 Alexander Hartmann1†
Alexander Hartmann1† Susanne Krasemann
Susanne Krasemann Markus Glatzel
Markus Glatzel- 1Center of Diagnostics, Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Center for Applied Nanotechnology, GmbH, Hamburg, Germany
Exosomes are involved in the progression of neurodegenerative diseases. The cellular prion protein (PrPC) is highly expressed on exosomes. In neurodegenerative diseases, PrPC has at least two functions: It is the substrate for the generation of pathological prion protein (PrPSc), a key player in the pathophysiology of prion diseases. On the other hand, it binds neurotoxic amyloid-beta (Aß) oligomers, which are associated with initiation and progression of Alzheimer's disease (AD). This has direct consequences for the role of exosomal expressed PrPC. In prion diseases, exosomal PrP leads to efficient dissemination of pathological prion protein, thus promoting spreading and transmission of the disease. In AD, exosomal PrPC can bind and detoxify Aß oligomers thus acting protective. In both scenarios, assessment of the state of PrPC on exosomes derived from blood or cerebrospinal fluid (CSF) may be useful for diagnostic workup of these diseases. This review sums up current knowledge of the role of exosomal PrPC on different aspects of Alzheimer's and prion disease.
Introduction
Due to demographic shift in modern societies, neurodegenerative diseases are increasingly prevalent mainly among the elderly population (Prince et al., 2013). The progressive and irreversible degeneration of synapses and neurons leading to nervous system dysfunction is a hallmark of these diseases. In the vast majority of neurodegenerative conditions neuronal decay associates with generation and aggregation of specific proteins in the brain, thus these diseases are termed cerebral proteinopathies (Jellinger, 2003). A rare, yet well studied example of this group of diseases are prion diseases, where misfolding and deposition of PrPC into its pathogenic counterpart PrPSc plays a key role in disease initiation and progression (Prusiner, 1982; Aguzzi and Lakkaraju, 2016). Alzheimer's disease, on the other hand, represents a highly prevalent example of a cerebral proteinopathy (Walsh and Selkoe, 2016). Here, generation and deposition of aggregation-prone Aß peptide is involved in disease pathogenesis. The two diseases are linked not only by a considerable overlap regarding clinical presentation but also by shared risk genes and shared molecular pathways underlying neurodegeneration (Uchiyama et al., 2013; Udayar et al., 2013). Accordingly, it was not surprising to see that PrPC specifically binds ß-sheet rich proteins such as aggregated Aß or aggregated PrPSc (Lauren et al., 2009; Resenberger et al., 2011; Um et al., 2012; Falker et al., 2016). Consequences of this binding are less clear with some studies identifying PrPC as a high affinity receptor for oligomeric Aß transducing neurotoxic signaling (Walsh et al., 2002; Lesne et al., 2006), whereas others favor a role of this binding in clearance of neurotoxic proteins (Pflanzner et al., 2012). Furthermore, PrPC participates in Aß transcytosis across the blood-brain barrier (Devraj et al., 2016).
How misfolded protein species spread from neuron to neuron in the central nervous system (CNS) in neurodegenerative diseases is a matter of debate. Direct cell-cell contact might play a role, but in recent years other mechanisms such as vesicular transport have been proposed (Kalani et al., 2014). Exosomes are small extracellular vesicles that may facilitate spreading of disease pathology in dementia (Fevrier et al., 2004). Neuronal exosomes are highly enriched in PrPC. Because of the multiple roles of PrP in neurodegenerative diseases, a closer look is warranted. Thus, here we focus on the multiple roles that exosomal PrPC might play in the pathophysiology of neurodegenerative diseases.
The Cellular Prion Protein (PrPC)
The membrane-associated glycoprotein PrPC is involved in diverse processes including neuronal differentiation, neuroprotection, signal transduction, and cell adhesion (Vassallo and Herms, 2003; Aguzzi and Lakkaraju, 2016). It is highly expressed on neurons and present to a lower extent on other cell types such as lymphoid cells and myocytes (Vassallo and Herms, 2003; Aguzzi and Lakkaraju, 2016). The prion protein is composed of an unstructured N-terminal domain and a globular structured C-terminus, comprising mostly alpha-helices. PrPC is C-terminally anchored to the outer leaflet of the plasma membrane via its glycosylphosphatidylinositol (GPI) anchor and positioned in lipid rafts (Riek et al., 1997; Biasini et al., 2012).
In prion diseases, PrPC is converted into a disease-associated isoform of itself termed PrPSc. Seeds of multiple PrPSc-molecules promote further conversion of PrPC into PrPSc in a self-propagating mechanism thereby multiplying the amount of PrPSc and leading to aggregation and deposition of PrPSc in the brain (Telling et al., 1996). Whereas PrPC displays an alpha-helical structure, PrPSc is richer in beta-sheets. The latter renders PrPSc more stable against proteolytic digestion. Its partial resistance to proteinase K digestion is used as a diagnostic tool in prion diseases. However, it should be noted that protease sensitive prion species exist and that the relation between PK-resistant PrPSc and prion infectivity is not linear (Manson et al., 1999; Krasemann et al., 2013). A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy (Manson et al., 1999) and it is hypothesized that more than one prion species exist.
Exosomes in Physiology
Exosomes are small membranous vesicles found in a variety of body fluids and the extracellular space, with a diameter ranging from 50 to 150 nm (Raposo et al., 2011). They are generated by invagination of endosomal membranes to form intraluminal vesicles (ILVs) within multivesicular bodies (MVBs). Sorting of ILVs to lysosomes leads to degradation, whereas fusion of MVBs with the plasma membrane and subsequent exocytosis leads to release of these extracellular vesicles (Raposo et al., 2011; Guo et al., 2016). Thus, the distinction of exosomes from other extracellular vesicles such as microvesicles, which directly bud from the plasma membrane, is not trivial with an obvious morphological overlap between the two. Nevertheless, for terms of clarity and to facilitate integration of existing literature, in this review we will use the term “exosomes” to describe a subset of small extracellular vesicles defined by their size, their separation on sucrose density gradients, their protein composition and their shape (Falker et al., 2016). Most commonly, differential centrifugation is used for isolating exosomes from cell culture media or body fluids (Leblanc et al., 2017). Most of the papers cited in this article used modified versions of this ultracentrifugation protocol for exosome isolation. Further characterization using marker proteins for vesicles derived from intracellular multivesicular bodies (CD9, CD63, CD81, and TSG101) are used to ensure presence of exosomes. However, relative ratios of these “exosomal markers” differ between exosomes derived from different cell types (Haraszti et al., 2016) and even further purification with sucrose gradient does not lead to uniform vesicle populations regarding size and marker profile (Bobrie et al., 2012). Thus, it is prudent to point out that extracellular vesicles isolated using current protocols for exosome-isolation, lead to vesicle populations, which are enriched in exosomes, but certainly also contain vesicles of non-exosomal origin (Bobrie et al., 2012; Lotvall et al., 2014; Chernyshev et al., 2015; Kreimer et al., 2015; Abramowicz et al., 2016; Gardiner et al., 2016; Leblanc et al., 2017).
Exosomes transfer cargo such as proteins, lipids and nucleic acids from donor to recipient cells over long distances in a seemingly targeted fashion (Yanez-Mo et al., 2015). This is why they may play important roles in intercellular communication in CNS, where they might mediate neuronal and glia communication (Kramer-Albers and Hill, 2016), promote neuronal repair and growth, regulate the immune response and present antigens (Fröhlich et al., 2014; Yanez-Mo et al., 2015; Guo et al., 2016).
The protein signature of exosomes with a relative overrepresentation of plasma membrane proteins, cytosolic proteins and proteins involved in vesicle trafficking reflects their membranous origin. Interestingly, although PrPC is not considered a marker for exosomes, it is highly expressed on these and seems to be actively sorted into exosomes (Hill et al., 2008; Vella et al., 2008a; Falker et al., 2016).
The Role of Exosomes in Neurodegeneration
Exosomes are of interest in neurodegenerative disease for a number of reasons (Table 1). Firstly, due to their release into the extracellular space, they are attractive targets for diagnostic procedures (Liu et al., 2014; Goetzl et al., 2015).
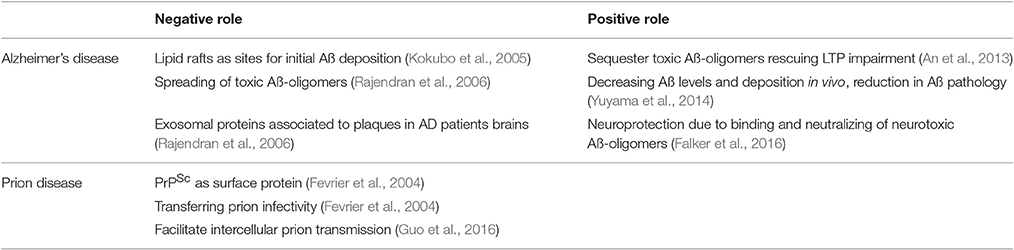
Table 1. Comparison of the function of exosomal PrP in different diseases.
Furthermore, exosomes play key roles in the pathophysiology of neurodegenerative diseases. Obviously, involvement of exosomes has been documented in prion-diseases such as Creutzfeldt-Jakob disease but also other more common forms of dementias such as Huntington-disease, tauopathies, amyotrophic lateral sclerosis, and Alzheimer's disease (Rajendran et al., 2006; Saa et al., 2014; Asai et al., 2015; Jeon et al., 2016; Polanco et al., 2016; Silverman et al., 2016).
Exosomal PrP in the Pathophysiology of Prion Disease: Spreading the Disease
Although there is only limited research on the role of exosomal PrP in prion disease, it is assumed, that exosomal PrPSc promotes dissemination of the disease within the CNS and in the lymphoreticular system.
The first studies suggesting an involvement of exosomal PrP in prion diseases came from cell culture based studies, which revealed an association between exosomes and PrPSc in media of prion infected cells (Fevrier et al., 2004). Further studies showed a link between release of retrovirus particles, presence of PrPSc, and prion infectivity on both, exosomes and retroviral particles, thus it was proposed that retroviral infection could be a cofactor in the spreading of prion disease (Alais et al., 2012). However, subsequent in vivo studies by our group and others did not provide evidence for this, but rather showed that subclinical retroviral infection acts as a disease modifier, but does not enhance spreading of the disease (Alais et al., 2012; Krasemann et al., 2012; Muth et al., 2016).
Further studies focusing on release of PrPSc from cells not only provided evidence that PrPSc associates with exosomes but also showed that release of PrPSc and prion infectivity could be attenuated by interfering with exosome biogenesis through inhibition of the endosomal sorting complex required for transport (ESCRT; Alais et al., 2008; Vilette et al., 2015). In line with this study, further research showed that pharmacological stimulation of exosome release by treatment with the ionophore Monensin increased release of infectious exosomes. Moreover, exosomes isolated from these in vitro experiments were able to transmit prion disease in vivo (Guo et al., 2016). The group of Vella described similar effects in vitro and in vivo. They showed that exosomes from prion-infected neuronal cell lines are capable to initiate prion propagation in uninfected non-neuronal cells and that these exosomes also provoke prion disease after inoculation in mice (Vella et al., 2007). In peripheral tissues, exosomes are released from prion infected dendritic cells possibly explaining the rapid colonization of prions in the lymphoreticular system (Klohn et al., 2013).
Further evidence for a role of exosomes in transmitting prion disease came from studies on blood-derived exosomes from prion-infected mice. Here, it was shown that those exosomes contained prion infectivity and injection into animals lead to successful transmission of disease (Cervenakova et al., 2016).
Exosomal PrP in the Diagnosis of Prion Disease: Potentially Useful
Human prion diseases show a wide spectrum of clinical presentations with disease durations ranging from months to several years (Geissen et al., 2007). Specific isoforms of PrPSc are related to specific human prion strains, which show differences in deposition pattern and disease manifestation (Wadsworth and Collinge, 2011). Since the definite diagnosis of human prion disease is only possible in a post mortem exanimation, current protocols to establish a probable diagnosis involve assessment of medical history, clinical symptoms and auxiliary tests such as analysis of body fluids e.g., cerebrospinal fluid (Glatzel et al., 2005; Collins et al., 2006). With current approaches assessing markers of neuronal death as surrogates for prion-caused neurodegeneration it is not possible to differentiate between prion strains. Recent data showed differences in the sorting of different prion strains into exosomes (Arellano-Anaya et al., 2015). Additionally, new techniques enable detection of minimal amounts of exosome associated PrPSc in blood (Berrone et al., 2015; Properzi et al., 2015). Thus, it is possible that the function of exosomes to spread prion infectivity and PrPSc in the body, may in fact be advantageous if exploited in a diagnostic setting.
Exosomal PrP in the Pathophysiology of AD: Neurotoxicity vs. Protection
In AD, Aß-oligomers rather than Aß aggregates cause synaptic dysfunction and network failure (Lesne et al., 2006). In advanced AD stages the Aß-oligomer equilibrium in the brain is shifted to more fibrillary Aß occurring as Aß-plaques. Nevertheless, there are still soluble oligomeric forms of Aß present, potentially leading to neurotoxicity.
In 2005 it was described that membrane-bound Aß is associated with lipid rafts within senile plaques, suggesting rafts as sites for initial Aß deposition (Kokubo et al., 2005). Hereon, it could be shown that exosomal proteins likewise accumulated in plaques of AD patient brains, suggesting a role of exosomes in AD (Rajendran et al., 2006). Interestingly, the inhibition of exosome secretion in an AD mouse model resulted in the reduction of Aß and Aß plaque formation, suggesting a disease promoting role for exosomes in AD (Dinkins et al., 2014).
In contrast, exosomes have been shown to induce Aß aggregation into non-toxic fibrils thus decreasing synaptotoxicity by clearance of potentially neurotoxic small Aß-oligomers in the extracellular space, possibly by enabling more efficient Aß uptake into microglia (Yuyama et al., 2012, 2014). In order to achieve these effects, Aß has to associate with exosomes. In principle, exosomes may be directly capable to process the amyloid precursor protein into Aß peptides (Vella et al., 2008b). However, it is more likely that cell derived Aß binds to exosomes. How this binding is achieved is a matter of debate and will be discussed below.
Exosomes have been shown to bind toxic Aß-oligomers and favor accelerated conversion into nontoxic Aß-fibrils resulting in protection from Aß-oligomer induced toxicity (Yuyama et al., 2012). We found that exosomal PrPC specifically binds to Aß-oligomers (Falker et al., 2016). Of note, binding of Aß-oligomers to neuronal PrPC is known to cause synaptic dysfunction (Lauren et al., 2009; Benilova et al., 2012). Thus, our finding opens up new facets for the Aß-receptor hypothesis and may explain why the enrichment of PrPC on exosomes correlates with the ability to sequester Aß-oligomers. Highest binding affinities to cell membrane PrPC where shown for small Aß42 species (dimers to pentamers) representing the major neurotoxic Aß-entities in AD (Lauren et al., 2009; Benilova et al., 2012). Binding of these Aß-species to exosomal PrPC resulted in fibrillization of neurotoxic Aß-entities into non-neurotoxic Aß-fibrils (Falker et al., 2016). The association of Aß and exosomes in the context of AD was also described in several publications by the group of Igaraschi. They showed that exosomes injected to the brain of AD mice associated with Aß which resulted in reduction of Aß pathology. Based on their findings they suggested that exosomes released from brain cells play a role in modulating Aß metabolism (Yuyama et al., 2014). The main difference to our studies is that Yuyama et al. suggested glycosphingolipids (GSLs) as the binding partner of Aß oligomers which are also enriched on exosomes (Yuyama et al., 2015), whereas we could identify PrPC on exosomes as a modulator of Aß binding and maybe detoxification (Falker et al., 2016). Since both data sets convincingly showed reduction of Aß toxicity, exosomal PrPC and GSLs may have complementary functions.
In conclusion we suggest a dual role for PrPC in AD. On the one hand, binding of Aß-oligomers to PrPC on neuronal plasma membranes may act neurotoxic possibly by inducing downstream neurotoxic signaling cascades (Figure 1). On the other hand, PrPC present on membranes of exosomes may act neuroprotective by accelerating conversion to non-toxic Aß-species. If this binding leads to enhanced degradation by microglia cells (An et al., 2013) or promotes deposition of Aß in non-toxic aggregation states such as-plaques (Rajendran et al., 2006) remains to be studied in more detail.
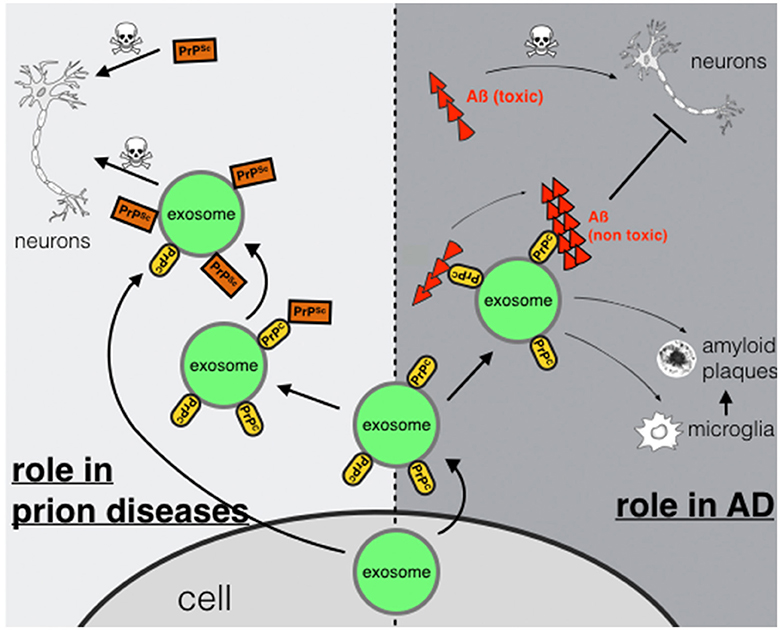
Figure 1. Exosomal PrPC: spreading or trapping of neurotoxic proteins in neurodegeneration. Role in prion diseases (left): transport of PrPSc via exosomes secreted from a prion-infected cell or binding of PrPSc to exosomal PrPC may enhance transmission and spreading. Role in AD (right): capturing and detoxifying of neurotoxic Aß-peptides by exosomal PrPC may act neuroprotective. Aß-fibrils bound to exosomes may contribute to Aß plaque formation or may enable uptake and degradation by microglia.
The Future of Exosomes and Dementia
Studying the functions of PrPC on exosomes in neurodegenerative diseases is tedious. This is in part due to the small size and extreme mobility of exosomes. The reliable tracking of exosomes allowing to determine the fate of exosomes on the cellular level is indispensable for this line of research. Current exosome labeling methods are not stable enough (e.g., pkh-membrane labeling dyes) or demand difficult pre-analytical steps (e.g., WGA-conjugates, transfection). New approaches generating persistent labels on exosomes without altering their properties would open up new perspectives in this line of research.
Furthermore, exosomes may be used in therapeutic contexts with the aim to deliver cargo specifically to the CNS due to their ability to cross the blood brain barrier (Record et al., 2011; Tominaga et al., 2015). To this respect, recent data revealed a possibility to target specific cellular populations e.g., in the brain, using exosomes to deliver therapeutic reagents (Alvarez-Erviti et al., 2011; Liu et al., 2015).
Conclusion
Exosomal PrP has at least two functions in neurodegenerative diseases. In prion diseases, the role of exosomal PrP is mostly disease-promoting, by spreading PrPSc and transferring prion infectivity. In AD, protective effects may be predominant, by sequestering toxic Aß-oligomers in the extracellular space leading to reduced neurotoxicity. Exosomal PrPC plays a central role in both aspects, as visualized in Figure 1 and summerized in Table 1. Further, work into the mechanism of this dual role is warranted. For this, novel reliable tools for exosome tracking at cellular level in vitro and in vivo are required.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Euronanomed Program and by grants of the Deutsche Forschungsgemeinschaft (SFB877, GRK1459). We thank the core facility for mouse pathology of the UKE for valuable support.
Abbreviations
PrP, prion protein; PrPC, cellular prion protein; PrPSc, scrapie prion protein; Aß, amyloid-beta; AD, Alzheimer's disease; CNS, central nervous system; GPI, glycosylphosphatidylinositol; nm, nanometer; ILVs, intraluminal vesicles; MVBs, multivesicular bodies; e.g., for example; ESCRT, endosomal sorting complex required for transport; vs, versus; GSLs, glycosphingolipids; PKH, Paul Karl Horan; WGA, Wheat Germ Aggluthinin; kDA, Kilodalton; GFP, green fluorescent protein; CSF, cerebrospinal fluid.
References
Abramowicz, A., Widlak, P., and Pietrowska, M. (2016). Proteomic analysis of exosomal cargo: the challenge of high purity vesicle isolation. Mol. Biosyst. 12, 1407–1419. doi: 10.1039/C6MB00082G
Aguzzi, A., and Lakkaraju, A. K. (2016). Cell biology of prions and prionoids: a status report. Trends Cell Biol. 26, 40–51. doi: 10.1016/j.tcb.2015.08.007
Alais, S., Simoes, S., Baas, D., Lehmann, S., Raposo, G., Darlix, J. L., et al. (2008). Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 100, 603–615. doi: 10.1042/BC20080025
Alais, S., Soto-Rifo, R., Balter, V., Gruffat, H., Manet, E., Schaeffer, L., et al. (2012). Functional mechanisms of the cellular prion protein (PrP(C)) associated anti-HIV-1 properties. Cell Mol. Life Sci. 69, 1331–1352. doi: 10.1007/s00018-011-0879-z
Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., and Wood, M. J. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345. doi: 10.1038/nbt.1807
An, K., Klyubin, I., Kim, Y., Jung, J. H., Mably, A. J., O'Dowd, S. T., et al. (2013). Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 6:47. doi: 10.1186/1756-6606-6-47
Arellano-Anaya, Z. E., Huor, A., Leblanc, P., Lehmann, S., Provansal, M., Raposo, G., et al. (2015). Prion strains are differentially released through the exosomal pathway. Cell Mol. Life Sci. 72, 1185–1196. doi: 10.1007/s00018-014-1735-8
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Berrone, E., Corona, C., Mazza, M., Vallino Costassa, E., Faro, M. L., Properzi, F., et al. (2015). Detection of cellular prion protein in exosomes derived from ovine plasma. J. Gen. Virol. 96, 3698–3702. doi: 10.1099/jgv.0.000291
Biasini, E., Turnbaugh, J. A., Unterberger, U., and Harris, D. A. (2012). Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103. doi: 10.1016/j.tins.2011.10.002
Bobrie, A., Colombo, M., Krumeich, S., Raposo, G., and Théry, C. (2012). Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 1:18397. doi: 10.3402/jev.v1i0.18397
Cervenakova, L., Saa, P., Yakovleva, O., Vasilyeva, I., de Castro, J., Brown, P., et al. (2016). Are prions transported by plasma exosomes? Transfus Apher. Sci. 55, 70–83. doi: 10.1016/j.transci.2016.07.013
Chernyshev, V. S., Rachamadugu, R., Tseng, Y. H., Belnap, D. M., Jia, Y., Branch, K. J., et al. (2015). Size and shape characterization of hydrated and desiccated exosomes. Anal Bioanal. Chem. 407, 3285–3301. doi: 10.1007/s00216-015-8535-3
Collins, S. J., Sanchez-Juan, P., Masters, C. L., Klug, G. M., van Duijn, C., Poleggi, A., et al. (2006). Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 129, 2278–2287. doi: 10.1093/brain/awl159
Devraj, K., Poznanovic, S., Spahn, C., Schwall, G., Harter, P. N., Mittelbronn, M., et al. (2016). BACE-1 is expressed in the blood-brain barrier endothelium and is upregulated in a murine model of Alzheimer's disease. J. Cereb. Blood Flow Metab. 36, 1281–1294. doi: 10.1177/0271678X15606463
Dinkins, M. B., Dasgupta, S., Wang, G., Zhu, G., and Bieberich, E. (2014). Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging 35, 1792–1800. doi: 10.1016/j.neurobiolaging.2014.02.012
Falker, C., Hartmann, A., Guett, I., Dohler, F., Altmeppen, H., Betzel, C., et al. (2016). Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 137, 88–100. doi: 10.1111/jnc.13514
Fevrier, B., Vilette, D., Archer, F., Loew, D., Faigle, W., Vidal, M., et al. (2004). Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688. doi: 10.1073/pnas.0308413101
Fröhlich, D., Kuo, W. P., Frühbeis, C., Sun, J.-J., Zehendner, C. M., Luhmann, H. J., et al. (2014). Multifaceted effects of oligodendroglial exosomes on neurons: impact on neuronal firing rate, signal transduction and gene regulation. Philos Trans. R. Soc. Lond. B Biol. Sci. 369:20130510. doi: 10.1098/rstb.2013.0510
Gardiner, C., Di Vizio, D., Sahoo, S., Thery, C., Witwer, K. W., Wauben, M., et al. (2016). Techniques used for the isolation and characterization of extracellular vesicles: results of a worldwide survey. J. Extracell. Vesicles 5:32945. doi: 10.3402/jev.v5.32945
Geissen, M., Krasemann, S., Matschke, J., and Glatzel, M. (2007). Understanding the natural variability of prion diseases. Vaccine 25, 5631–5636. doi: 10.1016/j.vaccine.2007.02.041
Glatzel, M., Stoeck, K., Seeger, H., Luhrs, T., and Aguzzi, A. (2005). Human prion diseases: molecular and clinical aspects. Arch. Neurol. 62, 545–552. doi: 10.1001/archneur.62.4.545
Goetzl, E. J., Boxer, A., Schwartz, J. B., Abner, E. L., Petersen, R. C., Miller, B. L., et al. (2015). Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85, 40–47. doi: 10.1212/WNL.0000000000001702
Guo, B. B., Bellingham, S. A., and Hill, A. F. (2016). Stimulating the release of exosomes increases the intercellular transfer of prions. J. Biol. Chem. 291, 5128–5137. doi: 10.1074/jbc.M115.684258
Haraszti, R. A., Didiot, M. C., Sapp, E., Leszyk, J., Shaffer, S. A., Rockwell, H. E., et al. (2016). High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 5:32570. doi: 10.3402/jev.v5.32570
Hill, A. F., Vella, L. J., Greenwood, D. L. V., Cappai, R., and Scheerlinck, J. P. Y. (2008). Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 124, 385–393. doi: 10.1016/j.vetimm.2008.04.002
Jellinger, K. A. (2003). General aspects of neurodegeneration. J. Neural Transm. Suppl (65), 101–144. doi: 10.1007/978-3-7091-0643-3_7
Jeon, I., Cicchetti, F., Cisbani, G., Lee, S., Li, E., Bae, J., et al. (2016). Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 132, 577–592. doi: 10.1007/s00401-016-1582-9
Kalani, A., Tyagi, A., and Tyagi, N. (2014). Exosomes: mediators of neurodegeneration, neuroprotection and therapeutics. Mol. Neurobiol. 49, 590–600. doi: 10.1007/s12035-013-8544-1
Klohn, P. C., Castro-Seoane, R., and Collinge, J. (2013). Exosome release from infected dendritic cells: a clue for a fast spread of prions in the periphery? J. Infect. 67, 359–368. doi: 10.1016/j.jinf.2013.07.024
Kokubo, H., Saido, T. C., Iwata, N., Helms, J. B., Shinohara, R., and Yamaguchi, H. (2005). Part of membrane-bound Aβ exists in rafts within senile plaques in Tg2576 mouse brain. Neurobiol. Aging 26, 409–418. doi: 10.1016/j.neurobiolaging.2004.04.008
Kramer-Albers, E. M., and Hill, A. F. (2016). Extracellular vesicles: interneural shuttles of complex messages. Curr. Opin. Neurobiol. 39, 101–107. doi: 10.1016/j.conb.2016.04.016
Krasemann, S., Neumann, M., Luepke, J. P., Grashorn, J., Wurr, S., Stocking, C., et al. (2012). Persistent retroviral infection with MoMuLV influences neuropathological signature and phenotype of prion disease. Acta Neuropathol. 124, 111–126. doi: 10.1007/s00401-012-0944-1
Krasemann, S., Neumann, M., Szalay, B., Stocking, C., and Glatzel, M. (2013). Protease-sensitive prion species in neoplastic spleens of prion-infected mice with uncoupling of PrP(Sc) and prion infectivity. J. Gen. Virol. 94(Pt 2), 453–463. doi: 10.1099/vir.0.045922-0
Kreimer, S., Belov, A. M., Ghiran, I., Murthy, S. K., Frank, D. A., and Ivanov, A. R. (2015). Mass-spectrometry-based molecular characterization of extracellular vesicles: lipidomics and proteomics. J. Proteome Res. 14, 2367–2384. doi: 10.1021/pr501279t
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Leblanc, P., Arellano-Anaya, Z. E., Bernard, E., Gallay, L., Provansal, M., Lehmann, S., et al. (2017). Isolation of exosomes and microvesicles from cell culture systems to study prion transmission. Methods Mol. Biol. 1545, 153–176. doi: 10.1007/978-1-4939-6728-5_11
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Liu, C. G., Song, J., Zhang, Y. Q., and Wang, P. C. (2014). MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer's disease. Mol. Med. Rep. 10, 2395–2400. doi: 10.3892/mmr.2014.2484
Liu, Y., Li, D., Liu, Z., Zhou, Y., Chu, D., Li, X., et al. (2015). Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci. Rep. 5:17543. doi: 10.1038/srep17543
Lotvall, J., Hill, A. F., Hochberg, F., Buzas, E. I., Di Vizio, D., Gardiner, C., et al. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 3:26913. doi: 10.3402/jev.v3.26913
Manson, J. C., Jamieson, E., Baybutt, H., Tuzi, N. L., Barron, R., McConnell, I., et al. (1999). A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. Embo J. 18, 6855–6864. doi: 10.1093/emboj/18.23.6855
Muth, C., Schröck, K., Madore, C., Hartmann, K., Fanek, Z., Butovsky, O., et al. (2016). Activation of microglia by retroviral infection correlates with transient clearance of prions from the brain but does not change incubation time. Brain Pathol. doi: 10.1111/bpa.12441. [Epub ahead of print].
Pflanzner, T., Petsch, B., Andre-Dohmen, B., Muller-Schiffmann, A., Tschickardt, S., Weggen, S., et al. (2012). Cellular prion protein participates in amyloid-beta transcytosis across the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 628–632. doi: 10.1038/jcbfm.2012.7
Polanco, J. C., Scicluna, B. J., Hill, A. F., and Gotz, J. (2016). Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 291, 12445–12466. doi: 10.1074/jbc.M115.709485
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63–75 e62. doi: 10.1016/j.jalz.2012.11.007
Properzi, F., Ferroni, E., Poleggi, A., and Vinci, R. (2015). The regulation of exosome function in the CNS: implications for neurodegeneration. Swiss Med. Wkly. 145:w14204. doi: 10.4414/smw.2015.14204
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144.
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Raposo, F., Fernandez-Cegri, V., De la Rubia, M. A., Borja, R., Beline, F., Cavinato, C., et al. (2011). Biochemical methane potential (BMP) of solid organic substrates: evaluation of anaerobic biodegradability using data from an international interlaboratory study. J. Chem. Technol. Biotechnol. 86, 1088–1098. doi: 10.1002/jctb.2622
Record, M., Subra, C., Silvente-Poirot, S., and Poirot, M. (2011). Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 81, 1171–1182. doi: 10.1016/j.bcp.2011.02.011
Resenberger, U. K., Winklhofer, K. F., and Tatzelt, J. (2011). Cellular prion protein mediates toxic signaling of amyloid beta. Neurodegener. Dis. 10, 298–300. doi: 10.1159/000332596
Riek, R., Hornemann, S., Wider, G., Glockshuber, R., and Wüthrich, K. (1997). NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 413, 282–288. doi: 10.1016/S0014-5793(97)00920-4
Saa, P., Yakovleva, O., de Castro, J., Vasilyeva, I., De Paoli, S. H., Simak, J., et al. (2014). First demonstration of transmissible spongiform encephalopathy-associated prion protein (PrPTSE) in extracellular vesicles from plasma of mice infected with mouse-adapted variant Creutzfeldt-Jakob disease by in vitro amplification. J. Biol. Chem. 289, 29247–29260. doi: 10.1074/jbc.M114.589564
Silverman, J. M., Fernando, S. M., Grad, L. I., Hill, A. F., Turner, B. J., Yerbury, J. J., et al. (2016). Disease mechanisms in ALS: misfolded SOD1 transferred through exosome-dependent and exosome-independent pathways. Cell. Mol. Neurobiol. 36, 377–381. doi: 10.1007/s10571-015-0294-3
Telling, G. C., Parchi, P., DeArmond, S. J., Cortelli, P., Montagna, P., Gabizon, R., et al. (1996). Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082.
Tominaga, N., Yoshioka, Y., and Ochiya, T. (2015). A novel platform for cancer therapy using extracellular vesicles. Adv. Drug Deliv. Rev. 95, 50–55. doi: 10.1016/j.addr.2015.10.002
Uchiyama, K., Muramatsu, N., Yano, M., Usui, T., Miyata, H., and Sakaguchi, S. (2013). Prions disturb post-Golgi trafficking of membrane proteins. Nat. Commun. 4, 1846. doi: 10.1038/ncomms2873
Udayar, V., Buggia-Prevot, V., Guerreiro, R. L., Siegel, G., Rambabu, N., Soohoo, A. L., et al. (2013). A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep. 5, 1536–1551. doi: 10.1016/j.celrep.2013.12.005
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Vassallo, N., and Herms, J. (2003). Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 86, 538–544. doi: 10.1046/j.1471-4159.2003.01882.x
Vella, L. J., Greenwood, D. L. V., Cappai, R., Scheerlinck, J.-P. Y., and Hill, A. F. (2008a). Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 124, 385–393. doi: 10.1016/j.vetimm.2008.04.002
Vella, L. J., Sharples, R. A., Lawson, V. A., Masters, C. L., Cappai, R., and Hill, A. F. (2007). Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590. doi: 10.1002/path.2145
Vella, L. J., Sharples, R. A., Nisbet, R. M., Cappai, R., and Hill, A. F. (2008b). The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 37, 323–332. doi: 10.1007/s00249-007-0246-z
Vilette, D., Laulagnier, K., Huor, A., Alais, S., Simoes, S., Maryse, R., et al. (2015). Efficient inhibition of infectious prions multiplication and release by targeting the exosomal pathway. Cell. Mol. Life Sci. 72, 4409–4427. doi: 10.1007/s00018-015-1945-8
Wadsworth, J. D., and Collinge, J. (2011). Molecular pathology of human prion disease. Acta Neuropathol. 121, 69–77. doi: 10.1007/s00401-010-0735-5
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Walsh, D. M., and Selkoe, D. J. (2016). A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 17, 251–260. doi: 10.1038/nrn.2016.13
Yanez-Mo, M., Siljander, P. R., Andreu, Z., Zavec, A. B., Borras, F. E., Buzas, E. I., et al. (2015). Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4:27066. doi: 10.3402/jev.v4.27066
Yuyama, K., Sun, H., Mitsutake, S., and Igarashi, Y. (2012). Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 287, 10977–10989. doi: 10.1074/jbc.M111.324616
Yuyama, K., Sun, H., Sakai, S., Mitsutake, S., Okada, M., Tahara, H., et al. (2014). Decreased amyloid-beta pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 289, 24488–24498. doi: 10.1074/jbc.M114.577213
Keywords: prion, exosome, micro vesicle, extracellular vesicle, neurodegeneration, beta-amyloid, prion disease, Alzheimer's disease
Citation: Hartmann A, Muth C, Dabrowski O, Krasemann S and Glatzel M (2017) Exosomes and the Prion Protein: More than One Truth. Front. Neurosci. 11:194. doi: 10.3389/fnins.2017.00194
Received: 22 December 2016; Accepted: 22 March 2017;
Published: 19 April 2017.
Edited by:
Diana K. Sarko, Southern Illinois University Carbondale, USAReviewed by:
Eun-Kyoung Choi, Hallym University, South KoreaRafael Linden, Federal University of Rio de Janeiro, Brazil
Alessandro Bertoli, University of Padua, Italy
Copyright © 2017 Hartmann, Muth, Dabrowski, Krasemann and Glatzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Glatzel, bS5nbGF0emVsQHVrZS5kZQ==
†Shared first authors.