 Mariana Vargas-Caballero1*†
Mariana Vargas-Caballero1*† Franziska Denk2,3†
Franziska Denk2,3† Heike J. Wobst2,4†
Heike J. Wobst2,4† Emily Arch2Chrysia-Maria Pegasiou1
Emily Arch2Chrysia-Maria Pegasiou1 Peter L. Oliver2Olivia A. Shipton2,5
Peter L. Oliver2Olivia A. Shipton2,5 Ole Paulsen5Richard Wade-Martins2*
Ole Paulsen5Richard Wade-Martins2*- 1Biological Sciences and Institute for Life Sciences, University of Southampton, Southampton, UK
- 2Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK
- 3Wolfson Centre for Age-Related Diseases, King's College London, London, UK
- 4AstraZeneca-Tufts Lab for Basic and Translational Neuroscience, Tufts University School of Medicine, Boston, MA, USA
- 5Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, UK
Microtubule associated protein tau (MAPT) is involved in the pathogenesis of Alzheimer's disease and many forms of frontotemporal dementia (FTD). We recently reported that Aβ-mediated inhibition of hippocampal long-term potentiation (LTP) in mice requires tau. Here, we asked whether expression of human MAPT can restore Aβ-mediated inhibition on a mouse Tau−/− background and whether human tau with an FTD-causing mutation (N296H) can interfere with Aβ-mediated inhibition of LTP. We used transgenic mouse lines each expressing the full human MAPT locus using bacterial artificial chromosome technology. These lines expressed all six human tau protein isoforms on a Tau−/− background. We found that the human wild-type MAPT H1 locus was able to restore Aβ42-mediated impairment of LTP. In contrast, Aβ42 did not reduce LTP in slices in two independently generated transgenic lines expressing tau protein with the mutation N296H associated with frontotemporal dementia (FTD). Basal phosphorylation of tau measured as the ratio of AT8/Tau5 immunoreactivity was significantly reduced in N296H mutant hippocampal slices. Our data show that human MAPT is able to restore Aβ42-mediated inhibition of LTP in Tau−/− mice. These results provide further evidence that tau protein is central to Aβ-induced LTP impairment and provide a valuable tool for further analysis of the links between Aβ, human tau and impairment of synaptic function.
Introduction
Multiple lines of evidence indicate an interaction between amyloid-beta (Aβ) and tau protein in Alzheimer's disease, and tau protein is required for the effect of Aβ in many experimental paradigms. Thus, cultured neurons from tau knockout (Tau−/−) mice are not susceptible to Aβ-induced synaptic damage and neurotoxicity (Rapoport et al., 2002; Nussbaum et al., 2012; Zempel et al., 2013) or to Aβ-induced defects in axonal transport (Vossel et al., 2011). Reducing tau levels prevents Aβ-induced cognitive deficits and premature mortality, reduces spontaneous and induced seizure activity, and prevents synaptic impairment in mutant APP-expressing mouse models (Roberson et al., 2007, 2011). Furthermore, Aβ plays a permissive role for the spread of tau pathology in vivo (Pooler et al., 2015). Similarly, tau oligomers produce an acute inhibition of hippocampal LTP and memory (Fá et al., 2016), and inhibition of the tau kinases CDK5 or GSK-3β reduces Aβ-induced neuronal cell death and malfunction (Llorens-Marítin et al., 2014) suggesting that abnormal tau mediates some of the effects of Aβ.
Previous reports consistently show that Aβ inhibits long-term potentiation (LTP) in the hippocampus (Ondrejcak et al., 2010), a widely accepted cellular model for learning and memory (Bliss and Collingridge, 1993), and we have previously shown that hippocampal slices from Tau−/− animals are not susceptible to human Aβ42-mediated LTP impairment (Shipton et al., 2011). Furthermore, our work and that of others have shown that inhibition of GSK-3β prevents LTP impairment following exposure to Aβ and prevents Aβ-mediated increase in tau phosphorylation at disease-relevant AT8 epitopes (Jo et al., 2011; Shipton et al., 2011). To further analyse the interaction of Aβ42 and human tau and build upon our previous work, here we asked whether expressing the human tau protein in mice on a Tau−/− background (Dawson et al., 2001) can restore the Aβ42-mediated inhibition of LTP. For this, we studied transgenic mice expressing one of two variants of the human tau protein: either a wild type form, or a mutant form carrying the disease-associated mutation, N296H, known to lead to frontotemporal dementia (FTD; Iseki et al., 2001). Both transgenes contain the complete MAPT genomic locus, which allows for the study of aberrant splicing, a phenomenon which has been observed in a variety of FTDs and tauopathies (Spillantini et al., 1998; Takanashi et al., 2002). The N296H mutation, in particular, is a splice site mutation that affects the isoform ratio detected in the insoluble fraction of tau fragments obtained from patient brains (Iseki et al., 2001) leading to an increase in tau isoforms harboring four microtubule-binding repeats (4R). Like many other familial FTD mutations (Denk and Wade-Martins, 2009), N296H causes increased inclusion of MAPT exon 10, leading to an overrepresentation of tau isoforms with four microtubule-binding repeats, known as 4R tau (Grover et al., 2002; Yoshida et al., 2002).
We hypothesized that the expression of wild type human tau would restore the inhibitory effect of Aβ42 on synaptic plasticity in Tau−/− mice. Further, we tested whether mutant N296H tau would either occlude or prevent the Aβ42-mediated inhibition of hippocampal LTP. Elucidating the extent to which normal or mutant human tau protein can restore Aβ42-mediated inhibition of LTP in Tau−/− mice should further enhance our understanding of the interaction between Aβ42 and tau protein in synapse dysfunction.
Materials and Methods
Ethical Statement
Animal care and experimental procedures were conducted in accordance with UK Home Office regulations under the Animals (Scientific Procedures) Act of 1986. The work was carried out under PPL 30/2757 and all efforts were made to optimize the number of animals used.
Transgenic MAPT Mice
Mice were generated using bacterial artificial chromosome (BAC) technology to express the 143 kb MAPT locus (Supplementary Methods) in mice with a genetic knock-out of endogenous tau (Tau−/−; Dawson et al., 2001) on a C57/BL6-J background. This allowed the physiological expression of all human tau isoforms. We chose to express the more common H1 haplotype given its association with increased risk of Alzheimer's disease. Mice expressed either MAPT-H1 or the same transgene with the point mutation N296H. Two lines of N296H P1-derived artificial chromosome (PAC) transgenic mice (N24 and N51) were generated to control for insertion effects. Samples for analysis of tau isoform expression with Western blot were subjected to alkaline phosphatase treatment (Lambda protein phosphatase NEV, 4,000 U per 80 μg lysate) and assessed with the human specific Tau-13 antibody (1:5000 Abcam). Both male and female mice were used in each experiment.
RNA In situ Hybridization
In situ hybridization for transgene expression was carried out on frozen 14 μm thick tissue sections using a DIG-labeled LNA probe (Exiqon) designed to be specific for human MAPT (5′ DIG-gctcagccatcctggttcaaa-DIG 3′). Hybridisation and signal detection were carried out as previously described (Jefferson and Volpi, 2010).
Exon 10 Splicing Measurements
Quantitative analysis of exon 10 splice ratios was obtained using the Sequenom MassARRAY Platform. Briefly, RT-PCR was performed to amplify exon 4 through to exon 11 of the MAPT gene. Sequenom analysis was performed by the Wellcome Trust Centre for Human Genetics, Oxford, to determine the relative amount of exon 10 inclusion vs. exclusion using mass-assisted laser desorption/ionization—time of flight (MALDI-TOF). Each MALDI-TOF assay was repeated eight times in both forward and reverse direction, and N = 2–3 mice were tested per group. A control construct expressing a known 1:1 ratio of 4R:3R tau was used for normalization.
Slice Preparation and Pharmacology
Following the procedure described in Shipton et al. (2011), parasagittal hippocampal slices (400 μm) were prepared after decapitation under deep isoflurane-induced anesthesia. After dissection in ice-cold artificial CSF (ACSF) containing (in mM) 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 25 NaHCO3, 10 glucose, pH 7.2–7.4, bubbled with carbogen gas (95% O2, 5% CO2), slices were maintained at room temperature (22–25°C) in a submerged-style holding chamber for at least 1 h and after that incubated in ACSF with or without freshly prepared 220 nM human Aβ42 (also referred to as Aβ1–42 Tocris, UK) in a submerged chamber for 1–3 h (Shipton et al., 2011; Um et al., 2012). Perfusion with a half-concentration of the drug continued after slices were transferred to the interface chamber for fEPSP recordings under the assumption that this reduction to 110 nM would not cause a washout of the Aβ42 effect on plasticity. LTP was induced with high frequency stimulation using a single 1 s long 100 Hz induction protocol. Mice used for electrophysiology were 4–7 months old. The electrophysiology data were obtained from 18 H1 mice, 15 N51 mice, and 6 N24 mice, Number of observations (N) represents the number of slices subject to each pharmacological treatment. Input-output, paired pulse and LTP data were obtained from the same slices. Only slices with a stable baseline (drift <10%) were used for LTP induction. A sample of concentrated Aβ42 (44 μM) was processed in the same way as above and collected at the time of LTP measurement for protein stain analysis (Supplementary Methods) to verify the presence of oligomeric Aβ assemblies in the solution (Supplementary Figure 1).
Data Analysis
Data were analyzed with one- or two-way ANOVA with genotype and treatment as independent variables. Data are represented as means ± SEM. Unless otherwise stated, post-hoc comparisons were corrected using Dunnett's method with H1 ACSF data as control.
To establish whether significant levels of LTP were obtained within each experimental group we performed a paired Student's t-test comparing baseline synaptic strength with synaptic strength at 40 ± 2.5 min post-HFS per individual experimental condition.
Western Blot
For analysis of slices incubated with or without addition of 220 nM Aβ42, following 1 h recovery at room temperature in ACSF, slices from 6 to 7 month old mice (N = 6 mice) were incubated for 2 h in either Aβ42 or control solutions. Slices were then snap frozen and collected for western blot analysis (Additional information in Supplementary Methods). Slices from the same mouse and treatment were pooled together resulting in N = 1 per mouse per treatment. AT8 and Tau-5 immunoreactivity was first normalized to GADPH. Immunoreactivity for both H1 and N51 was normalized to baseline H1 levels.
Results
Following our previous observation that early LTP in mice with a genetic knock out of tau protein (Tau−/−) is not affected by Aβ, we wanted to investigate whether transgenic expression of human tau (MAPT) genes would allow normal LTP on a Tau−/− background in mice and permit LTP inhibition by Aβ42.
We used three transgenic mouse lines. The first line carried the transgenic locus of wild-type human tau (line H1), and we used two independent lines carrying the tau N296H mutation linked to FTD with parkinsonism (lines N51 and N24). To identify the six tau isoforms we performed Western blot following a dephosphorylation treatment (Figure 1A) using a human tau specific antibody.
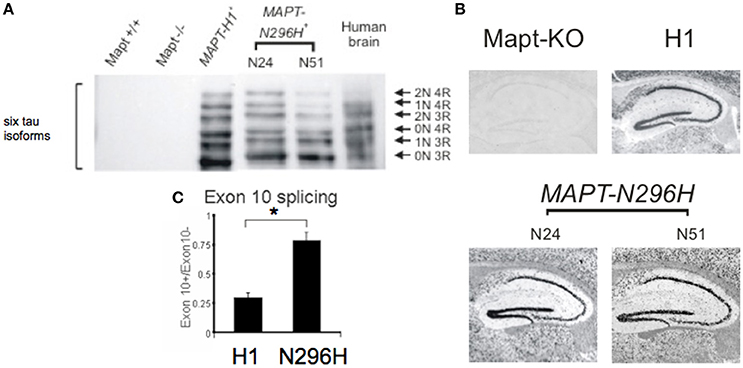
Figure 1. Six isoforms of human tau protein in transgenic mouse lines MAPT-H1, and MAPT-N296H expressed in a mouse Tau−/− (Mapt−/−) background. (A) Western blot with a human-specific tau antibody. MAPT-H1 and MAPT-N296H transgenic animals expressed all six isoforms of the human tau protein. (B) RNA in situ hybridization analyzing expression of MAPT in the hippocampus of Tau−/− and MAPT mice. (C) Quantitative analysis of splice ratios of MAPT exon 10+(4R)/exon 10−(3R) RNA transcript in the brains from H1 and N296H (line N24) mice showing an enhanced ratio of exon 10 inclusion in N296H mice, *p < 0.05.
We wanted to test the effect of expressing tau protein on hippocampal LTP; to confirm that human tau mRNA was expressed in the hippocampus we used in situ hybridisation. We observed an enrichment of our signal in the hippocampal cell body layers (Figure 1B) in a pattern similar to that observed in the Allen brain atlas for adult mouse tau (Allen Institute for Brain Science, 2015). To test whether expression of normal or mutant human tau caused changes at the mRNA expression level we measured the ratio of exon10 inclusion vs. exon10 exclusion in mRNA transcripts and we found that MAPT-N296H had a significantly higher inclusion of exon 10 (Line H1 0.30 ± 0.005 N = 3, N296H—line 24—0.78 ± 0.02 N = 2, P < 0.05, Figure 1C).
To test synaptic function and LTP in hippocampal CA3-CA1 synapses we obtained acute brain slices from these transgenic mice. We incubated slices in either 220 nM oligomeric Aβ42 (Supplementary Figure 1) or control ACSF for 1–3 h.
We have previously shown that basal synaptic transmission is not affected following acute Aβ42 incubation in WT or Tau−/− mice. To measure input-output relation, we evoked field excitatory postsynaptic potentials (fEPSPs) in CA1 with extracellular stimulation of Schaffer collaterals by delivering brief electrical pulses of increasing amplitude from 20 to 200 μA. We observed similar input-output curves for both ACSF and Aβ42 treatment in all three transgenic lines tested (Figures 2A–C). Repeated measures ANOVA between-subjects showed no significant effect of genotype [F(2, 68) = 0.33, P = 0.72] or treatment [F(1, 68) = 0.74, P = 0.39] and no genotype*treatment interaction [F(2, 68) = 0.51, P = 0.49].
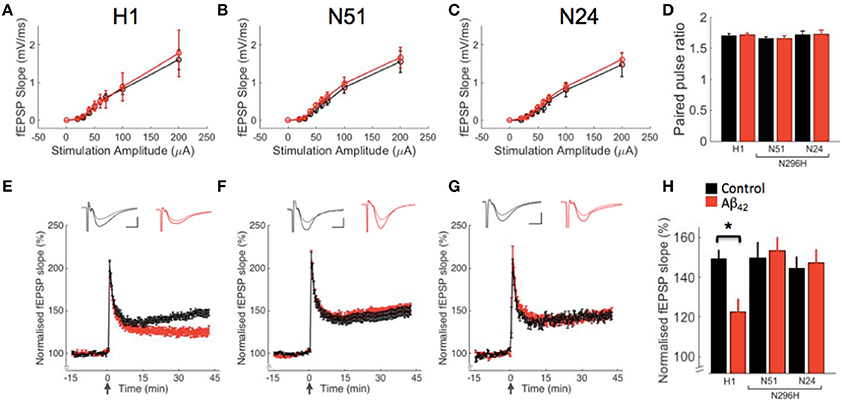
Figure 2. Hippocampal LTP in slices from mice expressing human tau on a Tau−/− background is impaired by Aβ42, however Aβ42 does not reduce LTP in slices from mice expressing tau protein N296H associated with FTD. Field recordings from CA3 to CA1 synapses in control ACSF (black) or after incubation with Aβ42 (red). (A–C) Synaptic input-output curves corresponding to genotypes above graph. (D) Paired-pulse data. (E–G) LTP in wild type human tau (H1) and two transgenic lines (N51, N24) expressing mutant tau (N296H). LTP was induced by delivering a high-frequency stimulation (HFS) train (100 Hz for 1 s). The insets show superimposed example traces before and 40 min after HFS for each condition. Scale bars: 5 ms/0.5 mV. (H) Summary of LTP results 40 min after HFS, *P < 0.05.
To characterize paired-pulse ratio as a measure of presynaptic function, we delivered paired stimuli 40 ms apart at CA3-CA1 synapses (Figure 2D). Two-way ANOVA on paired-pulse data showed no effect of genotype [F(2, 82) = 1.67, P = 0.19] or treatment [F(1, 82) = 0.06, P = 0.81] and no genotype*treatment interaction [F(2, 82) = 0.02, P = 0.98].
LTP in the hippocampus is widely assumed to be a cellular correlate of learning and memory. We previously showed that 220 nM Aβ42 inhibits tetanus-induced LTP in wild type mice and that it does not have an effect on LTP in Tau−/− mice (Shipton et al., 2011). We wanted to test whether transgenic expression of human tau restored the effect of Aβ42 on LTP in mice on a Tau−/− background; for this we used mice with transgenic expression of tau in its wild type form (line H1) or with a point mutation related to FTD (N296H, lines N24 and N51). To analyse the effects of Aβ42 on LTP we monitored fEPSPs by stimulating Schaffer collaterals and induced LTP with tetanic (HFS) stimulation (Figures 2E–G).
We found significant LTP following HFS in H1 mice (148.43 ± 5.47, N = 16, P < 1 × 10−8 compared to baseline) and, interestingly, also normal LTP levels in both lines expressing tau with the N296H mutation (line N51 149.82 ± 7.40, N = 18, P < 1 × 10−6, line N24 144.32 ± 5.80, N = 7, P < 1 × 10−5). We hypothesized that in H1 mice, incubation of acute slices with Aβ42 peptide would significantly impair hippocampal LTP owing to the wild type human tau expression. Furthermore, we wanted to test the effects of human tau with the N296H mutation on LTP under control conditions and following Aβ42 incubation. Following from pilot experiments on WT slices demonstrating an inhibitory effect of Aβ42 on LTP as described in Shipton et al. (2011) (not shown), we interleaved control and Aβ42 incubation experiments and compared the resulting LTP for the three transgenic lines with and without Aβ42. Following a two-way ANOVA analysis of LTP results, we found an effect of genotype F(2, 84) = 3.11, P < 0.05 on the induction of LTP and although no effect of treatment on LTP was observed F(1, 84) = 0.82, P = 0.37, importantly a genotype*treatment interaction was found F(2, 84) = 2.83, P < 0.05. fEPSP slopes in H1 slices with Aβ42 incubation were significantly different from baseline following HFS (LTP following Aβ42 incubation: 124.7 ± 6.2, N = 18, P < 1 × 10−4), however, post-hoc comparison between H1 in control conditions and H1 following incubation in Aβ42 revealed a significant difference between these treatments (P < 0.05, Figure 2H). Strikingly, there was no difference in LTP when comparing control and Aβ42 conditions in the N51 line with the N296H mutation (Aβ42 LTP, 153.6 ± 6.1, N = 20, P = 0.65 compared to control). These results showing no effect of Aβ42 on LTP in line N51 N296H mice are further supported by our data analyzing line N24, an independently generated line with the N296H mutation to control for transgene insertion effects. This line also showed normal LTP following Aβ42 incubation (Aβ42 LTP = 147.3 ± 6.3, N = 6, P = 0.50 compared to control levels, Figure 2H).
In contrast to N296H mice, Aβ42 significantly inhibited LTP in H1 mice. We have previously shown that tau phosphorylation measured with AT8 immunoreactivity was increased following incubation with Aβ42 in hippocampal slices from WT mice (Shipton et al., 2011). In order to investigate whether an increase in tau phosphorylation is associated with Aβ42-mediated LTP impairment, we assessed total tau levels and tau phosphorylation levels in acute slices from H1 and N296H mouse hippocampal slices. We first noted that in control samples total tau levels normalized to GAPDH were 5.4 ± 0.3 times higher in N51 animals compared to H1 values normalized to GAPDH (N = 6 H1, N = 6 N51, P < 1 × 10−5. Figures 3A,B). However, phosphorylated tau measured with the AT8 antibody as a proportion of total tau measured with the Tau5 antibody with Western blot was significantly reduced in N51 hippocampal slices in control ACSF conditions compared to H1 mice (0.43 ± 0.03 of normalized values from H1 mice, N = 6 H1, N = 6 N51, P < 1 × 10−5, Figure 3B).
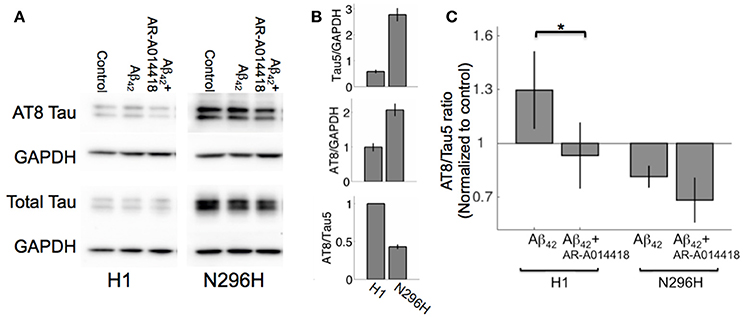
Figure 3. Tau protein phosphorylation measured as AT8 to Tau5 immunoreactivity ratio is lower for N296H hippocampal slices compared to H1. (A) Example Western blots showing immunoreactivity to AT8 (phosphorylated tau) and Tau5 (total tau) in slices from MAPT-H1 and MAPT-N256H (line N51) mice incubated in control conditions, with Aβ42, or with Aβ42 + GSK3 inhibitor. (B) Quantification of phosphorylated (AT8) tau (top), total (Tau5) tau (middle) immunoreactivity relative to housekeeping protein GAPDH for both strains, and quantification of AT8/Tau5 immunoreactivity ratio (bottom) for both strains in basal conditions (untreated slices) for N = 6 mice each. (C) Quantification of AT8/Tau5 immunoreactivity ratio in control conditions and following Aβ42 or Aβ42 + GSK3 inhibitor incubation for N = 6 mice, each normalized to basal levels in each genotype *P < 0.05.
We wanted to test whether a significant increase in phosphorylation would be observed in either H1 or N51 mouse hippocampus following incubation with Aβ42 as we previously reported in WT mice, and whether this could be prevented by using the specific GSK3 inhibitor AR-A014418 (Bhat et al., 2003). For this we normalized AT8/Tau5 level following Aβ42 or Aβ42 + AR-A014418 incubation to basal AT8/Tau5 levels as an internal control for each genotype. Two way ANOVA revealed an effect of genotype [F(1, 32) = 5.27, P < 0.05], however we did not see a main effect of treatment [F(2, 32) = 2, P = 0.15]. Post-hoc comparison of AT8/Tau5 ratios in control conditions between H1 and N296H resulted in a significant difference [H1 1.3 ± 0.2 (N = 6), N51 0.8 ± 0.1 (N = 6), P < 0.05], however, neither H1 nor N51 AT8/Tau5 ratio following Aβ42 incubation was significantly higher than normalized control levels within the same genotype (H1 P = 0.10; N51 P = 0.96; Figure 3C).
Discussion
We have shown that wild type human tau is able to restore Aβ42-mediated inhibition of LTP measured in mouse hippocampal slices on a mouse Tau−/− background. However, brain slices from transgenic mice expressing a version of tau protein with a point mutation associated with familial FTD (N296H, line N51) showed normal LTP both under control conditions and following Aβ42 incubation. We also assessed a second independently-derived mouse line expressing all six tau isoforms of N296H mutant tau (line N24) corroborating a mutation-dependent resistance to Aβ42-induced inhibition of LTP. Our data suggest that the N296H mutant tau acts like a functional knockout in the pathways that link Aβ42 to LTP impairment, since N296H mice like Tau−/− mice are protected from the effects of Aβ42 peptide on LTP.
The bacterial chromosome transgenic technology we used for generation of transgenic animals allows for anatomical expression patterns at physiological levels, however it is not possible to match exact level of protein expression in all transgenic lines. We measured protein expression levels in H1 and N51 mice, with H1 mice showing lower levels of tau protein. In all three lines we were able to observe the six tau isoforms produced by alternative splicing from the human tau locus. Wild type human tau in H1 mice was able to restore the Aβ42-mediated phenotype on a Tau−/− background, indicating sufficient protein levels to link Aβ42 with synaptic plasticity dysfunction. In N296H mice we observed normal basal levels of synaptic transmission and normal LTP. If N296H tau protein caused a gain of toxic function, a stronger phenotype would be expected (Roberson et al., 2007) from higher expression levels in line N296H-N51. However, we did not observe an enhanced effect under basal or Aβ42 conditions in slices with the N296H mutation and we therefore argue that expression levels did not confound our data.
We assessed phosphorylation levels of tau protein with the AT8 antibody compared to total levels of tau with the Tau5 antibody in H1 and N51 mice. A comparison of H1 and N51 lines showed that at basal levels the ratio of AT8 to Tau5 immunoreactivity was much higher in H1 hippocampi, however, these levels were not associated with dysfunction, as previously reported in WT mice (Shipton et al., 2011; Morris et al., 2015). We also did not observe tau aggregates in the H1 or N296H lines following AT8 immunohistochemistry in slices from up to 21 month old mice (data not shown). This is in contrast to a previously reported mouse line expressing the H1 haplotype under a P1-derived artificial chromosome with high expression levels on a Tau−/− background, which showed abnormal tau hyperphosphorylation from 3 months of age (Andorfer et al., 2003).
The AT8 phosphorylation levels, synaptic input-output curves, paired-pulse ratios, and hippocampal LTP data presented here are comparable to our previous data from wild type and Tau−/− mice (Shipton et al., 2011). Following our previous published observations and the electrophysiological recordings presented here we hypothesized that AT8 immunoreactivity would increase in H1 hippocampal slices incubated with Aβ42 compared to control conditions associated with inhibition of LTP. However, owing to low protein expression levels in H1 mice we had high variability in our Western blot analysis. Although we observed a trend of increased phosphorylation levels in H1 slices following Aβ42 incubation consistent with our previous findings, the results did not reach significance. It may be necessary to express H1 in homozygosity to yield a stronger phenotype for molecular analyses of tau phosphorylation and downstream events.
N51 mice expressing N296H mutant human tau, despite showing higher expression levels than H1 mice, showed starkly decreased levels of tau phosphorylation and were not susceptible to an Aβ42-induced increase in tau phosphorylation at the AT8 site. However, while these findings are congruent with a loss of function via a reduction in basal and Aβ42-induced tau phosphorylation, it is possible that the mutation, either in addition to or independent of its effects on tau phosphorylation, could lead to other changes in tau, such as other post-translational modifications. These could play a distinct role in the Aβ42 effects on hippocampal LTP that we do not rule out here.
There is mounting evidence for tau as a downstream mediator of Aβ effects (Rapoport et al., 2002; Roberson et al., 2007, 2011; Shipton et al., 2011; Vossel et al., 2011), however, the mechanisms by which tau mediates neuronal Aβ42-mediated dysfunction remains poorly understood. There is strong evidence that small soluble oligomers formed by phosphorylated forms of tau are highly toxic, however, it is unclear whether their early effects are driven by alterations in the cytoskeleton structure (Cowan and Mudher, 2013), by promoting downstream pathological cascades (De Strooper and Karran, 2016) or a combination of these. One potential set of mechanisms whereby the absence of functional tau in hippocampal neurons (i.e., Tau−/− or MAPT-N296H) could prevent the effect of amyloid beta on LTP is by modifying the cellular distribution of fyn kinase. Under normal conditions the physical tau/fyn association results in targeting of fyn kinase to postsynaptic sites (Ittner et al., 2010) leading to basal phosphorylation of synaptic NMDARs at Y1472 by fyn which is notably reduced in Tau−/− mice (Ittner et al., 2010). Aβ42 drives tau phosphorylation at GSK3β epitopes (Jo et al., 2011; Shipton et al., 2011; Mondragon-Rodriguez et al., 2012) by a variety of proposed mechanisms in the Wnt signaling pathway (Purro et al., 2012; Vargas et al., 2014). This tau phosphorylation results in enhanced dendritic fyn localization coupled to a disruption of the PSD-95/NMDAR interaction (Mondragon-Rodriguez et al., 2012) and abnormal postsynaptic density ultrastructure (Purro et al., 2012). The relationship between Aβ42-mediated disruption in glutamate uptake (Li et al., 2011) and tau phosphorylation is unclear, however this dysregulation has been reported to occur on the same time scale as in our present experiments and is well-placed to feed-back into the mechanism of Aβ42-mediated synaptic dysregulation on the time scale of hours. This suggests that the mechanisms described above could provide the basis of acute Aβ42-mediated LTP dysfunction. Here we show that normal but not mutant MAPT-N296H can restore Aβ42-mediated LTP dysfunction in H1 but not N296H mice. Our Western blot analysis demonstrates that MAPT-N296H is hypophosphorylated under basal conditions and that its phosphorylation measured at the AT8 epitope does not change following Aβ42 incubation in contrast to the phosphorylation increase observed in MAPT-H1 hippocampal slices. This suggests that the uncoupling of Aβ42 effects on LTP by MAPT-N296H occurs at the level of tau phosphorylation which could feed directly into the formation of small soluble tau oligomers (Cowan et al., 2010). This strengthens the link between Aβ42 signaling, tau phosphorylation at GSK3 epitope (detected by AT8 immunoreactivity) and LTP impairment observed in WT but not Tau−/− mice (Shipton et al., 2011). Nevertheless, our experiments do not address whether the MAPT-N296H mutation in tau affects its association with fyn or its subcellular localization.
The mechanisms by which FTD-related mutations lead to neuronal dysfunction and loss are not well understood. However, there is evidence that distinct tau gain or loss of function mutations or progranulin mutations (in the absence of tau mutations) can lead to FTD (Baker et al., 2006; De Silva et al., 2006). Our results indicate that the N296H mutation in MAPT leads to hypophosphorylation and to a loss of function in the mechanisms that link Aβ42 with impairment of LTP. Therefore, the mechanism that causes neuronal dysfunction in N296H mutant cells may differ from that leading to tau hyperphosphorylation and downstream mechanisms in AD. We suggest that the human tau transgenic models we present here—whereby wild type H1 human tau mediates Aβ42-inhibition of LTP and N296H does not—can be used to further explore Aβ42-tau interactions in disease.
Author Contributions
Scientific concept and experimental design: MVC, RW, and OP. Data analysis: MVC and HW. Creation of mouse lines and tau western blot: FD. In situ hybridisation: PO. LTP recording: MVC, EA, and OAS. Western blot analysis of AT8/Tau5 ratio: HW. Aβ protein stain: CP. Wrote the manuscript: MVC and HW. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MVC was supported by a Wellcome Trust OXION Training Fellowship and an equipment grant from Alzheimer's Research UK. MVC is funded by the Institute for Life Sciences University of Southampton. RW-M was supported by a Wellcome Trust Research Career Development Fellowship (073141/Z/03/Z), CurePSP and the Alzheimer's Society; FD held a Wellcome Trust DPhil in Neuroscience (075406/Z/04/A), and CMP is funded by the Gerald Kerkut Trust and IfLS. We thank Hana N. Dawson and Michael P. Vitek for Tau−/− mice. We thank Jenny Dworzak for her participation at an early phase of this project.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2017.00201/full#supplementary-material
Supplementary Figure 1. (A) Protein stain of ACSF with Aβ42 (at a concentration of 44 μM to allow detection) following 2 h from preparation showing oligomeric composition.
References
Allen Institute for Brain Science (2015). Allen Mouse Brain Atlas [Internet]. Available online at: http://mouse.brain-map.org
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., et al. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. doi: 10.1046/j.1471-4159.2003.01879.x
Baker, M., MacKenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
Bhat, R., Xue, Y., Berg, S., Hellberg, S., Ormo, M., Nilsson, Y., et al. (2003). Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 278, 45937–45945. doi: 10.1074/jbc.M306268200
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Cowan, C. M., and Mudher, A. (2013). Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 4:114. doi: 10.3389/fneur.2013.00114
Cowan, C. M., Bossing, T., Page, A., Shepherd, D., and Mudher, A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 120, 593–604. doi: 10.1007/s00401-010-0716-8
Dawson, H. N., Ferreira, A., Eyster, M. V., Ghoshal, N., Binder, L. I., and Vitek, M. P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187.
De Silva, R., Lashley, T., Strand, C., Shiarli, A. M., Shi, J., Tian, J., et al. (2006). An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 111, 329–340. doi: 10.1007/s00401-006-0048-x
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Denk, F., and Wade-Martins, R. (2009). Knock-out and transgenic mouse models of tauopathies. Neurobiol. Aging 30, 1–13. doi: 10.1016/j.neurobiolaging.2007.05.010
Fá, M., Puzzo, D., Piacentini, R., Staniszewski, A., Zhang, H., Baltrons, M. A., et al. (2016). Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep. 6:19393. doi: 10.1038/srep19393
Grover, A., DeTure, M., Yen, S. H., and Hutton, M. (2002). Effects on splicing and protein function of three mutations in codon N296 of tau in vitro. Neurosci. Lett. 323, 33–36. doi: 10.1016/S0304-3940(02)00124-6
Iseki, E., Matsumura, T., Marui, W., Hino, H., Odawara, T., Sugiyama, N., et al. (2001). Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 102, 285–292.
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jefferson, A., and Volpi, E. V. (2010). Fluorescence in situ hybridization (FISH) for genomic investigations in rat. Methods Mol. Biol. 659, 409–426. doi: 10.1007/978-1-60761-789-1_32
Jo, J., Whitcomb, D. J., Olsen, K. M., Kerrigan, T. L., Lo, S.-C., Bru-Mercier, G., et al. (2011). Aβ1–42 inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547. doi: 10.1038/nn.2785
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble A oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Llorens-Marítin, M., Jurado, J., Hernández, F., and Avila, J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046
Mondragon-Rodriguez, S., Trillaud-Doppia, E., Dudilot, A., Bourgeois, C., Lauzon, M., Leclerc, N., et al. (2012). Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 287, 32040–32053. doi: 10.1074/jbc.M112.401240
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1194. doi: 10.1038/nn.4067
Nussbaum, J. M., Schilling, S., Cynis, H., Silva, A., Swanson, E., Wangsanut, T., et al. (2012). Prion-like behavior and tau-dependent cytotoxicity of pyroglutamylated β-amyloid. Nature 485, 651–655. doi: 10.1038/nature11060
Ondrejcak, T., Klyubin, I., Hu, N.-W., Barry, A. E., Cullen, W. K., and Rowan, M. J. (2010). Alzheimer's disease amyloid β-protein and synaptic function. Neuromol. Med. 12, 13–26. doi: 10.1007/s12017-009-8091-0
Pooler, A. M., Polydoro, M., Eduardo, A. M., Nicholls, S. B., Reddy, S. M., Wegmann, S., et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta Neuropathol. Commun. 3, 14. doi: 10.1186/s40478-015-0199-x
Purro, S. A., Dickins, E. M., and Salinas, P. C. (2012). The secreted Wnt antagonist Dickkopf-1 is required for amyloid β-mediated synaptic loss. J. Neurosci. 32, 3492–3498. doi: 10.1523/JNEUROSCI.4562-11.2012
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369. doi: 10.1073/pnas.092136199
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., et al. (2011). Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's Disease. J. Neurosci. 31, 700–711. doi: 10.1523/JNEUROSCI.4152-10.2011
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Shipton, O. A., Leitz, J. R., Dworzak, J., Acton, C. E. J., Tunbridge, E. M., Denk, F., et al. (2011). Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J. Neurosci. 31, 1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U.S.A. 95, 7737–7741. doi: 10.1073/pnas.95.13.7737
Takanashi, M., Mori, H., Arima, K., Mizuno, Y., and Hattori, N. (2002). Expression patterns of tau mRNA isoforms correlate with susceptible lesions in progressive supranuclear palsy and corticobasal degeneration. Mol. Brain Res. 104, 210–219. doi: 10.1016/S0169-328X(02)00382-0
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Vargas, J. Y., Fuenzalida, M., and Inestrosa, N. C. (2014). In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer's disease model. J. Neurosci. 34, 2191–2202. doi: 10.1523/JNEUROSCI.0862-13.2014
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2011). Tau reduction prevents Aβ-induced defects in axonal transport. Science 330, 10–13. doi: 10.1126/science.1194653
Yoshida, H., Crowther, R. A., and Goedert, M. (2002). Functional effects of tau gene mutations deltaN296 and N296H. J. Neurochem. 80, 548–551. doi: 10.1046/j.0022-3042.2001.00729.x
Keywords: Alzheimer's disease, amyloid beta, frontotemporal dementia, tau, MAPT, N296H
Citation: Vargas-Caballero M, Denk F, Wobst HJ, Arch E, Pegasiou C-M, Oliver PL, Shipton OA, Paulsen O and Wade-Martins R (2017) Wild-Type, but Not Mutant N296H, Human Tau Restores Aβ-Mediated Inhibition of LTP in Tau−/− mice. Front. Neurosci. 11:201. doi: 10.3389/fnins.2017.00201
Received: 23 November 2016; Accepted: 24 March 2017;
Published: 24 April 2017.
Edited by:
Ottavio Arancio, Columbia University, USAReviewed by:
Claudio Grassi, Università Cattolica del Sacro Cuore, ItalyLuciano D'Adamio, AECOM, USA
Daniela Puzzo, University of Catania, Italy
Copyright © 2017 Vargas-Caballero, Denk, Wobst, Arch, Pegasiou, Oliver, Shipton, Paulsen and Wade-Martins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Vargas-Caballero, bS52YXJnYXMtY2FiYWxsZXJvQHNvdG9uLmFjLnVr
Richard Wade-Martins, cmljaGFyZC53YWRlLW1hcnRpbnNAZHBhZy5veC5hYy51aw==
†These authors have contributed equally to this work.