 Erwin Lemche
Erwin Lemche Oleg S. Chaban2
Oleg S. Chaban2- 1Section of Cognitive Neuropsychiatry, Department of Psychosis Studies, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, UK
- 2Section of Psychosomatic Medicine, Bogomolets National Medical University, Kiev, Ukraine
- 3Department of Medical Science, Institute of Clinical Research, Berlin, Germany
Impact of environmental stress upon pathophysiology of the metabolic syndrome (MetS) has been substantiated by epidemiological, psychophysiological, and endocrinological studies. This review discusses recent advances in the understanding of causative roles of nutritional factors, sympathomedullo-adrenal (SMA) and hypothalamic-pituitary adrenocortical (HPA) axes, and adipose tissue chronic low-grade inflammation processes in MetS. Disturbances in the neuroendocrine systems for leptin, melanocortin, and neuropeptide Y (NPY)/agouti-related protein systems have been found resulting directly in MetS-like conditions. The review identifies candidate risk genes from factors shown critical for the functioning of each of these neuroendocrine signaling cascades. In its meta-analytic part, recent studies in epigenetic modification (histone methylation, acetylation, phosphorylation, ubiquitination) and posttranscriptional gene regulation by microRNAs are evaluated. Several studies suggest modification mechanisms of early life stress (ELS) and diet-induced obesity (DIO) programming in the hypothalamic regions with populations of POMC-expressing neurons. Epigenetic modifications were found in cortisol (here HSD11B1 expression), melanocortin, leptin, NPY, and adiponectin genes. With respect to adiposity genes, epigenetic modifications were documented for fat mass gene cluster APOA1/C3/A4/A5, and the lipolysis gene LIPE. With regard to inflammatory, immune and subcellular metabolism, PPARG, NKBF1, TNFA, TCF7C2, and those genes expressing cytochrome P450 family enzymes involved in steroidogenesis and in hepatic lipoproteins were documented for epigenetic modifications.
Causative Problems in the Metabolic Syndrome
Metabolic Syndrome is a consensus construct with wide acceptance based on its clinical usefulness and growing epidemiologic importance. It is currently defined by concurrent appearance of risk ranges in lipid traits, progredient prediabetes, pronounced adiposity with emphasis on abdominal obesity, and subclinical cardiovascular conditions. The concept implicitly assumes that a malignantly degrading spiral increases the probability of a co-existence of pathophysiologically relevant risk ranges, mainly resulting in organ damage resulting from type 2 diabetes mellitus (T2DM), hepatosteatosis, and cardiovascular incidences. Because, in comparison, single MetS components tend to rarely incur alone, plausibility justifies in summary the employment of the MetS construct. It has in the meantime also become clear that the number of metabolic abnormalities is correlated with overall MetS risk, so emphasizing meaningfulness of its usage.
Acceleration in Incidences
In the last decade, several researchers have alarmed the public of an international epidemic of MetS (Zimmet et al., 2001; Ford et al., 2002; Caballero, 2007; Capoulade et al., 2012), which has its greatest acceleration rates in threshold and emerging market countries. MetS is present in only 12.5% of patients with normal glucose tolerance, in 55% of those with impaired fasting glucose, and in 81% of those with T2DM (Ginsberg and Stalenhoef, 2003). The presence of MetS increased the risk of T2DM manifestation 24-fold during a 5-year period (Sattar et al., 2003). Furthermore, CVD risk climbed toward 20% once T2DM had developed in MetS patients (Girman et al., 2004). MetS risk increases with aging, being 44% in the seventh decade of life having the MetS compared to 7% prevalence in the third decade (Ford et al., 2002). There are further implications of MetS for aging biology because of associations of MetS components with oxidative stress (Moreira et al., 2015), DNA damage (Chen et al., 2015), telomere attrition (Révész et al., 2015), and mitochondrial damage due to sirtuin depletion (Guarente, 2006).
Obesity as a Central Precondition for MetS
The overall rising incidence of MetS is to a certain degree a consequence of the obesity epidemic in developed and threshold populations. Although obesity figures in the U.S. were historically reported to be on a rise since the first decades of the twentieth century, dramatic accelerations worldwide have been described for the past four decades (Zimmet et al., 2001; Caballero, 2007). According to newest figures announced on 12 October 2015 from the World Obesity Federation, it is suggested that, if current trends continue, 2.7 billion adults worldwide will be overweight by 2025. This is a 35% increase from 2.0 billion in 2014. The developmental precursor of adulthood MetS is seen in childhood ectopic lipid storage interfering with hepatic insulin signal transduction (Nelson and Bremer, 2010; Melka et al., 2013) with similar acceleration rates, and presumably resulting from epigenetic modifications and regulatory programming of obesity genes, and likely induced by nutrition-caused alterations of the gut microbiome (Remely et al., 2014b; Chang and Neu, 2015).
Brief History: Syndrome X to MetS
Over the last 12 years, MetS criteria have seen five major revisions in details; however, all operationalized definitions address specific metabolic abnormalities, hypertension and obesity (Eckel et al., 2010). An early conceptualization of MetS is contained in the Observationes Medicae (1641; book II, section 46 on diabetes) of the Amsterdam anatomist Nicolaes Tulp (1593–1674). In the context of gout as diabetic complication spoke French rheumatologist Jean Pierre Camus 1966 of a “metabolic trisyndrome” with hyperlipidaemia. In 1975, Hans Haller of the Dresden Academy of Medicine Carl Gustav Carus, coined the term “metabolic syndrome,” unbeknownst to the West, to support his observation of a coincidence of obesity, dyslipidaemia, hepatosteatosis, and disturbed glucose metabolism above chance level (Haller and Hanefeld, 1975; Haller and Leonhardt, 1981). The first description of resistance against insulin-dependent glucose uptake was proclaimed by Gerald Reaven (Reaven, 1988), then named “syndrome X.” Reaven built on clinical observations in the 1920s, of the Swede Eskil Kylin (Kylin, 1921, 1923), the Spaniard Gregorio Marañón (Maranon, 1922), and the American Elliot Joslin (Joslin, 1921). These clinicians had postulated a syndromal cohesion of hypertension with hyperglycaemia in prediabetes, although currently hypertension is no longer seen as a necessary prerequisite for MetS. But the terminology “syndrome X” was soon abandoned, as there were also terms circulating such as cardiac X syndrome and fragile X syndrome. In efforts for clarification, the term was henceforth specified to metabolic syndrome X, and then eventually shortened to the Metabolic Syndrome; a current synonym is Reaven-syndrome in commemoration of its modern re-inceptor. The World Health Organization (WHO) defined MetS as a group of risk factors for CVD and T2DM (Alberti and Zimmet, 1998): renaming into MetS was promoted by Eckel et al. (2005); Grundy et al. (2005a,b); Turek et al. (2005), with new guidelines 2005 and 2006 of the American Heart Association and the International Diabetes Federation, respectively, representing current standards.
The Quest for Causation
Abnormalities in the anterior pituitary gland and other hypothalamic structures regulating hunger-satiety homeostasis through the polypeptides leptin and ghrelin (Turek et al., 2005), and the melanocortins MSH and ACTH (Iwen et al., 2008), are considered responsible for MetS. These lead to defects in the hypothalamic-pituitary-adrenal axis (HPA), which may progress to the onset of T2DM. Recent evidence suggests that imbalanced autonomic nervous system output causes the simultaneous occurrence of T2DM, dyslipidaemia, hypertension, and visceral obesity: MC4R neurons in amygdala, arcuate nucleus, paraventricular nucleus, nucleus suprachiasmaticus, and anterior pituitary regulate food intake (Turek et al., 2005; Buijs and Kreier, 2006), energy expenditure (Balthasar et al., 2005) or influence vasoconstriction via angiotensin mediated activity of the sympathetic nervous system (Greenfield et al., 2009). There is also evidence that hepatic cholesterol reuptake is steered through parasympathetic pathways by MC4R-expressing neurons (Perez-Tilve et al., 2010; Krashes et al., 2016). Of endocrinologic factors, elevated cortisol levels are suspected to contribute to insulin resistance (Lewis et al., 2010). Such associations between sympathetic hyperexcitability, HPA axis hyperactivation, and decreased vagally mediated (anti-)inflammatory reflex (Figure 1), and MetS features were found in T2DM sufferers, where elevated plasma cortisol predicts greater prevalence of CVD (Reynolds et al., 2010).
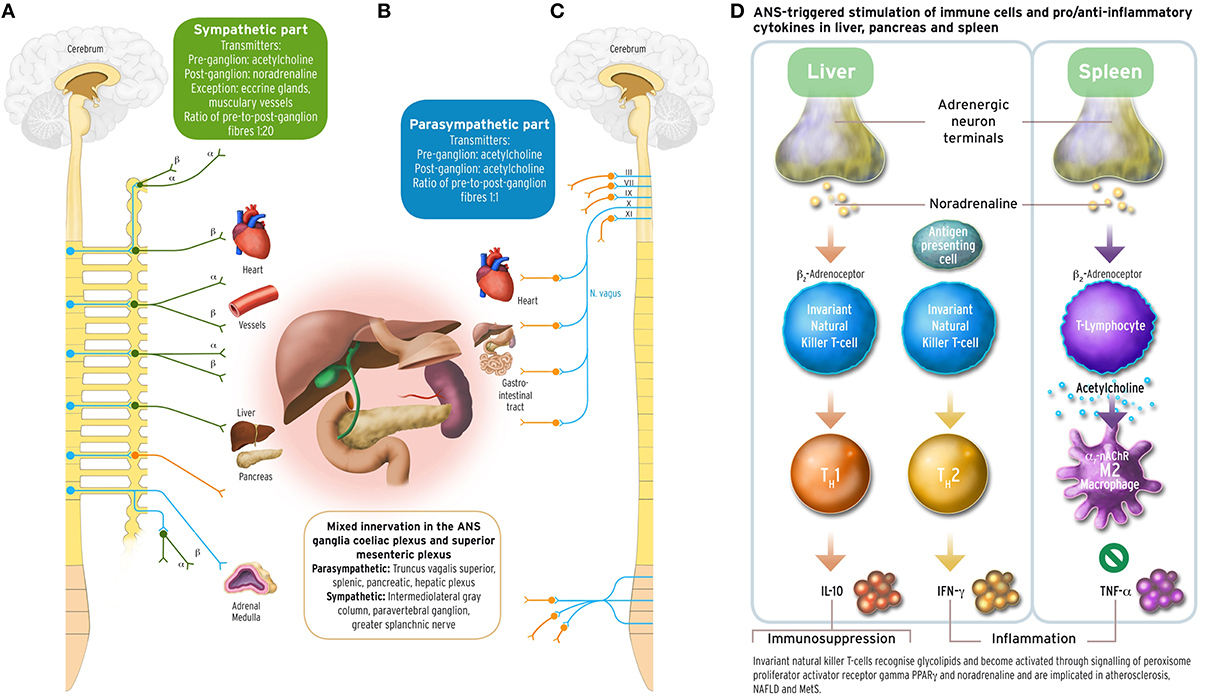
Figure 1. Sympathetic and parasympathetic innervation of the coeliac and superior mesenteric plexus ganglia, and immune and cytokine mechanisms in cholinergic anti-inflammatory pathway. The acetylcholinergic anti-inflammatory pathway, the efferent arc of the inflammatory reflex (Section Inflammation and Arterial Rigidity; Tracey, 2002) converging in the spleen, has been discussed under the aspect of being a target for possible interventions counteracting autonomic imbalance in the metabolic syndrome related to chronic inflammation. Schematically depicted are innervations from the sympathetic and parasympathetic branches of the ANS, with their transmitters, into organ systems relevant for MetS. Left part panel (A): efferent fibers in the sympathetic branch with adrenoceptor type. Middle part panel (B): mixed sympathetic and vagal fiber connections into the coeliac and the superior mesenteric plexus ganglia innervating liver, pancreas and spleen. Right part panel (C): efferent fibers in the parasympathetic branch of the ANS. The insert panel (D) to the outer right hand side illustrates schematically the role of vagus stimulation-derived, yet noradrenergic transmission into liver and spleen, and the acetylcholinergic transmission between CD4+ T helper cells and macrophages in the spleen. Vagus departs the brainstem as its cranial nerve X, and vagal efferent outflow regulates visceral organs by counterbalancing sympathoexcitation, inhibiting cytokine release, and safeguarding against inflammatory damage to liver, pancreas, spleen, lungs, or kidneys in endotoxaemic states. The outflow of the vagus nerve triggers adrenergic neurons in the coeliac ganglion innervating the spleen further to liver and pancreas. Vagal influence to spleen T lymphocytes stimulates the release of the neurotransmitter acetylcholine (ACh), and activation of the α7 subunit of the nicotinic ACh receptor (α7 nAChR; Section Inflammation and Arterial Rigidity) expressed on cell membranes of splenic macrophages and other cytokine secreting cells. Vagal tone attenuates here production of the inflammatory response cytokine tumor necrosis factor alpha (TNFα) reciprocally related to sympathoexcitation (Zhang et al., 2003; Kisiswa et al., 2013). In the liver, noradrenergic innervation signals hepatic innate natural killer T cells (iNKT) (Van Kaer et al., 2013) to exert systemic immunosuppression. Increasing the vagal tone there induces a shift from pro-inflammatory T helper cell type 1 (TH1) cytokines such as interferon-γ (IFN-γ) to anti-inflammatory TH2-type cytokines, such as interleukin-10 (IL-10) (Tracey, 2007, 2009; Rosas-Ballina et al., 2008, 2011; Trakhtenberg and Goldberg, 2011). PVNH hypothalamic insulin promoter expressing neurons downregulate postprandial inflammation through cholinergic signaling in the spleen mediated by vagal outflow to the spleen, whereas vagotomy results in T2DM (Carvalheira et al., 2014; Wang L. et al., 2014). Vagus stimulation approaches for increasing vagal tone would therefore aim to counterbalance prolonged sympathoexcitation in MetS by supporting parasympathetic output. These could comprise, but are not limited to, device-based, pharmacological, and/or psychotherapeutic intervention approaches. With the advent of wearable transcutaneous stimulation devices, vagus nerve stimulation has become a convenient neuropsychological intervention method (Van Leusden et al., 2015). Drug discovery is still required to identify non-steroidal anti-inflammatory substances targeting the cholinergic pathway either peripherally (such as nicotinic α7 nAChR agonist applications) and/or centrally (such as CNI-1493), or existing TNFα antagonists such as infliximab or etanercept. Possible behavioral interventions: Guided physical activity trainings, mindfulness-based psychotherapies, psychosomatic body-relaxation and/or balancing techniques, biofeedback training. Other: immunotherapy yet to be developed (Van Kaer et al., 2013). Medical illustrations by Corinna Naujok, Charité Media Centre Berlin, Virchow Campus.
Traditional Hypotheses on Aetiopathology
Malnutritional Factors
Because genetic susceptibility varies strongly amongst different ethnicities, and because the overall gene pool did not substantially change over the last decades, the obesity and diabetes epidemic was assumed resulting mainly from detrimental environment factors, such as food availabilities, diet habits, or sedentary lifestyles.
Carbohydrate Nutritional Factors
Several studies have described a nutritional dependence of MetS features such as hypertriglyceridaemia and other lipid traits (primarily low-density lipoprotein, LDL), and insulin secretion (Lofgren et al., 2005; Volek and Feinman, 2005; Volek et al., 2005; Forsythe et al., 2008). Caloric restriction exerted by means of very low carbohydrate diets also reduces a range of pro-inflammatory markers when a reduction in adipose tissue can be reached. In reverse direction, high-calorie diet is inductive of obesity within a one-week span, and also correlated with elevated sympathetic activity (da Silva et al., 2014). It has, from this perspective, been argued that MetS manifestation is a consequence of high carbohydrate intake, as it attenuates high fasting glucose levels, triggers insulin secretion, contributes to high plasma triglycerides, attenuates the HDL proportion and supports high blood pressure (Volek and Feinman, 2005; Westman et al., 2007). A further transitional state toward MetS is seen in the development of a fatty liver disease (NAFLD), including dyslipidaemia, associated with sedentary life style and lack of physical exercise (Pinto et al., 2015). High carbohydrate intake is typically associated with microbiotic fermentation of nondigestible polysaccharides, which are transformed into better manageable short chain fatty acids (SFCAs, acetate, propionate, and butyrate) that, however, increase reactive oxygen species (ROS), pro-inflammatory cytokines, decrease gut membrane integrity, and induce epigenetic modifications via inhibition of histone deacetylases, thus reducing respective gene expression (Tan et al., 2014).
Fatty Diet Consumption and Fat Uptake
Another theoretical approach assigns priority to evidence stating fatty diet in first place rather than carbohydrates, although there is on-going controversy as to which of the two is more detrimental. Animal evidence suggests that consumption of saturated fat and high-cholesterol diet resembling animal fats is able to induce NAFLD, consistent with hepatic fat accumulation and inflammation (non-alcoholic steatohepatitis, NASH) as manifestation of MetS (Pavlov and Tracey, 2012; Mells et al., 2015). Experimental results suggest that cholesterol is correlated with leptin, interleukin-6 (IL-6), liver weight and liver weight/body weight ratio, fibrosis and α-smooth muscle actin (α-SMA) (a marker of myofibroblast formation) in the liver. The alternative view (Mells et al., 2015) argues that a lower carbohydrate and higher fat diet is more favorable in terms of reduction of abdominal and intramuscular fat deposition and the prevention of T2DM. Intervention studies relate this to more favorable fasting insulin and glucose levels, as well as activation of pancreatic β-cells.
According to novel theoretical accounts and findings (Parekh et al., 2014; Grundy, 2015), play SFCAs fat metabolites (SFCAs, short chain fatty acids, acetate, propionate, and butyrate) produced by the gut microbiota a key role in arriving at insulin resistance and obesity. These SFCAs may better serve increase in energy expenditure, but at the cost of greater lipid storage. SFCAs affect satiety sensing, and contribute to downregulating neuropeptide Y and the glucagon-like peptides with the consequence of hyperphagia. According to this theorizing enter SFCAs more easily colonic epithelium, interact with high caloric nutrients with the consequence of epigenetic programming. Furthermore, this may lead to a bias of the immune system by activation of T cells in the gastrointestinal tract, their migration into adipose tissue, and to maintenance of low-level inflammation (Chang and Neu, 2015). The evaluation of existing research findings also suggests that dysbiosis due to high-fat diet and reduction of gut biodiversity increases permeability of the intestine (Winer et al., 2016), with the consequences of systemic circulation of gut hormones and pro-inflammatory cytokines, and of reduction of proper functioning of mesenteric lymph nodes.
Sodium and Natriuresis
Salt sensitive regulation of blood pressure is documented in animal and human studies (He et al., 2013), and more pronounced in specific populations in accordance with genotype and environmental interactions (Sanders, 2009). Specifically, genes regulating renal function toward sodium excretion with the cytochrome enzyme class P450 (CYP11B2, CYP4A11, CYP3A5, see also Section Obesity: Lipid Transport and Storage) via aldosterone synthesis, and dopamine-receptor-1-mediated salt excretion, have been associated to blood pressure lowering in hypertensive rodents. Salt consumption and sodium levels related to essential hypertension have been substantiated for several decades (Svetkey et al., 1987; Deter et al., 1996, 2002, 2007a,b; Buchholz et al., 1999), and been found to be influenced by emotional arousal (anger, anxiety) involving sympathetic outflow. A recent epidemiological study quantified the explained variance for trait anxiety upon high blood pressure to 6% (Lemche et al., 2016) in MetS. Analyses of population study databases (Oh et al., 2015) confirmed a linear dependence of sodium excretion, blood pressure measures, glucose and insulin levels, lipid traits, and several fat mass measures in MetS patients, even in the absence of clinical hypertension, thus making it a new nutritional risk factor of MetS. High sodium intake showed positive linear association with glucocorticoid secretion and metabolites, insulin resistance, and inverse association with adiponectin (Baudrand et al., 2014).
Whereas, nutrition-related risk factors are critical for all the metabolic causations (Szczepanska-Sadowska et al., 2010; Thorp and Schlaich, 2015) nominated for MetS in recent theoretical concepts on MetS, namely (a) overeating, (b) insulin resistance, (c) visceral adiposity, (d) arterial stiffness, for the one non-metabolic, namely (e) chronic stress, mechanisms are located in central and autonomic nervous systems.
Stress-Related Factors
Although stress has been implicated in the pathophysiology of MetS for decades (Brindley, 1995; Peeke and Chrousos, 1995), the best-regarded epidemiological evidence on this risk factor is commonly attributed to the Whitehall II study. Whitehall II is a prospective cohort study of >10 k British civil servants, which are being investigated stratified for employment grade and including diurnal cortisol probes. The overall finding from the stratified results Brunner et al. (2002); Chandola et al. (2006, 2008) is that psychological stress measures accounted for 37% explained variance of the correlation between MetS and normetanephrine, heart-rate variability, cortisol and interleukin-6, whereas health behaviors explained 18% in neuroendocrine responses. Cross-sectionally, there was a relation of work stress to cortisol secretion, and longitudinally, a dose-response relation between chronic stress and MetS.
The first reports on MetS as long-term sequela of posttraumatic stress disorder (PTSD) appeared in context of paediatrics and childcare. Reports investigating drug abuse in veterans stated strong obesity figures, hypertension, T2DM and dyslipidaemia, which were not attributable to drug consumption. Three recent meta-analyses (Edmondson et al., 2012; Bartoli et al., 2013; Rosenbaum et al., 2015) confirmed an increased risk (ORs 1.6–2.0) for MetS and CVD in PTSD sufferers, with 35–50% prevalence of single MetS components (whereby only 20% expectable). The occurrence of MetS was further related to accelerate T2DM with accelerated age-related cognitive decline in veterans suffering from PTSD (Green E. et al., 2015).
Vagal-Sympathetic Imbalance
There is increasing evidence for autonomic imbalance in MetS, the exact nature of this sympathetic hyperexcitability appearing to have primarily consequences for the development of obesity and insulin resistance. Known are age differences in the balance of vagal and sympathetic cardiac and vasomotor regulation (Hart et al., 2014), resulting in lesser sympathetic activity in elderly, which would thence bias toward prediabetes by increasing age. Blunting of SNS outflow toward glucose intake is a characteristic response of diabetic MetS sufferers (Straznicky et al., 2009). On the other hand, it is a replicated finding that the “vagal brake,” namely heart rate recovery after arousal, is absent in MetS, and that this prolonged sympathoexcitation is also associated with single MetS components (Deniz et al., 2007; Kim et al., 2009). Evidence for chronic sympathoexcitation in MetS consists in (a) elevated urinary and plasma noradrenaline levels, (b) enhanced efferent muscle nerve activity, but not necessarily essential hypertension (Thorp and Schlaich, 2015). SNS hyperreactivity decreases muscular blood flow and glucose uptake thus leading into insulin resistance. Heightened sympathetic outflow further acts toward β-adrenoceptor sensitization. Microneurography of postganglionic sympathetic responses in firing bursts is here the best feasible measurement. Consistently replicated evidence shows that peripheral sympathetic muscular firing rates in MetS are related to its obesity component (Grassi et al., 2005; Straznicky et al., 2008). Efferent sympathetic traffic correlated with abdominal obesity and was inversely correlated with baroreflex functioning. SNS muscular neurography rates covaried with plasma noradrenalin; this is in contrast to cardiac sympathetic components (Mancia et al., 1998), which are less so. However, it has been shown that high levels of fasting insulin, an index of insulin resistance, were positively associated with the low-to-high frequency (LF/HF) ratio of the heart rate variability (HRV)—an index of the sympathovagal balance at the heart level (Emdin et al., 2001). Recent experimentation has demonstrated that simple vagotomy leads quickly into insulin insensitivity and T2DM (Wang L. et al., 2014). Insulin promoter neurons in the paraventricular, arcuate, dorsomedial, ventromedial, and lateral hypothalamic nuclei signal to the phosphoinositide 3-kinase (PI3K)/mTOR pathway (Richard, 2015) critical for cell survival to exert control upon the vagus through efferent neurons in the nucleus tractus solitarius and dorsal motor root nucleus of vagus, ultimately terminating in the spleen (Wang L. et al., 2014).
Effects of Sympathetic Outflow
In the meantime have a number of studies also suggested that development of insulin resistance may be induced by sympathetic activity (Masuo et al., 1997; Julius et al., 2000; Esler et al., 2006; Rafiq et al., 2015) based on associated TNFα contribution (Pavlov and Tracey, 2012). However, a correlation between insulin secretion and peripheral sympathetic activity is not generally present (Masuo et al., 1997; Curry et al., 2014), specifically not in young healthy individuals. It may thus be concluded that additional factors, such as nutritional state and/or age range (see above), may be required to develop insulin resistance. It is also possible that insulin resistance and hyperinsulinaemia develop as consequence of an obesity-SNS interaction that then triggers elevated adipokine levels (Thorp and Schlaich, 2015) inducing T2DM (see Section Melanocortin Receptors and Binding Sites).
Current models of sympathetic effects on renal functions assume (Rafiq et al., 2015) that SNS hyperarousal leads to elevated plasma noradrenalin release levels and renal upregulation of glucose transporters (which then trigger insulin secretion from pancreatic β-cells). When tissue, skeletal, and hepatic glucose uptake decreases, blood glucose level and plasma insulin increase, and result in insulin resistance. In this model, SNS induces T2DM by hypertension, but it may therefore not suffice for hyperinsulinaemia in MetS. Essential hypertension may simply be a consequence of preferential fat mass accumulation around kidneys, thus physically interrupting normal functioning of the renin-angiotensin-aldosterone system (RAAS) (da Silva et al., 2014), with the consequence of diminished renal-pressure natriuresis. Typically, however, there is a certain interrelation between obesity and hypertension (da Silva et al., 2014), although in MetS, hypertension may be absent, and vice versa. Also, in many instances, obesity, and hypertension present as non-covarying, thus suggesting partly distinct pathophysiological mechanisms. Repeatedly have physiological parameters obesity and hypertension (Lemche et al., 2016) in MetS samples formed distinct latent factor-analytical clusters. Not peripheral SNS activation leading to general vasoconstriction, but specific renal-sympathetic effects on the RAAS involving sodium retention is experimentally documented to result in essential hypertension (da Silva et al., 2014).
Catecholamine System-Related
In 1986, Landsberg (Kaufman et al., 1986; Landsberg, 1986; Landsberg and Young, 1986) first described an association of sympathetic outflow and noradrenaline levels, and carbohydrate (but not protein) intake (Kaufman et al., 1986; Landsberg, 1986; Landsberg and Young, 1986), with subsequent insulin action and thermogenesis, finally resulting in hypertension. With respect to hypertension, angiotensin II release was found related to noradrenergic function (Taddei and Grassi, 2005). Because catecholamine release is directly related to SNS hyperreactivity (Section Malnutritional Factors), these two factors are usually considered as one system, primarily mediating short-term stress response [also termed sympathomedullo-adrenal (SMA) axis].
Glucocorticoid System Related
Cortisol secretion, in contrast, is a physiological response to cope with repeated anxiety triggers as a result of chronic stress (Brown et al., 1982; Fisher et al., 1982; Rivier et al., 1982). Because of striking metabolic parallelisms of MetS and Cushing's Syndrome, the latter is generally considered a clinical model disease for MetS (Anagnostis et al., 2009). While hypercortisolism was early suspected to be causative for MetS (Brindley, 1995; Peeke and Chrousos, 1995), first diachronic evidence that chronic stress is largest cause of MetS via HPA and glucagon upregulation (Brunner et al., 2002; Wang, 2005) was documented in the Whitehall II sample of middle aged (Anagnostis et al., 2009). In higher ages, then, there is direct synchronic association between cortisol levels and MetS criteria (Vogelzangs et al., 2007; Almadi et al., 2013). Whilst attempts to linking MetS components with diurnal cortisol secretion were not always successful (Abraham et al., 2013) (for methodological or endocrine reasons), controversies have finally been resolved by the corroboration that the extent of hair cortisone deposition is related to abdominal obesity and other MetS components (Stalder et al., 2013; Kuehl et al., 2015).
Inflammation and Arterial Rigidity
Originally, adiposity was assumed to be the mere cause of MetS (and hence MetS but the exaggeration of obesity), but it is a relatively novel notion (Grundy, 2015) that it is rather the array of agents secreted by white adipose tissue, amongst them adipokines and cytokines, inducing chronic latent inflammation in MetS and obesity (Tilg and Moschen, 2006; Pavlov and Tracey, 2012). Adipokines consisting of leptin, adiponectin and others, pro-inflammatory cytokines (ILs1~18, tumor-necrosis factor-α TNFα), metabolic factors (resistin, visfatin, adipocyte fatty acid binding protein 1, apolipoprotein E), acute phase and immune proteins (CD40, CD40L, C-reactive protein CRP, serum amyloid A3, plasminogen activator inhibitor-1 PAI-1, macrophage migration inhibitory protein 1 MIP1), several angiogenic and endothelial growth factors, and angiotensinogen (Ali et al., 2013). Adipokines influence satiety sensation and pancreatic insulin responses, while cytokines introduce a pro-inflammatory, and pro-thrombotic state with elevated levels of CRP, TNFα, and IL-6 regulated by immune transcription protein NF-κB activation (Jimenez-Gomez et al., 2013) of dependent cytokine gene loci. Nuclear factor NF-κB also is, along with other factors (IL-6, cytokine signaling suppressor proteins, and endoplasmic reticular oxidative stress) (Tilg and Moschen, 2006), implicated in inflammation-induced T2DM (Cyphert et al., 2015), however, its own two gene loci (NFKB1 4q24 and NFKB2 10q24) have not been found associated with MetS themselves. NF-κB activation is though indirectly crucial for mediation of leptin and insulin effects on hypothalamic POMC expression (Plagemann et al., 2009). Postprandial inflammatory and immune responses (particularly pronounced toward high-fat and sucrose diets) are currently seen as a normal transitional state during digestion, and hence the Metabolic Syndrome as an exaggerated and enduring postprandial inflammation state (Pavlov and Tracey, 2012). Clinical evidence suggests that increased inflammation markers could more closely be associated with MetS components (as seen in atypically depressed) than with dysregulated HPA axis and higher cortisol levels (as seen in melancholy) (Lamers et al., 2013). Pro-inflammatory markers CRP and IL-6 are associated with total bodily fat mass impairing physical performance in higher ages (Beavers et al., 2013). Inflammation marker CRP is also the mediatory variable linking MetS with later age-related reduction of cognitive capacities (Dik et al., 2007).
Excitation of Inflammatory Response
The exact mechanism of chronic low-level inflammation induced by action of adipose tissue is based on T-cell and macrophage accumulation (with expression of key adipogenic factors, PPARγ, CCAA-enhancer-binding protein C/ebpβ, insulin-like growth factor IGF, and toll-like receptors 2 and 4 TLR2/4s) in adipocytes (Sun et al., 2012; Ali et al., 2013; Lefterova et al., 2014), and a lack of vagally mediated downregulation of the “inflammatory response” in MetS (Pavlov and Tracey, 2012; Figure 1). Hepatic afferent stimulation of the vagus nerve via the nucleus tractus solitarius excites efferent vagal outflow counteracting excessive cytokine production and thus ameliorating inflammatory responses. This limiting mechanism to the inflammatory response, the (anti-)“inflammatory reflex,” requires the expression of α7 nicotinic acetylcholine receptors (α7 nAChR), a ligand-gated ion channel expressed on macrophages, lymphocytes, and neurons, in the cholinergic signaling pathway (Olofsson et al., 2012). Obesity is further characterized by decreased energy expenditure, and heightened food intake, while high levels of leptin are released (Barnes and McDougal, 2014). In this context, also SNS activation is involved: Adipose tissue is only innervated by sympathetic nerves (Thorp and Schlaich, 2015), and lipid storage (uptake of fatty acids as triacylglycerides) is also dependent of binding of catecholamines and the pancreatic hormone glucagon to β-adrenoceptors on the surface of adipocytes, thereby activating adenylate cyclase (resulting in cAMP intracellular signaling; Ali et al., 2013). Accumulation of white adipose tissue in obesity is accompanied with indicators of inflammation IL-6 and CRP, but recent evidence suggests that (a) muscle sympathetic activity, and (b) vasomotor activity influencing hypertension are independent of tissue inflammation (Barnes et al., 2014). However, there seems to be a specific interaction of pro-inflammatory cytokine CRP and cortisol in MetS, leading to inhibition of lipoprotein lipase activity and concentration of nonesterified fatty acids (NEFAs) (Perry et al., 2001) in adipose tissue.
Inhibition of Inflammatory Response: The Anti-Inflammatory Reflex
It has been found that agouti-related protein (AgRP, Section Neuropeptide Y and Agouti-Related Hormone Receptors and Binding Sites) stimulates the HPA axis to release ACTH, cortisol, and ACTH in response to IL-1β in adipose tissue (Xiao et al., 2003), suggesting that elevated cortisol secretion in MetS could be inhibitive to adipose tissue inflammation. It is yet unclear, however, whether this is a mechanism that could replace or override the vagally mediated cholinergic signaling as part of the efferent branch of the anti-inflammatory reflex arc (Figure 1 and above). This would then be a reinforcing effect of plasma cortisol as part of the inflammatory response, in which TNFα and sympathoexcitation are reciprocal (Zhang et al., 2003). However, there is novel evidence also suggesting a secondary CRH system outside the HPA in adipose tissue (Section Adiponectin and Genomic Bases) (Seres et al., 2004; Fahlbusch et al., 2012; Subbannayya et al., 2013; Dermitzaki et al., 2014).
Inflammatory and Oxidative Stress Related to CVD
Increased inflammatory markers are also a concomitant of insulin resistance onset (Moreira et al., 2015) induced with adiponectin, resistin, and TNFα. Leptin also induces the production of nitric-oxide synthase 2 (NOS2) and, thereby, ROS. Angiotensin II (see below) and superoxide or ROS are amongst the main endothelium-derived constriction factors (Kang, 2014; Young and Davisson, 2015). It is hereby assumed that hyperoxigenation of LDL is the source of endothelial plaque genesis. ROS generation is assumed being caused by excess nutrient processing in mitochondria, triggering even further ROS accumulation, and impairing endoplasmic reticulum (ER) function in protein folding (Hotamisligil, 2010a,b; Hummasti and Hotamisligil, 2010). Vagally induced relaxation of endothelium occurs by acetylcholine signaling (Figure 1) and is critical for release of the endothelium-derived hyperpolarizing factor (EDHF). Normal regulation of vascular tone is increasingly impaired by reduction of EDHFs leading to endothelial dysfunction associated with CVD (Young et al., 2015).
Endothelial Dysfunction and Angiotensin II Release
In addition to the immunological and inflammatory factors detailed above, adipose tissue produces also the peptide angiotensin II, a regulator of hydrolysis in the RAAS via hypothalamic AT1-receptors. In blood vessels, AT1-receptors induce vasoconstriction, and thus may promote thrombosis and vascular injury (Perry et al., 2001). Increasing imbalance between vasoconstrictors and vasodilators or relaxants (Moreira et al., 2015) then causes impairment of baroreflex functioning through atherosclerosis. Impairment of baroreflex functioning is considered being a main cause of MetS, although hypertension is not essentially necessary for MetS. Hypothalamic arcuate nucleus is the regulatory site for insulin and lumbar baroreflex action (Cassaglia et al., 2011), and efferents via the paraventricular nucleus pathway.
Genomic Foundation of Basic Regulatory Mechanisms in MetS
Obesity: Lipid Transport and Storage
With both obesity and hyperlipidaemia are two MetS features related to deposition and metabolism of lipids. There is accumulating evidence (Farmer et al., 2008; Rivera et al., 2012; Cole et al., 2014) that genetic and epigenomic mechanisms steering lipid transport, uptake, and catalysis are central to MetS pathophysiology. In this general context, mediation of SNS for lipid transport and storage is present, as α2-adrenoceptors inhibit lipolysis, and β2∕3-adrenoceptors trigger lipolysis in adipose tissue, in addition to regulatory functions for glucagon, insulin, renin, and ghrelin secretion.
Genomic Loci of Plasma Lipid Traits
Plasma lipid traits are regulated by genomic loci interacting. One hundred and eighty five common variants were described in two large-scale studies of the Global Lipids Consortium (Teslovich et al., 2010; Do et al., 2013), amongst them APOA1, APOB, APOE, and TRIB1. Many SNPs are in close vicinity to those 18 loci known to cause Mendelian lipid disorders, such as the TRIBAL locus downstream from TRIB1, and exhibiting reciprocal regulatory expression (Proudfoot, 2011; Douvris et al., 2014). Within these apolipoprotein genes, specific genetic factors for MetS (i.e., SNPs relevant for MetS risk) became evident in the Diabetic Heart Study (Adams et al., 2014), and others (Crosby et al., 2014; Gaio et al., 2014): rs3135506 (Ser19Trp, APOA5), rs651821 (5′UTR, APOA5), rs13832449 (splice donor, APOC3) (Crosby et al., 2014). The APOA5 on locus 11q23, has binding affinity with LDL-Receptor gene (Nilsson et al., 2008), and the APOA5-related HTG specifically risky for CHD (Zhou et al., 2013). APOA5, APOC3, APOA1, and APOA4 loci are closely clustered in 11q23 (Nilsson et al., 2008), with SNPs rs2972146 near IRS1 locus shown associated with increased risk of T2DM, insulin resistance and hyperinsulinaemia (Teslovich et al., 2010); rs1042034 of APOB related to HTG as main lipoprotein of chylomicrons and lipid-rich particles (Teslovich et al., 2010), located on chromosome 2. APOC3 is related to VLDL, and affects lipid levels by postponing triglyceride catabolism (Russo et al., 2001). In reverse: a missense mutation lowers plasma triglycerides and CVD risk (Crosby et al., 2014). APOA-V suppresses exuberance of triacylglycerides (Pennacchio et al., 2001) by creating a feedback-loop for downregulation of apolipoprotein A5 (Caussy et al., 2014).
Genomic Loci for Fat Mass Accumulation
The European multicentre study of Aulchenko had shown that further to cholesterol/lipoprotein metabolism also lipid transport/obesity is a second cluster in CVD risks. The major locus amongst lipolysis genes is the LIPE (19q13.2) gene encoding lipase, where carriers of the D-allele show distortion in lipid metabolism and insulin sensitivity (Albert et al., 2014), also in CYP2C19, a monooxydase protein of the cytochrome P450 family, involved in catalysis of lipids, cholesterol and other steroid hormones such as cortisol (Gaio et al., 2014). It is possible that, in the MetS constellation, additional risk genes are relevant, such as CYP2C19 (10q24) (Gaio et al., 2014). The major lipid storage genes are those for the peroxisome proliferator-activated receptor γ (PPARG 3p25) mainly found in adipose tissue (Gu et al., 2014), and furthermore the obesity-related FTO (16q12.2) (Yang et al., 2014). A common variant of the latter rs9939609 has been found relevant for MetS components, hypertension, dyslipidaemia and CVD (He et al., 2014; Liguori et al., 2014). Common and rare variants in the melanocortin-4 receptor MC4R locus 18q21.32 (rs74861148, rs483125, and rs11872992) or its promoter region were associated with triglycerides, obesity and T2DM (Bazzi et al., 2014; Katsuura-Kamano et al., 2014; Muller et al., 2014).
T2DM: Melanocortins and Insulin Resistance
Two hundred and twenty one million individuals were suffering from T2DM in 2010 worldwide, and expected are 300 million sufferers by the year 2025 (Zimmet et al., 2001). Regarding the heritability of T2DM, it is currently conceived that gene polymorphisms pertaining to obesity, insulin resistance, dyslipidaemia, glucose uptake and pancreatic β–cell dysfunction coact in a way that ultimately results in T2DM (Ridderstråle and Groop, 2009). Genetic susceptibility is a necessary prerequisite to develop T2DM: in monozygotic twins there is 70% concordance, and in parent-offspring there is a risk of more than 40% probability (Lyssenko et al., 2005). T2DM is both a monogenic and a polygenic condition with a multitude of genes involved (Stumvoll et al., 2005). Eighteen T2DM gene loci have been isolated in genome wide scans (GWAS), the best replicated of which are melanocortin receptor-4 (MC4R), T-cell factor 7-like 2 (TCF7L2), and peroxisome proliferator-activated receptor gamma (PPARG) (Stumvoll et al., 2005; Ridderstråle and Groop, 2009) genes, but each gene with relatively small effect size (ORs 0.8–1.3).
Characterization of Functions
In several populations has the TCF7L2 (10q25.3) locus shown association with T2DM (Assmann et al., 2014; Ouhaibi-Djellouli et al., 2014). This locus encodes a transcription factor implicated in blood glucose homeostasis. It also regulates total cholesterol, LDL and HDL, and was found contributing to MetS in the context of increased CHD mortality (Khoroshinina et al., 2014). Peroxisomes are normally small cytoplasmic vesicles oxidizing by their enzymes fatty acids, and are also related to glucose metabolism. The PPAR nuclear receptors, which include PPARα, PPARδ, but particularly PPARγ, are transcription factors that mediate effects of fatty acids and their derivatives on gene expression, are, together with the mutually co-expressed C/ebpβ, the “master regulators” of adipogenesis (Lefterova et al., 2014). The protein encoded by PPARG (3p25) regulates adipocyte differentiation (Ali et al., 2013). All three PPARs, but especially PPARγ, are expressed in macrophages and modulate adipose tissue inflammation. PPARG was associated with T2DM in several populations (Black et al., 2015; Katome et al., 2015). Specifically the MC4R, less so MC3R, is expressed in the CNS widely (Millington, 2007) and related to feeding (and other) behaviors. Defects in the MC4R (18q22) lead to infantile hyperphagia, childhood obesity, elevated plasma insulin levels, and growth acceleration. The MC4R was alone or in combination with the FTO gene related to T2DM and food intake (Huang et al., 2011; Statsenko et al., 2013), and CVD. As the MC4R is capable to bind to a larger array of melanocyte stimulating hormones (MSHs), or melanotropins, specific mechanisms of action lie in the different ligands and probably cerebral locations. MSHs are a group of peptide hormones synthesized in hypothalamus and pituitary, binding to the same group of melanocortin receptors (MC1-5Rs). Melanotropins modulate central energy expenditure and regulate hunger feelings. From the proprotein pro-opio-melanocortin (POMC), α-MSH, β-MSH, and two γ-MSH isoforms are derived by cleavage. In the group of melanocortins is also adrenocorticotropin (ACTH), a derivate of POMC. Another polypeptide (derived with ACTH from POMC) is lipotropin (β and γ), which has central functions in lipolysis, lipid transport, and in steroid genesis. Yet another group of fragments are the endorphins, with proenkephalins A and B, and met-enkephalin. MC3Rs and MC4Rs and POMC neuron activations, further to leptin receptors, have evolved as key components with triggering cardiovascular consequences in MetS (da Silva et al., 2014; Section Melanocortin Receptors and Binding Sites).
Catecholamine Genomic Bases and Regulation
Stress has long been implicated as a non-metabolic causation in the pathogenesis of MetS (Hjemdahl, 2002), and theoretical accounts of MetS pathophysiology list endocrine factors of stress regulation in reciprocity with factors of cardiovascular regulation, metabolic regulation, and inflammatory regulation (Szczepanska-Sadowska et al., 2010). Short-term stress is mediated in the autonomic nervous system directly by its sympathetic branch, and through transmission by catecholamines, specifically neurotransmitter noradrenaline, under regulation by the catechol-o-methyltransferase (COMT 22q11.2) gene (Kopin et al., 1978).
Catechol-o-methyltransferase
COMT has been considered repeatedly when investigating the frequent comorbidity of major psychiatric disorders and MetS. The microdeletion syndrome at locus 22q11.2 interrupts COMT expression, resulting into a neurodevelopmental schizophrenia phenotype (Napoli et al., 2015), in which increased glycolysis and higher plasma cholesterol and triglyceride concentrations were found. The low-activity allele COMT Val158Met polymorphism is related to a subclinical MetS-phenotype involving elevated heart rates, blood pressure, abdominal obesity (Annerbrink et al., 2008). During normal aging, COMT and brain-derived neurotrophic factor (BDNF) showed additive effects on decline in executive functioning in interaction with apolipoprotein E metabolism (Sapkota et al., 2015).
Noradrenaline Signaling
The SNS is triggered from the amygdala (central and basolateral nuclei) in the presence of stress signals, as human effective connectivity studies have indicated (Lemche et al., 2006). In the brainstem, the main synthesis site in the rostral pons for noradrenaline is the locus coeruleus, besides the adrenal medulla. The projections to the major midbrain and cortical regions are exerted by noradrenergic neurons. We therefore move the focus upon noradrenaline, as the principle neurotransmitter of the SNS. Experimental physiological evidence isolated sympathetic activity and noradrenaline action explaining blood pressure variance (Fossum et al., 2004) during acute stress, thereby potentiating hypertensive action is in MetS (Huggett et al., 2004; Grassi et al., 2007). The adrenoceptor types α1 and α2 are employed in noradrenaline uptake and signaling, thereby inducing vasoconstriction, and relaxation, respectively, in smooth muscles. Adrenoceptors β1−3 are more specifically involved in metabolic processes, binding triggers intracellular concentrations of cAMP as second messenger. Cardiac output, renin, ghrelin (β1), and lipolysis, insulin secretion, glycogenolysis (β2,3), are mediated by this receptor class, making them relevant for MetS.
Adrenoceptor Gene Loci
In their genomic bases, ADRB1 (10q25.3) releases heterodimers that influence BMI, body weight regulation, blood pressure, and basic metabolic rate. GWAS have revealed that heart rhythm problems, failure, and blood pressure dysregulation are correlated with this locus (Gao et al., 2014), and its Arg389Gly polymorphism with obesity (Dudchenko et al., 2014). Different polymorphic forms, other variants, and epigenetic modification of the ADRB2 (5q31-32) gene have been correlated with obesity and T2DM, hyperinsulinaemia, NAFLD, and hyperleptinaemia (Bulatova et al., 2015). These are related to its mediation of hepatic blood flow with glycogenolysis and gluconeogenesis, and insulin secretion from pancreas. ADRB3 (8p11.23) is mainly expressed in brown and white adipose tissue, and becomes activated in energy expenditure, thermogenesis and lipolysis. Diseases associated with ADRB3 include obesity based on MC4R deficiency. In addition, the gene is also expressed in the vascular endothelium where it is involved in lipolysis, glucose uptake, cardio-inhibition and relaxation. Hypermethylation of the ADRB3 gene promoter in blood and visceral tissue is associated with metabolic disturbances (Guay et al., 2014), such as dyslipidaemia. Polymorphisms in the β3-adrenoceptor gene were observed with insulin resistance and high lipid profiles related to T2DM and MetS (Burguete-Garcia et al., 2014).
Noradrenaline Transporter Gene
The noradrenaline transporter gene SLC6A2 (NET 16q12.2) is central to noradrenaline homeostasis and presynaptic reuptake. Its SNP rs2242446 has been correlated to anxious arousal and PTSD (Pietrzak et al., 2015). SLC6A2 expression is restricted to noradrenergic neurons that innervate the adrenal medulla. Further links to MetS are in its involvement in cardiovascular regulation, obesity/weight regulation hepatic regulation, which though has not been systematically investigated yet. There is evidence that a SNP (Ala457Pro) in the NET gene (SLC6A2) may be underlying orthostatic autonomic dysregulation (Tellioglu and Robertson, 2001). An epigenetic mechanism (hypermethylation of CpG islands in the NET gene promoter region) that results in reduced expression of noradrenaline has also been described for orthostatic tachycardiac dysregulation, but not been replicated for panic disorder (Bayles et al., 2013).
Glucocorticoid and 11β-HSD-1 Genomic Bases and Regulation
Two stress pathways, including both the hypothalamic–pituitary–adrenal axis (HPA), and the noradrenergic sympathetic nervous system, have been considered relevant to MetS (Lambert et al., 2010). Long-lasting stress, in specific, is believed being maintained by enduring imbalance of the HPA, through secretion of the corticotropin-releasing hormone (CRH) resulting in hypercortisolism. Short-term sympathetic-noradrenergic action has been linked to state anxiety (Ziegler et al., 2012), whereas trait anxiety and “anxious temperament” (AT; both terms are used interchangeably in neuroscience) is located in hyperactive anxiety midbrain circuits: Anterior hippocampus, amygdala, and ventral striatum have been found to elevate cortisol levels through HPA axis hyperreactivity (Oler et al., 2010; Dinel et al., 2011; Rogers et al., 2013).
Corticotropin-Releasing Hormone Gene
Synthesized in the hypothalamic paraventricular nucleus, CRH release is also triggered by TNFα and IL-6 resulting from inflammatory states in order to dampen the immune response, and to adjust endogenous cortisol release controlling the inflammatory response. The expression of CRH1 (8q13.1) has been related to hypoglycaemia (Nussey et al., 1993), and its activation has been found central to fetal programming of later obesity (Stout et al., 2015). In contrast to SMA activation, cortisol release in response to repeated stress habituates more quickly (Schommer et al., 2003), but leads eventually into immunodeficiency by impairing CD19-promoted B-cell generation (McGregor et al., 2015). Furthermore, a second adipose glucocorticoid system has been described besides to the CNS HPA signaling pathway recently. Here it has been found that also adipose tissue expresses the neuropeptide CRH as part of the inflammatory response (Section Inflammation and Arterial Rigidity), together with in the immune system toll-like receptor-4 (TLR4), the production of inflammatory cytokines IL-6, TNFα and IL-1β, chemokine IL-8, monocyte attractant protein-1 (MCP-1), and of the adipokines adiponectin, resistin, and leptin (Dermitzaki et al., 2014). A second vertebrate corticotropin-releasing hormone gene CRH2 has recently been discovered but there is still a lacuna in human research (Grone and Maruska, 2015). In humans, the glucocorticoid receptor protein is encoded by NR3C1 gene, which is located on chromosome 5 (5q31). NR3C1 mediates the regulatory response to glucocorticoid response elements in the promoters of glucocorticoid responsive genes to activate their transcription, and as a regulator of other transcription factors. It has been found linked to MetS through mechanisms of epigenetic modification by histone methylation in response to early life trauma (Martin-Blanco et al., 2014; Palma-Gudiel et al., 2015) in e.g., personality and eating disorders.
Corticotropin Receptors and ACTH signaling
There are two subtypes of CRH receptors, both of which express ACTH, when bound by CRH. Corticotropinergic neurons are mainly located in the anterior pituitary, also in amygdala, hippocampus and locus coeruleus. The HPA signaling pathway is mainly dependent on corticotropin-releasing hormone receptor type 1 (CRH1) polymorphism on exon 6 of CRHR1 (locus 17q12-q22 in humans) (Rogers et al., 2013). Within the hippocampus, the CRHR1s are most abundant, but also present in liver tissue. Both CRHR1 and CRHR2 genes are strongly expressed in adipose tissue. CRHR1 (17q21.31) association is frequent in depression, and thus relevant to MetS, since depression is a frequent MetS-comorbidity. The CRHR2 (7p14.3) gene has been described involved in cardiovascular homeostasis, PTSD, and thus general susceptibility toward stress (Wolf et al., 2013). Their activation product, ACTH, is synthesized in basophile neurons of the anterior pituitary under regulation by CRH from its precursor POMC. ACTH, in turn binds to melanocortin receptors (Section T2DM: Melanocortins and Insulin Resistance).
Regulation of Cortisol Biosynthesis
Cortisol biosynthesis needs as a coenzyme the heme-containing cytochrome P450, secreted after oxidation from cholesterol in adrenal mitochondria in the zona fasciculata to pregnenolon, and also co-regulated by ACTH. The final stage of cortisol synthesis is reached by 11β-monooxygenases (2 isoforms, both CYP11B1 and CYP11B2 are on 8q21-q22), adrenal members of the cytochrom-P450 family, and their malfunction will result in missing feedback signaling to ACTH. The gene locus P450 (cytochrome) oxidoreductase (POR 7q11.2) encodes the endoplasmic reticulum membrane oxidoreductase, involved in steroidogenesis as well. As the gene product of POR is required for the activation of the microsomal P450-enzymes, several hepatic CYP-enzymes are hampered in their activity through mutations pertaining to these functions. Under the aspect of steroidogenesis, are impaired acyl-carbon bond cleaving cytochrome enzymes CYP17A1, CYP21A2, and CYP19A1. Respective mutations in their gene loci were observed relevant to the regulation of lipid traits, in specific hypercholesterolaemia, and blood pressure (Lu et al., 2015).
Cortisone-Cortisol Interconversion
11β-dehydroxysteroid dehydrogenase (HSD-11β) is a catalytic enzyme and a membrane protein of the endoplasmic reticulum converting free cortisol to inactive cortisone (and in type 1 isozyme, also vice versa through 11-oxidoreductase activity), in two isoforms 1 and 2, and eight known structures. As mentioned, isoform 1 also performs the reduction of cortisone to active cortisol in CNS, liver and adipose tissue, and, as has been suspected, thereby amplifying cortisol action (Seckl and Walker, 2001).
Isoform 2 oxidizes free cortisol to cortisone, a process seen in placenta, testes, lungs, or kidneys. For this reason 11β-HSD-2 has been implicated in neurodevelopmental susceptibility for the programming of diathesis toward chronic stress (Sousa, 2016). By this conversion it is prevented that the abundant ligand cortisol binds to the mineralcorticoid receptor (MR) in addition to the glucocorticoid receptor, thus securing only aldosterone being able binding to MR. The HSD-11β conversion activity is mainly a process in adipose tissue, with its distinct CRH-system described above, but exerting feedback effects upon the CNS HPA axis. It has therefore been proposed counteracting HSD-11β in adipose tissue as a treatment for MetS by both pharmacological and psychological interventions on central HPA and peripheral cortisol systems (Anagnostis et al., 2009). Green tea has shown potency to prevent hepatic cortisol activation by type 1 isozyme HSD-11β (Hintzpeter et al., 2014). Because of its amplificatory action of active glucocorticoids, isoform 1 HSD-11β, has been assumed be a pathogenic factor in MetS, T2DM, and age-related cognitive decline (Seckl and Walker, 2001).
HSD-11β Gene Loci
The protein encoded by the HSD11B1 (1q32-q41) gene is a microsomal enzyme that catalyses the conversion of the stress hormone cortisol to the inactive metabolite cortisone and reverse (see above). Too much cortisol can lead to central obesity, and several variations (rs10082248, rs2298930, and rs4545339) in this gene have been associated with obesity and insulin resistance in children (Ruan et al., 2014). Cortisol interconversion is mainly relevant at the visceral tissue level (Kilgour et al., 2015). There is initial evidence that HSD11B1 expression predicts insulin resistance (Koska et al., 2006; Gyllenhammer et al., 2014), and also a linear relationship between BMI and 11β-HSD1 has been observed when pooling across samples (Wake and Walker, 2006). Initial evidence also suggests that HSD11B1 expression is likely to be regulated in a tissue-specific manner (Wake and Walker, 2006; Wake et al., 2006), i.e., present in different levels in adipose tissue and mainly liver (Stimson and Walker, 2013). The regulation of both 11β-HSD isoform-genes is dependent on NF-κB (Lee et al., 2013) in adipose tissue. Both isoforms have been found relevant to childhood obesity (Ruan et al., 2014), the antecedent of MetS. In interaction with adiponectin, HSD11B1 determines the metabolic rate already in utero and postnatal development (Muramatsu-Kato et al., 2014), in a form of ontogenetic programming. HSD11B2 (16q22) protects cells from the growth-inhibiting and/or pro-apoptotic effects of cortisol, particularly during embryonic development. Mutations in this latter locus cause the syndrome of apparent mineralocorticoid excess and hypertension. Polymorphisms can regulate maternal cortisol levels in utero and regulate postnatal weight gain (Rogers et al., 2014). Replicated in association studies were polymorphisms for salt sensitivity, RAAS, and essential hypertension. Negative findings were yielded for HSD11B2 SNPs with adolescent obesity (Ruan et al., 2014); similarly, gene regulation studies identified only microRNA relevant to renal functioning (Rezaei et al., 2014). However, the polymorphic CA-repeat polymorphism in the first intron of HSD11B2 was significantly related to insulin insensitivity (Mune et al., 2013).
Adiponectin and Genomic Bases
Adiponectin is a regulatory peptide hormone secreted by adipocytes (but also by myocytes)—less when emptied, more when filled with lipids—relevant to glucose flux and lipid catabolism. A low adiponectin level in obese persons attenuates insulin action in tissue by induction of insensitivity via modification of its uptake by adipocytes (Ali et al., 2013). Such a condition is therefore conducive to insulin resistance and T2DM, but at the same time protective for endothelial tissue (Fortuño et al., 2003). High adiponectin levels, in contrast, are known for insulin sensitization and anti-inflammatory reflexes, whereas low levels have, however, been shown to be an independent risk factor for Alzheimer's and other dementias (van Himbergen et al., 2012). In addition, inverse relations between adiponectin and cortisol secretion have been described, whilst plasma cortisol being the best predictor of insulin resistance (Lehrke et al., 2008), however, the precise physiological mechanism of this antagonism remains unclear.
Release of Adiponectin
The adiponectin gene is known being expressed only in adipose tissue: ADIPOQ (3q27) with five exons expresses collagen-like proteins and co-factors. It has been associated with rapid excessive weight gain, obesity, low-frequency heart rate variability and cardiac mortality (Alehagen et al., 2015; Riestra et al., 2015), and negatively with risk for T2DM and CVD incidences (Lindberg et al., 2015). Complementary functions with leptin (see Section Leptin and Ghrelin Receptors and Binding Sites) were described for vascular injury (Fortuño et al., 2003). Low level of adiponectin is demonstrated being a singular independent risk factor for developing MetS (Díez and Iglesias, 2003; Renaldi et al., 2009). Direct interactions of adiponectin have presently been documented mainly with the other adipokines (Raucci et al., 2013; Sitticharoon et al., 2014), but first of all leptin (Section Leptin and Ghrelin Receptors and Binding Sites).
Adiponectin Receptors
Adponectin binds to three receptor types receptors (AdipoR1, AdipoR2, T-cadherin CHD13), which activate the hepatic and pancreatic enzyme 5′ AMP-activated protein kinase, p38-MAPK, a mitogen-activated protein kinase sensitive to stress, to pro-inflammatory cytokines, hepatic PPARα related to triglycerides, and to NF-κB transcription factor related to stress and inflammatory response (Thundyil et al., 2012). It has recently been described that adiponectin and its receptors modulate analgesic effects in the central nervous system further to its anti-inflammatory properties (Iannitti et al., 2015). The adiponectin receptors are ubiquitous in the cerebrum, but more densely expressed in pituitary, hypothalamus, brainstem, cortical neurons and endothelial cells. Recent findings suggest that adiponectin has neuroprotective properties and counteracts cerebral apoptosis (Song J. et al., 2015). Adiponectin receptors and binding (Tilg and Moschen, 2006) differ between receptor types: whereas ADIPOR1 is expressed ubiquitously in muscular tissue, ADIPOR2 is restricted to hepatic tissue. The binding is dependent on molecular-weight homomultimer forms of the specific molecule. Both receptors are known to mediate fatty-acid oxidation and glucose uptake (Yamauchi et al., 2003). ADIPOR1 (1q32.1) was associated with insulin resistance, T2DM, and CAD, specified to different susceptibility SNPs (Jin et al., 2014). ADIPOR2 (12p13.31) exhibited correlations with CAD, stroke, and T2DM (Yuan and Teng, 2014). The CHD13 (16q23.3) protein regulates axon growth during neural differentiation and vascular endothelial cells from apoptosis due to oxidative stress, and is associated with resistance against atherosclerosis. The interaction of its SNPs and methylation has not yet been fully understood, but relations were found to diastolic blood pressure and HDL levels (Putku et al., 2015). Besides adiponectin, at least 15 other substances are subsumed to the category adipokines (Raucci et al., 2013), but outside the scope of this treatise.
Brain Regions of MetS Neuroendocrine Signaling Systems
Leptin and Ghrelin Receptors and Binding Sites
White adipose tissue regulates sympathetic output through release of peptides such as the proteohormone leptin, which have the ability to cross blood brain barrier and bind to receptors in higher ANS regions (Thorp and Schlaich, 2015). Leptin is a peptide hormone produced by adipose tissue in proportion to fat mass. The physiological function of leptin release is (a) to promote food intake, and (b) to increase energy expenditure (Barnes and McDougal, 2014). In obesity, leptin is assumed to drive sympathetic activity and to contribute to hypertension (Hall et al., 2010). Its central function is the maintenance of homeostasis in fat depot metabolism. Leptin functioning is, however, also dependent on the brain melanocortin system (da Silva et al., 2014; Bassi et al., 2015b). Leptin inhibits hunger sensations, and therefore, a lack of satiation feelings is induced by deficient cerebral leptin feedback signaling (Moreira et al., 2015). Such anorexigenic signals are mediated by leptin and insulin via hypothalamic POMC neurons activating MC4Rs. Leptin induced obesity states are linked to ROS increase in oxidative stress (Section Inflammation and Arterial Rigidity). A counterintuitive effect of leptin is present in its reinforcement of sympathetic action on the adrenocortical system, but an important link to hypertension. This accounts for its interrelations with the cardiovascular system, the sympathetic ANS branch, metabolism, and obesity, and with chemoreceptors in the carotid bodies (Bassi et al., 2015a; Zeng et al., 2015). Leptin depletion in both animals and humans results in a quasi-MetS condition, but lacking essential hypertension and sympathetic hyperexcitability. It is concluded from these depletion states in leptin levels that insulin resistance and hyperinsulinaemia, hyperglycaemia, dyslipidaemia and visceral adiposity could also be related to leptin action (Bassi et al., 2015a). As mentioned, this requires the POMC and MC4R neurons to be activated: Mice with MC4R deficiency are (a) unresponsive to leptin, and (b) also develop artificial MetS symptoms analogous to those in leptin depletion, despite high leptin levels (Kooijman et al., 2014). Adiponectin and leptin levels are typically positively associated (Singh et al., 2016), but this correlation is absent in obese persons: Adipose tissue from obese subjects has impaired leptin signaling, which probably prevents increases in adiponectin levels in obese.
Leptin Biosynthesis
The protein leptin is a synthesis product of white adipocytes, expressed by the LEP (7q31.3) gene, active in haematopoiesis, angiogenesis, healing processes, immune, and inflammatory responses. To the degree that lipolysis is exerted in fat depots, plasma concentration of leptin diminishes, thus raising appetite. Besides in adipocytes, leptin is also produced by neurons in hypothalamus and pituitary (Morash et al., 1999). Leptin deficiency caused by null mutations (Mark, 2013) induces components of MetS such as hyperinsulinaemia and hyperlipidaemia, but sympathetic hypoactivation. In contrast, chronic leptin infusion leads to elevated blood pressure, under engagement of adrenoceptor activity. It is likely that leptin upregulation is a biomarker for chronic obesity-related inflammation from childhood onward (Reyes et al., 2015). Association of the LEP gene with severe obesity and T2DM have been reported, also case studies in LEP gene malfunction leading to binding-inactive leptin with infantile hyperphagia (Wabitsch et al., 2015).
Leptin Receptors
The leptin receptor gene LEPR (also LPR, OB-R, or CD295 1p31.3) is crucial for the satiation signaling pathway (Clément et al., 1998; Rolland et al., 1998), and null mutations lead to early onset childhood obesity, disturbance of the somatotropic axis and loss of puberty. Leptin receptors were identified in distinct neuron populations of the arcuate and paraventricular nuclei of the hypothalamus. The first group produce the AgRP and neuropeptide Y, inhibited by leptin. The second group produce α-MSH, and are activated by leptin, which is alike inhibitive of appetite. Leptin receptors, which are in regulation of blood pressure, have also been discovered in the rostroventral lateral medulla (RVLM). Recent experiments demonstrate that specifically adrenal sympathetic activity was steered by RVLM through leptinergic neuronal projections into the kidneys (Barnes and McDougal, 2014), further to regulating general mean arterial pressure. It is possible that, in obesity and T2DM, a deficiency in leptin uptake and binding is present due to high levels of triglycerides then suppressing leptin action (Oswal and Yeo, 2010). Of the leptin-receptor polymorphisms (Gln223Arg, Lys656Asn, and Lys109Arg), the presence of the Arg223 homozygous or the Asn656 allele was associated with elevated plasma leptin, BMI, waist circumference, and waist-to-hip ratio, but less catecholamine presence and dampened sympathetic activity (Masuo et al., 2008). Other mutations in LEPR have been identified causative for childhood obesity (Huvenne et al., 2015). Loci coupled to LEPR are LEPROT (1p31.3) and LEPROTL1 (8p12), modifying leptin receptor signaling and triggering expression of hepatic growth hormone receptors (Touvier et al., 2009). Copy number variations in these loci are as well associated with obesity, T2DM, and metabolic rates (Couturier et al., 2007; Jeon et al., 2010).
Ghrelin Physiological Functions and Binding
Ghrelin is growth hormone release inducing, as it activates the growth hormone secretagogue receptor in the hypothalamus, which results in the secretion of growth hormone (somatotropin). Further to leptin, ghrelin is involved with hunger-satiation-regulation in the hypothalamic nucleus arcuatus. Ghrelin is the “hunger hormone” and in many ways antagonistic to leptin action. Orexigenic signals are mediated by ghrelin via NPY receptors, inhibiting MC4Rs. It is released from ghrelinergic cells in the gastric mucosa, ε-cells of the pancreas, and the hypothalamus and anterior pituitary. The peptide hormone ghrelin is derived from posttranslational modification by cleavage from its precursor obestatin (Zhang et al., 2005), a peptide with opposing effects. Hunger states or sleep deprivation increase ghrelin levels, which would decline postprandially. Ghrelin stimulates NPY and agouti-related hormone (AgRP) action in the arcuate nucleus.
The ghrelin/obestatin gene GHRL (3p26-p25) produces these two products. Ghrelin is involved in energy homeostasis, and hereby regulates pancreatic glucose-stimulated insulin secretion. Obestatin also has multiple metabolic functions, but is regulative for adipocyte and glucose metabolisms. Low ghrelin level confers CVD and other risks, whereas high plasma ghrelin levels have vasoproctective effects (Laurila et al., 2014). The ghrelin receptor (GHSR 3q26.31 producing two transcripts, of which only type 1a is a receptor for ligand ghrelin) is found on the same cells in the brain as the leptin receptor. It activates hedonic dopaminergic neurons in the mesolimbic cholinergic–dopaminergic pathway that processes reward-related activation between ventral tegmental area and nucleus accumbens; possibly intragastric metabolic nutrient-sensing signals toward dorsal striatum (Stuber and Wise, 2016). It is primarily involved in the modulation of glucose and lipid metabolism, digestion, neuroprotection, and regulation of immune functions. Ghrelin receptors have high density in the hypothalamus and pituitary, and on the vagus nerve (on both afferent cell bodies and afferent nerve endings) and throughout the gastrointestinal tract. Meta-analytic analyses on the polymorphisms in GHSR indicated that these are in regulation of blood glucose (Pabalan et al., 2014). The gene for the obestatin receptor GPR39 (2q21-q22) may have functions in repair and wound healing, and is upregulated by antidepressants (Mlyniec et al., 2015).
Melanocortin Receptors and Binding Sites
Two types of melanocortin receptors have increasingly been placed central to MetS pathophysiological understanding, but in specific the MC4R (Section T2DM: Melanocortins and Insulin Resistance). The reason for that is due to their capability to bind also other ligands: POMC and MC4R neurons are necessary for leptin signaling, on the one hand, and on the other hand suppresses AgRP as ligand in MC4Rs their respective activation. The brain melanocortin system is embedded in leptin, ghrelin, and agouti-related energy and feeding homeostatic systems, as well as glutamatergic eating-reward behavior circuitry between lateral hypothalamic area, amygdala and VTA (Stuber and Wise, 2016). Leptin binds to leptin receptors (LEPRs) on AgRP-secreting neurons and POMC-secreting neurons in the arcuate nucleus of the hypothalamus. Leptin binding suppresses AgRP synthesis and triggers the production of POMC, which is the precursor for α-, β-, and γ-MSH: AgRP is the main antagonist of MC4R (Fani et al., 2014). Accordingly were cardiovascular and metabolic actions of leptin abolished in obese and non-obese MC4R deficient mice (Tallam et al., 2006). Stimulation of the MC4R causes a decrease in appetite and an increase in metabolism of fat and lean body mass: According to current account are ARC POMC and PVNH MC4R, and LPBN neuron populations responsible for nutrient chemosensation and relay of satiety evaluations to visceroceptive forebrain regions (Krashes et al., 2016). Functionality of MR4R is also necessary to induce blood pressure changes and metabolic alterations (Tallam et al., 2005; da Silva et al., 2006). Morbid obesity is associated with silencing of MC4R activation. In contrast to AgRP-related blockade of MC4R activation, stimulates α-MSH binding MC4R activation toward steering food intake via BDNF signaling to higher cerebral centers (Walley et al., 2009). Input to POMC neurons to trigger the brain melanocortin system requires (a) afferent vagal input of hunger signals via its dorsal motor root nucleus and NTS (Krashes et al., 2016), (b) leptin adipostatic signals crossing the blood-brain barrier, and (c) gut-released peptides cholecystokinin, ghrelin and PYY uptake (Cone, 2005; Ellacott et al., 2006). Its telencephalic network has not yet been exactly mapped, but is quite likely to comprise dysgranular cortices in visceroceptive insula, operculum, and in the caudolateral orbital frontal regions (Batterham et al., 2007).
Hypothalamic and Brainstem Nuclei
Distributions of MC3Rs and MC4Rs are widespread, and in relation to energy homeostasis, but specifically MC4Rs in paraventricular hypothalamic nucleus (PVNH) and amygdala are related to food intake behaviors (Balthasar, 2006). Basically, there are three main melanocortin circuits with (a) hypothalamic POMC neurons in the arcuate nucleus, in relation with the agouti-related neuropeptide and neuropeptide Y expressing neuron populations, (b) brainstem POMC neuron population in the commissural nucleus of the tractus solitarius, and (c) the telencephalic target system of MC3R and MC4Rs. Driving of sympathetic output is exerted mainly by allostatic excitatory neurons in the subfornical organ (SFO) (Wei et al., 2013; Young et al., 2013; Oka et al., 2015), and by the caudal vasomotor brainstem through leptin receptors. Leptin receptors are there expressed by adrenergic/noradrenergic C1/A1 cells, which overlap in the rostral ventrolateral medulla (RVLM) with melanocortin neurons (Grassi et al., 1998).
MSH Signaling
α-, β-, and γ-MSH bind to the MC4R receptor, a G-coupled transmembrane receptor, and this activates the brain melatonin system (da Silva et al., 2014). MC3R and MC4R are widespread over the brain, but only MR4R blockade results in hyperphagia causing obesity, and MC4R binding triggers energy expenditure while diminishing appetite (Tallam et al., 2005). Other proteins activating the MC4R are ACTH and POMC. However, MC2R (18p11.2) is also target of ACTH, and related to familial glucocorticoid deficiency, and/or blunted cortisol responding. MC3R (20q13.2-q13.3) increased fat mass despite decreased food intake, energy homeostasis, and also binds MSH and ACTH. Defects in MC4R (18q22) lead into an ontogenetic obesity phenotype with autosomal dominant obesity (Farooqi et al., 2003a,b). In childhood obesity, significant SNPs were its Val95Ile, Val166Ile, and Val179Ala mutations, which were primarily related to plasma lipid levels (Song et al., 2015). With regards to MetS, MC4R is the largest known single risk factor for combined obesity and T2DM manifestation, as replicated by several GWAS studies (Chambers et al., 2008; Loos et al., 2008; Thorleifsson et al., 2009; Willer et al., 2009). However, until 2014 more than 80 distinct mutations in its locus have been described (Fani et al., 2014). This finding is distinct from those in non-syndromal obesity (Walley et al., 2009), where FTO and the ghrelin receptor GHSR exhibited the largest odds ratios.
Expression of Melanocortin Receptors
Brain regions with highest levels of MC3R (Begriche et al., 2011) are the midbrain structures. MC3Rs are densely expressed in hypothalamic and limbic regions of the brain and in peripheral tissues. The ventromedial hypothalamus (VMH), a critical node in the neural circuits regulating feeding-related behaviors and metabolic homeostasis, exhibits dense MC3R expression. MC3R has been related to increased fat mass, and accelerated diet-induced obesity (DIO). MC3R causes in accordance with biorhythmic cycling hyperinsulinaemia, glucose intolerance, increased expression of lipogenic genes, and increased ketogenesis. Rhythmic expression of MC3R transcription factors was found regulating liver clock activity (Sutton et al., 2010). The brain regions with highest levels of MC4R (Rossi et al., 2011) are brainstem neurons including those in the dorsal motor nucleus of the vagus, paraventricular nucleus of the hypothalamus (PVNH), the amygdala, nucleus tractus solitarius, intermediolateral medulla (IML) (Krashes et al., 2016). Brain regions critically subserving the hunger-satiation regulation were described to consist in MC4R neuron populations in the PVNH toward the lateral parabrachial nucleus (LPBN) pathway (Garfield et al., 2015). A distinct interaction between MC4Rs in the PVNH and amygdala has been found sufficient to control food intake behaviors (Balthasar, 2006). The central MC4R pathway is also crucial for control of cholesterol metabolism by the liver, specifically in determining the HDL/LDL ratio (Perez-Tilve et al., 2010), a process decisive for the MetS constellation and CVD, if derailed. Regarding energy expenditure, a crucial role has been assigned to the circumventricular structure organum vasculosum laminae terminalis lacking the blood-brain barrier (Oka et al., 2015). This structure adjacent to cerebral ventricles receives input by IL-1β signaling and triggers TNFα synthesis. It has anatomical connections with the hypothalamic nucleus praeopticus (Oka et al., 2015), which relays output to the periaquaeductal gray (PAG) and Raphe nuclei, all related to thermogenesis (Cone, 2005).
Neuropeptide Y and Agouti-Related Hormone Receptors and Binding Sites
The likely neurophysiological linkages between sympathetic stress reactivity, the glucocorticoid system, and the melanocortin system are the neuropeptide Y (NPY) and the AgRP systems. These hormones are the major orexigenic signals in the hypothalamus. Both sympathetic nerves and immune cells are capable to produce NPY, which has a protective buffering function against the immune challenges exerted by low-level inflammation (Farzi et al., 2015), and contributes to resilience against environmental, inflammatory, and oxidative stress.
Physiological Function of Neuropeptide Y
The prevalence of NPY is abundant in the cerebrum, but major sites of action are the hippocampal formation, the amygdala and septum (Thorsell, 2010), with highest prevalence rates in cortices, limbic system, and hypothalamus. Its effects are to increase cortical excitability, while alerting with cardiovascular responsiveness, to raise intracellular calcium levels, and thus activating potassium channels. These latter processes exert vasoconstriction, while at the same time lowering blood pressure. However, a somatotropic function of NPY is in promoting growth of adipose tissue (Kuo et al., 2007). In addition, it has been shown that NPY modulates neurogenesis. Interestingly, these latter effects are similar to those elicited by neurotrophins (Angelucci et al., 2014). In sum, the activity effects of NPY are in alertness, anxiolysis, analgesia, fat deposition, and orexigenesis. Obesity states are characterized by increases in NPY mRNA and NPY release (Dryden et al., 1995).
Release of NPY in the PVNH triggers CRH, as there is also a negative feedback loop of CRH on NPY synthesis and release. In this reciprocal circuit mechanism, NPY action is a direct stimulation of synthesis and release of CRH and vice versa. This reciprocal interaction of CRH-NPY with a tension-vs-anxiolysis effect was concluded to be an internuclear interaction mechanism within the amygdala, specifically the basolateral amygdalar nucleus (BLA) (Thorsell, 2010), which is known to trigger anxiety responses, and perhaps the central amygdaloid nucleus (CeA). Furthermore, NPY concentrations were significantly higher in the medial praeoptic area (MPO), paraventricular (PVNH), ventromedial (VMN), and dorsomedial (DMN) nuclei of the hypothalamus. It is therefore likely that NPY synthesis and release participates in the emergence of T2DM. High levels of glucocorticosteroids stimulate gluconeogenesis, which increases blood glucose, triggers release of insulin. Insulin action is to reuptake and store glucose as glycogen, while insulin resistance finally results in T2DM.
Biosynthesis and Gene
In general, neuropeptide Y is essential in controlling cortical electrophysiological excitation, and thus in suppressing epileptic processes. Its intranasal application reduces PTSD symptoms pharmacologically. The NPY gene (7p15.1) has just one documented SNP, the Leu7Pro7 mutation, associated with elevated cholesterol, so conferring specific risks for MetS and CVD. In that polymorphism, specifically, abdominal fat deposition correlated with the rs16147 C and T alleles (Lin et al., 2015). The NPY is expressed by SNS neurons as an ubiquitous cerebral neurotransmitter that is not at all restricted to hypothalamus. However, neuropeptide Y (NPY) is in particular synthesized in arcuate (ARC) neurons, which then project principally to the paraventricular nucleus (PVNH). Axon terminals in PVNH also contained the highest levels of NPY immunoreactivity, together with the arcuate nucleus. NPY directly injected into the PVNH caused hyperphagia, reduced energy expenditure, and eventually produced obesity.
Receptor Subtypes and their Genomic Associations
There are up to six subtypes of NPY receptors currently known, but only four of which are known to mediate neuroendocrine responses: NPY1R, NPY2R, NPY4R, and NPY5R (Thorsell, 2010), regulation of food intake is mediated only by NPY1R and NPY5R receptors. The NPY1R (4q32.2) exerts anxiolytic activation (Olesen et al., 2012b), triggers mobilization of intracellular calcium and inhibition of adenylate cyclase activity. Variants in NPY1R were found related to heritable autonomic traits in circulation, baroreflex functioning, and pressor response to environmental stress (Wang et al., 2009). Expression of NPY1R in adipose tissue was observed relating to the MetS components weight and insulin resistance (Sitticharoon et al., 2013). NPY2R (4q31) participates in modification of cardiometabolic traits (Wei et al., 2013), with its 21 known SNPs. Haplotypes of the proximal promoter variants G-1606A, C-599T, and A-224G disrupted predicted dependent genes, influenced the transcription of the interferon regulation factor related to IL-6, and the hepatocyte nuclear factor. NPY4R (10q11.2) gene products are the targets of the pancreatic polypeptide, which, in the adrenal medulla, locally enhances the secretion of catecholamines (Cavadas et al., 2001). NPY5R (4q32.2) activation results in behavioral hyperactivity (Olesen et al., 2012a), in epilepsy suppression (Gøtzsche et al., 2012), and in regulating food intake, while defects in this gene being associated with eating disorders and BMI (Li et al., 2014).
Rodent studies have identified the NPY2R activation as critical for an animal model of MetS. In this context it was possible to show that stress consisting of environmental or social stressors trigger NPY release from sympathetic nerves, which in turn sensitizes its NPY2Rs for glucocorticoid-dependent action in the abdominal fat depot (Kuo et al., 2007). Visceral fat accumulation is thus a consequence of NPY2R activation, which incites proliferation of adipocytes, triggers angiogenesis in adipose tissue, and promotes migration of macrophages into adipose tissue (Ali et al., 2013). In this accumulation of visceral fat were glucocorticoids described as central in stress-mediated exacerbation of DIO. Respective results suggested that glucocorticoids become modulators of NPY-NPY2R signaling. In the presence of high-calorie nutrition, plasma corticosterone levels, a precursor of aldosterone, raise, and in such a constellation, sympathetic excitation increases the conversion of the inactive steroid precursor to active cortisol by upregulating 11β-HSD-1 activity, specifically in the abdominal fat (Kuo et al., 2007).
The catecholamine hormone adrenaline has been shown to upregulate NPY and NPY2R expression in utero (Han et al., 2012) in dependence of the glucocorticoid system. Prenatal stress accelerates adipogenic programming of embryonic stem cells with adrenaline during their adipogenic differentiation.
Agouti-Related Neuropeptide
Agouti-related neuropeptide (AgRP) is only synthesized in neurons, which also contain NPY (Bäckberg et al., 2004; Krashes et al., 2016), mainly in the arcuate nucleus, but also PVNH, of the hypothalamus, and which also contain ghrelin receptors. Its main expression sites are, besides the hypothalamus, the subthalamic nucleus, and the adrenal medulla. Starvation sensations have been shown to be dependent on the output of AgRP populations in ARC toward PVNH pathway (Atasoy et al., 2012), which are abrogated by food stimuli in interplay of AgRP and SFO neurons (Betley et al., 2015). The AgRP neuron population, in turn, is inhibited by MC4R neurons in the PVNH, mediated by a circuit between PVNH and lateral zone LPNH (Garfield et al., 2015). AgRP also stimulates the HPA to release ACTH, cortisol and so counteracts pro-inflammatory cytokine IL-1β (Xiao et al., 2003). The expression of AGRP (16q22) in the adrenal gland blocks α-MSH-induced secretion of corticosterone (Dhillo et al., 2003). Acute stress response, in turn, downregulates AGRP expression as measured by its mRNA. High plasma AgRP levels are a further correlate of obesity. The main physiological function of AgRP is in the hypothalamic control of food intake by means of intracellular calcium levels. AgRP and NPY are secreted to increase appetite, triggered by pro-inflammatory cytokines to decrease metabolism and energy expenditure. It also enhances the ACTH response toward IL-1β, suggesting it may play a role in the modulation of neuroendocrine response against inflammation. It also serves as endogenous antagonist of the MC3Rs and MC4Rs, which blocks binding of α-MSH presumably through competitive ligand binding. An enduring AgRP-induced blockade of MC4R leads to hyperphagia and obesity. Polymorphisms in the AGRP (16q22) locus have been linked with anorexia nervosa (Vink et al., 2001), furthermore in cognitive functioning there, and to late onset obesity in its rs11575892 T allele (Kalnina et al., 2009). The polymorphism Ala67Thr is, however, protective against this obesity risk (Sözen et al., 2007).
Findings on Epigenetic Modification and Gene Regulation in MetS
The past decade has seen the conduction of studies elucidating the posttranscriptional and posttranslational activity of genes presumably involved in MetS, as specified in the preceding sections. Specific new evidence accumulated in the context of studies in “programming” of these genes in effect of interaction with environmental processes, early or later in life.
Epigenetic Modifications
The risk gene loci for the development of MetS identified in the previous parts were entered to a meta-analytic evaluation. Table 1 lists the findings describing epigenetic processes histone methylation, lysine acetylation, serine phosphorylation, and ubiquitination. As can be seen, most results are currently pertaining to POMC neurons in the hypothalamus. In summary, it can be said that several studies support the presence of early life stress (ELS) and DIO programming in this convergence region. Furthermore, the results suggest tissue-specific modification of risk gene expression in the midbrain, liver, and adipose tissue. With regards to neuroendocrine systems, epigenetic modifications were found in cortisol-related (here HSD11B1 expression), melanocortin, leptin, NPY, and adiponectin genes. With respect to adiposity genes, epigenetic modifications were documented for fat mass gene cluster APOA1/C3/A4/A5, and the lipolysis gene LIPE. With regard to inflammatory, immune and subcellular metabolism, PPARG, NKBF1, TNFA, TCF7C2, and those genes expressing members of the cytochrome P450 family that are involved in steroidogenesis and in relation to hepatic lipoproteins. These results on MetS are more consistent than those previously published on obesity (van Dijk et al., 2015). Studies directly pertaining to core HPA axis are very few up to present, with < 10 entries in PubMed: In this context, existing studies addressed the NR3C1 and CRH1 loci, with one study (Bockmühl et al., 2015) related to MetS. In sum, there is a lacuna of research in this area.
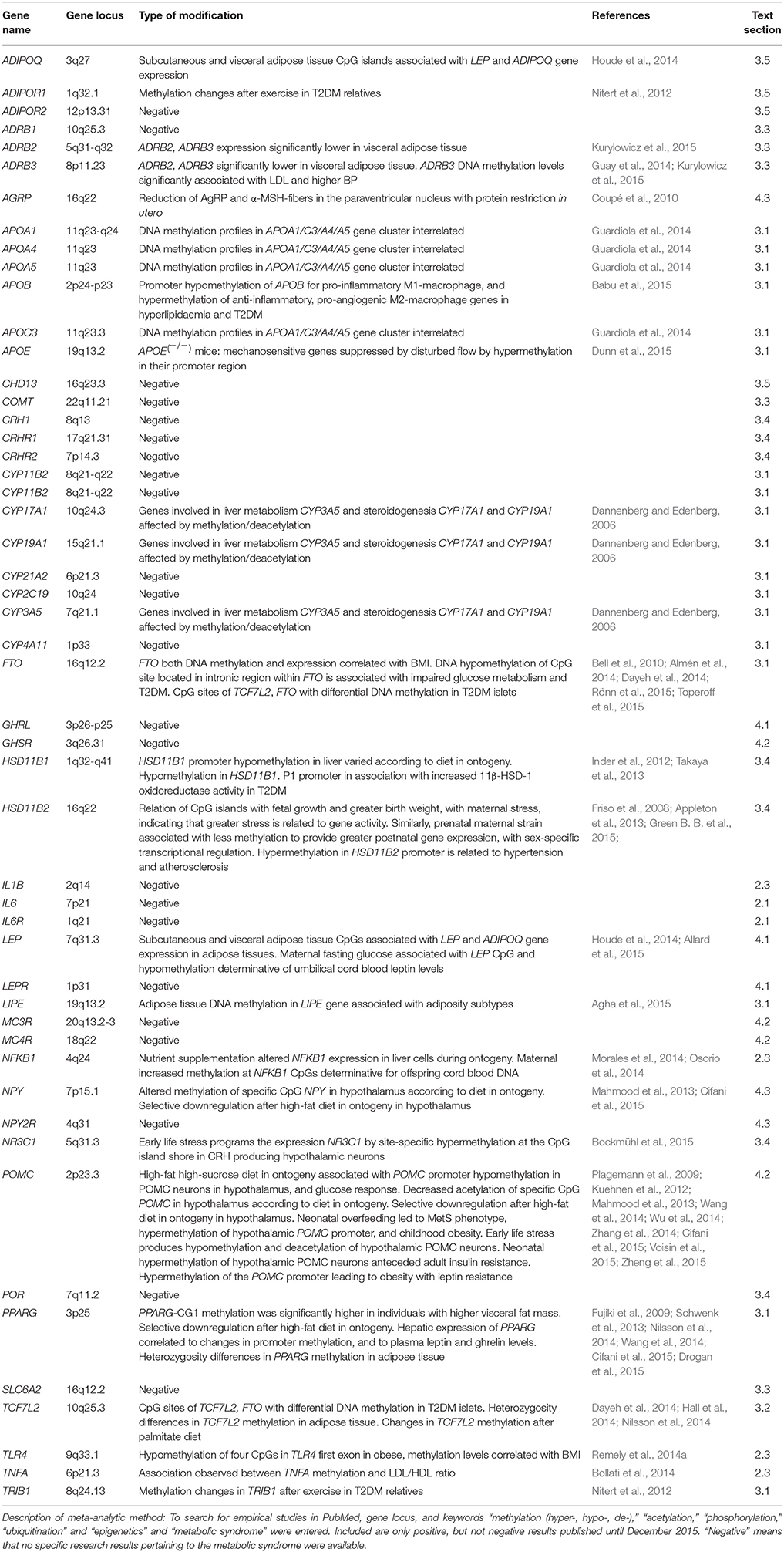
Table 1. Risk genes and findings on epigenetic modification in the metabolic syndrome.
Gene Regulation
Further on were the results of microRNA studies in relation to MetS relevant processes evaluated. MicroRNAs are small non-coding RNA protein pieces that function as post-transcriptional gene regulators. Although transcription factors work both as activators and repressors, however, all currently known microRNAs solely work as repressors (Chen and Rajewsky, 2007) to protein translation.
To date, about 40 microRNAs have been identified pertaining to MetS (Dehwah et al., 2012) and in lipoprotein metabolism, specifically in glucose uptake, insulin secretion and adipogenesis. It is assumed that these play central roles in diabetic complications and progression to chronic diabetes (Wegner et al., 2014). Here, miR-495, miR-432, and miR-376a at chromosome 14q32 expression were found critical for survival of islets in pancreatic ß-cells in T2DM sufferers (Kameswaran et al., 2014). Best studied are hitherto microRNAs during adipogenesis and obesity, where miR-146b regulates the proliferation of visceral pre-adipocytes and promote their differentiation, a target of Krüppel-like transcription factor KLF7 (Chen et al., 2014). MiR-27a and miR-27b abundance decreased PPARγ during adipogenesis of human pluripotent adipose-derived stem cells, thus counteracting PPARγ (Karbiener et al., 2009; Kim et al., 2010). MiR-122 decrease was described central in hepatic fatty-acid and cholesterol synthesis rate, by reducing hepatosteatosis (Esau et al., 2006), together with miR-223 in obesity (Kilic et al., 2015), also with miR-425, miR-126, miR-16, miR-634 (Li et al., 2015), miR-519d, miR-27 and miR-519d for target PPARγ (McGregor and Choi, 2011), miR-26 triglyceride accumulation (Song et al., 2014). MiR-370, miR-22, miR-758 also engaged in liver-adipocyte interaction (Flowers et al., 2013), furthermore miR-10b, miR-302a, miR-378, miR-613, miR-224 (Peng et al., 2013, 2014). Regarding to epigenetic effects, MiR-21, miR-17, miR-200, miR-221/222, miR-203 have been found altered by glucose, or polysaturated fatty acid diets (Palmer et al., 2014), hence suggesting programming effects across the life-span.
Much less well studied have been the three key neuroendocrine systems leptin, melanocortin, and NPY. Leptin uptake and insulin secretion were found controlled by miR-200a, miR-200b, and miR-429, which are related to obesity. Leptin treatment downregulating these miRNAs in hypothalamus increased leptin receptor and insulin receptor expression, also inducing weight loss and improving hepatic insulin responsiveness (Crépin et al., 2014). Leptin upregulation by miR-383, miR-384-3p, and miR-488 provided proper functioning of activity of pro-opiomelanocortin (POMC) neurons (Derghal et al., 2015). Then, inhibition of miR-375 in the intermediate lobe of the pituitary gland increased POMC secretion, whereas miR-375 overexpression down-regulated ACTH secretion stimulated by CRH (Zhang et al., 2013). Deletion of the microRNA-processing enzyme Dicer, modulated by nutrient availability in the hypothalamus, defective glucose metabolism, and alterations in the pituitary-adrenocortical axis, resulted in neurodegeneration of POMC-expressing neurons in early ontogeny (Schneeberger et al., 2012). Further to that, it was found that loss of miR-103 due to Dicer knock-out triggered activation of mammalian target of rapamycin (mTOR) pathway in the arcuate nucleus leading to imbalance in the levels of neuropeptide Y, will be resulting in severe hyperphagia (Vinnikov et al., 2014). MiR-7a expression was particularly prominent in the SFO, a circumventricular organ with ependymal bridges of the blood-brain barrier, as well as the suprachiasmatic, paraventricular, periventricular, supraoptic, dorsomedial and arcuate hypothalamic nuclei and constrained to neuropeptide Y/AgRP-containing-neurons located in the ventromedial aspect of the arcuate nucleus (Herzer et al., 2012).
Summary and Conclusion
Research in the past decade has considerably sharpened previous concepts on aetiopathology of the constellation of MetS. It has now become possible to encircle and narrow in key pathophysiological processes. At the level of causation, three domains, namely nutritional factors, environmental and psychological stress factors, and chronic low-grade inflammatory processes in visceral adipose tissue can be focussed on. Nutrient-induced enforcement of oxidative stress in subcellular organelles, mitochondria and endoplasmic reticula are likely to bias steroidogenesis and to cascade from oxidative stress to impaired glucose uptake and insulin resistance. The current state of findings also justifies the assumption that stress susceptibility mainly in the central HPA may be reinforced by 11β-HSD-1 action in visceral adipose and hepatic tissues.
Main findings of the present meta-analytic investigation on the effects of posttranslational modifications in specific risk gene loci support the notion that psychological stress and nutrient impact lead to genotype-environmental interactions that shape the MetS phenotype. Recent evidence derived from studies of DNA methylation, histone acetylation, and other epigenetic processes have been able to support the central role of POMC neuron population in the hypothalamus (mostly in the arcuate nucleus). Summarising the convergent brain structures involved in the stress physiology of MetS, presently available evidence suggests that the processing of environmental stress is performed by basolateral amygdaloid and possibly central amygdaloid nuclei, which trigger sympathoexcitation. Central regulation of hunger-satiation homeostasis can, according to studies evaluated, be assigned to the interplay of arcuate and paraventricular nuclei. Less well investigated are the roles of circumventricular and SFOs in biasing the sympathetic output in interplay with leptin, MC4R, and NPY systems.
Novel aetiopathological and treatment concepts could arise from the fact that HPA dysfunction in MetS could be originated at the synthesis of glucocorticoids in a second peripheral CRH system that may be active in adipose tissue and liver thus biasing the stress susceptibility of the central hypothalamic-pituitary regulation. Future research should further investigate interactions of risk genotypes, environmental factors, and epigenetic programming in the pathophysiology of MetS.
Author Contributions
Drafting manuscript EL, OC, AL; Revising the manuscript for important intellectual content EL, OC, AL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, S. B., Rubino, D., Sinaii, N., Ramsey, S., and Nieman, L. K. (2013). Cortisol, obesity, and the metabolic syndrome: a cross-sectional study of obese subjects and review of the literature. Obesity (Silver Spring) 21, E105–E117. doi: 10.1002/oby.20083
Adams, J. N., Raffield, L. M., Freedman, B. I., Langefeld, C. D., Ng, M. C., Carr, J. J., et al. (2014). Analysis of common and coding variants with cardiovascular disease in the Diabetes Heart Study. Cardiovasc. Diabetol. 13:77. doi: 10.1186/1475-2840-13-77
Agha, G., Houseman, E. A., Kelsey, K. T., Eaton, C. B., Buka, S. L., and Loucks, E. B. (2015). Adiposity is associated with DNA methylation profile in adipose tissue. Int. J. Epidemiol. 44, 1277–1287. doi: 10.1093/ije/dyu236
Albert, J. S., Yerges-Armstrong, L. M., Horenstein, R. B., Pollin, T. I., Sreenivasan, U. T., Chai, S., et al. (2014). Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315. doi: 10.1056/NEJMoa1315496
Alberti, K. G., and Zimmet, P. Z. (1998). Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 15, 539–553.
Alehagen, U., Vorkapic, E., Ljungberg, L., Länne, T., and Wågsäter, D. (2015). Gender difference in adiponectin associated with cardiovascular mortality. BMC Med. Genet. 16:37. doi: 10.1186/s12881-015-0187-9
Ali, A. T., Hochfeld, W. E., Myburgh, R., and Pepper, M. S. (2013). Adipocyte and adipogenesis. Eur. J. Cell Biol. 92, 229–236. doi: 10.1016/j.ejcb.2013.06.001
Allard, C., Desgagné, V., Patenaude, J., Lacroix, M., Guillemette, L., Battista, M. C., et al. (2015). Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics 10, 342–351. doi: 10.1080/15592294.2015.1029700
Almadi, T., Cathers, I., and Chow, C. M. (2013). Associations among work-related stress, cortisol, inflammation, and metabolic syndrome. Psychophysiology 50, 821–830. doi: 10.1111/psyp.12069
Almén, M. S., Nilsson, E. K., Jacobsson, J. A., Kalnina, I., Klovins, J., Fredriksson, R., et al. (2014). Genome-wide analysis reveals DNA methylation markers that vary with both age and obesity. Gene 548, 61–67. doi: 10.1016/j.gene.2014.07.009
Anagnostis, P., Athyros, V. G., Tziomalos, K., Karagiannis, A., and Mikhailidis, D. P. (2009). Clinical review: the pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J. Clin. Endocrinol. Metab. 94, 2692–2701. doi: 10.1210/jc.2009-0370
Angelucci, F., Gelfo, F., Fiore, M., Croce, N., Mathé, A. A., Bernardini, S., et al. (2014). The effect of neuropeptide Y on cell survival and neurotrophin expression in in-vitro models of Alzheimer's disease. Can. J. Physiol. Pharmacol. 92, 621–630. doi: 10.1139/cjpp-2014-0099
Annerbrink, K., Westberg, L., Nilsson, S., Rosmond, R., Holm, G., and Eriksson, E. (2008). Catechol O-methyltransferase val158-met polymorphism is associated with abdominal obesity and blood pressure in men. Metab. Clin. Exp. 57, 708–711. doi: 10.1016/j.metabol.2008.01.012
Appleton, A. A., Armstrong, D. A., Lesseur, C., Lee, J., Padbury, J. F., Lester, B. M., et al. (2013). Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS ONE 8:e74691. doi: 10.1371/journal.pone.0074691
Assmann, T. S., Duarte, G. C., Rheinheimer, J., Cruz, L. A., Canani, L. H., and Crispim, D. (2014). The TCF7L2 rs7903146 (C/T) polymorphism is associated with risk to type 2 diabetes mellitus in Southern-Brazil. Arq. Bras. Endocrinol. Metabol. 58, 918–925. doi: 10.1590/0004-2730000003510
Atasoy, D., Betley, J. N., Su, H. H., and Sternson, S. M. (2012). Deconstruction of a neural circuit for hunger. Nature 488, 172–177. doi: 10.1038/nature11270
Babu, M., Durga Devi, T., Mäkinen, P., Kaikkonen, M., Lesch, H. P., Junttila, S., et al. (2015). Differential promoter methylation of macrophage genes is associated with impaired vascular growth in ischemic muscles of hyperlipidemic and type 2 diabetic mice: genome-wide promoter methylation study. Circ. Res. 117, 289–299. doi: 10.1161/CIRCRESAHA.115.306424
Bäckberg, M., Madjid, N., Ögren, S. O., and Meister, B. (2004). Down-regulated expression of agouti-related protein (AGRP) mRNA in the hypothalamic arcuate nucleus of hyperphagic and obese tub/tub mice. Brain Res. Mol. Brain Res. 125, 129–139. doi: 10.1016/j.molbrainres.2004.03.012
Balthasar, N. (2006). Genetic dissection of neuronal pathways controlling energy homeostasis. Obesity (Silver Spring) 14(Suppl. 5), 222S–227S. doi: 10.1038/oby.2006.313
Balthasar, N., Dalgaard, L. T., Lee, C. E., Yu, J., Funahashi, H., Williams, T., et al. (2005). Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505. doi: 10.1016/j.cell.2005.08.035
Barnes, J. N., Charkoudian, N., Matzek, L. J., Johnson, C. P., Joyner, M. J., and Curry, T. B. (2014). Acute cyclooxygenase inhibition does not alter muscle sympathetic nerve activity or forearm vasodilator responsiveness in lean and obese adults. Physiol. Rep. 2:e12079. doi: 10.14814/phy2.12079
Barnes, M. J., and McDougal, D. H. (2014). Leptin into the rostral ventral lateral medulla (RVLM) augments renal sympathetic nerve activity and blood pressure. Front. Neurosci. 8:232. doi: 10.3389/fnins.2014.00232
Bartoli, F., Carrà, G., Crocamo, C., Carretta, D., and Clerici, M. (2013). Metabolic syndrome in people suffering from posttraumatic stress disorder: a systematic review and meta-analysis. Metab. Syndr. Relat. Disord. 11, 301–308. doi: 10.1089/met.2013.0010
Bassi, M., Furuya, W. I., Zoccal, D. B., Menani, J. V., Colombari, E., Hall, J. E., et al. (2015a). Control of respiratory and cardiovascular functions by leptin. Life Sci. 125, 25–31. doi: 10.1016/j.lfs.2015.01.019
Bassi, M., Nakamura, N. B., Furuya, W. I., Colombari, D. S., Menani, J. V., do Carmo, J. M., et al. (2015b). Activation of the brain melanocortin system is required for leptin-induced modulation of chemorespiratory function. Acta Physiol. (Oxf.) 213, 893–901. doi: 10.1111/apha.12394
Batterham, R. L., Ffytche, D. H., Rosenthal, J. M., Zelaya, F. O., Barker, G. J., Withers, D. J., et al. (2007). PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature 450, 106–109. doi: 10.1038/nature06212
Baudrand, R., Campino, C., Carvajal, C. A., Olivieri, O., Guidi, G., Faccini, G., et al. (2014). High sodium intake is associated with increased glucocorticoid production, insulin resistance and metabolic syndrome. Clin. Endocrinol. (Oxf.) 80, 677–684. doi: 10.1111/cen.12225
Bayles, R., Baker, E. K., Jowett, J. B., Barton, D., Esler, M., El-Osta, A., et al. (2013). Methylation of the SLC6a2 gene promoter in major depression and panic disorder. PLoS ONE 8:e83223. doi: 10.1371/journal.pone.0083223
Bazzi, M. D., Nasr, F. A., Alanazi, M. S., Alamri, A., Turjoman, A. A., Moustafa, A. S., et al. (2014). Association between FTO, MC4R, SLC30A8, and KCNQ1 gene variants and type 2 diabetes in Saudi population. Genet. Mol. Res. 13, 10194–10203. doi: 10.4238/2014.December.4.14
Beavers, K. M., Hsu, F. C., Houston, D. K., Beavers, D. P., Harris, T. B., Hue, T. F., et al. (2013). The role of metabolic syndrome, adiposity, and inflammation in physical performance in the Health ABC Study. J. Gerontol. A Biol. Sci. Med. Sci. 68, 617–623. doi: 10.1093/gerona/gls213
Begriche, K., Levasseur, P. R., Zhang, J., Rossi, J., Skorupa, D., Solt, L. A., et al. (2011). Genetic dissection of the functions of the melanocortin-3 receptor, a seven-transmembrane G-protein-coupled receptor, suggests roles for central and peripheral receptors in energy homeostasis. J. Biol. Chem. 286, 40771–40781. doi: 10.1074/jbc.M111.278374
Bell, C. G., Finer, S., Lindgren, C. M., Wilson, G. A., Rakyan, V. K., Teschendorff, A. E., et al. (2010). Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS ONE 5:e14040. doi: 10.1371/journal.pone.0014040
Betley, J. N., Xu, S., Cao, Z. F., Gong, R., Magnus, C. J., Yu, Y., et al. (2015). Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature 521, 180–185. doi: 10.1038/nature14416
Black, M. H., Wu, J., Takayanagi, M., Wang, N., Taylor, K. D., Haritunians, T., et al. (2015). Variation in PPARG is associated with longitudinal change in insulin resistance in Mexican Americans at risk for type 2 diabetes. J. Clin. Endocrinol. Metab. 100, 1187–1195. doi: 10.1210/jc.2014-3246
Bockmühl, Y., Patchev, A. V., Madejska, A., Hoffmann, A., Sousa, J. C., Sousa, N., et al. (2015). Methylation at the CpG island shore region upregulates NR3C1 promoter activity after early-life stress. Epigenetics 10, 247–257. doi: 10.1080/15592294.2015.1017199
Bollati, V., Favero, C., Albetti, B., Tarantini, L., Moroni, A., Byun, H. M., et al. (2014). Nutrients intake is associated with DNA methylation of candidate inflammatory genes in a population of obese subjects. Nutrients 6, 4625–4639. doi: 10.3390/nu6104625
Brindley, D. N. (1995). Role of glucocorticoids and fatty acids in the impairment of lipid metabolism observed in the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 19(Suppl. 1), S69–S75.
Brown, M. R., Fisher, L. A., Spiess, J., Rivier, C., Rivier, J., and Vale, W. (1982). Corticotropin-releasing factor: actions on the sympathetic nervous system and metabolism. Endocrinology 111, 928–931. doi: 10.1210/endo-111-3-928
Brunner, E. J., Hemingway, H., Walker, B. R., Page, M., Clarke, P., Juneja, M., et al. (2002). Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: nested case-control study. Circulation 106, 2659–2665. doi: 10.1161/01.CIR.0000038364.26310.BD
Buchholz, K., Schorr, U., Turan, S., Sharma, A. M., and Deter, H. C. (1999). [Emotional irritability and anxiety in salt-sensitive persons at risk for essential hypertension]. Psychother. Psychosom. Med. Psychol. 49, 284–289.
Buijs, R. M., and Kreier, F. (2006). The metabolic syndrome: a brain disease? J. Neuroendocrinol. 18, 715–716. doi: 10.1111/j.1365-2826.2006.01456.x
Bulatova, I. A., Shchekotova, A. P., Krivtsov, A. V., Shchekotov, V. V., Nasibullina, N. I., Suzdal'tseva, K. N., et al. (2015). [Metabolic disorder and polymorphism of the genes encoding for beta-2-adrenergic receptor and apolipoproteins B in chronic hepatitis C and non-alcoholic fatty liver diseases]. Klin. Med. (Mosk) 93, 35–41.
Burguete-Garcia, A. I., Martinez-Nava, G. A., Valladares-Salgado, A., Bermudez Morales, V. H., Estrada-Velasco, B., Wacher, N., et al. (2014). Association of beta1 and beta3 adrenergic receptors gene polymorphisms with insulin resistance and high lipid profiles related to type 2 diabetes and metabolic syndrome. Nutr. Hosp. 29, 1327–1334. doi: 10.3305/nh.2014.29.6.7367
Caballero, B. (2007). The global epidemic of obesity: an overview. Epidemiol. Rev. 29, 1–5. doi: 10.1093/epirev/mxm012
Capoulade, R., Clavel, M. A., Dumesnil, J. G., Chan, K. L., Teo, K. K., Tam, J. W., et al. (2012). Impact of metabolic syndrome on progression of aortic stenosis: influence of age and statin therapy. J. Am. Coll. Cardiol. 60, 216–223. doi: 10.1016/j.jacc.2012.03.052
Carvalheira, J. B., Odegaard, J. I., and Chawla, A. (2014). A new role for the brain in metabolic control. Nat. Med. 20, 472–473. doi: 10.1038/nm.3556
Cassaglia, P. A., Hermes, S. M., Aicher, S. A., and Brooks, V. L. (2011). Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J. Physiol. 589(Pt 7), 1643–1662. doi: 10.1113/jphysiol.2011.205575
Caussy, C., Charrière, S., Marçais, C., Di Filippo, M., Sassolas, A., Delay, M., et al. (2014). An APOA5 3′ UTR variant associated with plasma triglycerides triggers APOA5 downregulation by creating a functional miR-485-5p binding site. Am. J. Hum. Genet. 94, 129–134. doi: 10.1016/j.ajhg.2013.12.001
Cavadas, C., Silva, A. P., Mosimann, F., Cotrim, M. D., Ribeiro, C. A., Brunner, H. R., et al. (2001). NPY regulates catecholamine secretion from human adrenal chromaffin cells. J. Clin. Endocrinol. Metab. 86, 5956–5963. doi: 10.1210/jcem.86.12.8091
Chambers, J. C., Elliott, P., Zabaneh, D., Zhang, W., Li, Y., Froguel, P., et al. (2008). Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 40, 716–718. doi: 10.1038/ng.156
Chandola, T., Britton, A., Brunner, E., Hemingway, H., Malik, M., Kumari, M., et al. (2008). Work stress and coronary heart disease: what are the mechanisms? Eur. Heart J. 29, 640–648. doi: 10.1093/eurheartj/ehm584
Chandola, T., Brunner, E., and Marmot, M. (2006). Chronic stress at work and the metabolic syndrome: prospective study. BMJ 332, 521–525. doi: 10.1136/bmj.38693.435301.80
Chang, L., and Neu, J. (2015). Early factors leading to later obesity: interactions of the microbiome, epigenome, and nutrition. Curr. Probl. Pediatr. Adolesc. Health Care 45, 134–142. doi: 10.1016/j.cppeds.2015.03.003
Chen, K., and Rajewsky, N. (2007). The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 8, 93–103. doi: 10.1038/nrg1990
Chen, L., Dai, Y. M., Ji, C. B., Yang, L., Shi, C. M., Xu, G. F., et al. (2014). MiR-146b is a regulator of human visceral preadipocyte proliferation and differentiation and its expression is altered in human obesity. Mol. Cell. Endocrinol. 393, 65–74. doi: 10.1016/j.mce.2014.05.022
Chen, Y. W., Harris, R. A., Hatahet, Z., and Chou, K. M. (2015). Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc. Natl. Acad. Sci. U.S.A. 112, E4556–E4564. doi: 10.1073/pnas.1506954112
Cifani, C., Micioni Di Bonaventura, M. V., Pucci, M., Giusepponi, M. E., Romano, A., Di Francesco, A., et al. (2015). Regulation of hypothalamic neuropeptides gene expression in diet induced obesity resistant rats: possible targets for obesity prediction? Front. Neurosci. 9:187. doi: 10.3389/fnins.2015.00187
Clément, K., Vaisse, C., Lahlou, N., Cabrol, S., Pelloux, V., Cassuto, D., et al. (1998). A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392, 398–401. doi: 10.1038/32911
Cole, C. B., Nikpay, M., Lau, P., Stewart, A. F., Davies, R. W., Wells, G. A., et al. (2014). Adiposity significantly modifies genetic risk for dyslipidemia. J. Lipid Res. 55, 2416–2422. doi: 10.1194/jlr.P052522
Cone, R. D. (2005). Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578. doi: 10.1038/nn1455
Coupé, B., Amarger, V., Grit, I., Benani, A., and Parnet, P. (2010). Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology 151, 702–713. doi: 10.1210/en.2009-0893
Couturier, C., Sarkis, C., Séron, K., Belouzard, S., Chen, P., Lenain, A., et al. (2007). Silencing of OB-RGRP in mouse hypothalamic arcuate nucleus increases leptin receptor signaling and prevents diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 104, 19476–19481. doi: 10.1073/pnas.0706671104
Crépin, D., Benomar, Y., Riffault, L., Amine, H., Gertler, A., and Taouis, M. (2014). The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol. Cell. Endocrinol. 384, 1–11. doi: 10.1016/j.mce.2013.12.016
Crosby, J., Peloso, G. M., Auer, P. L., Crosslin, D. R., Stitziel, N. O., Lange, L. A., et al. (2014). Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 371, 22–31. doi: 10.1056/NEJMoa1307095
Curry, T. B., Hines, C. N., Barnes, J. N., Somaraju, M., Basu, R., Miles, J. M., et al. (2014). Relationship of muscle sympathetic nerve activity to insulin sensitivity. Clin. Auton. Res. 24, 77–85. doi: 10.1007/s10286-014-0235-0
Cyphert, T. J., Morris, R. T., House, L. M., Barnes, T. M., Otero, Y. F., Barham, W. J., et al. (2015). NF-kappaB-dependent airway inflammation triggers systemic insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R1144–R1152. doi: 10.1152/ajpregu.00442.2014
da Silva, A. A., do Carmo, J. M., Wang, Z., and Hall, J. E. (2014). The brain melanocortin system, sympathetic control, and obesity hypertension. Physiology (Bethesda) 29, 196–202. doi: 10.1152/physiol.00061.2013
da Silva, A. A., Kuo, J. J., Tallam, L. S., Liu, J., and Hall, J. E. (2006). Does obesity induce resistance to the long-term cardiovascular and metabolic actions of melanocortin 3/4 receptor activation? Hypertension 47, 259–264. doi: 10.1161/01.HYP.0000198458.70351.e0
Dannenberg, L. O., and Edenberg, H. J. (2006). Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics 7:181. doi: 10.1186/1471-2164-7-181
Dayeh, T., Volkov, P., Salö, S., Hall, E., Nilsson, E., Olsson, A. H., et al. (2014). Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 10:e1004160. doi: 10.1371/journal.pgen.1004160
Dehwah, M. A., Xu, A., and Huang, Q. (2012). MicroRNAs and type 2 diabetes/obesity. J. Genet. Genomics 39, 11–18. doi: 10.1016/j.jgg.2011.11.007
Deniz, F., Katircibasi, M. T., Pamukcu, B., Binici, S., and Sanisoglu, S. Y. (2007). Association of metabolic syndrome with impaired heart rate recovery and low exercise capacity in young male adults. Clin. Endocrinol. (Oxf.) 66, 218–223. doi: 10.1111/j.1365-2265.2006.02711.x
Derghal, A., Djelloul, M., Airault, C., Pierre, C., Dallaporta, M., Troadec, J. D., et al. (2015). Leptin is required for hypothalamic regulation of miRNAs targeting POMC 3′UTR. Front. Cell. Neurosci. 9:172. doi: 10.3389/fncel.2015.00172
Dermitzaki, E., Liapakis, G., Androulidaki, A., Venihaki, M., Melissas, J., Tsatsanis, C., et al. (2014). Corticotrophin-Releasing Factor (CRF) and the urocortins are potent regulators of the inflammatory phenotype of human and mouse white adipocytes and the differentiation of mouse 3T3L1 pre-adipocytes. PLoS ONE 9:e97060. doi: 10.1371/journal.pone.0097060
Deter, H. C., Blecher, A., and Weber, C. S. (2007a). Cardiovascular reactivity of patients with essential and renal hypertension in an emotion-triggering interview. Behav. Med. 32, 117–125. doi: 10.3200/BMED.32.4.117-125
Deter, H. C., Blum, B., and Schwarz, U. (2002). [Psychophysiological and psychological aspects of mild hypertension]. Psychother. Psychosom. Med. Psychol. 52, 265–274. doi: 10.1055/s-2002-32247
Deter, H. C., Klepper, A., and Schulte, K. H. (1996). Preliminary results of a differentiated emotion-stimulating interview in patients with essential hypertension as compared with inpatients of a psychosomatic unit and normal controls. Psychother. Psychosom. 65, 262–271. doi: 10.1159/000289086
Deter, H. C., Wolf, C., Blecher, A., Thomas, A., Zimmermann, F., and Weber, C. (2007b). Cardiovascular reactivity in patients with essential or renal hypertension under standardized mental stress. Clin. Exp. Hypertens. 29, 301–310. doi: 10.1080/10641960701500414
Dhillo, W. S., Small, C. J., Gardiner, J. V., Bewick, G. A., Whitworth, E. J., Jethwa, P. H., et al. (2003). Agouti-related protein has an inhibitory paracrine role in the rat adrenal gland. Biochem. Biophys. Res. Commun. 301, 102–107. doi: 10.1016/S0006-291X(02)02991-1
Díez, J. J., and Iglesias, P. (2003). The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol. 148, 293–300. doi: 10.1530/eje.0.1480293
Dik, M. G., Jonker, C., Comijs, H. C., Deeg, D. J., Kok, A., Yaffe, K., et al. (2007). Contribution of metabolic syndrome components to cognition in older individuals. Diabetes Care 30, 2655–2660. doi: 10.2337/dc06-1190
Dinel, A.-L., André, C., Aubert, A., Ferreira, G., Layé, S., and Castanon, N. (2011). Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS ONE 6:e24325. doi: 10.1371/journal.pone.0024325
Do, R., Willer, C. J., Schmidt, E. M., Sengupta, S., Gao, C., Peloso, G. M., et al. (2013). Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 45, 1345–1352. doi: 10.1038/ng.2795
Douvris, A., Soubeyrand, S., Naing, T., Martinuk, A., Nikpay, M., Williams, A., et al. (2014). Functional analysis of the TRIB1 associated locus linked to plasma triglycerides and coronary artery disease. J. Am. Heart Assoc. 3, e000884. doi: 10.1161/JAHA.114.000884
Drogan, D., Boeing, H., Janke, J., Schmitt, B., Zhou, Y., Walter, J., et al. (2015). Regional distribution of body fat in relation to DNA methylation within the LPL, ADIPOQ and PPARgamma promoters in subcutaneous adipose tissue. Nutr. Diabetes 5, e168. doi: 10.1038/nutd.2015.19
Dryden, S., Pickavance, L., Frankish, H. M., and Williams, G. (1995). Increased neuropeptide Y secretion in the hypothalamic paraventricular nucleus of obese (fa/fa) Zucker rats. Brain Res. 690, 185–188. doi: 10.1016/0006-8993(95)00628-4
Dudchenko, I. A., Pristupa, L. N., Ataman, A. V., and Garbuzova, V. (2014). Genetic dependency of blood pressure and heart rate in patients with arterial hypertension and obesity. Vestn. Ross. Akad. Med. Nauk. 40–46. doi: 10.15690/vramn.v69i5-6.1042
Dunn, J., Simmons, R., Thabet, S., and Jo, H. (2015). The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int. J. Biochem. Cell Biol. 67, 167–176. doi: 10.1016/j.biocel.2015.05.001
Eckel, R. H., Alberti, K. G., Grundy, S. M., and Zimmet, P. Z. (2010). The metabolic syndrome. Lancet 375, 181–183. doi: 10.1016/S0140-6736(09)61794-3
Eckel, R. H., Grundy, S. M., and Zimmet, P. Z. (2005). The metabolic syndrome. Lancet 365, 1415–1428. doi: 10.1016/S0140-6736(05)66378-7
Edmondson, D., Richardson, S., Falzon, L., Davidson, K. W., Mills, M. A., and Neria, Y. (2012). Posttraumatic stress disorder prevalence and risk of recurrence in acute coronary syndrome patients: a meta-analytic review. PLoS ONE 7:e38915. doi: 10.1371/journal.pone.0038915
Ellacott, K. L., Halatchev, I. G., and Cone, R. D. (2006). Interactions between gut peptides and the central melanocortin system in the regulation of energy homeostasis. Peptides 27, 340–349. doi: 10.1016/j.peptides.2005.02.031
Emdin, M., Gastaldelli, A., Muscelli, E., Macerata, A., Natali, A., Camastra, S., et al. (2001). Hyperinsulinemia and autonomic nervous system dysfunction in obesity: effects of weight loss. Circulation 103, 513–519. doi: 10.1161/01.CIR.103.4.513
Esau, C., Davis, S., Murray, S. F., Yu, X. X., Pandey, S. K., Pear, M., et al. (2006). miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3, 87–98. doi: 10.1016/j.cmet.2006.01.005
Esler, M., Straznicky, N., Eikelis, N., Masuo, K., Lambert, G., and Lambert, E. (2006). Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 48, 787–796. doi: 10.1161/01.HYP.0000242642.42177.49
Fahlbusch, F. B., Ruebner, M., Volkert, G., Offergeld, R., Hartner, A., Menendez-Castro, C., et al. (2012). Corticotropin-releasing hormone stimulates expression of leptin, 11beta-HSD2 and syncytin-1 in primary human trophoblasts. Reprod. Biol. Endocrinol. 10, 80. doi: 10.1186/1477-7827-10-80
Fani, L., Bak, S., Delhanty, P., van Rossum, E. F., and van den Akker, E. L. (2014). The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int. J. Obes. (Lond.) 38, 163–169. doi: 10.1038/ijo.2013.80
Farmer, A., Korszun, A., Owen, M. J., Craddock, N., Jones, L., Jones, I., et al. (2008). Medical disorders in people with recurrent depression. Br. J. Psychiatry 192, 351–355. doi: 10.1192/bjp.bp.107.038380
Farooqi, I. S., Keogh, J. M., Yeo, G. S., Lank, E. J., Cheetham, T., and O'Rahilly, S. (2003a). Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095. doi: 10.1056/NEJMoa022050
Farooqi, I. S., Yeo, G. S., and O'Rahilly, S. (2003b). Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N. Engl. J. Med. 349, 606–609; author reply -9. doi: 10.1056/NEJMoa021971
Farzi, A., Reichmann, F., and Holzer, P. (2015). The homeostatic role of neuropeptide Y in immune function and its impact on mood and behaviour. Acta Physiol. (Oxf.) 213, 603–627. doi: 10.1111/apha.12445
Fisher, L. A., Rivier, J., Rivier, C., Spiess, J., Vale, W., and Brown, M. R. (1982). Corticotropin-releasing factor (CRF): central effects on mean arterial pressure and heart rate in rats. Endocrinology 110, 2222–2224. doi: 10.1210/endo-110-6-2222
Flowers, E., Froelicher, E. S., and Aouizerat, B. E. (2013). MicroRNA regulation of lipid metabolism. Metab. Clin. Exp. 62, 12–20. doi: 10.1016/j.metabol.2012.04.009
Ford, E. S., Giles, W. H., and Dietz, W. H. (2002). Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287, 356–359. doi: 10.1001/jama.287.3.356
Forsythe, C. E., Phinney, S. D., Fernandez, M. L., Quann, E. E., Wood, R. J., Bibus, D. M., et al. (2008). Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 43, 65–77. doi: 10.1007/s11745-007-3132-7
Fortuño, A., Rodríguez, A., Gómez-Ambrosi, J., Frühbeck, G., and Díez, J. (2003). Adipose tissue as an endocrine organ: role of leptin and adiponectin in the pathogenesis of cardiovascular diseases. J. Physiol. Biochem. 59, 51–60. doi: 10.1007/BF03179868
Fossum, E., Høieggen, A., Reims, H. M., Moan, A., Rostrup, M., Eide, I., et al. (2004). High screening blood pressure is related to sympathetic nervous system activity and insulin resistance in healthy young men. Blood Press. 13, 89–94. doi: 10.1080/08037050310031008
Friso, S., Pizzolo, F., Choi, S. W., Guarini, P., Castagna, A., Ravagnani, V., et al. (2008). Epigenetic control of 11-beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 199, 323–327. doi: 10.1016/j.atherosclerosis.2007.11.029
Fujiki, K., Kano, F., Shiota, K., and Murata, M. (2009). Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 7:38. doi: 10.1186/1741-7007-7-38
Gaio, V., Nunes, B., Fernandes, A., Mendonça, F., Horta Correia, F., Beleza, A., et al. (2014). Genetic variation at the CYP2C19 gene associated with metabolic syndrome susceptibility in a South Portuguese population: results from the pilot study of the European Health Examination Survey in Portugal. Diabetol. Metab. Syndr. 6, 23. doi: 10.1186/1758-5996-6-23
Gao, Y., Lin, Y., Sun, K., Wang, Y., Chen, J., Wang, H., et al. (2014). Orthostatic blood pressure dysregulation and polymorphisms of beta-adrenergic receptor genes in hypertensive patients. J. Clin. Hypertens. (Greenwich) 16, 207–213. doi: 10.1111/jch.12272
Garfield, A. S., Li, C., Madara, J. C., Shah, B. P., Webber, E., Steger, J. S., et al. (2015). A neural basis for melanocortin-4 receptor-regulated appetite. Nat. Neurosci. 18, 863–871. doi: 10.1038/nn.4011
Ginsberg, H. N., and Stalenhoef, A. F. (2003). The metabolic syndrome: targeting dyslipidaemia to reduce coronary risk. J. Cardiovasc. Risk 10, 121–128. doi: 10.1177/174182670301000207
Girman, C. J., Rhodes, T., Mercuri, M., Pyörälä, K., Kjekshus, J., Pedersen, T. R., et al. (2004). The metabolic syndrome and risk of major coronary events in the Scandinavian Simvastatin Survival Study (4S) and the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Am. J. Cardiol. 93, 136–141. doi: 10.1016/j.amjcard.2003.09.028
Gøtzsche, C. R., Nikitidou, L., Sørensen, A. T., Olesen, M. V., Sørensen, G., Christiansen, S. H. O., et al. (2012). Combined gene overexpression of neuropeptide Y and its receptor Y5 in the hippocampus suppresses seizures. Neurobiol. Dis. 45, 288–296. doi: 10.1016/j.nbd.2011.08.012
Grassi, G., Cattaneo, B. M., Seravalle, G., Lanfranchi, A., and Mancia, G. (1998). Baroreflex control of sympathetic nerve activity in essential and secondary hypertension. Hypertension 31, 68–72. doi: 10.1161/01.HYP.31.1.68
Grassi, G., Dell'Oro, R., Quarti-Trevano, F., Scopelliti, F., Seravalle, G., Paleari, F., et al. (2005). Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia 48, 1359–1365. doi: 10.1007/s00125-005-1798-z
Grassi, G., Quarti-Trevano, F., Seravalle, G., and Dell'Oro, R. (2007). Cardiovascular risk and adrenergic overdrive in the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 17, 473–481. doi: 10.1016/j.numecd.2007.01.004
Green, B. B., Armstrong, D. A., Lesseur, C., Paquette, A. G., Guerin, D. J., Kwan, L. E., et al. (2015). The role of placental 11-beta hydroxysteroid dehydrogenase type 1 and type 2 methylation on gene expression and infant birth weight. Biol. Reprod. 92:149. doi: 10.1095/biolreprod.115.128066
Green, E., Fairchild, J. K., Kinoshita, L. M., Noda, A., and Yesavage, J. (2015). Effects of posttraumatic stress disorder and metabolic syndrome on cognitive aging in veterans. Gerontologist 56, 72–81. doi: 10.1093/geront/gnv040
Greenfield, J. R., Miller, J. W., Keogh, J. M., Henning, E., Satterwhite, J. H., Cameron, G. S., et al. (2009). Modulation of blood pressure by central melanocortinergic pathways. N. Engl. J. Med. 360, 44–52. doi: 10.1056/NEJMoa0803085
Grone, B. P., and Maruska, K. P. (2015). A second corticotropin-releasing hormone gene (CRH2) is conserved across vertebrate classes and expressed in the hindbrain of a basal neopterygian fish, the spotted gar (Lepisosteus oculatus). J. Comp. Neurol. 523, 1125–1143. doi: 10.1002/cne.23729
Grundy, S. M. (2015). Adipose tissue and metabolic syndrome: too much, too little or neither. Eur. J. Clin. Invest. 45, 1209–1217. doi: 10.1111/eci.12519
Grundy, S. M., Cleeman, J. I., Daniels, S. R., Donato, K. A., Eckel, R. H., Franklin, B. A., et al. (2005b). Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement: executive summary. Crit. Pathw. Cardiol. 4, 198–203. doi: 10.1097/00132577-200512000-00018
Grundy, S. M., Cleeman, J. I., Daniels, S. R., Donato, K. A., Eckel, R. H., Franklin, B. A., et al. (2005a). Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112, 2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404
Gu, S. J., Guo, Z. R., Zhou, Z. Y., Hu, X. S., Wu, M., and Zhang, N. (2014). Peroxisome proliferatoractivated receptor gamma polymorphisms as risk factors for dyslipidemia. Mol. Med. Rep. 10, 2759–2763. doi: 10.3892/mmr.2014.2553
Guardiola, M., Oliva, I., Guillaumet, A., Martín-Trujillo, A., Rosales, R., Vallvé, J. C., et al. (2014). Tissue-specific DNA methylation profiles regulate liver-specific expression of the APOA1/C3/A4/A5 cluster and can be manipulated with demethylating agents on intestinal cells. Atherosclerosis 237, 528–535. doi: 10.1016/j.atherosclerosis.2014.10.029
Guarente, L. (2006). Sirtuins as potential targets for metabolic syndrome. Nature 444, 868–874. doi: 10.1038/nature05486
Guay, S. P., Brisson, D., Lamarche, B., Biron, S., Lescelleur, O., Biertho, L., et al. (2014). ADRB3 gene promoter DNA methylation in blood and visceral adipose tissue is associated with metabolic disturbances in men. Epigenomics 6, 33–43. doi: 10.2217/epi.13.82
Gyllenhammer, L. E., Alderete, T. L., Mahurka, S., Allayee, H., and Goran, M. I. (2014). Adipose tissue 11betaHSD1 gene expression, betacell function and ectopic fat in obese African Americans versus Hispanics. Obesity (Silver Spring) 22, 14–18. doi: 10.1002/oby.20571
Hall, E., Volkov, P., Dayeh, T., Bacos, K., Rönn, T., Nitert, M. D., et al. (2014). Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med. 12:103. doi: 10.1186/1741-7015-12-103
Hall, J. E., da Silva, A. A., do Carmo, J. M., Dubinion, J., Hamza, S., Munusamy, S., et al. (2010). Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J. Biol. Chem. 285, 17271–17276. doi: 10.1074/jbc.R110.113175
Haller, H., and Hanefeld, M. (1975). “Synoptische Betrachtung metabolischer Risikofaktoren,” in Lipidstoffwechselstörungen, eds H. Haller, M. Hanefeld, and W. Jaross (Jena: Gustav Fischer Verlag), 254–64.
Han, R., Kitlinska, J. B., Munday, W. R., Gallicano, G. I., and Zukowska, Z. (2012). Stress hormone epinephrine enhances adipogenesis in murine embryonic stem cells by up-regulating the neuropeptide Y system. PLoS ONE 7:e36609. doi: 10.1371/journal.pone.0036609
Hart, E. C., Wallin, B. G., Barnes, J. N., Joyner, M. J., and Charkoudian, N. (2014). Sympathetic nerve activity and peripheral vasodilator capacity in young and older men. Am. J. Physiol. Heart Circ. Physiol. 306, H904–H909. doi: 10.1152/ajpheart.00181.2013
He, D., Fu, M., Miao, S., Hotta, K., Chandak, G. R., and Xi, B. (2014). FTO gene variant and risk of hypertension: a meta-analysis of 57,464 hypertensive cases and 41,256 controls. Metab. Clin. Exp. 63, 633–639. doi: 10.1016/j.metabol.2014.02.008
He, F. J., Li, J., and Macgregor, G. A. (2013). Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ 346:f1325. doi: 10.1136/bmj.f1325
Herzer, S., Silahtaroglu, A., and Meister, B. (2012). Locked nucleic acid-based in situ hybridisation reveals miR-7a as a hypothalamus-enriched microRNA with a distinct expression pattern. J. Neuroendocrinol. 24, 1492–1504. doi: 10.1111/j.1365-2826.2012.02358.x
Hintzpeter, J., Stapelfeld, C., Loerz, C., Martin, H. J., and Maser, E. (2014). Green tea and one of its constituents, epigallocatechine-3-gallate, are potent inhibitors of human 11beta-hydroxysteroid dehydrogenase type 1. PLoS ONE 9:e84468. doi: 10.1371/journal.pone.0084468
Hjemdahl, P. (2002). Stress and the metabolic syndrome: an interesting but enigmatic association. Circulation 106, 2634–2636. doi: 10.1161/01.CIR.0000041502.43564.79
Hotamisligil, G. S. (2010a). Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 16, 396–399. doi: 10.1038/nm0410-396
Hotamisligil, G. S. (2010b). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. doi: 10.1016/j.cell.2010.02.034
Houde, A.-A., Légaré, C., Hould, F.-S., Lebel, S., Marceau, P., Tchernof, A., et al. (2014). Cross-tissue comparisons of leptin and adiponectin: DNA methylation profiles. Adipocyte 3, 132–140. doi: 10.4161/adip.28308
Huang, W., Sun, Y., and Sun, J. (2011). Combined effects of FTO rs9939609 and MC4R rs17782313 on obesity and BMI in Chinese Han populations. Endocrine 39, 69–74. doi: 10.1007/s12020-010-9413-6
Huggett, R. J., Burns, J., Mackintosh, A. F., and Mary, D. A. (2004). Sympathetic neural activation in nondiabetic metabolic syndrome and its further augmentation by hypertension. Hypertension 44, 847–852. doi: 10.1161/01.HYP.0000147893.08533.d8
Hummasti, S., and Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 107, 579–591. doi: 10.1161/CIRCRESAHA.110.225698
Huvenne, H., Le Beyec, J., Pépin, D., Alili, R., Kherchiche, P. P., Jeannic, E., et al. (2015). Seven novel deleterious LEPR mutations found in early-onset obesity: a DeltaExon6-8 shared by subjects from Reunion Island, France, suggests a founder effect. J. Clin. Endocrinol. Metab. 100, E757–E766. doi: 10.1210/jc.2015-1036
Iannitti, T., Graham, A., and Dolan, S. (2015). Adiponectin-mediated analgesia and anti-inflammatory effects in rat. PLoS ONE 10:e0136819. doi: 10.1371/journal.pone.0136819
Inder, W. J., Obeyesekere, V. R., Jang, C., and Saffery, R. (2012). Evidence for transcript-specific epigenetic regulation of glucocorticoid-stimulated skeletal muscle 11beta-hydroxysteroid dehydrogenase-1 activity in type 2 diabetes. Clin. Epigenetics 4:24. doi: 10.1186/1868-7083-4-24
Iwen, K. A., Senyaman, O., Schwartz, A., Drenckhan, M., Meier, B., Hadaschik, D., et al. (2008). Melanocortin crosstalk with adipose functions: ACTH directly induces insulin resistance, promotes a pro-inflammatory adipokine profile and stimulates UCP-1 in adipocytes. J. Endocrinol. 196, 465–472. doi: 10.1677/JOE-07-0299
Jeon, J. P., Shim, S. M., Nam, H. Y., Ryu, G. M., Hong, E. J., Kim, H. L., et al. (2010). Copy number variation at leptin receptor gene locus associated with metabolic traits and the risk of type 2 diabetes mellitus. BMC Genomics 11:426. doi: 10.1186/1471-2164-11-426
Jimenez-Gomez, Y., Mattison, J. A., Pearson, K. J., Martin-Montalvo, A., Palacios, H. H., Sossong, A. M., et al. (2013). Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 18, 533–545. doi: 10.1016/j.cmet.2013.09.004
Jin, Z., Pu, L., Sun, L., Chen, W., Nan, N., Li, H., et al. (2014). Identification of susceptibility variants in ADIPOR1 gene associated with type 2 diabetes, coronary artery disease and the comorbidity of type 2 diabetes and coronary artery disease. PLoS ONE 9:e100339. doi: 10.1371/journal.pone.0100339
Joslin, E. P. (1921). The prevention of diabetes mellitus. JAMA 76, 79–84. doi: 10.1001/jama.1921.02630020001001
Julius, S., Valentini, M., and Palatini, P. (2000). Overweight and hypertension: a 2-way street? Hypertension 35, 807–813. doi: 10.1161/01.HYP.35.3.807
Kalnina, I., Kapa, I., Pirags, V., Ignatovica, V., Schiöth, H. B., and Klovins, J. (2009). Association between a rare SNP in the second intron of human Agouti related protein gene and increased BMI. BMC Med. Genet. 10:63. doi: 10.1186/1471-2350-10-63
Kameswaran, V., Bramswig, N. C., McKenna, L. B., Penn, M., Schug, J., Hand, N. J., et al. (2014). Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 19, 135–145. doi: 10.1016/j.cmet.2013.11.016
Kang, K. T. (2014). Endothelium-derived relaxing factors of small resistance arteries in hypertension. Toxicol. Res. 30, 141–148. doi: 10.5487/TR.2014.30.3.141
Karbiener, M., Fischer, C., Nowitsch, S., Opriessnig, P., Papak, C., Ailhaud, G., et al. (2009). microRNA miR-27b impairs human adipocyte differentiation and targets PPARgamma. Biochem. Biophys. Res. Commun. 390, 247–251. doi: 10.1016/j.bbrc.2009.09.098
Katome, T., Namekata, K., Mitamura, Y., Semba, K., Egawa, M., Naito, T., et al. (2015). Expression of intraocular peroxisome proliferator-activated receptor gamma in patients with proliferative diabetic retinopathy. J. Diabetes Complicat. 29, 275–281. doi: 10.1016/j.jdiacomp.2014.10.010
Katsuura-Kamano, S., Uemura, H., Arisawa, K., Yamaguchi, M., Hamajima, N., Wakai, K., et al. (2014). A polymorphism near MC4R gene (rs17782313) is associated with serum triglyceride levels in the general Japanese population: the J-MICC Study. Endocrine 47, 81–89. doi: 10.1007/s12020-014-0306-y
Kaufman, L. N., Young, J. B., and Landsberg, L. (1986). Effect of protein on sympathetic nervous system activity in the rat. Evidence for nutrient-specific responses. J. Clin. Invest. 77, 551–558. doi: 10.1172/JCI112336
Khoroshinina, L. P., Tur'eva, L. V., Glotov, O. S., Poliakova, I. V., and Baranov, V. S. (2014). [Frequency of some diseases and conditions in geriatric patients with coronary heart disease and various genotypes of transcription factor 7-like 2 protein]. Adv. Gerontol. 27, 704–709.
Kilgour, A. H., Semple, S., Marshall, I., Andrews, P., Andrew, R., and Walker, B. R. (2015). 11beta-hydroxysteroid dehydrogenase activity in the brain does not contribute to systemic interconversion of cortisol and cortisone in healthy men. J. Clin. Endocrinol. Metab. 100, 483–489. doi: 10.1210/jc.2014-3277
Kilic, I. D., Dodurga, Y., Uludag, B., Alihanoglu, Y. I., Yildiz, B. S., Enli, Y., et al. (2015). MicroRNA -143 and -223 in obesity. Gene 560, 140–142. doi: 10.1016/j.gene.2015.01.048
Kim, M. K., Tanaka, K., Kim, M. J., Matsuo, T., and Ajisaka, R. (2009). Exercise training-induced changes in heart rate recovery in obese men with metabolic syndrome. Metab. Syndr. Relat. Disord. 7, 469–476. doi: 10.1089/met.2008.0086
Kim, S. Y., Kim, A. Y., Lee, H. W., Son, Y. H., Lee, G. Y., Lee, J. W., et al. (2010). miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochem. Biophys. Res. Commun. 392, 323–328. doi: 10.1016/j.bbrc.2010.01.012
Kisiswa, L., Osório, C., Erice, C., Vizard, T., Wyatt, S., and Davies, A. M. (2013). TNFalpha reverse signaling promotes sympathetic axon growth and target innervation. Nat. Neurosci. 16, 865–873. doi: 10.1038/nn.3430
Kooijman, S., Boon, M. R., Parlevliet, E. T., Geerling, J. J., van de Pol, V., Romijn, J. A., et al. (2014). Inhibition of the central melanocortin system decreases brown adipose tissue activity. J. Lipid Res. 55, 2022–2032. doi: 10.1194/jlr.M045989
Kopin, I. J., Lake, R. C., and Ziegler, M. (1978). Plasma levels of norepinephrine. Ann. Intern. Med. 88, 671–680. doi: 10.7326/0003-4819-88-5-671
Koska, J., de Courten, B., Wake, D. J., Nair, S., Walker, B. R., Bunt, J. C., et al. (2006). 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue and prospective changes in body weight and insulin resistance. Obesity (Silver Spring) 14, 1515–1522. doi: 10.1038/oby.2006.175
Krashes, M. J., Lowell, B. B., and Garfield, A. S. (2016). Melanocortin-4 receptor-regulated energy homeostasis. Nat. Neurosci. 19, 206–219. doi: 10.1038/nn.4202
Kuehl, L. K., Hinkelmann, K., Muhtz, C., Dettenborn, L., Wingenfeld, K., Spitzer, C., et al. (2015). Hair cortisol and cortisol awakening response are associated with criteria of the metabolic syndrome in opposite directions. Psychoneuroendocrinology 51, 365–370. doi: 10.1016/j.psyneuen.2014.09.012
Kuehnen, P., Mischke, M., Wiegand, S., Sers, C., Horsthemke, B., Lau, S., et al. (2012). An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet. 8:e1002543. doi: 10.1371/journal.pgen.1002543
Kuo, L. E., Kitlinska, J. B., Tilan, J. U., Li, L., Baker, S. B., Johnson, M. D., et al. (2007). Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 13, 803–811. doi: 10.1038/nm1611
Kurylowicz, A., Jonas, M., Lisik, W., Jonas, M., Wicik, Z. A., Wierzbicki, Z., et al. (2015). Obesity is associated with a decrease in expression but not with the hypermethylation of thermogenesis-related genes in adipose tissues. J. Transl. Med. 13, 31. doi: 10.1186/s12967-015-0395-2
Kylin, E. (1921). Hypertonie u. Zuckerkrankheit: Beitrag zur Symptomatologie des Altersdiabetes. Zentralbl Inn Med 42, 873–877.
Kylin, E. (1923). Studien über das Hypertonie-, Hyperglycämie-, und Hyperurikämiesyndrom. Zentralbl Inn Med 44, 105–125.
Lambert, E., Dawood, T., Straznicky, N., Sari, C., Schlaich, M., Esler, M., et al. (2010). Association between the sympathetic firing pattern and anxiety level in patients with the metabolic syndrome and elevated blood pressure. J. Hypertens. 28, 543–550. doi: 10.1097/HJH.0b013e3283350ea4
Lamers, F., Vogelzangs, N., Merikangas, K. R., de Jonge, P., Beekman, A. T., and Penninx, B. W. (2013). Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol. Psychiatry 18, 692–699. doi: 10.1038/mp.2012.144
Landsberg, L. (1986). Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q. J. Med. 61, 1081–1090.
Landsberg, L., and Young, J. B. (1986). Caloric intake and sympathetic nervous system activity. Implications for blood pressure regulation and thermogenesis. J. Clin. Hypertens. 2, 166–171.
Laurila, M., Santaniemi, M., Kesäniemi, Y. A., and Ukkola, O. (2014). High plasma ghrelin protects from coronary heart disease and Leu72Leu polymorphism of ghrelin gene from cancer in healthy adults during the 19 years follow-up study. Peptides 61, 122–129. doi: 10.1016/j.peptides.2014.09.012
Lee, J. H., Gao, Z., and Ye, J. (2013). Regulation of 11beta-HSD1 expression during adipose tissue expansion by hypoxia through different activities of NF-kappaB and HIF-1alpha. Am. J. Physiol. Endocrinol. Metab. 304, E1035–E1041. doi: 10.1152/ajpendo.00029.2013
Lefterova, M. I., Haakonsson, A. K., Lazar, M. A., and Mandrup, S. (2014). PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 25, 293–302. doi: 10.1016/j.tem.2014.04.001
Lehrke, M., Broedl, U. C., Biller-Friedmann, I. M., Vogeser, M., Henschel, V., Nassau, K., et al. (2008). Serum concentrations of cortisol, interleukin 6, leptin and adiponectin predict stress induced insulin resistance in acute inflammatory reactions. Crit. Care 12, R157. doi: 10.1186/cc7152
Lemche, A. V., Chaban, O. S., and Lemche, E. (2016). Trait anxiety but not state anxiety level associates with biomarkers for hypertension in the metabolic syndrome. Psychophysiology. doi: 10.1111/psyp.12623. [Epub ahead of print].
Lemche, E., Giampietro, V. P., Surguladze, S. A., Amaro, E. J., Andrew, C. M., Williams, S. C., et al. (2006). Human attachment security is mediated by the amygdala: evidence from combined fMRI and psychophysiological measures. Hum. Brain Mapp. 27, 623–635. doi: 10.1002/hbm.20206
Lewis, J. G., Borowski, K. K., Shand, B. I., George, P. M., and Scott, R. S. (2010). Plasma sex hormone-binding globulin, corticosteroid-binding globulin, cortisol, and free cortisol levels in outpatients attending a lipid disorders clinic: a cross-sectional study of 1137 subjects. Horm. Metab. Res. 42, 274–279. doi: 10.1055/s-0029-1243260
Li, J., Zhou, C., Li, J., Su, Z., Sang, H., Jia, E., et al. (2015). Global correlation analysis for microRNA and gene expression profiles in human obesity. Pathol. Res. Pract. 211, 361–368. doi: 10.1016/j.prp.2014.11.014
Li, P., Tiwari, H. K., Lin, W. Y., Allison, D. B., Chung, W. K., Leibel, R. L., et al. (2014). Genetic association analysis of 30 genes related to obesity in a European American population. Int. J. Obes. (Lond.) 38, 724–729. doi: 10.1038/ijo.2013.140
Liguori, R., Labruna, G., Alfieri, A., Martone, D., Farinaro, E., Contaldo, F., et al. (2014). The FTO gene polymorphism (rs9939609) is associated with metabolic syndrome in morbidly obese subjects from southern Italy. Mol. Cell. Probes 28, 195–199. doi: 10.1016/j.mcp.2014.03.004
Lin, X., Qi, Q., Zheng, Y., Huang, T., Lathrop, M., Zelenika, D., et al. (2015). Neuropeptide Y genotype, central obesity, and abdominal fat distribution: the POUNDS LOST trial. Am. J. Clin. Nutr. 102, 514–519. doi: 10.3945/ajcn.115.107276
Lindberg, S., Jensen, J. S., Bjerre, M., Pedersen, S. H., Frystyk, J., Flyvbjerg, A., et al. (2015). Adiponectin, type 2 diabetes and cardiovascular risk. Eur. J. Prev. Cardiol. 22, 276–283. doi: 10.1177/2047487313514894
Lofgren, I., Zern, T., Herron, K., West, K., Sharman, M. J., Volek, J. S., et al. (2005). Weight loss associated with reduced intake of carbohydrate reduces the atherogenicity of LDL in premenopausal women. Metab. Clin. Exp. 54, 1133–1141. doi: 10.1016/j.metabol.2005.03.019
Loos, R. J., Lindgren, C. M., Li, S., Wheeler, E., Zhao, J. H., Prokopenko, I., et al. (2008). Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 40, 768–775. doi: 10.1038/ng.140
Lu, X., Wang, L., Lin, X., Huang, J., Charles Gu, C., He, M., et al. (2015). Genome-wide association study in Chinese identifies novel loci for blood pressure and hypertension. Hum. Mol. Genet. 24, 865–874. doi: 10.1093/hmg/ddu478
Lyssenko, V., Almgren, P., Anevski, D., Orho-Melander, M., Sjögren, M., Saloranta, C., et al. (2005). Genetic prediction of future type 2 diabetes. PLoS Med. 2:e345. doi: 10.1371/journal.pmed.0020345
Mahmood, S., Smiraglia, D. J., Srinivasan, M., and Patel, M. S. (2013). Epigenetic changes in hypothalamic appetite regulatory genes may underlie the developmental programming for obesity in rat neonates subjected to a high-carbohydrate dietary modification. J. Dev. Orig. Health Dis. 4, 479–490. doi: 10.1017/S2040174413000238
Mancia, G., Daffonchio, A., Di Rienzo, M., Ferrari, A. U., and Grassi, G. (1998). Methods to quantify sympathetic cardiovascular influences. Eur. Heart J. 19(Suppl. F), F7–F13.
Mark, A. L. (2013). Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R566–R581. doi: 10.1152/ajpregu.00180.2013
Martín-Blanco, A., Ferrer, M., Soler, J., Salazar, J., Vega, D., Andión, O., et al. (2014). Association between methylation of the glucocorticoid receptor gene, childhood maltreatment, and clinical severity in borderline personality disorder. J. Psychiatr. Res. 57, 34–40. doi: 10.1016/j.jpsychires.2014.06.011
Masuo, K., Mikami, H., Ogihara, T., and Tuck, M. L. (1997). Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am. J. Hypertens. 10, 77–83. doi: 10.1016/S0895-7061(96)00303-2
Masuo, K., Straznicky, N. E., Lambert, G. W., Katsuya, T., Sugimoto, K., Rakugi, H., et al. (2008). Leptin-receptor polymorphisms relate to obesity through blunted leptin-mediated sympathetic nerve activation in a Caucasian male population. Hypertens. Res. 31, 1093–1100. doi: 10.1291/hypres.31.1093
McGregor, B. A., Murphy, K. M., Albano, D. L., and Ceballos, R. M. (2015). Stress, cortisol, and B-lymphocytes: a novel approach to understanding academic stress and immune function. Stress. doi: 10.3109/10253890.2015.1127913. [Epub ahead of print].
McGregor, R. A., and Choi, M. S. (2011). microRNAs in the regulation of adipogenesis and obesity. Curr. Mol. Med. 11, 304–316. doi: 10.2174/156652411795677990
Melka, M. G., Abrahamowicz, M., Leonard, G. T., Perron, M., Richer, L., Veillette, S., et al. (2013). Clustering of the metabolic syndrome components in adolescence: role of visceral fat. PLoS ONE 8:e82368. doi: 10.1371/journal.pone.0082368
Mells, J. E., Fu, P. P., Kumar, P., Smith, T., Karpen, S. J., and Anania, F. A. (2015). Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J. Nutr. Biochem. 26, 285–292. doi: 10.1016/j.jnutbio.2014.11.002
Millington, G. W. (2007). The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. (Lond.) 4:18. doi: 10.1186/1743-7075-4-18
Mlyniec, K., Singewald, N., Holst, B., and Nowak, G. (2015). GPR39 Zn(2+)-sensing receptor: a new target in antidepressant development? J. Affect. Disord. 174, 89–100. doi: 10.1016/j.jad.2014.11.033
Morales, E., Groom, A., Lawlor, D. A., and Relton, C. L. (2014). DNA methylation signatures in cord blood associated with maternal gestational weight gain: results from the ALSPAC cohort. BMC Res. Notes 7:278. doi: 10.1186/1756-0500-7-278
Morash, B., Li, A., Murphy, P. R., Wilkinson, M., and Ur, E. (1999). Leptin gene expression in the brain and pituitary gland. Endocrinology 140, 5995–5998. doi: 10.1210/endo.140.12.7288
Moreira, M. C. S., Pinto, I. S., Mourão, A. A., Fajemiroye, J. O., Colombari, E., Reis, Â. A. S., et al. (2015). Does the sympathetic nervous system contribute to the pathophysiology of metabolic syndrome? Front. Physiol. 6:234. doi: 10.3389/fphys.2015.00234
Muller, Y. L., Thearle, M. S., Piaggi, P., Hanson, R. L., Hoffman, D., Gene, B., et al. (2014). Common genetic variation in and near the melanocortin 4 receptor gene (MC4R) is associated with body mass index in American Indian adults and children. Hum. Genet. 133, 1431–1441. doi: 10.1007/s00439-014-1477-6
Mune, T., Suwa, T., Morita, H., Isomura, Y., Takada, N., Yamamoto, Y., et al. (2013). Longer HSD11B2 CA-repeat in impaired glucose tolerance and type 2 diabetes. Endocr. J. 60, 671–678. doi: 10.1507/endocrj.EJ12-0108
Muramatsu-Kato, K., Itoh, H., Kobayashi-Kohmura, Y., Murakami, H., Uchida, T., Suzuki, K., et al. (2014). Comparison between placental gene expression of 11beta-hydroxysteroid dehydrogenases and infantile growth at 10 months of age. J. Obstet. Gynaecol. Res. 40, 465–472. doi: 10.1111/jog.12200
Napoli, E., Tassone, F., Wong, S., Angkustsiri, K., Simon, T. J., Song, G., et al. (2015). Mitochondrial citrate transporter-dependent metabolic signature in the 22q11.2 deletion syndrome. J. Biol. Chem. 290, 23240–23253. doi: 10.1074/jbc.M115.672360
Nelson, R. A., and Bremer, A. A. (2010). Insulin resistance and metabolic syndrome in the pediatric population. Metab. Syndr. Relat. Disord. 8, 1–14. doi: 10.1089/met.2009.0068
Nilsson, E., Jansson, P. A., Perfilyev, A., Volkov, P., Pedersen, M., Svensson, M. K., et al. (2014). Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63, 2962–2976. doi: 10.2337/db13-1459
Nilsson, S. K., Christensen, S., Raarup, M. K., Ryan, R. O., Nielsen, M. S., and Olivecrona, G. (2008). Endocytosis of apolipoprotein A-V by members of the low density lipoprotein receptor and the VPS10p domain receptor families. J. Biol. Chem. 283, 25920–25927. doi: 10.1074/jbc.M802721200
Nitert, M. D., Dayeh, T., Volkov, P., Elgzyri, T., Hall, E., Nilsson, E., et al. (2012). Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61, 3322–3332. doi: 10.2337/db11-1653
Nussey, S. S., Soo, S. C., Gibson, S., Gout, I., White, A., Bain, M., et al. (1993). Isolated congenital ACTH deficiency: a cleavage enzyme defect? Clin. Endocrinol. (Oxf.) 39, 381–385.
Oh, S. W., Han, K. H., Han, S. Y., Koo, H. S., Kim, S., and Chin, H. J. (2015). Association of sodium excretion with metabolic syndrome, insulin resistance, and body fat. Medicine (Baltimore) 94:e1650. doi: 10.1097/MD.0000000000001650
Oka, Y., Ye, M., and Zuker, C. S. (2015). Thirst driving and suppressing signals encoded by distinct neural populations in the brain. Nature 520, 349–352. doi: 10.1038/nature14108
Oler, J. A., Fox, A. S., Shelton, S. E., Rogers, J., Dyer, T. D., Davidson, R. J., et al. (2010). Amygdalar and hippocampal substrates of anxious temperament differ in their heritability. Nature 466, 864–868. doi: 10.1038/nature09282
Olesen, M. V., Christiansen, S. H., Gøtzsche, C. R., Holst, B., Kokaia, M., and Woldbye, D. P. D. (2012a). Y5 neuropeptide Y receptor overexpression in mice neither affects anxiety- and depression-like behaviours nor seizures but confers moderate hyperactivity. Neuropeptides 46, 71–79. doi: 10.1016/j.npep.2012.01.002
Olesen, M. V., Christiansen, S. H., Gøtzsche, C. R., Nikitidou, L., Kokaia, M., and Woldbye, D. P. D. (2012b). Neuropeptide Y Y1 receptor hippocampal overexpression via viral vectors is associated with modest anxiolytic-like and proconvulsant effects in mice. J. Neurosci. Res. 90, 498–507. doi: 10.1002/jnr.22770
Olofsson, P. S., Katz, D. A., Rosas-Ballina, M., Levine, Y. A., Ochani, M., Valdés-Ferrer, S. I., et al. (2012). alpha7 nicotinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol. Med. 18, 539–543. doi: 10.2119/molmed.2011.00405
Osorio, J. S., Ji, P., Drackley, J. K., Luchini, D., and Loor, J. J. (2014). Smartamine, M., and MetaSmart supplementation during the peripartal period alter hepatic expression of gene networks in 1-carbon metabolism, inflammation, oxidative stress, and the growth hormone-insulin-like growth factor 1 axis pathways. J. Dairy Sci. 97, 7451–7464. doi: 10.3168/jds.2014-8680
Oswal, A., and Yeo, G. (2010). Leptin and the control of body weight: a review of its diverse central targets, signaling mechanisms, and role in the pathogenesis of obesity. Obesity (Silver Spring) 18, 221–229. doi: 10.1038/oby.2009.228
Ouhaibi-Djellouli, H., Mediene-Benchekor, S., Lardjam-Hetraf, S. A., Hamani-Medjaoui, I., Meroufel, D. N., Boulenouar, H., et al. (2014). The TCF7L2 rs7903146 polymorphism, dietary intakes and type 2 diabetes risk in an Algerian population. BMC Genet. 15:134. doi: 10.1186/s12863-014-0134-3
Pabalan, N. A., Seim, I., Jarjanazi, H., and Chopin, L. K. (2014). Associations between ghrelin and ghrelin receptor polymorphisms and cancer in Caucasian populations: a meta-analysis. BMC Genet. 15:118. doi: 10.1186/s12863-014-0118-3
Palma-Gudiel, H., Córdova-Palomera, A., Leza, J. C., and Fañanás, L. (2015). Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci. Biobehav. Rev. 55, 520–535. doi: 10.1016/j.neubiorev.2015.05.016
Palmer, J. D., Soule, B. P., Simone, B. A., Zaorsky, N. G., Jin, L., and Simone, N. L. (2014). MicroRNA expression altered by diet: can food be medicinal? Ageing Res. Rev. 17, 16–24. doi: 10.1016/j.arr.2014.04.005
Parekh, P. J., Arusi, E., Vinik, A. I., and Johnson, D. A. (2014). The role and influence of gut microbiota in pathogenesis and management of obesity and metabolic syndrome. Front. Endocrinol. (Lausanne) 5:47. doi: 10.3389/fendo.2014.00047
Pavlov, V. A., and Tracey, K. J. (2012). The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat. Rev. Endocrinol. 8, 743–754. doi: 10.1038/nrendo.2012.189
Peeke, P. M., and Chrousos, G. P. (1995). Hypercortisolism and obesity. Ann. N.Y. Acad. Sci. 771, 665–676. doi: 10.1111/j.1749-6632.1995.tb44719.x
Peng, Y., Xiang, H., Chen, C., Zheng, R., Chai, J., Peng, J., et al. (2013). MiR-224 impairs adipocyte early differentiation and regulates fatty acid metabolism. Int. J. Biochem. Cell Biol. 45, 1585–1593. doi: 10.1016/j.biocel.2013.04.029
Peng, Y., Yu, S., Li, H., Xiang, H., Peng, J., and Jiang, S. (2014). MicroRNAs: emerging roles in adipogenesis and obesity. Cell. Signal. 26, 1888–1896. doi: 10.1016/j.cellsig.2014.05.006
Pennacchio, L. A., Olivier, M., Hubacek, J. A., Cohen, J. C., Cox, D. R., Fruchart, J. C., et al. (2001). An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 294, 169–173. doi: 10.1126/science.1064852
Perez-Tilve, D., Hofmann, S. M., Basford, J., Nogueiras, R., Pfluger, P. T., Patterson, J. T., et al. (2010). Melanocortin signaling in the CNS directly regulates circulating cholesterol. Nat. Neurosci. 13, 877–882. doi: 10.1038/nn.2569
Perry, C., Sattar, N., and Petrie, J. (2001). Adipose tissue: passive sump or active pump? Br. J. Diabetes Vasc. Dis. Res. 1, 110–114. doi: 10.1177/14746514010010020801
Pietrzak, R. H., Sumner, J. A., Aiello, A. E., Uddin, M., Neumeister, A., Guffanti, G., et al. (2015). Association of the rs2242446 polymorphism in the norepinephrine transporter gene SLC6A2 and anxious arousal symptoms of posttraumatic stress disorder. J. Clin. Psychiatry 76, e537–e538. doi: 10.4088/JCP.14l09346
Pinto, C. G., Marega, M., Carvalho, J. A., Carmona, F. G., Lopes, C. E., Ceschini, F. L., et al. (2015). Physical activity as a protective factor for development of non-alcoholic fatty liver in men. Einstein (Sao Paulo) 13, 34–40. doi: 10.1590/S1679-45082015AO2878
Plagemann, A., Harder, T., Brunn, M., Harder, A., Roepke, K., Wittrock-Staar, M., et al. (2009). Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587(Pt 20), 4963–4976. doi: 10.1113/jphysiol.2009.176156
Proudfoot, N. J. (2011). Ending the message: poly(A) signals then and now. Genes Dev. 25, 1770–1782. doi: 10.1101/gad.17268411
Putku, M., Kals, M., Inno, R., Kasela, S., Org, E., Koˇich, V., et al. (2015). CDH13 promoter SNPs with pleiotropic effect on cardiometabolic parameters represent methylation QTLs. Hum. Genet. 134, 291–303. doi: 10.1007/s00439-014-1521-6
Rafiq, K., Fujisawa, Y., Sherajee, S. J., Rahman, A., Sufiun, A., Kobori, H., et al. (2015). Role of the renal sympathetic nerve in renal glucose metabolism during the development of type 2 diabetes in rats. Diabetologia 58, 2885–2898. doi: 10.1007/s00125-015-3771-9
Raucci, R., Rusolo, F., Sharma, A., Colonna, G., Castello, G., and Costantini, S. (2013). Functional and structural features of adipokine family. Cytokine 61, 1–14. doi: 10.1016/j.cyto.2012.08.036
Reaven, G. M. (1988). Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37, 1595–1607. doi: 10.2337/diab.37.12.1595
Remely, M., Aumueller, E., Jahn, D., Hippe, B., Brath, H., and Haslberger, A. G. (2014a). Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef. Microbes 5, 33–43. doi: 10.3920/BM2013.006
Remely, M., Aumueller, E., Merold, C., Dworzak, S., Hippe, B., Zanner, J., et al. (2014b). Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene 537, 85–92. doi: 10.1016/j.gene.2013.11.081
Renaldi, O., Pramono, B., Sinorita, H., Purnomo, L. B., Asdie, R. H., and Asdie, A. H. (2009). Hypoadiponectinemia: a risk factor for metabolic syndrome. Acta Med. Indones. 41, 20–24.
Révész, D., Milaneschi, Y., Verhoeven, J. E., Lin, J., and Penninx, B. W. (2015). Longitudinal associations between metabolic syndrome components and telomere shortening. J. Clin. Endocrinol. Metab. 100, 3050–3059. doi: 10.1210/JC.2015-1995
Reyes, M., Quintanilla, C., Burrows, R., Blanco, E., Cifuentes, M., and Gahagan, S. (2015). Obesity is associated with acute inflammation in a sample of adolescents. Pediatr. Diabetes 16, 109–116. doi: 10.1111/pedi.12129
Reynolds, R. M., Labad, J., Strachan, M. W., Braun, A., Fowkes, F. G., Lee, A. J., et al. (2010). Elevated fasting plasma cortisol is associated with ischemic heart disease and its risk factors in people with type 2 diabetes: the Edinburgh type 2 diabetes study. J. Clin. Endocrinol. Metab. 95, 1602–1608. doi: 10.1210/jc.2009-2112
Rezaei, M., Andrieu, T., Neuenschwander, S., Bruggmann, R., Mordasini, D., Frey, F. J., et al. (2014). Regulation of 11beta-hydroxysteroid dehydrogenase type 2 by microRNA. Hypertension 64, 860–866. doi: 10.1161/HYPERTENSIONAHA.114.00002
Richard, D. (2015). Cognitive and autonomic determinants of energy homeostasis in obesity. Nat. Rev. Endocrinol. 11, 489–501. doi: 10.1038/nrendo.2015.103
Ridderstråle, M., and Groop, L. (2009). Genetic dissection of type 2 diabetes. Mol. Cell. Endocrinol. 297, 10–17. doi: 10.1016/j.mce.2008.10.002
Riestra, P., Gebreab, S. Y., Xu, R., Khan, R. J., Bidulescu, A., Correa, A., et al. (2015). Gender-specific associations between ADIPOQ gene polymorphisms and adiponectin levels and obesity in the Jackson Heart Study cohort. BMC Med. Genet. 16:65. doi: 10.1186/s12881-015-0214-x
Rivera, M., Cohen-Woods, S., Kapur, K., Breen, G., Ng, M. Y., Butler, A. W., et al. (2012). Depressive disorder moderates the effect of the FTO gene on body mass index. Mol. Psychiatry 17, 604–611. doi: 10.1038/mp.2011.45
Rivier, C., Brownstein, M., Spiess, J., Rivier, J., and Vale, W. (1982). In vivo corticotropin-releasing factor-induced secretion of adrenocorticotropin, beta-endorphin, and corticosterone. Endocrinology 110, 272–278. doi: 10.1210/endo-110-1-272
Rogers, J., Raveendran, M., Fawcett, G. L., Fox, A. S., Shelton, S. E., Oler, J. A., et al. (2013). CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol. Psychiatry 18, 700–707. doi: 10.1038/mp.2012.152
Rogers, S. L., Hughes, B. A., Jones, C. A., Freedman, L., Smart, K., Taylor, N., et al. (2014). Diminished 11beta-hydroxysteroid dehydrogenase type 2 activity is associated with decreased weight and weight gain across the first year of life. J. Clin. Endocrinol. Metab. 99, E821–E831. doi: 10.1210/jc.2013-3254
Rolland, V., Clement, K., Dugail, I., Guy-Grand, B., Basdevant, A., Froeuel, P., et al. (1998). Leptin receptor gene in a large cohort of massively obese subjects: no indication of the fa/fa rat mutation. Detection of an intronic variant with no association with obesity. Obes. Res. 6, 122–127. doi: 10.1002/j.1550-8528.1998.tb00325.x
Rönn, T., Volkov, P., Gillberg, L., Kokosar, M., Perfilyev, A., Jacobsen, A. L., et al. (2015). Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 24, 3792–3813. doi: 10.1093/hmg/ddv124
Rosas-Ballina, M., Ochani, M., Parrish, W. R., Ochani, K., Harris, Y. T., Huston, J. M., et al. (2008). Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. U.S.A. 105, 11008–11013. doi: 10.1073/pnas.0803237105
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdés-Ferrer, S. I., Levine, Y. A., Reardon, C., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. doi: 10.1126/science.1209985
Rosenbaum, S., Stubbs, B., Ward, P. B., Steel, Z., Lederman, O., and Vancampfort, D. (2015). The prevalence and risk of metabolic syndrome and its components among people with posttraumatic stress disorder: a systematic review and meta-analysis. Metab. Clin. Exp. 64, 926–933. doi: 10.1016/j.metabol.2015.04.009
Rossi, J., Balthasar, N., Olson, D., Scott, M., Berglund, E., Lee, C. E., et al. (2011). Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 13, 195–204. doi: 10.1016/j.cmet.2011.01.010
Ruan, L. L., Xu, J., Wang, C. L., and Zou, C. C. (2014). Variants of 11beta-hydroxysteroid dehydrogenase (HSD11B) gene type 1 and 2 in Chinese obese adolescents. J. Endocrinol. Invest. 37, 565–573. doi: 10.1007/s40618-014-0075-8
Russo, G. T., Meigs, J. B., Cupples, L. A., Demissie, S., Otvos, J. D., Wilson, P. W., et al. (2001). Association of the Sst-I polymorphism at the APOC3 gene locus with variations in lipid levels, lipoprotein subclass profiles and coronary heart disease risk: the Framingham offspring study. Atherosclerosis 158, 173–181. doi: 10.1016/S0021-9150(01)00409-9
Sanders, P. W. (2009). Dietary salt intake, salt sensitivity, and cardiovascular health. Hypertension 53, 442–445. doi: 10.1161/HYPERTENSIONAHA.108.120303
Sapkota, S., Vergote, D., Westaway, D., Jhamandas, J., and Dixon, R. A. (2015). Synergistic associations of catechol-O-methyltransferase and brain-derived neurotrophic factor with executive function in aging are selective and modified by apolipoprotein E. Neurobiol. Aging 36, 249–256. doi: 10.1016/j.neurobiolaging.2014.06.020
Sattar, N., Gaw, A., Scherbakova, O., Ford, I., O'Reilly, D. S., Haffner, S. M., et al. (2003). Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Circulation 108, 414–419. doi: 10.1161/01.CIR.0000080897.52664.94
Schneeberger, M., Altirriba, J., García, A., Esteban, Y., Castaño, C., García-Lavandeira, M., et al. (2012). Deletion of miRNA processing enzyme Dicer in POMC-expressing cells leads to pituitary dysfunction, neurodegeneration and development of obesity. Mol. Metab. 2, 74–85. doi: 10.1016/j.molmet.2012.10.001
Schommer, N. C., Hellhammer, D. H., and Kirschbaum, C. (2003). Dissociation between reactivity of the hypothalamus-pituitary-adrenal axis and the sympathetic-adrenal-medullary system to repeated psychosocial stress. Psychosom. Med. 65, 450–460. doi: 10.1097/01.PSY.0000035721.12441.17
Schwenk, R. W., Jonas, W., Ernst, S. B., Kammel, A., Jähnert, M., and Schürmann, A. (2013). Diet-dependent alterations of hepatic Scd1 expression are accompanied by differences in promoter methylation. Horm. Metab. Res. 45, 786–794. doi: 10.1055/s-0033-1348263
Seckl, J. R., and Walker, B. R. (2001). Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 142, 1371–1376. doi: 10.1210/endo.142.4.8114
Seres, J., Bornstein, S. R., Seres, P., Willenberg, H. S., Schulte, K. M., Scherbaum, W. A., et al. (2004). Corticotropin-releasing hormone system in human adipose tissue. J. Clin. Endocrinol. Metab. 89, 965–970. doi: 10.1210/jc.2003-031299
Singh, P., Sharma, P., Sahakyan, K. R., Davison, D. E., Sert-Kuniyoshi, F. H., Romero-Corral, A., et al. (2016). Differential effects of leptin on adiponectin expression with weight gain versus obesity. Int. J. Obes. (Lond.) 40, 266–274. doi: 10.1038/ijo.2015.181
Sitticharoon, C., Chatree, S., and Churintaraphan, M. (2013). Expressions of neuropeptide Y and Y1 receptor in subcutaneous and visceral fat tissues in normal weight and obese humans and their correlations with clinical parameters and peripheral metabolic factors. Regul. Pept. 185, 65–72. doi: 10.1016/j.regpep.2013.06.015
Sitticharoon, C., Nway, N. C., Chatree, S., Churintaraphan, M., Boonpuan, P., and Maikaew, P. (2014). Interactions between adiponectin, visfatin, and omentin in subcutaneous and visceral adipose tissues and serum, and correlations with clinical and peripheral metabolic factors. Peptides 62, 164–175. doi: 10.1016/j.peptides.2014.10.006
Song, G., Xu, G., Ji, C., Shi, C., Shen, Y., Chen, L., et al. (2014). The role of microRNA-26b in human adipocyte differentiation and proliferation. Gene 533, 481–487. doi: 10.1016/j.gene.2013.10.011
Song, J. Y., Wang, D., Ma, J., and Wang, H. J. (2015). [Mutation screening and function prediction of melanocortin-4 receptor gene in obese children]. Zhongguo Dang Dai Er Ke Za Zhi 17, 356–361.
Song, J., Kang, S. M., Kim, E., Kim, C. H., Song, H. T., and Lee, J. E. (2015). Adiponectin receptor-mediated signaling ameliorates cerebral cell damage and regulates the neurogenesis of neural stem cells at high glucose concentrations: an in vivo and in vitro study. Cell Death Dis. 6, e1844. doi: 10.1038/cddis.2015.220
Sousa, N. (2016). The dynamics of the stress neuromatrix. Mol. Psychiatry 21, 302–312. doi: 10.1038/mp.2015.196
Sözen, M. A., de Jonge, L. H., Greenway, F., Ravussin, E., Smith, S. R., and Argyropoulos, G. (2007). A rare mutation in AgRP, +79G>A, affects promoter activity. Eur. J. Clin. Nutr. 61, 809–812. doi: 10.1038/sj.ejcn.1602585
Stalder, T., Kirschbaum, C., Alexander, N., Bornstein, S. R., Gao, W., Miller, R., et al. (2013). Cortisol in hair and the metabolic syndrome. J. Clin. Endocrinol. Metab. 98, 2573–2580. doi: 10.1210/jc.2013-1056
Statsenko, M. E., Turkina, S. V., and Kosivtseva, M. A. (2013). [Additional advantages of mexicor used in combined therapy of coronary heat disease and diabetes mellitus of 2nd type]. Klin. Med. (Mosk.) 91, 59–64.
Stimson, R. H., and Walker, B. R. (2013). The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 15, 37–48. doi: 10.1515/hmbci-2013-0015
Stout, S. A., Espel, E. V., Sandman, C. A., Glynn, L. M., and Davis, E. P. (2015). Fetal programming of children's obesity risk. Psychoneuroendocrinology 53, 29–39. doi: 10.1016/j.psyneuen.2014.12.009
Straznicky, N. E., Eikelis, N., Lambert, E. A., and Esler, M. D. (2008). Mediators of sympathetic activation in metabolic syndrome obesity. Curr. Hypertens. Rep. 10, 440–447. doi: 10.1007/s11906-008-0083-1
Straznicky, N. E., Lambert, G. W., Masuo, K., Dawood, T., Eikelis, N., Nestel, P. J., et al. (2009). Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am. J. Clin. Nutr. 89, 27–36. doi: 10.3945/ajcn.2008.26299
Stuber, G. D., and Wise, R. A. (2016). Lateral hypothalamic circuits for feeding and reward. Nat. Neurosci. 19, 198–205. doi: 10.1038/nn.4220
Stumvoll, M., Goldstein, B. J., and van Haeften, T. W. (2005). Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365, 1333–1346. doi: 10.1016/S0140-6736(05)61032-X
Subbannayya, T., Balakrishnan, L., Sudarshan, G., Advani, J., Kumar, S., Mahmood, R., et al. (2013). An integrated map of corticotropin-releasing hormone signaling pathway. J. Cell Commun. Signal. 7, 295–300. doi: 10.1007/s12079-013-0197-3
Sun, S., Ji, Y., Kersten, S., and Qi, L. (2012). Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutr. 32, 261–286. doi: 10.1146/annurev-nutr-071811-150623
Sutton, G. M., Begriche, K., Kumar, K. G., Gimble, J. M., Perez-Tilve, D., Nogueiras, R., et al. (2010). Central nervous system melanocortin-3 receptors are required for synchronizing metabolism during entrainment to restricted feeding during the light cycle. FASEB J. 24, 862–872. doi: 10.1096/fj.09-142000
Svetkey, L. P., Weinberger, M. H., Gavras, H., Gavras, I., Brown, T. S., Deterding, J., et al. (1987). Double-blind, placebo-controlled trial of twice-daily nifedipine as a step-2 agent in mild essential hypertension. J. Clin. Hypertens. 3, 579–588.
Szczepanska-Sadowska, E., Cudnoch-Jedrzejewska, A., Ufnal, M., and Zera, T. (2010). Brain and cardiovascular diseases: common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J. Physiol. Pharmacol. 61, 509–521.
Taddei, S., and Grassi, G. (2005). Angiotensin II as the link between nitric oxide and neuroadrenergic function. J. Hypertens. 23, 935–937. doi: 10.1097/01.hjh.0000166831.48065.06
Takaya, J., Iharada, A., Okihana, H., and Kaneko, K. (2013). A calcium-deficient diet in pregnant, nursing rats induces hypomethylation of specific cytosines in the 11beta-hydroxysteroid dehydrogenase-1 promoter in pup liver. Nutr. Res. 33, 961–970. doi: 10.1016/j.nutres.2013.07.015
Tallam, L. S., da Silva, A. A., and Hall, J. E. (2006). Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension 48, 58–64. doi: 10.1161/01.HYP.0000227966.36744.d9
Tallam, L. S., Stec, D. E., Willis, M. A., da Silva, A. A., and Hall, J. E. (2005). Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension 46, 326–332. doi: 10.1161/01.HYP.0000175474.99326.bf
Tan, J., McKenzie, C., Potamitis, M., Thorburn, A. N., Mackay, C. R., and Macia, L. (2014). The role of short-chain fatty acids in health and disease. Adv. Immunol. 121, 91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
Tellioglu, T., and Robertson, D. (2001). Genetic or acquired deficits in the norepinephrine transporter: current understanding of clinical implications. Expert Rev. Mol. Med. 2001, 1–10. doi: 10.1017/s1462399401003878
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713. doi: 10.1038/nature09270
Thorleifsson, G., Walters, G. B., Gudbjartsson, D. F., Steinthorsdottir, V., Sulem, P., Helgadottir, A., et al. (2009). Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24. doi: 10.1038/ng.274
Thorp, A. A., and Schlaich, M. P. (2015). Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J. Diabetes Res. 2015:341583. doi: 10.1155/2015/341583
Thorsell, A. (2010). Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp. Biol. Med. (Maywood) 235, 1163–1167. doi: 10.1258/ebm.2010.009331
Thundyil, J., Pavlovski, D., Sobey, C. G., and Arumugam, T. V. (2012). Adiponectin receptor signalling in the brain. Br. J. Pharmacol. 165, 313–327. doi: 10.1111/j.1476-5381.2011.01560.x
Tilg, H., and Moschen, A. R. (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6, 772–783. doi: 10.1038/nri1937
Toperoff, G., Kark, J. D., Aran, D., Nassar, H., Ahmad, W. A., Sinnreich, R., et al. (2015). Premature aging of leukocyte DNA methylation is associated with type 2 diabetes prevalence. Clin. Epigenetics 7, 35. doi: 10.1186/s13148-015-0069-1
Touvier, T., Conte-Auriol, F., Briand, O., Cudejko, C., Paumelle, R., Caron, S., et al. (2009). LEPROT and LEPROTL1 cooperatively decrease hepatic growth hormone action in mice. J. Clin. Invest. 119, 3830–3838. doi: 10.1172/JCI34997
Tracey, K. J. (2007). Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Invest. 117, 289–296. doi: 10.1172/JCI30555
Tracey, K. J. (2009). Reflex control of immunity. Nat. Rev. Immunol. 9, 418–428. doi: 10.1038/nri2566
Trakhtenberg, E. F., and Goldberg, J. L. (2011). Neuroimmune communication. Science 334, 47–48. doi: 10.1126/science.1213099
Turek, F. W., Joshu, C., Kohsaka, A., Lin, E., Ivanova, G., McDearmon, E., et al. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045. doi: 10.1126/science.1108750
van Dijk, S. J., Molloy, P. L., Varinli, H., Morrison, J. L., Muhlhausler, B. S., and Members of Epi, S. (2015). Epigenetics and human obesity. Int. J. Obes. (Lond.) 39, 85–97. doi: 10.1038/ijo.2014.34
van Himbergen, T. M., Beiser, A. S., Ai, M., Seshadri, S., Otokozawa, S., Au, R., et al. (2012). Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: results from the Framingham Heart Study. Arch. Neurol. 69, 594–600. doi: 10.1001/archneurol.2011.670
Van Kaer, L., Parekh, V. V., and Wu, L. (2013). Invariant natural killer T cells as sensors and managers of inflammation. Trends Immunol. 34, 50–58. doi: 10.1016/j.it.2012.08.009
Van Leusden, J. W., Sellaro, R., and Colzato, L. S. (2015). Transcutaneous Vagal Nerve Stimulation (tVNS): a new neuromodulation tool in healthy humans? Front. Psychol. 6:102. doi: 10.3389/fpsyg.2015.00102
Vink, T., Hinney, A., van Elburg, A. A., van Goozen, S. H., Sandkuijl, L. A., Sinke, R. J., et al. (2001). Association between an agouti-related protein gene polymorphism and anorexia nervosa. Mol. Psychiatry 6, 325–328. doi: 10.1038/sj.mp.4000854
Vinnikov, I. A., Hajdukiewicz, K., Reymann, J., Beneke, J., Czajkowski, R., Roth, L. C., et al. (2014). Hypothalamic miR-103 protects from hyperphagic obesity in mice. J. Neurosci. 34, 10659–10674. doi: 10.1523/JNEUROSCI.4251-13.2014
Vogelzangs, N., Suthers, K., Ferrucci, L., Simonsick, E. M., Ble, A., Schrager, M., et al. (2007). Hypercortisolemic depression is associated with the metabolic syndrome in late-life. Psychoneuroendocrinology 32, 151–159. doi: 10.1016/j.psyneuen.2006.11.009
Voisin, S., Almén, M. S., Zheleznyakova, G. Y., Lundberg, L., Zarei, S., Castillo, S., et al. (2015). Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 7, 103. doi: 10.1186/s13073-015-0225-4
Volek, J. S., and Feinman, R. D. (2005). Carbohydrate restriction improves the features of metabolic syndrome. metabolic syndrome may be defined by the response to carbohydrate restriction. Nutr. Metab. (Lond.) 2:31. doi: 10.1186/1743-7075-2-31
Volek, J. S., Sharman, M. J., and Forsythe, C. E. (2005). Modification of lipoproteins by very low-carbohydrate diets. J. Nutr. 135, 1339–1342.
Wabitsch, M., Funcke, J. B., Lennerz, B., Kuhnle-Krahl, U., Lahr, G., Debatin, K. M., et al. (2015). Biologically inactive leptin and early-onset extreme obesity. N. Engl. J. Med. 372, 48–54. doi: 10.1056/NEJMoa1406653
Wake, D. J., and Walker, B. R. (2006). Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 in obesity. Endocrine 29, 101–108. doi: 10.1385/ENDO:29:1:101
Wake, D. J., Homer, N. Z., Andrew, R., and Walker, B. R. (2006). Acute in vivo regulation of 11beta-hydroxysteroid dehydrogenase type 1 activity by insulin and intralipid infusions in humans. J. Clin. Endocrinol. Metab. 91, 4682–4688. doi: 10.1210/jc.2006-0819
Walley, A. J., Asher, J. E., and Froguel, P. (2009). The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet. 10, 431–442. doi: 10.1038/nrg2594
Wang, L., Opland, D., Tsai, S., Luk, C. T., Schroer, S. A., Allison, M. B., et al. (2014). Pten deletion in RIP-Cre neurons protects against type 2 diabetes by activating the anti-inflammatory reflex. Nat. Med. 20, 484–492. doi: 10.1038/nm.3527
Wang, L., Rao, F., Zhang, K., Mahata, M., Rodriguez-Flores, J. L., Fung, M. M., et al. (2009). Neuropeptide Y(1) Receptor NPY1R discovery of naturally occurring human genetic variants governing gene expression in cella as well as pleiotropic effects on autonomic activity and blood pressure in vivo. J. Am. Coll. Cardiol. 54, 944–954. doi: 10.1016/j.jacc.2009.05.035
Wang, M. (2005). The role of glucocorticoid action in the pathophysiology of the Metabolic Syndrome. Nutr. Metab. (Lond.) 2:3. doi: 10.1186/1743-7075-2-3
Wang, X., Lacza, Z., Sun, Y. E., and Han, W. (2014). Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia 57, 236–245. doi: 10.1007/s00125-013-3072-0
Wegner, M., Neddermann, D., Piorunska-Stolzmann, M., and Jagodzinski, P. P. (2014). Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res. Clin. Pract. 105, 164–175. doi: 10.1016/j.diabres.2014.03.019
Wei, S. G., Zhang, Z. H., Beltz, T. G., Yu, Y., Johnson, A. K., and Felder, R. B. (2013). Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62, 118–125. doi: 10.1161/HYPERTENSIONAHA.113.01404
Wei, Z., Zhang, K., Wen, G., Balasubramanian, K., Shih, P. A., Rao, F., et al. (2013). Heredity and cardiometabolic risk: naturally occurring polymorphisms in the human neuropeptide Y(2) receptor promoter disrupt multiple transcriptional response motifs. J. Hypertens. 31, 123–133. doi: 10.1097/HJH.0b013e32835b053d
Westman, E. C., Feinman, R. D., Mavropoulos, J. C., Vernon, M. C., Volek, J. S., Wortman, J. A., et al. (2007). Low-carbohydrate nutrition and metabolism. Am. J. Clin. Nutr. 86, 276–284.
Willer, C. J., Speliotes, E. K., Loos, R. J., Li, S., Lindgren, C. M., Heid, I. M., et al. (2009). Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 41, 25–34. doi: 10.1038/ng.287
Winer, D. A., Luck, H., Tsai, S., and Winer, S. (2016). The intestinal immune system in obesity and insulin resistance. Cell Metab. 23, 413–426. doi: 10.1016/j.cmet.2016.01.003
Wolf, E. J., Mitchell, K. S., Logue, M. W., Baldwin, C. T., Reardon, A. F., Humphries, D. E., et al. (2013). Corticotropin releasing hormone receptor 2 (CRHR-2) gene is associated with decreased risk and severity of posttraumatic stress disorder in women. Depress. Anxiety 30, 1161–1169. doi: 10.1002/da.22176
Wu, Y., Patchev, A. V., Daniel, G., Almeida, O. F., and Spengler, D. (2014). Early-life stress reduces DNA methylation of the Pomc gene in male mice. Endocrinology 155, 1751–1762. doi: 10.1210/en.2013-1868
Xiao, E., Xia-Zhang, L., Vulliémoz, N. R., Ferin, M., and Wardlaw, S. L. (2003). Agouti-related protein stimulates the hypothalamic-pituitary-adrenal (HPA) axis and enhances the HPA response to interleukin-1 in the primate. Endocrinology 144, 1736–1741. doi: 10.1210/en.2002-220013
Yamauchi, T., Kamon, J., Ito, Y., Tsuchida, A., Yokomizo, T., Kita, S., et al. (2003). Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769. doi: 10.1038/nature01705
Yang, M., Xu, Y., Liang, L., Fu, J., Xiong, F., Liu, G., et al. (2014). The effects of genetic variation in FTO rs9939609 on obesity and dietary preferences in Chinese Han children and adolescents. PLoS ONE 9:e104574. doi: 10.1371/journal.pone.0104574
Young, C. N., and Davisson, R. L. (2015). Angiotensin-II, the brain, and hypertension: an update. Hypertension 66, 920–926. doi: 10.1161/HYPERTENSIONAHA.115.03624
Young, C. N., Li, A., Dong, F. N., Horwath, J. A., Clark, C. G., and Davisson, R. L. (2015). Endoplasmic reticulum and oxidant stress mediate nuclear factor-kappaB activation in the subfornical organ during angiotensin II hypertension. Am. J. Physiol. Cell Physiol. 308, C803–C812. doi: 10.1152/ajpcell.00223.2014
Young, C. N., Morgan, D. A., Butler, S. D., Mark, A. L., and Davisson, R. L. (2013). The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension 61, 737–744. doi: 10.1161/HYPERTENSIONAHA.111.00405
Yuan, B., and Teng, J. F. (2014). Association between adiponectin receptor 2 gene polymorphisms and cerebral infarction. Genet. Mol. Res. 13, 7808–7814. doi: 10.4238/2014.September.26.19
Zeng, W., Pirzgalska, R. M., Pereira, M. M., Kubasova, N., Barateiro, A., Seixas, E., et al. (2015). Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 163, 84–94. doi: 10.1016/j.cell.2015.08.055
Zhang, J. V., Ren, P. G., Avsian-Kretchmer, O., Luo, C. W., Rauch, R., Klein, C., et al. (2005). Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science 310, 996–999. doi: 10.1126/science.1117255
Zhang, N., Lin, J. K., Chen, J., Liu, X. F., Liu, J. L., Luo, H. S., et al. (2013). MicroRNA 375 mediates the signaling pathway of corticotropin-releasing factor (CRF) regulating pro-opiomelanocortin (POMC) expression by targeting mitogen-activated protein kinase 8. J. Biol. Chem. 288, 10361–10373. doi: 10.1074/jbc.M112.425504
Zhang, X., Yang, R., Jia, Y., Cai, D., Zhou, B., Qu, X., et al. (2014). Hypermethylation of Sp1 binding site suppresses hypothalamic POMC in neonates and may contribute to metabolic disorders in adults: impact of maternal dietary CLAs. Diabetes 63, 1475–1487. doi: 10.2337/db13-1221
Zhang, Z. H., Wei, S. G., Francis, J., and Felder, R. B. (2003). Cardiovascular and renal sympathetic activation by blood-borne TNF-alpha in rat: the role of central prostaglandins. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R916–R927. doi: 10.1152/ajpregu.00406.2002
Zheng, J., Xiao, X., Zhang, Q., Yu, M., Xu, J., Wang, Z., et al. (2015). Maternal and post-weaning high-fat, high-sucrose diet modulates glucose homeostasis and hypothalamic POMC promoter methylation in mouse offspring. Metab. Brain Dis. 30, 1129–1137. doi: 10.1007/s11011-015-9678-9
Zhou, J., Xu, L., Huang, R. S., Huang, Y., Le, Y., Jiang, D., et al. (2013). Apolipoprotein A5 gene variants and the risk of coronary heart disease: a casecontrol study and metaanalysis. Mol. Med. Rep. 8, 1175–1182. doi: 10.3892/mmr.2013.1642
Ziegler, M. G., Elayan, H., Milic, M., Sun, P., and Gharaibeh, M. (2012). Epinephrine and the metabolic syndrome. Curr. Hypertens. Rep. 14, 1–7. doi: 10.1007/s11906-011-0243-6
Keywords: metabolic syndrome, sympathetic autonomic nervous system, stress neuropsychobiology, hypothalamic-pituitary adrenocortical axis, epigenetic programming, gene regulation, microRNA, pathophysiology
Citation: Lemche E, Chaban OS and Lemche AV (2016) Neuroendocrinological and Epigenetic Mechanisms Subserving Autonomic Imbalance and HPA Dysfunction in the Metabolic Syndrome. Front. Neurosci. 10:142. doi: 10.3389/fnins.2016.00142
Received: 15 December 2015; Accepted: 21 March 2016;
Published: 14 April 2016.
Edited by:
Yvette France Taché, University of California, Los Angeles, USAReviewed by:
Bruno Bonaz, Grenoble Faculty of Medicine and Hospital, FranceDe-Pei Li, University of Texas MD Anderson Cancer Center, USA
Copyright © 2016 Lemche, Chaban and Lemche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erwin Lemche, ZXJ3aW4ubGVtY2hlQGtjbC5hYy51aw==