 Edward Beamer1
Edward Beamer1 Wolfgang Fischer
Wolfgang Fischer Tobias Engel
Tobias Engel- 1Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland
- 2Medical Faculty, Rudolf-Boehm-Institute of Pharmacology and Toxicology, University of Leipzig, Leipzig, Germany
Despite the progress made in the development of new antiepileptic drugs (AEDs), the biggest challenges that epilepsy presents to drug development have remained unchanged for the last 80 years: finding a treatment with potential for modifying disease progression and reducing the percentage of patients resistant to all pharmacological interventions. The mechanism of action of the majority of AEDs is based on blocking Na+ and/or Ca2+ channels, promotion of GABA or inhibition of glutamate signaling. In order for further progress to be made, however, a fuller picture of epilepsy will need to be considered, including changes to blood–brain barrier permeability, synaptic plasticity, network reorganization, and gliosis. In particular, brain inflammation has attracted much attention over recent years. Emerging evidence demonstrates a causal role for brain inflammation in lowering seizure thresholds and driving epileptogenesis. Consistent with this, intervening in pro-inflammatory cascades has shown promise in animal models of epilepsy, with clinical trials of anti-inflammatory agents already underway. The ATP-gated purinergic P2X7 receptor (P2X7) has been proposed as a novel drug target for a host of neurological conditions, including epilepsy. Constitutive expression of P2X7 in the CNS is mainly on microglia, but neuronal and astroglial expression has also been suggested. Its function as a gatekeeper of inflammation is most clearly understood, however, it also plays a number of other important roles pertinent to icto- and epileptogenesis: depolarization of the cell membrane, release of macromolecules, induction of apoptosis and synaptic reorganization. Changes in P2X7 expression have been reported following prolonged seizures (status epilepticus) and during chronic epilepsy in both experimental models and patients. While much of the early work focused on the study of P2X7 during status epilepticus, there is now mounting data showing involvement of this receptor during epilepsy. The present short review will discuss the most recent findings concerning P2X7 expression and function during epilepsy and the clinical potential for P2X7 antagonists as novel AEDs.
Epilepsy
Epilepsy encompasses a complex group of chronic neurological diseases, characterized by the manifestation of recurrent seizures and has an incidence of ~1% with over 60 million people worldwide suffering from the disease (Moshé et al., 2015). Epilepsy affects people of all ages, with incidence highest in the young and elderly (Bialer and White, 2010). Patients with epilepsy have a life expectancy reduced by 2–10% compared with the general population, a death rate 2–3 times higher, and a 4-fold risk of a host of co-morbidities such as depression and anxiety which impact upon quality of life (Moshé et al., 2015). Epilepsy can be acquired as a result of a brain insult, such as head trauma, stroke, or an episode of status epilepticus (SE). Equally, it can result from genetic polymorphisms, copy number variants, or de novo mutations, often involving changes in ion channel function (Rees, 2010; Pitkänen and Lukasiuk, 2011). The most common form of acquired epilepsy in adults is temporal lobe epilepsy (TLE), in which seizures arise from brain structures such as the hippocampus and amygdala (Chang and Lowenstein, 2003). Hippocampal sclerosis, characterized by a pattern of selective neuronal loss and reactive gliosis (Chang and Lowenstein, 2003) is the most common pathological finding in the brain of TLE patients.
Epileptogenesis is the process of structural and functional changes which transforms the normal brain to one that can generate the abnormal neuronal activity underlying seizures. Classically, the process of epileptogenesis is understood to occur during the latent period between an initial insult and first spontaneous seizure. More recent concepts of epileptogenesis, however, describe an ongoing process continuing to drive progression of the disease beyond its emergence (Pitkänen and Engel, 2014). Epileptogenesis is characterized by a number of pathological changes, such as delayed, ongoing neurodegeneration, synaptic plasticity, increased blood–brain barrier (BBB) permeability, extracellular matrix reorganization, neurogenesis, and chronic inflammation (Pitkänen and Lukasiuk, 2011). Currently available treatments, as discussed below, act to suppress seizures, but have little impact on the process of epileptogenesis.
Current Treatment
For newly diagnosed patients suffering from epilepsy, antiepileptic drugs (AEDs) are the frontline treatment (Wiebe and Jette, 2012). While the model of epilepsy as an imbalance between excitation and inhibition has served as the backbone for rational drug design, new AEDs have continued to enter the market, improving control of seizures, limiting adverse effects and broadening the available pharmacological armamentarium. This has offered greater scope for physicians to prescribe drugs tailored to the particular needs of a patient, such as avoiding complications with pregnancy (Patel and Pennell, 2016) or the exacerbation of co-morbidities, such as depression (Blond et al., 2016). Across a span of almost 80 years, however, the percentage of patients refractory to all available AEDs has steadfastly refused to move from 30% (Moshé et al., 2015), with TLE particularly resistant to treatment (Pitkänen and Lukasiuk, 2011). While currently there are more than 25 AEDs available on prescription, despite the superficial diversity, all current treatments rely on three main mechanisms of action based on rebalancing excitatory/inhibitory drive: blockade of Na+ and/or Ca2+ channels, promotion of GABAergic neurotransmission, or accessory antagonism of glutamate receptors (Bialer and White, 2010). Where AEDs do successfully control seizures, they may exacerbate co-morbidities or cause severe adverse effects (Elger, 2016). Further, available AEDs act mainly symptomatically, controlling seizures but having no effect on disease progression. Typically, patients are dependent on drugs for the entire duration of their life. Where they become refractory to treatment, surgical intervention remains, in the majority of cases, the only available avenue (Wiebe and Jette, 2012). There is therefore a pressing need for the development of new treatment strategies with a non-classical mechanism of action, which show efficacy in refractory patients, have a reduced burden of adverse effects, impact upon associated co-morbidities and retard disease progression. In recent years, the paradigm of epilepsy research has broadened, taking into account the importance of brain inflammation as a possible driver of hyperexcitability and neurodegeneration during epileptogenesis (Vezzani et al., 2016).
Brain Inflammation in Icto- and Epileptogenesis
Brain inflammation, implicated in a host of neurological disorders (Ransohoff, 2016), has been shown to drive increases in BBB permeability (Rochfort and Cummins, 2015), facilitate glutamatergic neurotransmission (Vezzani and Viviani, 2015), initiate pro-apoptotic signaling pathways, promote selective neuronal death and subsequent rewiring of networks, and stimulate astro- and microgliosis (Ransohoff, 2016). Release of pro-inflammatory cytokines, such as interleukin-1ß (IL-1β) and tumor necrosis factor-alpha (TNFα) increases both in experimental models of epilepsy and in patients (Vezzani et al., 2016). Further, numerous experimental and clinical findings demonstrate that brain inflammation plays a key role in the generation of seizures and the pathogenesis of epilepsy (Vezzani et al., 2016). Experiments showing pro-convulsive effects of pro-inflammatory molecules, such as IL-1β (Balosso et al., 2008) or high mobility group box 1 protein (HMGB1; Maroso et al., 2010) provide evidence that inflammation is capable of driving hyperexcitability. Conversely, anticonvulsive effects of drugs which interfere with inflammatory signaling (Vezzani et al., 2000; Balosso et al., 2008; Marchi et al., 2009; Maroso et al., 2010; Bedner et al., 2015) demonstrate the potential for targeting inflammatory signaling pathways in epilepsy.
ATP and Purinergic Signaling
ATP, besides its well-established role in cellular energy transfer, also functions as an important intercellular signaling molecule (Burnstock, 2013). ATP signaling is mediated by “purinergic” P2 receptors (P2Rs), classified into two subfamilies: the P2X homo- or heterotrimeric ionotropic receptors and the P2Y seven-transmembrane-spanning metabotropic receptors. P2X receptors are fast acting and have a lower affinity for ATP, whereas P2Y receptors are slower acting and respond to nanomolar ATP concentrations. To date, seven mammalian P2X subunits and eight P2Y receptors have been discovered (Abbracchio et al., 2009). Under physiological conditions, ATP mediates communication between glial cells and neurons, regulating processes such as synaptic transmission, glial Ca2+ waves (Burnstock, 2013), and sleep cycles (Chikahisa and Séi, 2011). Following an insult, ATP is released into the extracellular space at higher concentrations. This occurs either through the compromised cell membrane of damaged and apoptotic cells, or through exocytotic or non-exocytotic release (Dale and Frenguelli, 2009; Idzko et al., 2014), where it initiates inflammatory signaling cascades, principally via P2X7 (Volonté et al., 2012; see more details below). Once released, ATP is rapidly metabolized by ectonucleotidases into different breakdown products including ADP, AMP, and adenosine, each provoking different cellular responses through their activation of different purinergic receptors (Abbracchio et al., 2009).
P2X7 Expression and Function in the CNS
Constitutive expression of P2X7 in the CNS is largely restricted to microglia, ependymal cells and oligodendrocytes (Skaper, 2011), although, expression in neurons and astrocytes has also been reported (Armstrong et al., 2002; Engel et al., 2012). The principal site for neuronal P2X7 expression seems to be at presynaptic terminals (Miras-Portugal et al., 2003) where it may contribute to the regulation of neurotransmitter release, including GABA and glutamate (Sperlágh et al., 2002). The picture is not entirely clear, however, with contradictory results regarding P2X7 expression and function in both neurons and astrocytes (Sim et al., 2004; Jabs et al., 2007). Recent studies have demonstrated a role for post-transcriptional regulation in mediating cell-type specific changes in P2X7 expression in response to cues in the cellular environment. Jimenez-Mateos et al. (2015) report that microRNA-22 inhibits the translation of P2rx7 mRNA into protein in response to mild, non-cell death causing seizures.
While thought mainly to be activated under pathological conditions, P2X7 is believed to be important in cytokine release during normal brain functioning, with P2X7-deficient mice showing reduced cytokine production (Solle et al., 2001). Three distinct features of P2X7 equip it for responding to injury or stress in the CNS: relatively low affinity for ATP, slow desensitization dynamics, and the ability to permeablize the cell membrane to molecules up to 900 Daltons in size (Sperlágh and Illes, 2014). The low sensitivity of P2X7, depending on the extracellular Ca2+ and Mg2+ concentrations, make it less responsive to the micromolar fluctuations in ATP concentration associated with signaling under physiological conditions (Jiang, 2009). This allows the receptor to function mainly in response to the millimolar concentrations of ATP associated with cell death and excitotoxic stress (Fiebich et al., 2014). Following prolonged activation, P2X7 permeabilizes the cell membrane. It is currently unclear whether this mechanism is via the recruitment of other channels, such as pannexin 1 or the dilation of the P2X7 ion channel itself (Idzko et al., 2014). The consequences however, include a reduction in membrane potential, facilitation of glutamate release via exocytotic or non-exocytotic mechanisms, permeability of the membrane to macromolecules, activation of pro-apoptotic signaling cascades (Beamer et al., 2016), reduction of the action potential threshold via molecular changes at the axon initial segment (Del Puerto et al., 2015), and, as will be described in detail below, initiation of inflammatory cascades (Beamer et al., 2016). Increases in membrane permeability also facilitate the release of ATP itself (Suadicani et al., 2006), though the extent of the contribution of the P2X7 to the release of its own ligand is yet to be determined. All of these processes have the potential to contribute to an epileptic phenotype. Interestingly, P2X7 expression seems to be closely associated with ectonucleotidase tissue-non-specific alkaline phosphatase (TNAP) activity, with mice deficient in TNAP showing decreased P2X7 expression. Furthermore, deficiency in TNAP leads to the development of recurrent seizures which are partially mediated by P2X7 (Sebastián-Serrano et al., 2016).
P2X7 in Brain Inflammation
P2X7 regulates a variety of signaling pathways contributing to inflammation, with downstream effectors likely to be cell-type dependent. Much of the work performed delineating P2X7-signaling has been carried out on peripheral immune cells and in transfected human embryonic kidney 293 (HEK-293) cell lines, but evidence for P2X7 involvement in inflammatory pathways in the CNS is increasing (Beamer et al., 2016; Burnstock, 2016). The principal, and best elucidated, mechanism by which P2X7 contributes to neuroinflammation is via the activation of the NLRP3 inflammasome; a protein complex consisting of caspase-1, apoptosis-associated speck-like protein containing a CARD (ASC), and nod-like receptor protein 3 (NLRP3; Volonté et al., 2012). Mechanisms for P2X7-mediated inflammasome activation include facilitating increases in cell membrane permeability which may trigger inflammasome activation via the resulting decrease in intracellular K+ concentration (Muñoz-Planillo et al., 2013). The inflammasome initiates the cleavage of precursor interleukin molecules into the mature leaderless cytokines, IL-1β, and IL-18, prior to their release into the extracellular space (Beamer et al., 2016). Evidence has also accumulated for ATP-driven, P2X7-dependent release of reactive oxygen species (ROS) from mitochondria, particularly in microglia (Apolloni et al., 2013). Other pathways in which P2X7 activation contributes to the neuroinflammatory response may include inflammasome-independent release of prostaglandin E2 (PGE2) or the activation of membrane metalloproteinases, such as activity of disintegrin-like metalloproteinase 10 (ADAM10) and ADAM17, leading to the removal of chemokine (C-X-C motif) ligand 16 (CXCL16), cluster of differentiation 44 (CD44), soluble amyloid precursor protein (APP), and the IL-6 receptor from the cell membrane, thereby decreasing the sensitivity of the cell to anti-inflammatory mediators (Beamer et al., 2016).
P2X7 as a Drug Target in Epilepsy
While interest in the importance of purinergic signaling during seizures and epilepsy is increasing, the majority of studies, to date, have focused on the contribution of the ionotropic P2X subfamily, in particular, P2X7 (Engel et al., 2016; Rassendren and Audinat, 2016). Much of the early work trying to establish the role of P2X7 in ictogenesis and epileptogenesis was limited to the description of expressional changes of the receptor during and after seizures using techniques such as Western blotting and immunohistochemistry (Vianna et al., 2002; Rappold et al., 2006; Doná et al., 2009). Since 2010, however, there has been an explosion of data re-examining expressional changes of the receptor after SE and during epilepsy and to determine the functional contribution of P2X7 to seizures (Engel et al., 2016). To establish whether P2X7 blockade alters seizure severity and seizure-induced pathology, the animal models of choice have been mouse models of chemically-induced SE [e.g., by kainic acid (KA) or pilocarpine; Engel et al., 2016]. More recent studies have now also evaluated the effects of P2X7 antagonists in traditional seizure models such as the pentylenetetrazol (PTZ)- and maximal electroshock (MES) seizure threshold test as well as the PTZ-kindling model in rats (Fischer et al., 2016). There is now ample data available demonstrating the impact of P2X7 antagonism on seizure pathology during SE. This has already been extensively reviewed and will not be discussed here (Engel et al., 2016). Over recent years, the focus of research has shifted toward the study of P2X7 signaling during the process of epileptogenesis and epilepsy. This has mainly been due to technical advances in the field, such as the development of more specific, BBB-penetrating, and more brain stable P2X7 antagonists (Rech et al., 2016).
P2X7 Expression in Epilepsy
Data showing increased expression of P2X7 during epilepsy has been obtained from both experimental models of epilepsy and patients. Furthermore, the recent use of transgenic P2rx7-GFP reporter mice coupled with GFP-guided patch-clamp (Engel et al., 2012; Jimenez-Pacheco et al., 2016) has made it possible to establish the cell-specific expression pattern of P2X7 without relying solely on the use of antibodies (Sim et al., 2004). In one of the first studies using a rat model of pilocarpine-induced epilepsy, Vianna et al. (2002) provided evidence for an increase in hippocampal P2X7 expression, with strong P2X7 immunoreactivity in mossy fibers. These results were later added to by the same group in a second study showing increased hippocampal P2X7 immunoreactivity in microglia and glutamatergic nerve terminals (Doná et al., 2009). Elevated P2X7 protein levels have also been shown in the hippocampus and cortex in the intra-amygdala (i.a.) KA mouse model of epilepsy (Jimenez-Pacheco et al., 2013, 2016), in surgically resected hippocampus and neocortex from drug-refractory TLE patients (Jimenez-Pacheco et al., 2016) and in neocortical nerve terminals of TLE and non-TLE epilepsy patients (Barros-Barbosa et al., 2016). By using P2rx7-GFP reporter mice, our group has also now determined cell-specific patterns of P2rx7 transcription during epilepsy. In the i.a. KA model, cortical and hippocampal P2rx7 induction was mainly restricted to neurons and microglia. In the cortex, GFP induction was predominantly present in cortical layers V and VI (Jimenez-Pacheco et al., 2013), while in the hippocampus, increased GFP was most prominent in the hippocampal subfield CA1 followed by the dentate gyrus and subfield CA3 (Jimenez-Pacheco et al., 2016). These results have been confirmed by GFP-guided patch-clamp showing increased P2X7 currents in GFP-positive cells when compared to GFP-negative cells from the same animal (Jimenez-Pacheco et al., 2016). This step rules out possible artifacts due to the genetic approach used. In the same study, P2X7 expression was increased in synaptosomes from epileptic mice and showed altered calcium responses when challenged with P2X7 agonists (Jimenez-Pacheco et al., 2016). As observed previously in the i.a. KA model (Engel et al., 2012), GFP reporter activity in epileptic mice was absent in astrocytes, suggesting P2X7 is not increased in these glial cells during epilepsy (Jimenez-Pacheco et al., 2013, 2016).
P2X7 During Epileptogenesis
What is the mechanistic link between P2X7 activation and epileptogenesis? ATP, released in high concentrations from damaged cells after an initial brain insult such as stroke, brain trauma, ischemia, infection, or seizures themselves, may act as an acute “danger signal,” activating microglia (and possibly astrocytes and/or neurons) leading to the cleavage and release of mature, leaderless pro-inflammatory cytokines, in particular IL-1β, and other inflammatory molecules (Dale and Frenguelli, 2009; Rodrigues et al., 2015). Continuing brain inflammation may then promote astro- and microgliosis enhancing release of ATP and other gliotransmitters associated with a further increase (possibly via P2X7 upregulation) of the production of various pro-inflammatory cytokines (including IL-1ß, IL-6, and TNFα), danger molecules (e.g., HMGB1, S100ß protein), and other inflammatory mediators (e.g., nitric oxide, ROS, and PGE2). Alterations in membrane permeability, modifications of ion channel function, changes in glutamate receptor subunit expression or reduction of GABA-mediated inhibition, may, in turn, promote neuronal hyperexcitability and, as a consequence, rhythmic burst firing and epileptiform discharges, finally culminating in focal or generalized seizures (Di Maio, 2014). Injury and/or loss of neurons and synaptic remodeling (mossy fiber sprouting), breakdown of the BBB, lymphocyte accumulation and angiogenesis, all processes associated with an increase in P2X7 activity (Sperlágh and Illes, 2014), may also contribute to epileptogenesis and seizure initiation (Friedman and Dingledine, 2011). Chronic inflammation and recurrent seizures per se may promote the release of ATP and pro-inflammatory cytokines and activate immune responses which sustain seizure recurrence. This leads to a vicious cycle of P2X7 activation, inflammation, and lowering of the seizure threshold (Librizzi et al., 2012; Figure 1).
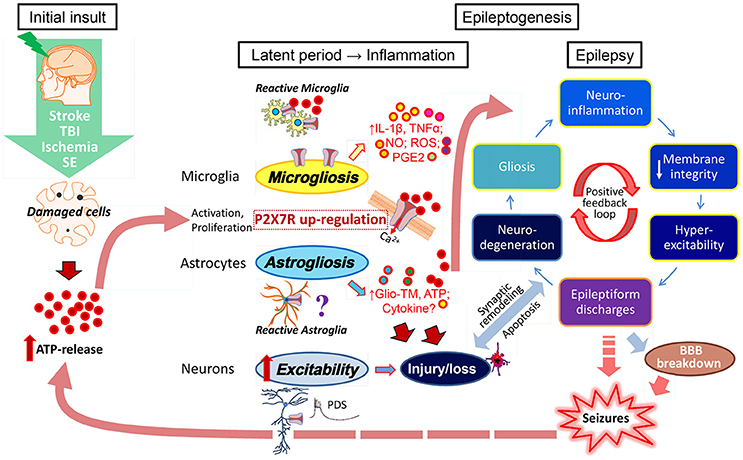
Figure 1. ATP-driven P2X7 activation as possible contributor to epileptogenesis and epilepsy. Brain inflammation has been suggested as a crucial etiopathogenic mechanism of epilepsy contributing to seizure generation and the development of epilepsy (Vezzani et al., 2016). Brain injury (stroke, trauma, ischemia)-induced cell damage and/or cell death associated with an up-regulation of P2X7 on microglia and possibly neurons, leads to a massive release of ATP which then acts as a danger signal resulting in the activation of astrocytes and microglia. The P2X7 in particular has been described as a major regulator of the synthesis and secretion of cytokines (IL-1ß, TNFα) and other inflammatory mediators (NO, ROS, PGE2) via microglial cells. However, the P2X7 may also trigger the release of gliotransmitters (Glio-TM) and ATP from astrocytes and neuronal terminals. Continuing brain inflammation is characterized by astro- and microgliosis with enhanced release of ATP and pro-inflammatory mediators/molecules causing changes in neuronal membrane integrity, modifications of ion channels and consequently neuronal hyperexcitability. This will lead to increased brain susceptibility to seizures initiated by paroxysmal depolarization shifts (PDS), rhythmic burst firing, and epileptiform discharges finally producing focal or generalized seizures. Recurrent seizures per se promote the release of ATP and pro-inflammatory cytokines and activate immune response that sustains seizure recurrence leading to a vicious cycle of increased inflammation and hyperexcitability. Other pathological mechanism may be involved in epileptogenesis and seizure initiation, such as injury and/or loss of neurons, synaptic remodeling (mossy fiber sprouting), BBB breakdown, and lymphocyte accumulation.
The first evidence for the antiepileptogenic potential of P2X7 antagonism was produced using the PTZ-kindling model in rats, a well-established model of epileptogenesis (Soni et al., 2015). In this model, repeated injections of PTZ, with an initial sub-convulsive dose, induce a progressive increase in seizure activity, culminating in the development of generalized tonic-clonic seizures (Fischer and Kittner, 1998; Dhir, 2012). Treatment with the P2X7 antagonist BBG significantly decreased the mean kindling score and restored behavioral deficits, including cognition and motor coordination (Soni et al., 2015). This effect was potentiated by the co-administration of ceftriaxone via up-regulation of the glutamate transporter, GLT-1. Drug washout experiments, however, were not performed. In a more recent study, also using the PTZ-kindling model in rats, the two potent and brain-permeable P2X7 antagonists, JNJ-47965567 and AFC-5128 (15 mg/kg s.c. 30 min, or 30 mg/kg i.p. 45 min before PTZ, respectively), significantly delayed kindling development. This effect was long-lasting, however, the compounds were unable to prevent or reverse the process of epileptogenesis (Fischer et al., 2016). In another study using the i.a. KA model, we found that hippocampal P2X7 up-regulation, achieved through the inhibition of a P2X7-suppressing microRNA (microRNA-22), resulted in a more severe epileptic phenotype (Jimenez-Mateos et al., 2015). Increased P2X7 expression was accompanied by increased cytokine levels (IL-1β and TNFα), astrogliosis and cognitive impairment. Interestingly, microgliosis was reduced in these mice (Jimenez-Mateos et al., 2015). In contrast to these results, the blockade of P2X7 by AZ10606120 (3 μg i.c.v. 30 min post-pilocarpine) or BBG (50 mg/kg i.p. 30 min post-pilocarpine; repeated once per day for 4 days) increased the number of seizures and their severity in rats observed for the 28 following days post-pilocarpine-induced SE (Rozmer et al., 2016), but P2X7 brain occupancy studies were not performed. This is somewhat surprising as AZ10606120 administered shortly after the induction of seizures revealed marked neuroprotective effects in hippocampal neurons (Rozmer et al., 2016), and previous studies by our group using the i.a. KA model have shown that protecting the brain from seizure-induced cell death resulted in a less severe epileptic phenotype (Engel et al., 2010). We do not know what the reasons for the observed differences are. As seen before for SE studies (Kim and Kang, 2011), however, P2X7 antagonism seems to provoke the opposite effect when applied to the KA or pilocarpine model, likely due to differences in the mechanism of induction. Moreover, differences in cell death, limited in the pilocarpine-induced SE mouse model (Engel et al., 2016), may result in differences in the availability of extracellular ATP. Differences in drugs, doses of drugs and route of delivery may also add to variation between studies.
P2X7 During Epilepsy
While increased P2X7 protein levels during epilepsy have been widely reported (Engel et al., 2016), until recently, no evidence existed for an impact of P2X7 antagonism on the epileptic phenotype at all and, therefore, whether P2X7 blockade represents a possible new treatment strategy for epilepsy. Two new studies however, both published in 2016 (Amhaoul et al., 2016; Jimenez-Pacheco et al., 2016), have now attempted to shed some light on this unresolved issue (Table 1). Both studies used KA as a trigger for epilepsy; while one study used the multiple low-dose KA model in rats (Amhaoul et al., 2016), the other, using the i.a. KA model in mice (Jimenez-Pacheco et al., 2016). To investigate the impact of P2X7 antagonism in chronically epileptic rats, the specific P2X7 antagonist JNJ-42253432 (0.6 g/kg/2 ml) was administered via subcutaneous (s.c.) injection for 1 week via mini-pumps in rats, 3 months following KA-induced SE. Epileptic rats treated with JNJ-42253432 experienced the same number of seizure episodes during treatment, however, the severity of these episodes was reduced. The authors concluded that rather than suppressing seizures, P2X7 antagonism leads to a shift in seizure severity, resulting in a milder epileptic phenotype. P2X7 blockade did not alter microgliosis in their study and no drug washout was performed to assess whether changes in the seizure phenotype persists following drug withdrawal (Amhaoul et al., 2016). In the second study, to test whether P2X7 blockade could ameliorate the epileptic phenotype in mice, epileptic mice were treated with another specific P2X7 antagonist, JNJ-47965567 (30 mg/kg i.p.), twice daily for 5 days, followed by a 5-day washout period. In this study, JNJ-47965567 treatment reduced the total amount of epileptic seizures by over 50% during the drug phase. Seizure severity, however, was unchanged. Remarkably, rather than returning to baseline, seizure rates in treated mice continued to decrease during the washout period implying that P2X7 antagonism could modify disease progression (Jimenez-Pacheco et al., 2016). This is even more outstanding as seizure rates in epileptic animals treated with conventional AEDs or anti-inflammatory drugs, if effective, returned to baseline immediately after drug cessation (Grabenstatter et al., 2007; Maroso et al., 2010; Klein et al., 2015). In addition, P2X7 antagonist-treated mice demonstrated a strong reduction in astrogliosis and microgliosis, even when analyzed after washout (Jimenez-Pacheco et al., 2016). We do not know how P2X7 blockade reduces epileptic seizures during and beyond treatment. While effects during treatment may be related to the reduction of neuronal excitability by changes in intracellular ion concentrations or changes in neurotransmitter release (Barros-Barbosa et al., 2016), the most likely explanation for the observed disease-modifying effect is a reduction in astrogliosis and microgliosis. P2X7 has been shown to activate astrocytes. Astrogliosis itself can contribute to a lowering of the seizure threshold via dysregulation of extracellular ionic balance, impaired neurotransmitter reuptake, and release of pro-inflammatory cytokines and purines, including ATP and adenosine (Bedner et al., 2015; Robel et al., 2015). P2X7 has also been shown to drive microglial activation directly, thereby increasing the release of cytokines such as IL-1β (Monif et al., 2009). Chronic brain inflammation in turn leads to increased extracellular concentrations of ATP and P2X7 activity. P2X7 antagonists may therefore act as a break interrupting the vicious cycle of increased neuroinflammation and hyperexcitability (Figure 1).
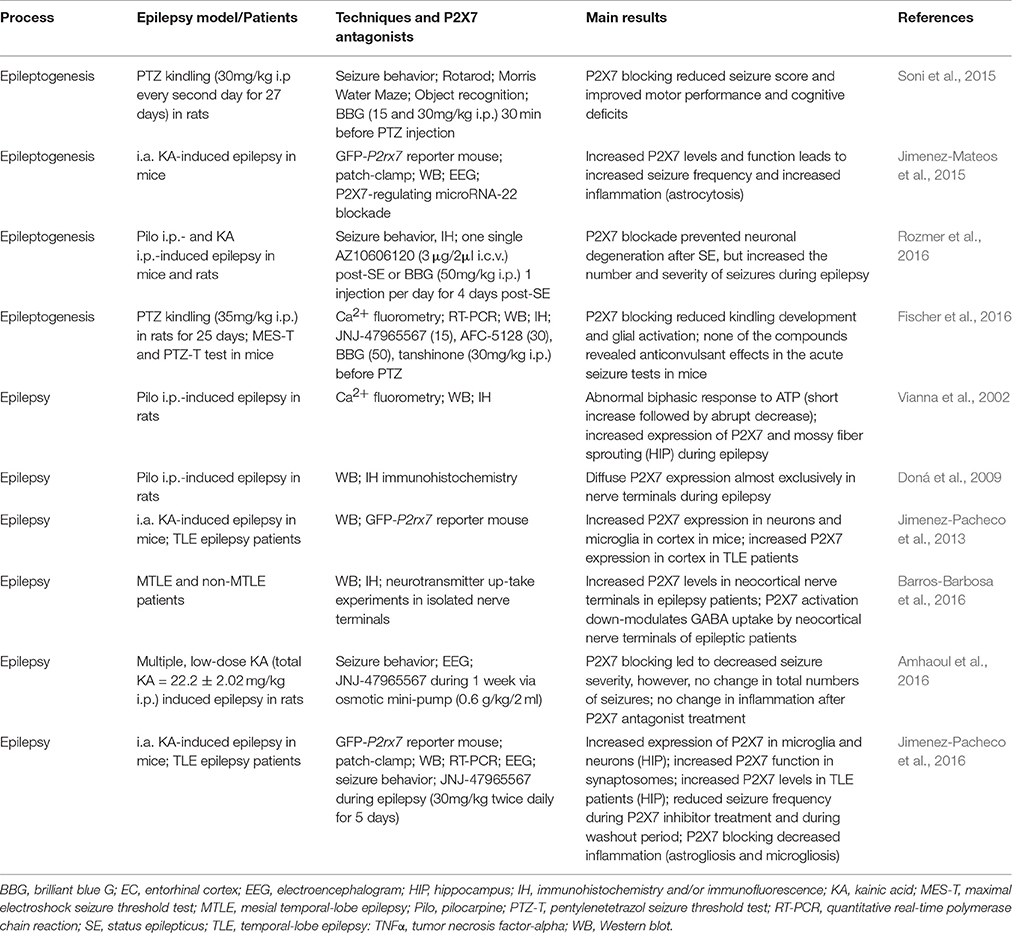
Table 1. Summary of the main findings related to P2X7 expression and function during epileptogenesis and epilepsy in experimental models of epilepsy and patients.
Conclusions and Future Directions
Mounting data obtained from both experimental animal models and patients has now convincingly demonstrated a causal role for P2X7 signaling during seizures and epilepsy. Whether P2X7 antagonism has an anticonvulsive or neuroprotective effect, however, is still not completely understood and seems to depend, at least in part, on the animal model used. While mixed results have been obtained regarding the effect of P2X7 antagonists when administered prior to or soon after the induction of SE, the effect of administration following the emergence of chronic epilepsy seems to consistently ameliorate the epileptic phenotype. How to bring P2X7 antagonists further toward a possible clinical application? The development of BBB permeable, brain stable and highly specific P2X7 antagonists has been an important step forward, however, several urgent questions remain to be resolved. (i) Adverse effects of P2X7 antagonism seem to be pertinent to the pilocarpine model, however, we must dissect why P2X7 antagonism results in a different phenotype according to the animal model used. (ii) We still do not know when ATP is released and what local concentrations are reached. Attempts have been made (Doná et al., 2016), however, the techniques used may lack sufficient temporal resolution to detect seizure-induced ATP release in the brain. (iii) What are the cell-specific contributions of P2X7 to disease progression? Various publications have suggested an upregulation of the receptor in neurons, however, the consequences of this up-regulation are unknown. (iv) Does P2X7 blockade lead to neuroprotection or is the reduction in cell death observed during SE a mere consequence of their anticonvulsive properties? (v) How can patients who could potentially benefit from P2X7 blockade be identified? Are there any biomarkers which would predict a pathological P2X7 activation in the brain? (vi) What is the optimal treatment regimen, dose and time window for application? Previous studies have suggested that a positive outcome may depend on the time-point of intervention (Roth et al., 2014; Kaiser et al., 2016). (vii) Finally, do P2X7 antagonists work where other AEDs fail, or show synergistic effects with these compounds? Could they have utility as adjunctive treatment in conjunction with current AEDs as demonstrated in the i.a. KA model and MES seizure threshold test (Engel et al., 2012; Fischer et al., 2016)?
In conclusion, while a possible benefit for P2X7 antagonism during acute seizures and epileptogenesis remains controversial, recent research has shown the potential of P2X7 antagonists for the treatment of epilepsy, thereby providing a much needed new target with a new mechanism of action distinct from currently used AEDs. The availability of new tools and drugs will hopefully shed light on outstanding unanswered questions and accelerate progress toward possible future clinical use.
Author Contributions
EB, wrote the manuscript; WF, designed the figure and edited the manuscript; TE, wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funding from Science Foundation Ireland (13/SIRG/2098 to TE) and from the Health Research Board (HRA-POR-2015-1243 to TE).
Abbreviations
BBB, blood–brain barrier; IL-1ß, interleukin-1ß; NO, nitric oxide; PGE2, prostaglandin E2; ROS, reactive oxygen species; SE, status epilepticus; TBI, traumatic brain injury; TNFα, tumor necrosis factor-alpha.
References
Abbracchio, M. P., Burnstock, G., Verkhratsky, A., and Zimmermann, H. (2009). Purinergic signalling in the nervous system: an overview. Trends Neurosci. 32, 19–29. doi: 10.1016/j.tins.2008.10.001
Amhaoul, H., Ali, I., Mola, M., Van Eetveldt, A., Szewczyk, K., Missault, S., et al. (2016). P2X7 receptor antagonism reduces the severity of spontaneous seizures in a chronic model of temporal lobe epilepsy. Neuropharmacology 105, 175–185. doi: 10.1016/j.neuropharm.2016.01.018
Apolloni, S., Amadio, S., Montilli, C., Volonté, C., and D'ambrosi, N. (2013). Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4102–4116. doi: 10.1093/hmg/ddt259
Armstrong, J. N., Brust, T. B., Lewis, R. G., and Macvicar, B. A. (2002). Activation of presynaptic P2X7-like receptors depresses mossy fiber-CA3 synaptic transmission through p38 mitogen-activated protein kinase. J. Neurosci. 22, 5938–5945.
Balosso, S., Maroso, M., Sanchez-Alavez, M., Ravizza, T., Frasca, A., Bartfai, T., et al. (2008). A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain 131, 3256–3265. doi: 10.1093/brain/awn271
Barros-Barbosa, A. R., Fonseca, A. L., Guerra-Gomes, S., Ferreirinha, F., Santos, A., Rangel, R., et al. (2016). Up-regulation of P2X7 receptor-mediated inhibition of GABA uptake by nerve terminals of the human epileptic neocortex. Epilepsia 57, 99–91 doi: 10.1111/epi.13263
Beamer, E., Gölöncsér, F., Horváth, G., Beko, K., Otrokocsi, L., Koványi, B., et al. (2016). Purinergic mechanisms in neuroinflammation: an update from molecules to behavior. Neuropharmacology 104, 94–104. doi: 10.1016/j.neuropharm.2015.09.019
Bedner, P., Dupper, A., Hüttmann, K., Müller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. doi: 10.1093/brain/awv067
Bialer, M., and White, H. S. (2010). Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 9, 68–82. doi: 10.1038/nrd2997
Blond, B. N., Detyniecki, K., and Hirsch, L. J. (2016). Assessment of treatment side effects and quality of life in people with epilepsy. Neurol. Clin. 34, 395–410. doi: 10.1016/j.ncl.2015.11.002
Burnstock, G. (2013). Introduction to purinergic signalling in the brain. Adv. Exp. Med. Biol. 986, 1–12. doi: 10.1007/978-94-007-4719-7_1
Burnstock, G. (2016). P2X ion channel receptors and inflammation. Purinergic Signal. 12, 59–67. doi: 10.1007/s11302-015-9493-0
Chang, B. S., and Lowenstein, D. H. (2003). Epilepsy. N. Engl. J. Med. 349, 1257–1266. doi: 10.1056/NEJMra022308
Chikahisa, S., and Séi, H. (2011). The role of ATP in sleep regulation. Front. Neurol. 2:87. doi: 10.3389/fneur.2011.00087
Dale, N., and Frenguelli, B. G. (2009). Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 7, 160–179. doi: 10.2174/157015909789152146
Del Puerto, A., Fronzaroli-Molinieres, L., Perez-Alvarez, M. J., Giraud, P., Carlier, E., Wandosell, F., et al. (2015). ATP-P2X7 receptor modulates axon initial segment composition and function in physiological conditions and brain injury. Cereb. Cortex 25, 2282–2294. doi: 10.1093/cercor/bhu035
Dhir, A. (2012). Pentylenetetrazol (PTZ) kindling model of epilepsy. Curr. Protoc. Neurosci. Chapter 9, Unit 9 37. doi: 10.1002/0471142301.ns0937s58
Di Maio, R. (2014). Neuronal mechanisms of epileptogenesis. Front. Cell Neurosci. 8:29. doi: 10.3389/fncel.2014.00029
Doná, F., Conceicao, I. M., Ulrich, H., Ribeiro, E. B., Freitas, T. A., Nencioni, A. L., et al. (2016). Variations of ATP and its metabolites in the hippocampus of rats subjected to pilocarpine-induced temporal lobe epilepsy. Purinergic Signal. 12, 295–302. doi: 10.1007/s11302-016-9504-9
Doná, F., Ulrich, H., Persike, D. S., Conceicao, I. M., Blini, J. P., Cavalheiro, E. A., et al. (2009). Alteration of purinergic P2X4 and P2X7 receptor expression in rats with temporal-lobe epilepsy induced by pilocarpine. Epilepsy Res. 83, 157–167. doi: 10.1016/j.eplepsyres.2008.10.008
Elger, C. E. (2016). Epilepsy in 2015: classic antiepileptic drugs under fire, and new options emerge. Nat. Rev. Neurol. 12, 72–74. doi: 10.1038/nrneurol.2016.1
Engel, T., Alves, M., Sheedy, C., and Henshall, D. C. (2016). ATPergic signalling during seizures and epilepsy. Neuropharmacology 104, 140–153. doi: 10.1016/j.neuropharm.2015.11.001
Engel, T., Gomez-Villafuertes, R., Tanaka, K., Mesuret, G., Sanz-Rodriguez, A., Garcia-Huerta, P., et al. (2012). Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 26, 1616–1628. doi: 10.1096/fj.11-196089
Engel, T., Murphy, B. M., Hatazaki, S., Jimenez-Mateos, E. M., Concannon, C. G., Woods, I., et al. (2010). Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. 24, 853–861. doi: 10.1096/fj.09-145870
Fiebich, B. L., Akter, S., and Akundi, R. S. (2014). The two-hit hypothesis for neuroinflammation: role of exogenous ATP in modulating inflammation in the brain. Front. Cell. Neurosci. 8:260. doi: 10.3389/fncel.2014.00260
Fischer, W., Franke, H., Krügel, U., Müller, H., Dinkel, K., Lord, B., et al. (2016). Critical evaluation of P2X7 receptor antagonists in selected seizure models. PLoS ONE 11:e0156468. doi: 10.1371/journal.pone.0156468
Fischer, W., and Kittner, H. (1998). Influence of ethanol on the pentylenetetrazol-induced kindling in rats. J. Neural. Transm. 105, 1129–1142. doi: 10.1007/s007020050117
Friedman, A., and Dingledine, R. (2011). Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 52(Suppl. 3), 33–39. doi: 10.1111/j.1528-1167.2011.03034.x
Grabenstatter, H. L., Clark, S., and Dudek, F. E. (2007). Anticonvulsant effects of carbamazepine on spontaneous seizures in rats with kainate-induced epilepsy: comparison of intraperitoneal injections with drug-in-food protocols. Epilepsia 48, 2287–2295. doi: 10.1111/j.1528-1167.2007.01263.x
Idzko, M., Ferrari, D., Riegel, A. K., and Eltzschig, H. K. (2014). Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 124, 1029–1037. doi: 10.1182/blood-2013-09-402560
Jabs, R., Matthias, K., Grote, A., Grauer, M., Seifert, G., and Steinhauser, C. (2007). Lack of P2X receptor mediated currents in astrocytes and GluR type glial cells of the hippocampal CA1 region. Glia 55, 1648–1655. doi: 10.1002/glia.20580
Jiang, L. H. (2009). Inhibition of P2X(7) receptors by divalent cations: old action and new insight. Eur. Biophys. J. 38, 339–346. doi: 10.1007/s00249-008-0315-y
Jimenez-Mateos, E. M., Arribas-Blazquez, M., Sanz-Rodriguez, A., Concannon, C., Olivos-Ore, L. A., Reschke, C. R., et al. (2015). MicroRNA targeting of the P2X7 purinoceptor opposes a contralateral epileptogenic focus in the hippocampus. Sci. Rep. 5:17486. doi: 10.1038/srep17486
Jimenez-Pacheco, A., Diaz-Hernandez, M., Arribas-Blazquez, M., Sanz-Rodriguez, A., Olivos-Ore, L. A., Artalejo, A. R., et al. (2016). Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J. Neurosci. 36, 5920–5932. doi: 10.1523/JNEUROSCI.4009-15.2016
Jimenez-Pacheco, A., Mesuret, G., Sanz-Rodriguez, A., Tanaka, K., Mooney, C., Conroy, R., et al. (2013). Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079. Epilepsia 54, 1551–1561. doi: 10.1111/epi.12257
Kaiser, M., Penk, A., Franke, H., Krügel, U., Nörenberg, W., Huster, D., et al. (2016). Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal. 12, 453–463. doi: 10.1007/s11302-016-9511-x
Kim, J. E., and Kang, T. C. (2011). The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J. Clin. Invest. 121, 2037–2047. doi: 10.1172/JCI44818
Klein, S., Bankstahl, M., and Löscher, W. (2015). Inter-individual variation in the effect of antiepileptic drugs in the intrahippocampal kainate model of mesial temporal lobe epilepsy in mice. Neuropharmacology 90, 53–62. doi: 10.1016/j.neuropharm.2014.11.008
Librizzi, L., Noè, F., Vezzani, A., De Curtis, M., and Ravizza, T. (2012). Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann. Neurol. 72, 82–90. doi: 10.1002/ana.23567
Marchi, N., Fan, Q., Ghosh, C., Fazio, V., Bertolini, F., Betto, G., et al. (2009). Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol. Dis. 33, 171–181. doi: 10.1016/j.nbd.2008.10.002
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., et al. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419. doi: 10.1038/nm.2127
Miras-Portugal, M. T., Díaz-Hernandez, M., Giráldez, L., Hervás, C., Gómez-Villafuertes, R., Sen, R. P., et al. (2003). P2X7 receptors in rat brain: presence in synaptic terminals and granule cells. Neurochem. Res. 28, 1597–1605 doi: 10.1023/A:1025690913206
Monif, M., Reid, C. A., Powell, K. L., Smart, M. L., and Williams, D. A. (2009). The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J. Neurosci. 29, 3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009
Moshé, S. L., Perucca, E., Ryvlin, P., and Tomson, T. (2015). Epilepsy: new advances. Lancet 385, 884–898. doi: 10.1016/S0140-6736(14)60456-6
Muñoz-Planillo, R., Kuffa, P., Martínez-Colón, G., Smith, B. L., Rajendiran, T. M., and Nunez, G. (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. doi: 10.1016/j.immuni.2013.05.016
Patel, S. I., and Pennell, P. B. (2016). Management of epilepsy during pregnancy: an update. Ther. Adv. Neurol. Disord. 9, 118–129. doi: 10.1177/1756285615623934
Pitkänen, A., and Engel, J. Jr. (2014). Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics 11, 231–241. doi: 10.1007/s13311-014-0257-2
Pitkänen, A., and Lukasiuk, K. (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186. doi: 10.1016/S1474-4422(10)70310-0
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Rappold, P. M., Lynd-Balta, E., and Joseph, S. A. (2006). P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 1089, 171–178. doi: 10.1016/j.brainres.2006.03.040
Rassendren, F., and Audinat, E. (2016). Purinergic signaling in epilepsy. J. Neurosci. Res. 94, 781–793. doi: 10.1002/jnr.23770
Rech, J. C., Bhattacharya, A., Letavic, M. A., and Savall, B. M. (2016). The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg. Med. Chem. Lett. 26, 3838–3845. doi: 10.1016/j.bmcl.2016.06.048
Rees, M. I. (2010). The genetics of epilepsy–the past, the present and future. Seizure 19, 680–683. doi: 10.1016/j.seizure.2010.10.029
Robel, S., Buckingham, S. C., Boni, J. L., Campbell, S. L., Danbolt, N. C., Riedemann, T., et al. (2015). Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci. 35, 3330–3345. doi: 10.1523/JNEUROSCI.1574-14.2015
Rochfort, K. D., and Cummins, P. M. (2015). The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem. Soc. Trans. 43, 702–706. doi: 10.1042/BST20140319
Rodrigues, R. J., Tomé, A. R., and Cunha, R. A. (2015). ATP as a multi-target danger signal in the brain. Front. Neurosci. 9:148. doi: 10.3389/fnins.2015.00148
Roth, T. L., Nayak, D., Atanasijevic, T., Koretsky, A. P., Latour, L. L., and McGavern, D. B. (2014). Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. doi: 10.1038/nature12808
Rozmer, K., Gao, P., Araujo, M. G., Khan, M. T., Liu, J., Rong, W., et al. (2016). Pilocarpine-induced status epilepticus increases the sensitivity of P2X7 and P2Y1 receptors to nucleotides at neural progenitor cells of the juvenile rodent hippocampus. Cereb. Cortex. doi: 10.1093/cercor/bhw178. [Epub ahead of print].
Sebastián-Serrano, Á., Engel, T., De Diego-García, L., Olivos-Oré, L. A., Arribas-Blázquez, M., Martínez-Frailes, C., et al. (2016). Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum. Mol. Genet. 25, 4143–4156. doi: 10.1093/hmg/ddw248
Sim, J. A., Young, M. T., Sung, H. Y., North, R. A., and Surprenant, A. (2004). Reanalysis of P2X7 receptor expression in rodent brain. J. Neurosci. 24, 6307–6314. doi: 10.1523/JNEUROSCI.1469-04.2004
Skaper, S. D. (2011). Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol. Disord. Drug Targets 10, 44–56. doi: 10.2174/187152711794488638
Solle, M., Labasi, J., Perregaux, D. G., Stam, E., Petrushova, N., Koller, B. H., et al. (2001). Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 276, 125–132. doi: 10.1074/jbc.M006781200
Soni, N., Koushal, P., Reddy, B. V., Deshmukh, R., and Kumar, P. (2015). Effect of GLT-1 modulator and P2X7 antagonists alone and in combination in the kindling model of epilepsy in rats. Epilepsy Behav. 48, 4–14. doi: 10.1016/j.yebeh.2015.04.056
Sperlágh, B., and Illes, P. (2014). P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol. Sci. 35, 537–547. doi: 10.1016/j.tips.2014.08.002
Sperlágh, B., Kofalvi, A., Deuchars, J., Atkinson, L., Milligan, C. J., Buckley, N. J., et al. (2002). Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J. Neurochem. 81, 1196–1211. doi: 10.1046/j.1471-4159.2002.00920.x
Suadicani, S. O., Brosnan, C. F., and Scemes, E. (2006). P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 26, 1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006
Vezzani, A., Lang, B., and Aronica, E. (2016). Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 6:a022699. doi: 10.1101/cshperspect.a022699
Vezzani, A., Moneta, D., Conti, M., Richichi, C., Ravizza, T., De Luigi, A., et al. (2000). Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 11534–11539. doi: 10.1073/pnas.190206797
Vezzani, A., and Viviani, B. (2015). Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96, 70–82. doi: 10.1016/j.neuropharm.2014.10.027
Vianna, E. P., Ferreira, A. T., Naffah-Mazzacoratti, M. G., Sanabria, E. R., Funke, M., Cavalheiro, E. A., et al. (2002). Evidence that ATP participates in the pathophysiology of pilocarpine-induced temporal lobe epilepsy: fluorimetric, immunohistochemical, and Western blot studies. Epilepsia 43(Suppl. 5), 227–229. doi: 10.1046/j.1528-1157.43.s.5.26.x
Volonté, C., Apolloni, S., Skaper, S. D., and Burnstock, G. (2012). P2X7 receptors: channels, pores and more. CNS Neurol. Disord. Drug Targets 11, 705–721. doi: 10.2174/187152712803581137
Keywords: epilepsy, seizures, drug-refractory, inflammation, ATP, P2X7 receptor
Citation: Beamer E, Fischer W and Engel T (2017) The ATP-Gated P2X7 Receptor As a Target for the Treatment of Drug-Resistant Epilepsy. Front. Neurosci. 11:21. doi: 10.3389/fnins.2017.00021
Received: 14 November 2016; Accepted: 11 January 2017;
Published: 02 February 2017.
Edited by:
Leanne Stokes, University of East Anglia, UKReviewed by:
Robert Wykes, UCL Institute of Neurology, UKThomas Grutter, University of Strasbourg, CNRS, France
Copyright © 2017 Beamer, Fischer and Engel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Engel, dGVuZ2VsQHJjc2kuaWU=