 Yuho Okita1
Yuho Okita1 Alexandre N. Rcom-H'cheo-Gauthier1
Alexandre N. Rcom-H'cheo-Gauthier1 Michael Goulding1
Michael Goulding1 Roger S. Chung2
Roger S. Chung2 Peter Faller3,4
Peter Faller3,4 Dean L. Pountney1*
Dean L. Pountney1*- 1Menzies Health Institute Queensland, Griffith University, Gold Coast, QLD, Australia
- 2Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, NSW, Australia
- 3Centre National de la Recherche Scientifique, Institut de Chimie UMR 7177, Université de Strasbourg, Strasbourg, France
- 4University of Strasbourg Institute for Advanced Study, Strasbourg, France
Metallothioneins (MTs) are proteins that function by metal exchange to regulate the bioavailability of metals, such as zinc and copper. Copper functions in the brain to regulate mitochondria, neurotransmitter production, and cell signaling. Inappropriate copper binding can result in loss of protein function and Cu(I)/(II) redox cycling can generate reactive oxygen species. Copper accumulates in the brain with aging and has been shown to bind alpha-synuclein and initiate its aggregation, the primary aetiological factor in Parkinson's disease (PD), and other alpha-synucleinopathies. In PD, total tissue copper is decreased, including neuromelanin-bound copper and there is a reduction in copper transporter CTR-1. Conversely cerebrospinal fluid (CSF) copper is increased. MT-1/2 expression is increased in activated astrocytes in alpha-synucleinopathies, yet expression of the neuronal MT-3 isoform may be reduced. MTs have been implicated in inflammatory states to perform one-way exchange of copper, releasing free zinc and recent studies have found copper bound to alpha-synuclein is transferred to the MT-3 isoform in vitro and MT-3 is found bound to pathological alpha-synuclein aggregates in the alpha-synucleinopathy, multiple systems atrophy. Moreover, both MT and alpha-synuclein can be released and taken up by neural cells via specific receptors and so may interact both intra- and extra-cellularly. Here, we critically review the role of MTs in copper dyshomeostasis and alpha-synuclein aggregation, and their potential as biomarkers and therapeutic targets.
Introduction
There is considerable interest in understanding the role of copper in the etiology of PD. Copper homeostasis is disrupted in the PD brain. Furthermore, in vitro studies demonstrate that copper can exacerbate alpha-synuclein aggregation (Carboni and Lingor, 2015; Davies et al., 2016). Recent studies have indicated that MTs, one of the major copper regulatory proteins in mammalian cells, may influence alpha-synuclein aggregation in a copper-dependent manner (Adam et al., 2016). This review provides an overview of recent literature describing the role of copper dyshomeostasis in PD and the potential role of MTs in this process.
Several neurodegenerative diseases, including PD, multiple system atrophy (MSA), and dementia with Lewy bodies (DLB), show abnormal folding and aggregation of the alpha-synuclein protein correlating with death of neuronal cells (Barnham et al., 2004; Barnham and Bush, 2008; Wong and Krainc, 2017). The pathogenesis of these disorders is complex; a result of the interplay between genetic susceptibility and environmental factors (Dauer and Prezedborski, 2003; Chai and Lim, 2013). PD is a chronic disease caused by the progressive degeneration of dopaminergic neurons, affecting both motor and non-motor functions (Lees et al., 2009; Jenner et al., 2013). Degeneration progresses from the brain stem to the locus coeruleus, dorsal and raphe nuclei, and ventral tegmental area to the prefrontal cortex (Wirdefeldt et al., 2011; Jucker and Walker, 2013). Age is the most significant risk factor (Dauer and Prezedborski, 2003), but other factors include head trauma or injury and obesity (Lees et al., 2009; Jenner et al., 2013). There are no diagnostic biomarkers for PD, yet it is estimated that the pre-symptomatic disease can extend over 10 years. PD can arise from autosomal dominant mutations associated with the alpha-synuclein gene, SNCA, resulting in A53T, H50Q, E46K, A53E, G51D, and A30P variants (Wirdefeldt et al., 2011; Nussbaum, 2017) and has also been linked to alpha-synuclein overexpression, accumulation and aggregation (Shin et al., 2009; Anandhan et al., 2015; Van der Perren et al., 2015). Recently, a point mutation within the Vps35 subunit of the retromer was linked to PD which results in disrupted trafficking of cathepsin D, a protease important for the degradation of alpha-synuclein (Follett et al., 2014).
DLB is characterized by progressive dementia, hallucinations, delusions and memory loss, rigidity, and postural disturbance (Noe et al., 2004), with both PD and DLB characterized by mostly neuronal alpha-synuclein deposits. A third prominent alpha-synucleinopathy is MSA a condition which shows alpha-synuclein inclusions mostly affecting glial cells (Spillantini and Goedert, 2000). MSA is rapidly progressive and shares most of its symptoms with PD, although degeneration occurs both to the central nervous system and periphery (Jellinger, 2014; Radford et al., 2014).
Current treatments for PD do not cure the underlying condition but alleviate symptoms (Dauer and Prezedborski, 2003; Jankovic, 2012; Jenner et al., 2013). Dopamine replacement therapy via the precursor L-DOPA (L-3,4-dihydroxyphenylalanine) is the most prevalent treatment (Jankovic, 2012). However, patients develop resistance to L-DOPA over time (Connolly and Lang, 2014) and prolonged treatment may eventually accelerate deterioration due to generation of reactive oxygen species (Barnham et al., 2004; Thanvi and Lo, 2004), whereas, pre-treatment with non-steroidal anti-inflammatories was found to be effective in animal PD models (McCarty, 2006; Pinto et al., 2016). The development of mechanism-based therapeutics and neuroprotective strategies, including metal chelation, is a focus of on-going research (Hart et al., 2009).
Lewy Bodies and Alpha-Synuclein
In PD pathology there is an accumulation of alpha-synuclein in the brain stem, spinal cord, and cortex (Lees et al., 2009), primarily in the form of intra-neuronal aggregates (Lewy bodies, LB) composed mostly of alpha-synuclein. Other aggregate proteins including Parkin, PINK-1, DJ-1, and LRRK-2 gene products relate to pathways such as autophagy/mitophagy processes, oxidative stress mechanisms, and mitochondrial survival (Shin et al., 2009). Alpha-synuclein makes up 1% of total brain soluble proteins in humans (Stefanis, 2012), with putative functions in dopamine production, uptake and storage as well as recycling of neurotransmitter vesicles (Sidhu et al., 2004; Marques and Outeiro, 2012; Stefanis, 2012). It is a small protein (14 kDa) existing as an unfolded monomer (Vekrellis et al., 2004; Cremades et al., 2012), although recent work has revealed that it is also present in oligomeric forms (Yates, 2011). Studies on alpha-synuclein knockout mice implicate its role in neurotransmitter regulation (Yavich et al., 2004). The protein may also act as a calcium channel activator and mediator of calcium entry and may increase cell toxicity from oxidative stress (Dryanovski et al., 2013; Rcom-H'cheo-Gauthier et al., 2016).
Current evidence shows alpha-synuclein oligomerizes and forms aggregates within the neuronal cytoplasm that have a deleterious outcome for the neuron and provide a link to neurotoxicity (Vekrellis et al., 2004; Chen and Feany, 2005; Stefanis, 2012). The inhibition of normal cellular function by alpha-synuclein aggregates leads to increased oxidative stress (Cremades et al., 2012), and overexpression of alpha-synuclein caused protein degradation malfunction phenotype in rats (Kirik et al., 2002; Vekrellis et al., 2004). It is unclear whether LB are neurotoxic or neuroprotective. They may be formed to sequester aberrant proteins that would otherwise be neurotoxic. Drosophila melanogaster models of PD have demonstrated that increased inclusion body formation is associated with a reduction in overall toxicity to neuronal cells (Chen and Feany, 2005). Similarly, rat models with inhibition of proteasome function show a rise in aggregates but cessation of neuronal death (Sawada et al., 2004). Although several recent reports indicate that LBs are neurotoxic, contributing to cell death in vivo with decreased mitochondrial and proteosomal function which causes increased oxidative stress and cell death (Branco et al., 2010). Alternatively, other studies suggest an inverse correlation between cell viability and the quantity of LB-like alpha-synuclein inclusion bodies (Wan and Chung, 2012). Recently, alpha-synuclein has been observed to propagate between neurons, whereby alpha-synuclein spread mimicked the progression of neurodegeneration observed in PD from the substantia nigra toward the frontal cortex (Luk et al., 2012). This propagation of alpha-synuclein resembles prion disease spread, where infectious proteins travel between host cells (Jucker and Walker, 2013; Valdinocci et al., 2017).
Copper and α-Synuclein Disease
Copper is an essential element, mainly as a catalytic center of several enzymes involved in mitochondrial energy transformation, the synthesis of neurotransmitters, cell signaling, and other crucial processes. It accumulates in the brain with aging (Zatta et al., 2008; Vasudevaraju et al., 2010; Pushkar et al., 2013) and has been shown to bind alpha-synuclein and initiate its aggregation (Paik et al., 1999; Rose et al., 2011; Carboni and Lingor, 2015). At physiological pH, two copper(II) binding sites exist on alpha-synuclein corresponding to M1-D2, and H50. However, at pH 5.0, the H50 binding is strongly diminished and alpha-synuclein additionally showed a completely different site for copper binding at D119–E123 (Drew et al., 2008; De Ricco et al., 2015a). Two regions have been shown to bind to copper(I): M1–M5 and M116–M127 (Camponeschi et al., 2013) with M1–M5 being the stronger and thus more relevant (Figure 1). Within the cell, both copper species co-exist and the transition from copper(II) to copper(I) in amyloid aggregates was suggested to contribute to the presence of ROS leading to cell damage (Miotto et al., 2014). The capacity of copper to bind to alpha-synuclein at H50 could also be highly relevant for disease pathology in humans. Indeed, the point mutation H50Q which leads to a familial form of PD corresponds to the major copper(II) binding site (Proukakis et al., 2013) and binding of copper(II) to this mutant results in a structurally different species compared to the copper-WT species (Villar-Piqué et al., 2016). The ability of copper to accept or donate electrons implicates it in the production of ROS in PD (Barnham et al., 2004; Barnham and Bush, 2008; Valensin et al., 2016), and it has more recently been shown that in the SN, total tissue copper is decreased (Montes et al., 2014). Moreover, some studies have found an increase of serum copper, and indeed a positive correlation between high concentration levels and severity of the disease (Brewer, 2009; Arnal et al., 2010). Furthermore, free copper levels, both in PD cases and DLB cases, were found to have increased in the cerebrospinal fluid (CSF) (Pall et al., 1987; Magaki et al., 2007; Hozumi et al., 2011).
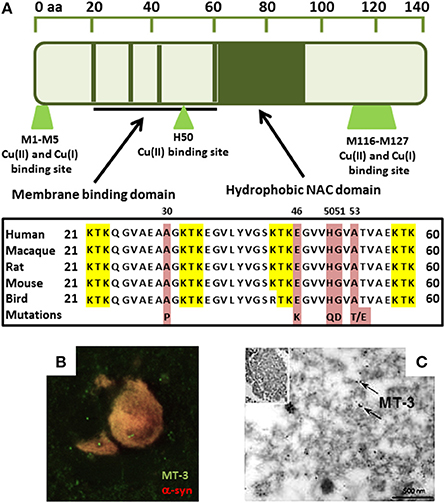
Figure 1. (A) Sequence alignment showing alpha-synuclein copper binding ligands overlap with PD-linked H50 mutation in membrane binding domain. Red represents the known point mutations in familial Parkinson's disease in alpha-synuclein. Yellow represents the KTK repeats in the N-terminus which are involved in lipid interaction. (B) Immunofluorescence shows MT-3 co-localizes with alpha-synuclein in glial cytoplasmic inclusions in MSA brain tissue. (C) MT-3 associates with alpha-synuclein filaments in isolated alpha-synuclein inclusions by immunogold-labeling (black dots; arrows) (Pountney et al., 2011).
Copper in extracellular fluid bound to copper-binding proteins, such as ceruloplasmin, is less harmful than the free, or loosely bound ions (Squitti et al., 2016). These metal-binding proteins act to sequester the reactive metal and most copper present in brain tissue is bound to transport carriers or other proteins (Davies et al., 2013; Squitti et al., 2016). Davies et al. found that neuromelanin-bound copper in the SN of PD patients decreased, suggesting increased free copper linked to neuronal death. It was also found that the copper transporter CTR-1 becomes drastically reduced in the SN of PD brain before regional cell loss begins (Davies et al., 2014). Intracellular copper levels are regulated by the action of membrane copper-ATPases (Cu-ATPases) that pump copper. Genetic mutation of Cu-ATPase (ATP7A) has been found to cause diseases of copper deficiency in the brain and the specific mutation of ATP7B results in diseases of copper accumulation (Lutsenko et al., 2007; Choi and Zheng, 2009; Davies et al., 2016). Recent work has examined copper chelation as a means to reduce free-radical damage, with clioquinol (a metal chelator) shown to reduce aggregation of beta-amyloid and alpha-synuclein in transgenic mice (Cherny et al., 2001; Adlard et al., 2008; Finkelstein et al., 2016). Metallothioneins (MT), contribute to copper regulation and may also act as ROS absorbers (Carter et al., 1984; Otsuka, 2004; Lutsenko et al., 2007). Additionally, MTs have been implicated in MSA (Hozumi, 2013) and the MT-3 isoform was found to co-localize with alpha-synuclein and coat alpha-synuclein filaments in the hallmark glial alpha-synuclein deposits (Figures 1B,C; Pountney et al., 2011). Furthermore, MT-3 was found to inhibit ROS production by copper-alpha-synuclein by a metal-swap mechanism (Meloni and Vašák, 2011).
Metallothioneins
MTs, are small (6–7 KDa) native Zn/Cd binding proteins (Margoshes and Vallee, 1957; Kägi and Vallee, 1960), composed of cysteine-metal cores that have a high affinity for copper (Pountney et al., 1995; Nordberg, 1998; Hozumi et al., 2004; Carpenè et al., 2007; Dong et al., 2015). MT-knockout mice become extremely susceptible to the effects of environmental stress, including copper-exposure (West et al., 2008; Manso et al., 2011; Petro et al., 2016). MTs occur in two major isoforms: MT-1/2 and MT-3 (Nordberg, 1998), with MT-1/2 present throughout the body, while MT-3 is present primarily in the brain (Palmiter et al., 1992). MT-3 differs by a unique acidic insert comprised of six amino acids and two Pro substitutions and has an observed ability to inhibit cell growth in cultured neurons (Palmiter et al., 1992; Chung et al., 2003; Howells et al., 2010). Recent work showed MT to be neuroprotective against proteinopathies in neurodegenerative disease, a finding which is reinforced by low MT-3 levels in neurons of Alzheimer's patients compared to healthy individuals (Kimura and Itoh, 2008; Howells et al., 2010; Uchida, 2010). Interestingly, this same deficiency is seen in PD, although this finding may be somewhat controversial (Hozumi et al., 2004). Previous work showed that MTs were more highly expressed in Parkinsonian astrocytes (Michael et al., 2011) and also in astrocytes in MSA (Pountney et al., 2011).
MTs bind copper with high affinity, suggesting a role in copper homeostasis (Chung et al., 2008, 2010), and mice displaying copper overload crossed with an MT 1/2 knockout did not survive past infancy (Kelly and Palmiter, 1996). Exchange between protein and metal has been observed in studies that found MTs prevented copper-induced aggregation of amyloid beta peptide or alpha-synuclein (Meloni et al., 2008; Meloni and Vašák, 2011; De Ricco et al., 2015b). The MT gene promoter contains a metal response element (Richards et al., 1984; Sadhu and Gedamu, 1989; West et al., 1990; Pountney et al., 1995), although the MT-3 isoform does not show metal inducibility (Faller, 2010). The cysteine-rich structure and metal-response element of MTs implicate a possible target for attenuating copper-induced aggregation of alpha-synuclein (West et al., 2008; Waldron et al., 2009). Copper-MT may also represent a potential biomarker for Parkinson's or Alzheimer's disease as MT levels increase in response to zinc, copper, or cadmium and physiological stress (Richards et al., 1984). MTs have been found to be recruited to alpha-synuclein aggregates in MSA (Figures 1B,C; Pountney et al., 2011). Other studies have shown that exogenous MT is readily taken up by neuronal cells and identified that neurons take up MT through the megalin receptor (Fitzgerald et al., 2007; Chung et al., 2008). Indeed, alpha-synuclein may also be taken up by neural cells or may interact extracellularly with MT.
Oxidative Stress and Neuroinflammation
Oxidative stress results from incomplete reduction of oxygen resulting in the formation of reactive oxygen species (ROS) and/or a decrease in the degradation of these species by endogenous anti-oxidant systems (Halliwell, 2001; Sas et al., 2007). It is unknown whether oxidative stress damage is a prerequisite of PD pathogenesis, or the result of the disease itself (Blesa et al., 2015). Autosomal recessive mutations involving the Parkin gene inhibit its function as a ubiquitin ligase, affecting protein degradation and turnover mechanisms, as well as mitophagy. This may be an important factor as mitochondrial dysfunction leads to oxidative stress (Kitada et al., 1998; Siddiqui et al., 2016). Additionally, PINK1 disruption results in loss-of-function of mitophagic systems. Mutations in DJ-1 similarly disrupt cellular oxidative stress responses and protein degradation systems (Shin et al., 2009). Moreover, MTs were found as effective ROS scavengers in a PD animal model (Ebadi et al., 2005; Ebadi and Sharma, 2006). MTs are capable of self-protection through maintenance of copper and zinc levels and they also directly protect against ROS (Shiraga et al., 1993). Recently, MTs were shown to inhibit the formation of inclusion body-like structures, Charnoly bodies, mitochondrially formed electron-dense membrane stacks found in neurons, leading to a lower observed incidence of alpha-synucleinopathies (Sharma et al., 2013b; Sharma and Ebadi, 2014a,b). The scavenging ability of MTs to combat ROS leads to a lowering of oxidative stress and therefore a potential role in neuroprotection against PD (Ebadi et al., 2005; Ebadi and Sharma, 2006; Hozumi, 2013).
Neuroinflammation has been linked to alpha-synucleinopathies and there is evidence of a role of MTs in inflammation in brain injury (Chung et al., 2009; Pedersen et al., 2009; Shastri et al., 2013; Vieira et al., 2015). MTs have been implicated in inflammatory states and act as exchangeable zinc stores which can swap with toxic metals, such as copper and cadmium (Rofe et al., 1992; Meloni et al., 2008). Astrocytes, the primary cell type expressing MT-1/2 in the brain, up-regulate MT-1/2 expression as a physiological response which may promote neuroregeneration as well as survival (Chung et al., 2008; Landowski et al., 2016). Treatment with MT-2 attenuated the immune response of neurotoxic quinolinic acid production in brain injury (Chung et al., 2009). More recently, overexpression of MT-1 in a mouse model of AD promoted reversal in behavioral symptoms of disease and more inert soluble amyloid species (Manso et al., 2016). Indeed, extracellular alpha-synuclein inhibits β-amyloid plaque formation (Bachhuber et al., 2015), a process that may also be influenced by extracellular MT binding (De Ricco et al., 2015b). Further, in a mouse model of AD, MT-3- and MT-1/2-deficient mice had lower plaque burdens and mortality, and performed better in behavioral tests (Manso et al., 2012a,b). Figure 2 summarizes the interplay between MT, alpha-synuclein and copper in different cell types in PD.
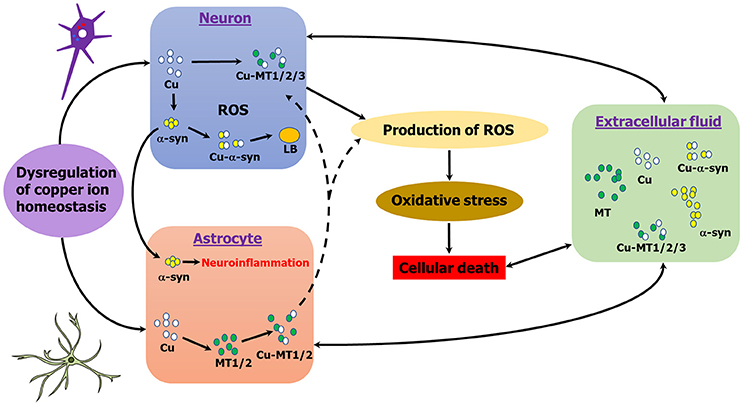
Figure 2. Schematic representation of copper, MT, α-syn interactions in PD. MT-1/2 expression is primarily astrocytic in the brain, whereas MT-3 is neuronal. MTs and alpha-synuclein may interact either intra- or extracellularly, either directly or indirectly. Copper induces alpha-synuclein aggregation, whereas MT's chelate copper. Copper binding to alpha-synuclein generates ROS, whereas MTs are powerful anti-oxidants.
Biomarker and Therapeutic Potential of Metallothionein
The presence of LB post-mortem is the only diagnostic marker for PD (Beach et al., 2013; Sharma et al., 2013a; Espay et al., 2017), although symptoms, such as tremor and rigidity appear after significant damage has already occurred (Jankovic, 2012; Connolly and Lang, 2014). Serum MT levels are significantly increased in pathological conditions including various cancer types (Krizkova et al., 2010) and following traumatic brain injury (Kukacka et al., 2006) and MT can traverse the glomerular apparatus into the urine from the blood supply, with the megalin receptor for MT-1/2 present in the choroid plexus (Lewis et al., 2012). The potential of MT as a biomarker has been speculated in lysosomal storage disorders with nervous system involvement and leukodystrophy (LSDs-LD), where increased white matter MT expression correlated to severity and progression of LSDs-LD and may be related to inflammation and oxidative stress (Cesani et al., 2014). Increased astrocytic MT expression in MSA may similarly point to the biomarker potential of MT in alpha-synuclein disease (Pountney et al., 2011). Indeed, MT is internalized by neural cells via the low-density lipoprotein receptor-related protein receptor, LRP-1, one of the primary trans-membrane transporters involved in the transfer of molecules out of the brain, suggesting that MT might be actively shuttled into the blood (Landowski et al., 2016). Exposure of rodents to heavy metals, such as copper resulted in substantial accumulation of Cu-MT in the liver and kidneys (Kurasaki et al., 1998) suggesting that copper-loaded rather than the natural Cu/Zn-loaded protein in serum or urine could provide a biomarker of copper-imbalance.
Glucocorticoids are regulators of MTs, particularly during fetal development (Quaife et al., 1986) and have been shown to produce increased MT levels in animals (Klaassen, 1981). Dexamethasone is a potent inducer of MT (Karin et al., 1980; Palmiter et al., 1992; Méndez-Armenta et al., 2003), although less effective than heavy metals (Kobayashi et al., 1985), increasing MT-1 mRNA synthesis five-fold (Mayo and Palmiter, 1981) by acting on the glucocorticoid response element in the promoter (Karin et al., 1984; Richards et al., 1984). Dexamethasone, is also neuroprotective via MT induction in an ALS model (Tokuda et al., 2015) and conserved dopamine content by 20% in a PD model (Kurkowska-Jastrzebska et al., 2004). It is tempting to speculate that MT induction could have therapeutic potential in alpha-synuclein diseases.
Conclusion
MTs may offer neuroprotection in alpha-synucleinopathies, however, further studies are needed to determine if the neuroprotective action is mediated via copper chelation or is associated with ROS scavenging, neuroinflammation or neurotrophic effects, if MT and alpha-synuclein interact directly or indirectly and if this is intra- or extracellular.
Author Contributions
All authors contributed equally to the writing. DP coordinated the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by Griffith University and Menzies Health Institute Queensland.
Abbreviations
AD, Alzheimer's disease; ALP, autophagy lysosomal pathway; ANS, autonomic nervous system; CSF, cerebrospinal fluid; DLB, Dementia with Lewy bodies; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (chemical); MSA, Multiple systems atrophy; MT, metallothionein; PD, Parkinson's disease; ROS, reactive oxygen species; SNpc, substantic nigra, pars compacta; UPS, ubiquitin proteasome system.
References
Adam, P., Krížková, S., Heger, Z., Babula, P., Pekarík, V., Vaculovičoá, M., et al. (2016). Metallothioneins in prion- and amyloid-related diseases. J. Alzheimers. Dis. 51, 637–656. doi: 10.3233/JAD-150984
Adlard, P. A., Cherny, R. A., Finkelstein, D. I., Gautier, E., Robb, E., Cortes, M., et al. (2008). Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59, 43–55. doi: 10.1016/j.neuron.2008.06.018
Anandhan, A., Rodriguez-Rocha, H., Bohovych, I., Griggs, A. M., Zavala-Flores, L., Reyes-Reyes, E. M., et al. (2015). Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 81, 76–93. doi: 10.1016/j.nbd.2014.11.018
Arnal, N., Cristalli, D. O., de Alaniz, M. J., and Marra, C. A. (2010). Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Res. 1319, 118–130. doi: 10.1016/j.brainres.2009.11.085
Bachhuber, T., Katzmarski, N., McCarter, J. F., Loreth, D., Tahirovic, S., Kamp, F., et al. (2015). Inhibition of amyloid-[beta] plaque formation by [alpha]-synuclein. Nat. Med. 21, 802–807. doi: 10.1038/nm.3885
Barnham, K. J., and Bush, A. I. (2008). Metals in alzheimer's and parkinsons diseases. Curr. Opin. Chem. Biol. 12, 222–228. doi: 10.1016/j.cbpa.2008.02.019
Barnham, K. J., Masters, C. L., and Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress - a report. Nat. Rev. Drug Discov. 3, 205–210. doi: 10.1038/nrd1330
Beach, T. G., Adler, C. H., Dugger, B. N., Serrano, G., Hidalgo, J., Henry-Watson, J., et al. (2013). Submandibular gland biopsy for the diagnosis of Parkinson disease. J. Neuropathol. Exp. Neurol. 72, 130–136. doi: 10.1097/NEN.0b013e3182805c72
Blesa, J., Trigo-Damas, I., Quiroga-Varela, A., and Jackson-Lewis, V. R. (2015). Oxidative stress and Parkinson's disease. Front Neuroanat. 9:91. doi: 10.3389/fnana.2015.00091
Branco, D. M., Arduino, D. M., Esteves, A. R., Silva, D. F., Cardoso, S. M., and Oliveira, C. (2010). Cross-talk between mitochondria and proteasome in Parkinson's disease pathogenesis. Front. Aging Neurosci. 2:17. doi: 10.3389/fnagi.2010.00017
Brewer, G. J. (2009). Risks of copper and iron toxicity during aging in humans. Chem. Res. Toxicol. 23, 319–326. doi: 10.1021/tx900338d
Camponeschi, F., Valensin, D., Tessari, I., Bubacco, L., Dell'Acqua, S., Casella, L., et al. (2013). Copper (I)-α-synuclein interaction: structural description of two independent and competing metal binding sites. Inorg. Chem. 52, 1358–1367. doi: 10.1021/ic302050m
Carboni, E., and Lingor, P. (2015). Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson's disease. Metallomics 7, 395–404. doi: 10.1039/c4mt00339j
Carpenè, E., Andreani, G., and Isani, G. (2007). Metallothionein functions and structural characteristics. J. Trace Elem. Med. Biol. 21, 35–39. doi: 10.1016/j.jtemb.2007.09.011
Carter, A. D., Felber, B. K., Walling, M., Jubier, M. F., Schmidt, C. J., and Hamer, D. H. (1984). Duplicated heavy metal control sequences of the mouse metallothionein-I gene. Proc. Natl. Acad. Sci. U.S.A. 81, 7392–7396. doi: 10.1073/pnas.81.23.7392
Cesani, M., Cavalca, E., Macco, R., Leoncini, G., Terreni, M. R., Lorioli, L., et al. (2014). Metallothioneins as dynamic markers for brain disease in lysosomal disorders. Ann. Neurol. 75, 127–137. doi: 10.1002/ana.24053
Chai, C., and Lim, K.-L. (2013). Genetic insights into sporadic Parkinson's disease pathogenesis. Curr. Genomics 14, 486–501. doi: 10.2174/1389202914666131210195808
Chen, L. I., and Feany, M. B. (2005). α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663. doi: 10.1038/nn1443
Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., McLean, C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676. doi: 10.1016/s0896-6273(01)00317-8
Choi, B.-S., and Zheng, W. (2009). Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 1248, 14–21. doi: 10.1016/j.brainres.2008.10.056
Chung, R. S., Howells, C., Eaton, E. D., Shabala, L., Zovo, K., Palumaa, P., et al. (2010). The native copper-and zinc-binding protein metallothionein blocks copper-mediated Aβ aggregation and toxicity in rat cortical neurons. PLoS ONE 5:e12030. doi: 10.1371/journal.pone.0012030
Chung, R. S., Leung, Y. K., Butler, C. W., Chen, Y., Eaton, E. D., Pankhurst, M. W., et al. (2009). Metallothionein treatment attenuates microglial activation and expression of neurotoxic quinolinic acid following traumatic brain injury. Neurotox. Res. 15, 381–389. doi: 10.1007/s12640-009-9044-y
Chung, R. S., Penkowa, M., Dittmann, J., King, C. E., Bartlett, C., Asmussen, J. W., et al. (2008). Redefining the role of metallothionein within the injured brain Extracellular metallothioneins play an important role in the astrocyte-neuron response to injury. J. Biol. Chem. 283, 15349–15358. doi: 10.1074/jbc.M708446200
Chung, R. S., Vickers, J. C., Chuah, M. I., and West, A. K. (2003). Metallothionein-IIA promotes initial neurite elongation and postinjury reactive neurite growth and facilitates healing after focal cortical brain injury. J. Neurosci. 23, 3336–3342.
Connolly, B. S., and Lang, A. E. (2014). Pharmacological treatment of Parkinson disease: a review. JAMA 311, 1670–1683. doi: 10.1001/jama.2014.3654
Cremades, N., Cohen, S. I. A., Deas, E., Abramov, A. Y., Chen, A. Y., Orte, A., et al. (2012). Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059. doi: 10.1016/j.cell.2012.03.037
Dauer, W., and Prezedborski, S. (2003). Parkinson's disease: mechanism and models. Neuron 39, 889–909. doi: 10.1016/S0896-6273(03)00568-3
Davies, K. M., Bohic, S., Carmona, A., Ortega, R., Cottam, V., Hare, D. J., et al. (2014). Copper pathology in vulnerable brain regions in Parkinson's disease. Neurobiol. Aging 35, 858–866. doi: 10.1016/j.neurobiolaging.2013.09.034
Davies, K. M., Hare, D. J., Cottam, V., Chen, N., Hilgers, L., Halliday, G., et al. (2013). Localization of copper and copper transporters in the human brain. Metallomics 5, 43–51. doi: 10.1039/C2MT20151H
Davies, K. M., Mercer, J. F., Chen, N., and Double, K. L. (2016). Copper dyshomoeostasis in Parkinson's disease: implications for pathogenesis and indications for novel therapeutics. Clin. Sci. 130, 565–574. doi: 10.1042/CS20150153
De Ricco, R., Valensin, D., Dell'Acqua, S., Casella, L., Dorlet, P., Faller, P., et al. (2015a). Remote His50 acts as a coordination switch in the high-affinity N-terminal centered copper (II) site of α-synuclein. Inorg. Chem. 54, 4744–4751. doi: 10.1021/acs.inorgchem.5b00120
De Ricco, R., Valensin, D., Dell'Acqua, S., Casella, L., Hureau, C., and Faller, P. (2015b). Copper (I/II), α/β-Synuclein and Amyloid-β: menage à Trois? ChemBioChem 16, 2319–2328. doi: 10.1002/cbic.201500425
Dong, G., Chen, H., Qi, M., Dou, Y., and Wang, Q. (2015). Balance between metallothionein and metal response element binding transcription factor 1 is mediated by zinc ions (Review). Mol. Med. Rep. 11, 1582–1586. doi: 10.3892/mmr.2014.2969
Drew, S. C., Ling Leong, S., Pham, C. L., Tew, D. J., Masters, C. L., Miles, L. A., et al. (2008). Cu2+ binding modes of recombinant α-synuclein− insights from EPR spectroscopy. J. Am. Chem. Soc. 130, 7766–7773. doi: 10.1021/ja800708x
Dryanovski, D. I., Guzman, J. N., Xie, Z., Galteri, D. J., Volpicelli-Daley, L. A., Lee, V. M. Y., et al. (2013). Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 33, 10154–10164. doi: 10.1523/jneurosci.5311-12.2013
Ebadi, M., and Sharma, S. (2006). Metallothioneins 1 and 2 attenuate peroxynitrite-induced oxidative stress in Parkinson disease. Exp. Biol. Med. 231, 1576–1583. doi: 10.1177/153537020623100919
Ebadi, M., Brown-Borg, H., El Refaey, H., Singh, B. B., Garrett, S., Shavali, S., et al. (2005). Metallothionein-mediated neuroprotection in genetically engineered mouse models of Parkinson's disease. Brain Res. Mol. Brain Res. 134, 67–75. doi: 10.1016/j.molbrainres.2004.09.011
Espay, A. J., Schwarzschild, M. A., Tanner, C. M., Fernandez, H. H., Simon, D. K., Leverenz, J. B., et al. (2017). Biomarker-driven phenotyping in Parkinson's disease: a translational missing link in disease-modifying clinical trials. Mov. Disord. 32, 319–324. doi: 10.1002/mds.26913
Faller, P. (2010). Neuronal growth inhibitory factor (metallothionein-3): reactivity and structure of metal–thiolate clusters. FEBS J. 277, 2921–2930. doi: 10.1111/j.1742-4658.2010.07717.x
Finkelstein, D. I., Hare, D. J., Billings, J. L., Sedjahtera, A., Nurjono, M., Arthofer, E., et al. (2016). Clioquinol improves cognitive, motor function, and microanatomy of the Alpha-synuclein hA53T transgenic mice. ACS Chem. Neurosci. 7, 119–129. doi: 10.1021/acschemneuro.5b00253
Fitzgerald, M., Nairn, P., Bartlett, C. A., Chung, R. S., West, A. K., and Beazley, L. D. (2007). Metallothionein-IIA promotes neurite growth via the megalin receptor. Exp. Brain Res. 183, 171–180. doi: 10.1007/s00221-007-1032-y
Follett, J., Norwood, S. J., Hamilton, N. A., Mohan, M., Kovtun, O., Tay, S., et al. (2014). The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic 15, 230–244. doi: 10.1111/tra.12136
Halliwell, B. (2001). Role of free radicals in the neurodegenerative diseases. Drugs Aging 18, 685–716. doi: 10.2165/00002512-200118090-00004
Hart, R. G., Pearce, L. A., Ravina, B. M., Yaltho, T. C., and Marler, J. R. (2009). Neuroprotection trials in Parkinson's disease: systematic review. Mov. Disord. 24, 647–654. doi: 10.1002/mds.22432
Howells, C., West, A. K., and Chung, R. S. (2010). Neuronal growth-inhibitory factor (metallothionein-3): evaluation of the biological function of growth-inhibitory factor in the injured and neurodegenerative brain. FEBS J. 277, 2931–2939. doi: 10.1111/j.1742-4658.2010.07718.x
Hozumi, I. (2013). Roles and therapeutic potential of metallothioneins in neurodegenerative diseases. Curr. Pharm. Biotechnol. 14, 408–413. doi: 10.2174/1389201011314040004
Hozumi, I., Asanuma, M., Yamada, M., and Uchida, Y. (2004). Metallothioneins and neurodegenerative diseases. J. Health Sci. 50, 323–331. doi: 10.1248/jhs.50.323
Hozumi, I., Hasegawa, T., Honda, A., Ozawa, K., Hayashi, Y., Hashimoto, K., et al. (2011). Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 303, 95–99. doi: 10.1016/j.jns.2011.01.003
Jankovic, J. J. (2012). Current concepts in parkinson's disease and other movement disorders. Curr. Opin. Neurol. 25, 429–432. doi: 10.1097/WCO.0b013e3283550cdd
Jellinger, K. A. (2014). Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov. Disord. 29, 1720–1741. doi: 10.1002/mds.26052
Jenner, P., Morris, H. R., Robbins, T. W., Goedert, M., Hardy, J., Ben-Shlomo, Y., et al. (2013). Parkinson's disease – the debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J. Parkinson's Dis. 3, 1–11. doi: 10.3233/JPD-130175
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kägi, J. H., and Vallee, B. L. (1960). Metallothionein: a cadmium-and zinc-containing protein from equine renal cortex. J. Biol. Chem. 235, 3460–3465.
Karin, M., Haslinger, A., Holtgreve, H., Richards, R. I., Krauter, P., Westphal, H. M., et al. (1984). Characterization of DNA sequences through which cadmium and glucocorticoid hormones induce human metallothionein-IIA gene. Nature 308, 513–519. doi: 10.1038/308513a0
Karin, M., Herschman, H. R., and Weinstein, D. (1980). Primary induction of metallothionein by dexamethasone in cultured rat hepatocytes. Biochem. Biophys. Res. Commun. 92, 1052–1059. doi: 10.1016/0006-291X(80)90808-6
Kelly, E. J., and Palmiter, R. D. (1996). A murine model of Menkes disease reveals a physiological function of metallothionein. Nat. Genet. 13, 219–222. doi: 10.1038/ng0696-219
Kimura, T., and Itoh, N. (2008). Function of metallothionein in gene expression and signal transduction: newly found protective role of metallothionein. J. Health Sci. 54, 251–260. doi: 10.1248/jhs.54.251
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., et al. (2002). Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791.
Kitada, T., Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Klaassen, C. D. (1981). Induction of metallothionein by adrenocortical steroids. Toxicology 20, 275–279. doi: 10.1016/0300-483X(81)90034-2
Kobayashi, S., Okada, T., and Kimura, M. (1985). Effects of dexamethasone on metallothionein induction by Zn, Cu and Cd in chang liver cells. Chem. Biol. Interact. 55, 347–356. doi: 10.1016/S0009-2797(85)80141-1
Krizkova, S., Masarik, M., Majzlik, P., Kukacka, J., Kruseova, J., Adam, V., et al. (2010). Serum metallothionein in newly diagnosed patients with childhood solid tumours. Acta Biochim. Pol. 57, 561.
Kukacka, J., Vajtr, D., Huska, D., Prusa, R., Houstava, L., Samal, F., et al. (2006). Blood metallothionein, neuron specific enolase, and protein S100B in patients with traumatic brain injury. Neuroendocrinol. Lett. 27, 116–120.
Kurasaki, M., Okabe, M., Saito, S., and Suzuki-Kurasaki, M. (1998). Copper metabolism in the kidney of rats administered copper and copper-metallothionein. Am. J. Physiol. 274, F783–F790.
Kurkowska-Jastrzebska, I., Litwin, T., Joniec, I., Ciesielska, A., Przybyłkowski, A., Członkowski, A., et al. (2004). Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson's disease. Int. Immunopharmacol. 4, 1307–1318. doi: 10.1016/j.intimp.2004.05.006
Landowski, L. M., Pavez, M., Brown, L. S., Gasperini, R., Taylor, B. V., West, A. K., et al. (2016). Low-density lipoprotein receptor-related proteins in a novel mechanism of axon guidance and peripheral nerve regeneration. J. Biol. Chem. 291, 1092–1102. doi: 10.1074/jbc.M115.668996
Lees, A. J., Hardy, J., and Revesz, T. (2009). Parkinson's disease. Lancet 373, 2055–2066. doi: 10.1016/S0140-6736(09)60492-X
Lewis, K. E., Chung, R. S., West, A. K., and Chuah, M. I. (2012). Distribution of exogenous metallothionein following intraperitoneal and intramuscular injection of metallothionein-deficient mice. Histol. Histopathol. 27, 1459–1470. doi: 10.14670/HH-27.1459
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O'Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Lutsenko, S., Barnes, N. L., Bartee, M. Y., and Dmitriev, O. Y. (2007). Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011–1046. doi: 10.1152/physrev.00004.2006
Magaki, S., Raghavan, R., Mueller, C., Oberg, K. C., Vinters, H. V., and Kirsch, W. M. (2007). Iron, copper, and iron regulatory protein 2 in Alzheimer's disease and related dementias. Neurosci. Lett. 418, 72–76. doi: 10.1016/j.neulet.2007.02.077
Manso, Y., Adlard, P. A., Carrasco, J., Vašák, M., and Hidalgo, J. (2011). Metallothionein and brain inflammation. JBIC J. Biol. Inorg. Chem. 16, 1103. doi: 10.1007/s00775-011-0802-y
Manso, Y., Carrasco, J., Comes, G., Meloni, G., Adlard, P. A., Bush, A. I., et al. (2012a). Characterization of the role of metallothionein-3 in an animal model of Alzheimer's disease. Cell. Mol. Life Sci. 69, 3683–3700. doi: 10.1007/s00018-012-1047-9
Manso, Y., Carrasco, J., Comes, G., Adlard, P. A., Bush, A. I., and Hidalgo, J. (2012b). Characterization of the role of the antioxidant proteins metallothioneins 1 and 2 in an animal model of Alzheimer's disease. Cell. Mol. Life Sci. 69, 3665–3681. doi: 10.1007/s00018-012-1045-y
Manso, Y., Comes, G., López-Ramos, J. C., Belfiore, M., Molinero, A., Giralt, M., et al. (2016). Overexpression of metallothionein-1 modulates the phenotype of the Tg2576 mouse model of Alzheimer's disease. J. Alzheimers Dis. 51, 81–95. doi: 10.3233/JAD-151025
Margoshes, M., and Vallee, B. L. (1957). A cadmium protein from equine kidney cortex. J. Am. Chem. Soc. 79, 4813–4814. doi: 10.1021/ja01574a064
Marques, O., and Outeiro, T. F. (2012). Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 3, e350. doi: 10.1038/cddis.2012.94
Mayo, K. E., and Palmiter, R. D. (1981). Glucocorticoid regulation of metallothionein-I mRNA synthesis in cultured mouse cells. J. Biol. Chem. 256, 2621–2624.
McCarty, M. F. (2006). Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Med. Hypotheses 67, 251–269. doi: 10.1016/j.mehy.2006.01.013
Meloni, G., and Vašák, M. (2011). Redox activity of α-synuclein–Cu is silenced by Zn 7-metallothionein-3. Free Radic. Biol. Med. 50, 1471–1479. doi: 10.1016/j.freeradbiomed.2011.02.003
Meloni, G., Sonois, V., Delaine, T., Guilloreau, L., Gillet, A., Teissié, J., et al. (2008). Metal swap between Zn7-metallothionein-3 and amyloid-β–Cu protects against amyloid-β toxicity. Nat. Chem. Biol. 4, 366–372. doi: 10.1038/nchembio.89
Méndez-Armenta, M., Villeda-Hernández, J., Barroso-Moguel, R., Nava-Ruíz, C., Jiménez-Capdeville, M. E., and Ríos, C. (2003). Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicol. Lett. 144, 151–157. doi: 10.1016/S0378-4274(03)00199-1
Michael, G. J., Esmailzadeh, S., Moran, L. B., Christian, L., Pearce, R. K., and Graeber, M. B. (2011). Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 12, 295–305. doi: 10.1007/s10048-011-0294-5
Miotto, M. C., Rodriguez, E. E., Valiente-Gabioud, A. A., Torres-Monserrat, V., Binolfi, A., Quintanar, L., et al. (2014). Site-specific copper-catalyzed oxidation of α-synuclein: tightening the link between metal binding and protein oxidative damage in Parkinson's disease. Inorg. Chem. 53, 4350–4358. doi: 10.1021/ic4031377
Montes, S., Rivera-Mancia, S., Diaz-Ruiz, A., Tristan-Lopez, L., and Rios, C. (2014). Copper and copper proteins in Parkinson's disease. Oxid. Med. Cell. Longev. 2014:147251. doi: 10.1155/2014/147251
Noe, E., Marder, K., Bell, K. L., Jacobs, D. M., Manly, J. J., and Stern, Y. (2004). Comparison of dementia with Lewy bodies to Alzheimer's disease and Parkinson's disease with dementia. Mov. Disord. 19, 60–67. doi: 10.1002/mds.10633
Nordberg, M. (1998). Metallothioneins: historical review and state of knowledge. Talanta 46, 243–254. doi: 10.1016/s0039-9140(97)00345-7
Nussbaum, R. L. (2017). Genetics of synucleinopathies. Cold Spring Harb. Perspect. Med. doi: 10.1101/cshperspect.a024109. [Epub ahead of print].
Otsuka, F. (2004). Transcriptional regulation of the metallothionein genes. J. Health Sci. 50, 332–335. doi: 10.1248/jhs.50.332
Paik, S. R., Shin, H. J., Lee, J. H., Chang, C. S., and Kim, J. (1999). Copper (II)-induced self-oligomerization of α-synuclein. Biochem. J. 340, 821–828. doi: 10.1042/bj3400821
Pall, H. S., Blake, D. R., Gutteridge, J. M., Williams, A. C., Lunec, J., Hall, M., et al. (1987). Raised cerebrospinal-fluid copper concentration in Parkinson's disease. Lancet 330, 238–241. doi: 10.1016/S0140-6736(87)90827-0
Palmiter, R. D., Findley, S. D., Whitmore, T. E., and Durnam, D. M. (1992). MT-III, a brain-specific member of the metallothionein gene family. Proc. Natl. Acad. Sci. U.S.A. 89, 6333–6337. doi: 10.1073/pnas.89.14.6333
Pedersen, M. Ø., Larsen, A., Stoltenberg, M., and Penkowa, M. (2009). Cell death in the injured brain: roles of metallothioneins. Prog. Histochem. Cytochem. 44, 1–27. doi: 10.1016/j.proghi.2008.10.002
Petro, A., Sexton, H. G., Miranda, C., Rastogi, A., Freedman, J. H., and Levin, E. D. (2016). Persisting neurobehavioral effects of developmental copper exposure in wildtype and metallothionein 1 and 2 knockout mice. BMC Pharmacol. Toxicol. 17:55. doi: 10.1186/s40360-016-0096-3
Pinto, M., Nissanka, N., Peralta, S., Brambilla, R., Diaz, F., and Moraes, C. T. (2016). Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol. Neurodegener. 11:25. doi: 10.1186/s13024-016-0090-7
Pountney, D. L., Dickson, T. C., Power, J. H. T., Vickers, J. C., West, A. J., and Gai, W. P. (2011). Association of metallothionein-III with oligodendroglial cytoplasmic inclusions in multiple system atrophy. Neurotox. Res. 19, 115–122. doi: 10.1007/s12640-009-9146-6
Pountney, D. L., Kägi, J. H. R., and Vašák, M. (1995). “Metallothioneins,” in Handbook on Metal-Ligand Interaction in Biological Fluids, Vol. 1, ed G. Berthon (New York, NY: Marcel Dekker), 431–442.
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Pushkar, Y., Robison, G., Sullivan, B., Fu, S. X., Kohne, M., Jiang, W., et al. (2013). Aging results in copper accumulations in glial fibrillary acidic protein-positive cells in the subventricular zone. Aging Cell 12, 823–832. doi: 10.1111/acel.12112
Quaife, C., Hammer, R. E., Mottet, N. K., and Palmiter, R. D. (1986). Glucocorticoid regulation of metallothionein during murine development. Dev. Biol. 118, 549–555. doi: 10.1016/0012-1606(86)90025-4
Radford, R., Wong, M. B., and Pountney, D. L. (2014). “Neurodegenerative aspects of multiple system atrophy,” in Handbook of Neurotoxicity, Vol. 110, ed R. M. Kostrzewa (New York, NY: Springer Science+Business Media), 2157–2180.
Rcom-H'cheo-Gauthier, A. N., Osborne, S. L., Meedeniya, A. C. B., and Pountney, D. L. (2016). Calcium: Alpha-synuclein interactions in Alpha-Synucleinopathies. Front. Neurosci. 10:570. doi: 10.3389/fnins.2016.00570
Richards, R. I., Heguy, A., and Karin, M. (1984). Structural and functional analysis of the human metallothionein-IA gene: differential induction by metal ions and glucocorticoids. Cell 37, 263–272. doi: 10.1016/0092-8674(84)90322-2
Rofe, A. M., Philcox, J. C., Haynes, D. R., Whitehouse, M. W., and Coyle, P. (1992). Changes in plasma zinc, copper, iron, and hepatic metallothionein in adjuvant-induced arthritis treated with cyclosporin. Biol. Trace Elem. Res. 34, 237–248. doi: 10.1007/BF02783679
Rose, F., Hodak, M., and Bernholc, J. (2011). Mechanism of copper(II)-induced misfolding of Parkinson's disease protein. Sci. Rep. 1:11. doi: 10.1038/srep00011
Sadhu, C., and Gedamu, L. (1989). Metal-specific posttranscriptional control of human metallothionein genes. Mol. Cell. Biol. 9, 5738–5741. doi: 10.1128/MCB.9.12.5738
Sas, K., Robotka, H., Toldi, J., and Vécsei, L. (2007). Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 257, 221–239. doi: 10.1016/j.jns.2007.01.033
Sawada, H., Kohno, R., Kihara, T., Izumi, Y., Sakka, N., Ibi, M., et al. (2004). Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes α-synuclein inclusions. J. Biol. Chem. 279, 10710–10719. doi: 10.1074/jbc.M308434200
Sharma, S., and Ebadi, M. (2014a). Significance of metallothioneins in aging brain. Neurochem. Int. 65, 40–48. doi: 10.1016/j.neuint.2013.12.009
Sharma, S., and Ebadi, M. (2014b). The Charnoly body as a universal biomarker of cell injury. Biomarkers Genomic Med. 6, 89–98. doi: 10.1016/j.bgm.2014.03.004
Sharma, S., Moon, C. S., Khogali, A., Haidous, A., Chabenne, A., Ojo, C., et al. (2013a). Biomarkers in Parkinson's disease (recent update). Neurochem. Int. 63, 201–229. doi: 10.1016/j.neuint.2013.06.005
Sharma, S., Rais, A., Sandhu, R., Nel, W., and Ebadi, P. M. (2013b). Clinical significance of metallothioneins in cell therapy and nanomedicine. Int. J. Nanomed. 8, 1477–1488. doi: 10.2147/IJN.S42019
Shastri, A., Bonifati, D. M., and Kishore, U. (2013). Innate immunity and neuroinflammation. Mediators Inflamm. 2013:342931. doi: 10.1155/2013/342931
Shin, J. H., Dawson, V. L., and Dawson, T. M. (2009). Snapshot: pathogenesis of Parkinson's Disease. Cell 139, 440.e1–440.e2. doi: 10.1016/j.cell.2009.09.026
Shiraga, H., Pfeiffer, R. F., and Ebadi, M. (1993). The effects of 6-hydroxydopamine and oxidative stress on the level of brain metallothionein. Neurochem. Int. 23, 561–566. doi: 10.1016/0197-0186(93)90104-D
Siddiqui, A., Rane, A., Rajagopalan, S., Chinta, S. J., and Andersen, J. K. (2016). Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson's disease are attenuated by restoration of PGC1alpha. Neurobiol. Dis. 93, 115–120. doi: 10.1016/j.nbd.2016.05.009
Sidhu, A., Wersinger, C., and Vernier, P. (2004). Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 18, 637–647. doi: 10.1096/fj.03-1112rev
Spillantini, M. G., and Goedert, M. (2000). The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 920, 16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x
Squitti, R., Siotto, M., Arciello, M., and Rossi, L. (2016). Non-ceruloplasmin bound copper and ATP7B gene variants in Alzheimer's disease. Metallomics 8, 863–873. doi: 10.1039/c6mt00101g
Stefanis, L. L. (2012). α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2:9399. doi: 10.1101/cshperspect.a009399
Thanvi, B. R., and Lo, T. C. (2004). Long term motor complications of levodpa: clinical features, mechanisms, and management strategies. Postgrad. Med. J. 80, 452–458. doi: 10.1136/pgmj.2003.013912
Tokuda, E., Watanabe, S., Okawa, E., and Ono, S. I. (2015). Regulation of intracellular copper by induction of endogenous metallothioneins improves the disease course in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics 12, 461–476. doi: 10.1007/s13311-015-0346-x
Uchida, Y. (2010). Molecular mechanisms of regeneration in Alzheimer's disease brain. Geriatr. Gerontol. Int. 10(Suppl. 1), S158–S168. doi: 10.1111/j.1447-0594.2010.00607.x
Valdinocci, D., Radford, R. A., Siow, S. M., Chung, R. S., and Pountney, D. L. (2017). Potential modes of intercellular α-Synuclein transmission. Int. J. Mol. Sci. 18:e469. doi: 10.3390/ijms18020469
Valensin, D., Dell'Acqua, S., Kozlowski, H., and Casella, L. (2016). Coordination and redox properties of copper interaction with α-synuclein. J. Inorg. Biochem. 163, 292–300. doi: 10.1016/j.jinorgbio.2016.04.012
Van der Perren, A., Van den Haute, C., and Baekelandt, V. (2015). Viral vector-based models of Parkinson's disease. Curr. Top. Behav. Neurosci. 22, 271–301. doi: 10.1007/7854_2014_310
Vasudevaraju, P., Jyothsna, T., Shamasundar, N. M., Rao, S. K., Balaraj, B. M., Rao, K. S. J., et al. (2010). New evidence on iron, copper accumulation and zinc depletion and its correlation with DNA integrity in aging human brain regions. Indian J. Psychiatry 52, 140. doi: 10.4103/0019-5545.64590
Vekrellis, K., Rideout, H. J., and Stefanis, L. (2004). Neurobiology of α-synuclein. Mol. Neurobiol. 30, 1–21. doi: 10.1385/MN:30:1:001
Villar-Piqué, A., da Fonseca, T. L., Sant'Anna, R., Szegö, É. M., Fonseca-Ornelas, L., Pinho, R., et al. (2016). Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity. Proc. Natl. Acad. Sci. U.S.A. 113, E6506–E6515. doi: 10.1073/pnas.1606791113
Vieira, B. D., Radford, R. A., Chung, R. S., Guillemin, G. J., and Pountney, D. L. (2015). Neuroinflammation in multiple system atrophy: response to and cause of α-synuclein aggregation. Front. Cell. Neurosci. 9:437. doi: 10.3389/fncel.2015.00437
Waldron, K. J., Rutherford, J. C., Ford, D., and Robinson, N. J. (2009). Metalloproteins and metal sensing. Nature 460, 823–830. doi: 10.1038/nature08300
Wan, O. W., and Chung, K. K. (2012). The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson's disease. PLoS ONE 7:e38545. doi: 10.1371/journal.pone.0038545
West, A. K., Hidalgo, J., Eddins, D., Levin, E. D., and Aschner, M. (2008). Metallothionein in the central nervous system: roles in protection, regeneration and cognition. Neurotoxicology 29, 489–503. doi: 10.1016/j.neuro.2007.12.006
West, A. K., Stallings, R., Hildebrand, C. E., Chiu, R., Karin, M., and Richards, R. I. (1990). Human metallothionein genes: structure of the functional locus at 16q13. Genomics 8, 513–518. doi: 10.1016/0888-7543(90)90038-v
Wirdefeldt, K., Adami, H., Cole, P., Trichopoulos, D., and Mandel, J. (2011). Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 26:1. doi: 10.1007/s10654-011-9581-6
Wong, Y. C., and Krainc, D. (2017). α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Yates, D. (2011). Neurodegenerative disease: α-synuclein gets a new look. Nat. Rev. Neurosci. 12, 550. doi: 10.1038/nrn3106
Yavich, L., Tanila, H., Vepsäläinen, S., and Jäkälä, P. (2004). Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 24, 11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004
Keywords: Parkinson's disease, α-synuclein, copper, multiple system atrophy, dementia with lewy bodies, metallothionein
Citation: Okita Y, Rcom-H'cheo-Gauthier AN, Goulding M, Chung RS, Faller P and Pountney DL (2017) Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 11:114. doi: 10.3389/fnins.2017.00114
Received: 28 October 2016; Accepted: 22 February 2017;
Published: 04 April 2017.
Edited by:
Federico Benetti, Scuola Internazionale di Studi Superiori Avanzati, ItalyReviewed by:
Éva M. Szegö, University of Göttingen, GermanyNicola B. Mercuri, University of Rome Tor Vergata, Italy
Copyright © 2017 Okita, Rcom-H'cheo-Gauthier, Goulding, Chung, Faller and Pountney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dean L. Pountney, ZC5wb3VudG5leUBncmlmZml0aC5lZHUuYXU=