 Denise M. Inman
Denise M. Inman Mohammad Harun-Or-Rashid
Mohammad Harun-Or-Rashid- Department of Pharmaceutical Sciences, Northeast Ohio Medical University, Rootstown, OH, USA
Axons can be several orders of magnitude longer than neural somas, presenting logistical difficulties in cargo trafficking and structural maintenance. Keeping the axon compartment well supplied with energy also presents a considerable challenge; even seemingly subtle modifications of metabolism can result in functional deficits and degeneration. Axons require a great deal of energy, up to 70% of all energy used by a neuron, just to maintain the resting membrane potential. Axonal energy, in the form of ATP, is generated primarily through oxidative phosphorylation in the mitochondria. In addition, glial cells contribute metabolic intermediates to axons at moments of high activity or according to need. Recent evidence suggests energy disruption is an early contributor to pathology in a wide variety of neurodegenerative disorders characterized by axonopathy. However, the degree to which the energy disruption is intrinsic to the axon vs. associated glia is not clear. This paper will review the role of energy availability and utilization in axon degeneration in glaucoma, a chronic axonopathy of the retinal projection.
Metabolic Vulnerability in Glaucoma
Glaucoma is the leading cause of irreversible blindness worldwide (Tham et al., 2014). It blinds through the dysfunction and degeneration of retinal ganglion cell (RGC) axons that carry visual information from the eye to the brain (Calkins and Horner, 2012; Casson et al., 2012). There is emerging evidence for a critical role for energy management in the axon degeneration observed in this disease. In our work, we have been using the DBA/2J mouse model of glaucoma, an inbred strain that develops increased intraocular pressure (IOP) secondary to an iris pigment dispersion disease (John et al., 1998; Anderson et al., 2002). IOP is the main modifiable risk factor in primary open angle glaucoma, and lowering IOP is the mainstay of treatment. Increased IOP in the DBA/2J (D2) leads to optic neuropathy and eventual RGC death in a manner that emulates human patients with the most common forms of glaucoma, with pathology developing slowly over time (John et al., 1998), and occurring in contiguous retinal regions (Jakobs et al., 2005). IOP elevations in D2 mice are similar in scale to human patients, and the magnitude of these increases correlates with axon degeneration (Inman et al., 2006).
The potential role of energy availability and utilization in glaucoma became apparent through optic nerve physiology experiments. We measured compound action potential (CAP), the summation of all action potentials after stimulation, in freshly isolated D2 optic nerves. CAP amplitude decreased significantly by 6 months of age (Baltan et al., 2010), prior to measurable axon structural pathology (Inman et al., 2006) or axon transport deficit (Dengler-Crish et al., 2014) in this model. The reduction in elicited neural activity was inversely correlated with IOP elevation in the D2; the higher the IOP exposure within an age group, the lower the CAP amplitude (Baltan et al., 2010). Notably, there was no difference in CAP latency or duration in the D2 at 6 or 10 months, and no impact of the K+ channel blocker 4-aminopyridine. Both observations argue against significant changes in myelination as observed in multiple sclerosis-associated optic neuropathy in humans (Chan, 2002). However, when D2 optic nerves were subjected to oxygen-glucose deprivation and then allowed to recover, nerves from 6-month-old mice in the high IOP group demonstrated impaired recovery when compared to their low IOP companions. By 10 months of age, however, D2 optic nerves could not sustain recovery from oxygen-glucose deprivation in any mice, independent of IOP history. These results suggested that both IOP and age influence energy reserves in this pathology. To corroborate energy depletion, ATP measurements from D2 optic nerve showed significantly decreased ATP in the high IOP groups within each age, and significantly depleted ATP levels when comparing 6 to 10-month-olds within IOP group (see Figure 1 for summary) (Baltan et al., 2010). The low ATP and clear lack of energy reserve in the D2 optic nerve with increased IOP suggested glaucoma-associated metabolic dysfunction. Strikingly, the metabolic dysfunction in the axon occurs quite early in the D2, prior to overt structural changes in the optic nerve. This suggests that resolution of the metabolic dysfunction could prevent axon degeneration and preserve visual function.
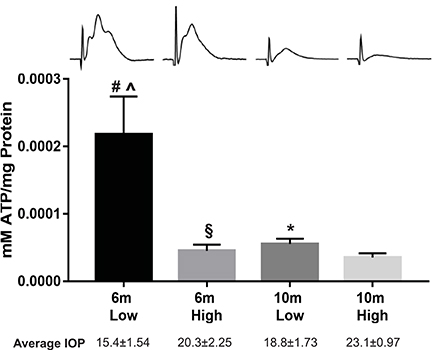
Figure 1. Optic nerve ATP levels in the DBA/2J model of glaucoma at 6 and 10 months of age, separated into low and high intraocular pressure (IOP) groups. Optic nerves were rapidly isolated then flash frozen in liquid nitrogen until assay. Upon thaw, the optic nerve was cut into small pieces while in 10% HClO4, then homogenized by sonication, and centrifuged at 4,500 rpm for 10 min at 4°C. The supernatant was collected, neutralized with 30 μl of 2.5 M KOH, then centrifuged at 14,000 rpm for 10 min at 4°C. The precipitate was removed, and supernatant kept on ice. Total ATP concentration was measured using the ATP bioluminescence assay kit (Roche), based on the ATP dependence of luciferase catalyzed oxidation of luciferin. Samples were diluted and mixed with the luciferase reagent and then absorbance was measured at 560 nm. Blank values were subtracted from the raw data, and ATP concentrations were calculated from a log–log plot of the standard curve data and normalized by the protein concentration. The values are expressed as millimoles of ATP per milligrams of protein. There was a significant difference in ATP levels in low vs. high IOP groups (t-test, #p = 0.012 for 6 m; *p = 0.049 for 10 m), and a significant difference in the ATP levels across 6 m vs. 10 m at low (t-test, ∧p = 0.014) and high (t-test, §p = 0.028) IOP. Above: representative compound action potential traces from each group; below: average IOP for each group. Adapted from Baltan et al. (2010).
The observations of a potential metabolic dysfunction in glaucoma raise a number of interesting possibilities about the mechanism of pathophysiology. Several fundamental concepts have implications for how glaucoma could be managed and what might be fruitful therapeutic approaches to combat the disease: (1) how energy is produced and utilized in axons, (2) how energy is associated with axon degeneration, (3) the nature of axon degeneration in glaucoma, and (4) how axon degeneration might be prevented. As discussed below, the entry points for the final destruction of the axon vary between neurodegenerative disorders but have much in common beyond initiation. The focus here is energy depletion, which arises from a number of circumstances, including ischemia, mitochondrial dysfunction, age-related changes in the NAD+-sirtuin-PGC-1α axis, loss of axon support factors, and activation of axon degeneration factors. Below we review the current understanding of energy production in white matter and possible intervention points in glaucoma and, by extension, other chronic axonopathies.
Energy in Axons
Energy is produced both within axons and around them using substrates delivered by the circulation and glial cells. Axons obtain most of their energy from ATP through oxidative phosphorylation in mitochondria. Glial cells that contact axons (oligodendrocytes and astrocytes) are capable of providing energy substrates to axons as well. This energy is necessary for axon function.
Within Axons: Oxidative Phosphorylation and Glycolysis
The central nervous system prefers glucose as its substrate; glucose enters the brain (and retina and optic nerve) through glucose transporters. Neurons express GLUT3, a high-affinity glucose transporter (Maher et al., 1991), while astrocytes and endothelial cells express GLUT1 (Garcia-Caceres et al., 2016). Both GLUT3 and GLUT1 can be upregulated through insulin signaling (Simpson et al., 2008). Glucose is converted through glycolysis to pyruvate; the resultant pyruvate is fed into the Krebs cycle in mitochondria for the generation of ATP. Glucose in axons can also be used to produce reducing equivalents like NADPH via the pentose-phosphate pathway in order to maintain redox balance (Stincone et al., 2015). Neurons are highly oxidative and dependent on their mitochondria. ATP produced by mitochondria in the average ON axon can comfortably sustain the observed firing rate of action potentials, the maintenance of the resting potential, and maintenance processes, according to a computational model that drew on published values for rat CNS (Harris and Attwell, 2012). The model indicated mitochondria could support all optic nerve axons using roughly 50% of the ATP generation capacity. There is only a calculated ATP shortfall for small axons, those below 0.8 μm in diameter (Harris and Attwell, 2012). In humans, normal optic nerve axon mean diameter ranges from 0.72 ± 0.07 μm (Mikelberg et al., 1989), to 0.96 ± 0.07 μm (Quigley et al., 1988), and 1.00 ± 0.06 μm (Jonas et al., 1990). Even within these ranges, a significant portion of the axons in the human optic nerve are likely to experience ATP shortfall if the model accurately predicts energy availability. The same is true for mouse, with a frequency distribution of optic nerve fiber diameter that peaks at 0.7–0.9 μm; 50.4% of axons are 0.9 um in diameter or less (Honjin et al., 1977), suggesting that there could also be a mitochondrial-derived ATP shortfall in a large subset of axons. The energy sufficiency calculations also depend upon the axon having enough access to glucose.
In general, there are adequate concentrations of GLUT3, the primary glucose transporter in neuronal membranes (Maher et al., 1991), to provide the necessary glucose levels to axons (Harris and Attwell, 2012). These estimates assume glucose diffusion to the middle of an internode quickly (see Figure 2); rat internodes average 240 μm (Ransom et al., 1991) while internode lengths in mouse ON average 110 μm (O'Meara et al., 2013). This internode length suggests glucose will readily diffuse; however, the predicted ATP shortfall in axons below 0.8 μm in diameter indicates a potential vulnerability of small axons. Small axons have light myelination, shorter internodes, slow conduction rates (Hursh, 1939), and more sparse mitochondria (Perge et al., 2009; Ohno et al., 2011). It has recently been demonstrated that glucose transporters are upregulated in response to activity in mouse myelinated axons (Saab et al., 2016), a potential solution to the possible lack of glucose in small axons. There are two caveats to this interpretation, however: The glucose transporters were upregulated in oligodendrocytes, not axons; and protein upregulation is not a dynamic response to momentary need. Axons likely rely on alternatives to glucose for maintenance of function.
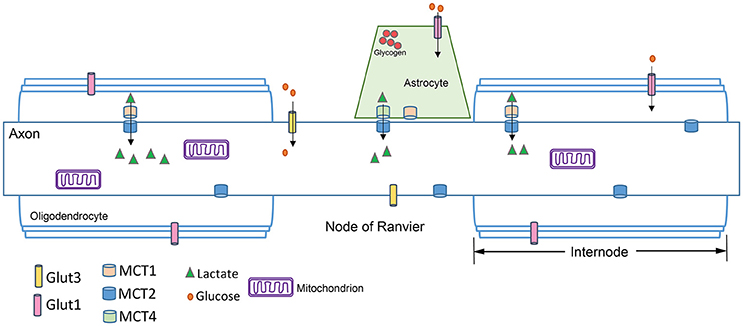
Figure 2. Axon schematic showing the distribution of glucose transporters (GLUT), and monocarboxylate transporters (MCT) in the axon, astrocytes and oligodendrocytes. GLUTs move glucose into the cell, while MCTs transport monocarboxylates such as lactate, pyruvate, and β-hydroxybutyrate. Glia express MCT1 and MCT4, while axons express MCT2. The kinetics of the MCTs are such that MCT1 is best suited for lactate export and MCT2 for lactate uptake because of its higher affinity for lactate (see text). The node of Ranvier is concentrated with ion channels (not shown) necessary for saltatory conduction. Astrocytes can interface with the axon at the node of Ranvier. Oligodendrocytes myelinate the axon; the region between nodes of Ranvier is the internode.
Around Axons: Glial-Derived Energy
Axons may be supplied with metabolic intermediates from astrocytes and oligodendrocytes to address potential shortfalls from glucose transport and diffusion. Several early studies demonstrated metabolic coupling between neuronal and glial compartments (Hamberger and Hyden, 1963; Pevzner, 1971, 1972). Glia-to-neuron energy substrate transfer was also revealed in early work on metabolic coupling between glia and neurons in the honeybee retina, indicating that glial cells released alanine that was taken up and metabolized by photoreceptor neurons (Tsacopoulos and Magistretti, 1996). In the mammalian CNS, glia mainly transfer monocarboxylates—L-lactate, pyruvate, or ketone bodies—rather than alanine, to neurons (Brown et al., 2003). The initial support for energy transfer from glia to axons came from studies of optic nerve explants that could maintain CAPs for approximately 30 min in the absence of glucose (Stys et al., 1991; Wender et al., 2000). Further study of optic nerve showed that CAP maintenance was dependent on transport of lactate (Wender et al., 2000; Tekkök et al., 2005). The critical evidence supporting intracellular metabolic substrate transfer is based on a number of observations, including: (1) the ability of glial cells to release L-lactate from their glycogen stores (Dringen et al., 1993), (2) the ability of neurons to take up L-lactate (Bouzier-Sore et al., 2003), (3) the cellular distribution of lactate dehydrogenase enzyme that converts lactate to pyruvate (Bittar et al., 1996), and (4) the cellular localization of monocarboxylate transporters (MCTs) within the CNS (Koehler-Stec et al., 1998; Pierre et al., 2002). Mounting evidence from different experimental studies indicates that oligodendrocytes and astrocytes significantly contribute to metabolic substrate transfer for axonal metabolic support.
Lactate, a glycolytic end product, can be converted to pyruvate for utilization in the Krebs cycle (Figure 3). Evidence from many different experiments indicates that lactate is an efficient oxidative energy substrate for neurons (Pellerin et al., 1998). The conversion of lactate to pyruvate is catalyzed by lactate dehydrogenase (LDH), and requires NAD+ as a cofactor. Cells that produce lactate glycolytically, like astrocytes, are abundant in one LDH isozyme, while cells that use lactate as a substrate, including neurons, are enriched in another (Tsacopoulos and Magistretti, 1996). Lactate is transported between glia and neurons through the monocarboxylate transporters (MCTs). MCTs are bidirectional extracellular membrane channels that transport lactate, pyruvate, and ketone bodies across the membrane depending on their concentration gradient (Pierre and Pellerin, 2005). The MCTs work by first binding a proton then one molecule of lactate; the transporter undergoes a conformational change that releases the proton and the lactate on the other side of the membrane (Dubinsky and Racker, 1978). MCT isoforms differ in their substrate-binding affinities and kinetics, making MCT1 best suited for lactate export and MCT2 for lactate uptake. Perhaps unsurprisingly, MCT1 is expressed preferentially in oligodendrocytes and astrocytes (Lee et al., 2012), while MCT2 is expressed in neurons and axons (Pierre et al., 2002; Simpson et al., 2007), and MCT4 is expressed in astrocytes (Rafiki et al., 2003). MCT2 has a much greater affinity than MCT1 or MCT4 for lactate and pyruvate (Simpson et al., 2007).
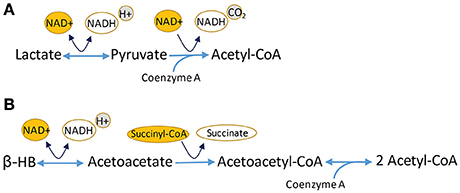
Figure 3. Conversion of ketone bodies, including lactate and β-hydroxybutyrate (β-HB), intermediates that can be utilized in Krebs cycle. (A) Lactate conversion to pyruvate is catalyzed by lactate dehydrogenase and requires NAD+. The reaction is reversible. Pyruvate is oxidized to acetyl-CoA, using NAD+ and giving off NADH and CO2. (B) β-HB can be utilized in Krebs cycle after a multi-step conversion reaction that results in the formation of two molecules of acetyl-CoA for every molecule of β-HB.
Glial Glycolysis Supports Axons
Glial cells and neurons form a metabolic unit in the CNS, with glial cells providing energy substrates that ensure neural function. In Drosophila, neurons die when glial cell glycolysis is impaired, indicating that glycolysis is essential to neuron survival (Volkenhoff et al., 2015). The metabolic unit is supported by communication among cells; axon firing can lead to upregulation of glucose transporter-1 in oligodendrocytes in mouse (Saab et al., 2016). Once fortified with additional glucose, oligodendrocytes release lactate, thereby maintaining energy supplies to myelinated axons in the optic nerve. Moreover, evidence from multiple sclerosis patient tissue suggests that oligodendrocytes in low glucose conditions decrease their lactate release (Rone et al., 2016), possibly imperiling myelin maintenance but allowing cell survival. This suggests that glia are responsive to axon activity through greater release of metabolic substrate, but they have limits.
Glycogen
Astrocytes have glycogen stores that are capable of being mobilized to provide glucose or lactate to axons (Pellerin et al., 1998; Erlichman et al., 2008). Mobilized glycogen allows for continued optic nerve CAP generation when mouse optic nerves are deprived of glucose (Wender et al., 2000). In vitro evidence suggests that lactate derived from glycogen breakdown is preferentially exported (Sickmann et al., 2005). The glycogen stores get broken down into lactate, which gets transferred to and used by axons, as shown by CAP failure when lactate transporters are blocked in periods of aglycemia in mouse optic nerve (Wender et al., 2000; Tekkök et al., 2005). Blocking glycogen breakdown accelerated CAP failure in optic nerve subjected to high-frequency stimulation (Brown et al., 2005). Furthermore, neuronal function can be maintained and neuronal death averted during hypoglycemia by increasing astrocytic glycogen stores in rat brain (Suh et al., 2007). These data demonstrate that lactate from glial glycogen stores can and will be used by axons under stress. In brain parenchyma, astrocytes have unique morphological structures and phenotypical features, which ideally position them to sense neuronal activity (Lundgaard et al., 2014) and respond with the suitable metabolic substrate to their surrounding microenvironment (Rouach et al., 2008). Is something similar at work in white matter, between astrocytes and axons? Gap junctions known to connect astrocytes and oligodendrocytes (Orthmann-Murphy et al., 2007) could facilitate movement of glucose or lactate from astrocyte to oligodendrocyte, thereby providing lactate or other substrates directly through the axon-facing MCT-1 on oligodendrocytes. Alternatively, astrocytes are known to contact nodes of Ranvier (Serwanski et al., 2017) and may deliver metabolic substrates directly.
Energy and Axon Degeneration
Axon degeneration is a complex process that can initiate from different neuronal compartments, e.g., the soma, terminal boutons, or the axon itself. Regardless, the initial stages of neurodegeneration often manifest in the axon compartment. Axon degeneration mechanisms can share three common elements: intra-axonal Ca++ dysregulation, axon transport deficits, and mitochondrial dysfunction (Fagiolini et al., 1997; Stys, 2004). Both axon transport deficits and mitochondrial dysfunction are energy-related pathologies. For example, molecular motors require energy for axon transport. Earlier work on this subject posited that mitochondrial dysfunction would preclude axon transport until it was demonstrated that molecular motors can use extra-mitochondrial sources of energy (Zala et al., 2013). This may dissociate axon transport deficit from mitochondria with regards to ATP demand; however, pathological change in mitochondrial dynamics can alter transport through other mechanisms (Crish and Calkins, 2011; Misko et al., 2012). Metabolic dysfunction could contribute to axon transport deficit, independent of mitochondria, through lack of substrate delivery to the axon. Anterograde axon transport deficit is known to be an important alteration in the pathology of glaucoma (Crish et al., 2010; Dengler-Crish et al., 2014). As will be discussed below, axon transport is required to maintain axon structure and function as well as traffic survival factors for the neuron (see Vrabec and Levin, 2007; Milde et al., 2013).
Mitochondria and Degeneration
Investigation into the mechanisms of axon degeneration underscores the central role of energy availability and mitochondria in keeping the axon compartment alive. For example, injured axons in zebrafish rapidly degenerate without mitochondria (Campbell et al., 2014). In C. elegans, mitochondrial targeting to axons can protect them from degeneration (Rawson et al., 2014). Similarly, releasing mitochondria from their syntaphilin tethers in axotomizedcultured cortical mouse axons can rescue energy deficits (Zhou et al., 2016), demonstrating the importance of ATP homeostasis to axon survival after injury. Some have determined mitochondria to be the site of axon protection (Avery et al., 2012), though preventing mitochondrial transport into axons prior to injury slowed, but did not stop, the course of axon degeneration in a Drosophila axotomy model (Kitay et al., 2013). Depolarizing mitochondria does not increase the rate of Wallerian degeneration in superior cervical ganglion axons in vitro, indicating that degeneration-associated Ca++ increases are extra-mitochondrial (Loreto et al., 2015). Axon regeneration, quite separate from degeneration, in mouse RGC axons (Cartoni et al., 2016) and C. elegans nerve cord axons (Han et al., 2016), does require mitochondria.
Glial Lactate Critical for Axon Survival
Recent experimental data demonstrates that MCTs are critical for maintaining axon health and integrity. Mice with 50% lower than normal MCT1 expression developed axonopathy in the optic nerve, suggesting that axons are quite sensitive to reduction in the provision of lactate from oligodendrocytes (Lee et al., 2012). Neurons in organotypic cultures could not survive without MCT1, and axons without it degenerated. MCT1 was also shown to be reduced in patients with amyotrophic lateral sclerosis (ALS) and in a mouse model of ALS (Lee et al., 2012). Deletion of MCT1 in oligodendrocytes substantially reduces the availability of local energy metabolites to the axon (Lee et al., 2012), potentially affecting the axonal energy-dependent processes such as axonal transport (Nave, 2010).
The crucial role of MCT1, a mover of energy substrate from the oligodendroglial compartment to axons, in axon function and survival spurred investigation into the regulation of energy exchange between oligodendrocyte and neuron (Saab et al., 2016). Stimulation of oligodendroglial NMDA receptors in response to axonal glutamate release upregulates glucose transporter GLUT1 in the oligodendrocyte. Increased uptake of glucose in the myelin as a result of GLUT1 expression ensures greater glucose delivery to the oligodendrocytes at a time when the axon is engaged in high spiking activity, thereby establishing a path to greater lactate release by the oligodendrocyte to the myelinated axon. Axons recovered from oxygen-glucose deprivation better with lactate than with glucose, indicating the oligodendrocytes were not merely passing along glucose to the axons (Saab et al., 2016).
A multi-pronged approach to supplying axons with energy, as outlined in the section on energy described above, is necessary because of the dire consequences of axonal energy depletion. Axons expend the bulk of their energy budget on the Na+-K+ ATPase which moves these ions across the axolemma during and after action potential firing (Ritchie, 1967). Without sufficient energy for Na+-K+ ATPase function, axons experience Na+ and Ca++ overload and then structural decline when Ca++-dependent proteases such as calpain (and potentially others) lay waste to the cytoskeleton, disrupting axon transport, physiology, and other vital axon maintenance processes. Extracellular Ca++ influx is both necessary and sufficient for axon degeneration (Court and Coleman, 2012); it is part of a “final common pathway” for various mechanisms of axon degeneration such as developmental axon pruning and Wallerian degeneration (Yang et al., 2013); reviewed in (Stirling and Stys, 2010; Tsutsui and Stys, 2013; Conforti et al., 2014).
Energy and Mechanisms of Axon Degeneration: Wallerian
A role for energy in the mechanism of axon degeneration emerged from research into the mechanism of axon protection in the Wallerian degeneration-slow (Wlds) mouse. Wlds mice carry a naturally occurring mutation that greatly slows axon degeneration, allowing axons that are separated from the cell body to maintain structural and conduction integrity for 2 weeks after transection in mice (Lunn et al., 1989). In a series of follow-up studies, it was demonstrated that the slow degeneration phenotype was intrinsic to the axon (Perry et al., 1990a) and was controlled by a single autosomal dominant gene (Perry et al., 1990b; Lyon et al., 1993). It was ultimately determined that the Wlds gene encodes a chimeric protein, a fusion of the N-terminal 70 amino acids of ubiquitination factor E4B (Ube4b) with nicotinamide mononucleotide adenylyltransferase-1 (NMNAT1) (Conforti et al., 2000; Mack et al., 2001). Subsequent research activity has indicated that the NMNAT1 portion of the fusion protein is primarily responsible for the Wlds phenotype, as shown in in dorsal root ganglion culture (Araki et al., 2004). It is the ATP-dependent activity of NMNAT that converts nicotinamide mononucleotide (NMN) to NAD+ (Figure 4). NAD+ is an essential coenzyme in four steps of the Krebs cycle, an essential cofactor in the GAPDH step of glycolysis, and the conversion of lactate into pyruvate requires NAD+, reducing it to NADH. Determining that NMNAT activity contributes to axon survival after axotomy placed energy as a central issue to the mechanism of axon degeneration.
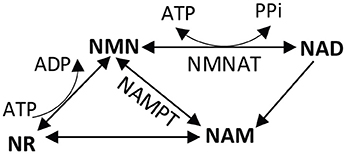
Figure 4. Nicotinamide dinucleotide (NAD+) salvage pathway. NAD+ can be synthesized de novo using tryptophan (not shown), or through a salvage pathway using vitamin precursors niacin (vitamin B3), nicotinamide (NAM), or nicotinamide riboside (NR). In the salvage pathway, NMNAT catalyzes the formation of NAD+ from NMN and ATP. The reverse reaction converts NAD+ into NMN, giving off ATP. NMN, nicotinamide mononucleotide; NAD, nicotinamide dinucleotide; NMNAT, nicotinamide mononucleotide adenylyltransferase; NAMPT, nicotinamide phosphoribosyltransferase.
Given the critical role of NMNAT in axon survival after injury, logic would dictate that the product of NMNAT activity, NAD+, is the currency of axon protection. Mammals use a variant of vitamin B3, nicotinamide, as the precursor for NAD+ synthesis (Galli et al., 2013) (Figure 4). Roughly 50% of the pyridine nucleotide NAD+ pool is in mitochondria, and 50% is in the cytoplasm in cultured neurons (Alano et al., 2010). NADH and NAD+ cannot themselves be transported into or out of the mitochondria. Cytosolic NAD+ depletion has been shown to block glucose utilization, consistent with its requirement in glycolysis. NAD+ is regenerated in cells by the activity of lactate dehydrogenase, through the electron transport chain in mitochondria, or the movement of malate and/or aspartate transport across the inner mitochondrial membrane. The major consumers of NAD+ in a cell include poly (ADP-ribose) polymerases (PARPs), and sirtuins (class III histone deacetylases) (Blander and Guarente, 2004; Belenky et al., 2007). Strikingly, eliminating two major NAD+ consuming enzymes (PARP1 and CD38) does not prevent axon degeneration in mouse sciatic nerve transection (Sasaki et al., 2009). In vivo, NMNAT1 overexpression was not sufficient for axon protection, but NMNAT3 targeted to the mitochondrial matrix protected sciatic nerves after axotomy (Yahata et al., 2009). NMNAT3 likely regenerates NAD+ in the mitochondria for cellular energetics (Lau et al., 2009). Since the mitochondrial NAD+ pool is segregated from the cytoplasmic, NMNAT3-based protection from degeneration implicates mitochondrial dysfunction in the energy depletion that contributes to axon degeneration. The mitochondria isolated from these axons had normal respiratory chain components but were making more ATP (Yahata et al., 2009). The centrality of NAD+ in energy production and utilization suggest a mechanism linking loss of mitochondrial function to axon degeneration (Di Lisa and Ziegler, 2001).
Mitochondria isolated from Wlds -expressing, protected mouse axons exhibit increased ATP production (Yahata et al., 2009) and enhanced Ca++ buffering (Avery et al., 2012). Importantly, Wlds can ameliorate decreases in ATP after axon injury in vitro (Wang et al., 2005). There are multiple NMNATs, each one localized to a cellular compartment. NMNAT1 is nuclear, NMNAT2 is cytoplasmic and associated with Golgi-derived vesicles, and NMNAT3 is found in the mitochondria (Ali et al., 2013). Each of these NAD+-synthesizing enzymes has been investigated individually and found to impact axon survival. Mutations in NMNAT1 cause Leber's congenital amaurosis, a retinal degeneration disease (Falk et al., 2012). Targeting NMNAT1 expression to the cytoplasm then inducing ocular hypertension protects RGCs from death (Zhu et al., 2013). The protective effect of a cytoplasmic version of NMNAT1 depended upon mitochondrial axon transport (Fang et al., 2014a). NMNAT2, however, has turned out to be the essential axon protection factor; when not transported to the axon, a degenerative program ensues (Gilley and Coleman, 2010). Mutations that extend the half-life of NMNAT2 increase its ability to delay axon degeneration, beyond that observed with the Wlds mutation (Milde et al., 2013). The Wlds protein can protect a cut axon for up to 4 h in vitro, while NMNAT2 depletion within 5 h commits the axon to degeneration (Gilley and Coleman, 2010; Wang et al., 2015). The longer half-life of Wlds over NMNAT2 may explain its ability to radically extend axon survival.
Identification of NMNAT2 as an axon protection factor has been accompanied by identification of two proteins that appear to be axon death factors (see Figure 5). PHR1, an E3 ubiquitin ligase, promotes axon degeneration after injury (Babetto et al., 2013) by influencing the turnover of NMNAT2. PHR1 deletion delays Wallerian degeneration. SARM1, a Toll-like receptor adaptor protein tethered to the mitochondria (Kim et al., 2007; Summers et al., 2014), contributes to axon degeneration such that its deletion significantly delays Wallerian degeneration after nerve transection (Osterloh et al., 2012). The mechanism of that delay includes increased NAD+ synthesis in the axon (Gerdts et al., 2015). SARM1 activation rapidly breaks down NAD+ after injury. SARM1 is required for activation of a post-injury MAPK cascade that disrupts energy homeostasis through ATP depletion in the axon (Yang et al., 2015). Cut neurites from SARM1−/− mice showed higher extracellular acidification rate (glycolysis) and higher maximal respiration than wildtype (Godzik and Coleman, 2015), suggesting a complex picture of the role of oxidative phosphorylation and glycolysis in maintaining ATP levels in injured axons. Isolated neurites show just a partial view of what is likely happening in vivo, where oligodendrocytes and astrocytes are capable of contributing to the overall energy scheme.
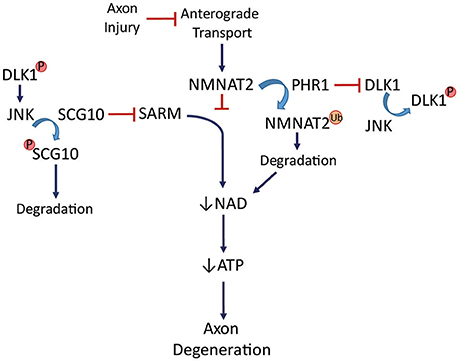
Figure 5. NMNAT2 is transported into the axon to support/protect axon integrity. PHR1, an E3 ubiquitin ligase, can ubiquitinate NMNAT2, promoting its degradation. This leads to decreases in NAD+, low ATP levels, and eventual axon degeneration. SARM activation also leads to axon degeneration through loss of NAD+ and ATP. Ubiquitination-deubiquitination of PHR1 keeps DLK1 in balance until it is phosphorylated by JNK. DLK1 can then activate JNK, which phosphorylates SCG10, leading to its degradation. Axonal degeneration can be blocked by SCG10. Loss of SARM or PHR1 delays degeneration. See text for details.
The prospect of axon survival has become quite complex with the further identification of elements capable of contributing to axon degeneration. Dual leucine zipper kinase (DLK), a MAP3K upstream of JNK, is necessary and sufficient for RGC death after optic nerve crush (Welsbie et al., 2013). Inhibiting the JNK cascade in DLK1−/− sensory neuron cultures delays Wallerian degeneration (Miller et al., 2009), suggesting that DLK1 acts through JNK to promote axon degeneration. There is some disagreement regarding the role of DLK1 in RGCs. Axon degeneration was not delayed after optic nerve crush in DLK1-deficient mice (Fernandes et al., 2014). While this suggests that kinases other than DLK1 may be capable of JNK activation for axon degeneration, a separate study showed that phosphorylation of c-Jun, a target of JNK, was dependent on DLK1 after optic nerve crush (Watkins et al., 2013). DLK1 was significantly upregulated in post-crush optic nerve and governed both the pro-apoptotic and pro-regenerative responses of RGCs (Watkins et al., 2013). JNK works in the commitment phase of the degeneration cascade since inhibiting it 3 h after axotomy of mouse or Drosophila axons was not sufficient to prevent axon loss (Miller et al., 2009). A protein phosphorylated early after axotomy in the distal segments of sensory nerve, SCG10, is a microtubule binding protein and JNK substrate whose phosphorylation targets it for degradation. SCG10 can delay degeneration of crushed mouse optic nerve if overexpressed (Shin et al., 2012). Loss of SCG10 alone, however, does not lead to axon degeneration. JNK regulates SCG10 turnover, linking it to DLK1. Interestingly, SCG10 is lost after axotomy even when NMNAT overexpression prevents axon degeneration (Shin et al., 2012).
After axotomy, PHR1 contributes to NMNAT2 degradation while SARM1 is similarly contributing to NAD+ depletion (Figure 5). DLK1 activates JNK, which ensures loss of SCG10. These events commit an axon to the degeneration cascade, and it is not yet known how the various elements interact or the precise timing. There are most certainly modifiers of the cascade as well as variations by mode of injury and type of neuron, as will be discussed below. Caveats to the general degeneration scheme include the fact that some of these mechanisms have been worked out in vitro, in isolated neurites. While this has established that Wlds, SARM, and DLK-1 are cell-autonomous in their impact on axon degeneration, the various unexplained aspects of degeneration mechanisms are being assiduously investigated.
Glaucomatous Axonopathy: What Form Does It Take?
The success of axon protection strategies that have emerged from investigation into the Wlds mutant applied to glaucoma may enable a determination about the mechanisms of axon degeneration in this disorder. In Wallerian degeneration, axons degenerate caudal to a lesion and in an asynchronous way over a population of axons within a nerve (Waller, 1850). The nature of the injury dictates its course since Wlds axons degenerate anterogradely (from the injury toward the synapse) after transection but retrogradely (from the synapse back toward the injury) after crush injury in peripheral nerve (Beirowski et al., 2005). Wallerian degeneration can be triggered even without physical axonal injury (Ferri et al., 2003; Gilley and Coleman, 2010). In distal axonopathy, for which there is evidence in mouse models of glaucoma (Schlamp et al., 2006; Crish et al., 2010), an axon degenerates from the distal-most point and moves retrogradely. It may be the case that “dying back,” or distal axonopathy is not separable from Wallerian degeneration in glaucoma, especially if glaucoma injury resembles a crush injury with retrograde degeneration. Support for shared mechanisms of distal axonopathy and Wallerian degeneration comes from models of Charcot-Marie-Tooth disease, a distal axonopathy that is significantly slowed by the expression of the Wlds (Meyer zu Horste et al., 2011). Wlds can also reduce the number of axon varicosities, enlarged portions of axons containing protein aggregates that are hallmarks of most neurodegenerative diseases. Despite this, Wlds cannot delay all axon degeneration (Vande Velde et al., 2004), and there may be age-related limits to axon degeneration delay (Samsam et al., 2003). Wlds has been shown to effectively delay the optic neuropathy of glaucoma in the mouse (Howell et al., 2007; Beirowski et al., 2008), though it did not prevent or delay RGC death. A comparison of pre-laminar and laminar axons (see Figure 6) in the optic nerve head of D2 mice with glaucoma show a significant decrease in axon number from pre-lamina to lamina, suggesting that axon damage initiates in the pre-lamina and distal axons degenerate (Howell et al., 2007). This would be consistent with an anterograde Wallerian degeneration as observed in transected axons (Beirowski et al., 2005). However, axon endbulbs can be observed throughout the optic nerve in the D2 mouse model of glaucoma (Buckingham et al., 2008; Crish et al., 2010; Dengler-Crish et al., 2014), suggesting a retrograde degeneration course that would be more in alignment with the response to crush injury (Beirowski et al., 2005). Consistent with this, one analysis of D2 optic nerve showed a distal to proximal pattern of degeneration that preceded loss of the corresponding RGCs (Schlamp et al., 2006). Without the ability to monitor the axons along their course, it cannot be ruled out that the end bulbs and axon fragmentation observed beyond the optic nerve head are distal portions of axons with an initiating injury at the optic nerve head.
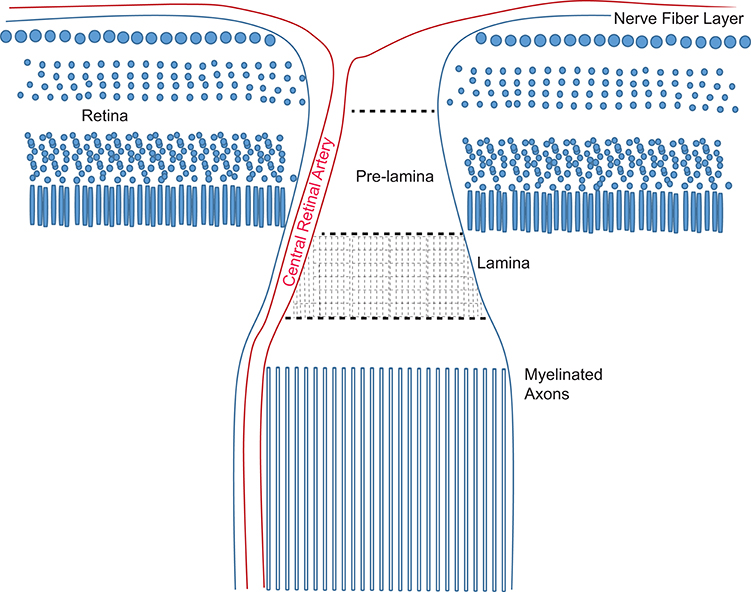
Figure 6. Schematic of the optic nerve head in the mouse. Retinal ganglion cell axons converge at the optic disk and then make a ~90° turn to form the optic nerve. The pre-lamina region is formed from the fasciculated axons as they traverse the retina. Columns of astrocytes maintain the fasciculated bundles of unmyelinated axons through the lamina. Beyond the lamina, retinal ganglion axons are myelinated by oligodendrocytes.
Neurotrophin deprivation has been considered an important potential mechanism of RGC decline and death. Manipulating receptor tyrosine kinase (Trk) signaling concomitant with growth factor provision has proven to be supportive of RGC survival (Cheng et al., 2002; Lebrun-Julien et al., 2009; Weber and Harman, 2013). Developmental axon pruning is an example of axon degeneration that can be triggered by loss of growth factor support from target areas. Axon pruning may share downstream mechanisms of degeneration with post-injury Wallerian degeneration, but the commitment phase differs. Wlds cannot prevent pruning in mice or Drosophila (Hoopfer et al., 2006). Since Wlds mice show normal CNS circuitry instead of a gross overabundance of unpruned connections, this is not unexpected. Evidence indicates there may be two mechanisms by which axons get pruned, one that is caspase-dependent and the other requiring loss of NAD+; the two may work together or run parallel to eliminate unneeded circuitry (Schoenmann et al., 2010). Growth factor deprivation initiates activation of caspases; Wlds and cytoplasmic NMNAT1, even SARM1 deletion (Gerdts et al., 2013), can protect against nerve growth factor (NGF) withdrawal-induced axon degeneration. DLK-1, after trophic deprivation, is JNK dependent and contributes to both axon degeneration and cell body apoptosis (Ghosh et al., 2011). Interestingly, DLK1 knockout in a optic nerve crush model did not protect RGC axon structure or function as measured by CAP amplitude (Fernandes et al., 2014), suggesting that degeneration in these axons is governed differently than those in development (Hoopfer et al., 2006) and potentially other sensory systems (Miller et al., 2009). Others determined that deficiency in DLK1 protected RGC somas from apoptosis and proximal axons from degeneration after optic nerve crush (Watkins et al., 2013). The pro-apoptotic factor BAX, when deleted in mice of the D2 background, preserved RGC somas but axons degenerated (Libby et al., 2005). These data indicate that degeneration of the optic nerve in the D2 model of glaucoma may be caspase-independent. The preponderance of evidence suggests that glaucomatous degeneration occurs through Wallerian degeneration rather than trophic deprivation-related pathways, though the initial kinase signaling has yet to be determined.
Energy and Glaucoma-Specific Axon Degeneration
RGCs Vulnerable to Energy Depletion
RGCs are not uniformly susceptible to cell death in glaucoma (Li et al., 2006; Della Santina et al., 2013), and whether specific axons are particularly vulnerable to degeneration is not known. In the cat optic nerve, 50% of the axons are in the category of small and lightly myelinated, and are therefore potentially susceptible to ATP shortfall. It is believed these axons correspond to γRGCs (Williams and Chalupa, 1983) which are the W cells by functional type. W cells include on- or off-tonic, on- or off-phasic, and on-off phasic cells (Fukuda et al., 1984; Watanabe et al., 1993). Axon caliber correlates relatively well with soma size (Huxlin and Goodchild, 1997; Coombs et al., 2006), suggesting that the sizable group comprising the slowest conducting axons should include RGCs with small to medium cell somas. A number of studies have determined that OFF transient RGCs show early morphological and functional changes in the microbead injection and laser photocoagulation models of glaucoma (Della Santina et al., 2013; El-Danaf and Huberman, 2015; Ou et al., 2016). These OFF transients are believed to be αRGCs, those with large somas and extensive dendritic arbors; however, there are a number of RGC morphological types with arbor stratification at 50% of inner plexiform layer (IPL) depth typical of OFF transient cells (Della Santina and Ou, 2016) that could have small to medium cell somas and axons possibly in the smaller ranges. The D2 CAP data (Figure 1) and OFF transient data are not necessarily in opposition since the traces clearly show that CAPs are lost from all axon calibers (Baltan et al., 2010); large αRGCs can be preferentially lost among other cells that also succumb. Interestingly, after optic nerve transection in mouse, αRGCs preferentially survive, including OFF transients; when treated with osteopontin and IGF-1 or by downregulating PTEN, regeneration only occurs in αRGCs (Duan et al., 2015). Though αRGCs are a diverse group, it would seem unlikely that cells particularly susceptible to raised IOP would be resistant to axon transection.
Mitochondria in Glaucoma
To understand the mechanism of metabolic dysfunction in D2 optic nerve, the mitochondria would be a reasonable starting point for investigation since axons obtain most of their energy from ATP through oxidative phosphorylation. One very relevant study determined that mitochondria isolated from patients resistant to optic neuropathy—exposed to high IOP but absent pathology—showed greater levels of systemic “mitochondrial efficiency,” including higher rates of ADP phosphorylation, hyperpolarized mitochondrial membrane potential, and enhanced Ca++ buffering capacity compared to control and glaucoma patient groups (Lascaratos et al., 2015). These observations single out the mitochondrial function as a potential biomarker for individuals susceptible to increased IOP. Our electron microscopic analysis of mitochondria in the D2 optic nerve is consistent with lower mitochondrial support in glaucoma patients. We observed a linear relationship between mitochondrial volume and axon volume in DBA/2J-Gpnmb+ (control strain that does not develop glaucoma) and pre-pathological D2 optic nerve. That relationship is abolished in the transport dysfunctional D2 optic nerve, for which we observed axon volumes that were not matched by appropriate mitochondrial volumes (Kleesattel et al., 2015). Interestingly, CAP amplitude measures suggest that the third peak of the CAP trace, corresponding to the slowest conducting axons, are lost earliest and in the greatest numbers, and in accordance to magnitude of IOP exposure, in the D2 mouse model of glaucoma (see Figure 1; Baltan et al., 2010).
What ties metabolism to susceptible RGCs? In short, very little is directly known about the specifics of OFF transient cell energy homeostasis, though mitochondrial distribution could be a factor. Whereas mitochondria take up greater axon volume in unmyelinated axons, lightly myelinated axons in mouse (Ohno et al., 2011) and guinea pig have slightly fewer mitochondria than predicted (Perge et al., 2009). Mitochondria in the D2 optic nerve are significantly smaller and possess reduced cristae with aging and increased IOP (Coughlin et al., 2015); this supports widespread fission of these organelles, as has been observed by others (Ju et al., 2008). Smaller mitochondria with reduced cristae have less machinery for oxidative phosphorylation, and therefore, lower energetic capacity. This would spell particular trouble if the optic nerve were ever to experience glucose shortage (for example, if glucose transporters were downregulated) because functional mitochondria are required for an axon to survive on lactate. Lactate conversion to pyruvate bypasses glycolysis. The resultant pyruvate is converted to acetyl-CoA for use in Krebs cycle (Figure 3). The intermediates produced there are used to establish the electron transport chain and the proton motive force for ATP Synthase (Complex V) and ATP production. Hence, survival on lactate requires functional mitochondria.
Unfortunately for axon survival, poorly functioning mitochondria are not being efficiently recycled in the D2 optic nerve (Coughlin et al., 2015). By not being replaced, these mitochondria can contribute to metabolic vulnerability by producing lower levels of ATP and comparatively more reactive oxygen species (ROS). Mitochondrial biosynthetic proteins such as PGC-1α decrease with age, including in the retina of D2 mice (Guo et al., 2014). Low PGC-1α levels likely limit mitochondrial biogenesis. With no prospect of generating new mitochondria, the lack of recycling of malfunctioning organelles is likely a survival mechanism.
Investigating the impact of upregulating mitophagy in glaucoma could resolve whether maintaining compromised mitochondria can ensure continued, albeit weakened, axon function. Some insight can be gleaned from a study in which autophagy was inhibited in glaucoma using 3-methyladenine (Seglen and Gordon, 1982). The authors observed significant axon degeneration after inhibiting autophagy, and axon protection when promoting autophagy through rapamycin treatment (Kitaoka et al., 2013). Conflicting results have accompanied those examining the role of autophagy in glaucoma, with autophagy induction detrimental to RGC soma survival in an ocular hypertension glaucoma model (Park et al., 2012), but rapamycin-induced autophagy protective of RGC somas after axotomy (Rodriguez-Muela and Boya, 2012).
Glial Cells
As outlined above, astrocytes and oligodendrocytes can exert significant control over the availability and utilization of energy in the optic nerve. Analyses of optic nerve head and axonal glia in glaucoma to date have focused on morphological change (Lampert et al., 1968; Dai et al., 2012; Sun and Jakobs, 2012; Lye-Barthel et al., 2013; Bosco et al., 2016; Cooper et al., 2016), or cell loss. In the D2 optic nerve, astrocyte hypertrophy occurred early in the disease process and was maintained; oligodendrocytes were lost only after axon loss (Son et al., 2010). Alternatively, a model of laser induced ocular hypertension led to significant oligodendrocyte loss within 1 week of IOP elevation (Nakazawa et al., 2006). Oligodendrocyte loss could be ameliorated with an antibody against TNFα, suggesting a role for microglial-associated inflammation in the loss of myelin. Specific attributes of oligodendrocytes in glaucoma have not been investigated, though NG2+ cells proliferate and generate new oligodendrocytes after axon degeneration in the optic nerve (Son et al., 2010). In considering what is understood about the other macroglial cell in the optic nerve during glaucoma, the relationship between astrocyte hypertrophy and metabolic support of axons is unknown. These knowledge gaps provide many fertile areas of investigation for the metabolic role of optic nerve glia in glaucomatous neurodegeneration.
Addressing Energy Failure in Glaucoma
Glaucoma is an asynchronous degeneration of the optic nerve. Does axon degeneration occur from loss of protection factors or activation of death factors, or is it both? Where and when does energy dysregulation occur? How does one support axons before they fail? One approach to prevent axon degeneration is to develop ways to target cells that are likely to succumb. As noted above, specific RGCs and compartments seem particularly susceptible. As research refines the susceptible populations, targeting strategies can also become more sophisticated, though certain protection strategies may show positive consequences to targeting all RGCs. Aging remains an important risk factor, as suggested by the significant decreases in ATP with age (Figure 1) as well as degree of IOP exposure. Various energy-related approaches have been used to prevent axon degeneration in models of neurodegeneration, including providing creatine, manipulating members of the NAD+-sirtuin-PGC1α axis, and upregulating the NMNATs. Strategies used to protect against axon degeneration in glaucoma in particular consist of providing glucose, upregulating NMNAT1 and NMNAT3, providing ketone bodies, and inhibiting histone deacetylases (HDACs).
Glucose
Protection from glaucoma-induced optic neuropathy can occur by increasing energy substrates such as glucose. In a rat laser photocoagulation model of glaucoma, induction of hyperglycemia 4 days prior to ocular hypertension was shown to decrease axon loss in the optic nerve by more than 50% and increase ganglion cell survival also by roughly 50% (Ebneter et al., 2011). While glial activation was decreased in the optic nerve, retinal glial showed comparable levels of reactivity (microglia and astrocytes/Müller glia). A caveat to this study included the relatively brief study period (2 weeks) (Ebneter et al., 2011). Longer periods of hyperglycemia (6 weeks to 3 months) after ocular hypertension did not show neuroprotection in the retina (Kanamori et al., 2004a), though a distinction between neuronal and glial cell death was not made. A critical issue with increasing blood glucose levels is that hyperglycemia does not necessarily translate into greater glucose availability to the CNS without matching levels of high affinity glucose transporters such as GLUT3, or GLUT1. Whereas GLUT1 appears to be upregulated by NMDA-R activation (Saab et al., 2016), insulin can promote translocation of GLUT1 and GLUT3 to the cell surface (Uemura and Greenlee, 2006). In chronic glaucoma models, the likelihood of NMDA-R activation sufficient to upregulate GLUT1 seems unlikely because RGCs generally fire at low rates (more than 90% fire at 10 Hz or less) (Koch et al., 2004), and axons are being lost. The ectopic glutamate release observed in the optic nerve head in one glaucoma model (Fu and Sretavan, 2012) would not be directed at oligodendrocytes because axons in the nerve head are unmyelinated, but might its target be laminar astrocytes in an attempt to upregulate glucose transporters? Episcleral cauterization to initiate ocular hypertension in rats led to increased insulin and insulin-like growth factor receptor levels, concomitant with phosphorylated Akt, in retina in the acute post-injury period (Kanamori et al., 2004b). In the R28 rat retina neural cell line subjected to serum deprivation, insulin has been shown to be a retinal neuron survival factor by acting through the PI3K-Akt signaling pathway to decrease caspase-3 activation (Barber et al., 2001). It is unknown if glucose transporters were upregulated by these insulin increases. Traumatic brain injury, known to result in an 8-fold increase in extracellular lactate (Nilsson et al., 1990), led to an increase in neural (GLUT3), but not glial (GLUT1) glucose transporters (Hamlin et al., 2001). This would be consistent with increased need for energy in the neural compartment.
Glycogen
Astrocytes are the primary storage location for glycogen (Cataldo and Broadwell, 1986). Glycogen is broken down to glucose, but studies have shown that glycogen mobilization in astrocytes provides lactate, not glucose, to the extracellular milieu (Dringen et al., 1993). During periods of aglycemia, astrocytic glycogen is rapidly depleted, able to sustain function for no more than a few minutes (Brown, 2004). Increasing glycogen stores, though, can maintain axon firing longer during periods of hypoglycemia in rat brain (Suh et al., 2007). Astrocytes form the glia lamina of the optic nerve head in rodents (Figure 6), and these cells undergo profound morphological change with ocular hypertension (Sun et al., 2009; Dai et al., 2012; Bosco et al., 2016; Cooper et al., 2016). Of note, the optic nerve head glia impacted by glaucoma no longer contact the basal lamina of the nerve (Dai et al., 2012), the ultrastructural location of glycogen granules (Brown, 2004). This suggests astrocyte glycogen might be a source of metabolic vulnerability in the glaucomatous optic nerve due to possible depletion. Increasing glycogen stores, however, may not make a significant difference to energy homeostasis in a chronic disease like glaucoma because of its rapid depletion. Glycogen has other potential roles though, such as support of glutamatergic neurotransmission (Obel et al., 2012). Glycogen production through glycogen synthase is regulated by three kinases, including glycogen synthase kinase 3 (GSK-3). GSK-3 in particular has probable implications for glaucoma through its targets and its promiscuity; GSK-3 has more predicted substrates than any other kinase (Linding et al., 2007). GSK-3 phosphorylation regulates transcription factors such as NF-κB, STAT3, Fos/Jun AP-1, p53 (Beurel et al., 2015); histone deacetylases; and proteins like the microtubule binding tau (Mandelkow et al., 1992), critical to Alzheimer's disease and neurodegenerative disease pathology (Stamer et al., 2002; Mandelkow et al., 2003). It is as yet unknown whether a connection exists between astrocyte glycogen production and neurodegenerative disease pathology through GSK-3.
Creatine
Ischemia is also postulated to occur as a result of poor blood flow at the optic nerve head, as observed in glaucoma patients (Schwartz, 1994; Grunwald et al., 1998); it also occurs in models of acute IOP increase (Sun et al., 2010). ATP turnover in cells is fast, but more so in tissues subjected to ischemia. ATP supply is maintained by a pool of phosphocreatine that exists at sites of high energy consumption because phosphocreatine can be converted by creatine kinase into ATP + creatine more quickly than ATP can be generated. Creatine is neuroprotective in many models of hypoxia, including for cerebral ischemia when delivered intercerebroventricularly (Lensman et al., 2006), and after experimental stroke when the creatine is in a formulation that extends half-life (Perasso et al., 2009). Pre-treating cortical axons with creatine prevented ischemia-induced damage as well as alleviating ATP depletion (Shen and Goldberg, 2012). This protection was independent of glial cells. Nevertheless, protection from ischemia corroborated previous studies demonstrating that creatine is not effective when delivered post-ischemia injury (Lensman et al., 2006; Shen and Goldberg, 2012). It may be the case that sufficient levels of creatine could not be delivered in a timely manner to counteract the depolarization and subsequent energy depletion in ischemia.
NMNATs
Similar to studies in sensory neurons, NMNATs have significant impact on RGCs and their axons. Embryonic mice (E18.5) nullizygous for NMNAT2 showed truncated RGC axons; the axons did not reach the optic chiasm, and there was no optic tract. RGC bodies appeared normal, with proximal axons that formed the optic nerve (Gilley et al., 2013). These results suggested a developmental role for NMNAT2 in axon extension. NMNAT3 upregulation in two different mouse models of glaucoma exerted significant protection of axons that could be reversed with treatment by 3-methyladenine, a PI3K inhibitor that reduces autophagy (Kitaoka et al., 2013). Cytoplasmic NMNAT1 upregulation protected RGC somas and axons in models of retinal ischemia and ocular hypertension (Zhu et al., 2013). The studies did not thoroughly examine the mechanism of NMNAT protection, though the apparent connection to autophagy in the Kitaoka study suggests that recycling of all but the most basic cellular components can help maintain energy homeostasis in RGCs subjected to pressure-induced insult.
NAD+-Sirtuin-PGC-1α Axis Manipulation
NAD+ levels decrease with aging (Guarente, 2014), and various studies of NAD+ replenishment (either direct or through precursors) demonstrated better metabolic health and mitochondrial function in diet-related diabetes (Yoshino et al., 2011), and in muscle stem cells in aged mice (Zhang et al., 2016). After axotomy in vitro, exogenous application of NAD+ was sufficient to slow the degeneration (Wang et al., 2005). Thus, a successful strategy for slowing or preventing axon degeneration has been to indirectly provide energy (Araki et al., 2004; Chung et al., 2013). Wallerian degeneration, the most likely mechanism by which glaucomatous RGC axons die, can be halted through increased NAD+ biosynthesis (Araki et al., 2004) and the action of a serine protease inhibitor that prevents ATP decrease (Ikegami et al., 2004). Despite NAD+ being an obvious candidate effector for axon protection, overexpression of Wlds and NMNAT prevents axon degeneration but does not increase basal NAD+ levels (Mack et al., 2001; Araki et al., 2004), and NAD+ levels are not essential to axon preservation (Sasaki et al., 2009). If NAD+ levels are dispensable, then perhaps it is ATP levels that are crucial to axon protection. Reducing axon ATP causes irreversible axon damage (Shen et al., 2013).
NMNAT overexpression has had some success in models of glaucoma (Kitaoka et al., 2013), though if energy support is the goal, a very promising candidate is the NAD+ precursor nicotinamide riboside (Figure 4). Nicotinamide riboside (NR), when orally delivered to humans and mice, exhibits superior bioavailability and pharmacokinetics than either nicotinamide or nicotinic acid. NR significantly increases NMN, NAM, and NAD+ levels in humans by the fourth dose of a daily 1,000 mg intake, and maintains those increases up to 24 h after the seventh dose (Trammell et al., 2016).
Sirtuins
The sirtuins, class III histone deacetylases (HDACs), have had variable impact on degeneration in the CNS, their activation effective at delaying neurodegeneration in some situations, but not all (Conforti et al., 2007). Indirect maintenance of NAD+ pools through HDAC inhibition has been demonstrated in neurodegenerative diseases (Langley et al., 2005). In a transgenic mouse model of Alzheimer's disease, nicotinamide treatment inhibited SIRT1 activity, reduced phosphorylated tau, and restored cognitive ability (Green et al., 2008). As was discussed above, PARP1 and SIRT1 are enzymes that require NAD+ for activity. PARP1 inhibition or elimination did not improve axon regeneration after optic nerve transection in mice (Wang et al., 2016), though treatment started 3 days after injury and there is only a few hours between injury and commencement of Wallerian degeneration.
Resveratrol, a SIRT1 activator, has been utilized as a tool to probe the complexities of SIRT1 and degeneration. Resveratrol delayed Wallerian degeneration in sensory neuron cultures and after sciatic nerve crush (Calliari et al., 2013). The degeneration delay could be blocked by eliminating an endogenous SIRT1 inhibitor, DBC-1. DBC-1 knockout nerves degenerated just as quickly as wildtype if NAD+ was not present, indicating that NAD+ was necessary for full SIRT1 activity and subsequent axon protection (Calliari et al., 2013). Resveratrol also induced phosphorylation of AMPK, a master regulator of energy homeostasis (Dasgupta and Milbrandt, 2007), which when activated, stimulates glucose uptake in the brain (Hardie et al., 2012).
Resveratrol increased mitochondrial number and improved mitochondrial function primarily through SIRT1 deacetylation of PGC-1α and subsequent increased PGC-1α activity in mice (Lagouge et al., 2006). These effects were observed in skeletal muscle and adipose tissue. In the CNS, resveratrol was found to increase brain lactate production and limit movement of radiolabeled pyruvate through Krebs cycle, thereby limiting oxidative phosphorylation (Rowlands et al., 2015). Is limiting oxidative phosphorylation a viable approach for preventing axon degeneration? The variable impact (positive and negative) of SIRT1 on Wallerian degeneration indicates the likelihood that the timing and overall energy state of the system dictate the response to SIRT1 activation. Blocking oxidative phosphorylation would limit ROS production, though blocking mitochondrial function, especially if axons are being supported by glial-derived lactate, would seem to be a risky approach.
SIRT1 manipulation has not been wholly favorable in glaucoma-related models. SIRT1 overexpression promoted RGC survival after optic nerve crush in mouse, an effect that was more pronounced than resveratrol treatment (Zuo et al., 2013). SIRT1 knockout did not increase RGC death over wildtype mice after optic nerve crush, suggesting complex interactions of cell death mechanisms. Moreover, functional testing of the RGCs showed no difference among the SIRT1 activation, overexpression, or knockout groups, suggesting that axon degeneration occurred regardless of SIRT1 status (Zuo et al., 2013). The SIRT1 downregulation that resulted from 60 min of retina ischemia in mice could be reversed with once daily mangiferin treatment, a xanthonoid antioxidant (Kim et al., 2015). Resolution of the potential impact of manipulating these key regulators of energy homeostasis in glaucoma will require closer investigation in order to determine if they might be reasonable therapeutic targets.
Mitochondria
Upregulating mitochondrial biogenesis would provide more energy-producing organelles to susceptible RGCs, but may place stressors on protein synthesis machinery, especially with aging. Greater numbers of mitochondria would still need to get trafficked along the axon, which may pose a significant problem depending on the stage of glaucoma. One regulatory complex important for mitochondrial biogenesis, mTOR, was no different in tissues between patients with ocular hypertension (OHT) and those with glaucoma (Lascaratos et al., 2015) even though the OHT patients demonstrated increased mitochondrial efficiency and no optic neuropathy. No change in mTOR suggests that the advantage conferred to OHT patients resistant to optic neuropathy did not include a drive toward making more mitochondria. Rather than make more mitochondria, a better strategy could be to improve mitochondrial function. Compensation for impaired oxidative phosphorylation in OPA1-linked autosomal dominant optic atrophy (ADOA) patients has been associated with preservation of vision (van Bergen et al., 2011). Complex II and III activity was higher in ADOA patients with better vision, and these patients had higher respiration rate. Improving mitochondrial function has been the asserted outcome for many of the abovementioned axon protection strategies such as insulin-associated PI3K/Akt activation, SIRT1 activation, and antioxidant treatment. The variable outcomes with these treatments suggest more specific changes as opposed to generalized improved mitochondrial function are necessary.
Mitochondrial Biogenesis
If unhealthy mitochondria contribute to glaucoma pathogenesis, ridding RGCs of the ROS-producing organelles would have to be coupled with mitochondrial biogenesis. A number of other neurodegenerative disease models have manipulated mitochondrial recycling or production to neuroprotective effect. Increased DNA damage response through hyperactivation of PARP-1 [poly (ADP-ribose) polymerase-1] can inhibit mitophagy by disrupting the NAD+-sirtuin-PGC-1α axis in cells developed from xeroderma pigmentosum group A patients (Fang et al., 2014b). Mitophagy in this context can be rescued by upregulating mitochondrial uncoupling protein 2 (UCP2) or providing the NAD+ precursors nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN). Resumption of mitophagy reduced mitochondrial mass, but also reactive oxygen species in cultured hippocampal neurons subjected to H2O2 or oxygen glucose deprivation (Fang et al., 2014a). Upregulation of UCP2, a target of PGC-1α, increases ATP levels, reduces ROS and can protect neurons from mitochondrial Complex I disruption in a Drosophila model of Parkinson's disease (Islam et al., 2012). However, UCP2 overexpression in the G93A SOD1 mouse model of ALS led to lower levels of ROS but acceleration of the disease course (Peixoto et al., 2013). The proton conductance function of UCP2 requires the presence of specific activators that include fatty acids and ROS-derived alkenals (Brand and Esteves, 2005), the lack of which might explain why UCP2 overexpression is not always protective. Necdin, a protein that binds SIRT1, can stabilize PGC-1α and inhibit its ubiquitin-dependent degradation, thereby enhancing mitochondrial biogenesis (Hasegawa et al., 2016). The neuroprotection against mitochondrial-related injury afforded by necdin resembles that shown by PGC-1α upregulation (Mudò et al., 2012).
Ketone Bodies
Insight into the potential role of metabolism in the pathogenesis of glaucoma has emerged from several recent studies. In the EAAC1−/− model of normal tension glaucoma, every-other-day-fasting (EODF) led to significant protection of FluoroGold-positive retrogradely labeled RGCs, as well as the b wave of multi-focal ERG (Guo et al., 2016). EODF appeared to exert its effects through elevated β-hydroxybutyrate (β-HB), a ketone and endogenous histone deacetylase (HDAC) inhibitor. Increased β-HB levels led to increased histone acetylation that occurred concomitantly with increased mRNA expression of BDNF, bFGF, and catalase (Guo et al., 2016). β-HB can inhibit the class I HDACs (HDAC1, 2, 3, and 8) and at least one class II HDAC (HDAC4) (Shimazu et al., 2013). Ketogenic or medium chain fatty acid diets that lead to high levels of β-HB have long been effective at controlling seizure in children with epilepsy (Yellen, 2008), and have also demonstrated neuroprotection in models of Parkinson's disease (Yang and Cheng, 2010), Alzheimer's disease (Henderson et al., 2009), and ALS (Zhao et al., 2006). An important question is whether increasing ketone levels and subsequent manipulation of energy pathways is responsible for the observed neuroprotection, or if the results can be ascribed particularly to the inhibition of histone acetylation. Inhibiting HDACs has been mostly effective at limiting damage in models of glaucoma. For example, inhibiting HDAC3 protected RGCs from death after optic nerve crush (Schmitt et al., 2014), and HDAC inhibition with valproic acid protected RGCs from death and preserved pERG amplitude in a rat model of ocular hypertension (Alsarraf et al., 2014). A broad HDAC inhibitor, trichostatin A, when delivered to D2 mice weekly for 4 months, did not protect the optic nerve from degeneration (Pelzel et al., 2012). What of the possibility that β-HB limits damage because it is a ketone body? Ketone bodies get converted into acetyl-CoA for utilization in the Krebs cycle (Figure 3), the biosynthetic hub whose intermediates provide the building blocks for fatty acids, nucleotides and the carbons for gluconeogenesis. In the near term, then, increased levels of monocarboxylates like ketone bodies would require the Krebs cycle and oxidative phosphorylation for energy generation. In mammals, more lactate could feed the production of glucose through gluconeogenesis, though this is an energy-expending process. L-lactate can protect cortical neurons from excitotoxic cell death in vitro (Llorente-Folch et al., 2016); this effect depends upon the aspartate-glutamate carrier in CNS mitochondria, ARALAR/AGC. This carrier is one half of the malate-aspartate shuttle that moves malate/aspartate and α-ketoglutarate/glutamate between the cytoplasm and the mitochondria in order to regenerate NAD+ from NADH in both compartments. Neuronal utilization of L-lactate depends on the this pathway (Llorente-Folch et al., 2016), underscoring the importance of NAD+ availability to managing the effects of too much NMDA receptor activation. This is one potential mechanism of ketone body neural protection that is separate from HDAC inhibition.
Resolving Metabolic Vulnerability
Aging is a primary risk factor in glaucoma. There is recognition across neurodegenerative diseases that glucoregulatory control decreases with aging, spawning clinical trials designed to ameliorate the effects of aging through the use of compounds like metformin (ClinicalTrials.gov Identifier: NCT02432287) or acarbose (Brewer et al., 2016). These trials do not address glaucoma in particular, but may impact the disease through improved glucoregulatory control. Serum citrate as a potential biomarker for glaucoma was the subject of a recently completed clinical trial that showed significantly decreased citrate in the serum of Caucasian glaucoma patients (Fraenkl et al., 2011). Citrate is a byproduct of the Krebs cycle, so decreased values support the idea that mitochondrial function is impaired in glaucoma patients. Mitochondrial function can be monitored in patient retinas using the green fluorescence emitted by oxidized flavoproteins, a byproduct of oxidative stress (Field et al., 2011). This development may improve diagnosis as well as provide a means to monitor future metabolic therapy.
The complexity of optic nerve degeneration in glaucoma—its chronic timing, its asynchronicity, its non-cell autonomous nature—presents many challenges to medicine. Having reviewed the various ways energy plays a role in axon degeneration, we have also summarized several metabolic-related mechanisms of protection. The evidence indicates energy plays a critical role in the timing and organization of axon degeneration, including in glaucoma.
Note Added in Proof
Significant RGC soma and axon protection from glaucoma-related degeneration in the D2 mouse was achieved through dietary supplementation of nicotinamide (Williams et al., 2017). The finding is quite robust, with significant protection at both low (550 mg/kg/day) and high levels (2,000 mg/kg/day) of nicotinamide delivery. The study used RNA-sequencing to show a reversion of gene expression to that demonstrated by the control strain, DBA/2-Gpnmb+, across clusters of genes that included those involved in oxidative phosphorylation; reactive oxygen species, glucose, and fatty acid metabolism; and DNA repair. These results support the existence of a metabolic vulnerability in glaucoma; the mechanism of protection is yet to be elucidated.
Author Contributions
DI conceived of the topic and outlined the review; DI and MH wrote the review.
Funding
This work was supported by NIH EY026662 (DI).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Samuel Crish for helpful comments and discussion.
References
Alano, C. C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T. M., and Swanson, R. A. (2010). NAD+ depletion is necessary and sufficient for poly (ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010
Ali, Y. O., Li-Kroeger, D., Bellen, H. J., Zhai, R. G., and Lu, H. C. (2013). NMNATs, evolutionarily conserved neuronal maintenance factors. Trends Neurosci. 36, 632–640. doi: 10.1016/j.tins.2013.07.002
Alsarraf, O., Fan, J., Dahrouj, M., Chou, C. J., Yates, P. W., and Crosson, C. E. (2014). Acetylation preserves retinal ganglion cell structure and function in a chronic model of ocular hypertension. Invest. Ophthalmol. Vis. Sci. 55, 7486–7493. doi: 10.1167/iovs.14-14792
Anderson, M. G., Smith, R. S., Hawes, N. L., Zabaleta, A., Chang, B., Wiggs, J. L., et al. (2002). Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet. 30, 81–85. doi: 10.1038/ng794
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi: 10.1126/science.1098014
Avery, M. A., Rooney, T. M., Pandya, J. D., Wishart, T. M., Gillingwater, T. H., Geddes, J. W., et al. (2012). WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr. Biol. 22, 596–600. doi: 10.1016/j.cub.2012.02.043
Babetto, E., Beirowski, B., Russler, E. V., Milbrandt, J., and DiAntonio, A. (2013). The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 3, 1422–1429. doi: 10.1016/j.celrep.2013.04.013
Baltan, S., Inman, D. M., Danilov, C. A., Morrison, R. S., Calkins, D. J., and Horner, P. J. (2010). Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 30, 5644–5652. doi: 10.1523/JNEUROSCI.5956-09.2010
Barber, A. J., Nakamura, M., Wolpert, E. B., Reiter, C. E., Seigel, G. M., Antonetti, D. A., et al. (2001). Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J. Biol. Chem. 276, 32814–32821. doi: 10.1074/jbc.M104738200
Beirowski, B., Adalbert, R., Wagner, D., Grumme, D. S., Addicks, K., Ribchester, R. R., et al. (2005). The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 6:6. doi: 10.1186/1471-2202-6-6
Beirowski, B., Babetto, E., Coleman, M. P., and Martin, K. R. (2008). The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 28, 1166–1179. doi: 10.1111/j.1460-9568.2008.06426.x
Belenky, P., Bogan, K. L., and Brenner, C. (2007). NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19. doi: 10.1016/j.tibs.2006.11.006
Beurel, E., Grieco, S. F., and Jope, R. S. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. doi: 10.1016/j.pharmthera.2014.11.016
Bittar, P. G., Charnay, Y., Pellerin, L., Bouras, C., and Magistretti, P. J. (1996). Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 16, 1079–1089. doi: 10.1097/00004647-199611000-00001
Blander, G., and Guarente, L. (2004). The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73, 417–435. doi: 10.1146/annurev.biochem.73.011303.073651
Bosco, A., Breen, K. T., Anderson, S. R., Steele, M. R., Calkins, D. J., and Vetter, M. L. (2016). Glial coverage in the optic nerve expands in proportion to optic axon loss in chronic mouse glaucoma. Exp. Eye Res. 150, 34–43. doi: 10.1016/j.exer.2016.01.014
Bouzier-Sore, A. K., Voisin, P., Canioni, P., Magistretti, P. J., and Pellerin, L. (2003). Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb. Blood Flow Metab. 23, 1298–1306. doi: 10.1097/01.WCB.0000091761.61714.25
Brand, M. D., and Esteves, T. C. (2005). Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2, 85–93. doi: 10.1016/j.cmet.2005.06.002
Brewer, R. A., Gibbs, V. K., and Smith, D. L. (2016). Targeting glucose metabolism for healthy aging. Nutr. Heal. Aging 4, 31–46. doi: 10.3233/NHA-160007
Brown, A. M. (2004). Brain glycogen re-awakened. J. Neurochem. 89, 537–552. doi: 10.1111/j.1471-4159.2004.02421.x
Brown, A. M., Sickmann, H. M., Fosgerau, K., Lund, T. M., Schousboe, A., Waagepetersen, H. S., et al. (2005). Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 79, 74–80. doi: 10.1002/jnr.20335
Brown, A. M., Tekkök, S. B., and Ransom, B. R. (2003). Glycogen regulation and functional role in mouse white matter. J. Physiol. 549, 501–512. doi: 10.1113/jphysiol.2003.042416
Buckingham, B. P., Inman, D. M., Lambert, W. S., Oglesby, E., Calkins, D. J., Steele, M. R., et al. (2008). Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J. Neurosci. 28, 2735–2744. doi: 10.1523/JNEUROSCI.4443-07.2008
Calkins, D. J., and Horner, P. J. (2012). The cell and molecular biology of glaucoma: axonopathy and the brain. Invest. Ophthalmol. Vis. Sci. 53, 2482–2484. doi: 10.1167/iovs.12-9483i
Calliari, A., Bobba, N., Escande, C., and Chini, E. N. (2013). Resveratrol delays Wallerian degeneration in a NAD(+) and DBC1 dependent manner. Exp. Neurol. 251C, 91–100. doi: 10.1016/j.expneurol.2013.11.013
Campbell, P. D., Shen, K., Sapio, M. R., Glenn, T. D., Talbot, W. S., and Marlow, F. L. (2014). Unique function of kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 34, 14717–14732. doi: 10.1523/JNEUROSCI.2770-14.2014
Cartoni, R., Norsworthy, M. W., Bei, F., Wang, C., Li, S., Zhang, Y., et al. (2016). The mammalian-specific protein Armcx1 regulates mitochondrial transport during axon regeneration. Neuron 92, 1294–1307. doi: 10.1016/j.neuron.2016.10.060
Casson, R. J., Chidlow, G., Wood, J. P., Crowston, J. G., and Goldberg, I. (2012). Definition of glaucoma: clinical and experimental concepts. Clin. Exp. Ophthalmol. 40, 341–349. doi: 10.1111/j.1442-9071.2012.02773.x
Cataldo, A. M., and Broadwell, R. D. (1986). Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J. Neurocytol. 15, 511–524. doi: 10.1007/BF01611733
Chan, J. W. (2002). Optic neuritis in multiple sclerosis. Ocul. Immunol. Inflamm. 10, 161–186. doi: 10.1076/ocii.10.3.161.15603
Cheng, L., Sapieha, P., Kittlerova, P., Hauswirth, W. W., and Di Polo, A. (2002). TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J. Neurosci. 22, 3977–3986.
Chung, S. H., Shen, W., and Gillies, M. C. (2013). Laser capture microdissection-directed profiling of glycolytic and mTOR pathways in areas of selectively ablated Müller cells in the murine retina. Invest. Ophthalmol. Vis. Sci. 54, 6578–6585. doi: 10.1167/iovs.13-12311
Conforti, L., Fang, G., Beirowski, B., Wang, M. S., Sorci, L., Asress, S., et al. (2007). NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 14, 116–127. doi: 10.1038/sj.cdd.4401944
Conforti, L., Gilley, J., and Coleman, M. P. (2014). Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409. doi: 10.1038/nrn3680
Conforti, L., Tarlton, A., Mack, T. G., Mi, W., Buckmaster, E. A., Wagner, D., et al. (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 11377–11382. doi: 10.1073/pnas.97.21.11377
Coombs, J., van der List, D., Wang, G. Y., and Chalupa, L. M. (2006). Morphological properties of mouse retinal ganglion cells. Neuroscience 140, 123–136. doi: 10.1016/j.neuroscience.2006.02.079
Cooper, M. L., Crish, S. D., Inman, D. M., Horner, P. J., and Calkins, D. J. (2016). Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Exp. Eye Res. 150, 22–33. doi: 10.1016/j.exer.2015.11.016
Coughlin, L., Morrison, R. S., Horner, P. J., and Inman, D. M. (2015). Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest. Ophthalmol. Vis. Sci. 56, 1437–1446. doi: 10.1167/iovs.14-16126
Court, F. A., and Coleman, M. P. (2012). Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 35, 364–372. doi: 10.1016/j.tins.2012.04.001
Crish, S. D., and Calkins, D. J. (2011). Neurodegeneration in glaucoma: progression and calcium- dependent intracellular mechanisms. Neuroscience 176, 1–11. doi: 10.1016/j.neuroscience.2010.12.036
Crish, S. D., Sappington, R. M., Inman, D. M., Horner, P. J., and Calkins, D. J. (2010). Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 107, 5196–5201. doi: 10.1073/pnas.0913141107
Dai, C., Khaw, P. T., Yin, Z. Q., Li, D., Raisman, G., and Li, Y. (2012). Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia 60, 13–28. doi: 10.1002/glia.21242
Dasgupta, B., and Milbrandt, J. (2007). Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 7217–7222. doi: 10.1073/pnas.0610068104
Della Santina, L., Inman, D. M., Lupien, C. B., Horner, P. J., and Wong, R. O. (2013). Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J. Neurosci. 33, 17444–17457. doi: 10.1523/JNEUROSCI.5461-12.2013
Della Santina, L., and Ou, Y. (2016). Who's lost first? Susceptibility of retinal ganglion cell types in experimental glaucoma. Exp. Eye Res. doi: 10.1016/j.exer.2016.06.006. [Epub ahead of print].
Dengler-Crish, C. M., Smith, M. A., Inman, D. M., Wilson, G. N., Young, J. W., and Crish, S. D. (2014). Anterograde transport blockade precedes deficits in retrograde transport in the visual projection of the DBA / 2J mouse model of glaucoma. Front. Neurosci. 8:290. doi: 10.3389/fnins.2014.00290
Di Lisa, F., and Ziegler, M. (2001). Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 492, 4–8. doi: 10.1016/S0014-5793(01)02198-6
Dringen, R., Gebhardt, R., and Hamprecht, B. (1993). Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 623, 208–214. doi: 10.1016/0006-8993(93)91429-V
Duan, X., Qiao, M., Bei, F., Kim, I. J., He, Z., and Sanes, J. R. (2015). Subtype-specific regeneration of retinal ganglion cells following axotomy: Effects of osteopontin and mtor signaling. Neuron 85, 1244–1256. doi: 10.1016/j.neuron.2015.02.017
Dubinsky, W. P., and Racker, E. (1978). The mechanism of lactate transport in human erythrocytes. J. Membr. Biol. 44, 25–36. doi: 10.1007/bf01940571
Ebneter, A., Chidlow, G., Wood, J. P., and Casson, R. J. (2011). Protection of retinal ganglion cells and the optic nerve during short-term hyperglycemia in experimental glaucoma. Arch. Ophthalmol. 129, 1337–1344. doi: 10.1001/archophthalmol.2011.269
El-Danaf, R. N., and Huberman, A. D. (2015). Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J. Neurosci. 35, 2329–2343. doi: 10.1523/JNEUROSCI.1419-14.2015
Erlichman, J. S., Hewitt, A., Damon, T. L., Hart, M., Kurascz, J., Li, A., et al. (2008). Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: a test of the astrocyte-neuron lactate-shuttle hypothesis. J. Neurosci. 28, 4888–4896. doi: 10.1523/JNEUROSCI.5430-07.2008
Fagiolini, M., Caleo, M., Strettoi, E., and Maffei, L. (1997). Axonal transport blockade in the neonatal rat optic nerve induces limited retinal ganglion cell death. J. Neurosci. 17, 7045–7052.
Falk, M., Zhang, Q., and Nakamaru-Ogiso, E. (2012). NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 44, 1040–1045. doi: 10.1038/ng.2361
Fang, C., Decker, H., and Banker, G. (2014a). Axonal transport plays a crucial role in mediating the axon-protective effects of NmNAT. Neurobiol. Dis. 68, 78–90. doi: 10.1016/j.nbd.2014.04.013
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., SenGupta, T., Nilsen, H., et al. (2014b). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896. doi: 10.1016/j.cell.2014.03.026
Fernandes, K. A., Harder, J. M., John, S. W., Shrager, P., and Libby, R. T. (2014). DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis. 69, 108–116. doi: 10.1016/j.nbd.2014.05.015
Ferri, A., Sanes, J. R., Coleman, M. P., Cunningham, J. M., and Kato, A. C. (2003). Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr. Biol. 13, 669–673. doi: 10.1016/S0960-9822(03)00206-9
Field, M. G., Yang, D., Bian, Z. M., Petty, H. R., and Elner, V. M. (2011). Retinal flavoprotein fluorescence correlates with mitochondrial stress, apoptosis, and chemokine expression. Exp. Eye Res. 93, 548–555. doi: 10.1016/j.exer.2011.06.023
Fraenkl, S. A., Muser, J., Groell, R., Reinhard, G., Orgul, S., Flammer, J., et al. (2011). Plasma citrate levels as a potential biomarker for glaucoma. J. Ocul. Pharmacol. Ther. 27, 577–580. doi: 10.1089/jop.2011.0062
Fu, C. T., and Sretavan, D. W. (2012). Ectopic vesicular glutamate release at the optic nerve head and axon loss in mouse experimental glaucoma. J. Neurosci. 32, 15859–15876. doi: 10.1523/JNEUROSCI.0038-12.2012
Fukuda, Y., Hsiao, C. F., Watanabe, M., and Ito, H. (1984). Morphological correlates of physiologically identified Y-, X-, and W-cells in cat retina. J. Neurophysiol. 52, 999–1013.
Galli, U., Travelli, C., Massarotti, A., Fakhfouri, G., Rahimian, R., Tron, G. C., et al. (2013). Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 56, 6279–6296. doi: 10.1021/jm4001049
Garcia-Caceres, C., Quarta, C., Varela, L., Gao, Y., Gruber, T., Legutko, B., et al. (2016). Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166, 867–880. doi: 10.1016/j.cell.2016.07.028
Gerdts, J., Brace, E. J., Sasaki, Y., Diantonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2013). Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33, 13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013
Ghosh, A. S., Wang, B., Pozniak, C. D., Chen, M., Watts, R. J., and Lewcock, J. W. (2011). DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol. 194, 751–764. doi: 10.1083/jcb.201103153
Gilley, J., Adalbert, R., Yu, G., and Coleman, M. P. (2013). Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 33, 13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Godzik, K., and Coleman, M. P. (2015). The axon-protective WLDS protein partially rescues mitochondrial respiration and glycolysis after axonal injury. J. Mol. Neurosci. 55, 865–871. doi: 10.1007/s12031-014-0440-2
Green, K. N., Steffan, J. S., Martinez-Coria, H., Sun, X., Schreiber, S. S., Thompson, L. M., et al. (2008). Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 28, 11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008
Grunwald, J. E., Piltz, J., Hariprasad, S. M., and DuPont, J. (1998). Optic nerve and choroidal circulation in glaucoma. Investig. Ophthalmol. Vis. Sci. 39, 2329–2336.
Guarente, L. (2014). Aging research - where do we stand and where are we going? Cell 159, 15–19. doi: 10.1016/j.cell.2014.08.041
Guo, X., Dason, E. S., Zanon-Moreno, V., Jiang, Q., Nahirny, A., Chan, D., et al. (2014). PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am. J. Pathol. 184, 1017–1029. doi: 10.1016/j.ajpath.2013.12.012
Guo, X., Kimura, A., Azuchi, Y., Akiyama, G., Noro, T., Harada, C., et al. (2016). Caloric restriction promotes cell survival in a mouse model of normal tension glaucoma. Sci. Rep. 6:33950. doi: 10.1038/srep33950
Hamberger, A., and Hyden, H. (1963). Inverse enzymatic changes in neurons and glia during increased function and hypoxia. J. Cell Biol. 16, 521–525. doi: 10.1083/jcb.16.3.521
Hamlin, G. P., Cernak, I., Wixey, J. A., and Vink, R. (2001). Increased expression of neuronal glucose transporter 3 but not glial glucose transporter 1 following severe diffuse traumatic brain injury in rats. J. Neurotrauma 18, 1011–1018. doi: 10.1089/08977150152693700
Han, S. M., Baig, H. S., and Hammarlund, M. (2016). Mitochondria localize to injured axons to support regeneration. Neuron 92, 1308–1323. doi: 10.1016/j.neuron.2016.11.025
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
Harris, J. J., and Attwell, D. (2012). The energetics of CNS white matter. J. Neurosci. 32, 356–371. doi: 10.1523/JNEUROSCI.3430-11.2012
Hasegawa, K., Yasuda, T., Shiraishi, C., Fujiwara, K., Przedborski, S., Mochizuki, H., et al. (2016). Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat. Commun. 7:10943. doi: 10.1038/ncomms10943
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond). 6, 1–25. doi: 10.1186/1743-7075-6-31
Honjin, R., Sakato, S., and Yamashita, T. (1977). Electron microscopy of the mouse optic nerve: a quantitative study of the total optic nerve fibers. Arch. Histol. Jpn. 40, 321–332. doi: 10.1679/aohc1950.40.321
Hoopfer, E. D., McLaughlin, T., Watts, R. J., Schuldiner, O., O'Leary, D. D., and Luo, L. (2006). Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 50, 883–895. doi: 10.1016/j.neuron.2006.05.013
Howell, G. R., Libby, R. T., Jakobs, T. C., Smith, R. S., Phalan, F. C., Barter, J. W., et al. (2007). Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol 179, 1523–1537. doi: 10.1083/jcb.200706181
Huxlin, K. R., and Goodchild, A. K. (1997). Retinal ganglion cells in the albino rat: revised morphological classification. J. Comp. Neurol. 385, 309–323.
Ikegami, K., Kato, S., and Koike, T. (2004). N-alpha-p-tosyl-L-lysine chloromethyl ketone (TLCK) suppresses neuritic degeneration caused by different experimental paradigms including in vitro Wallerian degeneration. Brain Res. 1030, 81–93. doi: 10.1016/j.brainres.2004.09.050
Inman, D. M., Sappington, R. M., Horner, P. J., and Calkins, D. J. (2006). Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest. Ophthalmol. Vis. Sci. 47, 986–996. doi: 10.1167/iovs.05-0925
Islam, R., Yang, L., Sah, M., Kannan, K., Anamani, D., Vijayan, C., et al. (2012). A neuroprotective role of the human uncoupling protein 2 (hUCP2) in a Drosophila Parkinson's Disease model. Neurobiol. Dis. 46, 137–146. doi: 10.1016/j.nbd.2011.12.055
Jakobs, T. C., Libby, R. T., Ben, Y., John, S. W., and Masland, R. H. (2005). Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol 171, 313–325. doi: 10.1083/jcb.200506099
John, S. W., Smith, R. S., Savinova, O. V., Hawes, N. L., Chang, B., Turnbull, D., et al. (1998). Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest. Ophthalmol. Vis. Sci. 39, 951–962.
Jonas, J. B., Muller-Bergh, J. A., Schlotzer-Schrehardt, U. M., and Naumann, G. O. (1990). Histomorphometry of the human optic nerve. Investig. Ophthalmol. Vis. Sci. 31, 736–744.
Ju, W. K., Kim, K. Y., Lindsey, J. D., Angert, M., Duong-Polk, K. X., Scott, R. T., et al. (2008). Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest. Ophthalmol. Vis. Sci. 49, 4903–4911. doi: 10.1167/iovs.07-1661
Kanamori, A., Nakamora, M., Mukuno, H., Maeda, H., and Negi, A. (2004a). Diabetes has an additive effect on neural apoptosis in rat retina with chronically elevated intraocular pressure. Curr. Eye Res. 28, 47–54. doi: 10.1076/ceyr.28.1.47.23487
Kanamori, A., Nakamura, M., Nakanishi, Y., Nagai, A., Mukuno, H., Yamada, Y., et al. (2004b). Akt is activated via insulin/IGF-1 receptor in rat retina with episcleral vein cauterization. Brain Res. 1022, 195–204. doi: 10.1016/j.brainres.2004.06.077
Kim, S.-J., Sung, M.-S., Heo, H., Lee, J.-H., and Park, S.-W. (2015). Mangiferin protects retinal ganglion cells in ischemic mouse retina via SIRT1. Curr. Eye Res. 0, 1–12. doi: 10.3109/02713683.2015.1050736
Kim, Y., Zhou, P., Qian, L., Chuang, J. Z., Lee, J., Li, C., et al. (2007). MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J. Exp. Med. 204, 2063–2074. doi: 10.1084/jem.20070868
Kitaoka, Y., Munemasa, Y., Kojima, K., Hirano, A., Ueno, S., and Takagi, H. (2013). Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 4:e860. doi: 10.1038/cddis.2013.391
Kitay, B. M., McCormack, R., Wang, Y., Tsoulfas, P., and Zhai, R. G. (2013). Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLD(S)-mediated axon protection independent of axonal mitochondria. Hum. Mol. Genet. 22, 1601–1614. doi: 10.1093/hmg/ddt009
Kleesattel, D., Crish, S. D., and Inman, D. M. (2015). Decreased energy capacity and increased autophagic activity in optic nerve axons with defective anterograde transport. Investig. Ophthalmol. Vis. Sci. 56, 8215–8227. doi: 10.1167/iovs.15-17885
Koch, K., Mclean, J., Berry, M., Sterling, P., Balasubramanian, V., and Freed, M. A. (2004). Efficiency of information transmission by retinal ganglion cells. 14, 1523–1530. doi: 10.1016/j.cub.2004.08.060
Koehler-Stec, E. M., Simpson, I. A., Vannucci, S. J., Landschulz, K. T., and Landschulz, W. H. (1998). Monocarboxylate transporter expression in mouse brain Monocarboxylate transporter expression in mouse brain. Am. J. Physiol. Endocrinol. Metab. 275, E516–E524.
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F., et al. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127, 1109–1122. doi: 10.1016/j.cell.2006.11.013
Lampert, P. W., Vogel, M. H., and Zimmerman, L. E. (1968). Pathology of the optic nerve in experimental acute glaucoma: electron microscopic studies. Invest. Ophthalmol. Vis. Sci. 7, 199–213.
Langley, B., Gensert, J. M., Beal, M. F., and Ratan, R. R. (2005). Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 4, 41–50. doi: 10.2174/1568007053005091
Lascaratos, G., Chau, K. Y., Zhu, H., Gkotsi, D., King, R., Gout, I., et al. (2015). Resistance to the most common optic neuropathy is associated with systemic mitochondrial efficiency. Neurobiol. Dis. 82, 78–85. doi: 10.1016/j.nbd.2015.05.012
Lau, C., Niere, M., and Ziegler, M. (2009). The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front. Biosci. 14, 410–431. doi: 10.2741/3252
Lebrun-Julien, F., Morquette, B., Douillette, A., Saragovi, H. U., and Di Polo, A. (2009). Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol. Cell. Neurosci. 40, 410–420. doi: 10.1016/j.mcn.2008.12.005
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lensman, M., Korzhevskii, D. E., Mourovets, V. O., Kostkin, V. B., Izvarina, N., Perasso, L., et al. (2006). Intracerebroventricular administration of creatine protects against damage by global cerebral ischemia in rat. Brain Res. 1114, 187–194. doi: 10.1016/j.brainres.2006.06.103
Li, R. S., Chen, B. Y., Tay, D. K., Chan, H. H., Pu, M. L., and So, K. F. (2006). Melanopsin-expressing retinal ganglion cells are more injury-resistant in a chronic ocular hypertension model. Investig. Ophthalmol. Vis. Sci. 47, 2951–2958. doi: 10.1167/iovs.05-1295
Libby, R. T., Li, Y., Savinova, O. V., Barter, J., Smith, R. S., Nickells, R. W., et al. (2005). Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet 1, 17–26. doi: 10.1371/journal.pgen.0010004
Linding, R., Jensen, L. J., Ostheimer, G. J., van Vugt, M. A., Jørgensen, C., Miron, I. M., et al. (2007). Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426. doi: 10.1016/j.cell.2007.05.052
Llorente-Folch, I., Rueda, C. B., Pérez-Liébana, I., Satrústegui, J., and Pardo, B. (2016). l-Lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J. Neurosci. 36, 4443–4456. doi: 10.1523/JNEUROSCI.3691-15.2016
Loreto, A., Di Stefano, M., Gering, M., and Conforti, L. (2015). Wallerian degeneration is executed by an NMN-SARM1-dependent Late Ca2+ influx but only modestly influenced by mitochondria. Cell Rep. 13, 2539–2552. doi: 10.1016/j.celrep.2015.11.032
Lundgaard, I., Osório, M. J., Kress, B. T., Sanggaard, S., and Nedergaard, M. (2014). White matter astrocytes in health and disease. Neuroscience 276, 161–173. doi: 10.1016/j.neuroscience.2013.10.050
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x
Lye-Barthel, M., Sun, D., and Jakobs, T. C. (2013). Morphology of astrocytes in a glaucomatous optic nerve. Invest. Ophthalmol. Vis. Sci. 54, 909–917. doi: 10.1167/iovs.12-10109
Lyon, M. F., Ogunkolade, B. W., Brown, M. C., Atherton, D. J., and Perry, V. H. (1993). A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proc. Natl. Acad. Sci. U.S.A. 90, 9717–9720. doi: 10.1073/pnas.90.20.9717
Mack, T. G., Reiner, M., Beirowski, B., Mi, W., Emanuelli, M., Wagner, D., et al. (2001). Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4, 1199–1206. doi: 10.1038/nn770
Maher, F., Davies-Hill, T. M., Lysko, P. G., Henneberry, R. C., and Simpson, I. A. (1991). Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron-specific expression of GLUT3. Mol. Cell. Neurosci. 2, 351–360. doi: 10.1016/1044-7431(91)90066-W
Mandelkow, E. M., Drewes, G., Biernat, J., Gustke, N., Van Lint, J., Vandenheede, J. R., et al. (1992). Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 314, 315–321. doi: 10.1016/0014-5793(92)81496-9
Mandelkow, E. M., Stamer, K., Vogel, R., Thies, E., and Mandelkow, E. (2003). Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007
Meyer zu Horste, G., Miesbach, T. A., Muller, J. I., Fledrich, R., Stassart, R. M., Kieseier, B. C., et al. (2011). The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiol. Dis. 42, 1–8. doi: 10.1016/j.nbd.2010.12.006
Mikelberg, F. S, Drance, S. M., Schulzer, M., Yidegiligne, H. M., and Weis, M. M. (1989). The normal human optic nerve. Axon count and axon diameter distribution. Ophthalmology 96, 1325–1328. doi: 10.1016/S0161-6420(89)32718-7
Milde, S., Gilley, J., and Coleman, M. P. (2013). Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 11:e1001539. doi: 10.1371/journal.pbio.1001539
Miller, B. R., Press, C., Daniels, R. W., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2009). A DLK-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 12, 387–389. doi: 10.1038/nn.2290
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012
Mudò, G., Mäkelä, J., Di Liberto, V., Tselykh, T. V., Olivieri, M., Piepponen, P., et al. (2012). Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell. Mol. Life Sci. 69, 1153–1165. doi: 10.1007/s00018-011-0850-z
Nakazawa, T., Nakazawa, C., Matsubara, A., Noda, K., Hisatomi, T., She, H., et al. (2006). Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26, 12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006
Nave, K. A. (2010). Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 11, 275–283. doi: 10.1038/nrn2797
Nilsson, P., Hillered, L., Pontén, U., and Ungerstedt, U. (1990). Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J. Cereb. Blood Flow Metab. 10, 631–637. doi: 10.1038/jcbfm.1990.115
O'Meara, R. W., Michalski, J. P., Anderson, C., Bhanot, K., Rippstein, P., and Kothary, R. (2013). Integrin-linked kinase regulates process extension in oligodendrocytes via control of actin cytoskeletal dynamics. J. Neurosci. 33, 9781–9793. doi: 10.1523/JNEUROSCI.5582-12.2013
Obel, L. F., Müller, M. S., Walls, A. B., Sickmann, H. M., Bak, L. K., Waagepetersen, H. S., et al. (2012). Brain glycogen - new perspectives on its metabolic function and regulation at the subcellular level. Front. Neuroenergetics 4:3. doi: 10.3389/fnene.2012.00003
Ohno, N., Kidd, G. J., Mahad, D., Kiryu-Seo, S., Avishai, A., Komuro, H., et al. (2011). Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J. Neurosci. 31, 7249–7258. doi: 10.1523/JNEUROSCI.0095-11.2011
Orthmann-Murphy, J. L., Freidin, M., Fischer, E., Scherer, S. S., and Abrams, C. K. (2007). Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27, 13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Ou, Y., Jo, R. E., Ullian, E. M., Wong, R. O., and Della Santina, L. (2016). Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension. J. Neurosci. 36, 9240–9252. doi: 10.1523/JNEUROSCI.0940-16.2016
Park, H. Y. L., Kim, J. H., and Park, C. K. (2012). Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis. 3:e290. doi: 10.1038/cddis.2012.26
Peixoto, P. M., Kim, H.-J., Sider, B., Starkov, A., Horvath, T. L., and Manfredi, G. (2013). UCP2 overexpression worsens mitochondrial dysfunction and accelerates disease progression in a mouse model of amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 57, 104–110. doi: 10.1016/j.mcn.2013.10.002
Pellerin, L., Pellegri, G., Bittar, P., Charnay, Y., Bouras, C., Martin, J., et al. (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 20, 291–299. doi: 10.1159/000017324
Pelzel, H. R., Schlamp, C. L., Waclawski, M., Shaw, M. K., and Nickells, R. W. (2012). Silencing of Fem1cR3 gene expression in the DBA/2J mouse precedes retinal ganglion cell death and is associated with histone deacetylase activity. Invest. Ophthalmol. Vis. Sci. 53, 1428–1435. doi: 10.1167/iovs.11-8872
Perasso, L., Adriano, E., Ruggeri, P., Burov, S. V., Gandolfo, C., and Balestrino, M. (2009). In vivo neuroprotection by a creatine-derived compound: phosphocreatine-Mg-complex acetate. Brain Res. 1285, 158–163. doi: 10.1016/j.brainres.2009.06.009
Perge, J. A., Koch, K., Miller, R., Sterling, P., and Balasubramanian, V. (2009). How the optic nerve allocates space, energy capacity, and information. J. Neurosci. 29, 7917–7928. doi: 10.1523/JNEUROSCI.5200-08.2009
Perry, V., Brown, M., Lunn, E., Tree, P., and Gordon, S. (1990a). Evidence that very slow Wallerian degeneration in the C57Bl/Ola mice is an intrinsic property of the peripheral nerve. Eur. J. Neurosci. 2, 802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x
Perry, V. H., Brown, M. C., Lunn, E. R., Tree, P., Gordon, S., and Cahusac, C. (1990b). Evidence that the rate of wallerian degeneration is controlled by a single autosomal dominant gene. Eur. J. Neurosci. 2, 408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x
Pevzner, L. Z. (1971). Topochemical aspects of nucleic acid and protein metabolism within the neuron-neuroglia unit of the spinal cord anterior horn. J. Neurochem. 18, 895–907. doi: 10.1111/j.1471-4159.1971.tb12019.x
Pevzner, L. Z. (1972). Topochemical aspects of nucleic acid metabolism within the neuronal-neuroglia unit of cerebellum Purkinje cells. Brain Res. 46, 329–339. doi: 10.1016/0006-8993(72)90023-6
Pierre, K., Magistretti, P. J., and Pellerin, L. (2002). MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J. Cereb. Blood Flow Metab. 22, 586–595. doi: 10.1097/00004647-200205000-00010
Pierre, K., and Pellerin, L. (2005). Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. 94, 1–14. doi: 10.1111/j.1471-4159.2005.03168.x
Quigley, H. A., Dunkelberger, G. R., and Green, W. R. (1988). Chronic human glaucoma causing selectively greater loss of large optic nerve fibers. Ophthalmology 95, 357–363. doi: 10.1016/S0161-6420(88)33176-3
Rafiki, A., Boulland, J. L., Halestrap, A. P., Ottersen, O. P., and Bergersen, L. (2003). Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience 122, 677–688. doi: 10.1016/j.neuroscience.2003.08.040
Ransom, B. R., Butt, A. M., and Black, J. A. (1991). Ultrastructural identification of HRP-injected oligodendrocytes in the intact rat optic nerve. Glia 4, 37–45. doi: 10.1002/glia.440040105
Rawson, R. L., Yam, L., Weimer, R. M., Bend, E. G., Hartwieg, E., Horvitz, H. R., et al. (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 24, 760–765. doi: 10.1016/j.cub.2014.02.025
Ritchie, J. (1967). The oxygen consumption of mammalian non-myelinated nerve fibers at rest and during activity. J. Physiol. 188, 309–329. doi: 10.1113/jphysiol.1967.sp008141
Rodriguez-Muela, N., and Boya, P. (2012). Axonal damage, autophagy and neuronal survival. Autophagy 8, 286–288. doi: 10.4161/auto.8.2.18982
Rone, M. B., Cui, Q.-L., Fang, J., Wang, L.-C., Zhang, J., Khan, D., et al. (2016). Oligodendrogliopathy in multiple sclerosis: low glycolytic metabolic rate promotes oligodendrocyte survival. J. Neurosci. 36, 4698–4707. doi: 10.1523/JNEUROSCI.4077-15.2016
Rouach, N., Koulakoff, A., Abudara, V., Willecke, K., and Giaume, C. (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. doi: 10.1126/science.1164022
Rowlands, B. D., Lau, C. L., Ryall, J. G., Thomas, D. S., Klugmann, M., Beart, P. M., et al. (2015). Silent information regulator 1 modulator resveratrol increases brain lactate production and inhibits mitochondrial metabolism, whereas SRT1720 increases oxidative metabolism. J. Neurosci. Res. 93, 1147–1156. doi: 10.1002/jnr.23570
Saab, A. S., Tzvetavona, I. D., Trevisiol, A., Baltan, S., Dibaj, P., Kusch, K., et al. (2016). Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119–132. doi: 10.1016/j.neuron.2016.05.016
Samsam, M., Mi, W., Wessig, C., Zielasek, J., Toyka, K. V., Coleman, M. P., et al. (2003). The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J. Neurosci. 23, 2833–2839.
Sasaki, Y., Vohra, B. P. S., Lund, F. E., and Milbrandt, J. (2009). Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009
Schlamp, C. L., Li, Y., Dietz, J. A., Janssen, K. T., and Nickells, R. W. (2006). Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 7:66. doi: 10.1186/1471-2202-7-66
Schmitt, H. M., Pelzel, H. R., Schlamp, C. L., and Nickells, R. W. (2014). Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol. Neurodegener. 9, 39–54. doi: 10.1186/1750-1326-9-39
Schoenmann, Z., Assa-Kunik, E., Tiomny, S., Minis, A., Haklai-Topper, L., Arama, E., et al. (2010). Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci. 30, 6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010
Schwartz, B. (1994). Circulatory defects of the optic disk and retina in ocular hypertension and high pressure open-angle glaucoma. Surv. Ophthalmol. 38(Suppl):S23–S34. doi: 10.1016/0039-6257(94)90044-2
Seglen, P. O., and Gordon, P. B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 1889–1892. doi: 10.1073/pnas.79.6.1889
Serwanski, D. R., Jukkola, P., and Nishiyama, A. (2017). Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J. Comp. Neurol. 525, 535–552. doi: 10.1002/cne.24083
Shen, H., and Goldberg, M. P. (2012). Creatine pretreatment protects cortical axons from energy depletion in vitro. Neurobiol. Dis. 47, 184–193. doi: 10.1016/j.nbd.2012.03.037
Shen, H., Hyrc, K. L., and Goldberg, M. P. (2013). Maintaining energy homeostasis is an essential component of Wld(S)-mediated axon protection. Neurobiol. Dis. 59, 69–79. doi: 10.1016/j.nbd.2013.07.007
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. doi: 10.1126/science.1227166
Shin, J. E., Miller, B. R., Babetto, E., Cho, Y., Sasaki, Y., Qayum, S., et al. (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U.S.A. 109, E3696–E3705. doi: 10.1073/pnas.1216204109
Sickmann, H. M., Schousboe, A., Fosgerau, K., and Waagepetersen, H. S. (2005). Compartmentation of lactate originating from glycogen and glucose in cultured astrocytes. Neurochem. Res. 30, 1295–1304. doi: 10.1007/s11064-005-8801-4
Simpson, I., A., Carruthers, A., and Vannucci, S. J. (2007). Supply and demand in cerebral energy metabolism: the role of nutrient transpoters. J. Cereb. Blood Flow Metab. 27, 1766–1791. doi: 10.1038/sj.jcbfm.9600521
Simpson, I. A., Dwyer, D., Malide, D., Moley, K. H., Travis, A., and Vannucci, S. J. (2008). The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 295, E242–E253. doi: 10.1152/ajpendo.90388.2008
Son, J. L., Soto, I., Oglesby, E., Lopez-Roca, T., Pease, M. E., Quigley, H. A., et al. (2010). Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia 58, 780–789. doi: 10.1002/glia.20962
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stincone, A., Prigione, A., Cramer, T., Wamelink, M. M. C., Campbell, K., Cheung, E., et al. (2015). The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 90, 927–963. doi: 10.1111/brv.12140
Stirling, D. P., and Stys, P. K. (2010). Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol. Med. 16, 160–170. doi: 10.1016/j.molmed.2010.02.002
Stys, P. K. (2004). White matter injury mechanisms. Curr. Mol. Med. 4, 113–130. doi: 10.2174/1566524043479220
Stys, P. K., Waxman, S. G., and Ransom, B. R. (1991). Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann. Neurol. 30, 375–380. doi: 10.1002/ana.410300309
Suh, S. W., Bergher, J. P., Anderson, C. M., Treadway, J. L., Fosgerau, K., and Swanson, R. A. (2007). Astrocyte glycogen sustains neuronal activity during hypoglycemia : studies with the glycogen phosphorylase inhibitor CP-316,819. J. Pharmacol. Exp. Ther. 321, 45–50. doi: 10.1124/jpet.106.115550
Summers, D. W., DiAntonio, A., and Milbrandt, J. (2014). Mitochondrial dysfunction induces sarm1-dependent cell death in sensory neurons. J. Neurosci. 34, 9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014
Sun, D., and Jakobs, T. C. (2012). Structural remodeling of astrocytes in the injured CNS. Neuroscientist 18, 567–588. doi: 10.1177/1073858411423441
Sun, D., Lye-Barthel, M., Masland, R. H., and Jakobs, T. C. (2009). The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J. Comp. Neurol. 516, 1–19. doi: 10.1002/cne.22058
Sun, M.-H., Pang, J.-H. S., Chen, S.-L., Han, W.-H., Ho, T.-C., Chen, K.-J., et al. (2010). Retinal protection from acute glaucoma-induced ischemia-reperfusion injury through pharmacologic induction of heme oxygenase-1. Invest. Ophthalmol. Vis. Sci. 51, 4798–4808. doi: 10.1167/iovs.09-4086
Tekkök, S. B., Brown, A. M., Westenbroek, R., Pellerin, L., and Ransom, B. R. (2005). Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 81, 644–652. doi: 10.1002/jnr.20573
Tham, Y. C., Li, X., Wong, T. Y., Quigley, H. A., Aung, T., and Cheng, C. Y. (2014). Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121, 2081–2090. doi: 10.1016/j.ophtha.2014.05.013
Trammell, S. A. J., Schmidt, M. S., Weidemann, B. J., Redpath, P., Jaksch, F., Dellinger, R. W., et al. (2016). Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7:12948. doi: 10.1038/ncomms12948
Tsacopoulos, M., and Magistretti, P. J. (1996). Metabolic coupling glia and neurons. J. Neurosci. 16, 877–885.
Tsutsui, S., and Stys, P. K. (2013). Metabolic injury to axons and myelin. Exp. Neurol. 246, 26–34. doi: 10.1016/j.expneurol.2012.04.016
Uemura, E., and Greenlee, H. W. (2006). Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 198, 48–53. doi: 10.1016/j.expneurol.2005.10.035
van Bergen, N. J., Crowston, J. G., Kearns, L. S., Staffieri, S. E., Hewitt, A. W., Cohn, A. C., et al. (2011). Mitochondrial oxidative phosphorylation compensation may preserve vision in patients with OPA1-linked autosomal dominant optic atrophy. PLoS ONE 6:e21347. doi: 10.1371/journal.pone.0021347
Vande Velde, C., Garcia, M. L., Yin, X., Trapp, B. D., and Cleveland, D. W. (2004). The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromol. Med. 5, 193–203. doi: 10.1385/NMM:5:3:193
Volkenhoff, A., Weiler, A., Letzel, M., Stehling, M., Klämbt, C., and Schirmeier, S. (2015). Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab. 22, 437–447. doi: 10.1016/j.cmet.2015.07.006
Vrabec, J. P., and Levin, L. A. (2007). The neurobiology of cell death in glaucoma. Eye (Lond). 21(Suppl. 1), S11–S14. doi: 10.1038/sj.eye.6702880
Waller, A. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. London 140, 423–429. doi: 10.1098/rstl.1850.0021
Wang, J. T., Medress, Z. A., Vargas, M. E., and Barres, B. A. (2015). Local axonal protection by WldS as revealed by conditional regulation of protein stability. Proc. Natl. Acad. Sci. U.S.A. 112, 10093–10100. doi: 10.1073/pnas.1508337112
Wang, J., Zhai, Q., Chen, Y., Lin, E., Gu, W., McBurney, M. W., et al. (2005). A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 170, 349–355. doi: 10.1083/jcb.200504028
Wang, X., Sekine, Y., Byrne, A. B., and Cafferty, W. B. J. (2016). Inhibition of poly-ADP-ribosylation fails to increase axonal regeneration or improve functional recovery after adult mammalian CNS injury. eNeuro 3, 1–10. doi: 10.1523/ENEURO.0270-16.2016
Watanabe, M., Sawai, H., and Fukuda, Y. (1993). Number, distribution, and morphology of retinal ganglion cells with axons regenerated into peripheral nerve graft in adult cats. J. Neurosci. 13, 2105–2117.
Watkins, T. A., Wang, B., Huntwork-Rodriguez, S., Yang, J., Jiang, Z., Eastham-Anderson, J., et al. (2013). DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U.S.A. 110, 4039–4044. doi: 10.1073/pnas.1211074110
Weber, A. J., and Harman, C. D. (2013). BDNF treatment and extended recovery from optic nerve trauma in the cat. Invest. Ophthalmol. Vis. Sci. 54, 6594–6604. doi: 10.1167/iovs.13-12683
Welsbie, D. S., Yang, Z., Ge, Y., Mitchell, K. L., Zhou, X., Martin, S. E., et al. (2013). Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. U.S.A. 110, 4045–4050. doi: 10.1073/pnas.1211284110
Wender, R., Brown, A. M., Fern, R., Swanson, R. A., Farrell, K., and Ransom, B. R. (2000). Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 20, 6804–6810.
Williams, P. A., Harder, J. M., Foxworth, N. E., Cochran, K. E., Philip, V. M., Porciatti, V., et al. (2017). Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 760, 756–760. doi: 10.1126/science.aal0092
Williams, R. W., and Chalupa, L. M. (1983). An analysis of axon caliber within the optic nerve of the cat: evidence of size groupings and regional organization. J. Neurosci. 3, 1554–1564.
Yahata, N., Yuasa, S., and Araki, T. (2009). Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 29, 6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009
Yang, J., Weimer, R. M., Kallop, D., Olsen, O., Wu, Z., Renier, N., et al. (2013). Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron 80, 1175–1189. doi: 10.1016/j.neuron.2013.08.034
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological axonal death through a Mapk cascade that triggers a local energy deficit. Cell 160, 161–176. doi: 10.1016/j.cell.2014.11.053
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153. doi: 10.1007/s12031-010-9336-y
Yellen, G. (2008). Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia 49, 80–82. doi: 10.1111/j.1528-1167.2008.01843.x
Yoshino, J., Mills, K. F., Yoon, M. J., and Imai, S. (2011). Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell. Metab. 14, 528–536. doi: 10.1016/j.cmet.2011.08.014
Zala, D., Hinckelmann, M.-V., Yu, H., Lyra da Cunha, M. M., Liot, G., Cordelières, F. P., et al. (2013). Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479–491. doi: 10.1016/j.cell.2012.12.029
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi: 10.1126/science.aaf2693
Zhao, Z., Lange, D. J., Voustianiouk, A., MacGrogan, D., Ho, L., Suh, J., et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7:29. doi: 10.1186/1471-2202-7-29
Zhou, B., Yu, P., Lin, M.-Y., Sun, T., Chen, Y., and Sheng, Z.-H. (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol. 214, 103–119. doi: 10.1083/jcb.201605101
Zhu, Y., Zhang, L., Sasaki, Y., Milbrandt, J., and Gidday, J. M. (2013). Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Invest. Ophthalmol. Vis. Sci. 54, 25–36. doi: 10.1167/iovs.12-10861
Keywords: mitochondria, lactate, axonopathy, Wallerian degeneration, optic neuropathy
Citation: Inman DM and Harun-Or-Rashid M (2017) Metabolic Vulnerability in the Neurodegenerative Disease Glaucoma. Front. Neurosci. 11:146. doi: 10.3389/fnins.2017.00146
Received: 20 January 2017; Accepted: 08 March 2017;
Published: 30 March 2017.
Edited by:
Irving E. Vega, Michigan State University, USAReviewed by:
Lee J. Martin, Johns Hopkins School of Medicine, USAAlberto Granzotto, Centro Scienze dell'Invecchiamento e Medicina Traslazionale, Italy
James C. Vickers, University of Tasmania, Australia
Copyright © 2017 Inman and Harun-Or-Rashid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denise M. Inman, ZGlubWFuQG5lb21lZC5lZHU=