 Tesfaye W. Tefera
Tesfaye W. Tefera Karin Borges
Karin Borges- Laboratory for Neurological Disorders and Metabolism, School of Biomedical Sciences, Department of Pharmacology, The University of Queensland, Brisbane, QLD, Australia
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease primarily characterized by loss of motor neurons in brain and spinal cord. The death of motor neurons leads to denervation of muscle which in turn causes muscle weakness and paralysis, decreased respiratory function and eventually death. Growing evidence indicates disturbances in energy metabolism in patients with ALS and animal models of ALS, which are likely to contribute to disease progression. Particularly, defects in glucose metabolism and mitochondrial dysfunction limit the availability of ATP to CNS tissues and muscle. Several metabolic approaches improving mitochondrial function have been investigated in vitro and in vivo and showed varying effects in ALS. The effects of metabolic approaches in ALS models encompass delays in onset of motor symptoms, protection of motor neurons and extension of survival, which signifies an important role of metabolism in the pathogenesis of the disease. There is now an urgent need to test metabolic approaches in controlled clinical trials. In addition, more detailed studies to better characterize the abnormalities in energy metabolism in patients with ALS and ALS models are necessary to develop metabolically targeted effective therapies that can slow the progression of the disease and prolong life for patients with ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a complex multi-pathogenic neurodegenerative disorder primarily characterized by the loss of upper motor neurons in the brain and lower motor neurons in the spinal cord and the brain stem (Robberecht and Philips, 2013). As a result of this neuronal loss, motor function declines progressively which then leads to impairments in respiratory function and eventually death within 2–5 years of diagnosis (Kiernan et al., 2011). The exact mechanisms underlying the selective loss of motor neurons in the disease are not yet known. However, several mechanisms appear to contribute, including glutamate excitotoxicity (Rothstein et al., 1990), oxidative stress (Barber et al., 2006), protein aggregation (Rosen et al., 1993; Bruijn et al., 1998), cytoskeletal and axonal transport abnormalities (Julien and Beaulieu, 2000), inflammation (Julien and Beaulieu, 2000), hyperexcitability (Zanette et al., 2002; Vucic and Kiernan, 2006; Vucic et al., 2008) and altered energy metabolism (Dupuis et al., 2004, 2011). It is believed that several of these mechanisms are interconnected and together are responsible for the progression of the disease. To date, there is only one approved medication available to treat ALS, namely riluzole, which only increases survival by 3 months (Bensimon et al., 1994). Therefore, it is imperative to look for other potential treatments that can halt the progression of the disease and extend life. In this review, we discuss impairments in energy metabolism in ALS and review the effects of metabolic therapies investigated so far in animal models of ALS and/or patients with ALS.
Energy Metabolism Disturbances in ALS
There are many lines of evidence for the role of disturbed energy metabolism in ALS pathogenesis. Energy homeostasis, the balance between energy intake and expenditure, appears to be compromised in patients with ALS (Dupuis et al., 2011). Besides, some patients exhibit hypermetabolism (an increased metabolism at rest) (Kasarskis et al., 1996; Desport et al., 2001; Bouteloup et al., 2009; Marin et al., 2011), body weight loss (Körner et al., 2013b) and abnormal metabolism of lipids (Dupuis et al., 2011), which are likely to contribute to disease. These and many studies discussed below reveal problems in energy metabolism in whole tissues and even the whole organism. It is not entirely clear to which extent the metabolic changes found in ALS patients and ALS models only arise as a result of neurodegeneration. Moreover, little is known about the specific metabolic changes within the motor neurons themselves and if they contribute to the degeneration. Other early reviews discuss reasons for the specific degeneration of motor neurons in ALS (Shaw and Eggett, 2000; Cleveland and Rothstein, 2001).
Specific Dysfunctions in Energy Producing Pathways in ALS
Under normal physiological conditions, glucose is the main energy substrate for neuronal tissue and a major fuel for muscles. However, there are indications of defects in the glucose metabolizing pathways in ALS namely in the glycolysis and the TCA cycle pathways in CNS and muscle (Palamiuc et al., 2015; Tefera et al., 2016). In addition, there is decreased glucose uptake in numerous brain regions and spinal cord of animal models and patients with ALS (Dalakas et al., 1987; Hatazawa et al., 1988; Ludolph et al., 1992; Browne et al., 2006; Miyazaki et al., 2012; Cistaro et al., 2014). In muscle, namely the tibialis anterior muscle of the superoxide dismutase 1 (SOD1G86R) mice, the activities of the enzyme phosphofructokinase 1 (PFK1), the rate limiting enzyme in glycolysis, were reduced (Palamiuc et al., 2015). Furthermore, in the gastrocnemius muscle of SOD1G93A mice, the activities of the TCA cycle enzyme 2-oxoglutarate dehydrogenase (OGDH) were found to be reduced in the late symptomatic phases of the disease, indicating impairments in the TCA cycle pathway (Tefera et al., 2016). Furthermore, changes in the expression of several genes involved in energy metabolism have been observed in muscle, brain and spinal cord tissues and cells isolated from them. Particularly, genes involved in the mitochondrial electron transport chain are significantly altered (Ferraiuolo et al., 2007, 2011; Lederer et al., 2007; Raman et al., 2015). Mitochondria are the main site of energy production and they are also the main sites for the generation of reactive oxygen species. Compromised mitochondrial function has been recognized for a long time in many neurodegenerative disorders, which is expected to lead to reduced production of ATP and increased oxidative stress (Cozzolino and Carrì, 2012). Similarly in ALS, numerous studies have shown functional and structural abnormalities in mitochondria that give rise to reduced oxidative phosphorylation and subsequent decreased generation of ATP (Jung et al., 2002; Mattiazzi et al., 2002; Menzies et al., 2002; Wiedemann et al., 2002; Browne et al., 2006). In addition, in vitro studies showed that mitochondrial dysfunction in astrocytes may lead to generation of free radicals and release of toxic factors that could contribute to motor neuron dysfunction (Nagai et al., 2007; Cassina et al., 2008). In conclusion, pathological changes in the energy metabolizing pathways that lead to deficiency in ATP production and/or utilization may contribute to progression of the disease.
Role of Glial Cells in Metabolism and ALS
Although ALS is mainly characterized by the selective loss of motor neurons, neighboring non-neuronal cells such as astrocytes (Clement et al., 2003; Nagai et al., 2007), microglia (Beers et al., 2006; Boillée et al., 2006b) and oligodendrocytes (Yamanaka et al., 2008; Lee et al., 2012; Kang et al., 2013) can contribute to the progression of the disease (reviewed by Boillée et al., 2006a). The metabolic coupling between neurons and astrocytes has long been known. During neurotransmission, neurons release glutamate into the synapse which can be taken up by astrocytes. It is then metabolized to glutamine, which can be supplied to neurons to resynthesize glutamate. Many immunohistochemical studies have shown the loss of glutamate transporters in astrocytes in spinal cord and brain of patients with ALS (Rothstein et al., 1995; Fray et al., 1998; Sasaki et al., 2000). Deficiencies in glutamate uptake and the glutamate-glutamine cycle can lead to problems in neuronal signaling and promotes excitotoxicity (reviewed by Danbolt, 2001; Van Den Bosch et al., 2006). In addition, according to the astrocyte-neuron lactate shuttle hypothesis, glutamate stimulated metabolism of glucose in astrocytes produces lactate which can be transferred to neurons to be utilized as fuel (Pellerin and Magistretti, 1994). Altered gene expression of the astrocytic lactate efflux transporter (monocarboxylate transporter 4, SLC16A4) together with reduced lactate levels found in the spinal cord of hSOD1G93A mice further suggest that metabolic interaction between neurons and astrocytes may be disrupted in ALS (Ferraiuolo et al., 2011).
The potential of metabolic coupling between neurons and oligodendrocytes has gained a lot of interest in recent years and it appears that oligodendrocytes also can release lactate. Recent studies displayed the involvement of oligodendrocytes in the pathogenesis of ALS (Lee et al., 2012; Kang et al., 2013; Philips et al., 2013). Healthy oligodendrocytes can metabolically support neurons by providing energy metabolites via monocarboxylate transporters (Fünfschilling et al., 2012; Lee et al., 2012; Morrison et al., 2013). In contrast, the transport of glycolytic substrates such as lactate from oligodendrocytes to neurons is found to be disrupted in SOD1G93A mice spinal cord (Lee et al., 2012; Kang et al., 2013). Consistent with this, the expressions of the oligodendrocytic lactate transporter monocarboxylate transporter 1 (SLC16A1) was decreased in the spinal cords of patients with ALS and SOD1G93A mice (Lee et al., 2012; Philips et al., 2013) suggesting abnormalities in lactate transfer.
In addition to astrocytes and oligodendrocytes, microglia have been shown to be involved in ALS pathogenesis (Beers et al., 2006; Boillée et al., 2006b). Microglia are immune cells within the CNS, similar to macrophages, which can either have a neuroprotective or a neurotoxic role during ALS progression. It is believed that microglia protect neurons early in the disease stage. However, as the disease progresses, activated microglia can be pro-inflammatory and neurotoxic (Appel et al., 2011; Dibaj et al., 2011). In vivo imaging studies demonstrated increased microglia activation and inflammatory activity in spinal cord during symptomatic stages in the SOD1G93A mouse model (Dibaj et al., 2011). Reduced expression of mutant SOD1 in microglia delayed late disease progression and increased survival of SOD1G37R mice (Boillée et al., 2006b). In conclusion, perturbations in glial cells can affect neurons and directly or indirectly can contribute to energy deficits in neurons. The studies discussed demonstrate the importance of non-neuronal cells in supporting neurons in various ways and indicate that abnormalities in glial cells can exacerbate the progression of the disease.
High Calorie Diets in ALS
A number of studies have tried to correct the energy deficit using metabolic therapies in animal models and patients with ALS, with the hope that an improved metabolism can delay symptoms and extend survival in ALS (Dupuis et al., 2004; Zhao et al., 2006, 2012; Mattson et al., 2007; Miquel et al., 2012; Ari et al., 2014; Wills et al., 2014; Palamiuc et al., 2015; Tefera et al., 2016). A study by Dupuis et al. (2004) in SOD1G86R mice found aberrations of energy metabolism such as reduction of adiposity, increased energy expenditure and lipolysis, and examined the effects of a high fat diet that consisted of regular chow supplemented with 21% (wt/wt) butter fat and 0.15% (wt/wt) cholesterol. A 20% increase in the mean survival of these mice compared to mice fed with control diet was demonstrated and motor neuron loss was reduced. Moreover, SOD1G93A mice fed either a high fat diet (containing 47% fat, 38% carbohydrates and 15% protein) or with control diet (constituting 17% fats, 64% carbohydrates and 19% protein) starting at 6 weeks of age also showed an increase in survival and the onset of motor symptoms and weight loss were delayed significantly. While mice treated with control diet died by 180 days, high fat died treated mice survived until 220 days and some of them survived more than 270 days (Mattson et al., 2007). On a similar note, several studies have alluded that body mass index is strongly correlated with survival of patients with ALS (reviewed in Ngo et al., 2014). A prospective study of over a million people for about 14–28 years demonstrated that obese people have a 30–40% lower risk of developing ALS compared to those with a healthy weight (O'Reilly et al., 2013). Given the strong link found between low body mass index and decreased survival with ALS (Paganoni et al., 2011), a randomized double blind phase 2 clinical study was performed in small number of patients with ALS to test the safety and tolerability of high caloric diets, which were either high in carbohydrates or high in fat. Compared to an isocaloric control diet, the high carbohydrate-high calorie diet was safe, tolerable and effective in delaying weight loss and prolonging survival in the patients (Wills et al., 2014). Further large scale trials are needed to corroborate these promising effects. Also due to the small sample size, a beneficial effect of a high fat high calorie diet cannot yet be ruled out completely. On the other hand, high fat diets are linked with side effects such as brain inflammation, cognitive deficits, and anxiety and depressive behaviors (Zhang et al., 2005; Pistell et al., 2010; Dutheil et al., 2016). Together, these studies show the importance of adequate energy supply to slow the progression of ALS.
Metabolic Treatments in ALS
Given that defects in energy metabolism are evident in ALS, addressing these disturbances is a reasonable approach to improve quality of life and prolong survival in patients with ALS. Because of the impairments found in glucose metabolism together with the increased energy demand in neurons and muscles, additional fuel appears to be important. To increase energy uptake, additional alternative metabolic fuels for the CNS and muscle also appear warranted and could include ketones and medium chain fats, but also metabolic intermediates, such as pyruvate, lactate, α-ketoglutarate and others. Moreover, compounds that can enhance mitochondrial function to generate more energy are expected to ameliorate disease progression. This includes substances that improve mitochondrial function by inhibiting oxidative stress or stabilizing mitochondrial membranes. There are also supplements that provide energy substrates that can refill deficient C4 carbon TCA cycle intermediates (anaplerosis), which thereby will improve oxidative phosphorylation of any fuel in addition to providing alternative fuel. This includes triheptanoin and α-ketoglutarate. Consistent with these theoretical notions, alternative fuels, metabolic supplements and mitochondrial function enhancers have been investigated with the purpose of improving ALS symptoms and delaying disease progression in mouse models of ALS, cell cultures and in patients with ALS. These metabolic potential treatments include triheptanoin, ketogenic diet, caprylic triglyceride, dichloroacetate, the Deanna protocol, pyruvate, lactate, creatine, coenzyme Q10, MitoQ, dexpramipexole, acetyl-L-carnitine and olesoxime, which we will discuss below. Their mechanisms and effects are also summarized in Figure 1.
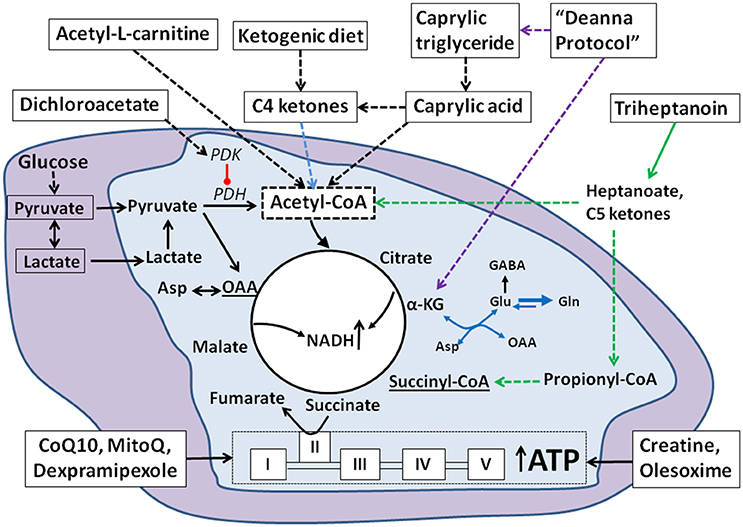
Figure 1. Summary of cellular mechanisms of metabolic approaches in ALS. Under normal physiological conditions, glucose is the main fuel for CNS or muscle cells and is metabolized via glycolysis into two molecules of pyruvate in the cytosol (purple background). Pyruvate can be converted by pyruvate dehydrogenase (PDH) into acetyl CoA in the mitochondria (light blue background), which enters the TCA cycle by condensing with oxaloacetate (OAA) to form citrate. Citrate gets further metabolized to generate NADH and ATP. Lactate is a glycolytic product that can be used to provide energy. According to the lactate shuttle hypothesis, lactate can also be transported into the mitochondria via monocarboxylate transporters and then get converted to pyruvate to enter the TCA cycle. In ALS, where energy demand is high and glucose metabolism is defective, alternative fuels can be provided by the ketogenic diet producing C4 ketones and the medium chain triglycerides (MCT). Among the MCTs, caprylic triglyceride is metabolized to C4 ketones and caprylic acid and triheptanoin provides heptanoate and C5 ketones, which all can be taken up from the blood and used by the CNS and muscle cells. The C4 and C5 ketones as well as caprylic acid are converted into acetyl-CoA and enter the TCA cycle. Increased C4 ketone levels can also activate astrocytes and facilitate conversion of glutamate into glutamine. This further provides more glutamine for the synthesis of GABA and less conversion of glutamate to aspartate (blue arrows, see section The Ketogenic Diet and Caprylic Triglyceride). Triheptanoin provides three molecules of heptanoate (green arrows), which can be converted into C5 ketones in the liver. Heptanoate and C5 ketones can enter the brain and muscle to be metabolized to acetyl-CoA and propionyl CoA. Propionyl CoA is an anaplerotic molecule that can be further converted into succinyl-CoA and enter into the TCA cycle. This restores (lost) C4 TCA cycle intermediate levels and promotes TCA cycling. Dichloroacetate is a PDH kinase inhibitor, which activates PDH thereby increasing the entry of pyruvate into the TCA cycle. Creatine provides high energy phosphates and stabilizes membranes and hence enhances mitochondrial function. The mitochondrial targeted antioxidants CoQ10, MitoQ and dexpramipexole promote mitochondrial energy generation. Olesoxime stabilizes mitochondrial membrane permeability transition pores. The Deanna protocol is a combination of supplements that can provide acetyl CoA and intermediates, such as α-ketoglutarate (α-KG) that feed into the TCA cycle (purple dashed arrows).
Triheptanoin
Triheptanoin is an alternative fuel, which can also boost the metabolism of other fuels by refilling deficient C4 TCA cycle metabolites and is therefore ideal for disorders with high energy needs. Triheptanoin is a medium chain triglyceride (MCT) containing the odd chain C7 fatty acid heptanoate. After hydrolysis in the gastro-intestinal tract, medium chain fatty acids diffuse into cells, the blood stream and into mitochondria. This enhances their availability to various organs including the CNS and muscles. In contrast, long chain fatty acids are bound to plasma proteins and require transporter proteins for uptake into cells and mitochondria (Schönfeld and Wojtczak, 2016). When heptanoate is metabolized by β-oxidation, it generates two molecules of acetyl-CoA and one molecule of propionyl-CoA. In the liver, heptanoate can form the C5 ketones, β-ketopentanoate and β-hydroxypentanoate, which are released into the blood and can be taken up by neuronal and other cells through monocarboxylate transporters. Similar to heptanoate, the C5 ketones give rise to acetyl-CoA and propionyl-CoA. Propionyl-CoA enters the TCA cycle after it is carboxylated by propionyl-CoA carboxylase and then metabolized to succinyl-CoA, thereby (re-) filling the (deficient) C4 TCA cycle intermediate pool (reviewed in Brunengraber and Roe, 2006). C4 TCA cycle intermediates contribute to the formation of oxaloacetate, which is needed to condense with acetyl-CoA to citrate. Thus, overall heptanoate and C5 ketones provide additional alternative fuel to form acetyl-CoA and by increasing the levels of oxaloacetate improve acetyl-CoA entry into the TCA cycle, required for aerobic ATP production. The ability of triheptanoin to provide both acetyl-CoA and propionyl-CoA makes it an ideal metabolic treatment for disorders with deficient levels of TCA cycle intermediates and impairments in energy production. For instance, triheptanoin alleviated metabolic disturbances associated with several neurological and neuromuscular disorders and improved symptoms in models of epilepsy (Willis et al., 2010; Thomas et al., 2012; Hadera et al., 2014), Canavan disease (Francis et al., 2014), autism disorders (Park et al., 2014) and Alzheimer's Disease (Aso et al., 2013) as well as patients and a mouse model of glucose transporter 1 deficiency (Marin-Valencia et al., 2013; Pascual et al., 2014; Mochel et al., 2016), and patients with Huntington's disease (Mochel et al., 2010; Adanyeguh et al., 2015). In chronically “epileptic” mice it was shown that triheptanoin partially restores reduced TCA cycle intermediate and metabolite levels (Willis et al., 2010; Hadera et al., 2014). Cognisant of the positive effects of triheptanoin in other brain and neuromuscular disorders and its mechanism of action, we hypothesized that treatment with triheptanoin, being an alternative substrate to glucose provides degenerating cells with much needed energy, which can delay cell death and thereby the onset of motor symptoms in the ALS mouse model. Thus we recently investigated the effects of triheptanoin in the hSOD1G93A mice (Tefera et al., 2016).
We first showed that in the gastrocnemius muscle of the hSOD1G93A mice, gene expression of enzymes involved in the glycolysis, TCA cycle and anaplerotic pathways such as glyceraldehyde-3-phosphate dehydrogenase, pyruvate, 2-oxoglutarate and succinate dehydrogenases, at earlier (10 weeks) and at later symptomatic stages (25 weeks) of the disease was reduced. Downregulation of these metabolic enzymes in muscle even at early stages where grip strength was still intact suggests that metabolic defects in the energy producing pathways could contribute to disease progression in ALS (Tefera et al., 2016). We also found reduced activities of 2-oxoglutarate dehydrogenase enzyme at the late stage of the disease (25 weeks) indicating abnormalities in the TCA cycle pathway. We then showed that treatment of hSOD1G93A mice with triheptanoin (where 35% of calories were from this oil, 17% w/diet w) from P35 until P70 prevented downregulation of the enzymes such as succinate dehydrogenase, glutamic pyruvic transaminase 2 and the propionyl carboxylase β subunit compared to mice treated with control diet. Similarly, treatment with triheptanoin protected against motor neuron loss by 33% in the lumbar spinal cord (Tefera et al., 2016). It also delayed the onset of motor symptoms which was shown by improvements in grip strength and rotarod tests. It is believed that the neuroprotective effects of triheptanoin due to increased ATP generation contribute to improvements in motor function. In addition, increased ATP generation in remaining neurons and muscle is likely to also benefit motor function. Please note that our study was underpowered to determine changes in survival. It remains to be seen if triheptanoin is effective in other rodent ALS models and if it can be tolerated by patients with ALS.
The Ketogenic Diet and Caprylic Triglyceride
Ketogenic diets consist of extremely high amounts of fats, while carbohydrates and proteins are restricted. They are successfully used for the treatment of childhood epilepsies, where several controlled clinical trials have now shown efficacy (Martin et al., 2016) and there is also some efficacy against seizures in adults. Ketogenic diets are now being tested in a variety of disorders, including some characterized by neurodegeneration, such as Alzheimer's Disease (Van der Auwera et al., 2005; Henderson et al., 2009) and traumatic brain (Prins et al., 2005; Appelberg et al., 2009; Hu et al., 2009b; Deng-Bryant et al., 2011) and spinal injury (Streijger et al., 2013). Administration of ketones after controlled cortical impact injury in young rats reduced lesion volume, attenuated motor and cognitive deficits, reduced cellular apoptotic markers, and improved cerebral ATP levels (Prins et al., 2005; Appelberg et al., 2009; Hu et al., 2009a,b; Deng-Bryant et al., 2011). Also, BHB was able to rescue neurons from degeneration, alleviate motor deficits against 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity, and improve mitochondrial ATP generation (Tieu et al., 2003). Similarly, it improved cerebral energy metabolism in ischemic rat brain (Suzuki et al., 2001) and prevented glutamate mediated neuronal toxicity and lipid peroxidation (Mejía-Toiber et al., 2006). While on a ketogenic diet, blood glucose levels remain consistently in the lower part of the normal range and the liver produces the C4 ketones, β-hydroxy butyrate (BHB) and aceto-acetate, which are secreted into the blood. If C4 ketones are available, the CNS will use C4 ketones as fuel preferably to glucose (Courchesne-Loyer et al., 2016).
In SOD1G93A mice, a ketogenic diet was shown to protect against the loss of motor neurons in the spinal cord and to delay the onset of behavioral symptoms (Zhao et al., 2006). BHB was also able to increase the rate of ATP generation in mitochondria isolated from the spinal cord of SOD1G93A mice as well as in cultured spinal cord neurons from these mice in the presence of rotenone (Zhao et al., 2006). Mechanisms by which the ketogenic diet modifies disease progression are probably multifold (Masino and Rho, 2012). Most likely the use of C4 ketones as alternative fuels as well as their anti-oxidant and anti-inflammatory effects plays important roles. Moreover, C4 ketones can alter metabolism in the brain. It has been hypothesized that ketones activate astrocytes and hence increases the conversion of glutamate to glutamine by glutamine synthetase thereby reducing the availability of glutamate in the synapse (Yudkoff et al., 2008). This reduces glutamate excitotoxicity, which is one of the mechanisms most likely involved in the pathogenesis of ALS (Rothstein et al., 1990). Moreover, according to the glutamate-glutamine-GABA cycle, astrocytes release glutamine to neurons and inhibitory neurons convert it to GABA. Thus, more glutamine in astrocytes may also lead to increased availability of glutamate for the synthesis of GABA, the primary inhibitory neurotransmitter in the brain (Yudkoff et al., 2008). It is also hypothesized that this leads to less glutamate available to produce aspartate, another excitatory neurotransmitter in the brain (Yudkoff et al., 2008). Cortical hyperexcitability due to reduced intracortical inhibition has been shown in patients with ALS and preceded the onset of ALS symptoms (Vucic and Kiernan, 2006; Vucic et al., 2008). Reductions in GABA levels found in patients with ALS in motor cortex compared to healthy controls (Foerster et al., 2012, 2013) may contribute to cortical hyperexcitability. Similarly, in the cortex of wobbler mouse model of ALS, reduced GABA mediated inhibition in the layer 5 pyramidal neurons as a result of loss of GABAergic interneurons was shown, resulting in cortical excitability (Nieto-Gonzalez et al., 2011).
The MCT of eight carbon octanoate, caprylic triglyceride, has also been investigated (Zhao et al., 2012). After hydrolysis of the ester bonds, octanoic (caprylic) acid can be metabolized quickly by β-oxidation to acetyl CoA and C4 ketone bodies to supply the TCA cycle. As mentioned above, one advantage of medium chain fatty acids is that they are able to easily diffuse through plasma as well as mitochondrial membranes and do not require transport proteins (Schönfeld and Wojtczak, 2016). Thus, MCTs can serve as a fast fuel especially in conditions with decreased uptake of glucose. In SOD1G93A mice, feeding 10% (w/w) caprylic triglyceride improved motor performance in the rotarod test, protected against motor neuron loss and increased oxygen consumption in mitochondria isolated from the spinal cord (Zhao et al., 2012). Please note that this MCT dose is not very high and blood ketone levels were unchanged in the presymptomatic phase and only about doubled in the postsymptomatic phase in SOD1G93A mice. In comparison while on ketogenic diet the levels quadrupled in the same mouse strain (Zhao et al., 2006). This may indicate that octanoate itself may be sufficient for neuroprotection and ketone formation may not be necessary.
Disappointingly, neither the ketogenic diet nor caprylic triglyceride improved the survival of SOD1G93A mice (Zhao et al., 2006, 2012). Also, it is important to note that in other studies octanoate limited glucose metabolism in rodents (Wlaz et al., 2012; McDonald et al., 2014). Given that glucose utilization is compromised in ALS, further inhibiting glucose metabolism may worsen disease progression. Last, ketogenic diets are associated with loss of body weight, which in ALS can worsen disease progression. Thus, these apparently successful metabolic approaches in this ALS mouse model cannot be fully recommended for ALS patients.
The Deanna Protocol
The Deanna protocol is a combination of nutritional supplements containing energy supplements, vitamins, and antioxidants such as glutathione, nicotinamide adenine dinucleotide (NADH), coenzyme Q10 (CoQ10) and ubiquinol in addition to MCTs if tolerated. It was claimed to have beneficial effects in anecdotal reports (Fournier et al., 2013). The effects of the main constituents of the supplement, including arginine α-ketoglutarate (AAKG), MCT rich in caprylic triglyceride and GABA, were tested in SOD1G93A mice (Ari et al., 2014). AAKG provides α-ketoglutarate, which is taken up by sodium-dependent transport carriers into synaptosomes and can be incorporated into the TCA cycle (Shank and Campbell, 1981, 1984). In addition, the medium chain fats can be rapidly metabolized and easily diffuse into mitochondria to provide acetyl CoA. Ari and colleagues showed that this simplified metabolic treatment delayed motor symptoms and increased survival in SOD1G93A mice (Ari et al., 2014). In our opinion, the true effects of the Deanna protocol in patients with ALS are worthwhile to be studied. However, this will be very difficult, because most patients take dietary supplements and vitamins.
Dichloroacetate
Dichloroacetate (DCA) is a pyruvate dehydrogenase (PDH) kinase inhibitor (Whitehouse et al., 1974). Inhibition of PDH kinase leads to less phosphorylation of PDH resulting in activation of this enzyme that catalyzes the conversion of pyruvate to acetyl CoA (McAllister et al., 1973). Thus, DCA facilitates oxidation of glucose and generation of ATP in the mitochondria (Itoh et al., 2003). The effects of DCA were examined in the SOD1G93A mouse model of ALS (Miquel et al., 2012). DCA prevented the toxicity of astrocytes to motor neurons in neuron-astrocyte co-cultures from SOD1G93A rat spinal cord. In addition, when DCA was administered to SOD1G93A mice starting at 10 weeks; it reduced motor neuron loss in the lumbar spinal cord by 25%, delayed onset of ALS motor symptoms in a grip strength test and improved survival (Miquel et al., 2012). The mechanism by which it modifies disease progression is believed to be improved astrocytic TCA cycling and mitochondrial function (Miquel et al., 2012). In another similar study by Palamiuc et al. (2015) in SOD1G86R mice, the protective effects of DCA were corroborated. In this study, DCA improved grip strength performance, prevented downregulation of mitochondrial genes, and protected against muscle denervation. At doses used in metabolic studies, DCA was well tolerated in patients with brain cancers (Dunbar et al., 2014). Its safety, tolerability and efficacy in ALS patients remain to be studied.
Pyruvate
Pyruvate is a glycolytic metabolite that can enter the TCA cycle following its conversion into acetyl CoA. It was shown to be neuroprotective in cultured mouse striatal neurons against N-methyl D-aspartate (NMDA) and H2O2 mediated toxicity (Desagher et al., 1997; Maus et al., 1999) and in cortical neuronal cultures subjected to oxygen-glucose deprivation by improving cellular energy metabolism, reducing reactive oxygen species levels and anti-inflammatory mechanisms (Shen et al., 2010; Pan et al., 2012). Pyruvate's ability to protect cortical neurons against H2O2 toxicity involves monocarboxylate transporters (Nakamichi et al., 2005). In juvenile rat hippocampal slices, pyruvate administration was able to restore ATP levels and rescue neurons against NMDA mediated excitotoxicity (Izumi and Zorumski, 2010). Pyruvate treatment reduced infarct size and improved neurological functions in a rat middle cerebral artery occlusion model of ischemia (Yu et al., 2005). It also improved cerebral oxidative metabolism and neurological outcomes in rats following cortical contusion injury (Fukushima et al., 2009; Moro and Sutton, 2010), protected against MPTP induced degeneration of neurons and reduced oxidative stress in a mouse model of Parkinson's disease (Huh et al., 2011; Satpute et al., 2013), and reduced cognitive deficits in a mouse model of Alzheimer's disease (Isopi et al., 2015).
In the SOD1G93A mouse model of ALS, pyruvate (1000 mg/kg/week i.p.) slowed disease progression and also improved motor symptoms in a rotarod test and survival by 10.5% when it was administered starting at the onset of disease (Park et al., 2007). However, in a similar study performed to evaluate the effects of pyruvate with increased frequency of dosing (500 mg/kg i.p., 6 days a week) in SOD1G93A mice, pyruvate was not able to alter the onset of the disease or survival of mice (Esposito et al., 2007). Esposito and colleagues hypothesized that their higher levels of pyruvate may not lead to larger benefit due to pyruvate's U-shaped dose-response curve (Esposito et al., 2007). The different outcomes may therefore be explained by the use of different dosing and different mouse strains.
Lactate
Lactate is the end product of the glycolytic pathway (Rogatzki et al., 2015) and also can be utilized as an energy fuel following its conversion into pyruvate and subsequent oxidation in the mitochondria (Schurr, 2008). Studies have demonstrated the role of lactate as a neuroprotective energy substrate in brain disorders such as cerebral ischemia (Schurr et al., 2001; Berthet et al., 2009; Horn and Klein, 2013; Castillo et al., 2015) and traumatic brain injury (TBI) (reviewed in Brooks and Martin, 2014; Carpenter et al., 2015; Patet et al., 2016). Early administration of lactate in rat organotypic hippocampal slices after oxygen-glucose deprivation protected against neuronal death, decreased lesion size and showed better neurologic outcomes in a mouse model of middle cerebral artery occlusion (Berthet et al., 2009, 2012). Lactate has also been shown to provide neuroprotection against glutamate mediated toxicity in rat hippocampal slices (Schurr and Gozal, 2011), in mouse cortical neuron cultures (Jourdain et al., 2016) and in rat cortex in vivo (Ros et al., 2001). In addition, exogenous administration of lactate reduced brain lesion size in a rat controlled cortical impact models of TBI (Alessandri et al., 2012) and in patients with severe TBI (Bouzat et al., 2014). Similarly, lactate infusion prevented intracranial hypertensive episodes in patients with severe TBI (Ichai et al., 2009, 2013; Bouzat et al., 2014), and attenuated cognitive deficits in rat lateral fluid percussion injury (Rice et al., 2002; Holloway et al., 2007) and in patients with mild TBI (Bisri et al., 2016).
The mechanism of lactate was linked to its ability to improve cerebral energy metabolism and preserve ATP levels in injured brain (Holloway et al., 2007; Gallagher et al., 2009; Bouzat et al., 2014; Jourdain et al., 2016). In addition to its effect as an energy fuel, lactate can function as a signaling molecule by activating hydrocarboxylic acid receptor 1 (HCA1) receptors (Castillo et al., 2015). ATP produced from lactate was also shown to act on the P2Y receptors (Jourdain et al., 2016). Lactate can be used either directly following its cerebral uptake or indirectly as a gluconeogenic precursor (Glenn et al., 2015a,b). Using lactate as an energy fuel for neurons with energy deficit in diseases such as ALS may improve symptoms, especially if reductions of astrocytic lactate transporters found in SOD1G93A mice (Ferraiuolo et al., 2011) are not affecting neuronal lactate uptake.
Creatine
Creatine is phosphorylated by ATP to phosphocreatine, which is another high energy phosphate in addition to ATP. Sufficient amounts of creatine are therefore important to prevent energy depletion. It also stabilizes mitochondrial membranes by binding to phospholipids (Persky and Brazeau, 2001). It has demonstrated beneficial effects in treating energy impairments in several neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease and Huntington's disease (reviewed by Adhihetty and Beal, 2008). Early studies in ALS showed that administration of creatine to SOD1G93A mice prevented loss of motor neurons, protected neurons from oxidative damage in the spinal cord, and improved motor performance and survival of mice in a dose dependent manner (Klivenyi et al., 1999). Also, long term supplementation of creatine in SOD1G93A mice was able to reduce NMDA induced release of glutamate in the cerebral cortex (Andreassen et al., 2001b). Similarly, treatment with creatine prevented depletion of ATP in spinal cord and cerebral cortex of SOD1G93A mice (Browne et al., 2006). Although preclinical studies were promising, several clinical trials failed to show significant beneficial effects in improving the progression of the disease or survival of patients (Groeneveld et al., 2003; Shefner et al., 2004; Rosenfeld et al., 2008).
Coenzyme Q
Coenzyme Q is an electron acceptor in the electron transport chain in mitochondria. It is a powerful antioxidant that scavenges free radicals. Because of its effect in mitochondrial disorders, coenzyme Q has been tried in the SOD1G93A mouse model of ALS (Matthews et al., 1998) and in patients with ALS (Kaufmann et al., 2009). The idea is to promote activation of enzymes involved in the electron transport chain which might be defected due to reduced coenzyme Q in ALS. Coenzyme Q was neuroprotective against 3-nitropropionic induced striatal lesions and it increased the life span of SOD1G93A mice (Matthews et al., 1998). However, the promising effects of Coenzyme Q in animal experiments were not replicated in phase II clinical trials (Kaufmann et al., 2009).
MitoQ
MitoQ is an antioxidant which accumulates in mitochondria (Kelso et al., 2001). It is a derivative of coenzyme Q that promotes the uptake of endogenous coenzyme Q and improves mitochondrial function (Tauskela, 2007). Cassina et al. (2008) showed that mitochondrial function is impaired in the spinal cords of symptomatic SOD1G93A rats, specifically mitochondrial oxygen consumption by the electron transport chain. In addition, SOD1G93A rat astrocytes showed signs of oxidative stress. Therefore, they used MitoQ to target antioxidants to the mitochondria. They demonstrated that pretreatment of cultured SOD1G93A astrocytes with MitoQ was able to reduce the formation of superoxide and peroxynitrite radicals, protect motor neurons against oxidative damage and improve the ability of mitochondria to generate ATP (Cassina et al., 2008). Consistent with this, in a similar study in the SOD1G93A mouse model, treatment with MitoQ at the onset of disease also delayed the decline of mitochondrial function in the spinal cord, protected against motor neuron loss and astrogliosis in the spinal cord. Moreover, it preserved the integrity of the neuromuscular junction, improved hind limb grip strength as well as the life span of mice (Miquel et al., 2014), raising the hope that MitoQ may be effective in ALS patients. On the other hand, in a study with 128 Parkinson's Disease patients, 40 and 80 mg MitoQ per day was associated with dose-dependent increased incidence of nausea and vomiting (Snow et al., 2010). Also, there was no change in progression of disease on a Parkinson Disease Rating Scale.
Dexpramipexole
Dexpramipexole, an enantiomer of pramipexole, is believed to increase ATP generation by improving efficiency of oxidative phosphorylation as well as reducing oxidative damage of mitochondria (Gribkoff and Bozik, 2008). In vitro studies showed that it is able to promote metabolic efficiency of injured cells and scavenging of free radicals in the mitochondria (Danzeisen et al., 2006; Alavian et al., 2012). In another study, dexpramipexole prevented H2O2 mediated cell death and prevented the generation of mitochondrial reactive oxygen species in neuroblastoma cells (Ferrari-Toninelli et al., 2010). In the SOD1G93A mouse model, dexpramipexole preserved motor function and prolonged life span (Danzeisen et al., 2006). Due to these beneficial effects in vitro and in vivo, dexpramipexole was investigated in a double blind randomized phase 2 clinical trial in patients with ALS, showing that treatment for 12 months was safe and well tolerated (Cudkowicz et al., 2011). Dexpramipexole also prevented functional decline in a dose dependent manner compared to placebo (Cudkowicz et al., 2011). However, despite the promising effects in this study, a phase 3 clinical trial failed to show improvements in disease symptoms (Cudkowicz et al., 2013). Dexpramipexole's failure was linked to lack of prior rigorous preclinical testing. Studies performed after the last clinical trial, in induced pluripotent stem cells (iPSCs) derived from patients with ALS (Yang et al., 2013), in rat primary cortical neurons transfected with human mutant TDP-43 as well as in the SOD1G93A mouse model (Vieira et al., 2014) showed no beneficial effect.
Acetyl-L-Carnitine
Acetyl-L-carnitine supplies L-carnitine, which is important in the transport of long chain fatty acids across mitochondrial membranes (Fritz and Yue, 1964). In addition, the acetyl moiety can be transferred to provide acetyl CoA. Therefore, acetyl-L-carnitine improves cerebral oxidative energy production (Zanelli et al., 2005) and fatty acid oxidation in muscles (Siliprandi et al., 1965). It has shown beneficial effects in models of traumatic brain (Scafidi et al., 2010) and spinal cord injury (Zhang et al., 2015), cerebral ischemia (Shuaib et al., 1995; Jalal et al., 2010), stroke (Lolic et al., 1997) and Alzheimer's disease (Virmani et al., 2001) and patients with Alzheimer's disease (Spagnoli et al., 1991; Pettegrew et al., 1995). In primary motor neuron cultures from rats, acetyl-L-carnitine was found to protect against kainate and NMDA-induced toxicity (Bigini et al., 2002). In light of this, a randomized double blind placebo-controlled trial was performed in small number of patients with ALS to investigate the effects of add-on acetyl-L-carnitine to riluzole vs. riluzole only treatment. Patients tolerated the combination well and had a better score in the ALS Functional Rating Scale (Beghi et al., 2013). More in vivo studies in ALS models and a larger phase III trial are now needed to prove efficacy.
Olesoxime
Olesoxime is an experimental compound believed to stabilize mitochondrial permeability transition pores (Bordet et al., 2007). In vitro studies showed increased motor neuron survival in a motor neuron degeneration model in rats (Bordet et al., 2007). In addition, treatment of SOD1G93A transgenic mice with olesoxime improved motor function in the hanging grid test, delayed the onset of body weight loss and prolonged survival by 10% (Bordet et al., 2007). Olesoxime was also shown to delay denervation of muscle and decrease activation of microglia and motor neuron death in the lumbar spinal cord (Sunyach et al., 2012). However, a phase II-III double blind, randomized, placebo-controlled clinical trial failed to show efficacy in patients with ALS (Lenglet et al., 2014). Similar to dexpramipexole, absence of rigorous evaluation in preclinical studies may have led to its failure. In iPSCs derived from patients with ALS, olesoxime showed inconsistent effects (Yang et al., 2013).
Others
Other compounds that directly or indirectly affect energy metabolism such as sirtuins prevent fragmentation of mitochondria and promote an increase in ketone levels and are potential therapeutic targets in ALS (Körner et al., 2013a; Pasinetti et al., 2013). Resveratrol, an antioxidant, which is believed to increase sirtuin 1 expression, was able to delay onset of symptoms, prevent motor neuron loss and extend survival when treatment was initiated at early stages of the disease in hSOD1G93A mice (Mancuso et al., 2014; Song et al., 2014). Moreover, dysregulation of AMP-activated protein kinase (AMPK), an energy sensor which is induced by energy depletion was also linked to ALS (reviewed in Perera and Turner, 2016). AMPK was shown to be activated in TDP-43 mice spinal cord (Perera et al., 2014) and reducing its activity prevented loss of motor neurons in Caenorhabditis elegans models of ALS (Lim et al., 2012). In general, substances that can counteract energy depletion through modulation of the energy producing pathways could help in delaying disease progression.
Conclusions
Of the metabolic approaches discussed above, creatine, dexpramipexole, olesoxime, MitoQ and Coenzyme Q have been investigated in randomized clinical trials (summarized in Table 1). All failed to deliver beneficial effects except acetyl-L-carnitine which showed improved ALS Functional Rating Scale in small number of patients in a phase II clinical trial. The effects of ketogenic diet, caprylic triglyceride, DCA, the Deanna protocol and triheptanoin in patients with ALS remains to be seen. It is critical to note that although numerous (around 50) compounds have been investigated in randomized clinical trials, almost all of them failed to show efficacy except riluzole, the only approved drug so far (Mitsumoto et al., 2014). On the other hand, analysis of preclinical therapeutic trials in hSOD1G93A mouse model of ALS revealed that publication bias and poorly designed experiments such as absence of randomization and blinding, lack of adequate statistical power, and variations within the animal models may lead to over reporting of false positive results (Benatar, 2007; Scott et al., 2008; Perrin, 2014). In addition, methodological flaws in clinical trials could also lead to failure to find efficacy of treatments (Mitsumoto et al., 2014). Therefore, it is of utmost importance to investigate future potential treatments in adequately designed preclinical and clinical trials.
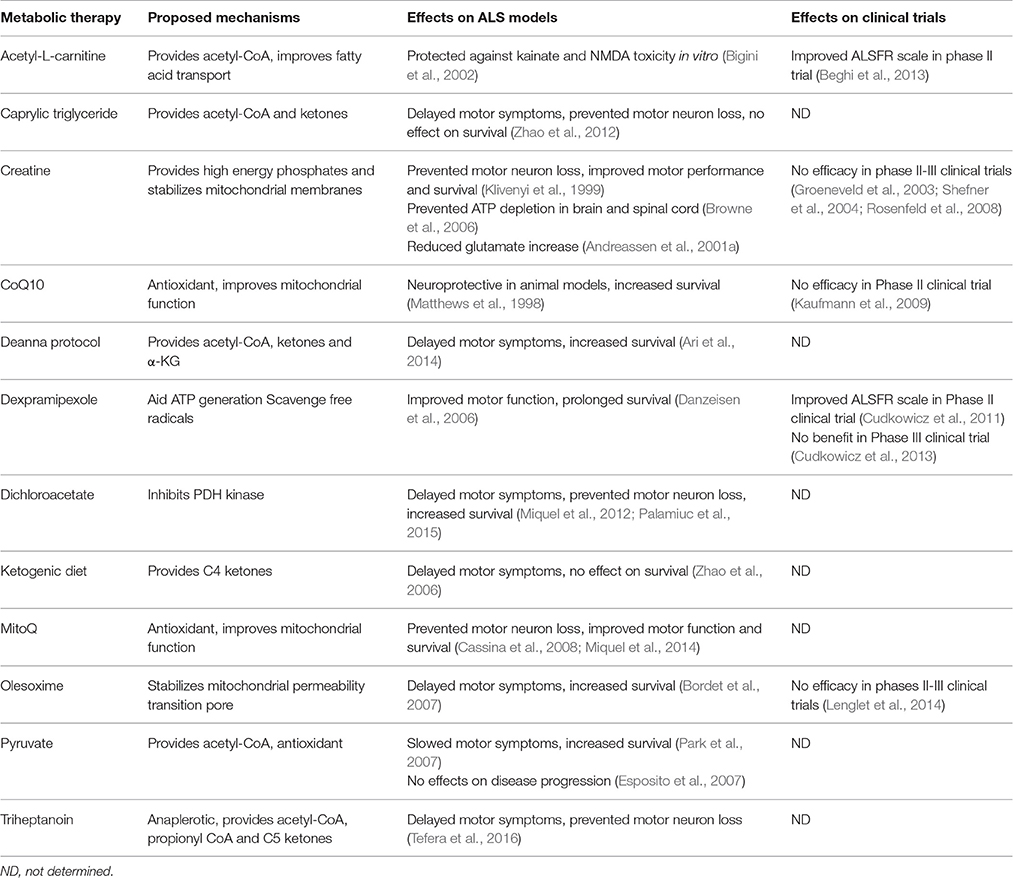
Table 1. Summary of metabolic approaches in ALS and their effects in models of ALS and patients with ALS.
In conclusion, many studies demonstrated that metabolic disturbances are evident in patients with ALS and animal models. Only, a few studies have investigated the effects of metabolic treatments in ALS disease progression and survival. The therapeutic effects of these compounds in animal models and in vitro studies either in delaying motor symptoms, protecting against motor neuron loss and/or improving survival suggest that further studies should be performed to characterize abnormalities in energy metabolism in ALS and explore potential metabolic therapies.
Author Contributions
TT and KB designed the review. TT drafted the review. KB revised critically for important intellectual content. TT and KB approved this to be published.
Funding
We are grateful for funding from NHMRC (KB, 014068) and UQ International (TT).
Conflict of Interest Statement
KB applied for a patent for use of triheptanoin in ALS.
The other author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adanyeguh, I. M., Rinaldi, D., Henry, P. G., Caillet, S., Valabregue, R., Durr, A., et al. (2015). Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84, 490–495. doi: 10.1212/WNL.0000000000001214
Adhihetty, P. J., and Beal, M. F. (2008). Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular Med. 10, 275–290. doi: 10.1007/s12017-008-8053-y
Alavian, K. N., Dworetzky, S. I., Bonanni, L., Zhang, P., Sacchetti, S., Mariggio, M. A., et al. (2012). Effects of dexpramipexole on brain mitochondrial conductances and cellular bioenergetic efficiency. Brain Res. 1446, 1–11. doi: 10.1016/j.brainres.2012.01.046
Alessandri, B., Schwandt, E., Kamada, Y., Nagata, M., Heimann, A., and Kempski, O. (2012). The neuroprotective effect of lactate is not due to improved glutamate uptake after controlled cortical impact in rats. J. Neurotrauma 29, 2181–2191. doi: 10.1089/neu.2011.2067
Andreassen, O. A., Dedeoglu, A., Ferrante, R. J., Jenkins, B. G., Ferrante, K. L., Thomas, M., et al. (2001a). Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol. Dis. 8, 479–491. doi: 10.1006/nbdi.2001.0406
Andreassen, O. A., Jenkins, B. G., Dedeoglu, A., Ferrante, K. L., Bogdanov, M. B., Kaddurah-Daouk, R., et al. (2001b). Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J. Neurochem. 77, 383–390. doi: 10.1046/j.1471-4159.2001.00188.x
Appel, S. H., Zhao, W., Beers, D. R., and Henkel, J. S. (2011). The microglial-motoneuron dialogue in ALS. Acta Myol. 30, 4–8.
Appelberg, K. S., Hovda, D. A., and Prins, M. L. (2009). The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma 26, 497–506. doi: 10.1089/neu.2008.0664
Ari, C., Poff, A. M., Held, H. E., Landon, C. S., Goldhagen, C. R., Mavromates, N., et al. (2014). Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS ONE 9:e103526. doi: 10.1371/journal.pone.0103526
Aso, E., Semakova, J., Joda, L., Semak, V., Halbaut, L., Calpena, A., et al. (2013). Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer's disease. Curr. Alzheimer Res. 10, 290–297. doi: 10.2174/15672050112099990128
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067. doi: 10.1016/j.bbadis.2006.03.008
Beers, D. R., Henkel, J. S., Xiao, Q., Zhao, W., Wang, J., Yen, A. A., et al. (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 16021–16026. doi: 10.1073/pnas.0607423103
Beghi, E., Pupillo, E., Bonito, V., Buzzi, P., Caponnetto, C., Chió, A., et al. (2013). Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 397–405. doi: 10.3109/21678421.2013.764568
Benatar, M. (2007). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13. doi: 10.1016/j.nbd.2006.12.015
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Eng. J. Med. 330, 585–591. doi: 10.1056/NEJM199403033300901
Berthet, C., Castillo, X., Magistretti, P. J., and Hirt, L. (2012). New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 34, 329–335. doi: 10.1159/000343657
Berthet, C., Lei, H., Thevenet, J., Gruetter, R., Magistretti, P. J., and Hirt, L. (2009). Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 29, 1780–1789. doi: 10.1038/jcbfm.2009.97
Bigini, P., Larini, S., Pasquali, C., Muzio, V., and Mennini, T. (2002). Acetyl-L-carnitine shows neuroprotective and neurotrophic activity in primary culture of rat embryo motoneurons. Neurosci. Lett. 329, 334–338. doi: 10.1016/S0304-3940(02)00667-5
Bisri, T., Utomo, B. A., and Fuadi, I. (2016). Exogenous lactate infusion improved neurocognitive function of patients with mild traumatic brain injury. Asian J. Neurosurg. 11, 151–159. doi: 10.4103/1793-5482.145375
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bordet, T., Buisson, B., Michaud, M., Drouot, C., Galéa, P., Delaage, P., et al. (2007). Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 322, 709–720. doi: 10.1124/jpet.107.123000
Bouteloup, C., Desport, J. C., Clavelou, P., Guy, N., Derumeaux-Burel, H., Ferrier, A., et al. (2009). Hypermetabolism in ALS patients: an early and persistent phenomenon. J. Neurol. 256, 1236–1242. doi: 10.1007/s00415-009-5100-z
Bouzat, P., Sala, N., Suys, T., Zerlauth, J. B., Marques-Vidal, P., Feihl, F., et al. (2014). Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 40, 412–421. doi: 10.1007/s00134-013-3203-6
Brooks, G. A., and Martin, N. A. (2014). Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front. Neurosci. 8:408. doi: 10.3389/fnins.2014.00408
Browne, S. E., Yang, L., DiMauro, J. P., Fuller, S. W., Licata, S. C., and Beal, M. F. (2006). Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 22, 599–610. doi: 10.1016/j.nbd.2006.01.001
Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., et al. (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. doi: 10.1126/science.281.5384.1851
Brunengraber, H., and Roe, C. R. (2006). Anaplerotic molecules: current and future. J. Inherit. Metab. Dis. 29, 327–331. doi: 10.1007/s10545-006-0320-1
Carpenter, K. L., Jalloh, I., and Hutchinson, P. J. (2015). Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 9:112. doi: 10.3389/fnins.2015.00112
Cassina, P., Cassina, A., Pehar, M., Castellanos, R., Gandelman, M., de León, A., et al. (2008). Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J. Neurosci. 28, 4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008
Castillo, X., Rosafio, K., Wyss, M. T., Drandarov, K., Buck, A., Pellerin, L., et al. (2015). A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J. Cereb. Blood Flow Metab. 35, 1561–1569. doi: 10.1038/jcbfm.2015.115
Cistaro, A., Pagani, M., Montuschi, A., Calvo, A., Moglia, C., Canosa, A., et al. (2014). The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur. J. Nucl. Med. Mol. Imaging 41, 844–852. doi: 10.1007/s00259-013-2667-5
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. doi: 10.1126/science.1086071
Cleveland, D. W., and Rothstein, J. D. (2001). From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819. doi: 10.1038/35097565
Courchesne-Loyer, A., Croteau, E., Castellano, C. A., St-Pierre, V., Hennebelle, M., and Cunnane, S. C. (2016). Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. doi: 10.1177/0271678X16669366. [Epub ahead of print].
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Cudkowicz, M., Bozik, M. E., Ingersoll, E. W., Miller, R., Mitsumoto, H., Shefner, J., et al. (2011). The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat. Med. 17, 1652–1656. doi: 10.1038/nm.2579
Cudkowicz, M. E., van den Berg, L. H., Shefner, J. M., Mitsumoto, H., Mora, J. S., Ludolph, A., et al. (2013). Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet. Neurol. 12, 1059–1067. doi: 10.1016/S1474-4422(13)70221-7
Dalakas, M. C., Hatazawa, J., Brooks, R. A., and Di Chiro, G. (1987). Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann. Neurol. 22, 580–586. doi: 10.1002/ana.410220504
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
Danzeisen, R., Schwalenstoecker, B., Gillardon, F., Buerger, E., Krzykalla, V., Klinder, K., et al. (2006). Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride]. J. Pharmacol. Exp. Ther. 316, 189–199. doi: 10.1124/jpet.105.092312
Deng-Bryant, Y., Prins, M. L., Hovda, D. A., and Harris, N. G. (2011). Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 28, 1813–1825. doi: 10.1089/neu.2011.1822
Desagher, S., Glowinski, J., and Prémont, J. (1997). Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J. Neurosci. 17, 9060–9067.
Desport, J. C., Preux, P. M., Magy, L., Boirie, Y., Vallat, J. M., Beaufrère, B., et al. (2001). Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 74, 328–334.
Dibaj, P., Steffens, H., Zschüntzsch, J., Nadrigny, F., Schomburg, E. D., Kirchhoff, F., et al. (2011). In vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS ONE 6:e17910. doi: 10.1371/journal.pone.0017910
Dunbar, E. M., Coats, B. S., Shroads, A. L., Langaee, T., Lew, A., Forder, J. R., et al. (2014). Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest. New Drugs 32, 452–464. doi: 10.1007/s10637-013-0047-4
Dupuis, L., Oudart, H., René, F., Gonzalez de Aguilar, J. L., and Loeffler, J. P. (2004). Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 101, 11159–11164. doi: 10.1073/pnas.0402026101
Dupuis, L., Pradat, P. F., Ludolph, A. C., and Loeffler, J. P. (2011). Energy metabolism in amyotrophic lateral sclerosis. Lancet. Neurol. 10, 75–82. doi: 10.1016/S1474-4422(10)70224-6
Dutheil, S., Ota, K. T., Wohleb, E. S., Rasmussen, K., and Duman, R. S. (2016). High-fat diet induced anxiety and anhedonia: impact on brain homeostasis and inflammation. Neuropsychopharmacology 41, 1874–1887. doi: 10.1038/npp.2015.357
Esposito, E., Capasso, M., di Tomasso, N., Corona, C., Pellegrini, F., Uncini, A., et al. (2007). Antioxidant strategies based on tomato-enriched food or pyruvate do not affect disease onset and survival in an animal model of amyotrophic lateral sclerosis. Brain Res. 1168, 90–96. doi: 10.1016/j.brainres.2007.06.095
Ferraiuolo, L., Heath, P. R., Holden, H., Kasher, P., Kirby, J., and Shaw, P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134(Pt 9), 2627–2641. doi: 10.1093/brain/awr193
Ferrari-Toninelli, G., Maccarinelli, G., Uberti, D., Buerger, E., and Memo, M. (2010). Mitochondria-targeted antioxidant effects of S(−) and R(+) pramipexole. BMC Pharmacol. 10:2. doi: 10.1186/1471-2210-10-2
Foerster, B. R., Callaghan, B. C., Petrou, M., Edden, R. A., Chenevert, T. L., and Feldman, E. L. (2012). Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 78, 1596–1600. doi: 10.1212/WNL.0b013e3182563b57
Foerster, B. R., Pomper, M. G., Callaghan, B. C., Petrou, M., Edden, R. A., Mohamed, M. A., et al. (2013). An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol. 70, 1009–1016. doi: 10.1001/jamaneurol.2013.234
Fournier, C., Bedlack, B., Hardiman, O., Heiman-Patterson, T., Gutmann, L., Bromberg, M., et al. (2013). ALS Untangled No. 20: the Deanna protocol. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 319–323. doi: 10.3109/21678421.2013.788405
Francis, J. S., Markov, V., and Leone, P. (2014). Dietary triheptanoin rescues oligodendrocyte loss, dysmyelination and motor function in the nur7 mouse model of Canavan disease. J. Inherit. Metab. Dis. 37, 369–381. doi: 10.1007/s10545-013-9663-6
Fray, A. E., Ince, P. G., Banner, S. J., Milton, I. D., Usher, P. A., Cookson, M. R., et al. (1998). The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur. J. Neurosci. 10, 2481–2489. doi: 10.1046/j.1460-9568.1998.00273.x
Fritz, I. B., and Yue, K. T. (1964). Effects of carnitine on acetyl-coa oxidation by heart muscle mitochondria. Am. J. Physiol. 206, 531–535.
Fukushima, M., Lee, S. M., Moro, N., Hovda, D. A., and Sutton, R. L. (2009). Metabolic and histologic effects of sodium pyruvate treatment in the rat after cortical contusion injury. J. Neurotrauma 26, 1095–1110. doi: 10.1089/neu.2008.0771
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi: 10.1038/nature11007
Gallagher, C. N., Carpenter, K. L., Grice, P., Howe, D. J., Mason, A., Timofeev, I., et al. (2009). The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 132(Pt 10), 2839–2849. doi: 10.1093/brain/awp202
Glenn, T. C., Martin, N. A., Horning, M. A., McArthur, D. L., Hovda, D. A., Vespa, P., et al. (2015a). Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J. Neurotrauma 32, 820–832. doi: 10.1089/neu.2014.3483
Glenn, T. C., Martin, N. A., McArthur, D. L., Hovda, D. A., Vespa, P., Johnson, M. L., et al. (2015b). Endogenous nutritive support after traumatic brain injury: peripheral lactate production for glucose supply via gluconeogenesis. J. Neurotrauma 32, 811–819. doi: 10.1089/neu.2014.3482
Gribkoff, V. K., and Bozik, M. E. (2008). KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2, 6-benzothiazole-diamine dihydrochloride monohydrate] for the treatment of amyotrophic lateral sclerosis. CNS Neurosci. Ther. 14, 215–226. doi: 10.1111/j.1755-5949.2008.00048.x
Groeneveld, G. J., Veldink, J. H., van der Tweel, I., Kalmijn, S., Beijer, C., de Visser, M., et al. (2003). A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann. Neurol. 53, 437–445. doi: 10.1002/ana.10554
Hadera, M. G., Smeland, O. B., McDonald, T. S., Tan, K. N., Sonnewald, U., and Borges, K. (2014). Triheptanoin partially restores levels of tricarboxylic acid cycle intermediates in the mouse pilocarpine model of epilepsy. J. Neurochem. 129, 107–119. doi: 10.1111/jnc.12610
Hatazawa, J., Brooks, R. A., Dalakas, M. C., Mansi, L., and Di Chiro, G. (1988). Cortical motor-sensory hypometabolism in amyotrophic lateral sclerosis: a PET study. J. Comput. Assist. Tomogr. 12, 630–636. doi: 10.1097/00004728-198807000-00019
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond). 6:31. doi: 10.1186/1743-7075-6-31
Holloway, R., Zhou, Z., Harvey, H. B., Levasseur, J. E., Rice, A. C., Sun, D., et al. (2007). Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta Neurochir. 149, 919–927. discussion: 927. doi: 10.1007/s00701-007-1241-y
Horn, T., and Klein, J. (2013). Neuroprotective effects of lactate in brain ischemia: dependence on anesthetic drugs. Neurochem. Int. 62, 251–257. doi: 10.1016/j.neuint.2012.12.017
Hu, Z. G., Wang, H. D., Jin, W., and Yin, H. X. (2009a). Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 39, 76–83.
Hu, Z. G., Wang, H. D., Qiao, L., Yan, W., Tan, Q. F., and Yin, H. X. (2009b). The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Inj. 23, 459–465. doi: 10.1080/02699050902788469
Huh, S. H., Chung, Y. C., Piao, Y., Jin, M. Y., Son, H. J., Yoon, N. S., et al. (2011). Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson's disease. J. Immunol. 187, 960–969. doi: 10.4049/jimmunol.1100009
Ichai, C., Armando, G., Orban, J. C., Berthier, F., Rami, L., Samat-Long, C., et al. (2009). Sodium lactate versus mannitol in the treatment of intracranial hypertensive episodes in severe traumatic brain-injured patients. Intensive Care Med. 35, 471–479. doi: 10.1007/s00134-008-1283-5
Ichai, C., Payen, J. F., Orban, J. C., Quintard, H., Roth, H., Legrand, R., et al. (2013). Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: a randomized controlled trial. Intensive Care Med. 39, 1413–1422. doi: 10.1007/s00134-013-2978-9
Isopi, E., Granzotto, A., Corona, C., Bomba, M., Ciavardelli, D., Curcio, M., et al. (2015). Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer's disease without reducing amyloid and tau pathology. Neurobiol. Dis. 81, 214–224. doi: 10.1016/j.nbd.2014.11.013
Itoh, Y., Esaki, T., Shimoji, K., Cook, M., Law, M. J., Kaufman, E., et al. (2003). Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4879–4884. doi: 10.1073/pnas.0831078100
Izumi, Y., and Zorumski, C. F. (2010). Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci. Lett. 478, 131–135. doi: 10.1016/j.neulet.2010.04.078
Jalal, F. Y., Böhlke, M., and Maher, T. J. (2010). Acetyl-L-carnitine reduces the infarct size and striatal glutamate outflow following focal cerebral ischemia in rats. Ann. N.Y. Acad. Sci. 1199, 95–104. doi: 10.1111/j.1749-6632.2009.05351.x
Jourdain, P., Allaman, I., Rothenfusser, K., Fiumelli, H., Marquet, P., and Magistretti, P. J. (2016). L-Lactate protects neurons against excitotoxicity: implication of an ATP-mediated signaling cascade. Sci. Rep. 6:21250. doi: 10.1038/srep21250
Julien, J. P., and Beaulieu, J. M. (2000). Cytoskeletal abnormalities in amyotrophic lateral sclerosis: beneficial or detrimental effects? J. Neurol. Sci. 180, 7–14. doi: 10.1016/S0022-510X(00)00422-6
Jung, C., Higgins, C. M., and Xu, Z. (2002). Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 83, 535–545. doi: 10.1046/j.1471-4159.2002.01112.x
Kang, S. H., Li, Y., Fukaya, M., Lorenzini, I., Cleveland, D. W., Ostrow, L. W., et al. (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579. doi: 10.1038/nn.3357
Kasarskis, E. J., Berryman, S., Vanderleest, J. G., Schneider, A. R., and McClain, C. J. (1996). Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am. J. Clin. Nutr. 63, 130–137.
Kaufmann, P., Thompson, J. L., Levy, G., Buchsbaum, R., Shefner, J., Krivickas, L. S., et al. (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 66, 235–244. doi: 10.1002/ana.21743
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi: 10.1074/jbc.M009093200
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Klivenyi, P., Ferrante, R. J., Matthews, R. T., Bogdanov, M. B., Klein, A. M., Andreassen, O. A., et al. (1999). Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med. 5, 347–350. doi: 10.1038/6568
Körner, S., Böselt, S., Thau, N., Rath, K. J., Dengler, R., and Petri, S. (2013a). Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: neuroprotective or neurotoxic properties of sirtuins in ALS? Neurodegener. Dis. 11, 141–152. doi: 10.1159/000338048
Körner, S., Hendricks, M., Kollewe, K., Zapf, A., Dengler, R., Silani, V., et al. (2013b). Weight loss, dysphagia and supplement intake in patients with amyotrophic lateral sclerosis (ALS): impact on quality of life and therapeutic options. BMC Neurol. 13:84. doi: 10.1186/1471-2377-13-84
Lederer, C. W., Torrisi, A., Pantelidou, M., Santama, N., and Cavallaro, S. (2007). Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics 8:26. doi: 10.1186/1471-2164-8-26
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lenglet, T., Lacomblez, L., Abitbol, J. L., Ludolph, A., Mora, J. S., Robberecht, W., et al. (2014). A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 21, 529–536. doi: 10.1111/ene.12344
Lim, M. A., Selak, M. A., Xiang, Z., Krainc, D., Neve, R. L., Kraemer, B. C., et al. (2012). Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 32, 1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012
Lolic, M. M., Fiskum, G., and Rosenthal, R. E. (1997). Neuroprotective effects of acetyl-L-carnitine after stroke in rats. Ann. Emerg. Med. 29, 758–765. doi: 10.1016/S0196-0644(97)70197-5
Ludolph, A. C., Langen, K. J., Regard, M., Herzog, H., Kemper, B., Kuwert, T., et al. (1992). Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol. Scand. 85, 81–89. doi: 10.1111/j.1600-0404.1992.tb04003.x
Mancuso, R., del Valle, J., Modol, L., Martinez, A., Granado-Serrano, A. B., Ramirez-Núñez, O., et al. (2014). Resveratrol improves motoneuron function and extends survival in SOD1G93A ALS mice. Neurotherapeutics 11, 419–432. doi: 10.1007/s13311-013-0253-y
Marin, B., Desport, J. C., Kajeu, P., Jesus, P., Nicolaud, B., Nicol, M., et al. (2011). Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J. Neurol. Neurosurg. Psychiatr. 82, 628–634. doi: 10.1136/jnnp.2010.211474
Marin-Valencia, I., Good, L. B., Ma, Q., Malloy, C. R., and Pascual, J. M. (2013). Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J. Cereb. Blood Flow Metab. 33, 175–182. doi: 10.1038/jcbfm.2012.151
Martin, K., Jackson, C. F., Levy, R. G., and Cooper, P. N. (2016). Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2:CD001903. doi: 10.1002/14651858.CD001903.pub3
Masino, S. A., and Rho, J. M. (2012). “Mechanisms of ketogenic diet action,” in Jasper's Basic Mechanisms of the Epilepsies, 4th Edn., eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information), 1483–1515.
Matthews, R. T., Yang, L., Browne, S., Baik, M., and Beal, M. F. (1998). Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. U.S.A. 95, 8892–8897. doi: 10.1073/pnas.95.15.8892
Mattiazzi, M., D'Aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.M203065200
Mattson, M. P., Cutler, R. G., and Camandola, S. (2007). Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. 9, 17–20. doi: 10.1385/NMM:9:1:17
Maus, M., Marin, P., Israël, M., Glowinski, J., and Prémont, J. (1999). Pyruvate and lactate protect striatal neurons against N-methyl-D-aspartate-induced neurotoxicity. Eur. J. Neurosci. 11, 3215–3224. doi: 10.1046/j.1460-9568.1999.00745.x
McAllister, A., Allison, S. P., and Randle, P. J. (1973). Effects of dichloroacetate on the metabolism of glucose, pyruvate, acetate, 3-hydroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem. J. 134, 1067–1081. doi: 10.1042/bj1341067
McDonald, T. S., Tan, K. N., Hodson, M. P., and Borges, K. (2014). Alterations of hippocampal glucose metabolism by even versus uneven medium chain triglycerides. J. Cereb. Blood Flow Metab. 34, 153–160. doi: 10.1038/jcbfm.2013.184
Mejía-Toiber, J., Montiel, T., and Massieu, L. (2006). D-beta-hydroxybutyrate prevents glutamate-mediated lipoperoxidation and neuronal damage elicited during glycolysis inhibition in vivo. Neurochem. Res. 31, 1399–1408. doi: 10.1007/s11064-006-9189-5
Menzies, F. M., Cookson, M. R., Taylor, R. W., Turnbull, D. M., Chrzanowska-Lightowlers, Z. M., Dong, L., et al. (2002). Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125(Pt 7), 1522–1533. doi: 10.1093/brain/awf167
Miquel, E., Cassina, A., Martinez-Palma, L., Bolatto, C., Trías, E., Gandelman, M., et al. (2012). Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS ONE 7:e34776. doi: 10.1371/journal.pone.0034776
Miquel, E., Cassina, A., Martínez-Palma, L., Souza, J. M., Bolatto, C., Rodríguez-Bottero, S., et al. (2014). Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 70, 204–213. doi: 10.1016/j.freeradbiomed.2014.02.019
Mitsumoto, H., Brooks, B. R., and Silani, V. (2014). Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. 13, 1127–1138. doi: 10.1016/s1474-4422(14)70129-2
Miyazaki, K., Masamoto, K., Morimoto, N., Kurata, T., Mimoto, T., Obata, T., et al. (2012). Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J. Cereb. Blood Flow Metab. 32, 456–467. doi: 10.1038/jcbfm.2011.155
Mochel, F., Duteil, S., Marelli, C., Jauffret, C., Barles, A., Holm, J., et al. (2010). Dietary anaplerotic therapy improves peripheral tissue energy metabolism in patients with Huntington's disease. Eur. J. Hum. Genet. 18, 1057–1060. doi: 10.1038/ejhg.2010.72
Mochel, F., Hainque, E., Gras, D., Adanyeguh, I. M., Caillet, S., Héron, B., et al. (2016). Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J. Neurol. Neurosurg. Psychiatr. 87, 550–553. doi: 10.1136/jnnp-2015-311475
Moro, N., and Sutton, R. L. (2010). Beneficial effects of sodium or ethyl pyruvate after traumatic brain injury in the rat. Exp. Neurol. 225, 391–401. doi: 10.1016/j.expneurol.2010.07.013
Morrison, B. M., Lee, Y., and Rothstein, J. D. (2013). Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 23, 644–651. doi: 10.1016/j.tcb.2013.07.007
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nakamichi, N., Kambe, Y., Oikawa, H., Ogura, M., Takano, K., Tamaki, K., et al. (2005). Protection by exogenous pyruvate through a mechanism related to monocarboxylate transporters against cell death induced by hydrogen peroxide in cultured rat cortical neurons. J. Neurochem. 93, 84–93. doi: 10.1111/j.1471-4159.2005.02999.x
Ngo, S. T., Steyn, F. J., and McCombe, P. A. (2014). Body mass index and dietary intervention: implications for prognosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 340, 5–12. doi: 10.1016/j.jns.2014.02.035
Nieto-Gonzalez, J. L., Moser, J., Lauritzen, M., Schmitt-John, T., and Jensen, K. (2011). Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb. Cortex 21, 625–635. doi: 10.1093/cercor/bhq134
O'Reilly, É. J., Wang, H., Weisskopf, M. G., Fitzgerald, K. C., Falcone, G., McCullough, M. L., et al. (2013). Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 205–211. doi: 10.3109/21678421.2012.735240
Paganoni, S., Deng, J., Jaffa, M., Cudkowicz, M. E., and Wills, A.-M. (2011). Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 44, 20–24. doi: 10.1002/mus.22114
Palamiuc, L., Schlagowski, A., Ngo, S. T., Vernay, A., Dirrig-Grosch, S., Henriques, A., et al. (2015). A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 7, 526–546. doi: 10.15252/emmm.201404433
Pan, R., Rong, Z., She, Y., Cao, Y., Chang, L. W., and Lee, W. H. (2012). Sodium pyruvate reduces hypoxic-ischemic injury to neonatal rat brain. Pediatr. Res. 72, 479–489. doi: 10.1038/pr.2012.107
Park, J. H., Hong, Y. H., Kim, H. J., Kim, S. M., Kim, M. J., Park, K. S., et al. (2007). Pyruvate slows disease progression in a G93A SOD1 mutant transgenic mouse model. Neurosci. Lett. 413, 265–269. doi: 10.1016/j.neulet.2006.11.058
Park, M. J., Aja, S., Li, Q., Degano, A. L., Penati, J., Zhuo, J., et al. (2014). Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 9:e109527. doi: 10.1371/journal.pone.0109527
Pascual, J. M., Liu, P., Mao, D., Kelly, D. I., Hernandez, A., Sheng, M., et al. (2014). Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 71, 1255–1265. doi: 10.1001/jamaneurol.2014.1584
Pasinetti, G. M., Bilski, A. E., and Zhao, W. (2013). Sirtuins as therapeutic targets of ALS. Cell Res. 23, 1073–1074. doi: 10.1038/cr.2013.94
Patet, C., Suys, T., Carteron, L., and Oddo, M. (2016). Cerebral Lactate Metabolism After Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 16, 31. doi: 10.1007/s11910-016-0638-5
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Perera, N. D., Sheean, R. K., Scott, J. W., Kemp, B. E., Horne, M. K., and Turner, B. J. (2014). Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS ONE 9:e95549. doi: 10.1371/journal.pone.0095549
Perera, N. D., and Turner, B. J. (2016). AMPK signalling and defective energy metabolism in amyotrophic lateral sclerosis. Neurochem. Res. 41, 544–553. doi: 10.1007/s11064-015-1665-3
Persky, A. M., and Brazeau, G. A. (2001). Clinical pharmacology of the dietary supplement creatine monohydrate. Pharmacol. Rev. 53, 161–176.
Pettegrew, J. W., Klunk, W. E., Panchalingam, K., Kanfer, J. N., and McClure, R. J. (1995). Clinical and neurochemical effects of acetyl-L-carnitine in Alzheimer's disease. Neurobiol. Aging 16, 1–4. doi: 10.1016/0197-4580(95)80001-8
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136(Pt 2), 471–482. doi: 10.1093/brain/aws339
Pistell, P. J., Morrison, C. D., Gupta, S., Knight, A. G., Keller, J. N., Ingram, D. K., et al. (2010). Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 219, 25–32. doi: 10.1016/j.jneuroim.2009.11.010
Prins, M. L., Fujima, L. S., and Hovda, D. A. (2005). Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 82, 413–420. doi: 10.1002/jnr.20633
Raman, R., Allen, S. P., Goodall, E. F., Kramer, S., Ponger, L. L., Heath, P. R., et al. (2015). Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 41, 201–226. doi: 10.1111/nan.12147
Rice, A. C., Zsoldos, R., Chen, T., Wilson, M. S., Alessandri, B., Hamm, R. J., et al. (2002). Lactate administration attenuates cognitive deficits following traumatic brain injury. Brain Res. 928, 156–159. doi: 10.1016/S0006-8993(01)03299-1
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rogatzki, M. J., Ferguson, B. S., Goodwin, M. L., and Gladden, L. B. (2015). Lactate is always the end product of glycolysis. Front. Neurosci. 9:22. doi: 10.3389/fnins.2015.00022
Ros, J., Pecinska, N., Alessandri, B., Landolt, H., and Fillenz, M. (2001). Lactate reduces glutamate-induced neurotoxicity in rat cortex. J. Neurosci. Res. 66, 790–794. doi: 10.1002/jnr.10043
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rosenfeld, J., King, R. M., Jackson, C. E., Bedlack, R. S., Barohn, R. J., Dick, A., et al. (2008). Creatine monohydrate in ALS: effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph. Lateral Scler. 9, 266–272. doi: 10.1080/17482960802028890
Rothstein, J. D., Tsai, G., Kuncl, R. W., Clawson, L., Cornblath, D. R., Drachman, D. B., et al. (1990). Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 28, 18–25. doi: 10.1002/ana.410280106
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi: 10.1002/ana.410380114
Sasaki, S., Komori, T., and Iwata, M. (2000). Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 100, 138–144. doi: 10.1007/s004019900159
Satpute, R., Lomash, V., Kaushal, M., and Bhattacharya, R. (2013). Neuroprotective effects of alpha-ketoglutarate and ethyl pyruvate against motor dysfunction and oxidative changes caused by repeated 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine exposure in mice. Hum. Exp. Toxicol. 32, 747–758. doi: 10.1177/0960327112468172
Scafidi, S., Racz, J., Hazelton, J., McKenna, M. C., and Fiskum, G. (2010). Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev. Neurosci. 32, 480–487. doi: 10.1159/000323178
Schönfeld, P., and Wojtczak, L. (2016). Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J. Lipid Res. 57, 943–954. doi: 10.1194/jlr.R067629
Schurr, A. (2008). Lactate: a major and crucial player in normal function of both muscle and brain. J. Physiol. 586(Pt 11), 2665–2666. doi: 10.1113/jphysiol.2008.155416
Schurr, A., and Gozal, E. (2011). Aerobic production and utilization of lactate satisfy increased energy demands upon neuronal activation in hippocampal slices and provide neuroprotection against oxidative stress. Front. Pharmacol. 2:96. doi: 10.3389/fphar.2011.00096
Schurr, A., Payne, R. S., Miller, J. J., Tseng, M. T., and Rigor, B. M. (2001). Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 895, 268–272. doi: 10.1016/S0006-8993(01)02082-0
Scott, S., Kranz, J. E., Cole, J., Lincecum, J. M., Thompson, K., Kelly, N., et al. (2008). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. 9, 4–15. doi: 10.1080/17482960701856300
Shank, R. P., and Campbell, G. L. (1981). Avid Na+-dependent, high-affinity uptake of alpha-ketoglutarate by nerve terminal enriched material from mouse cerebellum. Life Sci. 28, 843–850. doi: 10.1016/0024-3205(81)90045-X
Shank, R. P., and Campbell, G. L. (1984). Alpha-ketoglutarate and malate uptake and metabolism by synaptosomes: further evidence for an astrocyte-to-neuron metabolic shuttle. J. Neurochem. 42, 1153–1161. doi: 10.1111/j.1471-4159.1984.tb12724.x
Shaw, P. J., and Eggett, C. J. (2000). Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 247(Suppl. 1), I17–I27. doi: 10.1007/bf03161151
Shefner, J. M., Cudkowicz, M. E., Schoenfeld, D., Conrad, T., Taft, J., Chilton, M., et al. (2004). A clinical trial of creatine in ALS. Neurology 63, 1656–1661. doi: 10.1212/01.WNL.0000142992.81995.F0
Shen, H., Hu, X., Liu, C., Wang, S., Zhang, W., Gao, H., et al. (2010). Ethyl pyruvate protects against hypoxic-ischemic brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol. Dis. 37, 711–722. doi: 10.1016/j.nbd.2009.12.010
Shuaib, A., Waqaar, T., Wishart, T., Kanthan, R., and Howlett, W. (1995). Acetyl-L-carnitine attenuates neuronal damage in gerbils with transient forebrain ischemia only when given before the insult. Neurochem. Res. 20, 1021–1025. doi: 10.1007/BF00995555
Siliprandi, N., Siliprandi, D., and Ciman, M. (1965). Stimulation of oxidation of mitochondrial fatty acids and of acetate by acetylcarnitine. Biochem. J. 96, 777–780. doi: 10.1042/bj0960777
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'Sullivan, J. D., Fung, V., et al. (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 25, 1670–1674. doi: 10.1002/mds.23148
Song, L., Chen, L., Zhang, X., Li, J., and Le, W. (2014). Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1G93A mouse model of amyotrophic lateral sclerosis. Biomed Res. Int. 2014:483501. doi: 10.1155/2014/483501
Spagnoli, A., Lucca, U., Menasce, G., Bandera, L., Cizza, G., Forloni, G., et al. (1991). Long-term acetyl-L-carnitine treatment in Alzheimer's disease. Neurology 41, 1726–1732. doi: 10.1212/WNL.41.11.1726
Streijger, F., Plunet, W. T., Lee, J. H., Liu, J., Lam, C. K., Park, S., et al. (2013). Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS ONE 8:e78765. doi: 10.1371/journal.pone.0078765
Sunyach, C., Michaud, M., Arnoux, T., Bernard-Marissal, N., Aebischer, J., Latyszenok, V., et al. (2012). Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 62, 2346–2352. doi: 10.1016/j.neuropharm.2012.02.013
Suzuki, M., Sato, K., Dohi, S., Sato, T., Matsuura, A., and Hiraide, A. (2001). Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn. J. Pharmacol. 87, 143–150. doi: 10.1254/jjp.87.143
Tefera, T. W., Wong, Y., Barkl-Luke, M. E., Ngo, S. T., Thomas, N. K., McDonald, T. S., et al. (2016). Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 11:e0161816. doi: 10.1371/journal.pone.0161816
Thomas, N. K., Willis, S., Sweetman, L., and Borges, K. (2012). Triheptanoin in acute mouse seizure models. Epilepsy Res. 99, 312–317. doi: 10.1016/j.eplepsyres.2011.12.013
Tieu, K., Perier, C., Caspersen, C., Teismann, P., Wu, D. C., Yan, S. D., et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112, 892–901. doi: 10.1172/JCI200318797
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Van der Auwera, I., Wera, S., Van Leuven, F., and Henderson, S. T. (2005). A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr. Metab. (Lond). 2:28. doi: 10.1186/1743-7075-2-28
Vieira, F. G., LaDow, E., Moreno, A., Kidd, J. D., Levine, B., Thompson, K., et al. (2014). Dexpramipexole is ineffective in two models of ALS related neurodegeneration. PLoS ONE 9:e91608. doi: 10.1371/journal.pone.0091608
Virmani, M. A., Caso, V., Spadoni, A., Rossi, S., Russo, F., and Gaetani, F. (2001). The action of acetyl-L-carnitine on the neurotoxicity evoked by amyloid fragments and peroxide on primary rat cortical neurones. Ann. N.Y. Acad. Sci. 939, 162–178. doi: 10.1111/j.1749-6632.2001.tb03623.x
Vucic, S., and Kiernan, M. C. (2006). Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129(Pt 9), 2436–2446. doi: 10.1093/brain/awl172
Vucic, S., Nicholson, G. A., and Kiernan, M. C. (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131(Pt 6), 1540–1550. doi: 10.1093/brain/awn071
Whitehouse, S., Cooper, R. H., and Randle, P. J. (1974). Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 141, 761–774. doi: 10.1042/bj1410761
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Willis, S., Stoll, J., Sweetman, L., and Borges, K. (2010). Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol. Dis. 40, 565–572. doi: 10.1016/j.nbd.2010.07.017
Wills, A. M., Hubbard, J., Macklin, E. A., Glass, J., Tandan, R., Simpson, E. P., et al. (2014). Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet 383, 2065–2072. doi: 10.1016/S0140-6736(14)60222-1
Wlaz, P., Socala, K., Nieoczym, D., Luszczki, J. J., Zarnowska, I., Zarnowski, T., et al. (2012). Anticonvulsant profile of caprylic acid, a main constituent of the medium-chain triglyceride (MCT) ketogenic diet, in mice. Neuropharmacology 62, 1882–1889. doi: 10.1016/j.neuropharm.2011.12.015
Yamanaka, K., Boillee, S., Roberts, E. A., Garcia, M. L., McAlonis-Downes, M., Mikse, O. R., et al. (2008). Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 105, 7594–7599. doi: 10.1073/pnas.0802556105
Yang, Y. M., Gupta, S. K., Kim, K. J., Powers, B. E., Cerqueira, A., Wainger, B. J., et al. (2013). A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12, 713–726. doi: 10.1016/j.stem.2013.04.003
Yu, Y. M., Kim, J. B., Lee, K. W., Kim, S. Y., Han, P. L., and Lee, J. K. (2005). Inhibition of the cerebral ischemic injury by ethyl pyruvate with a wide therapeutic window. Stroke 36, 2238–2243. doi: 10.1161/01.STR.0000181779.83472.35
Yudkoff, M., Daikhin, Y., Horyn, O., and Nissim, I. (2008). Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia 49(Suppl. 8), 73–75. doi: 10.1111/j.1528-1167.2008.01841.x
Zanelli, S. A., Solenski, N. J., Rosenthal, R. E., and Fiskum, G. (2005). Mechanisms of ischemic neuroprotection by acetyl-L-carnitine. Ann. N.Y. Acad. Sci. 1053, 153–161. doi: 10.1196/annals.1344.013
Zanette, G., Tamburin, S., Manganotti, P., Refatti, N., Forgione, A., and Rizzuto, N. (2002). Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin. Neurophysiol. 113, 1688–1697. doi: 10.1016/S1388-2457(02)00288-2
Zhang, X., Dong, F., Ren, J., Driscoll, M. J., and Culver, B. (2005). High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp. Neurol. 191, 318–325. doi: 10.1016/j.expneurol.2004.10.011
Zhang, Z. Y., Fan, Z. K., Cao, Y., Jia, Z. Q., Li, G., Zhi, X. D., et al. (2015). Acetyl-L-carnitineameliorates mitochondrial damage and apoptosis following spinal cord injury in rats. Neurosci. Lett. 604, 18–23. doi: 10.1016/j.neulet.2015.06.021
Zhao, W., Varghese, M., Vempati, P., Dzhun, A., Cheng, A., Wang, J., et al. (2012). Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 7:e49191. doi: 10.1371/journal.pone.0049191
Keywords: amyotrophic lateral sclerosis, energy metabolism, medium chain fatty acids, mitochondria, metabolic treatment
Citation: Tefera TW and Borges K (2017) Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 10:611. doi: 10.3389/fnins.2016.00611
Received: 09 November 2016; Accepted: 26 December 2016;
Published: 10 January 2017.
Edited by:
Shaida A. Andrabi, University of Alabama School of Medicine, USAReviewed by:
Hirac Gurden, Paris Diderot University, FranceAvital Schurr, University of Louisville, USA
Copyright © 2017 Tefera and Borges. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Borges, ay5ib3JnZXNAdXEuZWR1LmF1