 Bevan S. Main
Bevan S. Main Myles R. Minter
Myles R. Minter- 1Laboratory for Brain Injury and Dementia, Department of Neuroscience, Georgetown University Medical Center, Washington, DC, USA
- 2Department of Neurobiology, University of Chicago, Chicago, IL, USA
- 3The Microbiome Center, University of Chicago, Chicago, IL, USA
Neuro-inflammation is a critical process by which the brain coordinates chemokine-regulated cellular recruitment, cytokine release, and cell-mediated removal of pathogenic material to protect against infection or brain injury. Dysregulation of this immune response is involved in multiple neurodegenerative disorders, however the precise contribution of neuro-inflammation to the exacerbation and progression of these diseases remains unclear. Evidence now suggests that commensal micro-organisms populating the host and their metabolites, collectively termed the microbiome, regulate innate immunity by influencing peripheral immune cell populations, and modulating microglial phenotype. Recent preclinical studies now demonstrate that perturbations in the host microbiome can induce alterations in pathological phenotypes associated with numerous neurodegenerative diseases. How perturbations in the host microbiome and subsequently altered peripheral immune status are communicated to the brain to influence neuro-inflammatory processes in these neurodegenerative disease settings is far from understood. This review provides insight into the regulation of neuro-inflammatory processes by the host microbiome in the context of neurodegenerative disease and highlights the potential importance of the blood-brain barrier and blood-cerebrospinal fluid-brain barrier, functioning as “immune barriers,” to communicate host immune status to the brain. Understanding the mechanisms by which the commensal microbiome communicates with the brain to influence neuro-inflammatory processes will be critical in the development of microbially-targeted therapeutics in the potential treatment of neurodegenerative disorders.
Introduction
Decades of research investigating neuro-inflammatory processes has supported the involvement of resident surveying CNS cells (microglia/astrocytes), in initiating innate immune responses that contribute to neurodegenerative disease pathology. Coordinated glial reactivity is required for efficient removal of pathogenic insults and cellular debris arising from tissue injury, as dysregulation of this CNS neuro-inflammatory response contributes to the progression of neurodegenerative disorders including traumatic brain injury (TBI) (Karve et al., 2016), stroke (Xiong et al., 2016), multiple sclerosis (MS) (Patejdl et al., 2016), Parkinson's disease (PD) (Taylor et al., 2013), and Alzheimer's disease (AD) (Minter et al., 2016b). Within these neurodegenerative states, microglia exhibit a biased pro-inflammatory activation state and great effort has been made to identify therapeutic means by which to correct this bias, promoting anti-inflammatory and neuroprotective microglial activities (reviewed in, Du et al., 2016). Recent evidence now suggests that complex bilateral communication between the peripheral immune status and central immunity plays a key role in influencing various neuro-inflammatory processes, including microglial activation.
A critical immunological compartment regulating general host immune homeostasis (reviewed in, Honda and Littman, 2016) and microglial development is the microbiome, the diverse commensal microbial community (and their metabolites) that populate host tissues. C57BL/6 mice treated with broad-spectrum combinatorial antibiotics (ABX), to deplete microbiota, or generated in gnotobiotic facilities (Germ-Free, GF) display elevated microglial number, marked hypertrophy and alterations in the transcriptome that governs basal microglial surveillance (Erny et al., 2015). Upon exposure to lipopolysaccharide (LPS) or lymphocytic choriomeningitis virus these microglia secreted less pro-inflammatory cytokines and chemokines, suggesting that GF microglia are either naïve to inflammatory stimuli or contain adaptations that shift their activity toward an anti-inflammatory state. Interestingly, supplementation with microbially-derived short-chain fatty acids (SCFAs) was sufficient to restore the microglial function of GF mice akin to specific-pathogen free (SPF)-housed mice. Using a transcriptomics approach, microglia isolated from GF mice displayed significantly down-regulated expression of gene clusters associated with microglial maturation and innate immune responses, specifically type-1 interferon (IFN) signaling (Matcovitch-Natan et al., 2016). These studies demonstrate an important role of commensal host microbiota in regulating microglial development and homeostasis, hence investigators are now interested in ascertaining the role of the microbiome in brain health and neurodegenerative disease.
The Commensal Microbiome in Neurodegenerative Diseases
The importance of exposure to diverse microbiota, from birth, in promoting healthy neuronal development, and connectivity has long been documented (Kim et al., 2003; Juarez et al., 2008). A recent study now demonstrates that ABX-induced microbial perturbations results in basal and exercise-induced hippocampal neurogenic defects (Mohle et al., 2016). This deficit is attributed to the reduction in infiltrating Ly6Chi monocytes, as adoptive transfer of these cells into ABX-treated mice is sufficient to restore neurogenesis. ABX-induced microbial dysbioisis also worsens recovery after spinal cord injury in mice, resulting in decreased white matter volume and enhanced pro-inflammatory macrophage, CD45R+ B-cell and CD3+ T-cell infiltration within the lesion epicenter (Kigerl et al., 2016). Interestingly, probiotic supplementation post-spinal cord injury enhances anti-inflammatory FoxP3+ T regulatory cell (T-reg) recruitment and protects against white matter deterioration.
The host commensal microbiome also regulates neuronal myelination whereby GF mice exhibit hyper-myelinated axons in the pre-frontal cortex and enhanced expression of myelinating gene clusters at the transcriptomics level (Hoban et al., 2016). MS is a complex autoimmune disorder resulting in progressive demyelination of CNS-residing neurons with sufferers possessing altered gut microbial composition (Chen et al., 2016; Jangi et al., 2016). Strikingly, Proteobacteria is readily detected within inflammatory demyelinating lesions of cerebral white matter (Branton et al., 2016). Prophylactic administration of polysaccharide A (PSA), produced by the commensal Bacteroides fragilis, alleviates experimental autoimmune encephalitis-induced clinical-like symptoms, protects against demyelination and reduces pro-inflammatory cytokine burden (Wang et al., 2014). These beneficial observations result from PSA-induced expansion of CD39+/CD4+ T-cell populations through toll-like receptor (TLR)-2 signaling that regulate FoxP3+ T-reg function within the cervical and mesenteric lymph nodes. These studies suggest the microbiota influence critical immune compartments and regulate neuronal development, myelination, and recovery after injury. Given this, the field is now focusing on the role of the microbiome in neurodegenerative diseases that exhibit hallmark neuro-inflammatory pathology.
Stroke patients develop a vessel occlusion that deprives downstream tissue of vital nutrients, triggering a primary area of cell death. Upon tissue reperfusion a neuro-inflammatory response is initiated that contributes to a secondary injury termed the penumbra. ABX-treated mice subjected to the middle cerebral artery occlusion (MCAO) model of ischemic stroke display a 60% reduction in brain infarct volume and improved sensorimotor function (Benakis et al., 2016). These mice display elevated levels of FoxP3+ T-reg cells that suppress IL-17+ γδ T-cell proliferation and trafficking to the meninges post-MCAO. The observed reduction in brain CXCL1 and CXCL2 expression then alleviates inflammatory leukocyte infiltration within the infarct and improves outcome after the ischemic event.
In AD, deposition of amyloid-beta (Aβ) confers a plaque-localized neuro-inflammatory response that contributes to disease progression. A recent study has now correlated specific up-regulated pro-inflammatory gut bacteria with elevated pro-inflammatory mediator expression in clinically diagnosed AD patients (Cattaneo et al., 2016). Interestingly, AD sufferers exhibit increased susceptibility to bacterial infection and patients display plaque deposition co-localized with E. coli-derived LPS or K99 pili proteins (Zhan et al., 2016). Furthermore, intra-cerebral injection of viable Salmonella Typhimurium triggers seeding and accelerated Aβ deposition in 5xFAD mice (Kumar et al., 2016). These findings were validated in transgenic nematodes and reinforce the known anti-microbial nature of Aβ (Soscia et al., 2010), but potentially highlight a novel mechanism for AD exacerbation. A recent study now provides evidence that GF Thy1-APPSWE/PS1L166P mice exhibit reductions in Aβ pathology and microglial reactivity (Harach et al., 2017). Additionally, ABX-induced perturbations in the microbiome of APPSWE/PS1ΔE9 mice alters peripherally circulating cytokine profiles, confers reductions in Aβ deposition and reduces plaque-localized gliosis (Minter et al., 2016c). Both the aforementioned MCAO and APPSWE/PS1ΔE9 mouse studies demonstrate that microbial abundance post-ABX treatment recovers rapidly, however community diversity is significantly reduced with both displaying dominant expansion of Lachnospiraceae and Verrucomicrobiaceae. This implies that it is not necessarily the sheer number of microbiota, but rather their population diversity, that impact on host physiology.
Similar to AD, alpha-synuclein (α-syn) deposition observed in PD patients triggers a deposit-localized neuro-inflammatory response that facilitates disease progression. One un-conventional PD exacerbation hypothesis suggests that aberrant gut-localized accumulation of α-syn propagates to the brain via vagal nerve retrograde transport and stimulates pathogenic α-syn seeding (Burke et al., 2008; Del Tredici and Braak, 2008). Whilst this proposal remains controversial, it is now documented that PD patients harbor distinct alterations in microbial diversity of the gastrointestinal (GI) tract (Scheperjans et al., 2015; Unger et al., 2016). A recent seminal study now demonstrates that GF α-syn overexpressing mice display improved motor function and attenuated α-syn aggregation within the caudate putamen and substantia nigra in comparison to SPF-housed mice (Sampson et al., 2016). These mice display altered microglial morphology and reduced pro-inflammatory TNFα and IL-6 cytokine load and these observations are corroborated in ABX-treated α-syn mice. Supplementation of GF α-syn mice with microbially-derived SCFAs exacerbated motor deficits and importantly, GF mice colonized with fecal matter from PD patients resulted in enhanced motor dysfunction compared to mice that received material from healthy controls.
Similar to Aβ, α-syn possesses anti-microbial activity against multiple bacterial, yeast, and fungal strains (Park et al., 2016). In subsequent AD and PD studies it will be critical to address whether the attenuated Aβ and α-syn deposition observed in microbially-perturbed mouse models is a direct result of less protein seeding, due to a reduction in microbial stimulus, or an altered host immune response that triggers active peptide clearance. Regardless, these studies highlight the importance of the commensal microbiome in regulating host immunity and pathology in multiple neurodegenerative disease settings.
Addressing Microbiome-Host Interactions: A Role for Immune Barriers?
The aforementioned preclinical studies suggest that altering microbial composition elicits distinct changes in host immune compartments, impacting pathology in neurodegenerative disease models. This may be through direct modulation of microglial development and function within the brain parenchyma or through alteration of peripheral immune cell populations that communicate with the brain. The underlying question remains how a perturbation in the microbiome is communicated to the brain to induce functional changes and confer phenotypic alterations observed in these preclinical studies?
TLR expression on epithelial cells of the GI tract permits the host to constantly survey and react to the luminal microbiome (Mu et al., 2015). This monitoring mechanism ensures gut health by modulating host-immune responses to ensure microbial homeostasis. The vagal nerve innervates both mucosal villi and epithelial layers of the GI tract and can be activated in response to inflammatory mediators, produced from TLR signaling, or microbially-derived metabolites to initiate communication of the peripheral immune state to brain (Forsythe et al., 2014). Subsequent activation of the hypothalamic-pituitary-adrenal axis and the anti-inflammatory cholinergic neuronal reflex in the CNS results in significant alterations of innate immunity (Petra et al., 2015, Figure 1B).
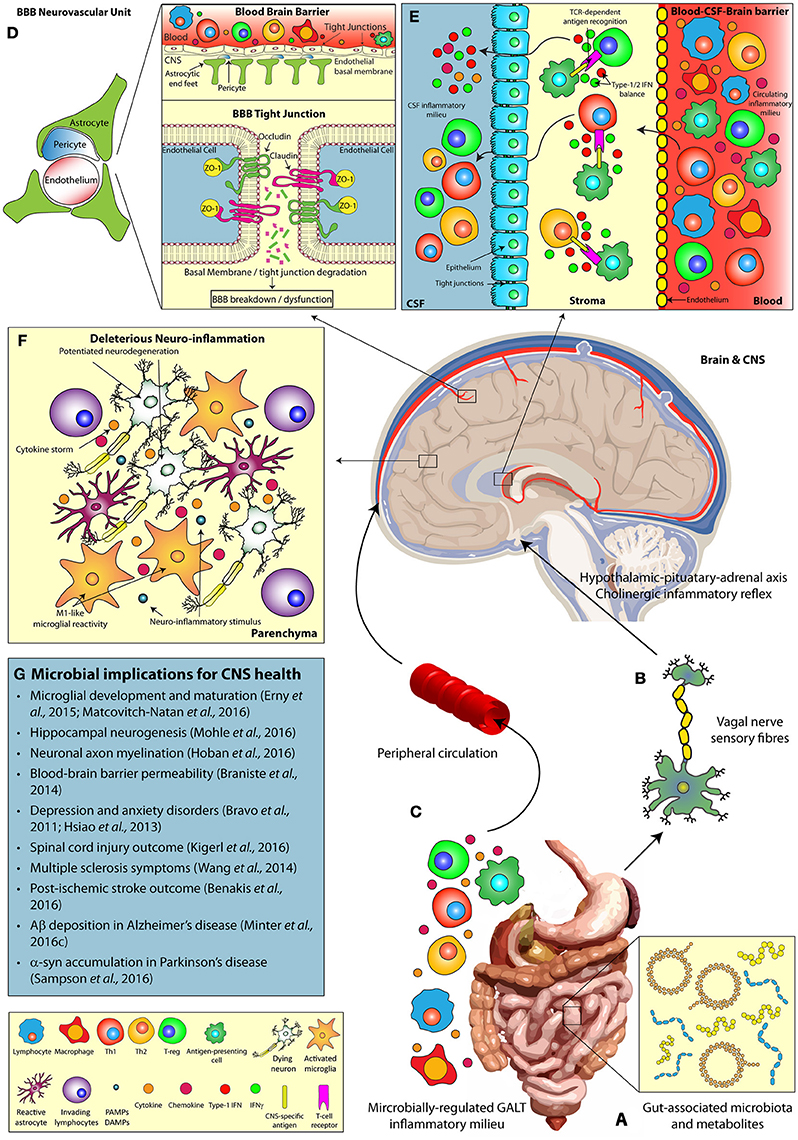
Figure 1. The microbiomes influence on host CNS health and neurodegenerative disease. Multiple pathways facilitate the bi-directional communication between the microbiome and the CNS, influencing brain function. (A) Alterations in gut microbial composition and metabolomics profiles can induce (B) direct stimulation of the vagal nerve, resulting in activation of the hypothalamic-pituitary-adrenal and regulation of innate CNS immune responses. (C) In addition, microbially-regulated GALT immune cell populations and the commensal microbiota themselves can alter the circulating peripheral inflammatory milieu (including cytokine profiles), which in turn play a key role in (D) the modulation of BBB integrity (via tight junction degradation) and (E) T cell infiltration at the BCSFBB. Alterations in the gut microbial diversity may also influence (F) deleterious neuro-inflammatory CNS responses, including modulations in microglial reactivity, and cytokine production, (G) impacting the progression of neurodegenerative disease states. α-syn, Alpha-synuclein; Aβ, Amyloid-beta; BBB, Blood-brain barrier; BCSFBB, Blood-cerebrospinal fluid-brain barrier; CD, Cluster of differentiation; CNS, Central nervous system; CSF, Cerebrospinal fluid; DAMP, Damage-associated molecular pattern; GALT, Gut-associated lymphoid tissue; IFN, Interferon; PAMP, Pattern-associated molecular pattern; TCR, T-cell receptor; Th, T-helper cell; T-reg, T-regulatory cell; ZO, Zonula occludens.
Vagal nerve activation in response to gut microbial dysbiosis is considered a major contributor to exacerbation of depressive and autism spectrum disorders. Mice populated with Lactobacillus rhamnosus displayed differential expression patterns of GABA receptors throughout the brain and attenuated depressive symptoms (Bravo et al., 2011). These beneficial probiotic effects were not observed in vagotomized mice and additional evidence describing similar beneficial effects of B. fragilis supplementation in a maternally-immune activated anxiety mouse model supports these findings (Hsiao et al., 2013). Alongside vagal activity an autonomic nervous mechanism conferring gut microbial perturbations following brain injury is also evident (Houlden et al., 2016).
Despite the importance of vagal nerve stimulation in gut-brain axis communication, there remains alternate immune barriers of the brain that are critical in sensing the microbially-regulated circulating peripheral inflammatory milieu (Figure 1C).
Blood Brain Barrier (BBB)
The BBB consists of capillary endothelial cells (connected by tight junctions), astrocytes, and pericytes that form the neurovascular unit responsible for barrier integrity (Sweeney et al., 2016). These barrier units separate the CNS and systemic circulation, thus regulating the brain microenvironment independently from the circulating inflammatory milieu. Critically, loss of BBB integrity significantly contributes to the progression of neurodegenerative disorders including TBI (Price et al., 2016), AD (Ryu and Mclarnon, 2009), and PD (Kortekaas et al., 2005). The commensal microbiota regulate peripheral immune cell compartments, implicated in these disorders, and secrete immunologically-active metabolites that circulate to the brain. Thus, the question remains of how the microbiome potentially regulates BBB permeability and stability (Figure 1D) and what implications this has for inflammatory responses in neurodegenerative diseases?
Indeed, gram positive bacterial components elicit pro-inflammatory cytokine responses which alter tight junction protein expression and disrupt BBB integrity in vitro (Boveri et al., 2006). Microbial perturbations induced by feeding rodents a “Western diet,” high in saturated fat and sugar (Claesson et al., 2012), triggers increased BBB permeability and cognitive deficits (Davidson et al., 2013). This observation suggests the microbiome may be a previously un-recognized regulator of BBB integrity. Indeed, a seminal study by Braniste et al. demonstrated elevated BBB permeability in fetal and adult GF mice independent of sex (Braniste et al., 2014). This BBB dysfunction was associated with reduced expression of the tight junction proteins occludin and claudin-5. Furthermore, colonization of GF mice with flora from SPF mice restored BBB integrity identified by reduced extravasation of Evans blue dye into brain parenchyma, alongside restored expression of occludin and claudin-5. Interestingly, these results were also replicated when GF mice were mono-colonized with the bacterial strains Clostridium tyrobutyricum and Bacteroides thetaiotaomicron, both of which produce the SCFAs butyrate, acetate and propionate. Collectively, these data highlight the importance of specific microbial populations, particularly SCFA-producing strains, in regulating BBB physiology.
The therapeutic benefits of butyrate, provided either as pure sodium butyrate or generated by SCFA-producing bacteria, has been recognized in neuropsychiatric disorders (Bourassa et al., 2016), yet the underlying mechanism(s) by which it influences BBB function remains unclear. In vivo evidence suggests that the protective effect of sodium butyrate relates to its ability to stabilize BBB integrity, with sodium butyrate treated GF mice displaying reduced Evans blue extravasation in multiple brain regions and increased occludin levels (Braniste et al., 2014). In a model of transient focal cerebral ischemia sodium butyrate attenuated BBB permeability, suppressed NFκB-mediated production of MMP9, and prevented tight junction degradation (Wang et al., 2011). In a mouse model of TBI, decreased expression of occludin and Zonula Occludens-1 (ZO-1) was identified 24 h post-injury, a response that was ameliorated with sodium butyrate treatment, resulting in restoration of BBB integrity and beneficial neurological outcomes (Li et al., 2016). Whether these changes in barrier permeability induced by circulating SCFAs involve direct effects on the brain endothelium or indirect signaling by the enteroendocrine cells of the gut warrants further investigation.
Microbial metabolites can also influence immune responses in the periphery, which in turn affect BBB integrity through activation of TLRs. Lipoteichoic acid (LTA), a cell wall component of gram-positive bacteria and TLR2 agonist, activates peripheral immune responses and downstream signaling cascades, resulting in BBB dysfunction and behavioral abnormalities. In this manner, intra-peritoneal administration of bacillus subtilis-derived LTA increases pro-inflammatory IL-1β, IL-6, TNFα, and IFNγ cytokine expression in the circulation (Mayerhofer et al., 2017). These LTA-induced immunological alterations are accompanied by decreased claudin-5, occludin and tight junction protein-1 expression in the amygdala, highlighting the capacity of LTA to influence BBB composition through peripheral-CNS communication.
In addition to the regulation of BBB integrity by the microbiome, astonishing new findings now suggest that the microbiome impacts transport mechanisms across an intact barrier. In this manner, microbially-derived peptidoglycan (PGN) crosses the BBB, activating various PGN-recognition proteins (Pglrp1, Pglrp2, Pglrp3, Pglrp4), as well as TLR2 and the NOD-like receptors in the developing brain (Arentsen et al., 2017). Manipulation of the microbiome through GF and/or ABX-treated mice reduces CNS expression of the aforementioned recognition proteins, as well as the PGN transporter (PepT1). Interestingly, genetic deletion of Pglrp2 changes synapse-related gene expression and induces sex-dependent variations in social behavior, similar to that observed in microbially-perturbed mice (Arentsen et al., 2017). These findings challenge traditional view that microbial products only translocate into the brain in situations of compromised BBB integrity. This discovery of independent transit across the BBB and subsequent CNS immune signaling presents a novel axis for future investigation into the mechanism underlying microbiota-brain communication.
Blood-Cerebrospinal Fluid-Brain Barrier (BCSFBB)
Diversification of the microbiota and their metabolites is a critical regulator of peripheral immune cell proliferation and activity (Dorrestein et al., 2014). In particular, the microbiome closely regulates the innate lymphoid cell populations of the gastro-intestinal tract (Gury-Benari et al., 2016) and these cells can, in turn, modulate T-cell populations residing in the lamina propia and mesenteric lymph nodes (Sonnenberg and Artis, 2015). T-cells circulate around the body and enter the brain via the choroid plexus (CP) (Engelhardt and Ransohoff, 2005), responsible for producing cerebrospinal fluid (CSF), whereby they contribute to neuro-inflammatory events and modulate microglial activity. Hence alterations in the commensal microbiome may be communicated to the brain via an altered immune status at the BCSFBB (Figure 1E).
The composition of immune cells populating the CP has important ramifications for neuro-inflammatory responses and brain health. The CP is populated with a subset of CD4+ memory T-cells that display T-cell receptor profiles unique to CNS-derived antigens. In aged mice these cells favor a Th2 response with elevated IL-4 and decreased type-2 IFNγ production that stimulates the CP epithelium to drive CCL11 production known to be deleterious to brain function (Villeda et al., 2011). Experimentally-induced proliferation of memory T cells restored the IL-4:IFNγ imbalance at the CP in these mice and prevented cognitive deficits (Baruch et al., 2013). Additional studies have now identified a CP-localized type-1 IFN response that drives deleterious aging. Importantly, aged mice supplemented with a type-1 IFN neutralizing antibody displayed reduced CCL11 expression, attenuated hippocampal glial reactivity, elevated BDNF production, and improved neurogenesis (Baruch et al., 2014). A dysregulated type-1 IFN signature has been observed in aging disorders, that appear to be regulated by the microbiome, including AD (Taylor et al., 2014; Minter et al., 2016a) and PD (Main et al., 2016) and it is now considered that a balance of type-1 and type-2 IFN signaling at the CP is required to impart healthy brain immunity and aging (Deczkowska et al., 2016).
Studies now suggest that intervening with “immune checkpoints” to re-correct immune cell phenotypes promotes clearance of pathogenic peptides (Aβ or α-syn) and cellular debris in neuro-degenerative disorders. Transient deletion of FoxP3+ T-regs triggers an IFNγ-mediated immune activation of the CP and results in recruitment of peripheral macrophages and repopulating T-regs, facilitating removal of Aβ plaques and restoring cognitive function in 5xFAD mice (Baruch et al., 2015). Similar beneficial IFNγ-dependent immune activity and Aβ clearance has also been demonstrated by programmed cell death protein-1 (PD-1) immune checkpoint blockade in the same animal model (Baruch et al., 2016). It is evident that re-programming, not dampening, of immune responses is critical in the treatment of neurodegenerative disorders. Considering perturbations in commensal microbial diversity alter peripheral T-cell compartments it will be critical to assess how these cell populations are affected at the CP immune barrier. Furthermore, it remains plausible that alterations in microbial diversity may trigger transient changes in T-cell phenotypes recruited to the CP that mimic the protective or deleterious phenotypes of CP-recruited cells observed in the aforementioned studies. Additional studies focusing on the BCSFBB in microbially-perturbed neurodegenerative disease models will provide much needed insight into how the commensal microbiome regulates brain immunity and identify novel therapeutic targets.
Conclusion
Evidence presented from numerous studies now indicates the commensal microbiome exhibits fine control over host immunity that may have implications in developmental and neurodegenerative disorders. The precise mechanisms by which a perturbation in the host microbiome triggers alterations in peripheral immune compartments and neuro-inflammatory responses remains far from understood. We propose that investigations focusing on “immune barriers” of the brain, namely the BBB and BCSFBB, will provide important insights into how the microbiome communicates with the brain and we encourage future studies targeting the microbiome in neurodegenerative diseases to address these barriers (Figure 1). These studies may identify novel microbially-derived therapies to induce host immunological alterations and treat these disorders.
Author Contributions
BM and MM conceptualized, intellectualized, wrote, reviewed, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Cure Alzheimer's Fund. MM would like to acknowledge Prof. Sangram S. Sisodia (University of Chicago, Chicago, IL, USA) for mentorship.
Abbreviations
α-syn, Alpha-synuclein; ABX, Antibiotics; AD, Alzheimer's disease; APPSWE, Amyloid precursor protein (Swedish mutant); Aβ, Amyloid-beta; BBB, Blood-brain barrier; BCSFBB, Blood-cerebrospinal fluid-brain barrier; BDNF, Brain-derived neurotrophic factor; CCL, Chemokine (C-C motif) ligand; CD, Cluster of differentiation; CNS, Central nervous system; CP, Choroid plexus; CSF, Cerebrospinal fluid; CXCL, Chemokine (C-X-C motif) ligand; DAMP, Damage-associated molecular pattern; DNA, Deoxyribonucleic acid; EAE, Experimental autoimmune encephalitis; FAD, Familial Alzheimer's disease; FoxP3, Forkhead box P3; GABA, Gamma-aminobutyric acid; GALT, Gut-associated lymphoid tissue; GF, Germ-free; GI, Gastrointestinal; HPA, Hypothalamic-pituitary axis; IFN, Interferon; IL, Interleukin; LPS, Lipopolysaccharide; LTA, Lipoteichoic acid; MCAO, Middle cerebral artery occlusion; MMP, Matrix metalloproteinase; MS, Multiple Sclerosis; NOD, Nucleotide-binding oligomerization domain; NFκB, Nuclear factor kappa B; PD, Parkinson's disease; PD-1, Programmed cell death protein-1; PGN, Peptidoglycan; PS1ΔE9, Presenilin-1 (exon 9 deletion); PSA, Polysaccharide A; SCFA, Short-chain fatty acid; SPF, Specific pathogen-free; TBI, Traumatic brain injury; TLR, Toll-like receptor; TNF, Tumour necrosis factor; T-reg, T-regulatory cell; ZO-1, Zona occludens-1.
References
Arentsen, T., Qian, Y., Gkotzis, S., Femenia, T., Wang, T., Udekwu, K., et al. (2017). The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol. Psychiatry 22, 257–266. doi: 10.1038/mp.2016.182
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Baruch, K., Deczkowska, A., Rosenzweig, N., Tsitsou-Kampeli, A., Sharif, A. M., Matcovitch-Natan, O., et al. (2016). PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat. Med. 22, 135–137. doi: 10.1038/nm.4022
Baruch, K., Ron-Harel, N., Gal, H., Deczkowska, A., Shifrut, E., Ndifon, W., et al. (2013). CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc. Natl. Acad. Sci. U.S.A. 110, 2264–2269. doi: 10.1073/pnas.1211270110
Baruch, K., Rosenzweig, N., Kertser, A., Deczkowska, A., Sharif, A. M., Spinrad, A., et al. (2015). Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat. Commun. 6:7967. doi: 10.1038/ncomms8967
Benakis, C., Brea, D., Caballero, S., Faraco, G., Moore, J., Murphy, M., et al. (2016). Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat. Med. 22, 516–523. doi: 10.1038/nm.4068
Bourassa, M. W., Alim, I., Bultman, S. J., and Ratan, R. R. (2016). Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci. Lett. 625, 56–63. doi: 10.1016/j.neulet.2016.02.009
Boveri, M., Kinsner, A., Berezowski, V., Lenfant, A. M., Draing, C., Cecchelli, R., et al. (2006). Highly purified lipoteichoic acid from gram-positive bacteria induces in vitro blood-brain barrier disruption through glia activation: role of pro-inflammatory cytokines and nitric oxide. Neuroscience 137, 1193–1209. doi: 10.1016/j.neuroscience.2005.10.011
Braniste, V., Al-Asmakh, M., Kowal, C., Anuar, F., Abbaspour, A., Toth, M., et al. (2014). The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 6:263ra158. doi: 10.1126/scitranslmed.3009759
Branton, W. G., Lu, J. Q., Surette, M. G., Holt, R. A., Lind, J., Laman, J. D., et al. (2016). Brain microbiota disruption within inflammatory demyelinating lesions in multiple sclerosis. Sci. Rep. 6:37344. doi: 10.1038/srep37344
Bravo, J. A., Forsythe, P., Chew, M. V., Escaravage, E., Savignac, H. M., Dinan, T. G., et al. (2011). Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. U.S.A. 108, 16050–16055. doi: 10.1073/pnas.1102999108
Burke, J. F., Gnall, E., Umrudden, Z., Kyaw, M., and Schick, P. K. (2008). Critical analysis of a computer-assisted tutorial on ECG interpretation and its ability to determine competency. Med. Teach. 30, e41–e48. doi: 10.1080/01421590801972471
Cattaneo, A., Cattane, N., Galluzzi, S., Provasi, S., Lopizzo, N., Festari, C., et al. (2016). Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68. doi: 10.1016/j.neurobiolaging.2016.08.019
Chen, J., Chia, N., Kalari, K. R., Yao, J. Z., Novotna, M., Soldan, M. M., et al. (2016). Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 6:28484. doi: 10.1038/srep28484
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O'connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi: 10.1038/nature11319
Davidson, T. L., Hargrave, S. L., Swithers, S. E., Sample, C. H., Fu, X., Kinzig, K. P., et al. (2013). Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience 253, 110–122. doi: 10.1016/j.neuroscience.2013.08.044
Deczkowska, A., Baruch, K., and Schwartz, M. (2016). Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol. 37, 181–192. doi: 10.1016/j.it.2016.01.006
Del Tredici, K., and Braak, H. (2008). A not entirely benign procedure: progression of Parkinson's disease. Acta Neuropathol. 115, 379–384. doi: 10.1007/s00401-008-0355-5
Dorrestein, P. C., Mazmanian, S. K., and Knight, R. (2014). Finding the missing links among metabolites, microbes, and the host. Immunity 40, 824–832. doi: 10.1016/j.immuni.2014.05.015
Du, L., Zhang, Y., Chen, Y., Zhu, J., Yang, Y., and Zhang, H. L. (2016). Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. doi: 10.1007/s12035-016-0245-0. [Epub ahead of print].
Engelhardt, B., and Ransohoff, R. M. (2005). The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 26, 485–495. doi: 10.1016/j.it.2005.07.004
Erny, D., Hrabe de Angelis, A. L., Jaitin, D., Wieghofer, P., Staszewski, O., David, E., et al. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977. doi: 10.1038/nn.4030
Forsythe, P., Bienenstock, J., and Kunze, W. A. (2014). Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 817, 115–133. doi: 10.1007/978-1-4939-0897-4_5
Gury-Benari, M., Thaiss, C. A., Serafini, N., Winter, D. R., Giladi, A., Lara-Astiaso, D., et al. (2016). The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231.e1213–1246.e1213. doi: 10.1016/j.cell.2016.07.043
Harach, T., Marungruang, N., Duthilleul, N., Cheatham, V., Mc Coy, K. D., Frisoni, G., et al. (2017). Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7:41802. doi: 10.1038/srep41802
Hoban, A. E., Stilling, R. M., Ryan, F. J., Shanahan, F., Dinan, T. G., Claesson, M. J., et al. (2016). Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry 6:e774. doi: 10.1038/tp.2016.42
Honda, K., and Littman, D. R. (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. doi: 10.1038/nature18848
Houlden, A., Goldrick, M., Brough, D., Vizi, E. S., Lenart, N., Martinecz, B., et al. (2016). Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun. 57, 10–20. doi: 10.1016/j.bbi.2016.04.003
Hsiao, E. Y., Mcbride, S. W., Hsien, S., Sharon, G., Hyde, E. R., Mccue, T., et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463. doi: 10.1016/j.cell.2013.11.024
Jangi, S., Gandhi, R., Cox, L. M., Li, N., von Glehn, F., Yan, R., et al. (2016). Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7:12015. doi: 10.1038/ncomms12015
Juarez, I., Gratton, A., and Flores, G. (2008). Ontogeny of altered dendritic morphology in the rat prefrontal cortex, hippocampus, and nucleus accumbens following Cesarean delivery and birth anoxia. J. Comp. Neurol. 507, 1734–1747. doi: 10.1002/cne.21651
Karve, I. P., Taylor, J. M., and Crack, P. J. (2016). The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 173, 692–702. doi: 10.1111/bph.13125
Kigerl, K. A., Hall, J. C., Wang, L., Mo, X., Yu, Z., and Popovich, P. G. (2016). Gut dysbiosis impairs recovery after spinal cord injury. J. Exp. Med. 213, 2603–2620. doi: 10.1084/jem.20151345
Kim, H. R., Jung, K. Y., Kim, S. Y., Ko, K. O., Lee, Y. M., and Kim, J. M. (2003). Delivery modes and neonatal EEG: spatial pattern analysis. Early Hum. Dev. 75, 35–53. doi: 10.1016/j.earlhumdev.2003.09.004
Kortekaas, R., Leenders, K. L., Van Oostrom, J. C., Vaalburg, W., Bart, J., Willemsen, A. T., et al. (2005). Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 57, 176–179. doi: 10.1002/ana.20369
Kumar, D. K., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 8:340ra372. doi: 10.1126/scitranslmed.aaf1059
Li, H., Sun, J., Wang, F., Ding, G., Chen, W., Fang, R., et al. (2016). Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 1642, 70–78. doi: 10.1016/j.brainres.2016.03.031
Main, B. S., Zhang, M., Brody, K. M., Ayton, S., Frugier, T., Steer, D., et al. (2016). Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia 64, 1590–1604. doi: 10.1002/glia.23028
Matcovitch-Natan, O., Winter, D. R., Giladi, A., Vargas Aguilar, S., Spinrad, A., Sarrazin, S., et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:aad8670. doi: 10.1126/science.aad8670
Mayerhofer, R., Frohlich, E. E., Reichmann, F., Farzi, A., Kogelnik, N., Frohlich, E., et al. (2017). Diverse action of lipoteichoic acid and lipopolysaccharide on neuroinflammation, blood-brain barrier disruption, and anxiety in mice. Brain Behav. Immun. 60, 174–187. doi: 10.1016/j.bbi.2016.10.011
Minter, M. R., Moore, Z., Zhang, M., Brody, K. M., Jones, N. C., Shultz, S. R., et al. (2016a). Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 4:72. doi: 10.1186/s40478-016-0341-4
Minter, M. R., Taylor, J. M., and Crack, P. J. (2016b). The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. J. Neurochem. 136, 457–474. doi: 10.1111/jnc.13411
Minter, M. R., Zhang, C., Leone, V., Ringus, D. L., Zhang, X., Oyler-Castrillo, P., et al. (2016c). Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6:30028. doi: 10.1038/srep30028
Mohle, L., Mattei, D., Heimesaat, M. M., Bereswill, S., Fischer, A., Alutis, M., et al. (2016). Ly6C(hi) Monocytes provide a link between antibiotic-induced changes in Gut microbiota and adult hippocampal neurogenesis. Cell Rep. 15, 1945–1956. doi: 10.1016/j.celrep.2016.04.074
Mu, C., Yang, Y., and Zhu, W. (2015). Crosstalk between the immune receptors and Gut microbiota. Curr. Protein Pept. Sci. 16, 622–631. doi: 10.2174/1389203716666150630134356
Park, S. C., Moon, J. C., Shin, S. Y., Son, H., Jung, Y. J., Kim, N. H., et al. (2016). Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem. Biophys. Res. Commun. 478, 924–928. doi: 10.1016/j.bbrc.2016.08.052
Patejdl, R., Penner, I. K., Noack, T. K., and Zettl, U. K. (2016). Multiple sclerosis and fatigue: a review on the contribution of inflammation and immune-mediated neurodegeneration. Autoimmun. Rev. 15, 210–220. doi: 10.1016/j.autrev.2015.11.005
Petra, A. I., Panagiotidou, S., Hatziagelaki, E., Stewart, J. M., Conti, P., and Theoharides, T. C. (2015). Gut-microbiota-brain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin. Ther. 37, 984–995. doi: 10.1016/j.clinthera.2015.04.002
Price, L., Wilson, C., and Grant, G. (2016). “Chapter 4: Blood-brain barrier pathophysiology following traumatic brain injury,” in Translational Research in Traumatic Brain Injury, eds D. Laskowitz and G. Grant (Boca Raton, FL: CRC Press/Taylor and Francis Group).
Ryu, J. K., and Mclarnon, J. G. (2009). A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J. Cell. Mol. Med. 13, 2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167, 1469.e12–1480.e12. doi: 10.1016/j.cell.2016.11.018
Scheperjans, F., Aho, V., Pereira, P. A., Koskinen, K., Paulin, L., Pekkonen, E., et al. (2015). Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov. Disord. 30, 350–358. doi: 10.1002/mds.26069
Sonnenberg, G. F., and Artis, D. (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 21, 698–708. doi: 10.1038/nm.3892
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 5:e9505. doi: 10.1371/journal.pone.0009505
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Taylor, J. M., Main, B. S., and Crack, P. J. (2013). Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson's disease. Neurochem. Int. 62, 803–819. doi: 10.1016/j.neuint.2012.12.016
Taylor, J. M., Minter, M. R., Newman, A. G., Zhang, M., Adlard, P. A., and Crack, P. J. (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol. Aging 35, 1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089
Unger, M. M., Spiegel, J., Dillmann, K. U., Grundmann, D., Philippeit, H., Burmann, J., et al. (2016). Short chain fatty acids and gut microbiota differ between patients with Parkinson's disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72. doi: 10.1016/j.parkreldis.2016.08.019
Villeda, S. A., Luo, J., Mosher, K. I., Zou, B., Britschgi, M., Bieri, G., et al. (2011). The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. doi: 10.1038/nature10357
Wang, Y., Telesford, K. M., Ochoa-Reparaz, J., Haque-Begum, S., Christy, M., Kasper, E. J., et al. (2014). An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat. Commun. 5:4432. doi: 10.1038/ncomms5432
Wang, Z., Leng, Y., Tsai, L. K., Leeds, P., and Chuang, D. M. (2011). Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab. 31, 52–57. doi: 10.1038/jcbfm.2010.195
Xiong, X. Y., Liu, L., and Yang, Q. W. (2016). Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 142, 23–44. doi: 10.1016/j.pneurobio.2016.05.001
Keywords: microbiome, immunity, blood brain barrier, blood cerebrospinal fluid-brain barrier, Alzheimer's disease, Parkinson's disease, stroke, neuro-inflammation
Citation: Main BS and Minter MR (2017) Microbial Immuno-Communication in Neurodegenerative Diseases. Front. Neurosci. 11:151. doi: 10.3389/fnins.2017.00151
Received: 13 January 2017; Accepted: 09 March 2017;
Published: 23 March 2017.
Edited by:
Tibor Hortobágyi, University of Debrecen, HungaryReviewed by:
Adam Denes, University of Manchester, UKPaola Bossù, Fondazione Santa Lucia (IRCCS), Italy
Egle Solito, Queen Mary University of London, UK
Copyright © 2017 Main and Minter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bevan S. Main, YnNtNTNAZ2VvcmdldG93bi5lZHU=
Myles R. Minter, bW1pbnRlckB1Y2hpY2Fnby5lZHU=