 Ester Aso1,2*
Ester Aso1,2* Isidro Ferrer1,2*
Isidro Ferrer1,2*- 1Institut de Neuropatologia, Servei d'Anatomia Patològica, Bellvitge Biomedical Research Institute (IDIBELL)-Hospital Universitari de Bellvitge, Universitat de Barcelona, L'Hospitalet de Llobregat, Spain
- 2CIBERNED - Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas, Instituto Carlos III, Madrid, Spain
The CB2 receptor is one of the components of the endogenous cannabinoid system, a complex network of signaling molecules and receptors involved in the homeostatic control of several physiological functions. Accumulated evidence suggests a role for CB2 receptors in Alzheimer's disease (AD) and indicates their potential as a therapeutic target against this neurodegenerative disease. Levels of CB2 receptors are significantly increased in post-mortem AD brains, mainly in microglia surrounding senile plaques, and their expression levels correlate with the amounts of Aβ42 and β-amyloid plaque deposition. Moreover, several studies on animal models of AD have demonstrated that specific CB2 receptor agonists, which are devoid of psychoactive effects, reduce AD-like pathology, resulting in attenuation of the inflammation associated with the disease but also modulating Aβ and tau aberrant processing, among other effects. CB2 receptor activation also improves cognitive impairment in animal models of AD. This review discusses available data regarding the role of CB2 receptors in AD and the potential usefulness of specific agonists of these receptors against AD.
Overview of Alzheimer's Disease
Alzheimer's disease (AD) is an age-dependent neurodegenerative disorder characterized by slowly progressive cognitive decline with fatal outcome. To date, no effective treatment is available. Dementia due to AD occurs in one in nine people aged 65 and in about one in four at the age of 85 (Hebert et al., 2013). Prevalence is expected to grow in coming decades as the size and proportion of the older population continue to increase due to the rise in life expectancy in developed countries (Hebert et al., 2013).
AD is morphologically distinguished by the presence in the brain of senile plaques, mainly composed of different species of fibrillar β-amyloid (Aβ) produced by the cleavage of the β-amyloid precursor protein (APP), and neurofibrillary tangles composed of various isoforms of hyper-phosphorylated and truncated tau protein. Senile plaques are surrounded by dystrophic neurites, reactive astrocytes, and microglia. Neurofibrillary tangles first appear in selected nuclei of the brain stem, and entorhinal and transentorhinal cortex, and then progress to the hippocampus and limbic system, and finally to most of the telencephalon (Braak and Braak, 1991). The development and progression of senile plaques does not parallel the evolution of tau pathology in sporadic AD (Thal et al., 2002). Aβ and tau misfolded proteins compromise neural activity due to an increase in toxic function and/or loss of their normal function, thus contributing to the decline of neuronal organization manifested as synaptic dysfunction and neuronal death (Duyckaerts and Dickson, 2011; Ferrer, 2012). A self-propagating process of misfolded proteins has been suggested to explain disease progression (Jucker and Walker, 2013). Aβ and misfolded tau aggregate into seeds that are able to modify native proteins causing them to aggregate and to form pathogenic assemblies in a prion-like way (Meyer-Luehmann et al., 2006; Clavaguera et al., 2009; Stöhr et al., 2012).
It is important to note that AD-related pathology begins more than 20 years before the onset of dementia. First stages of AD in which lesions are restricted to the brain stem and inner parts of the temporal lobe are usually asymptomatic. About 80% of individuals aged 65 present senile plaques and/or neurofibrillary tangles in specific brain areas but only about 5% of them suffer from dementia. This is an important point as AD-related pathology is common in the elderly but this does not inevitably lead to dementia. AD changes restricted to the inner temporal lobe can progress slowly and be well tolerated in some individuals. Only the accumulation of lesions in certain individuals determines a progression of the neurodegenerative disease, which leads to dementia once reached determinate threshold (Ferrer, 2012). The slow progression of the neurodegenerative process visualizes a putative temporal window for therapeutic intervention. However, to date most therapeutic interventions aimed at modifying a single pathological factor (e.g., cholinergic dysfunction, inflammation, Aβ and/or tau aberrant processing) have failed because of their limited benefit or for safety reasons (Scheltens et al., 2016). Considering that multiple alterations are concomitant to Aβ and tau aberrant processing in AD (Ferrer, 2012), compounds with pleiotropic activity which will target in parallel several processes that play key roles in AD are expected to yield greater benefits than those obtained by current therapies (Bolognesi et al., 2009; Frautschy and Cole, 2010). Inflammation, mitochondrial dysfunction, oxidative stress, and impaired function of degradation pathways are the most prominent concomitant pathological events (Keller et al., 2000; Ferrer, 2009; Sultana and Butterfield, 2010; López-González et al., 2015), as briefly described in the following paragraphs. These alterations are potential targets of therapeutic intervention.
Inflammation has been proposed as a key factor in the pathogenesis of AD. This is characterized by microglial activation, reactive astrocytes and elevated expression of cytokines and mediators of the inflammatory response. It has been proposed that microglial activation in AD can have beneficial and detrimental effects depending on the stage of the disease. Thus, the acute microglial reaction aims at removing the abnormal protein aggregates appearing at the early stages of the disease. However, cumulative formation of aberrant protein aggregates drives to chronic inflammation which has detrimental consequences due to the sustained exposure to chemokines, cytokines and other inflammatory mediators (Heneka et al., 2015). Conversion of microglia from detrimental (M1) to beneficial (M2) phenotype may be achieved by modulation of pro-inflammatory signaling pathways such as the NLRP3 inflammasome (Heneka et al., 2013). Similar to microglial cells, astrocytes contribute to inflammation in AD by releasing cytokines, interleukins, nitric oxide (NO), and other toxic molecules in response to Aβ exposure at the time they also participate in the internalization and degradation of Aβ (Heneka et al., 2015). However, anti-inflammatory treatments failed to produce beneficial effects in patients with severe cognitive impairment and dementia. This fact is probably due to the fact that inflammatory responses in AD differ not only depending on the stage of the disease but also on the region involved (López-González et al., 2015). That means that inflammatory responses in some regions have a beneficial phenotype whereas they have a deleterious phenotype in other regions in the same individual, thus stressing the need to identify new regulators or modulators of the inflammatory response that can be adapted to specific molecular targets (López-González et al., 2015).
Altered mitochondria are also key factors in the pathogenesis of AD. This includes impaired energy metabolism and increased production of free radicals with subsequent oxidative and nitrosative damage affecting lipids, proteins and nucleic acids (Sultana and Butterfield, 2010). These alterations are already observed in the entorhinal cortex at early stages of AD ultimately leading to neuron exhaustion (Ferrer, 2009). Several studies in AD transgenic mouse models support the potential beneficial effect of compounds targeting mitochondrial dysfunction although the clinical benefit of such drugs in humans is still not known (Onyango et al., 2016).
Finally, another prominent concomitant pathological event in AD is impaired function of degradation pathways (Keller et al., 2000). Oxidative damage and some other pathological events may alter protein structure and function in AD. These modified proteins have to be removed to prevent their toxic accumulation. However, the ubiquitin-proteasome system and autophagy mechanisms are impaired due to the toxic effects of Aβ and oxidative stress damage thus leading to the accumulation of oxidized/unfolded proteins that may contribute to neuronal loss (Tramutola et al., 2016).
Endogenous Cannabinoid System: A Role in Neurodegenerative Diseases
Among the candidates to fulfill the requirements for novel effective multi-target therapies against neurodegenerative diseases are newly emerging compounds that target the endogenous cannabinoid system (ECS; Aso and Ferrer, 2014; Fagan and Campbell, 2014; Fernández-Ruiz et al., 2015). Interest in the ECS derives from the pleiotropic activity of this complex network of lipid molecules and receptors, which is involved in homeostatic control of several physiological functions in brain and other organs (Iannotti et al., 2016). The ECS is composed of (i) at least two subtypes of cannabinoid Gi∕o-coupled receptors, CB1 and CB2 (Pertwee et al., 2010), (ii) certain endogenous ligands, mainly arachidonoylethanolamine or anandamide (AEA) and 2-arachidonoylglycerol (2-AG) derived from the membrane phospholipids (Pertwee, 2015), (iii) several enzymes responsible for endocannabinoid biosynthesis and metabolism (Ligresti et al., 2005), and (iv) molecules linked to the cellular uptake and transport of certain endocannabinoids (Fowler, 2013). CB1 receptors are the most abundant cannabinoid receptors and are located in brain, mainly in neurons but also in glial cells, and in peripheral tissues (Hu and Mackie, 2015). CB1 activity regulates important brain functions including cognition and memory, emotion, motor control, feeding, and pain perception, by modulating excitatory and inhibitory neurotransmission (Wilson and Nicoll, 2002; Howlett, 2005). Moreover, CB1 receptors mediate psychoactive effects of cannabis derivatives (Maldonado et al., 2011). In contrast, activation of CB2 receptors is not accompanied by psychoactive effects (Buckley et al., 2000). CB2 was initially considered a peripheral cannabinoid receptor because in situ hybridization analysis revealed high levels of CB2 mRNA in spleen but levels below the detection thresholds in brain. CB2 receptors were demonstrated to modulate immune cell migration and the release of cytokines in cells of the immune system (Cabral and Griffin-Thomas, 2009). However, more recent findings have shown that CB2 receptors are also present in other tissues including the central nervous system (Atwood and Mackie, 2010). CB2 receptors are highly inducible and under certain conditions are expressed in brain, mainly by microglia, with levels increasing as these immune cells are activated. CB2 modulates microglial migration and infiltration into brain areas with active neuroinflammation and degeneration (Walter et al., 2003; Fernández-Ruiz et al., 2008). Moreover, CB2 receptors are also present at detectable and functional levels in a subset of neurons with increasing expression levels following injury (Atwood and Mackie, 2010). Apart from the regulation of inflammatory processes, some experimental designs also suggest that CB2 receptors may play a role in nociception (Jhaveri et al., 2007; Whiteside et al., 2007), gastrointestinal function (Wright et al., 2008), neural progenitor cell proliferation and axon guidance (Palazuelos et al., 2012; Duff et al., 2013), and synaptic transmission (Kim and Li, 2015; Li and Kim, 2016), among other functions. Most of the evidence comes from pharmacological studies using specific CB2 agonists and antagonists, and from genetically manipulated mice. However, the location of CB2 receptors mediating such effects is not conclusively documented.
As mentioned before, the ECS has a pleiotropic activity and is able to modulate several alterations occurring during normal and pathological aging, including protein misfolding, inflammation, excitotoxicity, mitochondrial dysfunction, and oxidative stress (Bilkei-Gorzo, 2012; Aso and Ferrer, 2014; Fagan and Campbell, 2014; Fernández-Ruiz et al., 2015). Evidence about the role of ECS on aging derives from observations in genetic models. Thus, deficiency in CB1 receptors contributes to acceleration of aging (Bilkei-Gorzo et al., 2005, 2012) whereas deletion of the endocannabinoid degrading enzyme FAAH enhances age-related microglial activity and concomitant inflammatory responses in brain (Ativie et al., 2015). In contrast, stimulation of certain ECS components produces beneficial effects in experimental models of neurodegenerative diseases (Aso and Ferrer, 2014; Fagan and Campbell, 2014; Fernández-Ruiz et al., 2015). These findings demonstrate a role for ECS in normal and pathological aging that has sustained interest in developing therapies against neurodegenerative diseases based on ECS modulation. Major attention has been focused on the use of cannabinoid agonists, but the psychoactive effects elicited by compounds targeting CB1 receptors have served to limit their potential development in clinical practice. For this reason, the study of specific CB2 agonists which are devoid of psychoactive effects is promising, although detailed clinical evaluation is still needed (Atwood et al., 2012).
CB2 Receptors in AD Brains
A few studies have addressed the analysis of CB2 contents in AD brain but all of them have resulted in similar findings. A significant increase in CB2 receptor levels has been found in post-mortem AD brains mainly expressed in microglia surrounding senile plaques (Benito et al., 2003; Ramírez et al., 2005; Solas et al., 2013). Similarly, enhanced CB2 PET binding has been reported in the brain in an animal model of AD (Savonenko et al., 2015). In addition, CB2-specific staining is also observed in tangle-like bearing neurons and in dystrophic neurites from frontal cortex in AD (Ramírez et al., 2005). Interestingly, expression levels of CB2 receptors correlate with Aβ42 levels and plaque deposition although not with cognitive status (Solas et al., 2013), thus suggesting that these pathogenic events induce CB2 receptor expression. The strong induction of CB2 receptors in affected microglia is therapeutically advantageous since it would permit their selective activation in damaged tissues, thereby minimizing the possibility of deleterious side effects. However, CB2 receptors in AD brain are nitrosylated, probably as a consequence of microglial activation and peroxynitrite radical formation, and this may contribute to the impaired coupling of these receptors to downstream effector signaling molecules (Ramírez et al., 2005). Nevertheless, the functionality of CB2 receptors seems to be at least partially preserved in AD according to the results of pharmacological experiments carried out in AD models, as described in the following sections.
CB2 Receptor as a Therapeutic Target in AD: Evidence from Experimental Models
During the last decade, a number of studies have provided experimental evidence about the potential therapeutic properties of compounds targeting CB2 receptors in cellular and animal models that mimic a variety of AD-related changes. A summary of pharmacological findings supporting this hypothesis is shown in Table 1. Moreover, at least three different genetically manipulated murine models have recently been created to further demonstrate a role for CB2 receptors in this neurodegenerative disease (Table 2). Most of these assays are focused on the potential benefit derived from the well-known anti-inflammatory properties of CB2 agonists, but some of them also reveal the capacity of CB2 receptors to modulate Aβ and hyper-phosphorylated tau levels, among other molecular alterations.
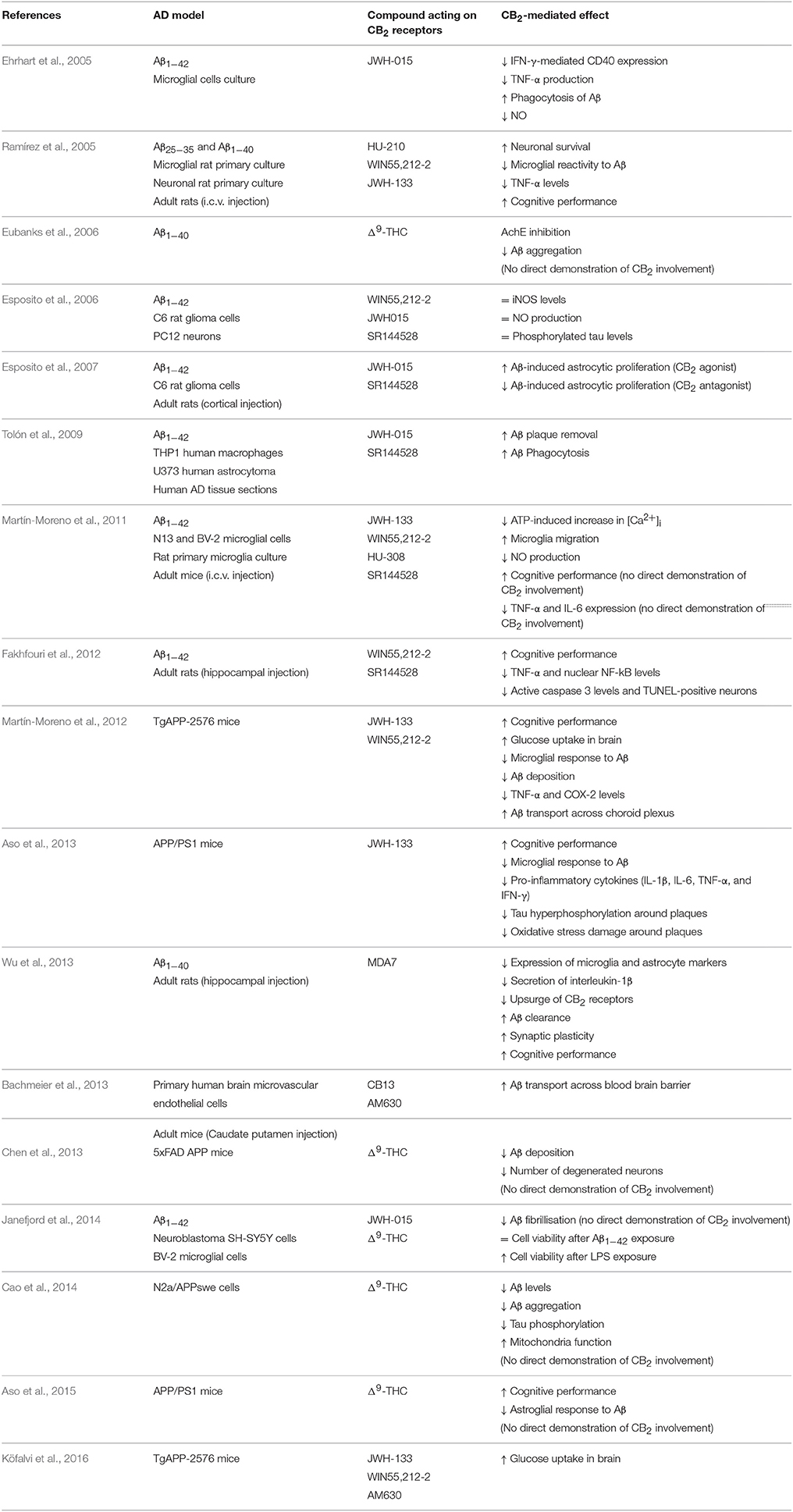
Table 1. Pharmacological evidence of CB2 receptor as a therapeutic target in AD.
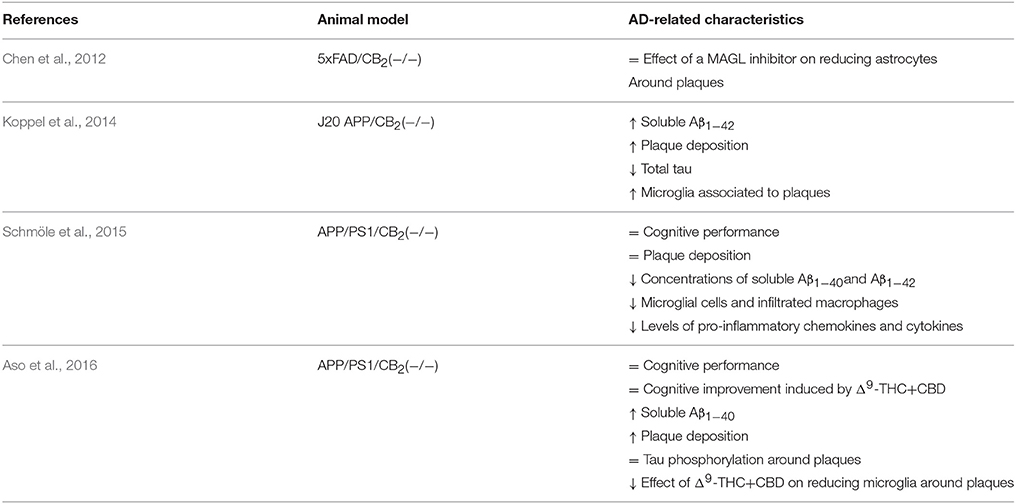
Table 2. Evidence about the role of CB2 receptors in AD obtained from genetically modified mice.
Anti-Inflammatory Effects of CB2 Receptor Activity
Inflammation is common in most neurodegenerative diseases including AD, and it may contribute to progressive neuronal damage. Microglia play a major role in neuroinflammation. Activated microglia produce cytokines and mediators of inflammatory response which, in combination with neurons and astrocytes, create a complex cytokine cycle with deleterious consequences in brain when sustained over time (Heneka et al., 2014; McGeer and McGeer, 2015). CB2 receptors, mainly expressed in microglia, inhibit microglia-mediated neurotoxicity by reducing the production of pro-inflammatory molecules and by modulating macrophage migration in several pathological conditions (Cabral and Griffin-Thomas, 2009). In addition, CB2 activity facilitates the transformation of microglial cells from the M1 to M2 phenotype which is suggested to favor phagocytosis and reparative mechanisms (Mecha et al., 2015). As summarized in Table 1, a number of studies have shown anti-inflammatory effects of CB2 agonists in different models of AD. Thus, in vitro experiments have demonstrated that the selective agonists JWH-015, JWH-133, and HU-308, and the mixed CB1–CB2 receptor agonists WIN55,212-2 and HU-210 reduce the release of pro-inflammatory cytokines in microglial cell cultures exposed to different species of the toxic Aβ peptide (Ehrhart et al., 2005; Ramírez et al., 2005; Martín-Moreno et al., 2011). These findings may be the result of CB2 agonists reducing microglial activation by decreasing intracellular calcium concentration, as demonstrated in microglial cell cultures (Martín-Moreno et al., 2011). Moreover, JWH-133 and WIN55,212-2 promote microglial migration, which facilitates the phagocytosis of aggregated Aβ (Martín-Moreno et al., 2011). CB2 agonist JWH-015 facilitates Aβ-induced astrocytic proliferation in cell culture which participates in the inflammatory process as well (Esposito et al., 2007). These findings have been corroborated in vivo by the administration of selective CB2 and mixed CB1–CB2 receptor agonists to rats and mice inoculated with Aβ into the brain, resulting in reduced levels of several pro-inflammatory cytokines and decreased microglia reactivity to the Aβ insult (Ramírez et al., 2005; Esposito et al., 2007; Martín-Moreno et al., 2011; Fakhfouri et al., 2012; Wu et al., 2013). In some cases, the specificity of CB2-induced effects has been demonstrated by the co-administration of the selective CB2 antagonist SR144528 (Esposito et al., 2007; Martín-Moreno et al., 2011; Fakhfouri et al., 2012). Moreover, transgenic mice bearing APP mutations linked to familial AD exhibit a reduction in the number of activated microglial cells surrounding Aβ deposits and in the levels of pro-inflammatory cytokines after chronic treatment with the selective CB2 receptor agonist JWH-133 (Martín-Moreno et al., 2012; Aso et al., 2013). Considering that systemic inflammation may exacerbate the progression of AD (Lim et al., 2015) and that CB2 receptor is highly expressed in the peripheral immune system (Atwood and Mackie, 2010), it can be speculated that systemic CB2-driven actions may be also beneficial in AD.
Genetic models designed to unravel the role of CB2 receptors in AD progression have produced divergent findings regarding inflammatory responses (Table 2). A significant increase in the number of activated microglia associated with plaques has been reported in J20 APP transgenic AD mice lacking, in addition, the CB2 receptor (Koppel et al., 2014). Knocking down CB2 receptor gene in APP/PS1 mice results in a reduction of microglia reactivity and in the levels of pro-inflammatory chemokines and cytokines (Schmöle et al., 2015; Aso et al., 2016). Inhibition of monoacylglycerol lipase (MAGL), one of the main enzymes responsible for endocannabinoids degradation, results effective at reducing astroglial reaction to amyloid plaques in 5xFAD mice lacking CB2 receptor (Chen et al., 2012). However, the effect induced by the combination of Δ9-THC+CBD is reduced in APP/PS1 mice knockout for CB2 receptor (Aso et al., 2016). Divergent results may be related to the differing genetic backgrounds of mouse models, but in any case they point to a role for CB2 receptors in the control of microglial and inflammatory responses to Aβ insults.
Modulation of Aβ and Hyper-Phosphorylated Tau Processing
A number of studies have proposed a direct role for CB2 receptors in the modulation of Aβ peptide levels in brain. Most of them suggest the participation of CB2 receptors in Aβ clearance rather than in Aβ production and aggregation. In this sense, activation of CB2 receptors with the specific agonist JWH-015 facilitates Aβ phagocytosis by human macrophages in brain sections obtained from AD cases (Tolón et al., 2009) and by microglia in cell culture (Ehrhart et al., 2005). Similarly, MDA7, another potent synthetic CB2 agonist, promotes Aβ clearance in the brains of Aβ-injected rats (Wu et al., 2013). JWH-133 and WIN55,212-2 favor Aβ transport through the choroid plexus in vitro (Martín-Moreno et al., 2012). The facilitation of Aβ clearance across the blood brain barrier has also been demonstrated using the synthetic CB1–CB2 receptor agonist CB13 in in vitro and in vivo models (Bachmeier et al., 2013). These findings may explain, at least in part, the reduction in Aβ levels in APP transgenic mice after chronic treatment with the agonists JWH-133 and WIN55,212-2 (Martín-Moreno et al., 2012). A few reports have also suggested a direct effect of the mixed CB1–CB2 agonist Δ9-THC on the reduction of Aβ aggregation (Eubanks et al., 2006; Cao et al., 2014; Janefjord et al., 2014) and on the promotion of Aβ degradation (Chen et al., 2013). However, demonstration of a direct involvement of CB1 or CB2 receptors is lacking in these studies.
Further evidence of CB2 participation in Aβ processing derives from the study of AD models with genetic deletion of this receptor (Table 2). Two of the three models had increased soluble Aβ levels and increased numbers of amyloid plaques in adult mouse brains (Koppel et al., 2014; Aso et al., 2016). In the case of APP/PS1 mice lacking CB2 receptors, the increased Aβ deposition observed may be related to the reduced microglial reaction in their brains (Aso et al., 2016), considering the role of CB2 activity in promoting microglial-induced Aβ phagocytosis (Ehrhart et al., 2005; Tolón et al., 2009). These observations reinforce the hypothesis that CB2 receptors facilitate Aβ clearance whereas their absence results in greater Aβ accumulation in brain. However, a slight reduction in soluble Aβ and plaque content has been reported in aged AD mice lacking CB2 receptors (Schmöle et al., 2015), suggesting that CB2 receptor participation in Aβ processing may vary along with the progression of the neurodegenerative process.
A role for CB2 receptors in the modulation of tau hyper-phosphorylation has also been proposed. Early studies performed in cell cultures demonstrated that the mixed CB1–CB2 agonist WIN55,212-2 inhibited tau protein hyper-phosphorylation in Aβ-stimulated PC12 neuronal cells, but that this effect was mediated mainly by CB1 receptors (Esposito et al., 2006). Moreover, a specific CB2 agonist failed to modify tau hyper-phosphorylation in the same experimental conditions (Esposito et al., 2006). Δ9-THC, a mixed CB1–CB2 agonist, is able to reduce tau phosphorylation in N2a/APPswe cells (Cao et al., 2014) but no direct evidence has been found about the specific involvement of CB2 receptors in such effect. It is worth noting that in vivo experiments have demonstrated that chronic treatment with the specific CB2 agonist JWH-133 significantly reduces tau hyper-phosphorylation at the Thr181 site in the vicinity of Aβ plaques in APP/PS1 mice (Aso et al., 2013). This effect may be explained by concomitant decreased expression of active forms of GSK3β, p38 and SAPK/JNK in the vicinity of Aβ plaques in JWH-133-treated APP/PS1 mice (Aso et al., 2013). In contrast, no difference in tau hyper-phosphorylation at site Thr181 was observed in APP/PS1 mice lacking CB2 receptors (Aso et al., 2016), suggesting that the activation of these receptors may avoid tau phosphorylation but their absence does not alter the process of tau phosphorylation. Yet J20 APP mice knocked out for the CB2 receptor gene show decreased levels of total tau protein without modifications of its phosphorylation state (Koppel et al., 2014). Considering all these observations, it is clear that the role of CB2 receptors in tau processing requires further investigation, as the available information is variegated and not conclusive.
Other Effects: Neuronal Survival, Anti-Oxidative, Glucose Metabolism, and Cognition
Targeting CB2 receptors produces additional benefits in AD. Thus, CB2 receptor agonists promote cell survival in the face of Aβ insults in in vitro and in vivo models (Ramírez et al., 2005; Fakhfouri et al., 2012; Chen et al., 2013; Janefjord et al., 2014). Moreover, anti-oxidant effects have been reported for compounds activating CB2 receptors. Specifically, two studies have demonstrated that specific CB2 agonists reduce the production of free radical NO induced by Aβ exposure in microglial cell culture (Ehrhart et al., 2005; Martín-Moreno et al., 2011), although these results have not been replicated in a glioma cell line (Esposito et al., 2006). In vivo experiments also show that activation of CB2 receptors reduces oxidative stress damage and promotes anti-oxidative stress responses; chronic treatment with JWH-133 reduces hydroxynonenal adducts derived from lipid peroxidation and enhances the levels of superoxide dismutase 1 and superoxide dismutase 2 in the vicinity of plaques in APP/PS1 mice (Aso et al., 2013). The mechanisms by which CB2 receptors mediate these anti-oxidant effects remain elusive. It has been reported that the CB1–CB2 agonist Δ9-THC improves mitochondrial function (Cao et al., 2014), thus presumably contributing to a reduction in the production of free radicals, but further study is needed to support this hypothesis. Additional benefits of the activation of CB2 receptors in AD may derive from the ability of these receptors to mediate glucose uptake in brain (Martín-Moreno et al., 2012; Köfalvi et al., 2016), which may counteract the well-known glucose metabolism deficit in AD brains (Mosconi et al., 2008; Cohen and Klunk, 2014).
More importantly, CB2 selective and CB1–CB2 mixed agonists prevent memory deficits in Aβ-injected rats and mice after chronic administration (Ramírez et al., 2005; Martín-Moreno et al., 2011; Fakhfouri et al., 2012; Wu et al., 2013) and improve cognitive performance in two different transgenic mouse models of AD (Martín-Moreno et al., 2012; Aso et al., 2013). The mechanisms of action underlying cognitive improvement are assumed to be multiple and likely related mainly to the capacity of CB2 receptors to mitigate the harmful effects of several molecules produced in AD brains. In fact, AD-like mice lacking CB2 receptors display the same cognitive performance as the corresponding transgenic control mice (Schmöle et al., 2015; Aso et al., 2016), suggesting that CB2 receptors may not play a direct role on cognition.
Conclusions and Future Perspectives
Taken together, the experimental observations discussed in the present review indicate that AD induces CB2 receptor expression and that targeting CB2 receptors has beneficial effects in AD. Specifically, CB2 receptor agonists reduce inflammatory responses linked to Aβ production and deposition, facilitate Aβ clearance, increase cell viability in the presence of Aβ, and promote glucose uptake in brain. Moreover, CB2 activity likely reduces tau hyper-phosphorylation and oxidative stress damage caused by Aβ peptides (Figure 1). As a result of the combination of these effects, among others, CB2 receptor agonists induce cognitive improvement in AD models.
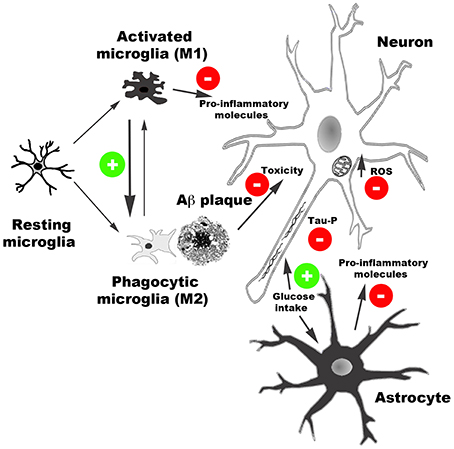
Figure 1. Schematic representation of main effects of CB2 receptor activation reported in AD models. CB2 receptor agonists reduce the release of pro-inflammatory molecules, facilitate Aβ clearance by promoting microglia phagocytic phenotype, reduce Aβ neurotoxicity, and facilitate glucose uptake. Moreover, CB2-mediated activity reduces oxidative stress damage produced by reactive oxidative species (ROS) and tau hyper-phosphorylation.
Considering the evidence of pleiotropic activity and lack of undesirable psychoactive effects of CB2 receptors, compounds acting on such cannabinoid receptors represent a promising therapy against AD. Nevertheless, there is still no information regarding the efficacy or toxicity in human beings of compounds specifically targeting CB2 receptors, which might exhibit some side effects such as immune suppression (Pertwee, 2005). For these reasons, progress toward clinical practice requires further investigation.
Author Contributions
EA and IF contributed equally to writing this review. Both authors give final approval of the text.
Funding
The authors' work is supported by CIBERNED, Institute of Health Carlos III (Spanish Ministry of Economy and Competitiveness) and confunded by FEDER funds/European Regional Development Fund (ERDF) - a way to build Europe (PI14/00757 to IF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank T. Yohannan for editorial assistance.
References
Aso, E., Andrés-Benito, P., Carmona, M., Maldonado, R., and Ferrer, I. (2016). Cannabinoid receptor 2 participates in amyloid-β processing in a mouse model of Alzheimer's disease but plays a minor role in the therapeutic properties of a cannabis-based medicine. J. Alzheimers Dis. 51, 489–500. doi: 10.3233/JAD-150913
Aso, E., and Ferrer, I. (2014). Cannabinoids for treatment of Alzheimer's disease: moving toward the clinic. Front. Pharmacol. 5:37. doi: 10.3389/fphar.2014.00037
Aso, E., Juvés, S., Maldonado, R., and Ferrer, I. (2013). CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AßPP/PS1 mice. J. Alzheimers Dis. 35, 847–858. doi: 10.3233/JAD-130137
Aso, E., Sánchez-Pla, A., Vegas-Lozano, E., Maldonado, R., and Ferrer, I. (2015). Cannabis-based medicine reduces multiple pathological processes in AßPP/PS1 mice. J. Alzheimers Dis. 43, 977–991. doi: 10.3233/JAD-141014
Ativie, F., Albayram, O., Bach, K., Pradier, B., Zimmer, A., and Bilkei-Gorzo, A. (2015). Enhanced microglial activity in FAAH(−/−) animals. Life Sci. 138, 52–56. doi: 10.1016/j.lfs.2014.12.016
Atwood, B. K., and Mackie, K. (2010). CB2: a cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 160, 467–479. doi: 10.1111/j.1476-5381.2010.00729.x
Atwood, B. K., Straiker, A., and Mackie, K. (2012). CB2: therapeutic target-in-waiting. Prog. Neuropsychopharmacol. Biol. Psychiatry 38, 16–20. doi: 10.1016/j.pnpbp.2011.12.001
Bachmeier, C., Beaulieu-Abdelahad, D., Mullan, M., and Paris, D. (2013). Role of the cannabinoid system in the transit of beta-amyloid across the blood-brain barrier. Mol. Cell. Neurosci. 56, 255–262. doi: 10.1016/j.mcn.2013.06.004
Benito, C., Núñez, E., Tolón, R. M., Carrier, E. J., Rábano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer's disease brains. J. Neurosci. 23, 11136–11141.
Bilkei-Gorzo, A., Drews, E., Albayram, Ö., Piyanova, A., Gaffal, E., Tueting, T., et al. (2012). Early onset of aging-like changes is restricted to cognitive abilities and skin structure in Cnr1(−/−) mice. Neurobiol. Aging 33, e11–e22. doi: 10.1016/j.neurobiolaging.2010.07.009
Bilkei-Gorzo, A., Racz, I., Valverde, O., Otto, M., Michel, K., Sastre, M., et al. (2005). Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc. Natl. Acad. Sci. U.S.A. 102, 15670–15675. doi: 10.1073/pnas.0504640102
Bilkei-Gorzo, A. (2012). The endocannabinoid system in normal and pathological brain ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 3326–3341. doi: 10.1098/rstb.2011.0388
Bolognesi, M. L., Matera, R., Minarini, A., Rosini, M., and Melchiorre, C. (2009). Alzheimer's disease: new approaches to drug discovery. Curr. Opin. Chem. Biol. 13, 303–308. doi: 10.1016/j.cbpa.2009.04.619
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Buckley, N. E., McCoy, K. L., Mezey, E., Bonner, T., Zimmer, A., Felder, C. C., et al. (2000). Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur. J. Pharmacol. 396, 141–149. doi: 10.1016/S0014-2999(00)00211-9
Cabral, G. A., and Griffin-Thomas, L. (2009). Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 11:e3. doi: 10.1017/S1462399409000957
Cao, C., Li, Y., Liu, H., Bai, G., Mayl, J., Lin, X., et al. (2014). The potential therapeutic effects of THC on Alzheimer's disease. J. Alzheimers Dis. 42, 973–984. doi: 10.3233/JAD-140093
Chen, R., Zhang, J., Wu, Y., Wang, D., Feng, G., Tang, Y. P., et al. (2012). Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep. 2, 1329–1339. doi: 10.1016/j.celrep.2012.09.030
Chen, R., Zhang, J., Fan, N., Teng, Z. Q., Wu, Y., Yang, H., et al. (2013). Δ(9)-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell 155, 1154–1165. doi: 10.1016/j.cell.2013.10.042
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Cohen, A. D., and Klunk, W. E. (2014). Early detection of Alzheimer's disease using PiB and FDG PET. Neurobiol. Dis. 72, 117–122. doi: 10.1016/j.nbd.2014.05.001
Duff, G., Argaw, A., Cecyre, B., Cherif, H., Tea, N., Zabouri, N., et al. (2013). Cannabinoid receptor CB2 modulates axon guidance. PLoS ONE 8:e70849. doi: 10.1371/journal.pone.0070849
Duyckaerts, C., and Dickson, D. (2011). Neuropathology of Alzheimer's disease and its variants, in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd Edn., eds D. Dickson and R. Weller (West Sussex: Wiley-Blackwell), 62–91.
Ehrhart, J., Obregon, D., Mori, T., Hou, H., Sun, N., Bai, Y., et al. (2005). Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2:29. doi: 10.1186/1742-2094-2-29
Esposito, G., De Filippis, D., Steardo, L., Scuderi, C., Savani, C., Cuomo, V., et al. (2006). CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci. Lett. 404, 342–346. doi: 10.1016/j.neulet.2006.06.012
Esposito, G., Iuvone, T., Savani, C., Scuderi, C., De Filippis, D., Papa, M., et al. (2007). Opposing control of cannabinoid receptor stimulation on amyloid-beta-induced reactive gliosis: in vitro and in vivo evidence. J. Pharmacol. Exp. Ther. 322, 1144–1152. doi: 10.1124/jpet.107.121566
Eubanks, L. M., Rogers, C. J., Beuscher, A. E. IV., Koob, G. F., Olson, A. J., Dickerson, T. J., et al. (2006). A molecular link between the active component of marijuana and Alzheimer's disease pathology. Mol. Pharm. 3, 773–777. doi: 10.1021/mp060066m
Fagan, S. G., and Campbell, V. A. (2014). The influence of cannabinoids on generic traits of neurodegeneration. Br. J. Pharmacol. 171, 1347–1360. doi: 10.1111/bph.12492
Fakhfouri, G., Ahmadiani, A., Rahimian, R., Grolla, A. A., Moradi, F., and Haeri, A. (2012). WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 63, 653–666. doi: 10.1016/j.neuropharm.2012.05.013
Fernández-Ruiz, J., Pazos, M. R., García-Arencibia, M., Sagredo, O., and Ramos, J. A. (2008). Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol. Cell. Endocrinol. 286, S91–S96. doi: 10.1016/j.mce.2008.01.001
Fernández-Ruiz, J., Romero, J., and Ramos, J. A. (2015). Endocannabinoids and neurodegenerative disorders: Parkinson's disease, Huntington's chorea, Alzheimer's disease, and others. Handb. Exp. Pharmacol. 231, 233–259. doi: 10.1007/978-3-319-20825-1_8
Ferrer, I. (2009). Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer's disease. J. Bioenerg. Biomembr. 41, 425–431. doi: 10.1007/s10863-009-9243-5
Ferrer, I. (2012). Defining Alzheimer as a common age-related neurodegenerative process not inevitably leading to dementia. Prog. Neurobiol. 397, 38–51. doi: 10.1016/j.pneurobio.2012.03.005
Fowler, C. J. (2013). Transport of endocannabinoids across the plasma membrane and within the cell. FEBS. J. 280, 1895–1904. doi: 10.1111/febs.12212
Frautschy, S. A., and Cole, G. M. (2010). Why pleiotropic interventions are needed for Alzheimer's disease. Mol. Neurobiol. 41, 392–409. doi: 10.1007/s12035-010-8137-1
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi: 10.1212/WNL.0b013e31828726f5
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Howlett, A. C. (2005). Cannabinoid receptor signaling. Handb. Exp. Pharmacol. 168, 53–79. doi: 10.1007/3-540-26573-2_2
Hu, S. S., and Mackie, K. (2015). Distribution of the endocannabinoid system in the central nervous system. Handb. Exp. Pharmacol. 231, 59–93. doi: 10.1007/978-3-319-20825-1_3
Iannotti, F. A., Di Marzo, V., and Petrosino, S. (2016). Endocannabinoids and endocannabinoid-related mediators: targets, metabolism and role in neurological disorders. Prog. Lipid Res. 62, 107–128. doi: 10.1016/j.plipres.2016.02.002
Janefjord, E., Mååg, J. L., Harvey, B. S., and Smid, S. D. (2014). Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 34, 31–42. doi: 10.1007/s10571-013-9984-x
Jhaveri, M. D., Sagar, D. R., Elmes, S. J., Kendall, D. A., and Chapman, V. (2007). Cannabinoid CB2 receptor-mediated anti-nociception in models of acute and chronic pain. Mol. Neurobiol. 36, 26–35. doi: 10.1007/s12035-007-8007-7
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer's disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Kim, J., and Li, Y. (2015). Chronic activation of CB2 cannabinoid receptors in the hippocampus increases excitatory synaptic transmission. J. Physiol. 593, 871–886. doi: 10.1113/jphysiol.2014.286633
Köfalvi, A., Lemos, C., Martín-Moreno, A. M., Pinheiro, B. S., García-García, L., Pozo, M. A., et al. (2016). Stimulation of brain glucose uptake by cannabinoid CB(2) receptors and its therapeutic potential in Alzheimer's disease. Neuropharmacology. doi: 10.1016/j.neuropharm.2016.03.015. [Epub ahead of print].
Koppel, J., Vingtdeux, V., Marambaud, P., d'Abramo, C., Jimenez, H., Stauber, M., et al. (2014). CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer's disease. Mol. Med. 20, 29–36. doi: 10.2119/molmed.2013.00140
Li, Y., and Kim, J. (2016). Deletion of CB2 cannabinoid receptors reduces synaptic transmission and long-term potentiation in the mouse hippocampus. Hippocampus 26, 275–281. doi: 10.1002/hipo.22558
Ligresti, A., Cascio, M. G., and Di Marzo, V. (2005). Endocannabinoid metabolic pathways and enzymes. Curr. Drug Targets CNS Neurol. Disord. 4, 615–623. doi: 10.2174/156800705774933104
Lim, S. L., Rodriguez-Ortiz, C. J., and Kitazawa, M. (2015). Infection, systemic inflammation, and Alzheimer's disease. Microbes Infect. 17, 549–556. doi: 10.1016/j.micinf.2015.04.004
López-González, I., Schlüter, A., Aso, E., Garcia-Esparcia, P., Ansoleaga, B., Llorens, F., et al. (2015). Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J. Neuropathol. Exp. Neurol. 74, 319–344. doi: 10.1097/NEN.0000000000000176
Maldonado, R., Berrendero, F., Ozaita, A., and Robledo, P. (2011). Neurochemical basis of cannabis addiction. Neuroscience 181, 1–17. doi: 10.1016/j.neuroscience.2011.02.035
Martín-Moreno, A. M., Brera, B., Spuch, C., Carro, E., García-García, L., Delgado, M., et al. (2012). Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 9:8. doi: 10.1186/1742-2094-9-8
Martín-Moreno, A. M., Reigada, D., Ramírez, B. G., Mechoulam, R., Innamorato, N., Cuadrado, A., et al. (2011). Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer's disease. Mol. Pharmacol. 79, 964–973. doi: 10.1124/mol.111.071290
McGeer, P. L., and McGeer, E. G. (2015). Targeting microglia for the treatment of Alzheimer's disease. Expert Opin. Ther. Targets 19, 497–506. doi: 10.1517/14728222.2014.988707
Mecha, M., Feliú, A., Carrillo-Salinas, F. J., Rueda-Zubiaurre, A., Ortega-Gutiérrez, S., de Sola, R. G., et al. (2015). Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 49, 233–245. doi: 10.1016/j.bbi.2015.06.002
Meyer-Luehmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E., et al. (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784. doi: 10.1126/science.1131864
Mosconi, L., Pupi, A., and De Leon, M. J. (2008). Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann. N.Y. Acad. Sci. 1147, 180–195. doi: 10.1196/annals.1427.007
Onyango, I. G., Dennis, J., and Khan, S. M. (2016). Mitochondrial dysfunction in Alzheimer's disease and the rationale for bioenergetics based therapies. Aging Dis. 7, 201–214. doi: 10.14336/AD.2015.1007
Palazuelos, J., Ortega, Z., Díaz-Alonso, J., Guzmán, M., and Galve-Roperh, I. (2012). CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 287, 1198–1209. doi: 10.1074/jbc.M111.291294
Pertwee, R. G., Howlett, A. C., Abood, M. E., Alexander, S. P., Di Marzo, V., Elphick, M. R., et al. (2010). International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol. Rev. 62, 588–631. doi: 10.1124/pr.110.003004
Pertwee, R. G. (2015). Endocannabinoids and their pharmacological actions. Handb. Exp. Pharmacol. 231, 1–37. doi: 10.1007/978-3-319-20825-1_1
Pertwee, R. G. (2005). Pharmacological actions of cannabinoids. Handb. Exp. Pharmacol. 168, 1–51. doi: 10.1007/b137831
Ramírez, B. G., Blázquez, C., Gómez del Pulgar, T., Guzmán, M., and de Ceballos, M. L. (2005). Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J. Neurosci. 25, 1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005
Savonenko, A. V., Melnikova, T., Wang, Y., Ravert, H., Gao, Y., Koppel, J., et al. (2015). Cannabinoid CB2 receptors in a mouse model of Aβ amyloidosis: immunohistochemical analysis and suitability as a PET biomarker of neuroinflammation. PLoS ONE 10:e0129618. doi: 10.1371/journal.pone.0129618
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer's disease. Lancet. doi: 10.1016/S0140-6736(15)01124-1. [Epub ahead of print].
Schmöle, A. C., Lundt, R., Ternes, S., Albayram, Ö., Ulas, T., Schultze, J. L., et al. (2015). Cannabinoid receptor 2 deficiency results in reduced neuroinflammation in an Alzheimer's disease mouse model. Neurobiol. Aging 36, 710–719. doi: 10.1016/j.neurobiolaging.2014.09.019
Solas, M., Francis, P. T., Franco, R., and Ramírez, M. J. (2013). CB2 receptor and amyloid pathology in frontal cortex of Alzheimer's disease patients. Neurobiol. Aging 34, 805–808. doi: 10.1016/j.neurobiolaging.2012.06.005
Stöhr, J., Watts, J. C., Mensinger, Z. L., Oehler, A., Grillo, S. K., DeArmond, S. J., et al. (2012). Purified and synthetic Alzheimer's amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. U.S.A. 109, 11025–11030. doi: 10.1073/pnas.1206555109
Sultana, R., and Butterfield, D. A. (2010). Role of oxidative stress in the progression of Alzheimer's disease. J. Alzheimers Dis. 19, 341–353. doi: 10.3233/JAD-2010-1222
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Tolón, R. M., Núñez, E., Pazos, M. R., Benito, C., Castillo, A. I., Martínez-Orgado, J. A., et al. (2009). The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 1283, 148–154. doi: 10.1016/j.brainres.2009.05.098
Tramutola, A., Di Domenico, F., Barone, E., Perluigi, M., and Butterfield, D. A. (2016). It is all about (U)biquitin: role of altered ubiquitin-proteasome system and UCHL1 in Alzheimer Disease. Oxid. Med. Cell. Longev. 2016:2756068. doi: 10.1155/2016/2756068
Walter, L., Franklin, A., Witting, A., Wade, C., Xie, Y., Kunos, G., et al. (2003). Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 23, 1398–1405.
Whiteside, G. T., Lee, G. P., and Valenzano, K. J. (2007). The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr. Med. Chem. 14, 917–936. doi: 10.2174/092986707780363023
Wilson, R. I., and Nicoll, R. A. (2002). Endocannabinoid signaling in the brain. Science 296, 678–682. doi: 10.1126/science.1063545
Wright, K. L., Duncan, M., and Sharkey, K. A. (2008). Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br. J. Pharmacol. 153, 263–270. doi: 10.1038/sj.bjp.0707486
Keywords: CB2 receptor, cannabinoids, Alzheimer, neuroinflammation, β-amyloid, tau, oxidative stress
Citation: Aso E and Ferrer I (2016) CB2 Cannabinoid Receptor As Potential Target against Alzheimer's Disease. Front. Neurosci. 10:243. doi: 10.3389/fnins.2016.00243
Received: 29 March 2016; Accepted: 17 May 2016;
Published: 31 May 2016.
Edited by:
Marialessandra Contino, Università degli Studi di Bari “Aldo Moro,” ItalyReviewed by:
Aurel Popa-Wagner, University of Medicine Rostock, GermanyAdam Denes, University of Manchester, UK
Andras Bilkei-Gorzo, University of Bonn, Germany
Julian Romero, Francisco de Vitoria University, Spain
Copyright © 2016 Aso and Ferrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ester Aso, YXNvQGJlbGx2aXRnZWhvc3BpdGFsLmNhdA==;
Isidro Ferrer, ODA4MmlmYUBnbWFpbC5jb20=