 Jianguo Li1,2,3
Jianguo Li1,2,3 Jun-Jie Koh1
Jun-Jie Koh1 Shouping Liu1
Shouping Liu1 Rajamani Lakshminarayanan1
Rajamani Lakshminarayanan1 Chandra S. Verma1,2,4,5
Chandra S. Verma1,2,4,5 Roger W. Beuerman1,3*
Roger W. Beuerman1,3*- 1Ocular Chemistry and Anti-Infectives, Singapore Eye Research Institute, Singapore, Singapore
- 2Agency for Science, Technology and Research (A*STAR), Bioinformatics Institute, Singapore, Singapore
- 3Duke-NUS Graduate Medical School, SRP Neuroscience and BD, Singapore, Singapore
- 4Department of Biological Sciences, National University of Singapore, Singapore, Singapore
- 5School of Biological Sciences, Nanyang Technological University, Singapore, Singapore
Antimicrobial peptides (AMPs) are promising next generation antibiotics that hold great potential for combating bacterial resistance. AMPs can be both bacteriostatic and bactericidal, induce rapid killing and display a lower propensity to develop resistance than do conventional antibiotics. Despite significant progress in the past 30 years, no peptide antibiotic has reached the clinic yet. Poor understanding of the action mechanisms and lack of rational design principles have been the two major obstacles that have slowed progress. Technological developments are now enabling multidisciplinary approaches including molecular dynamics simulations combined with biophysics and microbiology toward providing valuable insights into the interactions of AMPs with membranes at atomic level. This has led to increasingly robust models of the mechanisms of action of AMPs and has begun to contribute meaningfully toward the discovery of new AMPs. This review discusses the detailed action mechanisms that have been put forward, with detailed atomistic insights into how the AMPs interact with bacterial membranes. The review further discusses how this knowledge is exploited toward developing design principles for novel AMPs. Finally, the current status, associated challenges, and future directions for the development of AMP therapeutics are discussed.
Introduction
The continued emergence of resistant pathogens world-wide, particularly among Gram-negative bacteria, has become a leading health care challenge (McKenna, 2013). Resistant strains of bacteria/fungus/yeast are regularly being found for almost every antimicrobial used clinically. On the other hand, a decline in the approval of new antibiotics has further exacerbated the problem, leading to an “antibiotic crisis” (Livermore, 2004). Persistence of the current situation is driving clinicians to use drugs that are associated with significant toxicity. In contrast to small molecule antibiotics, antimicrobial peptides (AMPs) act on the bacterial membrane which is an evolutionarily conserved component of bacterial cells (Yeaman and Yount, 2003). Bacterial membranes define the phenotype and membrane mutations would likely be costly for bacteria, therefore AMPs are less likely to induce bacterial resistance (Zasloff, 2002). Moreover, AMPs are bactericidal and they kill bacteria much more rapidly than conventional antibiotics (Zasloff, 1987; Romeo et al., 1988; Mygind et al., 2005; Marr et al., 2006; Bai et al., 2012; Deslouches et al., 2013).
There are more than 2,000 naturally occurring and synthetic AMPs (Wang G. et al., 2016). Naturally occurring AMPs, including defensins, are produced in various living organisms including humans and are part of the innate immune system (Diamond et al., 2009). In humans, these molecules are found in lymphocytes and epithelial surfaces (e.g., skin, eye, lung, and intestines etc.). For example, Paneth cells, the specialized secretory epithelial cells in the small intestine, produce high levels of AMPs (e.g., alpha-defensins, lysozyme etc.), resulting in a controlled number of bacteria in the small intestine (Ayabe et al., 2004; Bevins and Salzman, 2011). The tear fluid of the eye is also a rich source of AMPs and include the defensins, lysozyme, and cathelicidins, protecting the eyes from infections (McDermott, 2013). Other AMPs can be either synthesized ribosomally or produced non-ribosomally in bacteria and fungi during cultivation on various carbon sources (Makovitzki et al., 2006). Lipopeptides such as polymyxins are produced as metabolites in bacteria or fungi. However, these are also membrane targeting, so, in this review, the general term, AMP will be used to refer to them. In addition to their antimicrobial-activity, some AMPs have other important roles in immune regulation, inflammation, anti-cancer, sepsis, and wound healing. Indeed, some AMPs demonstrate not only antimicrobial, but also anti-inflammatory and immune regulatory activities (Sjogren, 2004; Gordon et al., 2005). The non-antimicrobial functions of AMPs are out of the scope of this review, and the interested reader can refer to relevant references (Ganz, 2003a,b; Marta Guarna et al., 2006; Van Lenten et al., 2008; McCormick and Weinberg, 2010; Shin and Jo, 2011; Li et al., 2012; Schuerholz et al., 2012; Gaspar et al., 2013; Hilchie et al., 2013; Martin et al., 2015a,b; Brandenburg et al., 2016).
AMPs are generally short peptides with <50 amino acids. Although there are more than 2,000 naturally occurring or synthetic AMPs, their lengths, sequences and 3-dimensional (3-D) conformations differ significantly, making it difficult to relate the sequence/structure to antimicrobial activity. Moreover, AMPs can undergo conformational changes upon adsorption onto the bacterial membrane and have evolved several mechanisms of action, which further complicate the analysis of a structure-activity relationship (SAR). As there is insufficient detailed knowledge of the action mechanisms in bacteria, synthetic AMPs are generally designed by trial and error (Epand and Vogel, 1999). Overcoming this gap in understanding could make antibiotic design closer to what is the common structure based approach for drug design and the action mechanism is crucial to this goal. Although the action mechanism for AMPs have been discussed in earlier reviews, most of them focus on the description of the general features of empirical models (e.g., pore-forming and carpet models), few discuss the detailed atomistic events and the dynamic processes of AMP mechanisms of action (Epand and Vogel, 1999; Shai, 2002; Guilhelmelli et al., 2013; Lee et al., 2016). Recently, the employment of molecular dynamics (MD) simulations has deepened our understanding of the underlying action mechanism at a molecular level. Based on insights at atomic level, rational molecular principles have been put forward for the development of AMP therapeutics. Our primary focus in this review is to provide a fundamental understanding of the atomistic mechanisms of various AMPs and discuss molecular principles for practical AMP design. In addition, the current status of development of AMP antibiotics as well as challenges and future prospects for AMP therapeutics are discussed.
Modes of Action of AMPs
Structure of the Bacterial Membrane: The Target of AMPs
To understand the mode of action of AMPs, we begin with a discussion of the structure and physical properties of the bacterial membrane-the target of AMPs. Bacteria are broadly classified as either Gram-positive or Gram-negative, characterized by significant differences in their cell envelopes (Figure 1). The inner or cytoplasmic membranes of both bacteria groups are similar. However, the outer cell envelopes are significantly different. In Gram-positive bacteria, there is a layer of crosslinked peptidoglycan decorated with negatively charged teichoic acid surrounding the cytoplasmic membrane, forming a thick matrix which maintains the rigidity of the bacterial cell. Nano-sized pores penetrate into the peptidoglycan layers, allowing AMPs to diffuse through (Meroueh et al., 2006). In contrast, the peptidoglycan layer in Gram-negative bacteria is much thinner and less cross-linked. In addition, Gram-negative bacteria have an additional outer membrane outside the peptidoglycan layer. The inner leaflet consists purely of phosphate lipids while the outer leaflet is primarily a coat of lipopolysaccharide (LPS; Ruiz et al., 2006). LPS molecules are decorated with a high number of negatively charged phosphate groups that are engaged in salt-bridges with divalent cations (e.g., Ca2+ and Mg2+), resulting in an electrostatic network (Nikaido, 2003). This electrostatic region serves as a primary barrier to most hydrophobic antibiotics, resulting in low permeability. Therefore, the details as to how AMPs penetrate into Gram-positive and Gram-negative bacteria must vary in their atomistic interactions. In the case of Gram-positive bacteria, AMPs need to diffuse across the peptidoglycan matrix first and then act on the cytoplasmic membrane. In contrast, killing Gram-negative bacteria involves perturbation or disruption of both outer and cytoplasmic membranes. Inability to permeabilize or disrupt the outer membrane results in the loss of antimicrobial activity. Daptomycin is able to disrupt the cytoplasmic membrane, but not able to permeabilize/disrupt the outer membrane of Gram-negative bacteria. As such, it is highly active against Gram-positive bacteria such as methicillin-resistant Staphylococcus aureus (MRSA), but has no activity against Gram-negative bacteria (Tally and DeBruin, 2000).
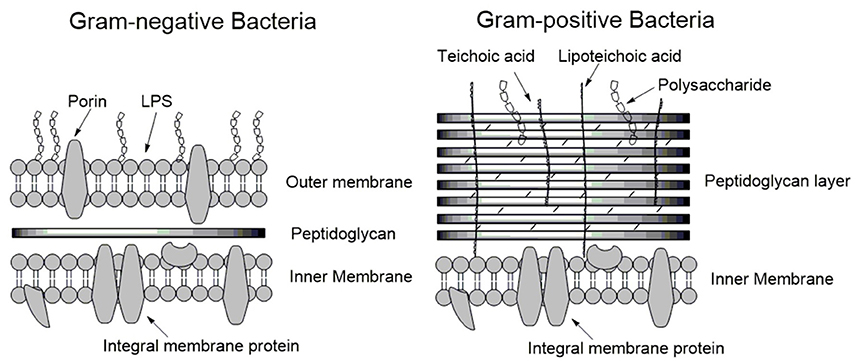
Figure 1. Schematic membrane structures of Gram-positive and Gram-negative bacteria. The cytoplasmic membranes of both types of bacteria are similar. Gram-positive bacteria have a thick layer of peptidoglycan surrounding the cytoplasmic membrane, protecting the bacterium. In contrast, the layer of peptidoglycan in Gram-negative bacteria is thin and an additional outer membrane is present. LPS forms a major part of the outer leaflet of the outer membrane/cell wall, the inner leaflet is comprised of phospholipids.
Parameters Affecting AMP Movement to the Cytoplasmic Membrane
The cytoplasmic membrane (also called as the inner membrane) of Gram-positive and Gram-negative bacteria are comprised of a mixture of zwitterionic and anionic phospholipids, such as phosphatidylethanolamine (POPE), phosphatidylglycerol (POPG), and cardiolipin (CL). Models of action for AMPs acting on the cytoplasmic membrane include pore formation (e.g., barrel stave or toroidal pores) and carpet mechanism (Brogden, 2005; Melo et al., 2009). For an AMP to disrupt the cytoplasmic membrane, the AMP molecules must first accumulate on the membrane surface up to a critical concentration. However, diffusion barriers in either the outer membrane or the periplasmic space affect their partition onto the membrane. This is a more direct path for Gram-positive bacteria as the AMPs only need to diffuse through nano-sized pores in the peptidoglycan, which in most cases is not a rate-limiting step (Vollmer et al., 2008). In fact, the peptidoglycan layer can facilitate AMP accumulation on the surface of the cytoplasmic membrane due to favorable interactions between teichoic acid and cationic AMPs (Malanovic and Lohner, 2016).
In the case of Gram-negative pathogens, AMPs need to permeabilize or disrupt both the outer and cytoplasmic membranes, resulting in a two-step process (Figure 2; Schwechheimer and Kuehn, 2015). Although the outer membrane of Gram-negative pathogens significantly modulates the antimicrobial activity of AMPs, in most cases, the inner membrane is the rate-limiting step. For example, polymyxin B has strong antimicrobial activity against Gram-negative pathogens due to its ability to disrupt both the outer and the cytoplasmic membranes. However, removing the lipid tail resulting in polymyxin B nonapeptide, and only the outer membrane is permeabilized. As a result, the nonapeptide also loses antimicrobial activity (Ofek et al., 1994).
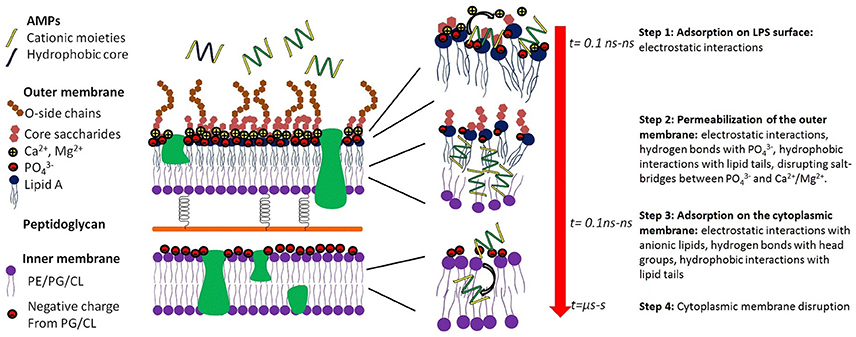
Figure 2. Key molecular events of the interaction of an AMP which results in killing Gram-negative baceria. Step 1, adsorption of the AMP to the surface of the outer membrane, which is very rapid (t = ns, nanosec) and is driven by electrostatic interactions. Step 2, permeabilization of the outer membrane, which is mediated by complex interactions including electrostatic interactions, hydrogen bonds, and hydrophobic interactions. In step 3, the AMP diffuses through the periplasmic space and reaches the surface of the inner membrane. Step 4 is the disruption of the cytoplasmic membrane, loss of the transmembrane potential, influx of water, and killing of the bacteria.
The detailed mode of interactions between AMPs and the outer membrane of Gram-negative bacteria is poorly understood due to the complex structure of the LPS molecules, which consists of a lipid A component, an inner and an outer core portion and a sugar portion (Figure 2). The initial event is the adsorption onto the outer membrane surface, which can occur within tens of nanoseconds and is largely mediated by electrostatic interactions between cationic AMPs and anionic LPS molecules. Reduction in the electrostatic driving force significantly affects the partition of AMPs to the outer membrane and may result in the loss of antimicrobial activity. For example, modification of the phosphate groups on the LPS molecules such as deacylation, hydroxylation, or addition of phosphoethanolamine endows bacteria resistance to colistin (polymyxin E; Olaitan et al., 2014). Upon adsorption onto the outer membrane, AMPs form hydrogen bonds with the phosphate groups, disrupting the salt-bridges between phosphate groups and divalent cations and destabilizing the outer membrane. In addition to the electrostatic forces, the hydrophobic moieties of an AMP can also interact with the lipid tails of the LPS molecule, further destabilizing the close packing of the outer membrane. After the AMP permeabilizes/disrupts the outer membrane, it diffuses inward to the periplasmic space and adsorbs onto the surface of the cytoplasmic membrane. Once a critical surface concentration on the cytoplasmic membrane is reached, the AMP induces significant perturbations and disorganizations of the cytoplasmic membrane, resulting in loss of the transmembrane potential, and eventual death of the bacterial cell.
Characteristics of the interactions of AMP with the outer and cytoplasmic membranes include: length of the AMP sequence, the total and density of cationic charges, the total number of hydrogen bonds donors and the 3-D conformation of the AMP in solution and at the membrane (Sitaram and Nagaraj, 1999; Bürck et al., 2008). The AMP structures that have been determined experimentally and deposited into the protein data bank provide little information for SAR as these structures are either in solution or in crystals rather than in the membrane environment. Due to the short sequences of most AMPs, their structures are sensitive to their environment. By comparing the CD spectrum of AMPs in solution and in the presence of liposomes, a large number of AMPs (e.g., melittin, magainin) have been found to undergo conformational transitions from random conformations in solution to helical or beta-sheet conformations on the membrane surface (Ramamoorthy et al., 2006; Hartings et al., 2008). For example, melittin adopts a helical conformation and protegrin adopts a beta-sheet conformation upon adsorption onto the membrane surface (Blazyk et al., 2001; Lu et al., 2005; Rausch et al., 2005, 2007; Dong et al., 2012). In silico modeling further reveals that both the alpha-helical and beta-sheet conformations are amphiphilic, with the cationic residues interacting with the anionic head groups and the hydrophobic residues penetrating into the lipid tail region of the membrane. Hence the electrostatic and hydrophobic interactions are two driving forces that steer an AMP toward and into the bacterial membrane.
Early Events in AMP Disruption of the Cytoplasmic Membrane
After adsorption onto the membrane surface, AMPs can induce a variety of membrane perturbations within tens of nanoseconds (Li et al., 2013). For most AMPs, an early event is the formation of hydrogen bonds between the basic residues (e.g., arginine and lysine) and the phosphate groups of the lipids. At physiological conditions, both lysine and arginine are hydrogen bond donors. However, arginine can form more stable bidentate hydrogen bonds with phosphate groups. Hydrophobic residues can further penetrate and disorganize the lipid tail region of the membrane. As more AMP molecules accumulate at the membrane-water interface, the membrane becomes thin, as observed for most AMPs (Sato and Feix, 2006; Ye et al., 2010). Moreover, as the total volume of the membrane is roughly constant, membrane thinning results in lateral expansion, affecting the mechanical properties of the membrane. For example, with the increase in the area per lipid, the surface tension increases and the bending modulus decreases dramatically, implying membrane deformation (Szleifer et al., 1988; Stevens, 2004). The expansion of the membrane also results in reduction in the packing of the lipid molecules leading to the formation of a large number of cavities, significantly reducing the translocation free energy of water molecules across the lipid tail region. As a result, a large number of water translocations occur, and the membrane becomes leaky with the collapse of the transmembrane potential and additional membrane dysfunction, such as inhibition of ATP production and loss of proton motive force (Dimroth et al., 2000) and rapid death of the bacterium.
Some AMPs, particularly the highly cationic ones can induce lipid re-arrangements and the formation of lipid rafts. B2088, an 18 residue peptide dimer with a high content of basic residues (12 positive charges), once adsorbed on the bacterial membrane, was found to recruit anionic lipids surrounding the peptide, resulting in micro-domains rich in anionic lipids in the outer leaflet of the membrane (Bai et al., 2012). Due to the binding to AMPs, lipid molecules surrounding AMP molecules display slow diffusivity, which not only affects the fluidity and structure of the membrane, but also results in significant tension, particularly along the domain boundaries (Guo et al., 2011). Clustering of anionic lipids in model bacterial membranes was found for other peptides as well (Oreopoulos et al., 2010; Polyansky et al., 2010; Wadhwani et al., 2012; Scheinpflug et al., 2015). Some AMPs were found to even induce flip-flop of anionic lipids from the inner leaflet to the outer leaflet, resulting in highly negatively charged outer leaflet and less negatively charged inner leaflet (Qian and Heller, 2011). This asymmetric distribution of charged lipids further destroys the membrane potential, resulting in membrane destabilization.
Membrane geometry as measured by the curvature is a fundamental property denoting stability with its basis in lipid organization and protein inclusions. Wong and coworkers suggested that negative Gaussian curvature is a prerequisite topology for the formation of membrane pores (Schmidt et al., 2010, 2011; Schmidt and Wong, 2013). It was found that POPE has the highest tendency to form a negative Gaussian curvature (“saddle-splay”) because of its small head group with respect to its large lipid tails (Yang et al., 2007; Schmidt et al., 2011). Interestingly, bacterial membranes consist of a high percentage of POPE while human membranes are mainly composed of POPC, implying that the preference of AMPS for POPE-rich membranes contributes significantly to the selectivity of AMPs. MD simulation studies have also shown that arginine residues can induce higher negative Gaussian curvature than lysine residues. The guanidinium group of arginine can form bidentate hydrogen bonds and coordinate two phosphate groups at <0.5 nm, while the distance is 0.7 nm for lysine (Schmidt et al., 2012a,b; Wu et al., 2013). Wu et al. employed both coarse-grained and atomistic models to simulate the interactions of poly-arginine and poly-lysine with model bacterial membranes (Wu et al., 2013). Their results revealed the different interactions of arginine and lysine residues with lipid molecules. The guanidinium group of arginine can simultaneously interact with both phosphate and glycerol groups, and thus induce greater negative Gaussian curvatures than lysine residues, as the latter can only engage interactions with phosphate groups. Wong and coworkers also found that incorporation of hydrophobic moieties further reinforces negative Gaussian curvature, which is consistent with the antimicrobial activity of various arginine-rich AMPs. For example, a series (RW)n peptides, which consists of only arginine (R) and tryptophan (W), display high antimicrobial activity (Strøm et al., 2002, 2003; Chan et al., 2006). In particular, an analog of (RW)n peptide LTX-109 has entered into clinical trials (Isaksson et al., 2011b).
Molecular Models of Pore Formation
A number of AMPs form membrane pores (Table 1). The timescale of pore formation ranges from microseconds to seconds. Membrane pores result in the loss of membrane potential and rapid release of intracellular components and death. Depending on the geometry of the pores as well as the interactions of AMPs with the pores, pores can be described by the barrel-stave or the toroidal model (Figure 3). The formation of barrel-stave pores is driven by hydrophobic match. In this model, the membrane does not display significant curvature and the hydration of the membrane remains unchanged. In the case of the toroidal model, as the AMP molecules penetrate deeper into the membrane, the head groups of the lipids are dragged into the lipid tail region to form toroidal shaped pores while the lipid tails are packed away from the surface of the pore, resulting in significant lipid disorder, and membrane curvature change (Sengupta et al., 2008). As a result, toroidal pores are also accompanied by enhanced membrane hydration, as evidenced by significant water penetration into the membrane (Sengupta et al., 2008; Manna and Mukhopadhyay, 2009). In toroidal pores, AMPs primarily interact with the pores electrostatically since the surface of toroidal pores are covered by the phosphate head groups and the AMP molecules have less hydrophobic contact with the lipid tails. In contrast, in barrel stave pores, both electrostatic and hydrophobic interactions are important since the AMP molecules interact with both the head groups and the lipid tails (Mihajlovic and Lazaridis, 2010; Bertelsen et al., 2012).
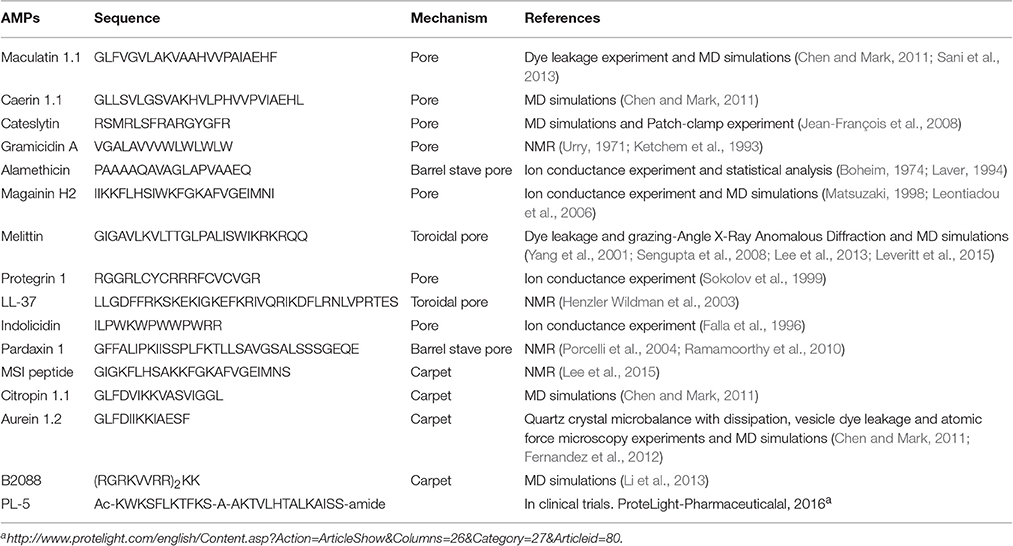
Table 1. Representative AMPs and their modes of action.
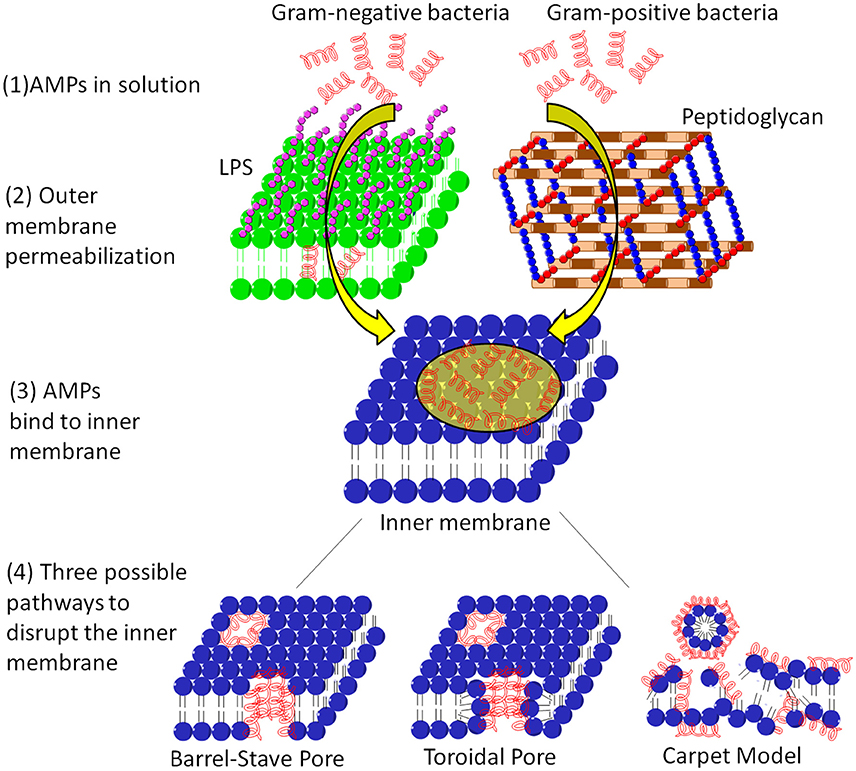
Figure 3. The membrane systems of Gram-positive and Gram-negative bacteria and the action mechanisms of AMPs on the cytoplasmic membrane. For Gram negative bacteria, the AMP needs to permeabilize the outer membrane before reach the cytoplasmic membrane, whereas for Gram positive bacteria, the AMP only needs to diffuse through the peptidoglycan layer via nano-sized pores. After adsorption onto the cytoplasmic membrane, the AMP can induce membrane pores such as barrel-stave pore and toroidal pore, or defragment the membrane.
Although induction of membrane pores is a general mechanism for a large number of AMPs, the morphology of these membrane pores can be quite diverse in terms of pore diameter, lipid conformation surrounding the pore, life time of the pore (e.g., transient pore or permanent pore) and the number of AMP molecules required to stabilize the pore. Regarding the shape of the pore, AMPs such as alamethicin, dermcidin, and pardaxin induce barrel stave pores (Laver, 1994; Porcelli et al., 2004; Song et al., 2013), while most other AMPs induce toroidal pores (Matsuzaki, 1998; Sokolov et al., 1999; Yang et al., 2001; Henzler Wildman et al., 2003; Sengupta et al., 2008; Lee et al., 2013). In terms of the pore size, magainin induces small toroidal pores of ~ 2–3 nm in diameter that only allow water and small intracellular molecules to leak out (Takeshima et al., 2003; Brogden, 2005), while lacticin Q and protegrin can form much larger pores of 4.6 and 9 nm in diameter, respectively, allowing the leakage of much large intracellular molecules (Yoneyama et al., 2009; Lam et al., 2012). In addition, the pore size depends on various factors such as the lipid composition and the peptide/lipid ratio. At nanomolar concentrations, melittin can only induce transient pores. As the melittin concentration increases beyond a critical value (e.g., peptide/lipid ratio of 1/45), it induces stable membrane pores of 2.5–3 nm in diameter, and the pore size further increases with the peptide/lipid ratio (Terwilliger et al., 1982; Matsuzaki et al., 1997; Lee et al., 2013). Another well-studied AMP, maculatin can also form an ensemble of structurally diverse pores (Wang Y. et al., 2016). Moreover, the formation of membrane pores depends on the lipid composition of the membrane as well. Evidence for this lies in the observation that most AMPs induce membrane pores at a much higher concentration in mammalian membranes than in bacterial membranes. The former mainly consists of zwitterionic lipids (e.g., POPC) and cholesterol, which are closely packed and thus have a low tendency to form membrane pores. In contrast, bacterial membranes consist of high percentage of POPE, which has a small head group and thus has higher tendency to form negative Gaussian curvatures, as discussed above (Schmidt and Wong, 2013).
Biophysical methods used to study the pore structure as well as AMP conformations in the pores, include small angle x-ray scattering (SAXS), oriented circular dichroism (OCD, solid-state NMR, quartz crystal microbalance (QCM; Yang et al., 2007; Kwon et al., 2013; Wang et al., 2015; Bürck et al., 2016). However, these methods typically give the early or final states of the AMP-membrane complex, e.g., the static structures of AMPs and membrane pores. The dynamics of the pore formation process is missing because the intermediate states of pore formation are either in non-equilibrium or metastable states and thus have life times that are too short to be observed. To obtain dynamic information on pore formation, MD simulations using atomistic and coarse-grained models have been carried out (Edit et al., 2007; Kirsch and Böckmann, 2016). Due to the complexity of the pore formation process, the sub-microsecond time scales that MD simulations can access and the fact that pore formation is a rare event, it is still difficult to observe the complete process of pore formation in conventional MD simulations. Nevertheless, Leontiadou et al. were the first to successfully simulate the process of toroidal pore formation by magainin (Leontiadou et al., 2006). In the simulations, the toroidal pore displays higher disorder than the normal toroidal pore model, reflecting the dynamic nature of pores in membranes. In another simulation from the same group, similar effects were observed for melittin. One or two melittin molecules were observed to line within the pore axis, while other peptide molecules located at the two ends of the pore (Sengupta et al., 2008). Removing the positive charges of melittin molecules failed to induce membrane pores, revealing the importance of electrostatic interactions. Interestingly, in both simulations, peptides lose their helical conformations when binding to the membrane pores, suggesting that the helical conformation is not necessary for stabilization of the membrane pores, at least for the case of toroidal pores. Coarse-grained MD simulations were also performed for melittin and transient toroidal pores were observed at high peptide/lipid ratios (Santo et al., 2013). It was shown that typically 3–5 melittin molecules are involved in each pore and similar to the atomistic simulations, not all the melittin molecules span across the membrane. The difference in the pores between atomistic and coarse-grained simulations suggests diverse morphologies of melittin induced membrane pores. These MD simulations, although only carried out at a micro-second time scale, nevertheless provide valuable molecular insights into the action mechanism of AMPs.
Carpet Mechanism
Not all AMPs induce membrane pores. Some AMPs adsorb onto the membrane surface and orient parallel to the membrane. As their surface concentrations reach a critical value, these AMPs disrupt the integrity of the super molecular structure of the membrane via membrane fragmentation, a process called carpet mechanism or detergent model (Figure 3; Gazit et al., 1996). Some examples of AMPs with carpet mechanism are listed in Table 1. In the carpet model, the outer leaflet is covered by a high surface concentration of AMP molecules, while the inner leaflet is free of AMP binding. The large imbalance of charge and surface tension across the membrane eventually leads to catastrophic collapse of membrane integrity and leakage of the cytoplasmic contents, ions, and biomolecules. In contrast, in the pore forming mechanism, only intracellular molecules smaller than the pore can exit. The size of the released molecules can be used to distinguish the pore forming mechanism from a carpet mechanism.
In the carpet mechanism, the complete picture of membrane collapse is not well-understood because (i) the process is highly non-equilibrium; (ii) multiple pathways of membrane lysis exist, making it difficult to be detected in biophysical experiments. However, at a peptide concentration lower than the critical concentration, the membrane undergoes some intrinsic perturbations such as membrane thinning, lateral expansion, clustering of anionic lipids, and membrane deformation, and high surface tension. Interestingly, these membrane perturbations can be used to correlate to the subsequent membrane lysis. For example, Wimley proposed an interfacial activity model to relate these membrane perturbations to the membrane activity of an AMP (Wimley, 2010). In the surface activity model, the membrane activity of an AMP is a function of the surface activity, which depends on two elements: (i) the partition of an AMP to the surface of the bacterial membrane and (ii) the ability to induce membrane perturbations. As the former is mostly driven by electrostatic interactions, AMPs with greater charge are more likely to achieve high concentrations on the membrane surface. The latter depends on many factors such as the 3-D conformation of the AMP, the physical properties of the AMP, and the actual interactions of the AMP with the lipids. Given the fact that the membrane is amphiphilic in nature, AMPs with facial amphiphilicity display high affinity for the membrane. The destabilization of the hydrophobic-water interface is critical for subsequent membrane lysis. In addition, membrane perturbations can be further enhanced via AMP aggregation, a mechanism similar to the toxic effects of an amyloid peptide (Milov et al., 2009; Di Scala et al., 2016). For example, LL-37, HAL-2, and the C-terminal segment of human beta-defensin 3, can achieve high surface concentration via self-aggregation upon adsorption onto the surface (Bai et al., 2009; Wang et al., 2014; Bonucci et al., 2015). The self-aggregation of maculatin 1.1, a membrane-active antimicrobial peptide (AMP) from the Australian tree frog leads to deeper penetration into the membrane core and significant change of curvature of the membrane (Bond et al., 2008). The self-aggregation of AMPs leads to an enhanced local concentration of AMPs around the membrane and has been used to design multimeric AMPs (Arnusch et al., 2007; Bai et al., 2012; Shin et al., 2013; Lakshminarayanan et al., 2014; Koh et al., 2015a).
Methods for AMP Design
Although research on AMP development has been highly active in the past years, few molecules have entered into the market. For an AMP to be used as a therapeutic agent, it needs to possess several druggable properties: (i) high antimicrobial activity; (ii) low toxicity to the mammalian membrane; (iii) high proteolytic stability; (iv) low serum binding; and (v) low cost. Most AMPs have minimum inhibitory concentration (MIC) values <10 μg/ml, while conventional antibiotics may have MICs in the range of 1–2 μg/ml or even sub μg/ml range, suggesting that significant effort is still needed to design new AMPs with higher activity. Various approaches have been employed to design new AMPs with low MIC, high stability, and low toxicity. These methods include the time-consuming mutation based empirical method, statistically based bioinformatics methods, and more sophisticated mechanism based methods such as MD simulations and biophysical experiments. Recently, multi-disciplinary approaches combining computational predictions, biophysical characterizations and biological validations have proved to be more promising (Bai et al., 2012; Koh et al., 2015a; Lakshminarayanan et al., 2016). This section discusses various experimental and computational methods that have been used for AMP design. Clearly, if the details of the membrane interactions were known design of an appropriate AMP would be facilitated.
Mutation Based Empirical Methods
Early attempts to design AMPs were largely carried out by trial and error due to poor understanding of the action mechanism. Many designed AMPs are actually based on the modifications of existing natural AMPs. A number of approaches can be tried to re-engineer a naturally occurring AMP, such as sequence shuffling and alanine scanning. Sequence shuffling, in which the positions of the amino acids in the sequence are changed can be the simple sequence reversal or via a combinatorial library. Since sequence shuffling does not change the hydrophobicity and the net positive charges, it has been used to optimize the antimicrobial activity of specific AMPs (Monroc et al., 2006; Cherkasov et al., 2009). The technique of alanine scanning mutates each residue or a group of residues to alanine and the antimicrobial activity is monitored for improvement. Alanine scanning has been widely used in AMP design and has proved to be very useful in identifying residues critical for antimicrobial activity (Grieco et al., 2011; Hänchen et al., 2013).
Statistically Based Bioinformatics Methods
AMP databases enhance computer aided AMP design (Fjell et al., 2007; Thomas et al., 2010; Wang G. et al., 2016). Various bioinformatics tools have been developed, including simple statistical modeling, SARs, neural networks, and machine learning. In general, these bioinformatics based tools need a database with known antimicrobial activity of certain peptides as the training set. By using different bioinformatics algorithms, key structural, and biophysical features are extracted from the training set and can be used to predict the antimicrobial activity of unknown peptides, which is referred to as the test set. For example, Mishra and Wang employed database filtering technology to determine the key parameters of AMPs (e.g., amino acid composition, peptide hydrophobic content, and net charge) and subsequently used these parameters for the ab initio design of potent AMPs against MRSA (Mishra and Wang, 2012). Besides the AMP database, design principles can also be obtained from other sources, such as genomes and proteomes (Li et al., 2007; Brand et al., 2012; Kim et al., 2016).
Mechanism Based Methods
Although the bioinformatics methods are simple and fast, they are still black boxes, as there is little information that reveals the action mechanisms. A more powerful and rational method would be to design new AMPs based on the mode of interaction with the membrane. To this purpose, detailed understanding of the action mechanisms of existing AMPs is required. MD simulation is a powerful method that can give atomistic information regarding the interactions of AMPs with bacterial membranes. If the time scale of the simulation is long enough, membrane disruption or pore formation can be directly observed (Wang Y. et al., 2016). MD simulations have been previously used to decipher the action mechanism of AMPs, and recently have been successfully applied to the design of new AMPs and antimicrobial peptidomimetics (Tsai et al., 2009; Tew et al., 2010a; Li et al., 2013, 2015; John Fox et al., 2016). For example, Bai et al. performed atomistic MD simulations of a short cationic AMP B1088 and found that the charge density plays an important role in its interactions with bacterial membrane mimics (Bai et al., 2009). Based on this information, they subsequently designed a covalent peptide dimer B2088 and a tetramer B4010 which demonstrated enhanced antimicrobial activity and proteolytic stability (Li et al., 2013; Lakshminarayanan et al., 2014). By using coarse-grained MD simulations, Tew et al. successfully designed a number of synthetic mimics of AMPs with high membrane selectivity (Tew et al., 2010a). One of their compounds brilacidin is currently under phase II clinical trials.
NMR can also provide mechanistic details for improved AMP design and in fact complements MD simulations. Similar to MD simulations, NMR, particularly solid state NMR, can provide information regarding the 3-D conformation of the peptide as well as the mode of interactions with model lipid membranes (Strandberg and Ulrich, 2004; Su et al., 2010). These include identification of the biophysical properties of critical residues that mediate the interactions with the membrane. For instance, Saravanan et al. used NMR combined with other biophysical experiments to design tryptophan and arginine rich decamer peptides and potent antimicrobial activity and low toxicity of the decamer peptide were found to arise from an optimal ratio between the positive charges and hydrophobicity (Saravanan et al., 2014). Later the same group used NMR to further design β-boomerang lipo-peptides that neutralized LPS (Mohanram and Bhattacharjya, 2014). Jeong et al. used NMR experiments not only to design a series of LPcin analogs with potent antimicrobial activities, but also elucidated the 3-D structure of a peptide-membrane complex (Jeong et al., 2016).
Principles for Practical Design of AMPs
Based on the methods discussed above, some general design principles have been proposed which can be directly used to guide AMP design and are briefly discussed below.
Amphiphilicity
Amphiphilicity is perhaps the most striking feature of AMPs, including facial amphiphilicity (Vandenburg et al., 2002), bola-amphiphilicity (Ali, 2007), radial amphiphilicity (Xiong et al., 2015), etc., all with at least one cationic moiety and one hydrophobic moiety. Considering the amphiphilic nature of membranes, amphiphilic peptides are expected to have high membrane affinity. Facially amphiphilic AMPs are usually helical with one side being cationic and the other side being hydrophobic. When adsorbed onto the bacterial membrane, these AMPs locate at the membrane surface, with the cationic face interacting with the head groups and the hydrophobic face penetrating into the lipid tail region, resulting in significant perturbations at the membrane-water interface. On the other hand, bola-amphiphilic peptides are expected to adopt transmembrane conformations, driven by hydrophobic match. In such a case, the two cationic moieties interact with the two head group regions, while the hydrophobic moiety interacts with the lipid tails. When several bola-amphiphilic peptide molecules oligomerize in the bacterial membrane, membrane pores can be formed (Matile et al., 2011).
Peptide Crosslinking
As both the pore forming mechanism and the carpet mechanism depend on the concentration of the peptide on the bacterial membrane surface, various methods have been proposed to enhance the surface concentration of AMPs. An effective way to enhance surface concentration of AMPs is through peptide self-aggregation, which leads to more effective membrane disruption compared to the monomeric peptide. For example, LL-37 self-aggregates on the bacterial membrane leading to the formation of toroidal pores (Bonucci et al., 2015). However, if the peptide is highly cationic, the self-aggregation is inhibited due to electrostatic repulsion between AMP molecules. In such a case, covalent linking can be used to generate covalent peptide aggregates, known as multimeric peptides. As discussed above, the covalent peptide dimer B2088 and tetramer B4010 displayed much higher antimicrobial activity than the peptide monomeric unit comprising these peptides (Bai et al., 2012; Lakshminarayanan et al., 2014, 2016).
Role of Arg
Most AMPs are cationic as a result of a high percentage of basic residues, Lys or Arg. Although both carry positive charges at neutral pH, the pKa values of Arg (12.45) and Lys (10.5) are different, and this will affect their protonation states in the membrane environment. Theoretical calculations found that Arg will retain its protonation state in the lipid tail region of the membrane, while Lys becomes deprotonated in the bilayer center (Yoo and Cui, 2008, 2010; Gleason et al., 2013). Due to the high pKa and multi-dentate hydrogen bonding property of Arg, Arg-rich peptides are thought to have stronger interactions with membranes. For example, a twin-arginine motif was found to assist peptide translocation and polyarginine itself is an efficient cell penetrating peptide (Chaddock et al., 1995; Bechara and Sagan, 2013). Studies also showed that Arg can induce more negative Gaussian curvatures than Lys due to its bidentate hydrogen bonding with PO4 groups (Schmidt et al., 2010, 2011; Schmidt and Wong, 2013; Wu et al., 2013). Accordingly, various Arg-rich AMPs have been designed. For example, (RW)n peptides display excellent antimicrobial activity (Liu et al., 2007). The side chain of Arg residue, the guanidine group was found to greatly enhance the action of antimicrobial peptidomimetics compared to the side chain of Lys residue (Andreev et al., 2014). Although many studies have shown the preference of Arg over Lys in terms of antimicrobial activity, some peptides prefer Lys over Arg residues. For example, arginine modified polymyxin B displayed reduced antimicrobial activity, suggesting that there appears not to be a general rule for selective preference of Arg and Lys by AMPs (Rabanal et al., 2015).
Peptide Truncation
Most of the classical antimicrobial peptides are fairly large and expensive to synthesize. This has led to the design of ultra-short peptides, with only 3–4 amino acids. The sequence of the peptidic moiety and the length of the hydrophobic moiety appear to determine the spectrum of antimicrobial activities. Despite their short lengths, their modes of action involves permeation and disintegration of the membrane organization, similar to that of many classical AMPs (Makovitzki et al., 2006) As electrostatic interactions with the bacterial membrane are still involved, almost all ultrashort peptides designed and synthesized to date contain cationic amino acids, such as arginine and lysine (Findlay et al., 2012; Domalaon et al., 2014; Mishra et al., 2015) For instance, KYR is an amino acid sequence of bovine hemoglobin alpha chain, which was part of the longer amino acid sequence that was obtained from hydrolysing the hemoglobin alpha chain. KYR is one of the shortest AMPs known (Nasompag et al., 2015). Most of the ultrashort peptides are conjugated with a fatty acid tail to provide additional hydrophobicity to kill bacteria efficiently (Makovitzki et al., 2006)
Incorporating Unnatural Amino Acids
The synthetic AMPs are not restricted to the 20 natural amino acids. Instead, they can incorporate various unnatural amino acids or have additional chemical modification. The direct advantage of AMPs containing unnatural amino acids is their high proteolytic stability. More importantly, as AMPs require a delicate balance of cationic and hydrophobic groups, chemical modifications enables easy fine-tuning of the hydrophobic balance. The commonly used approach for chemical modification includes use of more hydrophobic amino acids, lipid, and aromatic modifications. For example, lipid modifications of the above mentioned peptide dimer B2088 results in C8-B2088, which demonstrated enhanced antimicrobial activity (Koh et al., 2015a). Similarly, modification of the Phe residue with an additional benzene ring significantly enhances the antimicrobial activity of a short peptide FRFR-NH2, while maintaining its low toxicity to mammalian membranes (Lau et al., 2015). Multiple modifications have been used together to achieve high activity. LTX-109, a short synthetic AMP with both lipid and aromatic modifications, has been in clinical trials (Saravolatz et al., 2012b). Recently, a pharmacophore model has been proposed for the design of short AMP mimetics with the sequence of RXR, where X is a hydrophobic scaffold (Li et al., 2015). Derivatives of the pharmacophore model have shown excellent activity against resistant pathogens, low toxicity to mammalian membranes, and extremely high stability (Koh et al., 2015b).
Current Status: Examples of AMPs/Peptidomimetics in Clinical Trials
In the past 30 years, continuous efforts have been made to develop AMPs as clinically useful antimicrobials due to their advantages over conventional antibiotics such as a rapid bacterial killing, good selectivity toward the bacterial membrane, and a low propensity to give rise to bacterial resistance (Bai et al., 2012). However, to date, no designed AMP antibiotics have yet reached the clinic. Nevertheless, as described below, a number of AMPs and AMP derivatives are already at the pre-clinical stage and in clinical trials.
PL-5 is an α-helical AMP with a sequence of Ac-K-W-K-S-F-L-K-T-F-K-S-A-A-K-T-V-L-H-T-A-L-K-A-I-S-S-amide, where Ac = N-acetyl and amide = C-terminal amide. PL-5 is developed by ProteLight Pharmaceuticals and has recently obtained approval from the China Food and Drug Administration (CFDA) to enter clinical trials for skin infection in the year 2016. It is noteworthy that PL-5 is the first AMP to enter the clinical stage in China. PL-5 is a low toxicity and highly potent AMP against a broad spectrum of drug-resistant bacteria. In addition, PL-5 is able to synergize with conventional antibiotics to improve antibacterial activity in vitro and in vivo against both Gram-positive and Gram-negative bacteria. This may help prevent or delay the emergence of antibiotic resistance (Feng et al., 2015).
POL7080 is a synthetic cyclic peptide derived from protegrin I. POL7080 is active against Gram-negative bacteria and works by inhibiting a homolog of the β-barrel protein LptD. LptD is an outer-membrane protein widely distributed in Gram-negative bacteria that functions in the assembly of LPS in the outer leaflet of the outer membrane (Braun and Silhavy, 2002) LptD is involved in the outer-membrane biogenesis of lipopolysaccharide. Significantly, POL7080 is highly active on a broad panel of clinical isolates including multi-drug resistant Pseudomonas with outstanding in vivo efficacy in septicemia, lung and thigh infection models (Polyphor) POL7080 is developed by Polyphor Ltd and has competed a phase I clinical trial with its partner Roche. POL7080 has also completed a phase-II trial in 20 patients with exacerbation of non-cystic fibrosis bronchiectasis in 2015 (Butler et al., 2017). To date, the structure of POL7080 has not been revealed.
DPK-060 is a cationic peptide that has recently completed a Phase II study of topical application for atopic dermatitis. DPK-060 is a broad spectrum cationic peptide active against both Gram-positive and Gram-negative bacteria. Similar to other AMPs, DPK-060 is also membrane targeting (Harvey et al., 2015). DPK-060 is developed by Pergamum AB. The results from a Phase II clinical trial of DPK-060 in outer ear infections showed a statistically significant improvement in a 10-day cure rate compared to placebo and that DPK-060 is safe and tolerable (Lee et al., 2015) However, no recent reports of DPK-060 development have been forthcoming. Pergamum AB has also developed LL-37, a human cathelicidin subunit. Human cathelicidin is synthesized by numerous cells as an inactive precursor, hCAP18/LL-37. It consists of a highly conserved N-terminal signal sequence, a conserved cathelin domain, and a small antimicrobial C-terminal domain. The small antimicrobial C-terminal domain is known as LL-37 (Vandamme et al., 2012). The LL-37 domain is released by cleaving the proteolytic hCAP18/LL-37 precursor. This domain exhibits antimicrobial activities against both Gram-positive and Gram-negative bacteria (Overhage et al., 2008; Barlow et al., 2011) LL-37 has a peptide sequence of LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRT. LL-37 is developed for treatment of chronic leg ulcers. The clinical phase results show that LL-37 has a significantly improved healing rate compared to placebo (Lee et al., 2015).
Innate Defense Regulators (IDRs) are a novel class of synthetic peptides that enhance the control of microbial infections. IDRs do not impact the adaptive immune system and do not interfere with chemotherapy, radiation therapy or antibiotic treatments. SGX942 contains the active ingredient dusquetide (also referred to as SGX-94). Dusquetide is a fully synthetic, 5-amino acid peptide derived from Indolicidin with high aqueous solubility and stability (Soligenix) SGX94, has broad-spectrum activity against Gram-negative and Gram-positive bacterial infections caused by intracellular or extracellular bacteria and also complements the actions of standard of care antibiotics (North et al., 2016). Since SGX-94 acts through host pathways to provide both broad-spectrum anti-infective capability as well as control of inflammation, IDRs are unlikely to be impacted by resistance mechanisms. It also offers potential clinical advantages in the fight against emerging and antibiotic resistant bacterial infections (North et al., 2016) SGX-942 was first developed by Inimex and is currently being pursued in a Phase-II trial by Soligenix as treatment for oral mucositis (Butler et al., 2017). SGX942 has previously demonstrated safety and tolerability in a double-blind, placebo-controlled, healthy volunteer Phase I clinical trial. SGX942 has been awarded Fast Track designation from the FDA for the treatment of oral mucositis as a result of radiation and/or chemotherapy treatment in head and neck cancer patients.
There are also several AMPs that were not approved by FDA or failed at an earlier development stage, such as Iseganan, Omiganan, and XMP-629 and Locilex (Ahmad et al., 2012). Dipexium acknowledged that Locilex did not meet the primary clinical endpoint of superiority vs. placebo or “vehicle” namely the cream without its active ingredient of pexiganan. In order to overcome the major limitations of AMPs such as systemic toxicity and proteolytic instability, the development of small-molecule-based membrane-targeting antimicrobials that maintain the essential key characteristics of AMPs, has received considerable attention (Lohan and Bisht, 2013). Peptidomimetics are a new generation of small-molecule antimicrobials that mimic the structure and antibacterial action of AMPs. Design of peptidomimetics involves the introduction of amide bond isosteres or peptide backbone modifications via non-natural side chains to mimic a peptide structure or function (Niu et al., 2012; Lohan and Bisht, 2013).
Brilacidin is a small-molecule arylamide mimic of AMPs that shows potent antimicrobial activity against a wide range of drug-susceptible and multidrug-resistant Gram-negative and Gram-positive bacteria (Tew et al., 2002, 2010b; Liu et al., 2004) Brilacidin has a planar, conformationally restrained scaffold with four positive guanadinyl and pyridinyl substitutions and two trufluoromethane hydrophobic substitutions (Figure 4). Brilacidin was first developed by Polymedix Inc. and purchased by Cellceutix corp. in September 2013 (Butler et al., 2017). Brilacidin has completed phase IIa and phase IIb trials for the treatment of acute S. aureus skin and skin structure infections. Compared to daptomycin, the results show no serious adverse effects and the efficacy is similar to daptomycin across all brilacidin treatment groups in 215 patients Similar to other AMPs, brilacidin is a membrane targeting antimicrobial. It causes membrane disruption and shows efficacy in a MRSA keratitis model when applied topically. At 0.5% solution, brilacidin shows minimal irritation and is equally efficacious as vancomycin (Kowalski et al., 2016) In addition to brilacidin, Cellceutix is also developing CTIX-1278 (structure not revealed), a defensin mimetic-compound, against the drug resistant superbug Klebsiella pneumoniae. CTIX-1278 is efficacious in a thigh burden study using a mouse model. The results are encouraging as CTIX-1278 shows similar efficacy compared to carbapenem.
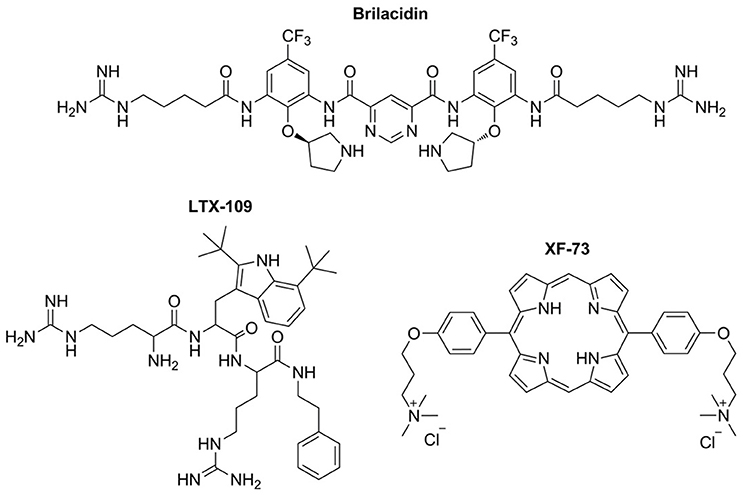
Figure 4. Structures of AMP mimetics in clinical studies. All the three molecules contain a large hydrophobic moiety and two cationic moieties, forming a cationic-hydrophobic-cationic motif, and mimicking the interactions of AMPs with the bacterial membrane.
LTX-109 is developed by Lytix Biopharma, which focus on topical treatment of skin infections and nasal eradication of staphylococcus. LTX 109 is a synthetic antimicrobial peptidomimetic, which has completed phase 2 trials for the treatment of impetigo in the year 2014 and uncomplicated skin and skin structure infection (uSSSI) in the year 2011 (Butler et al., 2017). LTX-109 has the chemical structure Arg-Tbt-Arg-NH-EtPh (Figure 4). Arg provides the cationic charge and the tertiary butyl group is important to increase the hydrophobicity. In general, LTX-109 is active against a broad range of bacteria including E. coli and S. aureus (Isaksson et al., 2011a) Significantly, LTX-109 is also active against a panel of drug-resistant Gram-positive bacteria, such as MRSA, vancomycin-intermediate resistant, daptomycin resistant, and linezolid-resistant strains (Saravolatz et al., 2012a) LTX-109 kills bacteria via the membrane targeting mechanism.
Exeporfinium chloride (XF-73) is a synthetic dicationic porphyrin derivative being developed by Destiny Pharma (Brighton, UK) that has been evaluated in phase-I/II trials for the prevention of post-surgical staphylococcal nasal infections (Figure 4; Butler et al., 2017). XF-73 has completed 5 Phase I/IIa clinical studies in Europe/US. XF-73 is a photosensitizer that has broad-spectrum antimicrobial activities against Gram-positive, Gram-negative and Candida albicans (Farrell et al., 2010; Gonzales et al., 2013). XF-73 exhibits potent, non-lytic, bactericidal activity against S. aureus. Similar to AMPs, interaction of XF-73 with the cytoplasmic membrane is lethal to S. aureus, leading to release of intracellular components and bacterial cell death (Ooi et al., 2009). In addition, XF-73 showed no drug resistance emergence from four common MRSA strains tested in a multi-step (55 passage) resistance study, as a ≤ 4-fold increase in MIC against the strains tested (Farrell et al., 2011). On 05 September 2016, Destiny Pharma announced that XF-73 can be delivered safely and is well-tolerated in a two-stage US clinical trial using intra-nasally applied exeporfinium chloride gels (DMID contract number HHSN27220800026C). In addition, no drug was detected in the bloodstream (Destiny Pharma, accessed on 12 January 2017).
Peptides have appeared in a wide range of applications in other clinical areas (Fosgerau and Hoffmann, 2015). For instance, thymalfasin, a short peptide with 28 amino acids, has been used in clinics for its immune regulatory function (Sjogren, 2004). With more and more emerging strategies to design new generation AMPs with improved efficacy, safety, and tolerability, we believe that peptide antibiotic still offer enormous growth potential to reach the clinic in the near future.
Challenges and Future Perspectives
Current AMP Development: Challenges and Solutions
AMP development has been an active research area in the past 30 years, but only recently has there been a positive outlook for commercial success. There are challenges that limit the design of potent AMPs, such as the poor understanding of the target-drug interaction and the lack of rational design principles. Besides activity, issues such as toxicity, serum binding, stability, and product cost are also practical considerations. Solutions to overcome these limitations have been proposed and have become hotspots of current AMP research and development. A side benefit of AMP research is that emphasis has changed from screening unknowns in a microbiology setting to defining the target bringing antibiotic development closer to conventional structure based drug development.
Toxicity of AMPs can occur at different levels, including membrane toxicity, cellular toxicity, and systemic toxicity. Membrane selectivity is a widely accepted parameter to characterize in vitro membrane toxicity and is defined as the ratio of HC50/MIC, where HC50 is the concentration needed to cause 50% hemolysis of human red blood cells. As stated earlier in this review molecular charge of bacterial membranes and membranes of human cells differ so that AMPs with higher positive charges show enhanced affinity for the bacterial membrane, resulting in higher antimicrobial activity (lower MIC) and conversely less toxicity to human cells (Zelezetsky and Tossi, 2006). Moreover, it is also proposed that AMP hydrophobicity can affect human membrane toxicity, a factor useful in the design of branched lipo-peptides with minimal toxicity (Koh et al., 2015a).
Cellular/metabolic toxicity and systemic toxicity are more difficult to predict as the underlying mechanisms are complex. Cellular toxicity refers to single cell toxicity, which can be measured for human cells using MTT assays, LDH release, and ATP synthesis (Fotakis and Timbrell, 2006). Systemic toxicity can arise from various effects such as activation of transcription factors, binding to macromolecular receptors in the body, alteration of metabolic pathways, and triggering immune response, making it challenging to predict. For example, polymyxin B, the last resort for the treatment of multi-drug resistant Gram-negative bacteria, although safe at the membrane level, can cause significant nephro- and neurotoxicity (Falagas and Kasiakou, 2006). To address and predict the issues surrounding systemic toxicity, several strategies have been employed. Computational toxicology uses machine learning algorithms to predict toxic outcomes (Valerio, 2009). Another strategy is via formulation (Carmona-Ribeiro and de Melo Carrasco, 2014). For example, Gramicidin, a topical AMP is effective against many Gram-positive bacteria, but has significant hemolysis. However, incorporating gramicidin in a dioctadecyldimethylammonium bromide (DODAB) bilayer not only results in reduced toxicity, but also leads to broader antimicrobial activity against both E. coli and S. aureus (Ragioto et al., 2014).
AMPs consisting of all natural amino acids may need to enhance their proteolytic stability. This limitation may not be a serious problem for topical applications, but results in significantly reduced half-life in systemic applications. Various approaches can be used to enhance the proteolytic stability of AMPs. The direct way is to mutate key amino acids at the cleavage site to D amino acids or similar analogs. For example, arginine can be replaced by D-Arg or homoarginine, while lysine can be replaced by D-Lys or ornithine. However, the effect of L-to-D mutation on the antimicrobial activity needs to be re-evaluated, although in most cases the L-to-D mutation does not alter the antimicrobial activity significantly (Hong et al., 1999; Berthold et al., 2013; Carmona et al., 2013). In addition, chemical modifications as discussed in Section Principles for Practical Design of AMPs, such as incorporation of unnatural amino acids and cross-linking can function to improve peptide stability.
Cationic AMPs tend to display high affinity for serum proteins, decreasing the available concentration of drug; however, at the same time this is a general issue with most antibiotics. For example, it was shown that AMPs can interact with drug site II of albumin via hydrophobic interactions (Sivertsen et al., 2014). In addition, the cationic residues of most cationic AMPs make them good substrates for the chymotrypsin family of endoproteases (Perona and Craik, 1997). The strong protein binding property significantly reduces the effective concentration of the AMP available to combat bacteria (Svenson et al., 2007). Moreover, host cells can also interfere with the activity of AMPs. Starr et al. pointed out that interactions with host cells can lead to significant loss of activity in vivo, in a way very similar to the effects of serum protein binding (Starr et al., 2016). To develop AMPs with less binding to serum proteins/host cells, detailed PK/PD studies are required. Compared to small molecule antibiotics, AMPs may be more expensive to produce; however, this limitation can be overcome by the use of synthetic biology (Cameron et al., 2014). Using genetically engineered microbial fermentation, large amounts of recombinant peptides can be produced. For example, a fusion protein containing the antimicrobial sequence at its C-terminus was successfully expressed in E. Coli., and subsequent cleavage released AMP P2 (Haught et al., 1998).
Future Perspectives
Importantly, AMPs probably represent the best option for the treatment of multi-drug resistant infections. Considerable effort has been expended in this area with progress and a number of AMP/peptidomimetics are in different phases of clinical trials. Since the MIC values for most AMPs are still higher than many conventional antibiotics, the primary task is to improve the antimicrobial activity, reduce the toxicity, and improve delivery efficiency. Another promising area is the design of membrane active peptidomimetics to mimic the action of existing AMPs, which can be achieved by chemical modification of existing AMPs or using unnatural amino acids. Compared to AMPs, peptidomimetics greatly expand the molecular space of membrane active antimicrobials and have the advantages of high proteolytic stability and optimizing the hydrophobicity. However, this needs to be coupled with the more detailed understanding of the molecular and atomistic interactions between AMPs/peptidomimetics and the molecular complex of the Gram-negative membrane system. Computer aided drug design, particularly the mechanism based in silico design approach such as MD simulations has a great potential to help overcome some of these limitations. When combined with other methods in a multi-disciplinary setting, translation of fundamental knowledge to practical clinical therapeutics can be greatly accelerated. This approach should also be activated to overcome the AMP resistant strains such as the recently appeared colistin (also known as polymyxin E) resistant strains (Fernández et al., 2013; Olaitan et al., 2014; Li et al., 2015). The advantages of combining the in silico simulations and NMR is that the approach is adaptable to the challenge of bacteria with modified LPS structure. If successful a new age of antibiotics could be forthcoming with less resistance, longer clinical utility and greater opportunities for special purpose design of antibiotics and other antimicrobials.
Author Contributions
JL, JK, and SL wrote the drafted the manuscript; RL, CV, and RB modified the manuscript. All authors discussed and contributed to the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the grant from NMRC/TCR/002-SERI/2008/R618, NMRC/TCR/R1018 and NMRC/BNIG/2016/2014, Singapore.
Abbreviations
AMP, Antimicrobial peptide; POPE, Phosphatidylethanolamine; POPG, Phosphatidylglycerol; POPC, Phosphatidylcholine; POPS, Phosphatidylserine; CL, Cardiolipin; LPS, Lipopolysaccharide; SAR, Structure-activity relationship; CD, Circular dichroism; MD, Molecular dynamics; MRSA, methicillin-resistant Staphylococcus aureus; MIC, minimum inhibitory concentration.
References
Ahmad, A., Ahmad, E., Rabbani, G., Haque, S., Arshad, M., and Khan, R. H. (2012). Identification and design of antimicrobial peptides for therapeutic applications. Curr. Protein Pept. Sci. 13, 211–223. doi: 10.2174/138920312800785076
Ali, H. E.-S. (2007). Synthesis, surface properties and antimicrobial activity of bolaamphiphile/oppositely charged conventional surfactant mixed systems. J. Surfactants Deterg. 10, 117–124. doi: 10.1007/s11743-007-1021-y
Andreev, K., Bianchi, C., Laursen, J. S., Citterio, L., Hein-Kristensen, L., Gram, L., et al. (2014). Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim. Biophys. Acta Biomembranes 1838, 2492–2502. doi: 10.1016/j.bbamem.2014.05.022
Arnusch, C. J., Branderhorst, H., de Kruijff, B., Liskamp, R. M. J., Breukink, E., and Pieters, R. J. (2007). Enhanced membrane pore formation by multimeric/oligomeric antimicrobial peptides. Biochemistry 46, 13437–13442. doi: 10.1021/bi7015553
Ayabe, T., Ashida, T., Kohgo, Y., and Kono, T. (2004). The role of Paneth cells and their antimicrobial peptides in innate host defense. Trends Microbiol. 12, 394–398. doi: 10.1016/j.tim.2004.06.007
Bai, Y., Liu, S., Jiang, P., Zhou, L., Li, J., Tang, C., et al. (2009). Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry 48, 7229–7239. doi: 10.1021/bi900670d
Bai, Y., Liu, S., Li, J., Lakshminarayanan, R., Sarawathi, P., Tang, C., et al. (2012). Progressive structuring of a branched antimicrobial peptide on the path to the inner membrane target. J. Biol. Chem. 287, 26606–26617. doi: 10.1074/jbc.M112.363259
Barlow, P. G., Svoboda, P., Mackellar, A., Nash, A. A., York, I. A., Pohl, J., et al. (2011). Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 6:e25333. doi: 10.1371/journal.pone.0025333
Bechara, C., and Sagan, S. (2013). Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587, 1693–1702. doi: 10.1016/j.febslet.2013.04.031
Bertelsen, K., Dorosz, J., Hansen, S. K., Nielsen, N. C., and Vosegaard, T. (2012). Mechanisms of peptide-induced pore formation in lipid bilayers investigated by oriented 31P solid-state NMR spectroscopy. PLoS ONE 7:e47745. doi: 10.1371/journal.pone.0047745
Berthold, N., Czihal, P., Fritsche, S., Sauer, U., Schiffer, G., Knappe, D., et al. (2013). Novel Apidaecin 1b analogs with superior serum stabilities for treatment of infections by gram-negative pathogens. Antimicrob. Agents Chemother. 57, 402–409. doi: 10.1128/AAC.01923-12
Bevins, C. L., and Salzman, N. H. (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368. doi: 10.1038/nrmicro2546
Blazyk, J., Wiegand, R., Klein, J., Hammer, J., Epand, R. M., Epand, R. F., et al. (2001). A novel linear amphipathic β-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 276, 27899–27906. doi: 10.1074/jbc.M102865200
Boheim, G. (1974). Statistical analysis of alamethicin channels in black lipid membranes. J. Membr. Biol. 19, 277–303. doi: 10.1007/BF01869983
Bond, P. J., Parton, D. L., Clark, J. F., and Sansom, M. S. P. (2008). Coarse-grained simulations of the membrane-active antimicrobial peptide maculatin 1.1. Biophys. J. 95, 3802–3815. doi: 10.1529/biophysj.108.128686
Bonucci, A., Caldaroni, E., Balducci, E., and Pogni, R. (2015). A spectroscopic study of the aggregation state of the human antimicrobial peptide LL-37 in bacterial versus host cell model membranes. Biochemistry 54, 6760–6768. doi: 10.1021/acs.biochem.5b00813
Brand, G. D., Magalhães, M. T. Q., Tinoco, M. L. P., Aragão, F. J. L., Nicoli, J., Kelly, S. M., et al. (2012). Probing protein sequences as sources for encrypted antimicrobial peptides. PLoS ONE 7:e45848. doi: 10.1371/journal.pone.0045848
Brandenburg, K., Heinbockel, L., Correa, W., and Lohner, K. (2016). Peptides with dual mode of action: killing bacteria and preventing endotoxin-induced sepsis. Biochim. Biophys. Acta Biomembranes 1858, 971–979. doi: 10.1016/j.bbamem.2016.01.011
Braun, M., and Silhavy, T. J. (2002). Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 45, 1289–1302. doi: 10.1046/j.1365-2958.2002.03091.x
Brogden, K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. doi: 10.1038/nrmicro1098
Bürck, J., Roth, S., Wadhwani, P., Afonin, S., Kanithasen, N., Strandberg, E., et al. (2008). Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys. J. 95, 3872–3881. doi: 10.1529/biophysj.108.136085
Bürck, J., Wadhwani, P., Fanghänel, S., and Ulrich, A. S. (2016). Oriented circular dichroism: a method to characterize membrane-active peptides in oriented lipid bilayers. Accounts Chem. Res. 49, 184–192. doi: 10.1021/acs.accounts.5b00346
Butler, M. S., Blaskovich, M. A. T., and Cooper, M. A. (2017). Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 70, 3–24. doi: 10.1038/ja.2016.72
Cameron, D. E., Bashor, C. J., and Collins, J. J. (2014). A brief history of synthetic biology. Nat. Rev. Microbiol. 12, 381–390. doi: 10.1038/nrmicro3239
Carmona, G., Rodriguez, A., Juarez, D., Corzo, G., and Villegas, E. (2013). Improved protease stability of the antimicrobial peptide pin2 substituted with D-amino acids. Protein J. 32, 456–466. doi: 10.1007/s10930-013-9505-2
Carmona-Ribeiro, A. M., and de Melo Carrasco, L. D. (2014). Novel formulations for antimicrobial peptides. Int. J. Mol. Sci. 15, 18040–18083. doi: 10.3390/ijms151018040
Chaddock, A. M., Mant, A., Karnauchov, I., Brink, S., Herrmann, R. G., Klösgen, R. B., et al. (1995). A new type of signal peptide: central role of a twin-arginine motif in transfer signals for the delta pH-dependent thylakoidal protein translocase. EMBO J. 14, 2715–2722.
Chan, D. I., Prenner, E. J., and Vogel, H. J. (2006). Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta Biomembranes 1758, 1184–1202. doi: 10.1016/j.bbamem.2006.04.006
Chen, R., and Mark, A. E. (2011). The effect of membrane curvature on the conformation of antimicrobial peptides: implications for binding and the mechanism of action. Eur. Biophys. 40, 545–553. doi: 10.1007/s00249-011-0677-4
Cherkasov, A., Hilpert, K., Jenssen, H., Fjell, C. D., Waldbrook, M., Mullaly, S. C., et al. (2009). Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 4, 65–74. doi: 10.1021/cb800240j
Deslouches, B., Steckbeck, J. D., Craigo, J. K., Doi, Y., Mietzner, T. A., and Montelaro, R. C. (2013). Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob. Agents Chemother. 57, 2511–2521. doi: 10.1128/AAC.02218-12
Di Scala, C., Yahi, N., Boutemeur, S., Flores, A., Rodriguez, L., Chahinian, H., et al. (2016). Common molecular mechanism of amyloid pore formation by Alzheimer's β-amyloid peptide and α-synuclein. Sci. Rep. 6:28781. doi: 10.1038/srep28781
Diamond, G., Beckloff, N., Weinberg, A., and Kisich, K. O. (2009). The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 15, 2377–2392. doi: 10.2174/138161209788682325
Dimroth, P., Kaim, G., and Matthey, U. (2000). Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J. Exp. Biol. 203, 51–59. doi: 10.1128/AAC.45.6.1799-1802.2001
Domalaon, R., Yang, X., O'Neil, J., Zhanel, G. G., Mookherjee, N., and Schweizer, F. (2014). Structure-activity relationships in ultrashort cationic lipopeptides: the effects of amino acid ring constraint on antibacterial activity. Amino Acids 46, 2517–2530. doi: 10.1007/s00726-014-1806-z
Dong, N., Ma, Q., Shan, A., Lv, Y., Hu, W., Gu, Y., et al. (2012). Strand length-dependent antimicrobial activity and membrane-active mechanism of arginine- and valine-rich β-hairpin-like antimicrobial peptides. Antimicrob. Agents Chemother. 56, 2994–3003. doi: 10.1128/AAC.06327-11
Edit, M., Christian, K., and Tieleman, D. P. (2007). Computer simulation of antimicrobial peptides. Curr. Med. Chem. 14, 2789–2798. doi: 10.2174/092986707782360105
Epand, R. M., and Vogel, H. J. (1999). Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembranes. 1462, 11–28. doi: 10.1016/S0005-2736(99)00198-4
Falagas, M. E., and Kasiakou, S. K. (2006). Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit. Care. 10:R27. doi: 10.1186/cc3995
Falla, T. J., Karunaratne, D. N., and Hancock, R. E. W. (1996). Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 271, 19298–19303. doi: 10.1074/jbc.271.32.19298
Farrell, D. J., Robbins, M., Rhys-Williams, W., and Love, W. G. (2010). In vitro activity of XF-73, a novel antibacterial agent, against antibiotic-sensitive and -resistant Gram-positive and Gram-negative bacterial species. Int. J. Antimicrob. Agents 35, 531–536. doi: 10.1016/j.ijantimicag.2010.02.008
Farrell, D. J., Robbins, M., Rhys-Williams, W., and Love, W. G. (2011). Investigation of the potential for mutational resistance to XF-73, retapamulin, mupirocin, fusidic acid, daptomycin, and vancomycin in methicillin-resistant Staphylococcus aureus isolates during a 55-passage study. Antimicrob. Agents Chemother. 55, 1177–1181. doi: 10.1128/AAC.01285-10
Feng, Q., Huang, Y., Chen, M., Li, G., and Chen, Y. (2015). Functional synergy of alpha-helical antimicrobial peptides and traditional antibiotics against Gram-negative and Gram-positive bacteria in vitro and in vivo. Eur. J. Clin. Microbiol. Infect. Dis. 34, 197–204. doi: 10.1007/s10096-014-2219-3
Fernandez, D. I., Le Brun, A. P., Whitwell, T. C., Sani, M.-A., James, M., and Separovic, F. (2012). The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 14, 15739–15751. doi: 10.1039/c2cp43099a
Fernández, L., Álvarez-Ortega, C., Wiegand, I., Olivares, J., Kocíncová, D., Lam, J. S., et al. (2013). Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57, 110–119. doi: 10.1128/AAC.01583-12
Findlay, B., Zhanel, G. G., and Schweizer, F. (2012). Investigating the antimicrobial peptide window of activity using cationic lipopeptides with hydrocarbon and fluorinated tails. Int. J. Antimicrob. Agents 40, 36–42. doi: 10.1016/j.ijantimicag.2012.03.013
Fjell, C. D., Hancock, R. E. W., and Cherkasov, A. (2007). AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics 23, 1148–1155. doi: 10.1093/bioinformatics/btm068
Fosgerau, K., and Hoffmann, T. (2015). Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128. doi: 10.1016/j.drudis.2014.10.003
Fotakis, G., and Timbrell, J. A. (2006). In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 160, 171–177. doi: 10.1016/j.toxlet.2005.07.001
Ganz, T. (2003a). Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720. doi: 10.1038/nri1180
Ganz, T. (2003b). The role of antimicrobial peptides in innate immunity. Integr. Comp. Biol. 43, 300–304. doi: 10.1093/icb/43.2.300
Gaspar, D., Veiga, A. S., and Castanho, M. A. (2013). From antimicrobial to anticancer peptides. a review. Front. Microbiol. 4:294. doi: 10.3389/fmicb.2013.00294
Gazit, E., Miller, I. R., Biggin, P. C., Sansom, M. S. P., and Shai, Y. (1996). Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J. Mol. Biol. 258, 860–870. doi: 10.1006/jmbi.1996.0293
Gleason, N. J., Vostrikov, V. V., Greathouse, D. V., and Koeppe, R. E. (2013). Buried lysine, but not arginine, titrates and alters transmembrane helix tilt. Proc. Natl. Acad. Sci. U.S.A. 110, 1692–1695. doi: 10.1073/pnas.1215400110
Gonzales, F. P., Felgenträger, A., Baumler, W., and Maisch, T. (2013). Fungicidal photodynamic effect of a twofold positively charged porphyrin against Candida albicans planktonic cells and biofilms. Future Microbiol. 8, 785–797. doi: 10.2217/fmb.13.44
Gordon, Y. J., Romanowski, E. G., and McDermott, A. M. (2005). A Review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 30, 505–515. doi: 10.1080/02713680590968637
Grieco, P., Luca, V., Auriemma, L., Carotenuto, A., Saviello, M. R., Campiglia, P., et al. (2011). Alanine scanning analysis and structure–function relationships of the frog-skin antimicrobial peptide temporin-1Ta. J. Pept. Sci. 17, 358–365. doi: 10.1002/psc.1350
Guilhelmelli, F., Vilela, N., Albuquerque, P., Derengowski, L. da S., Silva-Pereira, I., and Kyaw, C. M. (2013). Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 4:353. doi: 10.3389/fmicb.2013.00353
Guo, L., Smith-Dupont, K. B., and Gai, F. (2011). Diffusion as a probe of peptide-induced membrane domain formation. Biochemistry 50, 2291–2297. doi: 10.1021/bi102068j
Hänchen, A., Rausch, S., Landmann, B., Toti, L., Nusser, A., and Süssmuth, R. D. (2013). Alanine scan of the peptide antibiotic feglymycin: assessment of amino acid side chains contributing to antimicrobial activity. ChemBioChem 14, 625–632. doi: 10.1002/cbic.201300032
Hartings, M. R., Gray, H. B., and Winkler, J. R. (2008). Probing melittin helix−coil equilibria in solutions and vesicles. J. Phys. Chem. B 112, 3202–3207. doi: 10.1021/jp709866g
Harvey, A. L., Edrada-Ebel, R., and Quinn, R. J. (2015). The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 14, 111–129. doi: 10.1038/nrd4510
Haught, C., Davis, G. D., Subramanian, R., Jackson, K. W., and Harrison, R. G. (1998). Recombinant production and purification of novel antisense antimicrobial peptide in Escherichia coli. Biotechnol. Bioeng. 57, 55–61. doi: 10.1002/(SICI)1097-0290(19980105)57:1<55::AID-BIT7>3.0.CO;2-U
Henzler Wildman, K. A., Lee, D.-K., and Ramamoorthy, A. (2003). Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 42, 6545–6558. doi: 10.1021/bi0273563
Hilchie, A. L., Wuerth, K., and Hancock, R. E. W. (2013). Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 9, 761–768. doi: 10.1038/nchembio.1393
Hong, S. Y., Oh, J. E., and Lee, K.-H. (1999). Effect of d-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 58, 1775–1780. doi: 10.1016/S0006-2952(99)00259-2
Isaksson, J., Brandsdal, B. O., Engqvist, M., Flaten, G. E., Svendsen, J. S., and Stensen, W. (2011a). A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 54, 5786–5795. doi: 10.1021/jm200450h
Isaksson, J., Brandsdal, B. O., Engqvist, M., Flaten, G. E., Svendsen, J. S. M., and Stensen, W. (2011b). A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 54, 5786–5795. doi: 10.1021/jm200450h
Jean-François, F., Elezgaray, J., Berson, P., Vacher, P., and Dufourc, E. J. (2008). Pore formation induced by an antimicrobial peptide: electrostatic effects. Biophys. J. 95, 5748–5756. doi: 10.1529/biophysj.108.136655
Jeong, J.-H., Kim, J.-S., Choi, S.-S., and Kim, Y. (2016). NMR structural studies of antimicrobial peptides: LPcin analogs. Biophys. J. 110, 423–430. doi: 10.1016/j.bpj.2015.12.006
John Fox, S., Li, J., Tan, Y. S. N., Nguyen, M., Pal, A., Ouaray, Z., et al. (2016). The multifaceted roles of molecular dynamics simulations in drug discovery. Curr. Pharm. Des. 22, 3585–3600. doi: 10.2174/1381612822666160425120507
Ketchem, R., Hu, W., and Cross, T. (1993). High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 261, 1457–1460. doi: 10.1126/science.7690158
Kim, I.-W., Lee, J. H., Subramaniyam, S., Yun, E.-Y., Kim, I., Park, J., et al. (2016). De novo transcriptome analysis and detection of antimicrobial peptides of the American cockroach periplaneta Americana (Linnaeus). PLoS ONE 11:e0155304. doi: 10.1371/journal.pone.0155304
Kirsch, S. A., and Böckmann, R. A. (2016). Membrane pore formation in atomistic and coarse-grained simulations. Biochim. Biophys. Acta Biomembranes 1858, 2266–2277. doi: 10.1016/j.bbamem.2015.12.031
Koh, J.-J., Lin, H., Caroline, V., Chew, Y. S., Pang, L. M., Aung, T. T., et al. (2015a). N-lipidated peptide dimers: effective antibacterial agents against gram-negative pathogens through lipopolysaccharide permeabilization. J. Med. Chem. 58, 6533–6548. doi: 10.1021/acs.jmedchem.5b00628
Koh, J.-J., Lin, S., Aung, T. T., Lim, F., Zou, H., Bai, Y., et al. (2015b). Amino acid modified xanthone derivatives: novel, highly promising membrane-active antimicrobials for multidrug-resistant gram-positive bacterial infections. J. Med. Chem. 58, 739–752. doi: 10.1021/jm501285x
Kowalski, R. P., Romanowski, E. G., Yates, K. A., and Mah, F. S. (2016). An independent evaluation of a novel peptide mimetic, brilacidin (PMX30063), for ocular anti-infective. J. Ocul. Pharmacol. Ther. 32, 23–27. doi: 10.1089/jop.2015.0098
Kwon, B., Waring, A., and J., Hong, M. (2013). A 2H solid-state NMR study of lipid clustering by cationic antimicrobial and cell-penetrating peptides in model bacterial membranes. Biophys. J. 105, 2333–2342. doi: 10.1016/j.bpj.2013.08.020
Lakshminarayanan, R., Liu, S., Li, J., Nandhakumar, M., Aung, T. T., Goh, E., et al. (2014). Synthetic multivalent antifungal peptides effective against fungi. PLoS ONE 9:e87730. doi: 10.1371/journal.pone.0087730r
Lakshminarayanan, R., Tan, W. X., Aung, T. T., Goh, E. T. L., Muruganantham, N., Li, J., et al. (2016). Branched peptide, B2088, disrupts the supramolecular organization of lipopolysaccharides and sensitizes the gram-negative bacteria. Sci. Rep. 6:25905. doi: 10.1038/srep25905
Lam, K. L. H., Wang, H., Siaw, T. A., Chapman, M. R., Waring, A. J., Kindt, J. T., et al. (2012). Mechanism of structural transformations induced by antimicrobial peptides in lipid membranes. Biochim. Biophys. Acta Biomembranes 1818, 194–204. doi: 10.1016/j.bbamem.2011.11.002
Lau, Q. Y., Ng, F. M., Cheong, J. W. D., Yap, Y. Y. A., Tan, Y. Y. F., Jureen, R., et al. (2015). Discovery of an ultra-short linear antibacterial tetrapeptide with anti-MRSA activity from a structure–activity relationship study. Eur. J. Med. Chem. 105, 138–144. doi: 10.1016/j.ejmech.2015.10.015
Laver, D. R. (1994). The barrel-stave model as applied to alamethicin and its analogs reevaluated. Biophys. J. 66(Pt 1), 355–359. doi: 10.1016/S0006-3495(94)80784-2
Lee, D.-K., Bhunia, A., Kotler, S. A., and Ramamoorthy, A. (2015). Detergent-type membrane fragmentation by MSI-78, MSI-367, MSI-594, and MSI-843 antimicrobial peptides and inhibition by cholesterol: a solid-state nuclear magnetic resonance study. Biochemistry 54, 1897–1907. doi: 10.1021/bi501418m
Lee, M.-T., Sun, T.-L., Hung, W.-C., and Huang, H. W. (2013). Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. U.S.A. 110, 14243–14248. doi: 10.1073/pnas.1307010110
Lee, T. H., Hall, K. N., and Aguilar, M. I. (2016). Antimicrobial peptide structure and mechanism of action: a focus on the role of membrane structure. Curr. Top. Med. Chem. 16, 25–39. doi: 10.2174/1568026615666150703121700
Leontiadou, H., Mark, A. E., and Marrink, S. J. (2006). Antimicrobial peptides in action. J. Am. Chem. Soc. 128, 12156–12161. doi: 10.1021/ja062927q
Leveritt, J. M. III, John, M., Pino-Angeles, A., and Lazaridis, T. (2015). The structure of a melittin-stabilized pore. Biophys. J. 108, 2424–2426. doi: 10.1016/j.bpj.2015.04.006
Li, J., Liu, S., Koh, J.-J., Zou, H., Lakshminarayanan, R., Bai, Y., et al. (2015). A novel fragment based strategy for membrane active antimicrobials against MRSA. Biochim. Biophys. Acta Biomembranes 1848, 1023–1031. doi: 10.1016/j.bbamem.2015.01.001
Li, J., Liu, S., Lakshminarayanan, R., Bai, Y., Pervushin, K., Verma, C., et al. (2013). Molecular simulations suggest how a branched antimicrobial peptide perturbs a bacterial membrane and enhances permeability. Biochim. Biophys. Acta Biomembranes 1828, 1112–1121. doi: 10.1016/j.bbamem.2012.12.015
Li, J., Xu, X., Xu, C., Zhou, W., Zhang, K., Yu, H., et al. (2007). Anti-infection peptidomics of amphibian skin. Mol. Cell. Proteomics 6, 882–894. doi: 10.1074/mcp.M600334-MCP200
Li, Y., Xiang, Q., Zhang, Q., Huang, Y., and Su, Z. (2012). Overview on the recent study of antimicrobial peptides: origins, functions, relative mechanisms and application. Peptides 37, 207–215. doi: 10.1016/j.peptides.2012.07.001
Liu, D., Choi, S., Chen, B., Doerksen, R. J., Clements, D. J., Winkler, J. D., et al. (2004). Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem. Int. Ed. Engl. 43, 1158–1162. doi: 10.1002/anie.200352791
Liu, Z., Brady, A., Young, A., Rasimick, B., Chen, K., Zhou, C., et al. (2007). Length effects in antimicrobial peptides of the (RW)(n) series. Antimicrob. Agents Chemother. 51, 597–603. doi: 10.1128/AAC.00828-06
Livermore, D. M. (2004). The need for new antibiotics. Clin. Microbiol. Infect. 10, 1–9. doi: 10.1111/j.1465-0691.2004.1004.x
Lohan, S., and Bisht, G. S. (2013). Recent approaches in design of peptidomimetics for antimicrobial drug discovery research. Mini Rev. Med. Chem. 13, 1073–1088. doi: 10.2174/1389557511313070010
Lu, J.-X., Damodaran, K., Blazyk, J., and Lorigan, G. A. (2005). Solid-state nuclear magnetic resonance relaxation studies of the interaction mechanism of antimicrobial peptides with phospholipid bilayer membranes. Biochemistry 44, 10208–10217. doi: 10.1021/bi050730p
Makovitzki, A., Avrahami, D., and Shai, Y. (2006). Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. U.S.A. 103, 15997–16002. doi: 10.1073/pnas.0606129103
Malanovic, N., and Lohner, K. (2016). Gram-positive bacterial cell envelopes: the impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembranes 1858, 936–946. doi: 10.1016/j.bbamem.2015.11.004
Manna, M., and Mukhopadhyay, C. (2009). Cause and effect of melittin-induced pore formation: a computational approach. Langmuir 25, 12235–12242. doi: 10.1021/la902660q
Marr, A. K., Gooderham, W. J., and Hancock, R. E. W. (2006). Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol. 6, 468–472. doi: 10.1016/j.coph.2006.04.006
Marta Guarna, M., Coulson, R., and Rubinchik, E. (2006). Anti-inflammatory activity of cationic peptides: application to the treatment of acne vulgaris. FEMS Microbiol. Lett. 257, 1–6. doi: 10.1111/j.1574-6968.2006.00156.x
Martin, L., De Santis, R., Koczera, P., Simons, N., Haase, H., Heinbockel, L., et al. (2015a). The synthetic antimicrobial peptide 19-2.5 interacts with heparanase and heparan sulfate in murine and human sepsis. PLoS ONE 10:e0143583. doi: 10.1371/journal.pone.0143583
Martin, L., van Meegern, A., Doemming, S., and Schuerholz, T. (2015b). Antimicrobial peptides in human sepsis. Front. Immunol. 6:404. doi: 10.3389/fimmu.2015.00404
Matile, S., Vargas Jentzsch, A., Montenegro, J., and Fin, A. (2011). Recent synthetic transport systems. Chem. Soc. Rev. 40, 2453–2474. doi: 10.1039/c0cs00209g
Matsuzaki, K. (1998). Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta Biomembranes 1376, 391–400. doi: 10.1016/s0304-4157(98)00014-8
Matsuzaki, K., Yoneyama, S., and Miyajima, K. (1997). Pore formation and translocation of melittin. Biophys. J. 73, 831–838. doi: 10.1016/S0006-3495(97)78115-3
McCormick, T. S., and Weinberg, A. (2010). Epithelial cell-derived antimicrobial peptides are multi-functional agents that bridge innate and adaptive immunity. Periodontol. 2000 54, 195–206. doi: 10.1111/j.1600-0757.2010.00373.x
McDermott, A. M. (2013). Antimicrobial compounds in tears. Exp. Eye Res. 117, 53–61. doi: 10.1016/j.exer.2013.07.014
McKenna, M. (2013). Antibiotic resistance: the last resort. Nature 499, 394–396. doi: 10.1038/499394a
Melo, M. N., Ferre, R., and Castanho, M. A. (2009). Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 7, 245–250. doi: 10.1038/nrmicro2095
Meroueh, S. O., Bencze, K. Z., Hesek, D., Lee, M., Fisher, J. F., Stemmler, T. L., et al. (2006). Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 103, 4404–4409. doi: 10.1073/pnas.0510182103
Mihajlovic, M., and Lazaridis, T. (2010). Antimicrobial peptides in toroidal and cylindrical pores. Biochim. Biophys. Acta 1798, 1485–1493. doi: 10.1016/j.bbamem.2010.04.004
Milov, A. D., Samoilova, R. I., Tsvetkov, Y. D., De Zotti, M., Formaggio, F., Toniolo, C., et al. (2009). Structure of self-aggregated alamethicin in ePC membranes detected by pulsed electron-electron double resonance and electron spin echo envelope modulation spectroscopies. Biophys. J. 96, 3197–3209. doi: 10.1016/j.bpj.2009.01.026
Mishra, B., and Wang, G. (2012). Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 134, 12426–12429. doi: 10.1021/ja305644e
Mishra, B., Lushnikova, T., and Wang, G. (2015). Small lipopeptides possess anti-biofilm capability comparable to daptomycin and vancomycin. RSC Adv. 5, 59758–59769. doi: 10.1039/C5RA07896B
Mohanram, H., and Bhattacharjya, S. (2014). β-boomerang antimicrobial and antiendotoxic peptides: lipidation and disulfide bond effects on activity and structure. Pharmaceuticals 7, 482–501. doi: 10.3390/ph7040482
Monroc, S., Badosa, E., Besalú, E., Planas, M., Bardají, E., Montesinos, E., et al. (2006). Improvement of cyclic decapeptides against plant pathogenic bacteria using a combinatorial chemistry approach. Peptides 27, 2575–2584. doi: 10.1016/j.peptides.2006.05.001
Mygind, P. H., Fischer, R. L., Schnorr, K. M., Hansen, M. T., Sonksen, C. P., Ludvigsen, S., et al. (2005). Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 437, 975–980. doi: 10.1038/nature04051
Nasompag, S., Dechsiri, P., Hongsing, N., Phonimdaeng, P., Daduang, S., Klaynongsruang, S., et al. (2015). Effect of acyl chain length on therapeutic activity and mode of action of the CX-KYR-NH2 antimicrobial lipopeptide. Biochim. Biophys. Acta 1848(Pt A), 2351–2364. doi: 10.1016/j.bbamem.2015.07.004
Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656. doi: 10.1128/MMBR.67.4.593-656.2003
Niu, Y., Wang, R. E., Wu, H., and Cai, J. (2012). Recent development of small antimicrobial peptidomimetics. Future Med. Chem. 4, 1853–1862. doi: 10.4155/fmc.12.111
North, J. R., Takenaka, S., Rozek, A., Kielczewska, A., Opal, S., Morici, L. A., et al. (2016). A novel approach for emerging and antibiotic resistant infections: innate defense regulators as an agnostic therapy. J. Biotechnol. 226, 24–34. doi: 10.1016/j.jbiotec.2016.03.032
Ofek, I., Cohen, S., Rahmani, R., Kabha, K., Tamarkin, D., Herzig, Y., et al. (1994). Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 38, 374–377. doi: 10.1128/AAC.38.2.374
Olaitan, A. O., Morand, S., and Rolain, J.-M. (2014). Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 5:643. doi: 10.3389/fmicb.2014.00643
Ooi, N., Miller, K., Hobbs, J., Rhys-Williams, W., Love, W., and Chopra, I. (2009). XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J. Antimicrob. Chemother. 64, 735–740. doi: 10.1093/jac/dkp299
Oreopoulos, J., Epand, R. F., Epand, R. M., and Yip, C. M. (2010). Peptide-induced domain formation in supported lipid bilayers: direct evidence by combined atomic force and polarized total internal reflection fluorescence microscopy. Biophys. J. 98, 815–823. doi: 10.1016/j.bpj.2009.12.4327
Overhage, J., Campisano, A., Bains, M., Torfs, E. C., Rehm, B. H., and Hancock, R. E. (2008). Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 76, 4176–4182. doi: 10.1128/IAI.00318-08
Perona, J. J., and Craik, C. S. (1997). Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J. Biol. Chem. 272, 29987–29990. doi: 10.1074/jbc.272.48.29987
Polyansky, A. A., Ramaswamy, R., Volynsky, P. E., Sbalzarini, I. F., Marrink, S. J., and Efremov, R. G. (2010). Antimicrobial peptides induce growth of phosphatidylglycerol domains in a model bacterial membrane. J. Phys. Chem. Lett. 1, 3108–3111. doi: 10.1021/jz101163e
Porcelli, F., Buck, B., Lee, D.-K., Hallock, K. J., Ramamoorthy, A., and Veglia, G. (2004). Structure and orientation of pardaxin determined by NMR experiments in model membranes. J. Biol. Chem. 279, 45815–45823. doi: 10.1074/jbc.M405454200
Qian, S., and Heller, W. T. (2011). Peptide-induced asymmetric distribution of charged lipids in a vesicle bilayer revealed by small-angle neutron scattering. J. Phys. Chem. B 115, 9831–9837. doi: 10.1021/jp204045t
Rabanal, F., Grau-Campistany, A., Vila-Farrés, X., Gonzalez-Linares, J., Borràs, M., Vila, J., et al. (2015). A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci. Rep. 5:10558. doi: 10.1038/srep10558
Ragioto, D. A. M. T., Carrasco, L. D. M., and Carmona-Ribeiro, A. M. (2014). Novel gramicidin formulations in cationic lipid as broad-spectrum microbicidal agents. Int. J. Nanomedicine 9, 3183–3192. doi: 10.2147/IJN.S65289
Ramamoorthy, A., Lee, D.-K., Narasimhaswamy, T., and Nanga, R. P. R. (2010). Cholesterol reduces pardaxin's dynamics—a barrel-stave mechanism of membrane disruption investigated by solid-state NMR. Biochim. Biophys. Acta Biomembranes 1798, 223–227. doi: 10.1016/j.bbamem.2009.08.012
Ramamoorthy, A., Thennarasu, S., Lee, D.-K., Tan, A., and Maloy, L. (2006). Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 91, 206–216. doi: 10.1529/biophysj.105.073890
Rausch, J. M., Marks, J. R., and Wimley, W. C. (2005). Rational combinatorial design of pore-forming β-sheet peptides. Proc. Natl. Acad. Sci. U.S.A. 102, 10511–10515. doi: 10.1073/pnas.0502013102
Rausch, J. M., Marks, J. R., Rathinakumar, R., and Wimley, W. C. (2007). β-sheet pore-forming peptides selected from a rational combinatorial library: mechanism of pore formation in lipid vesicles and activity in biological membranes. Biochemistry 46, 12124–12139. doi: 10.1021/bi700978h
Romeo, D., Skerlavaj, B., Bolognesi, M., and Gennaro, R. (1988). Structure and bactericidal activity of an antibiotic dodecapeptide purified from bovine neutrophils. J. Biol. Chem. 263, 9573–9575.
Ruiz, N., Kahne, D., and Silhavy, T. J. (2006). Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4, 57–66. doi: 10.1038/nrmicro1322
Sani, M. A., Whitwell, T. C., Gehman, J. D., Robins-Browne, R. M., Pantarat, N., Attard, T. J., et al. (2013). Maculatin 1.1 disrupts Staphylococcus aureus lipid membranes via a pore mechanism. Antimicrob. Agents Chemother. 57, 3593–3600. doi: 10.1128/AAC.00195-13
Santo, K. P., Irudayam, S. J., and Berkowitz, M. L. (2013). Melittin creates transient pores in a lipid bilayer: results from computer simulations. J. Phys. Chem. B 117, 5031–5042. doi: 10.1021/jp312328n
Saravanan, R., Li, X., Lim, K., Mohanram, H., Peng, L., Mishra, B., et al. (2014). Design of short membrane selective antimicrobial peptides containing tryptophan and arginine residues for improved activity, salt-resistance, and biocompatibility. Biotechnol. Bioeng. 111, 37–49. doi: 10.1002/bit.25003
Saravolatz, L. D., Pawlak, J., Johnson, L., Bonilla, H., Saravolatz, L. D. II, Fakih, M. G., et al. (2012a). In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 4478–4482. doi: 10.1128/AAC.00194-12
Saravolatz, L. D., Pawlak, J., Johnson, L., Bonilla, H., Saravolatz, L. D., Fakih, M. G., et al. (2012b). In vitro Activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 4478–4482. doi: 10.1128/AAC.00194-12
Sato, H., and Feix, J. B. (2006). Peptide–membrane interactions and mechanisms of membrane destruction by amphipathic α-helical antimicrobial peptides. Biochim. Biophys. Acta Biomembranes 1758, 1245–1256. doi: 10.1016/j.bbamem.2006.02.021
Scheinpflug, K., Krylova, O., Nikolenko, H., Thurm, C., and Dathe, M. (2015). Evidence for a novel mechanism of antimicrobial action of a cyclic R-,W-rich hexapeptide. PLoS ONE 10:e0125056. doi: 10.1371/journal.pone.0125056
Schmidt, N. W., and Wong, G. C. L. (2013). Antimicrobial peptides and induced membrane curvature: geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mater. Sci. 17, 151–163. doi: 10.1016/j.cossms.2013.09.004
Schmidt, N. W., Lis, M., Zhao, K., Lai, G. H., Alexandrova, A. N., Tew, G. N., et al. (2012a). Molecular basis for nanoscopic membrane curvature generation from quantum mechanical models and synthetic transporter sequences. J. Am. Chem. Soc. 134, 19207–19216. doi: 10.1021/ja308459j
Schmidt, N. W., Mishra, A., Lai, G. H., Davis, M., Sanders, L. K., Tran, D., et al. (2011). Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 133, 6720–6727. doi: 10.1021/ja200079a
Schmidt, N. W., Tai, K. P., Kamdar, K., Mishra, A., Lai, G. H., Zhao, K., et al. (2012b). Arginine in α-defensins: differential effects on bactericidal activity correspond to geometry of membrane curvature generation and peptide-lipid phase behavior. J. Biol. Chem. 287, 21866–21872. doi: 10.1074/jbc.M112.358721
Schmidt, N., Mishra, A., Lai, G. H., and Wong, G. C. L. (2010). Arginine-rich cell-penetrating peptides. FEBS Lett. 584, 1806–1813. doi: 10.1016/j.febslet.2009.11.046
Schuerholz, T., Brandenburg, K., and Marx, G. (2012). Antimicrobial peptides and their potential application in inflammation and sepsis. Crit. Care 16, 207–207. doi: 10.1186/cc11220
Schwechheimer, C., and Kuehn, M. J. (2015). Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 13, 605–619. doi: 10.1038/nrmicro3525
Sengupta, D., Leontiadou, H., Mark, A. E., and Marrink, S.-J. (2008). Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta Biomembranes 1778, 2308–2317. doi: 10.1016/j.bbamem.2008.06.007
Shai, Y. (2002). Mode of action of membrane active antimicrobial peptides. Biopolymers 66, 236–248. doi: 10.1002/bip.10260
Shin, D.-M., and Jo, E.-K. (2011). Antimicrobial peptides in innate immunity against mycobacteria. Immune Netw. 11, 245–252. doi: 10.4110/in.2011.11.5.245
Shin, J. R., Lim, K. J., Kim, D. J., Cho, J. H., and Kim, S. C. (2013). Display of multimeric antimicrobial peptides on the Escherichia coli cell surface and its application as whole-cell antibiotics. PLoS ONE 8:e58997. doi: 10.1371/journal.pone.0058997
Sitaram, N., and Nagaraj, R. (1999). Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim. Biophys. Acta. Biomembranes 1462, 29–54. doi: 10.1016/S0005-2736(99)00199-6
Sivertsen, A., Isaksson, J., Leiros, H.-K. S., Svenson, J., Svendsen, J.-S., and Brandsdal, B. O. (2014). Synthetic cationic antimicrobial peptides bind with their hydrophobic parts to drug site II of human serum albumin. BMC Struct. Biol. 14, 4–4. doi: 10.1186/1472-6807-14-4
Sjogren, M. H. (2004). Thymalfasin: an immune system enhancer for the treatment of liver disease. J. Gastroenterol. Hepatol. 19, S69–S72. doi: 10.1111/j.1440-1746.2004.03635.x
Sokolov, Y., Mirzabekov, T., Martin, D. W., Lehrer, R. I., and Kagan, B. L. (1999). Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta Biomembranes 1420, 23–29. doi: 10.1016/S0005-2736(99)00086-3
Song, C., Weichbrodt, C., Salnikov, E. S., Dynowski, M., Forsberg, B. O., Bechinger, B., et al. (2013). Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. U.S.A. 12, 4586–4591. doi: 10.1073/pnas.1214739110
Starr, C. G., He, J., and Wimley, W. C. (2016). Host cell interactions are a significant barrier to the clinical utility of peptide antibiotics. ACS Chem. Biol. 11, 3391–3399. doi: 10.1021/acschembio.6b00843
Stevens, M. J. (2004). Coarse-grained simulations of lipid bilayers. J. Chem. Phys. 121, 11942–11948. doi: 10.1063/1.1814058
Strandberg, E., and Ulrich, A. S. (2004). NMR methods for studying membrane-active antimicrobial peptides. Concepts Magn. Reson. Part A 23A, 89–120. doi: 10.1002/cmr.a.20024
Strøm, M. B., Haug, B. E., Skar, M. L., Stensen, W., Stiberg, T., and Svendsen, J. S. (2003). The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 46, 1567–1570. doi: 10.1021/jm0340039
Strøm, M. B., Rekdal, Ø., and Svendsen, J. S. (2002). Antimicrobial activity of short arginine- and tryptophan-rich peptides. J. Pept. Sci. 8, 431–437. doi: 10.1002/psc.398
Su, Y., DeGrado, W. F., and Hong, M. (2010). Orientation, dynamics and lipid interaction of an antimicrobial arylamide investigated by (19)F and (31)P solid-state NMR spectroscopy. J. Am. Chem. Soc. 132, 9197–9205. doi: 10.1021/ja103658h
Svenson, J., Brandsdal, B.-O., Stensen, W., and Svendsen, J. S. (2007). Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J. Med. Chem. 50, 3334–3339. doi: 10.1021/jm0703542
Szleifer, I., Kramer, D., Ben-Shaul, A., Roux, D., and Gelbart, W. M. (1988). Curvature elasticity of pure and mixed surfactant films. Phys. Rev. Lett. 60, 1966–1969. doi: 10.1103/PhysRevLett.60.1966
Takeshima, K., Chikushi, A., Lee, K.-K., Yonehara, S., and Matsuzaki, K. (2003). Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J. Biol. Chem. 278, 1310–1315. doi: 10.1074/jbc.M208762200
Tally, F. P., and DeBruin, M. F. (2000). Development of daptomycin for Gram-positive infections. J. Antimicrob. Chemother. 46, 523–526. doi: 10.1093/jac/46.4.523
Terwilliger, T. C., Weissman, L., and Eisenberg, D. (1982). The structure of melittin in the form I crystals and its implication for melittin's lytic and surface activities. Biophys. J. 37, 353–361. doi: 10.1016/S0006-3495(82)84683-3
Tew, G. N., Liu, D., Chen, B., Doerksen, R. J., Kaplan, J., Carroll, P. J., et al. (2002). De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. U.S.A. 99, 5110–5114. doi: 10.1073/pnas.082046199
Tew, G. N., Scott, R. W., Klein, M. L., and Degrado, W. F. (2010a). De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43, 30–39. doi: 10.1021/ar900036b
Tew, G. N., Scott, R. W., Klein, M. L., and DeGrado, W. F. (2010b). De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43, 30–39. doi: 10.1021/ar900036b
Thomas, S., Karnik, S., Barai, R. S., Jayaraman, V. K., and Idicula-Thomas, S. (2010). CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 38, D774–D780. doi: 10.1093/nar/gkp1021
Tsai, C.-W., Hsu, N.-Y., Wang, C.-H., Lu, C.-Y., Chang, Y., Tsai, H.-H. G., et al. (2009). Coupling molecular dynamics simulations with experiments for the rational design of indolicidin-analogous antimicrobial peptides. J. Mol. Biol. 392, 837–854. doi: 10.1016/j.jmb.2009.06.071
Urry, D. W. (1971). The gramicidin A transmembrane Channel: a proposed π((L,D)) helix. Proc. Natl. Acad. Sci. U.S.A. 68, 672–676. doi: 10.1073/pnas.68.3.672
Valerio, L. G. Jr. (2009). In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharmacol. 241, 356–370. doi: 10.1016/j.taap.2009.08.022
Van Lenten, B. J., Navab, M., Anantharamaiah, G. M., Buga, G. M., Reddy, S. T., and Fogelman, A. M. (2008). Multiple indications for anti-inflammatory peptides. Curr. Opin. Investig. Drugs 9, 1157–1162.
Vandamme, D., Landuyt, B., Luyten, W., and Schoofs, L. (2012). A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. 280, 22–35. doi: 10.1016/j.cellimm.2012.11.009
Vandenburg, Y. R., Smith, B. D., Biron, E., and Voyer, N. (2002). Membrane disruption ability of facially amphiphilic helical peptides. Chem. Commun. 16, 1694–1695. doi: 10.1039/b204640g
Vollmer, W., Blanot, D., and De Pedro, M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. doi: 10.1111/j.1574-6976.2007.00094.x
Wadhwani, P., Epand, R. F., Heidenreich, N., Bürck, J., Ulrich, A. S., and Epand, R. M. (2012). Membrane-active peptides and the clustering of anionic lipids. Biophys. J. 103, 265–274. doi: 10.1016/j.bpj.2012.06.004
Wang, G., Li, X., and Wang, Z. (2016). APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093. doi: 10.1093/nar/gkv1278
Wang, J., Li, Y., Wang, X., Chen, W., Sun, H., and Wang, J. (2014). Lipopolysaccharide induces amyloid formation of antimicrobial peptide HAL-2. Biochim. Biophys. Acta Biomembranes 1838, 2910–2918. doi: 10.1016/j.bbamem.2014.07.028
Wang, K. F., Nagarajan, R., and Camesano, T. A. (2015). Differentiating antimicrobial peptides interacting with lipid bilayer: molecular signatures derived from quartz crystal microbalance with dissipation monitoring. Biophys Chem. 196, 53–67. doi: 10.1016/j.bpc.2014.09.003
Wang, Y., Chen, C. H., Hu, D., Ulmschneider, M. B., and Ulmschneider, J. P. (2016). Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 7:13535. doi: 10.1038/ncomms13535
Wimley, W. C. (2010). Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 5, 905–917. doi: 10.1021/cb1001558
Wu, Z., Cui, Q., and Yethiraj, A. (2013). Why do arginine and lysine organize lipids differently? insights from coarse-grained and atomistic simulations. J. Phys. Chem. B 117, 12145–12156. doi: 10.1021/jp4068729
Xiong, M., Lee, M. W., Mansbach, R. A., Song, Z., Bao, Y., Peek, R. M., et al. (2015). Helical antimicrobial polypeptides with radial amphiphilicity. Proc. Natl. Acad. Sci. U.S.A. 112, 13155–13160. doi: 10.1073/pnas.1507893112
Yang, L., Gordon, V. D., Mishra, A., Som, A., Purdy, K. R., Davis, M. A., et al. (2007). Synthetic antimicrobial oligomers induce a composition-dependent topological transition in membranes. J. Am. Chem. Soc. 129, 12141–12147. doi: 10.1021/ja072310o
Yang, L., Harroun, T. A., Weiss, T. M., Ding, L., and Huang, H. W. (2001). Barrel-stave model or toroidal model? a case study on melittin pores. Biophys. J. 81, 1475–1485. doi: 10.1016/S0006-3495(01)75802-X
Ye, G., Gupta, A., DeLuca, R., Parang, K., and Bothun, G. D. (2010). Bilayer disruption and liposome restructuring by a homologous series of small Arg-rich synthetic peptides. Colloids Surf. B Biointerfaces 76, 76–81. doi: 10.1016/j.colsurfb.2009.10.016
Yeaman, M. R., and Yount, N. Y. (2003). Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Res. 55, 27–55. doi: 10.1124/pr.55.1.2
Yoneyama, F., Imura, Y., Ohno, K., Zendo, T., Nakayama, J., Matsuzaki, K., et al. (2009). Peptide-lipid huge toroidal pore, a new antimicrobial mechanism mediated by a lactococcal bacteriocin, lacticin, Q. Antimicrob. Agents Chemother. 53, 3211–3217. doi: 10.1128/AAC.00209-09
Yoo, J., and Cui, Q. (2008). Does arginine remain protonated in the lipid membrane? insights from microscopic pK(a) calculations. Biophys. J. 94, L61–L63. doi: 10.1529/biophysj.107.122945
Yoo, J., and Cui, Q. (2010). Chemical versus mechanical perturbations on the protonation state of arginine in complex lipid membranes: insights from microscopic pK(a) calculations. Biophys. J. 99, 1529–1538. doi: 10.1016/j.bpj.2010.06.048
Zasloff, M. (1987). Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. U.S.A. 84, 5449–5453. doi: 10.1073/pnas.84.15.5449
Zasloff, M. (2002). Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. doi: 10.1038/415389a
Keywords: antimicrobial peptides, action mechanism, membrane, antibiotic resistance, peptide antibiotics
Citation: Li J, Koh J-J, Liu S, Lakshminarayanan R, Verma CS and Beuerman RW (2017) Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 11:73. doi: 10.3389/fnins.2017.00073
Received: 29 November 2016; Accepted: 31 January 2017;
Published: 14 February 2017.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Nils Lambrecht, Veterans Administration Long Beach and University of California Irvine, USAJohn Michael Conlon, Ulster University, UK
Copyright © 2017 Li, Koh, Liu, Lakshminarayanan, Verma and Beuerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger W. Beuerman, cndiZXVlcm1hbkBnbWFpbC5jb20=