 Ilia V. Baskakov
Ilia V. Baskakov Elizaveta Katorcha
Elizaveta Katorcha- Department of Anatomy and Neurobiology, Center for Biomedical Engineering and Technology, University of Maryland School of Medicine, Baltimore, MD, USA
Mammalian prion or PrPSc is a proteinaceous infectious agent that consists of a misfolded, self-replicating state of a sialoglycoprotein called the prion protein, or PrPC. Sialylation of the prion protein N-linked glycans was discovered more than 30 years ago, yet the role of sialylation in prion pathogenesis remains poorly understood. Recent years have witnessed extraordinary growth in interest in sialylation and established a critical role for sialic acids in host invasion and host-pathogen interactions. This review article summarizes current knowledge on the role of sialylation of the prion protein in prion diseases. First, we discuss the correlation between sialylation of PrPSc glycans and prion infectivity and describe the factors that control sialylation of PrPSc. Second, we explain how glycan sialylation contributes to the prion replication barrier, defines strain-specific glycoform ratios, and imposes constraints for PrPSc structure. Third, several topics, including a possible role for sialylation in animal-to-human prion transmission, prion lymphotropism, toxicity, strain interference, and normal function of PrPC, are critically reviewed. Finally, a metabolic hypothesis on the role of sialylation in the etiology of sporadic prion diseases is proposed.
Introduction
Prions or PrPSc are proteinaceous infectious agents that consist of misfolded, self-replicating states of a sialoglycoprotein called the prion protein or PrPC (Prusiner, 1982; Legname et al., 2004). Prions cause prion diseases, a family of transmissible neurodegenerative maladies that have no treatment and are 100% lethal (Prusiner, 1998). Prions replicate by recruiting and converting PrPC molecules expressed by a host into misfolded PrPSc states (Cohen and Prusiner, 1998). In the PrPSc state, the prion protein can acquire conformationally distinct self-replicating states referred to as prion strains, which elicit different, strain-specific disease phenotypes (Thomzig et al., 2004; Spassov et al., 2006; Morales et al., 2016). While the fact that PrPSc is sialylated has been known for more than 30 years (Bolton et al., 1985), little is known about the role sialylation plays in prion diseases. This review article summarizes current knowledge on the role of sialylation of the prion protein in prion diseases.
Many Amyloidogenic Proteins Exhibit Prion-Like Behavior, But Prions are Unique
In recent years, convincing evidence was put in place illustrating that prion-like propagation of misfolded protein states is not limited to the prion protein (Jucker and Walker, 2013; Walker and Jucker, 2015). A number of amyloidogenic proteins or peptides, including Aβ, α-synuclein, tau, huntingtin, which are associated with a range of age-dependent neurodegenerative diseases, can also spread from cell to cell or be transmitted from animal to animal or human to animal in a prion-like fashion (Soto et al., 2006; Walker and Jucker, 2015). Even more striking, these amyloidogenic proteins can acquire several alternative disease-associated self-replicating states within the same amino acid sequence that recapitulates the prion strain phenomenon (Aguzzi, 2014; Stöhr et al., 2014; Watts et al., 2014).
While non-prion amyloidogenic proteins display certain characteristics of prion-like replication, several aspects make PrPSc unique. First, only PrPSc can be transmitted between organisms or species via natural routes (Brown and Gajdusek, 1991; Miller and Williams, 2004). Second, like microbial or viral agents, PrPSc shows incredibly high titers of up to 1010.5 infectious units per g of tissues in animal assays or 1013 units per g using in vitro assays (Makarava et al., 2012b). Such titers exceed by far those reported for other amyloidogenic proteins. Because different hosts are used for establishing titers (wild type vs. transgenic mice), direct comparison of prion titers with those displayed by non-prion amyloidogenic proteins should be done with caution. Keeping this in mind, 106 was found to be the highest dilution of brain material with Aβ deposits formed in tg2576 mice that was able to seed Aβ misfolding in the same mouse line (Morales et al., 2015). Because Tg2576 mice is a transgenic line that overexpresses the Amyloid Precursor Protein harboring the Swedish mutation and shows spontaneous plaque formation with age, the titers established in Tg2576 might be overestimated. Third, PrPSc-infected animals typically show a very robust course of disease progression characterized by a well-defined set of clinical symptoms, precise incubation time to disease, and a strict dependence of incubation time on dose. Fourth, in addition to the CNS, PrPSc accumulates in peripheral tissues, including the lymphoreticular system (Hilton et al., 1998; Sigurdson et al., 1999; Andréoletti et al., 2000; Aguzzi et al., 2013). In fact, not only does PrPSc colonize secondary lymphoid organs (SLOs), it replicates in SLOs autonomously from the CNS (Brown et al., 1999; Montrasio et al., 2000; Kujala et al., 2011; McCulloch et al., 2011). More surprisingly, despite low expression levels of PrPC in SLOs, SLOs are more permissive to prions than the CNS (Béringue et al., 2012; Halliez et al., 2014). As such, SLOs represent silent reservoirs of infection, where prions could hide undetected in human populations while imposing a high risk of transmission through surgery, organ or blood donation (Hilton et al., 2004; Peden et al., 2004, 2010; Wroe et al., 2006; Bishop et al., 2013). The events triggered by peripheral prion infection sets prions aside from all other known types of pathogens as well. Whereas most bacteria, parasites, and viruses trigger innate and adaptive immune responses, the mammalian immune system appears to be remarkably tolerant to prions (Aguzzi et al., 2003).
Introduction to Sialylation
Sialic acids (Sias) are a family of 9-carbon containing acidic monosaccharides that are found in terminal positions of N- and O-linked glycans of glycoproteins or glycolipids (Figure 1A) (Varki, 1999). Glycan sialylation is controlled by two groups of enzymes: sialyltransferases (STs) and sialidases (NEUs) (Audry et al., 2011; Miyagi and Yamaguchi, 2012). STs transfer sialic acids to the terminal positions of glycans. This process takes place in the trans-Golgi and involves 20 mammalian STs (Audry et al., 2011). STs are divided into four families according to the type of linkages synthesized (α2-3, α2-6, α2-8, or α2-9) and the selectivity toward N- or O-linked glycans (Takashima, 2008; Audry et al., 2011). NEUs, on the other hand, remove Sias from glycans. Four NEUs are found in mammals, they are expressed in a tissue-specific manner and display differences in cellular localization (Monti et al., 2010; Miyagi and Yamaguchi, 2012; Pshezhetsky and Ashmarina, 2013).
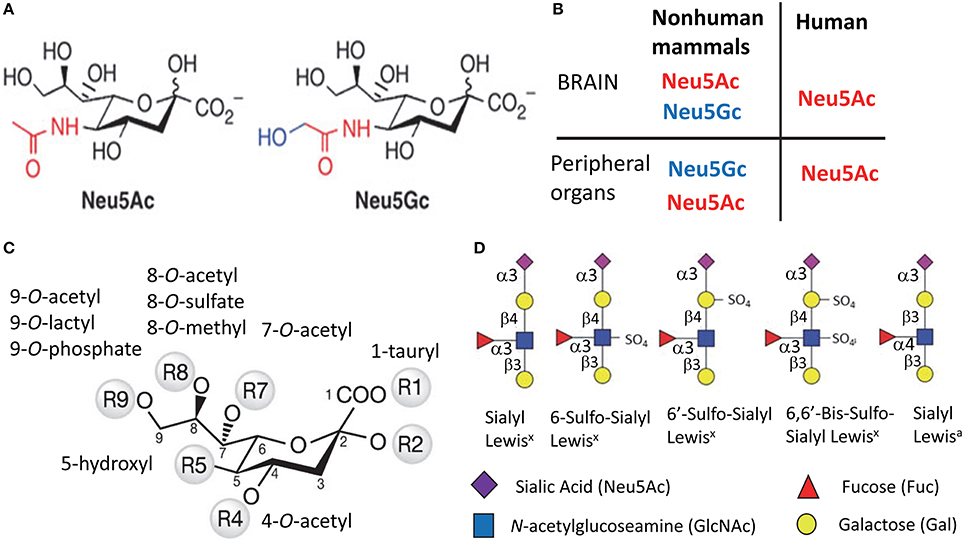
Figure 1. Structural diversity of Sias. Structures of two most common types of Sias, Neu5Ac, and Neu5Gc (A), and a diagram illustrating the differences in Sias synthesized in humans vs. non-human mammals (B). Structural diversity of Sias epitopes are achieved via naturally occurring modifications of Sias at 1-, 4-, 5-, 7-, 8-, or 9-carbon positions (C) and/or variations due to sulfation of galactose and N-acetylglucosamine that produce several Lewis glycoepitope families (D). Panel (D) shows only a small subset of possible sulfated variants.
Humans can synthesize only one type of Sias, which is N-acetylneuraminic acid (Neu5Ac) (Varki, 2010) (Figures 1A,B). With the exception of the ferret (Ng et al., 2014), the rest of mammalian species produce two types of Sias. Neu5Ac is the predominant type that is synthesized in a brain, whereas Neu5Ac and N-glycolylneuraminic acid (Neu5Gc) are synthesized by peripheral organs (Varki, 1999) (Figure 1B). The deficiency in synthesis of Neu5Gc in humans is due to an irreversible mutation in the gene encoding cytidine monophosphate N-acetylneuraminic acid hydroxylase (an enzyme that synthesize Neu5Gc from Neu5Ac) that occurred during evolution from primates to humans (Varki, 2010). Like humans, ferrets can produce only Neu5Ac (Ng et al., 2014). While humans lack the ability to synthesize Neu5Gc, it can be incorporated metabolically into human cells from diet (Samraj et al., 2015).
Sias on cell surface glycans and glycolipids form diverse structural epitopes that are involved in a number of cellular functions. The structural diversity of Sia epitopes is produced via several mechanisms. First, Sias can be attached to galactose or N-acetylgalactosamine of glycans via α2-3, α2-6, α2-8, or α2-9 linkages (Varki, 1999). Second, various natural substitutes including O-acetyl, N-glycolyl, O-lactyl, O-sulfate, O-phosphate, tauryl, hydroxyl, or O-methyl can be synthesized on carbons of Sias at 1-, 4-, 5-, 7-, 8-, or 9-positions, where O-acetyl being the most common substitute (Figure 1C). (Schauer et al., 2011). Third, in combination with Sias other groups including sulfate and fucose are involved in forming functional glycan epitopes including Sialyl Lewisx, Sialyl Lewisa, 6′Sulfo-Sialyl Lewisx (Figure 1D) (Fukuda et al., 1999). Fourth, complex glycans can exhibit several branching patterns that contribute to variations in density of Sia residues, a factor important for binding of multivalent ligands.
Sias are abundant on the surfaces of all mammalian cell types with an estimated local concentration on the cell surface glycocalyx approaching 100 mM (Collins et al., 2004). Recent years witnessed an extraordinary rise in interest to sialylation and established its role in host-pathogen interactions and communication between cells of immune system (Varki, 2008, 2010). Sias on the surface of mammalian cells act as a part of “self-associated molecular pattern” helping the immune system to recognize “self” from “altered self” or “non-self” (Varki, 2008; Brown and Neher, 2014). A decline in Sia content represents one of the molecular signatures of “apoptotic-cell-associated molecular patterns” found in apoptotic or aging cells (Savill et al., 2002; Brown and Neher, 2014). Removal of Sias from cell surface glycans exposes galactose residues that generate “eat me” signals for professional and non-professional macrophages. Examples include clearance of erythrocytes or platelets with reduced sialic acid residues by Kupffer cells (Aminoff et al., 1977; Jansen et al., 2012) or neurons by microglia (Linnartz et al., 2012; Linnartz-Gerlach et al., 2016). Lack of Sias on the cell surface is also a part of the “pathogen-associated molecular pattern” or PAMPs used by mammalian immune systems to recognize pathogens or asialoglycoproteins that need to be removed (Varki, 2008).
N-linked Glycans on PrPC and PrPSc are Sialylated
PrPC is posttranslationally modified with up to two N-linked glycans and a GPI anchor (Stahl et al., 1987; Endo et al., 1989). In PrPC, Sias are linked to the terminal positions of the two N-linked glycans via α2-3 or α2-6 linkages with the majority being linked via α2-6 (Turk et al., 1988; Endo et al., 1989; Stimson et al., 1999). Each of the two glycans has up to five terminal Sias (Endo et al., 1989; Rudd et al., 1999). Variation in structure and composition of N-linked glycans give rise to more than 400 different PrPC glycoforms (Endo et al., 1989; Stimson et al., 1999). Upon conversion of PrPC into PrPSc, the sialylated glycans and GPI are carried over, giving rise to sialylated PrPSc (Bolton et al., 1985; Stahl et al., 1993; Rudd et al., 1999).
In the absence of posttranslational modifications, the theoretical pI of the full-length mouse prion protein is expected to be 9.6 and the estimated charge at pH 7.5 is +9.5 (Katorcha et al., 2014). However, due to glycan sialylation, the actual pI of PrP molecules was found to be highly heterogeneous and spread from pH 9.6 to acidic pH (DeArmond et al., 1999; Katorcha et al., 2014). In intact PrPSc particles, the glycans are believed to be directed outwards, with the terminal sialic acid residues located at the interface with the extracellular environment or solvent (Wille et al., 2002; Govaerts et al., 2004; Requena and Wille, 2014).
Sialylation of GPI Anchor
In addition to sialylation of N-linked glycans, a single Sia could be also present on the GPI anchor of PrPC (Stahl et al., 1992). The question regarding sialylation status of GPIs within PrPSc has been controversial. As judged from mass-spectroscopy analysis of hamster-adapted prion strains Sc237 and 139H, approximately 30% of GPIs of brain-derived PrPSc were found to be sialylated (Stahl et al., 1992). Moreover, the composition of GPIs within PrPSc was found to be similar to that of PrPC (Stahl et al., 1992). In contrast, recent studies by Bate and coauthors claimed that PrPC with asialo-GPIs were not convertible into PrPSc and, even more, inhibited conversion of PrPC with sialo-GPIs into PrPSc (Bate et al., 2016b). To arrive at this conclusion, a cell painting technique was used for administering PrPC with sialo- or asialo-GPIs to cultured N2a neuroblastoma cells or primary neurons. Our recent studies that examined tissues from mice or hamsters infected with five prion strains or prion infected N2a cells or C2C12 myotube cells revealed that PrPC molecules with both sialo- and asialo-GPIs were recruited into PrPSc (Katorcha et al., 2016). Notably, the proportion of sialo- vs. asialo-GPIs within PrPSc was found to be controlled by host, tissue, and cell type, but not prion strain (Katorcha et al., 2016).
In a series of other studies that also employed cell painting techniques, Bate and coauthors suggested that toxicity triggered by PrPSc is dependent on the sialylation status of GPI anchor within PrPC, as clustering of PrPC molecules with sialo-GPIs led to activation of cytoplasmic phospholipase A2 and synapse damage (Bate and Williams, 2012b). Moreover, sialylation status of GPIs was found to modify the local environment of PrPC where the greater amounts of sialylated gangliosides and cholesterol were found in rafts surrounding PrPC with asialo-GPIs relative to PrPC with sialo-GPIs (Bate et al., 2016b). In addition, sialo-GPIs were found to target exogenous PrPC to synapses of neurons derived from the prion protein knockout mice (Bate et al., 2016a). Because cell painting technique was used in aforementioned studies, the questions whether the conclusions reached by using PrPC exogenously added to cells are valid for PrPC expressed by a cell or in animals have to be addressed.
The Effect of Sialylation of PrPSc Glycans on Prion Infectivity and Disease Outcome
Recent studies from our laboratory revealed that PrPSc with reduced sialylation levels does not induce prion disease in wild type animals (Katorcha et al., 2014). To produce PrPSc with reduced sialylation, Protein Misfolding Cyclic Amplification with beads (PMCAb) was conducted using PrPC as a substrate that was partially desialylated by treatment with sialidases (dsPMCAb). As a reference, PrPSc was also produced in PMCAb reactions conducted with non-treated PrPC. Both types of reactions were seeded with hamster scrapie strain 263K. All animals inoculated with brain-derived 263K developed clinical signs and showed substantial amounts of PrPSc in their brains (Figure 2). Animals inoculated with PMCAb-derived 263K developed disease at slightly longer incubation times relative to the control group that received brain-derived PrPSc. Such delay is attributed to a moderate shift in the sialylation pattern of PMCAb-derived 263K relative to that of brain-derived 263K (Figure 2). In the course of PMCAb, PrPC molecules with low sialylation status were preferentially recruited into PrPSc, producing a shift in sialylation status of PMCAb-derived material (Katorcha et al., 2014). Remarkably, no animals inoculated with dsPMCAb-derived material developed the disease (Katorcha et al., 2014) (Figure 2). Moreover, no PrPSc was detected in brains or spleens of animals from these groups by Western blot or serial PMCAb, arguing that the animals injected with dsPMCAb material were not subclinical carriers of scrapie. Because exposed galactose residues are believed to generate “eat me” signals, we propose that dsPMCAb-derived material is degraded rapidly due to an increase in amounts of terminal galactose as a result of partial removal of sialic acid residues. This hypothesis has to be tested in future studies.
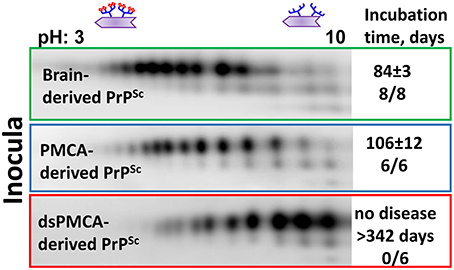
Figure 2. Analysis of sialylation status of brain-, PMCAb-, and dsPMCAb-derived PrPSc for 263K strain using 2D western blots. Incubation time to disease and number of animals that developed clinical disease out of total number of animals is shown on the right. The data represented here is a modification of the figure from previously published manuscript (Katorcha et al., 2014).
Factors that Control Sialylation of PrPSc
The sialylation status of PrPSc appears to be of paramount importance to prion infectivity; therefore, dissecting the mechanisms that control sialylation status of PrPSc is of great interest. Because PrPSc arises from PrPC via changes in its conformation, it is important to understand the mechanisms that control sialylation of PrPC.
Sialyltransferases and Sialidases
The steady-state level of sialylation in a cell is controlled by two groups of enzymes: STs and NEUs. Of the four NEUs expressed in mammals, NEU1 localizes to the lysosomes and cell surface, NEU2 is found in the cytoplasm and is expressed in muscles, NEU3 is at the plasma membrane, and NEU4 is associated with mitochondria, lysosomes, and ER, but can also be recruited to the cell surface (Monti et al., 2010; Miyagi and Yamaguchi, 2012; Pshezhetsky and Ashmarina, 2013). Because PrPC is localized at the cell surface and in endocytic/lysosomal compartments, three out of four NEUs (NEU1, NEU3, or NEU4) could be involved in regulating sialylation of PrPC. Surprisingly, brain materials from Neu1, Neu3, Neu4 knockout, or Neu3/Neu4 double knockout mice showed no differences in sialylation status of PrPC or its proteolytic fragment C1 in comparison to the corresponding wild type controls (Katorcha et al., 2015a). Moreover, suppressing NEU activity using the general inhibitor DANA did not change the sialylation of PrPC/C1 in neurobalstoma N2a cells, but did alter the global sialylation status (Katorcha et al., 2015a). These results suggested that upon removal of Sias from PrPC by cellular NEUs, PrPC molecules are degraded very fast and do not contribute to the steady-state pool of PrPC (Figure 3). If desialylation results in fast degradation, sialidase deficiency is expected to cause accumulation of PrPC and/or C1. Indeed, higher amounts of total PrP signal (PrPC plus C1) was observed in brains of Neu1, Neu3, and Neu4 knockout mice as expected (Katorcha et al., 2015a). An alternative hypothesis proposes that PrPC/C1 are not targeted by NEUs as a substrate.
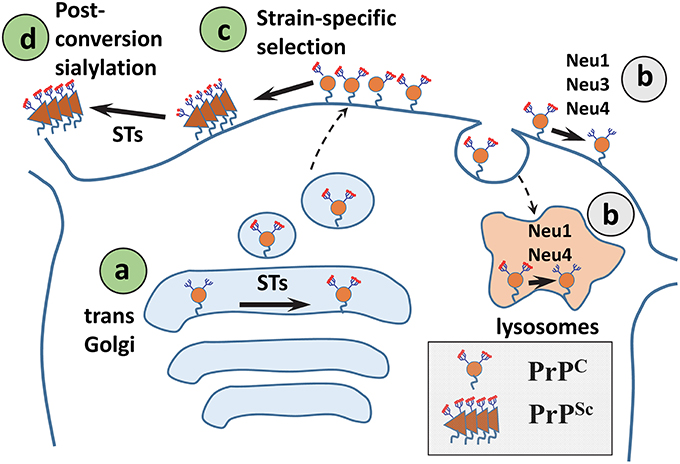
Figure 3. A diagram illustrating mechanisms that control sialylation of PrPSc. (A) The sialylation status of PrPC is controlled by STs in the trans-Golgi. (B) NEUs do not appear to affect the steady-state sialylation level of PrPC, presumably because desialylated PrPC is degraded rapidly. (C) Sialoglycoforms of PrPC are recruitment into PrPSc selectively according to their sialylation status and in a strain-specific manner. (D) In SLOs, PrPSc is a subject of post-conversion sialylation by STs. Sias are shown as red diamonds.
Modulating the activity of STs instead of NEUs may offer a more effective strategy for controlling the sialylation status of PrPC. A general inhibitor of STs 3Fax-Neu5Ac was found to reduce the sialylation level of PrPC in N2a cells (Katorcha et al., 2015a). Out of 20 mammalian STs, five STs exhibit substrate specificity for sialylation of N-linked glycans via α2-3 or α2-6 linkages, the type of linkages found in PrPC and PrPSc. Three out of the aforementioned five STs that supposedly have PrP-directed sialylation activity belong to the ST3 family (ST3Gal3, ST3Gal4, and ST3Gal6) and sialylate N-linked glycans via α2-3 linkages. The remaining two STs belong to the ST6 family (ST6Gal1 and ST6Gal2) and sialylate N-linked glycans via α2-6 linkages (Takashima, 2008; Audry et al., 2011). ST6Gal2 is found predominantly in fetal brain, whereas ST6Gal1 is expressed throughout the organism including the CNS (Takashima et al., 2002, 2003). Knocking out ST6Gal1 was found to reduce dramatically the amounts of α2-6 linked sialic acids in peripheral organs and the CNS (Martin et al., 2002), suggesting that ST6Gal1 is the main enzyme responsible for α2-6-linked sialylation and that its function is not redundant. In mice infected with prions expression of 165 glycosylation-related genes was analyzed (Guillerme-Bosselut et al., 2009). Among them, the expression levels of ST6Gal1 mRNA was found to be upregulated by ~3-fold in brain and spleen at the terminal stages of the disease that might reflect the pro-inflammatory response to the disease (Guillerme-Bosselut et al., 2009). It is not known whether the sialylation status of PrPC and/or PrPSc changes due to upregulation of ST6Gal1 in the course of prion infection.
Strain-Specific Selection of PrPC Sialoglycoforms
PrPC molecules are heterogeneous with respect to the sialylation levels of their N-linked glycans ranging from hyposialylated to hypersialylated (DeArmond et al., 1999; Katorcha et al., 2014; Schmitz et al., 2014). Using hamster strain 263K, Rudd and coauthors showed that in brain, the relative populations of sialyloglycoforms of PrPSc were very similar to those of PrPC (Rudd et al., 1999). This result led to the conclusion that PrPC sialoforms are recruited into PrPSc proportionally to their relative presentation in a cell (Rudd et al., 1999). Recent studies examined a panel of mouse and hamster strains and discovered a remarkable pattern: hypersialylated PrPC molecules were partially excluded from PrPSc (Katorcha et al., 2015b) (Figure 3). The degree to which hypersialylated PrPC were excluded was strain-specific and found to be minimal for 263K, explaining the findings by Rudd et al. Strain-specific exclusion suggests that some strains can accommodate heavily sialylated PrPC molecules better than others (Figure 4).
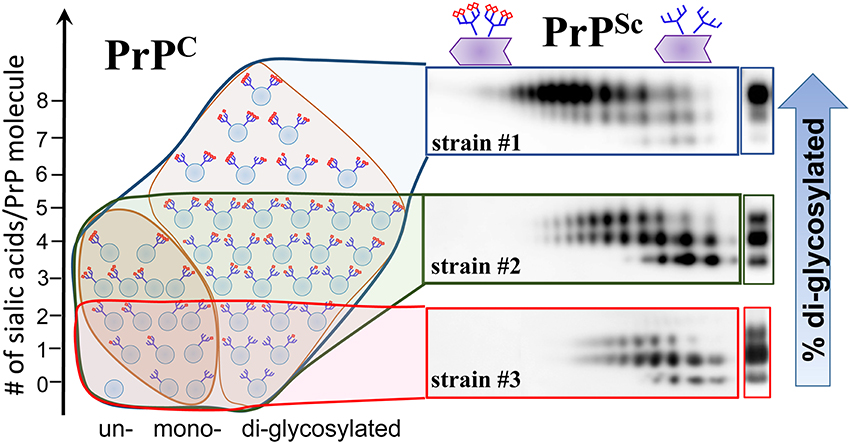
Figure 4. Schematic diagram illustrating that PrPSc strains recruit PrPC isoforms selectively according to PrPC sialylation status. While strain #1 recruits sialoglycoforms of PrPC without noticeable preferences, hypersialylated PrPC are preferentially excluded from the strain the #2 and even more so from strain #3. As a result of strain-specific exclusion of highly sialylated PrPC (illustrated by the 2D Western blots), the ratios of di- vs. mono-glycoforms within PrPSc changes in a strain-specific manner, as shown by 1D Western blots (right hand side). PrPC molecules are shown as blue circles and sialic acid residues—as red diamonds.
Tissue-Specific Post-conversion Sialylation of PrPSc
Upon prion transmission via peripheral routes, PrPSc is first sequestered by SLOs, including spleen and lymph nodes, prior to invasion of the CNS (Huang et al., 2002; Takakura et al., 2011; Castro-Seoane et al., 2012; Michel et al., 2012). Moreover, PrPSc replicates in SLOs independently of replication in the CNS (Brown et al., 1999; Montrasio et al., 2000; Kujala et al., 2011; McCulloch et al., 2011). Recent studies revealed that spleen-derived PrPSc is considerably more sialylated than brain-derived PrPSc (Srivastava et al., 2015). Enhanced sialylation of PrPSc in SLOs was observed regardless of prion strain, host species, or inoculation route (Srivastava et al., 2015). Remarkably, enhanced sialylation of PrPSc was not due to enhanced sialylation of PrPC expressed in SLOs, but appears to be due to post-conversion sialylation of PrPSc in SLOs by extracellular STs (Figure 3). While STs are traditionally believed to localize within the trans-Golgi (Harduin-Lepers et al., 2001), a number of studies reported ST activity in circulation or on surfaces of the cells of the immune system including polymorphonuclear leukocytes, monocyte-derived dendritic cells, lymphocytes, and T cells (Gross et al., 1996; Kaufmann et al., 1999; Schwartz-Albiez et al., 2004; Rifat et al., 2008; Cabral et al., 2010; Nasirikenari et al., 2014). Consistent with the hypothesis that extracellular STs are involved in enhancing sialylation of PrPSc, the sialylation status of foreign PrPSc acquired via peripheral exposure changed with colonization of SLOs (Srivastava et al., 2015). Moreover, enhanced sialylation of PrPSc was recapitulated in vitro by incubating brain-derived PrPSc with primary splenocytes or cultured macrophage RAW 264.7 cells (Srivastava et al., 2015). General inhibitors of STs suppressed enhanced sialylation of PrPSc (Srivastava et al., 2015). Thus, post-conversion sialylation is likely to camouflage PrPSc in SLOs. It would be interesting to test whether enhanced sialylation of PrPSc accounts for the high permissiveness of SLOs to prion infection.
Sialylation Contributes to Prion Replication Barrier
The conformational transition from PrPC into PrPSc is regulated by a large energy barrier that controls the prion conversion rate (Baskakov et al., 2001). Due to the large energy barrier, the spontaneous conversion of PrPC into PrPSc is very rare, explaining the low occurrence rates of sporadic prion diseases (Cohen and Prusiner, 1998). The magnitude of the energy barrier is attributed to the energy needed to unfold PrPC (Baskakov et al., 2001). Recent studies that employed PMCAb proposed that electrostatic repulsions between sialic acid residues also create structural constraints for PrPSc replication and contribute to the replication barrier (Katorcha et al., 2015b). In PrPSc particles, glycans are directed outward where the terminal sialic acid residues create a dense negative charge on the PrPSc surface (Wille et al., 2002; Govaerts et al., 2004; Requena and Wille, 2014). Because of strain-specific differences in PrPSc structures, the contribution of sialic residues to the barrier is expected to be strain-specific. Indeed, several lines of evidence support this hypothesis. First, heavily sialylated PrPC molecules were found to be partially excluded from conversion into PrPSc, and the degree of exclusion was found to be strain-specific (Katorcha et al., 2015b). Second, desialylation of PrPC by enzymatic treatment with sialidases removed the structural constraints and increased the rates of replication of PrPSc in PMCAb. The increase in replication rates was strain-specific, too, ranging from 20- to 106-fold (Katorcha et al., 2014, 2015b). Third, desialylation of PrPC was also found to considerably reduce the barrier in cross-seeded replication of PrPSc in PMCAb reactions (Katorcha et al., 2014). Together, these data suggest that the replication barrier attributable to glycan sialylation is universal, i.e., not only does it control the rate of prion replication within the same host but also the barrier associated with prion transmission between different species. The electrostatic repulsion between sialylated glycans of nascent PrPSc and the size of glycans are expected to impose a negative impact on the thermodynamic stability of PrPSc particles. This negative impact has to be counteracted by other forces that stabilize the packing of polypeptide chains within PrPSc particles. Notably, while thermodynamic stability of PrPSc varies depending on the strain-specific structure (Peretz et al., 2001; Colby et al., 2009; Ayers et al., 2011; Gonzalez-Montalban et al., 2011), the range of strain-specific thermodynamic stabilities of PrPSc is typically lower than those of amyloid fibrils generated in vitro from recombinant PrP (Sun et al., 2007, 2008). In part, such differences are likely due to electrostatic repulsion between sialylated glycans that recombinant PrP lacks.
Additional parameters have to be considered in discussing the effect of sialylation on the replication barrier in vivo. Because PrPC glycans could be bi-, tri- or tetra-antennary, PrPC molecules with the same number of sialic acid residues per molecule might have different number of terminal galactose that serves as “eat me” signal. PrPC molecules with substantial levels of terminal galactose are expected to be degraded quickly. While heavily sialylated PrPC molecules are excluded from conversion for conformational reasons, weakly sialylated PrPC with bulky glycans might not be involved in replication either due to their fast degradation. Therefore, in vivo the size of glycans, sialylation levels, and the number of exposed galactose are likely to define the availability and eligibility of PrPC as a substrate.
Sialylation and Strain-Specific Glycoform Ratio
The glycoform ratio within PrPSc is considered to be one of the primary intrinsic characteristics of prion strains or PrPSc subtypes (reviewed in Lawson et al., 2005). While the mechanisms behind strain-specific selectivity in recruitment of glycoforms remain unknown, the glycoform ratios have been used in the prion field for strain typing and classification of CJD type (Collinge et al., 1996; Somerville, 1999). Recent studies that analyzed strain-specific sialylation patterns of PrPSc revealed that the strain-specific glycoform ratio is due to exclusion of heavily sialylated PrPC molecules (Katorcha et al., 2015b) (Figure 4). Because diglycosylated PrPC carry more sialic acid residues per molecule on average than mono- or unglycosylated PrPC, the preferential exclusion of heavily sialylated PrPC is achieved via (i) selective recruitment of mono- and unglycosylated PrPC at the expense of diglycosylated PrPC, and (ii) preferential exclusion of hypersialylated diglycosylated PrPC (Katorcha et al., 2015b). In fact, a correlation between PrPSc sialylation status and the glycoform ratio exists (Katorcha et al., 2015b). Remarkably, when exposed to desialylated PrPC as a substrate, prion strains lose strain-specific selectivity toward PrPC glycoforms, and the glycoform ratio within PrPSc mirrors that of PrPC (Katorcha et al., 2015b).
N-linked Glycans and PrPSc Structure
The density of sialylation and size of N-linked glycans impose considerable structural constraints, limiting the range of plausible structures for PrPSc. Similar to amyloids formed by other amyloidogenic proteins or peptides, PrPSc exhibits a cross-β folding pattern (Wille et al., 2009; Ostapchenko et al., 2010), a key structural feature of amyloid states. However, the precise folding pattern of PrP molecules within PrPSc has been debated (reviewed in Requena and Wille, 2014). The recent PIRIBS model proposed that PrPSc consists of an in-register parallel β-sheet structure, in which each PrP molecule occupies a single layer within cross-β fibers (Groveman et al., 2014). This model is similar to those proposed earlier for amyloid fibrils formed by non-glycosylated recombinant PrP (Cobb et al., 2007; Tycko et al., 2010). Alternative models postulate that within PrPSc fibers each PrP molecule forms a multi-rung β-solenoid (Govaerts et al., 2004; Amenitsch et al., 2013). To discriminate between alternative models, we decided to determine the extent to which N-linked glycans can be accommodated within PrPSc folding patterns proposed by different models. According to the PIRIBS model, the glycans linked to the same amino acid residue on neighboring PrP molecules are separated by a distance of 4.7 Å (Figure 5A). For solenoid models, depending on the number of rungs formed by PrP molecules within the solenoid, the distance between glycans on neighboring PrP molecules could be 2 × 4.7 Å, 3 × 4.7 Å, or 4 × 4.7 Å for the solenoids consisted of 2, 3, or 4 rungs, respectively (Figures 5B–D). To model N-linked glycans of average size, we choose a tri-antennary glycan structure, since PrPC and PrPSc are known to carry bi-, tri-, and tetra-antennary glycans (Endo et al., 1989; Rudd et al., 1999; Stimson et al., 1999). Substantial spatial overlap was found between glycans of neighboring PrP molecules, if the glycan linkages were separated by distances 4.7 Å or 2 × 4.7 Å (Figures 5A,B). Such spatial constraints argue strongly against PIRIBS or 2-rung solenoids as plausible models of PrPSc. Minor spatial overlap was observed between neighboring glycans attached at the distance of 3 × 4.7 Å, and no overlap when the tri-antennary glycans were separated by 4 × 4.7 Å (Figures 5C,D). The minor overlap observed for 3-rung solenoid structures could be avoided if N-linked glycans are of smaller-sizes (bi-antennary) and/or oriented at various angles. The 2-rung solenoid structure would be still possible if glycosylated molecules alternated with non-glycosylated ones along PrPSc fibers. However, the percentage of non-glysoylated PrP molecules within PrPSc is known to be very small (Nishina et al., 2006; Katorcha et al., 2015b).
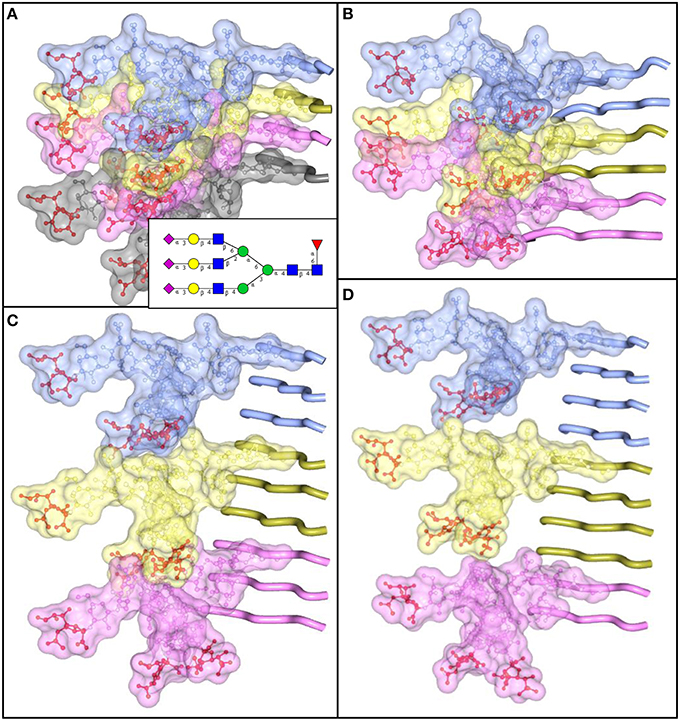
Figure 5. N-linked glycans impose spatial constraints on folding patterns of PrPSc. Cross beta-sheet structures carrying tri-antennary N-glycans (shown in inset) on each neighboring beta-strand (A), or every second (B), third (C), or fourth (D) beta-strand. Polypeptide chains are represented in the tube form, whereas N-glycans are represented in the ball-and-stick form. Each PrP molecule with corresponding N-glycan is of a different color. Sialic acid residues are colored in red; N-glycan electrostatic surfaces are semi-transparent. To model the dimension of cross-beta structures, the parallel beta-sheet model was adapted from PDB database entry 2RNM, an NMR structure for HET-s(218–289) prion in its amyloid form (Wasmer et al., 2008). Stretches of seven amino acid residues are shows for each beta strand without any change to the atomic coordinates. The structure of a tri-antennary N-linked glycan was taken from PDB entry 3QUM, a crystal structure of human prostate specific antigen (PSA) (Stura et al., 2011). Both calculations of electrostatic surfaces and generation of images were performed with CCP4MG software.
Metabolic Origin of Sporadic Prion Diseases
According to the Braak staging hypothesis, in Alzheimer's and Parkinson diseases amyloid deposits and pathology originate in certain areas of the CNS and spread in a prion-like manner through the brain in disease-specific patterns (Braak and Braak, 1991). It is not known from which brain area sporadic CJD originates and whether it spreads in a specific pattern. Bearing in mind that sialylation controls the height of the conformational transition barrier (Katorcha et al., 2015b), it is reasonable to propose that the first spontaneous PrPC-to-PrPSc conversion events have a higher chance of occurring in brain areas that express PrPC with glycans of small sizes and reduced sialylation levels and/or in individuals with deficient sialylation substrate. Consistent with the metabolic hypothesis, there is a decline in total sialic acid content as well as cell surface sialylation with age (Svennerholm et al., 1997). Notably, a polymorphism in β-secretase (BACE1), an enzyme that cleaves ST6Gal1, was recently shown to be a risk factor for sCJD suggesting that a link between sialylation and sCJD might exist (Calero et al., 2012). It would be interesting to test whether variations in BACE1 activity due to polymorphism contribute to stability and/or activity of ST6Gal1 in Golgi and affect sialylation status of PrPC.
Animal-to-Human Prion Transmission
Humans can only synthesize Neu5Ac, whereas Neu5Gc is the predominant type of Sias expressed in the periphery of mammals (Varki, 2010) (Figure 1B). The difference in the type of Sias expressed in the periphery of human and non-human mammals raises several important topics for discussion. First, it would be interesting to find out whether this difference contributes to the animal-to-human species barrier for prion transmission. Notably, in humans with high consumption of red meat, Neu5Gc incorporates metabolically into human cells and induces antibody responses against Neu5Gc (Samraj et al., 2015). While incorporation of Neu5Gc increases the likelihood of systemic inflammation (Samraj et al., 2015), antibodies against Neu5Gc might be beneficial for neutralizing prion infection of zoonic origin in humans. A second important aspect to consider is the functional consequences of enhanced sialylation of foreign PrPSc in SLOs (Srivastava et al., 2015). Enhanced sialylation in human SLOs could “humanize” prions of animal origin transmitted to humans by decorating them with Neu5Ac and helping to deceive the human immune system (Srivastava et al., 2015). A third interesting aspect is related to human-specific differences in the binding sites of human Siglecs for selective recognition of Neu5Ac over of Neu5Gc (Varki, 2010). Siglecs are a family of sialic acid-binding proteins with a number of important functions (reviewed in Rabinovich and Croci, 2012). While interactions between prions and Siglecs have not yet been documented (Bradford et al., 2014), such a possibility should not be excluded considering the large number of Siglecs expressed in humans and mice. Human-specific differences in Siglecs for selective recognition of Neu5Ac over of Neu5Gc are also important for the critical assessment of results obtained in humanized mice (mice expressing the human PrP gene). Humanized mice have often been used to assess susceptibility of humans to prion strains of animal origin or to model human-to-human transmission (Collinge et al., 1995; Asante et al., 2002; Wadsworth et al., 2004; Bishop et al., 2006; Cassard et al., 2014). Because humanized mice express mouse but not human Siglecs, interaction between Siglecs and PrPSc are expected to lead to different outcomes in humanized mice and in humans.
Sialylation of PrPSc and Lymphotropism
Prion strains show variable degrees of lymphotropism (Aguzzi et al., 2013). The molecular mechanism behind strain-specific lymphotropism is not known. It is also not known whether limited lymphotropism is due to deficient trafficking of certain strains to SLOs, impaired replication in SLOs, fast clearance in SLOs, or a combination of these factors. Recent studies revealed that sialylation of N-linked glycans at α2-6 linkages is responsible for directed trafficking and selective adhesion of hepatocarcinoma cells to SLOs (Zhang et al., 2013; Wang et al., 2015). Another work that employed synthetic glycoclusters demonstrated that in circulation the glycoclusters with α2-6 linked sialic residues were more stable and showed slower clearance rates in comparison to the glycoclusters with α2-3 linkages (Tanaka et al., 2010). It would be interesting to test whether sialylation and, in particular, α2-6 linkages also account for the lymphotropism of PrPSc.
Notably, the two types of human CJDs, sporadic and variant, show significant differences with respect to their lymphotropism, with variant CJD known to be much more lymphotropic than sCJD (Hill et al., 1999; Wadsworth et al., 2001; Halliez et al., 2014). PrPSc in variant CJD is predominantly diglycosylated and, as such, more sialylated than PrPSc in sCJD, which is predominantly monoglycosylated (Zanusso et al., 2004; Pan et al., 2005). The relative ranking of the two types of CJD with respect to sialylation is consistent with the hypothesis that sialylation is important for effective trafficking of PrPSc to SLOs.
Sialylation of PrPSc and Toxicity
Interaction of PrPSc particles with PrPC molecules anchored via GPI on the cell surface is believed to be important for triggering toxic signals (Solforosi et al., 2004; Sonati et al., 2012). Previous studies proposed that PrPSc-induced toxicity is mediated via PrPC molecules with sialylated GPI anchors (Bate and Williams, 2012a,b). Clustering of PrPC with sialo-GPIs was shown to trigger synapse damage via activating cytoplasmic phospholipase A2 in neurons cultured in vitro (Bate and Williams, 2012a,b).
It is not known whether the toxic potential of PrPSc depends on sialylation status of its N-linked glycans. Considering that two positively charged regions in PrP are involved in mediating toxic signals (Solomon et al., 2011; Westergard et al., 2011), it is plausible that the binding between PrPSc and PrPC involves electrostatic interactions between negatively charged sialic acid residues of PrPSc N-linked glycans and two solvent-exposed, positively charged regions of PrPC (Turnbaugh et al., 2012). In agreement with this hypothesis is the observation that colominic acid, which is a polymer of sialic acid, blocks neurotoxicity of PrPSc toward cortical neurons cultured in vitro (Ushijima et al., 1999). Other support of this hypothesis comes from studies where abnormal, self-replicating PrP states referred to as atypical PrPres were found to exhibit very low sialylation levels of N-glycans and lack of toxicity in animal studies (Kovacs et al., 2013; Makarava et al., 2015). Atypical PrPres was fully transmissible in animal bioassays and accumulated in the form of small synaptic deposits and large plaques. However, atypical PrPres alone, in the absence of PrPSc, did not cause neuronal death, pathological lesions or any clinical signs of prion diseases (Makarava et al., 2011, 2012a, 2015, 2016; Kovacs et al., 2013).
Sialylation and Strain Interference
Prion strain interference occurs when a host is infected with two or more prion strains (Dickinson et al., 1972; Kimberlin and Walker, 1985; Bartz et al., 2007; Schutt and Bartz, 2008; Shikiya et al., 2010). Strain interference refers to an extension of the incubation time to disease produced by a strain mixture relative to the incubation period produced alone by the strain with the shortest incubation time. Among factors that were previously discussed as main contributors to strain interference were competition between strains for substrate, cellular co-factors, or cellular replication sites (Bartz et al., 2007; Shikiya et al., 2010).
Strain-specific selection of PrPC sialoglycoforms adds an important dimension to the strain interference phenomenon (Katorcha et al., 2015b). Strains with substantial structural constraints rely on a narrow range of PrPC sialoglycoforms as substrates and are unlikely to be strong competitors (Figure 4). In contrast, strains capable of recruiting a broad range of PrPC sialoglycoforms have a greater chance of succeeding in competition for substrate. Evolution of prion diseases of synthetic origin and competition between two self-propagating states, atypical PrPres and PrPSc provides remarkable illustrations of how differences in selectivity toward PrPC sialoglycoforms determined the outcome of competition (Makarava et al., 2011, 2012a, 2013, 2016). Only a small fraction of PrPC sialoglycoforms that were acceptable as a substrate for PrPSc was found to be also a suitable substrate to atypical PrPres (Makarava et al., 2015). As a result, atypical PrPres replicated slower than PrPSc, and PrPSc outcompeted atypical PrPres (Makarava et al., 2011, 2012a, 2015, 2016). In conclusion, strain-specific selection of PrPC sialoglycoforms is an important factor that contributes to strain competition and interference.
Role of Sialylation in Normal Function of PrPC
The role of sialylation in the normal function of PrPC has yet to be explored. PrPC contains Lewis X [trisaccharide Galβ1-4(Fucα1-3)GlcNAc, abbreviated as Lex] and sialyl-Lewis X [tetrasaccharide NeuNAcα2-3Galβ1-4(Fucα1-3)GlcNAc, abbreviated as sLex] epitopes (Stimson et al., 1999) that are known to serve as ligands for selectins. PrPC containing Lex epitopes were found to bind E-, L-, and P-selectins with nanomolar affinities and in a Ca2+ dependent manner (Li et al., 2007). A variety of biological activities involving PrPC, including neurotrophic activities (Chen et al., 2003; Santuccione et al., 2005; Lima et al., 2007), involvement in cell adhesion (Schmitt-Ulms et al., 2001; Santuccione et al., 2005; Viegas et al., 2006; Málaga-Trillo et al., 2009), and cell proliferation and differentiation (Mouillet-Richard et al., 1999; Steele et al., 2006; Zhang et al., 2006; Lima et al., 2007; Lee and Baskakov, 2010, 2013; Panigaj et al., 2011; Santos et al., 2011), has been observed over the years. In particular, a growing number of studies have highlighted the role of PrPC in controlling self-renewal, proliferation and differentiation of stem cells, including human stem cells (Mouillet-Richard et al., 1999; Steele et al., 2006; Zhang et al., 2006; Lima et al., 2007; Lee and Baskakov, 2010, 2013, 2014; Panigaj et al., 2011; Santos et al., 2011). Considering that the proportion of di- vs. mono-, and unglycosylated PrPC glycoforms increases in the course of neuronal differentiation and with the density of cells cultured in vitro (Monnet et al., 2003; Novitskaya et al., 2007), it is plausible that PrPC glycosylation and sialylation is important for its function. Notably, recent studies revealed that deficiency in PrPC in a cell resulted in a loss of polysialylation of Neural Cell Adhesion Molecule 1 (NCAM1) (Mehrabian et al., 2015). The defect in polysialylation was found to be due to impairment in expression of sialyltransferase ST8Sia2, which is responsible for polysialylating glycoproteins.
Conclusions
Recent studies suggest that sialylation of PrPSc controls its fate in an organism and the outcomes of prion disease. PrPSc with reduced sialylation status did not cause prion disease presumably due to an increase in the amounts of terminal galactose that is believed to serve as “eat me” signal. The following mechanisms that define the sialylation of PrPSc have been identified: (i) sialylation of PrPC by STs, (ii) strain-specific selective recruitment of PrPC sialoglycoforms, and (ii) post-conversion enhanced sialylation of PrPSc in SLOs (Figure 3). In addition, sialylation of N-linked glycans was shown to contribute to the replication barrier that defines the rates of prion replication within the same host and prion transmission between different species. PrPC with glycans of small sizes and fewer sialic acid residues per PrPC molecule are expected to have a lower energy barrier for conversion relative to the heavily sialylated PrPC with bulky glycans. For explaining strain-specific differences in glycoform ratios, selective exclusion of heavily sialylated PrPC molecules from conversion due to strain-specific structural constraints was proposed. Nevertheless, because sialylation protects PrPSc against clearance and might be also important for prion transmission, lymphotropism and toxicity, to be highly infectious prion strains have to accommodate certain levels of sialylation. The precise role of sialylation in animal-to-human prion transmission, prion lymphotropism, toxicity, strain interference, and normal function of PrPC, have yet to be addressed and require future studies.
Author Contributions
IB wrote the manuscript; EK contributed to the section “N-linked glycans and PrPSc structure,” generated the models presented on Figure 3 and provided critical feedback.
Funding
Financial support for this study was provided by National Institute of Health Grants R01 NS045585 and R01 NS074998.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Saurabh Srivastava and Natallia Makarava for critical discussions and Pamela Wright for editing the manuscript.
References
Aguzzi, A. (2014). Neurodegeneration: alzheimer's disease under strain. Nature 512, 32–34. doi: 10.1038/512032a
Aguzzi, A., Heppner, F. L., Heikenwalder, M., Prinz, M., Mertz, K., Seeger, H., et al. (2003). Immune system and peripheral nerves in propagation of prions to CNS. Br. Med. Bull. 66, 141–159. doi: 10.1093/bmb/66.1.141
Aguzzi, A., Nuvolone, M., and Zhu, C. (2013). The immunology of prion diseases. Nat. Rev. Immunol. 13, 888–902. doi: 10.1038/nri3553
Amenitsch, H., Benetti, F., Ramos, A., Legname, G., and Requena, J. R. (2013). SAXS structural study of PrP(Sc) reveals ~11 nm diameter of basic double intertwined fibers. Prion 7, 496–500. doi: 10.4161/pri.27190
Aminoff, D., Bruegge, W. F., Bell, W. C., Sarpolis, K., and Williams, R. (1977). Role of sialic acid in survival of erythrocytes in the circulation: interaction of neuraminidase-treated and untreated erythrocytes with spleen and liver at the cellular level. Proc. Acad. Natl. Sci. U.S.A. 74, 1521–1524. doi: 10.1073/pnas.74.4.1521
Andréoletti, O., Berthon, P., Marc, D., Sarradin, P., Grosclaude, J., van Keulen, L., et al. (2000). Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 81, 3115–3126. doi: 10.1099/0022-1317-81-12-3115
Asante, E. A., Linehan, J. M., Desbruslais, M., Joiner, S., Gowland, I., Wood, A. L., et al. (2002). BSE prions propagate as either variant CJD-like or sporadic CJD-like prion stains in transgenic mice expressing human prion protein. EMBO J. 21, 6358–6366. doi: 10.1093/emboj/cdf653
Audry, M., Jeanneau, C., Imberty, A., Harduin-Lepers, A., Delannoy, P., and Breton, C. (2011). Current trend in the structure-activity relationships of sialylatransferases. Glycobiology 21, 716–726. doi: 10.1093/glycob/cwq189
Ayers, J. I., Schutt, C. R., Shikiya, R. A., Aguzzi, A., Kincaid, A. E., and Bartz, J. C. (2011). The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 7:e1001317. doi: 10.1371/journal.ppat.1001317
Bartz, J. C., Kramer, M. L., Sheehan, M. H., Hutter, J. A., Ayers, J. I., Bessen, R. A., et al. (2007). Prion interference is due to a reduction in strain-specific PrPSc levels. J. Virol. 81, 689–697. doi: 10.1128/JVI.01751-06
Baskakov, I. V., Legname, G., Prusiner, S. B., and Cohen, F. E. (2001). Folding of prion protein to its native à-helical conformation is under kinetic control. J. Biol. Chem. 276, 19687–19690. doi: 10.1074/jbc.C100180200
Bate, C., Nolan, W., McHale-Owen, H., and Williams, A. (2016a). Sialic acid within the glycosylphosphatidylinositol anchor targets the cellular prion protein to synapses. J. Biol. Chem. doi: 10.1074/jbc.M115.672394. [Epub ahead of print].
Bate, C., Nolan, W., and Williams, A. (2016b). Sialic acid on the glycosylphosphatidylinositol anchor regulates PrP-mediated cell signaling and prion formation. J. Biol. Chem. 291, 160–170. doi: 10.1074/jbc.M115.672394
Bate, C., and Williams, A. (2012a). Clustring of sialylated glycocylphosphatidylinositol anchors mediated PrP-induced activation of cytoplasmic phospholipase A2 and synapse damage. Prion 6, 350–353. doi: 10.4161/pri.21751
Bate, C., and Williams, A. (2012b). Neurodegeneration induced by clustering of sialylated glycosylphosphatidylinositols of prion proteins. J. Biol. Chem. 287, 7935–7944. doi: 10.1074/jbc.M111.275743
Béringue, V., Herzog, L., Jaumain, E., Reine, F., Sibille, P., Le Dur, A., et al. (2012). Facilitated cross-species transmission of prions in extraneural tissue. Science 335, 472–475. doi: 10.1126/science.1215659
Bishop, M. T., Diack, A. B., Ritchie, D. L., Ironside, J. W., Will, R. G., and Manson, J. C. (2013). Prion infectivity in the spleen of a PRNP heterozygous individual with subclinical variant Creutzfeldt-Jakob disease. Brain 136, 1139–1145. doi: 10.1093/brain/awt032
Bishop, M. T., Hart, P., Aitchison, L., Baybutt, H. N., Plinston, C., Thomson, V., et al. (2006). Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 5, 393–398. doi: 10.1016/S1474-4422(06)70413-6
Bolton, D. C., Meyer, R. K., and Prusiner, S. B. (1985). Scrapie PrP 27-30 is a sialoglycoprotein. J. Virol. 53, 596–606.
Braak, H., and Braak, E. (1991). Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Bradford, B. M., Crocker, P. R., and Mabbott, N. A. (2014). Peripheral prion disease pathogenesis is unaltered in the absence of sialoadhesin (Siglec-1/CD169). Immunology 143, 120–129. doi: 10.1111/imm.12294
Brown, G. C., and Neher, J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216. doi: 10.1038/nrn3710
Brown, K. L., Stewart, K., Ritchie, D. L., Mabbott, N. A., Williams, A., Fraser, H., et al. (1999). Scrapie replication in lyphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 5, 1308–1312. doi: 10.1038/15264
Brown, P., and Gajdusek, D. C. (1991). The human spongiform encephalopathies: kuru, Creutzfeldt-Jakob disease, and the Gerstmann-Sträussler-Scheinker syndrome. Curr. Top. Microbiol. Immunol. 172, 1–20. doi: 10.1007/978-3-642-76540-7_1
Cabral, M. G., Piteira, A. R., Silva, Z., Ligeiro, D., Brossmer, R., and Videira, P. A. (2010). Human dendritic cells contain cell surface sialyltransferase activity. Immunol. Lett. 131, 89–96. doi: 10.1016/j.imlet.2010.02.009
Calero, O., Bullido, M. J., Clarimón, J., Frank-García, A., Martínez-Martín, P., Lleó, A., et al. (2012). A common BACE1 polymorphism is a risk factor for sporadic Creutzfeldt-Jakob disease. PLoS ONE 7:0043926. doi: 10.1371/journal.pone.0043926
Cassard, H., Torres, J. M., Lacroux, C., Douet, J. Y., Benestad, S. L., Lantier, F., et al. (2014). Evidence for zoonotic potential of ovine scrapie prions. Nat. Commun. 5, 5821. doi: 10.1038/ncomms6821
Castro-Seoane, R., Hummerich, H., Sweeting, T., Tattum, M. H., Linehan, J. M., Fernandez de Marco, M., et al. (2012). Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog. 8:e1002538. doi: 10.1371/journal.ppat.1002538
Chen, S., Mangé, A., Dong, L., Lehmann, S., and Schachner, M. (2003). Prion protein as trans-ineracting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 22, 227–233. doi: 10.1016/S1044-7431(02)00014-3
Cobb, N. J., Sönnichsen, F. D., McHaourab, H., Surewicz, W.,and K. (2007). Molecular architecture of human prion protein amyloid: a parallel, in-register b-structure. Proc. Acad. Natl. Sci. U.S.A. 104, 18946–18951. doi: 10.1073/pnas.0706522104
Cohen, F. E., and Prusiner, S. B. (1998). Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67, 793–819. doi: 10.1146/annurev.biochem.67.1.793
Colby, D. W., Giles, K., Legname, G., Wille, H., Baskakov, I. V., DeArmond, S. J., et al. (2009). Design and construction of diverse mammalian prion strains. Proc. Acad. Natl. Sci. U.S.A. 106, 20417–20422. doi: 10.1073/pnas.0910350106
Collinge, J., Palmer, M. S., Sidle, K. C., Hill, A. F., Gowland, I., Meads, J., et al. (1995). Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 378, 779–783. doi: 10.1038/378779a0
Collinge, J., Sidle, K. C. L., Meads, J., Ironside, J., and Hill, A. F. (1996). Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383, 685–690. doi: 10.1038/383685a0
Collins, B. E., Blixt, O., DeSieno, A. R., Bovin, N., Marth, J. D., and Paulson, J. C. (2004). Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Acad. Natl. Sci. U.S.A. 101, 6104–6109. doi: 10.1073/pnas.0400851101
DeArmond, S. J., Qiu, Y., Sànchez, H., Spilman, P. R., Ninchak-Casey, A., Alonso, D., et al. (1999). PrPC glycoform heterogeneity as a function of brain region: implications for selective targeting of neurons by prion strains. J. Neuropathol. Exp. Neurol. 58, 1000–1009.
Dickinson, A. G., Fraser, H., Meikle, V. M. H., and Outram, G. W. (1972). Competition between different scrapie agents in mice. Nat. New Biol. 237, 244–245. doi: 10.1038/newbio237244a0
Endo, T., Groth, D., Prusiner, S. B., and Kobata, A. (1989). Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 28, 8380–8388. doi: 10.1021/bi00447a017
Fukuda, M., Hiraoka, N., and Yeh, J. C. (1999). C-Type lectins and sialyl lewis X Oligosaccharides versatile roles in cell–cell interaction. J. Cell Biol. 147, 467–470. doi: 10.1083/jcb.147.3.467
Gonzalez-Montalban, N., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2011). Relationship between conformational stability and amplification efficiency of prions. Biochemistry 50, 7933–7940. doi: 10.1021/bi200950v
Govaerts, C., Wille, H., Prusiner, S. B., and Cohen, F. E. (2004). Evidance for assembly of prions with left-handed b-helices into trimers. Proc. Acad. Natl. Sci. U.S.A. 101, 8342–8347. doi: 10.1073/pnas.0402254101
Gross, H. J., Merling, A., Moldenhauer, G., and Schwartz-Albiez, R. (1996). Ecto-sialyltransferase of human B lymphocytes reconstitutes differentiation markers in the presence of exogenous CMP-N-acetyl neuraminic acid. Blood 87, 5113–5126.
Groveman, B. R., Dolan, M. A., Taubner, L. M., Kraus, A., Wickner, R. B., and Caughey, B. (2014). Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 289, 24129–24142. doi: 10.1074/jbc.M114.578344
Guillerme-Bosselut, F., Forestier, L., Jayat-Vignoles, C., Vilotte, J. L., Popa, I., Portoukalian, J., et al. (2009). Glycosylation-related gene expression profiling in the brain and spleen of scrapie-affected mouse. Glycobiology 19, 879–889. doi: 10.1093/glycob/cwp062
Halliez, S., Reine, F., Herzog, L., Juamain, E., Haïk, S., Rezaei, H., et al. (2014). Accelerated, spleen-based titration of variant Creutzfeldt-Jakob disease infectivity in transgenic mice expressing human prion protein with sensitivity comparable to that of survival time bioassay. J. Virol. 88, 8678–8686. doi: 10.1128/JVI.01118-14
Harduin-Lepers, A., Vallejo-Ruiz, V., Krzewinski-Recchi, M. A., Samyn-Petit, B., Julien, S., and Delannoy, P. (2001). The human sialyltransferase family. Biochimie 83, 727–737. doi: 10.1016/S0300-9084(01)01301-3
Hill, A. F., Butterworth, R. J., Joiner, S., Jackson, G., Rossor, M. N., Thomas, D. J., et al. (1999). Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 353, 183–189. doi: 10.1016/S0140-6736(98)12075-5
Hilton, D. A., Fathers, E., Edwards, P., Ironside, J. W., and Zajicek, J. (1998). Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet 352, 703–704. doi: 10.1016/S0140-6736(98)24035-9
Hilton, D. A., Ghani, A. C., Conyers, L., Edwards, P., McCardle, L., Ritchie, D., et al. (2004). Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 203, 733–709. doi: 10.1002/path.1580
Huang, F. P., Farquhar, C. F., Mabbott, N. A., Bruce, M. E., and MacPherson, G. G. (2002). Migrating intestinal dendritic cells transport PrPSc from the gut. J. Gen. Virol. 83, 267–271. doi: 10.1099/0022-1317-83-1-267
Jansen, A. J. G., Josefsson, E. C., Rumjantseva, V., Liu, Q. P., Falet, H., Bergmeier, W., et al. (2012). Desialylation accelerates platelet clearance after refrigeration and initiates GPIba metalloproteinase-mediated cleavage in mice. Blood 119, 1263–1273. doi: 10.1182/blood-2011-05-355628
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Katorcha, E., Klimova, N., Makarava, N., Savtchenko, R., Pan, X., Annunziata, I., et al. (2015a). Knocking out of cellular neuraminidases Neu1, Neu3 or Neu4 does not affect sialylation status of the prion protein. PLoS ONE 10:e0143218. doi: 10.1371/journal.pone.0143218
Katorcha, E., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2015b). Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 5:16912. doi: 10.1038/srep16912
Katorcha, E., Makarava, N., Savtchenko, R., D'Azzo, A., and Baskakov, I. V. (2014). Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 10:e1004366. doi: 10.1371/journal.ppat.1004366
Katorcha, E., Srivastava, S., Klimova, N., and Baskakov, I. V. (2016). Sialylation of GPI anchors of mammalian prions is regulated in a host-, tissue- and cell-specific manner. J. Biol. Chem. doi: 10.1074/jbc.M116.732040. [Epub ahead of print].
Kaufmann, M., Blaser, C., Takashima, S., Schwartz-Albiez, R., Tsuji, S., and Pircher, H. (1999). Identification of an alpha2,6-sialyltransferase induced early after lymphocyte activation. Int. Immunol. 11, 731–738. doi: 10.1093/intimm/11.5.731
Kimberlin, R. H., and Walker, C. A. (1985). Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology 23, 74–81. doi: 10.1159/000149588
Kovacs, G. G., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2013). Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am. J. Pathol. 183, 1539–1547. doi: 10.1016/j.ajpath.2013.07.024
Kujala, P., Raymond, C. R., Romeijn, M., Godsave, S. F., van Kasteren, S. I., Wille, H., et al. (2011). Prion Uptake in the Gut: identification of the first uptake and replication sites. PLoS Pathog. 7:e1002449. doi: 10.1371/journal.ppat.1002449
Lawson, V. A., Collins, S. J., Masters, C. L., and Hill, A. F. (2005). Prion protein glycosylation. J. Neurochem. 93, 793–801. doi: 10.1111/j.1471-4159.2005.03104.x
Lee, Y. J., and Baskakov, I. V. (2010). Treatment with normal prion protein delays differentiation and helps to maintain proliferating activity in human embryonic stem cells. J. Neurochem. 114, 362–373. doi: 10.1111/j.1471-4159.2010.06601.x
Lee, Y. J., and Baskakov, I. V. (2013). The cellular form of the prion protein is involved in controlling cell cycle dynamics, self-renewal, and the fate of human embryonic stem cell differentiation. J. Neurochem. 124, 310–322. doi: 10.1111/j.1471-4159.2012.07913.x
Lee, Y. J., and Baskakov, I. V. (2014). The cellular form of the prion protein guides the differentiation of human embryonic stem cells into neuron-, oligodendrocyte-, and astrocyte-committed lineages. Prion 8, 266–275. doi: 10.4161/pri.32079
Legname, G., Baskakov, I. V., Nguyen, H. O. B., Riesner, D., Cohen, F. E., DeArmond, S. J., et al. (2004). Synthetic mammalian prions. Science 305, 673–676. doi: 10.1126/science.1100195
Li, C., Wong, P., Pan, T., Xiao, F., Yin, S., Chang, B., et al. (2007). Normal cellular prion protein is a ligand of selectins: binding requires Lex but is inhibited by sLex. Biochem. J. 406, 333–341. doi: 10.1042/BJ20061857
Lima, F. R., Arantes, C. P., Muras, A. G., Nomizo, R., Brentani, R. R., and Martins, V. R. (2007). Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 103, 2164–2176. doi: 10.1111/j.1471-4159.2007.04904.x
Linnartz, B., Kopatz, J., Tenner, A. J., and Neumann, H. (2012). Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 32, 946–952. doi: 10.1523/JNEUROSCI.3830-11.2012
Linnartz-Gerlach, B., Schuy, C., Shahraz, A., Tenner, A. J., and Neumann, H. (2016). Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia 64, 35–47. doi: 10.1002/glia.22901
Makarava, N., Kovacs, G. G., Savtchenko, R., Alexeeva, I., Budka, H., Rohwer, R. G., et al. (2011). Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 7:e1002419. doi: 10.1371/journal.ppat.1002419
Makarava, N., Kovacs, G. G., Savtchenko, R., Alexeeva, I., Ostapchenko, V. G., Budka, H., et al. (2012a). A new mechanism for transmissible prion diseases. J. Neurosci. 32, 7345–7355. doi: 10.1523/JNEUROSCI.6351-11.2012
Makarava, N., Savtchenko, R., Alexeeva, I., Rohwer, R. G., and Baskakov, I. V. (2012b). Fast and ultrasensitive method for quantitating prion infectivity titer. Nat. Commun. 3, 741. doi: 10.1038/ncomms1730
Makarava, N., Savtchenko, R., Alexeeva, I., Rohwer, R. G., and Baskakov, I. V. (2016). New molecular insight into mechanism of evolution of mammalian synthetic prions. Am. J. Pathol. 186, 1006–1014. doi: 10.1016/j.ajpath.2015.11.013
Makarava, N., Savtchenko, R., and Baskakov, I. V. (2013). Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification J. Biol. Chem. 288, 33–41. doi: 10.1074/jbc.M112.419531
Makarava, N., Savtchenko, R., and Baskakov, I. V. (2015). Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathol. Commun. 3:69. doi: 10.1186/s40478-015-0248-5
Málaga-Trillo, E., Solis, G. P., Schrock, Y., Geiss, C., Luncz, L., Thomanetz, V., et al. (2009). Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 7:e1000055. doi: 10.1371/journal.pbio.1000055
Martin, L. T., Marth, J. D., Varki, A., and Varki, N. M. (2002). Genetically altered mice with different sialyltransferase defficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetilation. J. Biol. Chem. 277, 32930–32938. doi: 10.1074/jbc.M203362200
McCulloch, L., Brown, K. L., Bradford, B. M., Hopkins, J., Bailey, M., Rajewsky, K., et al. (2011). Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 7:e1002402. doi: 10.1371/journal.ppat.1002402
Mehrabian, M., Brethour, D., Wang, H., Xi, Z., Rogaeva, E., and Schmitt-Ulms, G. (2015). The prion protein controls polysialylation of neural cell adhesion molecule 1 during cellular morphogenesis. PLoS ONE 10:e0133741. doi: 10.1371/journal.pone.0133741
Michel, B., Meyerett-Reid, C., Johnson, T., Ferguson, A., Wyckoff, C., Pulford, B., et al. (2012). Incunabular imminological events in prion trafficing. Sci. Rep. 2:440. doi: 10.1038/srep00440
Miller, M. W., and Williams, E. S. (2004). Chronic wasting disease of cervids. Curr. Top. Microbiol. Immunol. 284, 193–214. doi: 10.1007/978-3-662-08441-0_8
Miyagi, T., and Yamaguchi, K. (2012). Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology 22, 880–896. doi: 10.1093/glycob/cws057
Monnet, C., Marthiens, V., Enslen, H., Frobert, Y., Sobel, A., and Mège, R. M. (2003). Heterogeneity and regulation of cellular prion protein glycoforms in neuronal cell lines. Eur. J. Neurosci. 18, 542–548. doi: 10.1046/j.1460-9568.2003.02777.x
Monti, E., Bonten, E., D'Azzo, A., Bresciani, R., Venerando, B., Borsani, G., et al. (2010). Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Adv. Carbohydr. Chem. Biochem. 64, 403–479. doi: 10.1016/S0065-2318(10)64007-3
Montrasio, F., Frigg, R., Glatzel, M., Klein, M. A., Mackay, F., Aguzzi, A., et al. (2000). Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288, 1257–1259. doi: 10.1126/science.288.5469.1257
Morales, R., Bravo-Alegria, J., Duran-Aniotz, C., and Soto, C. (2015). Titration of biologically active amyloid-β seeds in a transgenic mouse model of Alzheimer's disease. Sci. Rep. 5:9343. doi: 10.1038/srep09349
Morales, R., Hu, P. P., Duran-Aniotz, C., Moda, F., Diaz-Espinoza, R., Chen, B., et al. (2016). Strain-dependent profile of misfolded prion protein aggregates. Sci. Rep. 6:20526. doi: 10.1038/srep20526
Mouillet-Richard, S., Laurendeau, I., Vidaud, M., Kellermann, O., and Laplamche, J. L. (1999). Prion protein and neuronal differentiation: quantitative analysis of prnp gene expression in a murine inducible neuroectodermal progenitor. Microbes Infect. 1, 969–976. doi: 10.1016/S1286-4579(99)80514-0
Nasirikenari, M., Veillon, L., Collins, C. C., Azadi, P., and Lau, J. T. (2014). Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 sialyltransferase. J. Biol. Chem. 289, 7178–7189. doi: 10.1074/jbc.M113.508457
Ng, P. S., Böhm, R., Hartley-Tassell, L. E., Steen, J. A., Wang, H., Lukowski, S. W., et al. (2014). Ferrets exclusively synthesize Neu5Ac and express naturally humanized influenza A virus receptors. Nat. Commun. 5, 5750. doi: 10.1038/ncomms6750
Nishina, K., Deleault, N. R., Mahal, S., Baskakov, I., Luhrs, T., Riek, R., et al. (2006). The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45, 14129–14139. doi: 10.1021/bi061526k
Novitskaya, V., Makarava, N., Sylvester, I., Bronstein, I. B., and Baskakov, I. V. (2007). Amyloid fibrils of mammalian prion protein induce axonal degeneration in NTERA2-derived terminally differentiated neurons. J. Neurochem. 102, 398–407. doi: 10.1111/j.1471-4159.2007.04537.x
Ostapchenko, V. G., Sawaya, M. R., Makarava, N., Savtchenko, R., Nilsson, K. P., Eisenberg, D., et al. (2010). Two amyloid states of the prion protein display significantly different folding patterns. J. Mol. Biol. 400, 908–921. doi: 10.1016/j.jmb.2010.05.051
Panigaj, M., Glier, H., Wildova, M., and Holada, K. (2011). Expression of prion protein in mouse erythroid progenitors and differentiating murine erythroleukemia cells. PLoS ONE 6:e24599. doi: 10.1371/journal.pone.0024599
Pan, T., Li, R., Kang, S. C., Pastore, M., Wong, B. S., Ironside, J., et al. (2005). Biochemical fingerprints of prion diseases: scrapie prion protein in human prion diseases that share prion genotype and type. J. Neurochem. 92, 132–142. doi: 10.1111/j.1471-4159.2004.02859.x
Peden, A. H., Head, M. W., Ritchie, D. L., Bell, J. E., and Ironside, J. W. (2004). Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364, 527–529. doi: 10.1016/S0140-6736(04)16811-6
Peden, A., McCardle, L., Head, M. W., Love, S., Ward, H. J., Cousens, S. N., et al. (2010). Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16, 296–304. doi: 10.1111/j.1365-2516.2009.02181.x
Peretz, D., Scott, M. R., Groth, D., Williamson, R. A., Burton, D. R., Cohen, F. E., et al. (2001). Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 10, 854–863. doi: 10.1110/ps.39201
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Pshezhetsky, A. V., and Ashmarina, L. I. (2013). Desialylation of Surface Receptors as a New Dimension in Cell Signaling. Biochemistry (Mosc) 78, 736–745. doi: 10.1134/S0006297913070067
Rabinovich, G. A., and Croci, D. O. (2012). Regulatori circuits mediated by lectin-glycan interaction in autoimmunity and cancer. Immunity 36, 322–335. doi: 10.1016/j.immuni.2012.03.004
Requena, J. R., and Wille, H. (2014). The Structure of the infectious prion protein: experimental data and molecular models. Prion 8, 60–66. doi: 10.4161/pri.28368
Rifat, S., Kang, T. J., Mann, D., Zhang, L., Puche, A. C., Stamatos, N. M., et al. (2008). Expression of sialyltransferase activity on intact human neutrophils. J. Leukoc. Biol. 84, 1075–1081. doi: 10.1189/jlb.0706462
Rudd, P. M., Endo, T., Colominas, C., Groth, D., Wheeler, S. F., Harvey, D. J., et al. (1999). Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U.S.A. 96, 13044–13049. doi: 10.1073/pnas.96.23.13044
Samraj, A. N., Pearce, O. M. T., Läubli, H., Crittenden, A. N., Bergfeld, A. K., Banda, K., et al. (2015). A red meat-derived glycan promotes inflammation and cancer progression. Proc. Acad. Natl. Sci. U.S.A. 112, 542–547. doi: 10.1073/pnas.1417508112
Santos, T. G., Silva, I. R., Costa-Silva, B., Lepigue, A. P., Martins, V. R., and Lopes, M. H. (2011). Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 29, 1126–1136. doi: 10.1002/stem.664
Santuccione, A., Syntyk, V., Leshchyns'ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169 341–354. doi: 10.1083/jcb.200409127
Savill, J., Dransfield, I., Gregory, C., and Haslett, C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975. doi: 10.1038/nri957
Schauer, R., Srinivasan, G. V., Wipfer, D., Kniep, B., and Schwartz-Albiez, R. (2011). “O-Acetylated sialic acids and their role in immune defence,” in The Molecular Immunology of Complex Carbohydrates-3, ed A. M. Wu (New York, NY; Dordrecht; Heidelberg; London: Springer Science), 525–548.
Schmitt-Ulms, G., Legname, G., Baldwin, M. A., Ball, H. L., Bradon, N., Bosque, P. J., et al. (2001). Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225. doi: 10.1006/jmbi.2000.5183
Schmitz, M., Lullmann, K., Zafar, S., Ebert, E., Wohlhage, M., Oikonomou, P., et al. (2014). Association of prion protein genotype and scrapie prion protein type with cellular prion protein charge isoform profiles in cerebrospinal fluid of humans with sporadic or familial prion diseases. Neurobiol. Aging 35, 1177–1188. doi: 10.1016/j.neurobiolaging.2013.11.010
Schutt, C. R., and Bartz, J. C. (2008). Prion interference with multiple prion isolates. Prion 2, 61–63. doi: 10.4161/pri.2.2.6806
Schwartz-Albiez, R., Merling, A., Martin, S., Haas, R., and Gross, H. J. (2004). Cell surface sialylation and ecto-sialyltransferase activity of human CD34 progenitors from peripheral blood and bone marrow. Glycoconj. J. 21, 451–459. doi: 10.1007/s10719-004-5535-5
Shikiya, R. A., Ayers, J. I., Schutt, C. R., Kincaid, A. E., and Bartz, J. C. (2010). Coinfecting prion strains compete for a limiting cellular resource. J. Virol. 84, 5706–5714. doi: 10.1128/JVI.00243-10
Sigurdson, C. J., Williams, E. S., Miller, M. W., Spraker, T. R., O'Rourke, K. I., and Hoover, E. A. (1999). Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 80, 2757–2764. doi: 10.1099/0022-1317-80-10-2757
Solforosi, L., Criado, J. R., McGavern, D. B., Wirz, S., Sánchez-Alavez, M., Sugama, S., et al. (2004). Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514–1516. doi: 10.1126/science.1094273
Solomon, I. H., Khatri, N., Biasini, E., Massignan, T., Huettner, J. E., and Harris, D. A. (2011). An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 286, 14724–14736. doi: 10.1074/jbc.M110.214973
Somerville, R. A. (1999). Host and transmissible spongiform encephalopathy agent strain control glycosylation of PrP. J. Gen. Virol. 80, 1865–1872. doi: 10.1099/0022-1317-80-7-1865
Sonati, T., Reimann, R. R., Falsig, J., Baral, P. K., O'Connor, T., Hornemann, S., et al. (2012). The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106. doi: 10.1038/nature12402
Soto, C., Estrada, L., and Castilla, J. (2006). Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 31, 150–155. doi: 10.1016/j.tibs.2006.01.002
Spassov, S., Beekes, M., and Naumann, D. (2006). Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim. Biophys. Acta 1760, 1138–1149. doi: 10.1016/j.bbagen.2006.02.018
Srivastava, S., Makarava, N., Katorcha, E., Savtchenko, R., Brossmer, R., and Baskakov, I. V. (2015). Post-conversion sialylation of prions in lymphoid tissues. Proc. Acad. Natl. Sci. U.S.A. 112, E6654–E6662. doi: 10.1073/pnas.1517993112
Stahl, N., Baldwin, M. A., Hecker, R., Pan, K. M., Burlingame, A. L., and Prusiner, S. B. (1992). Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 31, 5043–5053. doi: 10.1021/bi00136a600
Stahl, N., Baldwin, M. A., Teplow, D. B., Hood, L., Gibson, B. W., Burlingame, A. L., et al. (1993). Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32, 1991–2002. doi: 10.1021/bi00059a016
Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240. doi: 10.1016/0092-8674(87)90150-4
Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S., and Macklis, J. D. (2006). Prion protein (PrPC) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Acad. Natl. Sci. U.S.A. 103, 3416–3421. doi: 10.1073/pnas.0511290103
Stimson, E., Hope, J., Chong, A., and Burlingame, A. L. (1999). Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 38, 4885–4895. doi: 10.1021/bi982330q
Stöhr, J., Condello, C., Watts, C. J., Bloch, L., Oehler, A., Nick, M., et al. (2014). Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Acad. Natl. Sci. U.S.A. 111, 10329–10334. doi: 10.1073/pnas.1408968111
Stura, E. A., Muller, B. H., Bossus, M., Michel, S., Jolivet-Reynaud, C., and Ducancel, F. (2011). Crystal structure of human prostate-specific antigen in a sandwich antibody complex. J. Mol. Biol. 414, 530–544. doi: 10.1016/j.jmb.2011.10.007
Sun, Y., Breydo, L., Makarava, N., Yang, Q., Bocharova, O. V., and Baskakov, I. V. (2007). Site-specific conformational studies of PrP amyloid fibrils revealed two cooperative folding domain within amyloid structure. J. Biol. Chem. 282, 9090–9097. doi: 10.1074/jbc.M608623200
Sun, Y., Makarava, N., Lee, C. I., Laksanalamai, P., Robb, F. T., and Baskakov, I. V. (2008). Conformational stability of PrP amyloid firbils controls their smallest possible fragment size. J. Mol. Biol. 376, 1155–1167. doi: 10.1016/j.jmb.2007.12.053
Svennerholm, L., Bostrom, K., and Jungbjer, B. (1997). Changes in weight and compositions of major mambrane components oh human brain during the span of adult human life of Swedes. Acta Neuropathol. 94, 345–352. doi: 10.1007/s004010050717
Takakura, I., Muiyazawa, K., Kanaya, T., Itani, W., Watanabe, K., Ohwada, S., et al. (2011). Orally administered prion protein is incorporated by m cells and spreads into lymphoid tissues with macrophages in prion protein knockout mice. Am. J. Pathol. 179, 1301–1309. doi: 10.1016/j.ajpath.2011.05.058
Takashima, S. (2008). Characterization of mouse sialylatransferase gene: their evolution and diversity. Biosci. Biotechnol. Biochem. 72, 1155–1167. doi: 10.1271/bbb.80025
Takashima, S., Truji, S., and Tsujimoto, M. (2002). Characterization of the second type of human beta-galactoside alpha 2,6-sialyltransferase (ST6Gal II), which sialylates Galbeta 1,4GlcNAc structures on oligosaccharides preferentially. Genomic analysis of human sialyltransferase genes. J. Biol. Chem. 277, 45719–45728. doi: 10.1074/jbc.M206808200
Takashima, S., Tsuji, S., and Tsujimoto, M. (2003). Comparison of the enzymatic properties of mouse beta-galactoside alpha2,6-sialyltransferases, ST6Gal I and II. J. Biochem. 134, 287–296. doi: 10.1093/jb/mvg142
Tanaka, K., Siwu, E. R. O., Minami, K., Hasegawa, K., Nozaki, S., Kanayama, Y., et al. (2010). Noninvasive imaging of dendrimer-type N-glycan clusters: in vivo dynamics dependence on oligosaccharide structure. Angew. Chem. Int. Ed. Engl. 49, 8195–8200. doi: 10.1002/anie.201000892
Thomzig, A., Spassov, S., Friedrich, M., Naumann, D., and Beekes, M. (2004). Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. J. Biol. Chem. 279, 33854. doi: 10.1074/jbc.m403730200
Turk, E., Teplow, D. B., Hood, L. E., and Prusiner, S. B. (1988). Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176, 21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x
Turnbaugh, J. A., Unterberger, U., Saá, P., Massignan, T., Fluharty, B. R., Bowman, F. P., et al. (2012). The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J. Neurosci. 32, 8817–8830. doi: 10.1523/JNEUROSCI.1103-12.2012
Tycko, R., Savtchenko, R., Ostapchenko, V. G., Makarava, N., and Baskakov, I. V. (2010). The a-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel á-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry 49, 9488–9497. doi: 10.1021/bi1013134
Ushijima, H., Perovic, S., Leuck, J., Rytik, P. G., Müller, W. E., and Schröder, H. C. (1999). Suppression of PrP(Sc)- and HIV-1 gp120 induced neuronal cell death by sulfated colominic acid. J. Neurovirol. 5, 289–299. doi: 10.3109/13550289909015815
Varki, A. (1999). “Sialic acids,” in Essentials of Glycobiology, eds A. Varki, R. Cummings, J. Esko, H. Freeze, G. Hart, and J. Marth (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 195–210.
Varki, A. (2008). Sialic acids in human health and disease. Trends Mol. Med. 14, 351–360. doi: 10.1016/j.molmed.2008.06.002
Varki, A. (2010). Uniquely human evolution of sialic acid genetics and biology. Proc. Acad. Natl. Sci. U.S.A. 107, 8939–8946. doi: 10.1073/pnas.0914634107
Viegas, P., Chaverot, N., Enslen, H., Perrière, N., Couraud, P. O., and Cazaubon, S. (2006). Junctional expression of the prion protein PrPC by brain endothelial cells: a role in trans-andothelial migration of human monocytes. J. Cell Sci. 119, 4634–4643. doi: 10.1242/jcs.03222
Wadsworth, J. D., Asante, E. A., Desbruslais, M., Linehan, J. M., Joiner, S., Gowland, I., et al. (2004). Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306, 1793–1796. doi: 10.1126/science.1103932
Wadsworth, J. D., Joiner, S., Hill, A. F., Campbell, T. A., Desbruslais, M., Luthert, P. J., et al. (2001). Tissues distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunobloting assay. Lancet 358, 171–180. doi: 10.1016/S0140-6736(01)05403-4
Walker, L. C., and Jucker, M. (2015). Neurodegenerative diseases: expanding the prion concept. Annu. Rev. Neurosci. 38, 87–103. doi: 10.1146/annurev-neuro-071714-033828
Wang, S., Chen, X., Wei, A., Yu, X., Niang, B., and Zhang, J. (2015). α2,6-linked sialic acids on N-glycans modulate the adhesion of hepatocarcinoma cells to lymph nodes. Tumor Biol. 36, 885–892. doi: 10.1007/s13277-014-2638-x
Wasmer, C., Lange, A., Van Melckebeke, H., Siemer, A. B., Riek, R., and Meier, B. H. (2008). Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 319, 1523–1526. doi: 10.1126/science.1151839
Watts, C. J., Condello, C., Stöhr, J., Oehler, A., Lee, J., DeArmond, S. J., et al. (2014). Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc. Acad. Natl. Sci. U.S.A. 111, 10323–10328. doi: 10.1073/pnas.1408900111
Westergard, L., Turnbaugh, J. A., and Harris, D. A. (2011). A nine amino acid domain is essential for mutant prion protein toxicity. J. Neurosci. 31, 14005–14017. doi: 10.1523/JNEUROSCI.1243-11.2011
Wille, H., Bian, W., McDonald, M., Kendall, A., Colby, D. W., Bloch, L., et al. (2009). Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Acad. Natl. Sci. U.S.A. 106, 16990–16995. doi: 10.1073/pnas.0909006106
Wille, H., Michelitsch, M. D., Guenebaut, V., Supattapone, S., Serban, A., Cohen, F. E., et al. (2002). Structural studies of the scrapie prion protein by electron crystallography. Proc. Acad. Natl. Sci. U.S.A. 99, 3563–3568. doi: 10.1073/pnas.052703499
Wroe, S. J., Pal, S., Siddique, D., Hyare, H., Macfariane, R., Joiner, S., et al. (2006). Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368, 2061–2067. doi: 10.1016/S0140-6736(06)69835-8
Zanusso, G., Farinazzo, A., Prelli, F., Fiorini, F., Gelati, M., Ferrari, S., et al. (2004). Identification of distinct N-terminal truncated forms of prion protein in different Creutzfeldt-Jakob disease subtypes. J. Biol. Chem. 279, 38936–38942. doi: 10.1074/jbc.M405468200
Zhang, C. C., Steele, A. D., Lindquist, S., and Lodish, H. F. (2006). Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Acad. Natl. Sci. U.S.A. 103, 21814–21819. doi: 10.1073/pnas.0510577103
Keywords: prions, prion disease, amyloid, sialic acid, sialylation, species barrier, sialyltransferase, neuraminidase
Citation: Baskakov IV and Katorcha E (2016) Multifaceted Role of Sialylation in Prion Diseases. Front. Neurosci. 10:358. doi: 10.3389/fnins.2016.00358
Received: 23 May 2016; Accepted: 18 July 2016;
Published: 08 August 2016.
Edited by:
Wendy Noble, King's College London, UKReviewed by:
Maria Dolores Ledesma, Centro de Biología Molecular Severo Ochoa, SpainGerold Schmitt-Ulms, University of Toronto, Canada
Copyright © 2016 Baskakov and Katorcha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilia V. Baskakov, YmFza2Frb3ZAdW1hcnlsYW5kLmVkdQ==