callison, R. C., and Lau, Y.-S. (2002). Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res. 956, 156–165. doi: 10.1016/S0006-8993(02)03514-X
Meulener, M., Whitworth, A. J., Armstrong-Gold, C. E., Rizzu, P., Heutink, P., Wes, P. D., et al. (2005). Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr. Biol. 15, 1572–1577. doi: 10.1016/j.cub.2005.07.064
Milanese, C., Sager, J. J., Bai, Q., Farrell, T. C., Cannon, J. R., Greenamyre, J. T., et al. (2012). Hypokinesia and reduced dopamine levels in zebrafish lacking β- and γ1-synucleins. J. Biol. Chem. 287, 2971–2983. doi: 10.1074/jbc.M111.308312
Miller, G. W. (2007). Paraquat: the red herring of Parkinson's Disease research. Toxicol. Sci. 100, 1–2. doi: 10.1093/toxsci/kfm223
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Ng, C.-H., Mok, S. Z. S., Koh, C., Ouyang, X., Fivaz, M. L., Tan, E.-K., et al. (2009). Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 29, 11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009
Nieto, M., Gil-Bea, F. J., Dalfó, E., Cuadrado, M., Cabodevilla, F., Sánchez, B., et al. (2006). Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol. Aging 27, 848–856. doi: 10.1016/j.neurobiolaging.2005.04.010
Nuber, S., Petrasch-Parwez, E., Winner, B., Winkler, J., von Hörsten, S., Schmidt, T., et al. (2008). Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J. Neurosci. 28, 2471–2484. doi: 10.1523/JNEUROSCI.3040-07.2008
O'Donnell, K. C., Lulla, A., Stahl, M. C., Wheat, N. D., Bronstein, J. M., and Sagasti, A. (2014). Axon degeneration and PGC-1α-mediated protection in a zebrafish model of α-synuclein toxicity. Dis. Model. Mech. 7, 571–582. doi: 10.1242/dmm.013185
Oliveras-Salvá, M., Van der Perren, A., Casadei, N., Stroobants, S., Nuber, S., D'Hooge, R., et al. (2013). rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol. Neurodegener. 8:44. doi: 10.1186/1750-1326-8-44
Oueslati, A., Paleologou, K. E., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2012). Mimicking phosphorylation at serine 87 inhibits the aggregation of human -synuclein and protects against its toxicity in a rat model of Parkinson's Disease. J. Neurosci. 32, 1536–1544. doi: 10.1523/JNEUROSCI.3784-11.2012
Ovadia, A., Zhang, Z., and Gash, D. M. (1995). Increased susceptibility to MPTP toxicity in middle-aged rhesus monkeys. Neurobiol. Aging 16, 931–937. doi: 10.1016/0197-4580(95)02012-8
Paisán-Ruíz, C., Jain, S., Evans, E. W., Gilks, W. P., Simón, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., et al. (2004). Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 279, 18614–18622. doi: 10.1074/jbc.M401135200
Park, J., Kim, S. Y., Cha, G.-H., Lee, S. B., Kim, S., and Chung, J. (2005). Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 361, 133–139. doi: 10.1016/j.gene.2005.06.040
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Perez, F. A., and Palmiter, R. D. (2005). Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 102, 2174–2179. doi: 10.1073/pnas.0409598102
Perez, R. G., and Hastings, T. G. (2004). Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J. Neurochem. 89, 1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated -synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Pesah, Y., Burgess, H., Middlebrooks, B., Ronningen, K., Prosser, J., Tirunagaru, V., et al. (2005). Whole-mount analysis reveals normal numbers of dopaminergic neurons following misexpression of alpha-Synuclein in Drosophila. Genesis 41, 154–159. doi: 10.1002/gene.20106
Pesah, Y., Pham, T., Burgess, H., Middlebrooks, B., Verstreken, P., Zhou, Y., et al. (2004). Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131, 2183–2194. doi: 10.1242/dev.01095
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poulin, J.-F., Zou, J., Drouin-Ouellet, J., Kim, K.-Y. A., Cicchetti, F., and Awatramani, R. B. (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep. 9, 930–943. doi: 10.1016/j.celrep.2014.10.008
Priyadarshini, M., Tuimala, J., Chen, Y. C., and Panula, P. (2013). A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol. Dis. 54, 127–138. doi: 10.1016/j.nbd.2013.02.002
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Przedborski, S., Levivier, M., Jiang, H., Ferreira, M., Jackson-Lewis, V., Donaldson, D., et al. (1995). Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 67, 631–647. doi: 10.1016/0306-4522(95)00066-R
Quilty, M. C., King, A. E., Gai, W.-P., Pountney, D. L., West, A. K., Vickers, J. C., et al. (2006). Alpha-synuclein is upregulated in neurones in response to chronic oxidative stress and is associated with neuroprotection. Exp. Neurol. 199, 249–256. doi: 10.1016/j.expneurol.2005.10.018
Ramonet, D., Daher, J. P. L., Lin, B. M., Stafa, K., Kim, J., Banerjee, R., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 6:e18568. doi: 10.1371/journal.pone.0018568
Ramsay, R. R., and Singer, T. P. (1986). Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J. Biol. Chem. 261, 7585–7587.
Riachi, N. J., Dietrich, W. D., and Harik, S. I. (1990). Effects of internal carotid administration of MPTP on rat brain and blood-brain barrier. Brain Res. 533, 6–14. doi: 10.1016/0006-8993(90)91788-I
Richfield, E. K., Thiruchelvam, M. J., Cory-Slechta, D. A., Wuertzer, C., Gainetdinov, R. R., Caron, M. G., et al. (2002). Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 175, 35–48. doi: 10.1006/exnr.2002.7882
Rodriguez-Oroz, M. C., Jahanshahi, M., Krack, P., Litvan, I., Macias, R., Bezard, E., et al. (2009). Initial clinical manifestations of Parkinson's disease: features and pathophysiological mechanisms. Lancet Neurol. 8, 1128–1139. doi: 10.1016/S1474-4422(09)70293-5
Rousseaux, M. W. C., Marcogliese, P. C., Qu, D., Hewitt, S. J., Seang, S., Kim, R. H., et al. (2012). Progressive dopaminergic cell loss with unilateral-to-bilateral progression in a genetic model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 109, 15918–15923. doi: 10.1073/pnas.1205102109
Sakaguchi-Nakashima, A., Meir, J. Y., Jin, Y., Matsumoto, K., and Hisamoto, N. (2007). LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 17, 592–598. doi: 10.1016/j.cub.2007.01.074
Sämann, J., Hegermann, J., von Gromoff, E., Eimer, S., Baumeister, R., and Schmidt, E. (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 284, 16482–16491. doi: 10.1074/jbc.M808255200
Sang, T.-K., Chang, H.-Y., Lawless, G. M., Ratnaparkhi, A., Mee, L., Ackerson, L. C., et al. (2007). A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J. Neurosci. 27, 981–992. doi: 10.1523/JNEUROSCI.4810-06.2007
Sauer, H., and Oertel, W. H. (1994). Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience 59, 401–415. doi: 10.1016/0306-4522(94)90605-X
Schmidt, N., and Ferger, B. (2001). Neurochemical findings in the MPTP model of Parkinson's disease. J. Neural Transm. 108, 1263–1282. doi: 10.1007/s007020100004
Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. doi: 10.1371/journal.pbio.0020362
Sheng, D., Qu, D., Kwok, K. H. H., Ng, S. S., Lim, A. Y. M., Aw, S. S., et al. (2010). Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 6:e1000914. doi: 10.1371/journal.pgen.1000914
Sherer, T. B., Betarbet, R., Testa, C. M., Seo, B. B., Richardson, J. R., Kim, J. H., et al. (2003). Mechanism of toxicity in rotenone models of Parkinson's disease. J. Neurosci. 23, 10756–10764.
Simola, N., Morelli, M., and Carta, A. R. (2007). The 6-hydroxydopamine model of Parkinson's disease. Neurotox. Res. 11, 151–167. doi: 10.1007/BF03033565
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. doi: 10.1126/science.1090278
Springer, W., Hoppe, T., Schmidt, E., and Baumeister, R. (2005). A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 14, 3407–3423. doi: 10.1093/hmg/ddi371
Srivastava, P., and Panda, D. (2007). Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J. 274, 4788–4801. doi: 10.1111/j.1742-4658.2007.06004.x
St Martin, J. L., Klucken, J., Outeiro, T. F., Nguyen, P., Keller-McGandy, C., Cantuti-Castelvetri, I., et al. (2007). Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 100, 1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x
Talpade, D. J., Greene, J. G., Higgins, D. S., and Greenamyre, J. T. (2000). In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J. Neurochem. 75, 2611–2621. doi: 10.1046/j.1471-4159.2000.0752611.x
Thiele, S. L., Warre, R., and Nash, J. E. (2012). Development of a unilaterally-lesioned 6-OHDA mouse model of Parkinson's disease. J. Vis. Exp. 60:3234. doi: 10.3791/3234
Thiruchelvam, M. J., Powers, J. M., Cory-Slechta, D. A., and Richfield, E. K. (2004). Risk factors for dopaminergic neuron loss in human alpha-synuclein transgenic mice. Eur. J. Neurosci. 19, 845–854. doi: 10.1111/j.0953-816X.2004.03139.x
Thrash, B., Thiruchelvan, K., Ahuja, M., Suppiramaniam, V., and Dhanasekaran, M. (2009). Methamphetamine-induced neurotoxicity: the road to Parkinson's disease. Pharmacol. Rep. 61, 966–977. doi: 10.1016/S1734-1140(09)70158-6
Thrash, B., Uthayathas, S., Karuppagounder, S. S., Suppiramaniam, V., and Dhanasekaran, M. (2007). Paraquat and Maneb induced neurotoxicity. Proc. West. Pharmacol. Soc. 50, 31–42.
Tofaris, G. K., Garcia Reitböck, P., Humby, T., Lambourne, S. L., O'Connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tong, Y., Pisani, A., Martella, G., Karouani, M., Yamaguchi, H., Pothos, E. N., et al. (2009). R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. U. S. A. 106, 14622–14627. doi: 10.1073/pnas.0906334106
Tong, Y., Yamaguchi, H., Giaime, E., Boyle, S., Kopan, R., Kelleher, R. J., et al. (2010). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884. doi: 10.1073/pnas.1004676107
Tsika, E., Kannan, M., Foo, C. S.-Y., Dikeman, D., Glauser, L., Gellhaar, S., et al. (2014). Conditional expression of Parkinson's disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis. 71, 345–358. doi: 10.1016/j.nbd.2014.08.027
Ungerstedt, U. (1968). 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–110. doi: 10.1016/0014-2999(68)90164-7
van der Putten, H., Wiederhold, K.-H., Probst, A., Barbieri, S., Mistl, C., Danner, S., et al. (2000). Neuropathology in mice expressing human alpha -synuclein. J. Neurosci. 20, 6021–6029. doi: 10.1371/journal.pone.0024834
Van Rompuy, A.-S., Lobbestael, E., Van der Perren, A., Van den Haute, C., and Baekelandt, V. (2014). Long-term overexpression of human wild-type and T240R mutant Parkin in rat substantia nigra induces progressive dopaminergic neurodegeneration. J. Neuropathol. Exp. Neurol. 73, 159–174. doi: 10.1097/NEN.0000000000000039
Varastet, M., Riche, D., Maziere, M., and Hantraye, P. (1994). Chronic MPTP treatment reproduces in baboons the differential vulnerability of mesencephalic dopaminergic neurons observed in Parkinson's disease. Neuroscience 63, 47–56. doi: 10.1016/0306-4522(94)90006-X
Ved, R., Saha, S., Westlund, B., Perier, C., Burnam, L., Sluder, A., et al. (2005). Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of -synuclein, Parkin, and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 280, 42655–42668. doi: 10.1074/jbc.M505910200
Venderova, K., Kabbach, G., Abdel-Messih, E., Zhang, Y., Parks, R. J., Imai, Y., et al. (2009). Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson's disease. Hum. Mol. Genet. 18, 4390–4404. doi: 10.1093/hmg/ddp394
Von Coelln, R., Thomas, B., Savitt, J. M., Lim, K. L., Sasaki, M., Hess, E. J., et al. (2004). Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc. Natl. Acad. Sci. U.S.A. 101, 10744–10749. doi: 10.1073/pnas.0401297101
Wagner, G. C., Seiden, L. S., and Schuster, C. R. (1979). Methamphetamine-induced changes in brain catecholamines in rats and guinea pigs. Drug Alcohol Depend. 4, 435–438. doi: 10.1016/0376-8716(79)90076-0
Wakamatsu, M., Ishii, A., Iwata, S., Sakagami, J., Ukai, Y., Ono, M., et al. (2008). Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol. Aging 29, 574–585. doi: 10.1016/j.neurobiolaging.2006.11.017
Winklhofer, K. F., and Haass, C. (2010). Mitochondrial dysfunction in Parkinson's disease. Biochim. Biophys. Acta 1802, 29–44. doi: 10.1016/j.bbadis.2009.08.013
Xiong, N., Huang, J., Zhang, Z., Zhang, Z., Xiong, J., Liu, X., et al. (2009). Stereotaxical infusion of rotenone: a reliable rodent model for Parkinson's Disease. PLoS ONE 4:e7878. doi: 10.1371/journal.pone.0007878
Yamada, M., Iwatsubo, T., Mizuno, Y., and Mochizuki, H. (2004). Overexpression of a-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of a-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J. Neurochem. 91, 451–461. doi: 10.1111/j.1471-4159.2004.02728.x
Yamaguchi, H., and Shen, J. (2007). Absence of dopaminergic neuronal degeneration and oxidative damage in aged DJ-1-deficient mice. Mol. Neurodegener. 2:10. doi: 10.1186/1750-1326-2-10
Keywords: Parkinson's Disease, Parkinsonian Disorders, lewy bodies, neurodegeneration, ventral tegmental area (VTA), toxin models, genetic models, substantia nigra pars compacta (SNc)
Citation: Jagmag SA, Tripathi N, Shukla SD, Maiti S and Khurana S (2016) Evaluation of Models of Parkinson's Disease. Front. Neurosci. 9:503. doi: 10.3389/fnins.2015.00503
Received: 13 October 2015; Accepted: 21 December 2015;
Published: 19 January 2016.
Edited by:
Claudio Soto, University of Texas, USAReviewed by:
Ines Moreno-Gonzalez, University of Texas, USATakahiko Tokuda, Kyoto Prefectural University of Medicine, Japan
Copyright © 2016 Jagmag, Tripathi, Shukla, Maiti and Khurana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sukant Khurana, c3VrYW50a2h1cmFuYUBnbWFpbC5jb20=
†These authors have contributed equally to this work.