 Zachary Freyberg
Zachary Freyberg Despoina Aslanoglou
Despoina Aslanoglou Ripal Shah3
Ripal Shah3 Jacob S. Ballon
Jacob S. Ballon- 1Department of Psychiatry, University of Pittsburgh, Pittsburgh, PA, United States
- 2Department of Cell Biology, University of Pittsburgh, Pittsburgh, PA, United States
- 3Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, United States
For decades, there have been observations demonstrating significant metabolic disturbances in people with schizophrenia including clinically relevant weight gain, hypertension, and disturbances in glucose and lipid homeostasis. Many of these findings pre-date the use of antipsychotic drugs (APDs) which on their own are also strongly associated with metabolic side effects. The combination of APD-induced metabolic changes and common adverse environmental factors associated with schizophrenia have made it difficult to determine the specific contributions of each to the overall metabolic picture. Data from drug-naïve patients, both from the pre-APD era and more recently, suggest that there may be an intrinsic metabolic risk associated with schizophrenia. Nevertheless, these findings remain controversial due to significant clinical variability in both psychiatric and metabolic symptoms throughout patients' disease courses. Here, we provide an extensive review of classic and more recent literature describing the metabolic phenotype associated with schizophrenia. We also suggest potential mechanistic links between signaling pathways associated with schizophrenia and metabolic dysfunction. We propose that, beyond its symptomatology in the central nervous system, schizophrenia is also characterized by pathophysiology in other organ systems directly related to metabolic control.
Schizophrenia is a chronic psychiatric illness characterized by hallucinations, delusions, cognitive symptoms, and negative symptoms. Use of antipsychotic drugs (APDs) has been a mainstay of treatment. Though these medications effectively treat the hallucinations, APDs also cause significant metabolic side effects including insulin resistance (IR), dyslipidemia obesity and ultimately type II diabetes (T2D; Ballon et al., 2014). Consequently, a longstanding controversy in managing patients with schizophrenia has been the premorbid risk for adverse metabolic states. We therefore pose the question: is there an intrinsic metabolic risk inherent to schizophrenia or is the metabolic phenotype primarily driven by the impact of APDs?
Pre-Antipsychotic era
Prior to the APD era, cohort studies (Kooy, 1919; Kasanin, 1926) noted increased incidence of abnormal glucose metabolism in people with schizophrenia (Henneman et al., 1954). These observations corroborated cross-sectional results demonstrating that the prevalence of diabetes was greater in patients with schizophrenia compared to the general population (Kasanin, 1926). Examination of the data in pre-APD studies of schizophrenia patients suggests that abnormal glucose metabolism far preceded the confounding effects of pharmacologic intervention. Interestingly, a study in 1922 determined that increased fasting and post-prandial blood glucose levels in untreated patients with schizophrenia was in part correlated to the severity of their illness. Catatonic patients experienced more than twice the maximum post-prandial plasma levels of glucose than disorganized schizophrenia patients. Moreover, the fasting glucose level after a 12-h fast was uniformly higher in catatonic patients than in those with other forms of schizophrenia (Lorenz, 1922).
Yet, there is a wide heterogeneity among the metabolic sequelae associated with schizophrenia across all stages of the illness. In the literature preceding the era of APD treatment, Kasanin (1926) reported on a case series of 154 people with dementia praecox (later renamed schizophrenia), stating: “From this compilation it is evident that we could find no curve that is characteristic of the “dementia praecox” group as a whole. This is not unexpected in view of the heterogeneous character of the clinical conditions described under this head” (Kasanin, 1926). Further, even for those taking medications with the highest metabolic risk such as, olanzapine and clozapine, there is a notable percentage, more than 15%, that do not gain weight (Leadbetter et al., 1992). Rather, these medications may still produce significant metabolic disturbances including hyperglycemia and dyslipidemia that are independent of effects on body mass (Kang and Lee, 2015). While such wide-ranging effects make the study of a definitive mechanism more challenging, they nevertheless demonstrate the likely existence of a metabolic phenotype intrinsic to schizophrenia. Moreover, there is accumulating evidence suggesting the likelihood of multiple pathways responsible for the diversity of metabolic phenotypes in people with schizophrenia.
Contemporary Evidence of Intrinsic Metabolic Risk in Schizophrenia
More recent studies have confirmed the pre-medication era findings, and have continued to show impaired glucose tolerance, dyslipidemia, and related aspects of metabolic syndrome in drug-naïve patients with schizophrenia, as compared to healthy controls (Fernandez-Egea et al., 2009; Enez Darcin et al., 2015). In a 2016 case-control study of drug-naïve patients with schizophrenia, almost one-fourth of patients had baseline impaired glucose tolerance compared to none of the controls. The patients with schizophrenia also had higher fasting glucose levels and greater IR. However, compared to the controls, the patients with schizophrenia had higher BMIs, LDL levels, TG levels, waist circumference, and hip circumference. It is unclear to what extent those factors confounded the impaired glucose tolerance observed in patients with schizophrenia, though it is notable that these anthropometric parameters differed between cases and controls before the opportunity for pharmacologic intervention (Chen et al., 2016). Similar findings were found in youth through the recent Tolerability and Efficacy of Antipsychotics (TEA) trial. Drug-naïve first-episode psychosis patients ages 12–17 years had higher waist circumference, total cholesterol, LDL, and non-HDL levels compared to controls (Jensen et al., 2017). These suggestions have been challenged in meta-analyses that exclude patients with affective psychosis (which could lead to confounding changes in dietary or exercise habits). One meta-analysis found that the difference in LDL levels between first-episode, non-affective psychosis patients and controls was clinically insignificant. The systemic review did, however, find that first-episode, non-affective psychosis patients had higher triglyceride levels, and lower HDL levels than the controls. The study suggests that this may represent subclinical dyslipidemia, and hypothesizes that psychosis-specific mechanisms may be at play, perhaps by sharing overlapping genetic regions with metabolic phenotypes (Misiak et al., 2017).
Another case-control of drug-naïve, first-episode patients with psychosis showed higher insulin and C-peptide levels and lower HDL levels compared to the age- and BMI-matched controls. This suggests that even when accounting for the possible effect that schizophrenia itself may have on BMI, IR is still increased in patients with psychosis compared to healthy controls. Interestingly, when controlling for BMI, patients and controls were no different in their triglyceride, total cholesterol, and fasting glucose levels (Petrikis et al., 2015). Indeed, use of APDs certainly increases the risk of metabolic disturbance, but as discussed, there is a baseline elevated risk for metabolic syndrome in people with schizophrenia prior to antipsychotic use. Consistent with this, a meta-analysis of 48 studies showed a 10.2% prevalence of metabolic syndrome in antipsychotic-naïve patients, but a rate of 19.4% for patients on aripiprazole, and 47.2% for patients on clozapine. This further suggests that the disease state has an independent influence on adverse metabolic symptoms beyond the treatment-induced effect (Vancampfort et al., 2015; see Table 1).
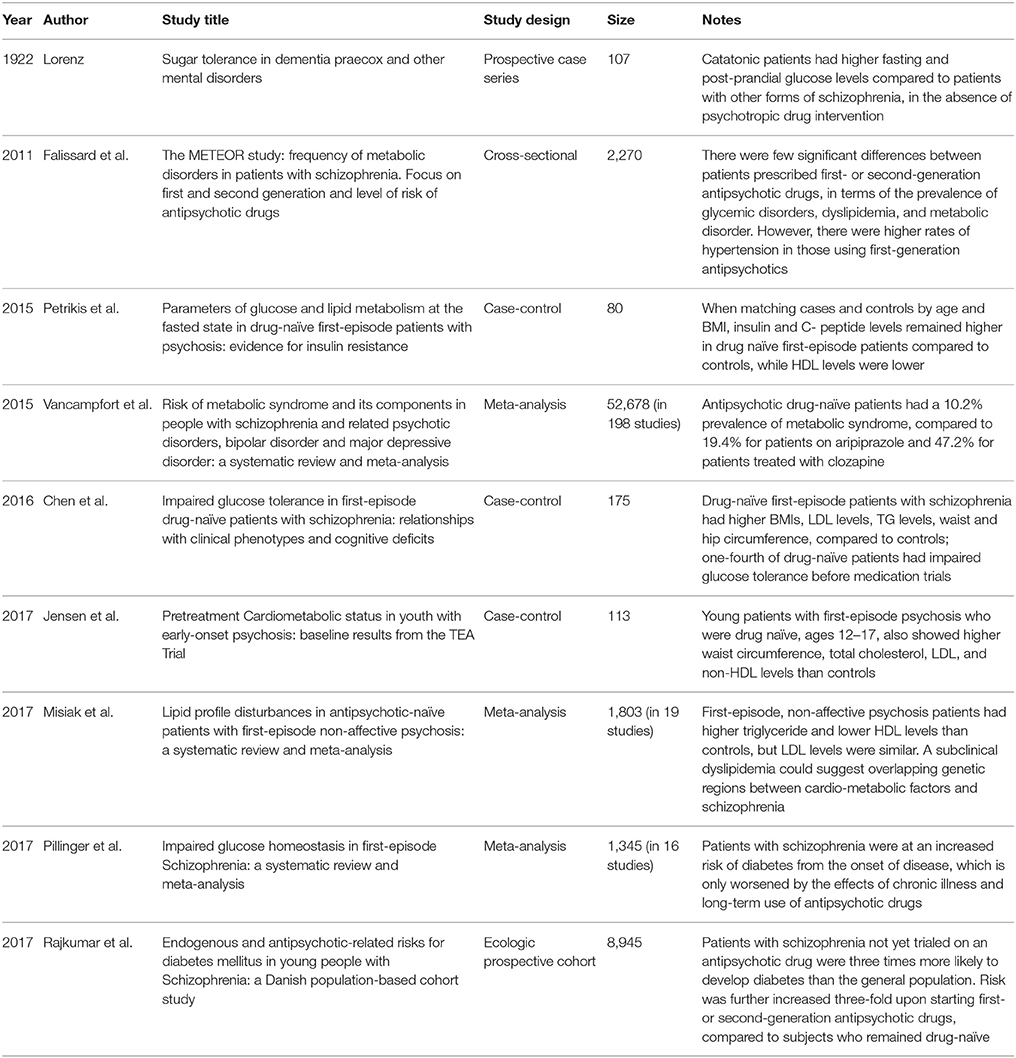
Table 1. Intrinsic metabolic risk in untreated patients with schizophrenia, amplified by use of antipsychotic drugs, as observed in multiple studies and meta-analyses.
In addition to individual smaller studies, two large meta-analyses of studies of first-episode and drug-naïve schizophrenia patients further showed a baseline increased risk for metabolic syndrome at the onset of treatment, which only worsened over progression of the illness (Mitchell et al., 2013). Glucose homeostasis was impaired from the onset of schizophrenia, despite patients having similar hemoglobin A1c levels relative to controls. The factors that were higher in patients included fasting plasma glucose levels, glucose levels after an oral glucose tolerance test, fasting insulin levels, and IR. The meta-analysis concluded that patients with schizophrenia were at an increased risk of diabetes, only exacerbated by the effects of chronic illness and long-term treatment (Pillinger et al., 2017).
A population-based study in Denmark tested the added risk of starting APDs. While APD-naïve patients with schizophrenia had a higher rate of diabetes by a factor of 3.07 compared with the general population, the risk of diabetes was 3.64 times higher in patients started on APDs compared to patients with schizophrenia who remained APD-naïve. Both first-generation and second-generation APDs increased this risk more than three-fold, with no statistically significant between the two drug classes in their capacity to cause these metabolic sequelae (Rajkumar et al., 2017).
Impaired glucose tolerance has also been demonstrated in non-psychotic, first-degree relatives of schizophrenia patients, further indicating a heritable phenotype that tracks with risk of psychosis, but is independent of the actual development of a psychotic disorder (Spelman et al., 2007). This lends credence to the concept of a novel metabolic endophenotype associated with schizophrenia with variable penetrance across both affected individuals and their unaffected relatives.
Significantly, the risk of metabolic abnormalities further increases significantly with duration of illness. Chronically-ill subjects exhibit increased rates of metabolic dysfunction compared to first-episode and drug-naïve patients (Mitchell et al., 2013; Correll et al., 2014). Additional lifestyle factors including sedentary lifestyle, poor diet, and increased smoking further add to the metabolic risks associated with schizophrenia (Brown et al., 1999).
Contribution of Antipsychotic Medications to Metabolic Risk
In addition to the intrinsic risk factors for metabolic disease associated with schizophrenia including genetics and lifestyle (Ballon et al., 2014; Heald et al., 2017; Rado, 2017), there is a consensus that APDs can further exacerbate these metabolic disturbances (Kane et al., 2004). Many APDs currently available in the United States have been associated with metabolic side effects to varying degrees (Kendall, 2011). Clozapine and olanzapine, though the most clinically effective APDs available, have also shown the greatest risk for inducing IR and significantly elevate risks for major cardiovascular events (e.g., acute coronary syndrome, ischemic stroke, and peripheral artery disease) by up to 2.8-fold (Szmulewicz et al., 2017). Indeed, 32% of patients taking olanzapine develop IR, in addition to gaining at least 15% of their baseline bodyweight (Citrome et al., 2011). Such APD-induced IR is a critical factor in the increased risk for coronary vascular disease, and thus is one of the principal causes of morbidity and premature mortality in this population (Hennekens et al., 2005; Laursen et al., 2012). Other APDs such as, ziprasidone and lurasidone have lesser cardiometabolic risks (Allison et al., 1999), though still elevate risk for other metabolic abnormalities such as, non-alcoholic fatty liver disease (Morlan-Coarasa et al., 2016).
Significantly, while much of the field's focus on metabolic side effects has been on newer medications (second-generation or atypical APDs), even first-generation APDs, such as, haloperidol or chlorpromazine, have been associated with weight gain and IR (Gordon et al., 1960; Fleischhacker et al., 2012). Consistent with this, in the large EUFEST trial (European First Episode Schizophrenia Trial), while olanzapine produced a 13.9 kg weight gain in subjects compared to baseline, haloperidol still caused a significant 7.3 kg weight gain as well (Kahn et al., 2008; Fleischhacker et al., 2012). Similarly, the “Evaluation of METabolic disordErs in schizOphRenic patients” (METEOR) study found no significant differences between patients prescribed first- or second-generation APDs in terms of the prevalence of glycemic disorders, dyslipidemia, and metabolic disorder (Falissard et al., 2011). These findings argue that most, if not all, APDs may cause or exacerbate metabolic symptoms and associated morbidity (Kahn et al., 2008). Data from the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) study also showed substantial effects on metabolic in subjects taking APDs (McEvoy et al., 2005). When compared with a matched sample of people without psychiatric illness from the National Health and Nutrition Examination Survey (NHANES), people with schizophrenia treated with APDs had the highest metabolic risk of any patient group (McEvoy et al., 2005). These results reaffirm that, independent of medication effects, schizophrenia appears to confer an intrinsic risk of metabolic dysfunction, and APD treatment exacerbates this preexisting susceptibility to metabolic disease (Spelman et al., 2007). Therefore, the combination of inherent metabolic risks, both genetics and lifestyle, when combined with APD treatment, leads to the greatest possible metabolic risk category in medicine.
Central Nervous System Mechanisms of Intrinsic and APD-Induced Metabolic Disturbances
Most studies examining either APD-induced metabolic side effects or intrinsic metabolic risk in schizophrenia have focused primarily on regions of the central nervous system (CNS) associated with metabolic control (e.g., hypothalamus). Consequently, numerous neurotransmitter and neuropeptide systems in the brain have been implicated in mediating these metabolic phenomena including the monoamines dopamine, serotonin, and histamine (Nasrallah, 2008).
Dopamine
The single unifying property of all APDs is their ability to act on dopamine receptors including dopamine D2 and D3 receptors (D2R and D3R). Consequently, increasing evidence suggests that central D2R/D3R play important roles in mediating both APDs' therapeutic actions as well as their metabolic side effects (Beaulieu et al., 2005; Karam et al., 2010; Ballon et al., 2014). Moreover, network and pathway-based analyses also suggest that these receptors are jointly associated with both schizophrenia and T2D, further implicating these receptors as mediators of intrinsic metabolic risk in schizophrenia (Liu et al., 2013). Indeed, D2R is expressed in the pituitary gland in lactotroph cells that produce and release of prolactin, a powerful hormone regulator of systemic glucose homeostasis (Lopez Vicchi et al., 2016); D2R also regulates proliferation of the lactotrophs themselves (Ben-Jonathan and Hugo, 2015; Lopez Vicchi et al., 2016). Additionally, D2R is involved in central regulation of appetite via signaling through the striatal reward pathways. Mutations or polymorphisms of D2R associated with diminished D2R levels in the CNS have been implicated with increased feeding motivation, food intake and development of overweight states (Wang et al., 2001, 2002; Palmiter, 2007). Lastly, dopamine and D2R signaling in hypothalamic regions such as, the suprachiasmatic nucleus may mediate the circadian rhythms responsible for metabolic control including systemic insulin sensitivity (Landgraf et al., 2014, 2016; Barandas et al., 2015).
Serotonin
In addition to dopamine, serotonin and serotonin receptors have been implicated in both intrinsic and APD-induced metabolic disturbances in schizophrenia (Kroeze et al., 2003; Tang et al., 2014). Single-nucleotide polymorphisms in 5HT2a and 5HT2c serotonin receptors are associated with several sequelae of metabolic dysfunction including obesity, glucose intolerance, and weight gain (Kring et al., 2009). Similarly, in rodent models, loss of function 5HT2c receptor mutations produce insulin-resistant and hyperphagic mice (Nonogaki et al., 1998). As the case with dopamine, these studies suggest that perturbation of brain serotonin signaling may play roles in both T2D and schizophrenia. Furthermore, CNS serotonin receptors are important targets for APDs, especially for atypical antipsychotics such as, clozapine and olanzapine (Reynolds and Kirk, 2010; Arranz et al., 2011). Though individual APDs have different respective binding affinities at the serotonin receptors, it has been suggested that APD actions at 5HT2a and 5HT2c receptors in the hypothalamus contribute significantly to iatrogenic weight gain (Ballon et al., 2014). In contrast, a recent study showed that polymorphisms in serotonin receptor genes HTR3A and HTR3B were not associated with predicting APD-induced weight gain (Zai et al., 2016).
Histamine
Histaminergic neurons are localized to the posterior hypothalamus and project throughout the brain including striatum. Increasing evidence suggests that brain histaminergic signaling plays important roles in feeding behaviors in part through its modulatory actions on the reward circuitry (Bolam and Ellender, 2016). Studies have demonstrated that histamine has an anorectic effect on food intake via its actions on histamine H1 receptors (H1R; Ishizuka and Yamatodani, 2012). Indeed, intracerebroventricular infusion of histamine in rodents reduces food intake likely through its actions on H1R (Ishizuka et al., 2006). Consistent with these observations, APDs with the greatest antagonist H1R affinity, clozapine, and olanzapine, also stimulate hypothalamic AMP-activated protein kinase (AMPK) that may culminate in increased appetite and feeding (Kim et al., 2007). Hypothalamic histamine H1R is also associated with modulation of systemic energy balance (He et al., 2013). Consequently, blockade of these histamine receptors by APDs leads to augmented activation of downstream AMPK-carnitine palmitoyltransferase 1 signaling of the receptors which culminates in increased appetite (He et al., 2013). Further, long term histamine receptor blockade causes fat accumulation by decreasing lipolysis in adipose tissue (He et al., 2013). Nevertheless, the roles of histamine receptors, including H1 and H3 receptors, in mediating metabolic risk in schizophrenia remain controversial (Shams and Muller, 2014). Relatively few genetic polymorphisms have been identified for these receptors and the results have been equivocal. For example, recent work examining SNPs in the genes encoding H1 and H3 receptors (HRH1 and HRH3, respectively) did not yield significant results for either receptor in mediating APD-induced weight gain (Godlewska et al., 2013; Tiwari et al., 2016).
APD-Induced Effects on Appetite and Energy Consumption
Weight gain is fundamentally due to an energy imbalance where there is a surplus of energy intake over energy consumption. This leads to storage of the excess energy typically leading to increased weight (Muller and Kennedy, 2006). Most commonly, these increases in energy intake are due to increased caloric intake secondary to enhanced appetite. In addition to the above evidence for APD actions on the histamine system in causing appetite changes that lead to obesity (Deng, 2013), second-generation APDs also act on several neuropeptides that modulate appetite and food intake. For example, olanzapine and risperidone have both been shown to increase levels of appetite-stimulating neuropeptides including neuropeptide Y (NPY) and agouti-related peptide (AgRP), along with elevations in H1R expression to further potentiate these effects (Lian et al., 2015). Olanzapine may also increase appetite through actions on ghrelin, an orexigenic peptide, where ghrelin receptor signaling is enhanced by the drug (Tagami et al., 2016). Likewise, APDs may further increase appetite by decreasing levels of proopiomelanocortin (POMC), an appetite-inhibiting neuropeptide (Lian et al., 2015). Importantly, POMC and its precursor pre-POMC are differentially processed to yield several other metabolically-relevant derivative molecules including β-endorphin, melanocyte-stimulating hormones (MSHs), and adrenocorticotropic hormone (ACTH; Millington, 2007; Mountjoy, 2015; Anderson et al., 2016). Recent work suggests that one of these derivatives, α-MSH, may play a role in appetite regulation (Vehapoglu et al., 2016). Consequently, decreased levels of α-MSH, an anorexigenic hormone, in response to risperidone treatment likely further contribute to the increases in appetite observed clinically (Baltatzi et al., 2008; Yanik et al., 2013). Evidence also suggests that one of the MSH receptors, melanocortin 4 receptor (MC4R), is strongly linked with both weight regulation and APD-induced obesity (Zhang et al., 2016). Indeed, MC4R function is implicated in energy expenditure as well as regulation of food intake independently of changes in body weight (Xu et al., 2013; Mountjoy, 2015). Furthermore, a single nucleotide polymorphism (SNP) in MC4R, rs17782313, has been associated with overeating as well as APD-induced weight gain (Yilmaz et al., 2015; Macneil and Muller, 2016). Two additional SNPs, rs8087522, and rs1801133, have also been linked to APD-induced weight gain (Malhotra et al., 2012; Macneil and Muller, 2016), suggesting that the MC4R locus may be an attractive candidate for predicting APD-induced weight gain and metabolic disruption.
In terms of energy expenditure, APDs such as olanzapine reduce locomotor activity in rodent models, which further disrupts the balance between energy intake and consumption and leads to weight gain (van der Zwaal et al., 2014). Moreover, these olanzapine-induced decreases in locomotor activity occur at doses that do not affect eating behavior (Weston-Green et al., 2011). It has been suggested that APDs' sedative properties play an important role in causing this diminished energy consumption (van der Zwaal et al., 2014). Consistent with this, there is significant incidence of somnolence in people treated with many second-generation APDs (Gao et al., 2008). Furthermore, thermogenesis is another measure of body energy consumption and olanzapine administration was shown to cause reductions in body core temperature in animal model (van der Zwaal et al., 2012, 2014). Therefore, these drug-induced decreases in body core temperature suggest overall decreases in energy expenditure. Nevertheless, it is unclear to date if this is the case. Several trials have shown either no significant effect of APDs on resting energy expenditure (REE; Graham et al., 2005; Vidarsdottir et al., 2010), decreased REE (Sharpe et al., 2005, 2006; Nilsson et al., 2006) or even elevated REE (Fountaine et al., 2010). Intriguingly, unmedicated people with schizophrenia exhibited decreased REE (Nilsson et al., 2006). Overall, these studies suggest that the relationships between changes in thermogenesis and REE are likely complex and APD effects on body core temperature may cause compensatory changes in REE.
The ability of APDs to differentially target the various respective receptor signaling systems may have profound effects on both appetite and energy balance. Differences in the respective APDs' actions on both arms of energy balance regulation is likely responsible both for differences in the magnitude of significant metabolic side effects caused by these medications. However, this is complicated clinically by the observation that there is relatively high overall variability in the phenotypes of APD-induced metabolic dysfunction. Of the people who develop metabolic side effects, some gain weight with no IR; some develop IR without weight gain, and some experience both. Based on the discrepancy between weight gain and IR, an alternate treatment approach has been to focus on effects of APDs on appetite (Mizuno et al., 2014). Therapeutically, this has led to studies of appetite suppressants like topiramate and dextroamphetamine, to target CNS appetite centers. Nevertheless, appetite suppressants, including topiramate, have little to no effect in treating either intrinsic or APD-induced metabolic dysfunction, and cause significant cognitive and/or psychotic side effects that limit their overall safety (Narula et al., 2010; Muscatello et al., 2011; Mizuno et al., 2014). This highlights the conundrum concerning the roles of monoamine signaling in schizophrenia's intrinsic effects on metabolism and APD-induced metabolic effects: despite significant strides in understanding the physiology and pharmacology of monoamine signaling in the CNS, there has been a paucity of major mechanistic breakthroughs or new treatments that have mitigated these metabolic disturbances (Mizuno et al., 2014).
Peripheral Mechanisms of Intrinsic and APD-Induced Metabolic Disturbances
A potential explanation for the limited efficacy of drugs targeting APD-induced metabolic dysfunction is their focus primarily on CNS targets. Increasing evidence suggests that the same molecular targets of APDs, such as, histamine, serotonin, and dopamine receptors, also exist in peripheral organs critical for metabolic control, including the pancreas, adipose tissue, and skeletal muscle (Garcia-Tornadu et al., 2010; Rubi and Maechler, 2010; Ballon et al., 2014; see Figure 1). These peripheral targets, together with the CNS, jointly regulate both body weight and glucose/insulin homeostasis, key factors in APD-induced metabolic dysfunction (Ballon et al., 2014). For example, insulin-secreting pancreatic beta cells not only produce their own dopamine, but also express D2R, an important APD target (Rubi et al., 2005; Simpson et al., 2012). There is further precedent for CNS neurotransmitter action in the periphery, including in the gastrointestinal tract and adipose tissue (Gershon, 2013). Moreover, there is crosstalk between the CNS and these peripheral organs since hypothalamic and brain stem neurons have been shown to influence beta cell, hepatocyte and adipocyte metabolism (Elmquist et al., 2005) and vice versa.
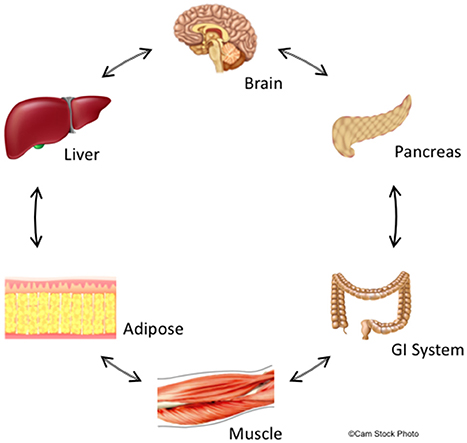
Figure 1. Summary of central and peripheral mechanisms contributing to deleterious effects of schizophrenia and APDs on glucose and lipid metabolism. The central nervous system,including metabolically-relevant areas in the brain such as hypothalamus, receive feedback from peripheral organs that regulate metabolism and appetite. Likewise, metabolic centers in the brain regulate metabolism throughout the body via actions on peripheral organs including GI tract, liver, pancreas, adipose tissue, and skeletal muscle. This feedback is bidirectional though non-sequential, and creates a delicate metabolic balance that is disturbed by biological changes intrinsic to schizophrenia and is further perturbed by APDs. We hypothesize that these central and peripheral target organs are linked through common molecular signaling networks involving dopaminergic, serotoninergic, histaminergic, and adipokine signaling. Since APDs act at receptors for these signaling systems, these drugs may have synergistic properties that significantly increase the risk of developing metabolic disturbances including insulin resistance and adiposity.
Glucose and insulin-sensitive tissues in the periphery, including pancreatic beta cells and adipose cells, express D2-like receptors (D2R, D3R, and D4R), which are key targets of APDs (Wilson et al., 1998). In addition, insulin-secreting pancreatic beta cells also express the CNS-specific isoform of the vesicular monoamine transporter, VMAT2, which is responsible for vesicular dopamine loading and storage (Anlauf et al., 2003). We have shown that dopamine acts as an autocrine or paracrine negative regulator of glucose-stimulated insulin secretion (GSIS) to dampen further insulin release (Figure 2A; Simpson et al., 2012). Moreover, we have also demonstrated that the addition of sulpiride, one of the most D2R/D3R-selective APDs (Newman et al., 2012), increases GSIS by 40% in pancreatic beta cells. Importantly, this APD-induced increase in insulin secretion is consistent with the chronic hyperinsulinemia found in APD-induced IR clinically (Henderson et al., 2005). Conversely, D2R and D3R agonism may have therapeutic effects on IR. Indeed, a quick-release formulation of the D2R/D3R agonist bromocriptine is FDA-approved to treat T2D (Mikhail, 2011; Lamos et al., 2016). Although the majority of work studying bromocriptine's metabolic effects has focused on its actions in the CNS, there is robust evidence that bromocriptine also targets the same peripheral dopamine signaling pathways altered by APDs, as described above (Holt et al., 2010; Farino et al., 2016; Lamos et al., 2016). Bromocriptine lowers both elevated glucose and insulin levels in humans (Liang et al., 1998; Holt et al., 2010). Based on this evidence, bromocriptine may be effective in treating APD-associated IR by targeting peripheral dopamine pathways disrupted by APDs. Consistent with this, data from small studies is validating this hypothesis, having demonstrated reestablishment of euglycemia in the context of APD treatment (Naguy and Al-Tajali, 2016). On a cellular level, we have now demonstrated that bromocriptine treatment diminishes GSIS comparably to dopamine in a dose-dependent manner in a beta cell-derived cell line (Figure 2B). These data suggest that dopamine D2-like receptors may play an important role in the periphery to modulate secreted insulin. Furthermore, the disruption of this regulatory signaling either through genetic changes intrinsic to the disease process in schizophrenia or via drug action by APDs may lead to many of the metabolic disturbances observed clinically.
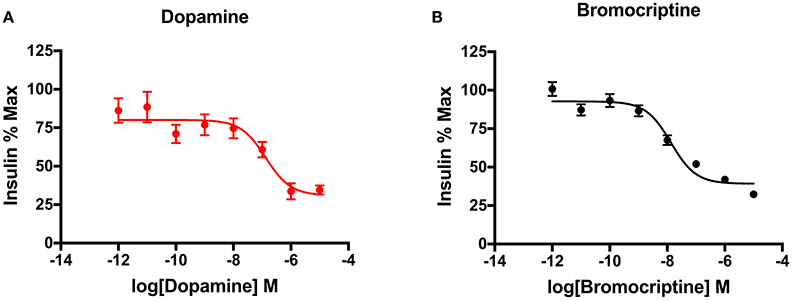
Figure 2. Dopaminergic modulation of insulin secretion in pancreatic beta cells. (A) Treatment of INS-IE cells, an established rat pancreatic beta cell-derived cell line, with dopamine potently inhibited glucose-stimulated insulin secretion in a dose-dependent manner (pIC50 = 7.83). (B) Agonism of dopamine D2 and D3 receptors by bromocriptine produced a decrease in glucose-stimulated insulin secretion comparable to dopamine (pIC50 = 6.87), suggesting that these receptors are important in mediating this effect in beta cells. Insulin secretion was measured via homogenous time-resolved fluorescence (HTRF) as described earlier (Farino et al., 2016). All experiments were performed in triplicate on n ≥ 3 separate experimental days. Data are represented as % maximal insulin secretion based on mean HTRF values ± SEM.
Existing Treatment Strategies
Given the importance of peripheral metabolic targets, initial attempts at treating APD-associated IR focused on current T2D medications already known to act in the periphery (Mizuno et al., 2014). The widely-used class of T2D drugs, thiazolidinediones, increase insulin sensitivity (IS) in T2D by acting on peripheral targets including adipose tissue, skeletal muscle, and liver to increase glucose utilization and decrease glucose production (Hauner, 2002). Besides T2D, these drugs have been applied to treatment of metabolic dysfunction associated with psychiatric disorders. However, when rosiglitazone was studied in people with schizophrenia on clozapine, it was ineffective (Henderson et al., 2009). Given these results with thiazolidinediones, further trials looked to other classes of insulin sensitizing drugs. Metformin, a biguanide, improves blood glucose levels in T2D through decreased hepatic glucose production and increased peripheral glucose utilization (Zhou et al., 2001). Though metformin modestly decreases APD-induced weight gain, like the thiazolidinediones, it has also failed to meet expectations for improving APD-induced IR (Jarskog et al., 2013; Rado and Von Ammon Cavanaugh, 2016). Based on the relative failure for using these otherwise well-established mechanistic strategies to treat IR in the setting of ongoing APD treatment, a new therapeutic strategy is clearly needed. To date, such attempts have been hampered by the relative paucity of knowledge regarding the biological modulators of metabolic dysfunction both in the cases of intrinsic and APD-induced metabolic dysfunction. Below we will review the roles of several such modulators.
Adiponectin and Metabolic Dysfunction
Adiponectin is an adipokine hormone, produced primarily by fat cells in adipose tissue that makes tissues more sensitive to insulin (Diez and Iglesias, 2003). Adiponectin levels are inversely related to obesity (Di Chiara et al., 2012). Further, low levels of circulating adiponectin are associated with IR and may provide a link between obesity and overall metabolic syndrome (Di Chiara et al., 2012). Increasing adiponectin leads to improvements in insulin sensitivity (IS) through enhanced tissue fat oxidation in adipose tissue, lowering circulating fatty acid levels and reducing intracellular triglyceride contents in liver and muscle (Diez and Iglesias, 2003). It further lowers inflammation and thus reduces risk of atherosclerosis, by suppressing the expression of adhesion molecules in vascular endothelial cells and cytokine production from macrophages. By suppressing the initial inflammatory process associated with early atherosclerosis, increasing levels of adiponectin lower the risk for cardiovascular sequelae of metabolic dysfunction (Diez and Iglesias, 2003). The combination of improving IS while lowering inflammation has generated strong interest in metabolic treatments that increase adiponectin.
The relationship between drug-naïve schizophrenia and adiponectin is unclear. An initial study found lower serum adiponectin levels in normal-weight, first-episode, drug-naïve, people with schizophrenia (Song et al., 2013). A recent meta-analysis was unable to confirm the finding, though did show a trend level of significance (p = 0.09) for decreased adiponectin in drug-naïve individuals with schizophrenia (Bartoli et al., 2015). The result was notable for significant heterogeneity within the sample, which indicates that there is a subset of people with schizophrenia who have abnormally low baseline adiponectin levels and may be at greater risk for APD-induced metabolic side effects. The introduction of APDs has been consistently shown to lower adiponectin levels (Bai et al., 2009; Tanyanskiy et al., 2015). The drugs most associated with decreased adiponectin are also the drugs most associated with metabolic side effects, clozapine (p < 0.001) and olanzapine (p = 0.04; Bartoli et al., 2015). Furthermore, dopamine receptors in adipocytes appear to regulate adiponectin levels, which suggests a putative link between the D2R/D3R polymorphisms implicated in the pathogenesis of schizophrenia and adiponectin's role in mediating intrinsic metabolic disturbances in people with the illness. Likewise, D2R/D3R antagonism ubiquitous to APDs may also connect adiponectin to APD-induced metabolic dysfunction (Borcherding et al., 2011).
Given the above evidence that the machinery for monoamine synthesis and dopamine receptor signaling is present in insulin-sensitive peripheral organs, it suggests that: (1) APDs target the same monoamine receptors in the periphery as they do in the CNS, and (2) monoamines are not only involved in APD-induced IR, but in overall insulin homeostasis (Bailey, 2000). Increasing evidence points to a potentially important contribution by monoamines to APD-induced metabolic dysfunction through direct action of APDs on peripheral monoaminergic targets including those found in insulin-secreting pancreatic beta cells (Kalra et al., 2011). This may therefore be consistent with D2R/D3R agonist bromocriptine's efficacy in the treatment of T2D based on findings that it reduces plasma glucose, triglyceride, and free fatty acid levels, and significantly decreases hemoglobin A1C levels as compared to placebo (Holt et al., 2010; Valiquette, 2011). (2) Robust metabolic findings have also been seen in obese, non-diabetic people comparable at least by metabolic markers to people with schizophrenia (Kamath et al., 1997; Kok et al., 2006).
In sum, it is increasingly evident that effects on metabolism caused by APD treatment and/or those intrinsic to schizophrenia are due to convergent effects on signaling through monoamines or neuromodulators in both the CNS and periphery. Metabolically-relevant tissues in the CNS and periphery share many of the same receptor signaling systems including the dopamine, serotonin, and histamine systems. Moreover, changes in central brain metabolic regulation propagate to target organs in the periphery including the gastrointestinal system (GI), pancreas, muscle, liver, and adipose tissues and these tissues respond in a reciprocal manner. Because APDs act concurrently on multiple receptor systems in all these tissues (e.g., dopamine, serotonin, histamine), the effects of these drugs' disruptions on metabolic regulation and appetite are likely cumulative and lead to the metabolic imbalances observed clinically (Figure 1).
Genes Implicated in Both Schizophrenia and Metabolism
To date, there is little definitive evidence of genes associated with schizophrenia having a causative role in the development of metabolic sequelae associated with either APD-induced metabolic disturbances or with the illness' intrinsic metabolic risk. Nevertheless, two genes associated with schizophrenia, AKT1 and neuregulin 1 (NRG1), may also play roles in systemic metabolic regulation. This suggests that mutations or polymorphisms in these genes may have roles not only in neuronal function within the CNS associated with schizophrenia's neuropsychiatric symptoms, but that these disruptions of function may also affect more global metabolic functions in other pathways including within insulin signaling in organs such as, the pancreas and liver.
AKT1
The gene encoding the serine-threonine kinase AKT1 has long been implicated in schizophrenia across multiple studies (Xu et al., 2007; Thiselton et al., 2008; Karam et al., 2010). AKT1 protein levels were diminished both in peripheral lymphocytes and in the brains of people with schizophrenia (Emamian et al., 2004; Arnold et al., 2005). Moreover, in rodent models, treatment with the APD haloperidol changed the phosphorylation responsible for regulating this kinase's ability to signal through its actions on another kinase, glycogen synthase kinase 3β (GSK3β) which was attributed as a potential mechanism for haloperidol's therapeutic efficacy (Emamian et al., 2004; Beaulieu et al., 2005). AKT1 is also especially relevant to intrinsic metabolic risk in schizophrenia since it is an important regulator of insulin signaling. Insulin receptor activity triggers phosphatidylinositol 3 kinase (PI3K)-dependent recruitment of AKT1 to the cell surface where it is subsequently activated (Guo, 2014). Conversely, loss of function AKT1 mutations are implicated in IR, suggesting a key role for the enzyme in metabolic control (Bernal-Mizrachi et al., 2004). Consistent with this, AKT phosphorylates downstream transcriptional activators including Foxo1 and SREBP1c that regulate glucose transport and insulin sensitivity in tissues throughout the body (Gonzalez et al., 2011; Guo, 2014). AKT1 signaling has also been implicated in APD-induced metabolic dysfunction, potentially through its effects on Wnt and beta catenin-mediated transcriptional changes (Freyberg et al., 2010). Significantly, the activity of AKT1 is modulated by D2R signaling where D2R activation leads to AKT1 inactivation (Beaulieu et al., 2007; Beaulieu and Gainetdinov, 2011). By this logic, it would be expected that D2R blockade by APDs would lead to increased levels of active AKT1 and therefore more effective insulin action rather than increased IR. A potential explanation of this apparent discrepancy is that AKT1 protein levels are diminished in people with schizophrenia (Emamian et al., 2004). Therefore, APD blockade of D2R receptors may be ineffective in enhancing insulin signaling since there is already a deficit of AKT1 signaling downstream of D2R and may even lead to changes in AKT-mediated regulation of metabolism (Freyberg et al., 2010).
NRG1
NRG1 encodes a growth factor that belongs to the larger family of epidermal growth factor (EGF) family critical for development of multiple organ systems including the nervous system, liver, heart, and skeletal muscle (Britsch, 2007; Guma et al., 2010; Mei and Nave, 2014). Of the four neuregulin isoforms described, polymorphisms in the NRG1 gene have been repeatedly associated with schizophrenia (Banerjee et al., 2010; Karam et al., 2010; Caillaud et al., 2016; Mostaid et al., 2017). Moreover, NRG1 SNPs were recently associated as predictors of APD clinical response (Li et al., 2017). Though data from computational pathway analyses have implicated interactions between NRG1 and Src signaling pathways which play roles in both schizophrenia and T2D (Liu et al., 2013), to date, the links between NRG1 and the metabolic disturbances intrinsic to schizophrenia are not well-established. However, in rodent models, NRG1 function has been shown to be important in glucose metabolism and insulin sensitivity both in liver and muscle (Caillaud et al., 2016; Lopez-Soldado et al., 2016). This may be attributable to NRG1's role in regulating hepatic glucose utilization and gluconeogenesis (Arai et al., 2017). Taken together, these data suggest that NRG1 may play a modulatory role in both in the CNS and systemically and disruption of its signaling either intrinsic to the disease processes in schizophrenia and/or with APDs may lead to the neuropsychiatric and metabolic dysfunction observed clinically.
Conclusions
We suggest a model, based on the above evidence, in which the disease processes underlying schizophrenia also confer a significant intrinsic risk for development of metabolic disturbances including IR and T2D. Consequently, these processes not only affect regions of the CNS implicated in the neuropsychiatric symptoms classically associated with the disorder including cognition, executive function and sensory perception, but likely target metabolically-relevant areas as well. Changes in neurotransmission within CNS regions such as, the hypothalamus in schizophrenia may also feedback upon metabolic signaling in the periphery including the endocrine pancreas, liver, and adipose tissue (Figure 1). Moreover, because these central and peripheral targets likely rely on conserved signaling pathways and molecules (e.g., dopamine, AKT1, NRG1), disruption or changes in these pathways in one organ may also be found in others and consequently may explain the reciprocal connections between multiple systems. Furthermore, APDs may further alter metabolism through their actions at these same CNS and peripheral sites of action. This results in APD-induced exacerbation of the preexisting metabolic risks intrinsic to schizophrenia. Along with additional lifestyle and environmental factors, the combination of APD-induced and intrinsic risk factors leads to the serious metabolic dysfunctions described clinically (Karam et al., 2010; Ballon et al., 2014). Therefore, improving our understanding of both the processes responsible for metabolic regulation and schizophrenia may elucidate the common mechanisms between both and ultimately inform new drug development strategies.
Author Contributions
ZF and JB: Conception and design, manuscript writing, editing and figure design. DA: Experimental work and conception and figure design. RS: Manuscript writing, editing, synthesis of previous literature and figure design.
Funding
This work is supported by a Department of Defense PRMRP Investigator Initiated Award PR141292, and the John F. and Nancy A. Emmerling Fund of The Pittsburgh Foundation (to ZF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Robin Freyberg for invaluable assistance in the preparation of this manuscript.
References
Allison, D. B., Mentore, J. L., Heo, M., Chandler, L. P., Cappelleri, J. C., Infante, M. C., et al. (1999). Antipsychotic-induced weight gain: a comprehensive research synthesis. Am. J. Psychiatry 156, 1686–1696.
Anderson, E. J., Cakir, I., Carrington, S. J., Cone, R. D., Ghamari-Langroudi, M., Gillyard, T., et al. (2016). 60 YEARS OF POMC: regulation of feeding and energy homeostasis by alpha-MSH. J. Mol. Endocrinol. 56, T157–T174. doi: 10.1530/JME-16-0014
Anlauf, M., Eissele, R., Schafer, M. K., Eiden, L. E., Arnold, R., Pauser, U., et al. (2003). Expression of the two isoforms of the vesicular monoamine transporter (VMAT1 and VMAT2) in the endocrine pancreas and pancreatic endocrine tumors. J. Histochem. Cytochem. 51, 1027–1040. doi: 10.1177/002215540305100806
Arai, T., Ono, Y., Arimura, Y., Sayama, K., Suzuki, T., Shinjo, S., et al. (2017). Type I neuregulin1alpha is a novel local mediator to suppress hepatic gluconeogenesis in mice. Sci. Rep. 7:42959. doi: 10.1038/srep42959
Arnold, S. E., Talbot, K., and Hahn, C. G. (2005). Neurodevelopment, neuroplasticity, and new genes for schizophrenia. Prog. Brain Res. 147, 319–345. doi: 10.1016/S0079-6123(04)47023-X
Arranz, M. J., Rivera, M., and Munro, J. C. (2011). Pharmacogenetics of response to antipsychotics in patients with schizophrenia. CNS Drugs 25, 933–969. doi: 10.2165/11595380-000000000-00000
Bai, Y. M., Chen, T. T., Yang, W. S., Chi, Y. C., Lin, C. C., Liou, Y. J., et al. (2009). Association of adiponectin and metabolic syndrome among patients taking atypical antipsychotics for schizophrenia: a cohort study. Schizophr. Res. 111, 1–8. doi: 10.1016/j.schres.2009.03.014
Bailey, C. J. (2000). Potential new treatments for type 2 diabetes. Trends Pharmacol. Sci. 21, 259–265. doi: 10.1016/S0165-6147(00)01506-6
Ballon, J. S., Pajvani, U., Freyberg, Z., Leibel, R. L., and Lieberman, J. A. (2014). Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol. Metab. 25, 593–600. doi: 10.1016/j.tem.2014.07.004
Baltatzi, M., Hatzitolios, A., Tziomalos, K., Iliadis, F., and Zamboulis, C. (2008). Neuropeptide Y and alpha-melanocyte-stimulating hormone: interaction in obesity and possible role in the development of hypertension. Int. J. Clin. Pract. 62, 1432–1440. doi: 10.1111/j.1742-1241.2008.01823.x
Banerjee, A., Macdonald, M. L., Borgmann-Winter, K. E., and Hahn, C. G. (2010). Neuregulin 1-erbB4 pathway in schizophrenia: from genes to an interactome. Brain Res. Bull. 83, 132–139. doi: 10.1016/j.brainresbull.2010.04.011
Barandas, R., Landgraf, D., McCarthy, M. J., and Welsh, D. K. (2015). Circadian clocks as modulators of metabolic comorbidity in psychiatric disorders. Curr. Psychiatry Rep. 17:98. doi: 10.1007/s11920-015-0637-2
Bartoli, F., Lax, A., Crocamo, C., Clerici, M., and Carra, G. (2015). Plasma adiponectin levels in schizophrenia and role of second-generation antipsychotics: a meta-analysis. Psychoneuroendocrinology 56, 179–189. doi: 10.1016/j.psyneuen.2015.03.012
Beaulieu, J. M., and Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217. doi: 10.1124/pr.110.002642
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2007). The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172. doi: 10.1016/j.tips.2007.02.006
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. doi: 10.1016/j.cell.2005.05.012
Ben-Jonathan, N., and Hugo, E. (2015). Prolactin (PRL) in adipose tissue: regulation and functions. Adv. Exp. Med. Biol. 846, 1–35. doi: 10.1007/978-3-319-12114-7_1
Bernal-Mizrachi, E., Fatrai, S., Johnson, J. D., Ohsugi, M., Otani, K., Han, Z., et al. (2004). Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet β cells. J. Clin. Invest. 114, 928–936. doi: 10.1172/JCI200420016
Bolam, J. P., and Ellender, T. J. (2016). Histamine and the striatum. Neuropharmacology 106, 74–84. doi: 10.1016/j.neuropharm.2015.08.013
Borcherding, D. C., Hugo, E. R., Idelman, G., De Silva, A., Richtand, N. W., Loftus, J., et al. (2011). Dopamine receptors in human adipocytes: expression and functions. PLoS ONE 6:e25537. doi: 10.1371/journal.pone.0025537
Britsch, S. (2007). The neuregulin-I/ErbB signaling system in development and disease. Adv. Anat. Embryol. Cell Biol. 190, 1–65. doi: 10.1007/978-3-540-37107-6
Brown, S., Birtwistle, J., Roe, L., and Thompson, C. (1999). The unhealthy lifestyle of people with schizophrenia. Psychol. Med. 29, 697–701. doi: 10.1017/S0033291798008186
Caillaud, K., Boisseau, N., Ennequin, G., Chavanelle, V., Etienne, M., Li, X., et al. (2016). Neuregulin 1 improves glucose tolerance in adult and old rats. Diabetes Metab. 42, 96–104. doi: 10.1016/j.diabet.2015.08.003
Chen, D. C., Du, X. D., Yin, G. Z., Yang, K. B., Nie, Y., Wang, N., et al. (2016). Impaired glucose tolerance in first-episode drug-naïve patients with schizophrenia: relationships with clinical phenotypes and cognitive deficits. Psychol. Med. 46, 3219–3230. doi: 10.1017/S0033291716001902
Citrome, L., Holt, R. I., Walker, D. J., and Hoffmann, V. P. (2011). Weight gain and changes in metabolic variables following olanzapine treatment in schizophrenia and bipolar disorder. Clin. Drug Investig. 31, 455–482. doi: 10.2165/11589060-000000000-00000
Correll, C. U., Robinson, D. G., Schooler, N. R., Brunette, M. F., Mueser, K. T., Rosenheck, R. A., et al. (2014). Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP study. JAMA Psychiatry 71, 1350–1363. doi: 10.1001/jamapsychiatry.2014.1314
Deng, C. (2013). Effects of antipsychotic medications on appetite, weight, and insulin resistance. Endocrinol. Metab. Clin. North Am. 42, 545–563. doi: 10.1016/j.ecl.2013.05.006
Di Chiara, T., Argano, C., Corrao, S., Scaglione, R., and Licata, G. (2012). Hypoadiponectinemia: a link between visceral obesity and metabolic syndrome. J. Nutr. Metab. 2012:175245. doi: 10.1155/2012/175245
Diez, J. J., and Iglesias, P. (2003). The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol. 148, 293–300. doi: 10.1530/eje.0.1480293
Elmquist, J. K., Coppari, R., Balthasar, N., Ichinose, M., and Lowell, B. B. (2005). Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J. Comp. Neurol. 493, 63–71. doi: 10.1002/cne.20786
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 36, 131–137. doi: 10.1038/ng1296
Enez Darcin, A., Yalcin Cavus, S., Dilbaz, N., Kaya, H., and Dogan, E. (2015). Metabolic syndrome in drug-naïve and drug-free patients with schizophrenia and in their siblings. Schizophr. Res. 166, 201–206. doi: 10.1016/j.schres.2015.05.004
Falissard, B., Mauri, M., Shaw, K., Wetterling, T., Doble, A., Giudicelli, A., et al. (2011). The METEOR study: frequency of metabolic disorders in patients with schizophrenia. Focus on first and second generation and level of risk of antipsychotic drugs. Int. Clin. Psychopharmacol. 26, 291–302. doi: 10.1097/YIC.0b013e32834a5bf6
Farino, Z. J., Morgenstern, T. J., Vallaghe, J., Gregor, N., Donthamsetti, P., Harris, P. E., et al. (2016). Development of a rapid insulin assay by homogenous time-resolved fluorescence. PLoS ONE 11:e0148684. doi: 10.1371/journal.pone.0148684
Fernandez-Egea, E., Bernardo, M., Donner, T., Conget, I., Parellada, E., Justicia, A., et al. (2009). Metabolic profile of antipsychotic-naive individuals with non-affective psychosis. Br. J. Psychiatry 194, 434–438. doi: 10.1192/bjp.bp.108.052605
Fleischhacker, W. W., Siu, C. O., Boden, R., Pappadopulos, E., Karayal, O. N., Kahn, R. S., et al. (2012). Metabolic risk factors in first-episode schizophrenia: baseline prevalence and course analysed from the European first-episode Schizophrenia trial. Int. J. Neuropsychopharmacol. 16, 987–995. doi: 10.1017/S1461145712001241
Fountaine, R. J., Taylor, A. E., Mancuso, J. P., Greenway, F. L., Byerley, L. O., Smith, S. R., et al. (2010). Increased food intake and energy expenditure following administration of olanzapine to healthy men. Obesity 18, 1646–1651. doi: 10.1038/oby.2010.6
Freyberg, Z., Ferrando, S. J., and Javitch, J. A. (2010). Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 167, 388–396. doi: 10.1176/appi.ajp.2009.08121873
Gao, K., Ganocy, S. J., Gajwani, P., Muzina, D. J., Kemp, D. E., and Calabrese, J. R. (2008). A review of sensitivity and tolerability of antipsychotics in patients with bipolar disorder or schizophrenia: focus on somnolence. J. Clin. Psychiatry 69, 302–309. doi: 10.4088/JCP.v69n0217
Garcia-Tornadu, I., Perez-Millan, M. I., Recouvreux, V., Ramirez, M. C., Luque, G., Risso, G. S., et al. (2010). New insights into the endocrine and metabolic roles of dopamine D2 receptors gained from the Drd2 mouse. Neuroendocrinology 92, 207–214. doi: 10.1159/000321395
Gershon, M. D. (2013). 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr. Opin. Endocrinol. Diabetes Obes. 20, 14–21. doi: 10.1097/MED.0b013e32835bc703
Godlewska, B. R., Olajossy-Hilkesberger, L., Olajossy, M., Limon, J., and Landowski, J. (2013). Polymorphisms of the histamine receptor (H1HR) gene are not associated with olanzapine-induced weight gain. J. Clin. Psychopharmacol. 33, 436–437. doi: 10.1097/JCP.0b013e3182900c9e
Gonzalez, E., Flier, E., Molle, D., Accili, D., and McGraw, T. E. (2011). Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 108, 10162–10167. doi: 10.1073/pnas.1019268108
Gordon, H., Law, A., Hohman, K., and Groth, C. (1960). The problem of overweight in hospitalized psychotic patients. Psychiatr. Q. 34, 69–82. doi: 10.1007/BF01675229
Graham, K. A., Perkins, D. O., Edwards, L. J., Barrier, R. C. Jr., Lieberman, J. A., and Harp, J. B. (2005). Effect of olanzapine on body composition and energy expenditure in adults with first-episode psychosis. Am. J. Psychiatry 162, 118–123. doi: 10.1176/appi.ajp.162.1.118
Guma, A., Martinez-Redondo, V., Lopez-Soldado, I., Canto, C., and Zorzano, A. (2010). Emerging role of neuregulin as a modulator of muscle metabolism. Am. J. Physiol. Endocrinol. Metab. 298, E742–E750. doi: 10.1152/ajpendo.00541.2009
Guo, S. (2014). Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220, T1–T23. doi: 10.1530/JOE-13-0327
Hauner, H. (2002). The mode of action of thiazolidinediones. Diabetes Metab. Res. Rev. 18(Suppl. 2), S10–S15. doi: 10.1002/dmrr.249
He, M., Deng, C., and Huang, X. F. (2013). The role of hypothalamic H1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs 27, 423–434. doi: 10.1007/s40263-013-0062-1
Heald, A., Pendlebury, J., Anderson, S., Narayan, V., Guy, M., Gibson, M., et al. (2017). Lifestyle factors and the metabolic syndrome in Schizophrenia: a cross-sectional study. Ann. Gen. Psychiatry 16:12. doi: 10.1186/s12991-017-0134-6
Henderson, D. C., Cagliero, E., Copeland, P. M., Borba, C. P., Evins, E., Hayden, D., et al. (2005). Glucose metabolism in patients with schizophrenia treated with atypical antipsychotic agents: a frequently sampled intravenous glucose tolerance test and minimal model analysis. Arch. Gen. Psychiatry 62, 19–28. doi: 10.1001/archpsyc.62.1.19
Henderson, D. C., Fan, X., Sharma, B., Copeland, P. M., Borba, C. P., Boxill, R., et al. (2009). A double-blind, placebo-controlled trial of rosiglitazone for clozapine-induced glucose metabolism impairment in patients with schizophrenia. Acta Psychiatr. Scand. 119, 457–465. doi: 10.1111/j.1600-0447.2008.01325.x
Hennekens, C. H., Hennekens, A. R., Hollar, D., and Casey, D. E. (2005). Schizophrenia and increased risks of cardiovascular disease. Am. Heart J. 150, 1115–1121. doi: 10.1016/j.ahj.2005.02.007
Henneman, D. H., Altschule, M. D., and Goncz, R. (1954). Carbohydrate metabolism in brain disease: II. Glucose metabolism in schizophrenic, manic-depressive, and involutional psychoses. AMA Arch. Intern. Med. 94, 402–416. doi: 10.1001/archinte.1954.00250030072008
Holt, R. I., Barnett, A. H., and Bailey, C. J. (2010). Bromocriptine: old drug, new formulation and new indication. Diabetes Obes. Metab. 12, 1048–1057. doi: 10.1111/j.1463-1326.2010.01304.x
Ishizuka, T., Nomura, S., Hosoda, H., Kangawa, K., Watanabe, T., and Yamatodani, A. (2006). A role of the histaminergic system for the control of feeding by orexigenic peptides. Physiol. Behav. 89, 295–300. doi: 10.1016/j.physbeh.2006.05.049
Ishizuka, T., and Yamatodani, A. (2012). Integrative role of the histaminergic system in feeding and taste perception. Front. Syst. Neurosci. 6:44. doi: 10.3389/fnsys.2012.00044
Jarskog, L. F., Hamer, R. M., Catellier, D. J., Stewart, D. D., Lavange, L., Ray, N., et al. (2013). Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am. J. Psychiatry 170, 1032–1040. doi: 10.1176/appi.ajp.2013.12010127
Jensen, K. G., Correll, C. U., Rudå, D., Klauber, D. G., Stentebjerg-Olesen, M., Fagerlund, B., et al. (2017). Pretreatment cardiometabolic status in youth with early-onset psychosis: baseline results from the TEA trial. J. Clin. Psychiatry. doi: 10.4088/JCP.15m10479. [Epub ahead of print].
Kahn, R. S., Fleischhacker, W. W., Boter, H., Davidson, M., Vergouwe, Y., Keet, I. P., et al. (2008). Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet 371, 1085–1097. doi: 10.1016/S0140-6736(08)60486-9
Kalra, S., Kalra, B., Agrawal, N., and Kumar, S. (2011). Dopamine: the forgotten felon in type 2 diabetes. Recent Pat. Endocr. Metab. Immune Drug Discov. 5, 61–65. doi: 10.2174/187221411794351842
Kamath, V., Jones, C. N., Yip, J. C., Varasteh, B. B., Cincotta, A. H., Reaven, G. M., et al. (1997). Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care 20, 1697–1701. doi: 10.2337/diacare.20.11.1697
Kane, J. M., Barrett, E. J., Casey, D. E., Correll, C. U., Gelenberg, A. J., Klein, S., et al. (2004). Metabolic effects of treatment with atypical antipsychotics. J. Clin. Psychiatry 65, 1447–1455. doi: 10.4088/JCP.v65n1102
Kang, S. H., and Lee, J. I. (2015). Metabolic disturbances independent of body mass in patients with schizophrenia taking atypical antipsychotics. Psychiatry Investig. 12, 242–248. doi: 10.4306/pi.2015.12.2.242
Karam, C. S., Ballon, J. S., Bivens, N. M., Freyberg, Z., Girgis, R. R., Lizardi-Ortiz, J. E., et al. (2010). Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol. Sci. 31, 381–390. doi: 10.1016/j.tips.2010.05.004
Kasanin, J. (1926). The blood sugar curve in mental disease: II. The schizophrenic (dementia praecox) groups. Arch. Neurol. Psychiatry 16, 414–419. doi: 10.1001/archneurpsyc.1926.02200280022002
Kendall, T. (2011). The rise and fall of the atypical antipsychotics. Br. J. Psychiatry 199, 266–268. doi: 10.1192/bjp.bp.110.083766
Kim, S. F., Huang, A. S., Snowman, A. M., Teuscher, C., and Snyder, S. H. (2007). From the cover: antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc. Natl. Acad. Sci. U.S.A. 104, 3456–3459. doi: 10.1073/pnas.0611417104
Kok, P., Roelfsema, F., Frolich, M., van Pelt, J., Stokkel, M. P., Meinders, A. E., et al. (2006). Activation of dopamine D2 receptors simultaneously ameliorates various metabolic features of obese women. Am. J. Physiol. Endocrinol. Metab. 291, E1038–E1043. doi: 10.1152/ajpendo.00567.2005
Kooy, F. H. (1919). Hyperglycemia in mental disorders. Brain 42, 214–290. doi: 10.1093/brain/42.3.214
Kring, S. I., Werge, T., Holst, C., Toubro, S., Astrup, A., Hansen, T., et al. (2009). Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS ONE 4:e6696. doi: 10.1371/journal.pone.0006696
Kroeze, W. K., Hufeisen, S. J., Popadak, B. A., Renock, S. M., Steinberg, S., Ernsberger, P., et al. (2003). H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28, 519–526. doi: 10.1038/sj.npp.1300027
Lamos, E. M., Levitt, D. L., and Munir, K. M. (2016). A review of dopamine agonist therapy in type 2 diabetes and effects on cardio-metabolic parameters. Prim. Care Diabetes 10, 60–65. doi: 10.1016/j.pcd.2015.10.008
Landgraf, D., Joiner, W. J., McCarthy, M. J., Kiessling, S., Barandas, R., Young, J. W., et al. (2016). The mood stabilizer valproic acid opposes the effects of dopamine on circadian rhythms. Neuropharmacology 107, 262–270. doi: 10.1016/j.neuropharm.2016.03.047
Landgraf, D., McCarthy, M. J., and Welsh, D. K. (2014). Circadian clock and stress interactions in the molecular biology of psychiatric disorders. Curr. Psychiatry Rep. 16:483. doi: 10.1007/s11920-014-0483-7
Laursen, T. M., Munk-Olsen, T., and Vestergaard, M. (2012). Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr. Opin. Psychiatry 25, 83–88. doi: 10.1097/YCO.0b013e32835035ca
Leadbetter, R., Shutty, M., Pavalonis, D., Vieweg, V., Higgins, P., and Downs, M. (1992). Clozapine-induced weight gain: prevalence and clinical relevance. Am. J. Psychiatry 149, 68–72. doi: 10.1176/ajp.149.1.68
Li, J., Yoshikawa, A., Brennan, M. D., Ramsey, T. L., and Meltzer, H. Y. (2017). Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr Res. S0920–S9964, 30196–30192. doi: 10.1016/j.schres.2017.04.009
Lian, J., De Santis, M., He, M., and Deng, C. (2015). Risperidone-induced weight gain and reduced locomotor activity in juvenile female rats: the role of histaminergic and NPY pathways. Pharmacol. Res. 95–96, 20–26. doi: 10.1016/j.phrs.2015.03.004
Liang, Y., Lubkin, M., Sheng, H., Scislowski, P. W., and Cincotta, A. H. (1998). Dopamine agonist treatment ameliorates hyperglycemia, hyperlipidemia, and the elevated basal insulin release from islets of ob/ob mice. Biochim. Biophys. Acta 1405, 1–13. doi: 10.1016/S0167-4889(98)00092-5
Liu, Y., Li, Z., Zhang, M., Deng, Y., Yi, Z., and Shi, T. (2013). Exploring the pathogenetic association between schizophrenia and type 2 diabetes mellitus diseases based on pathway analysis. BMC Med. Genomics 6(Suppl. 1), S17. doi: 10.1186/1755-8794-6-S1-S17
Lopez-Soldado, I., Niisuke, K., Veiga, C., Adrover, A., Manzano, A., Martinez-Redondo, V., et al. (2016). Neuregulin improves response to glucose tolerance test in control and diabetic rats. Am. J. Physiol. Endocrinol. Metab. 310, E440–E451. doi: 10.1152/ajpendo.00226.2015
Lopez Vicchi, F., Luque, G. M., Brie, B., Nogueira, J. P., Garcia Tornadu, I., and Becu-Villalobos, D. (2016). Dopaminergic drugs in type 2 diabetes and glucose homeostasis. Pharmacol. Res. 109, 74–80. doi: 10.1016/j.phrs.2015.12.029
Lorenz, W. F. (1922). Sugar tolerance in dementia praecox and other mental disorders. Arch. Neurol. Psychiatry 8, 184–196. doi: 10.1001/archneurpsyc.1922.02190140075007
Macneil, R. R., and Muller, D. J. (2016). Genetics of common antipsychotic-induced adverse effects. Mol. Neuropsychiatry 2, 61–78. doi: 10.1159/000445802
Malhotra, A. K., Correll, C. U., Chowdhury, N. I., Muller, D. J., Gregersen, P. K., Lee, A. T., et al. (2012). Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch. Gen. Psychiatry 69, 904–912. doi: 10.1001/archgenpsychiatry.2012.191
McEvoy, J. P., Meyer, J. M., Goff, D. C., Nasrallah, H. A., Davis, S. M., Sullivan, L., et al. (2005). Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr. Res. 80, 19–32. doi: 10.1016/j.schres.2005.07.014
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
Mikhail, N. (2011). Quick-release bromocriptine for treatment of type 2 diabetes. Curr. Drug Deliv. 8, 511–516. doi: 10.2174/156720111796642255
Millington, G. W. (2007). The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 4:18. doi: 10.1186/1743-7075-4-18
Misiak, B., Stańczykiewicz, B., Łaczmański, Ł., and Frydecka, D. (2017). Lipid profile disturbances in antipsychotic-naive patients with first-episode non-affective psychosis: a systematic review and meta-analysis. Schizophr. Res. 2017:S0920–9964(17)30169-X. doi: 10.1016/j.schres.2017.03.031
Mitchell, A. J., Vancampfort, D., De Herdt, A., Yu, W., and De Hert, M. (2013). Is the prevalence of metabolic syndrome and metabolic abnormalities increased in early schizophrenia? A comparative meta-analysis of first episode, untreated and treated patients. Schizophr. Bull. 39, 295–305. doi: 10.1093/schbul/sbs082
Mizuno, Y., Suzuki, T., Nakagawa, A., Yoshida, K., Mimura, M., Fleischhacker, W. W., et al. (2014). Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr. Bull. 40, 1385–1403. doi: 10.1093/schbul/sbu030
Morlan-Coarasa, M. J., Arias-Loste, M. T., Ortiz-Garcia De La Foz, V., Martinez-Garcia, O., Alonso-Martin, C., Crespo, J., et al. (2016). Incidence of non-alcoholic fatty liver disease and metabolic dysfunction in first episode schizophrenia and related psychotic disorders: a 3-year prospective randomized interventional study. Psychopharmacology 233, 3947–3952. doi: 10.1007/s00213-016-4422-7
Mostaid, M. S., Mancuso, S. G., Liu, C., Sundram, S., Pantelis, C., Everall, I. P., et al. (2017). Meta-analysis reveals associations between genetic variation in the 5′ and 3′ regions of Neuregulin-1 and schizophrenia. Transl. Psychiatry 7:e1004. doi: 10.1038/tp.2016.279
Mountjoy, K. G. (2015). Pro-Opiomelanocortin (POMC) neurones, POMC-derived peptides, melanocortin receptors and obesity: how understanding of this system has changed over the last decade. J. Neuroendocrinol. 27, 406–418. doi: 10.1111/jne.12285
Muller, D. J., and Kennedy, J. L. (2006). Genetics of antipsychotic treatment emergent weight gain in schizophrenia. Pharmacogenomics 7, 863–887. doi: 10.2217/14622416.7.6.863
Muscatello, M. R., Bruno, A., Pandolfo, G., Mico, U., Bellinghieri, P. M., Scimeca, G., et al. (2011). Topiramate augmentation of clozapine in schizophrenia: a double-blind, placebo-controlled study. J. Psychopharmacol. 25, 667–674. doi: 10.1177/0269881110372548
Naguy, A., and Al-Tajali, A. (2016). Bromocriptine mitigated paliperidone metabolic and neuro-hormonal side effects and improved negative domain in a case of early onset schizophrenia. Nord. J. Psychiatry 70, 318–319. doi: 10.3109/08039488.2015.1094127
Narula, P. K., Rehan, H. S., Unni, K. E., and Gupta, N. (2010). Topiramate for prevention of olanzapine associated weight gain and metabolic dysfunction in schizophrenia: a double-blind, placebo-controlled trial. Schizophr. Res. 118, 218–223. doi: 10.1016/j.schres.2010.02.001
Nasrallah, H. A. (2008). Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol. Psychiatry 13, 27–35. doi: 10.1038/sj.mp.4002066
Newman, A. H., Beuming, T., Banala, A. K., Donthamsetti, P., Pongetti, K., Labounty, A., et al. (2012). Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 55, 6689–6699. doi: 10.1021/jm300482h
Nilsson, B. M., Forslund, A. H., Olsson, R. M., Hambraeus, L., and Wiesel, F. A. (2006). Differences in resting energy expenditure and body composition between patients with schizophrenia and healthy controls. Acta Psychiatr. Scand. 114, 27–35. doi: 10.1111/j.1600-0447.2005.00700.x
Nonogaki, K., Strack, A. M., Dallman, M. F., and Tecott, L. H. (1998). Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat. Med. 4, 1152–1156. doi: 10.1038/2647
Palmiter, R. D. (2007). Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 30, 375–381. doi: 10.1016/j.tins.2007.06.004
Petrikis, P., Tigas, S., Tzallas, A. T., Papadopoulos, I., Skapinakis, P., and Mavreas, V. (2015). Parameters of glucose and lipid metabolism at the fasted state in drug-naive first-episode patients with psychosis: evidence for insulin resistance. Psychiatry Res. 229, 901–904. doi: 10.1016/j.psychres.2015.07.041
Pillinger, T., Beck, K., Gobjila, C., Donocik, J. G., Jauhar, S., and Howes, O. D. (2017). Impaired glucose homeostasis in first-episode Schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry 74, 261–269. doi: 10.1001/jamapsychiatry.2016.3803
Rado, J. (2017). The complex inter-relationship between diabetes and Schizophrenia. Curr. Diabetes Rev. 13, 195–212. doi: 10.2174/1573399812666161220144740
Rado, J., and Von Ammon Cavanaugh, S. (2016). A naturalistic randomized placebo-controlled trial of extended-release metformin to prevent weight gain associated with olanzapine in a US community-dwelling population. J. Clin. Psychopharmacol. 36, 163–168. doi: 10.1097/JCP.0000000000000469
Rajkumar, A. P., Horsdal, H. T., Wimberley, T., Cohen, D., Mors, O., Borglum, A. D., et al. (2017). Endogenous and antipsychotic-related risks for diabetes mellitus in young people with Schizophrenia: a Danish population-based cohort study. Am. J. Psychiatry 174, 686–694. doi: 10.1176/appi.ajp.2016.16040442
Reynolds, G. P., and Kirk, S. L. (2010). Metabolic side effects of antipsychotic drug treatment–pharmacological mechanisms. Pharmacol. Ther. 125, 169–179. doi: 10.1016/j.pharmthera.2009.10.010
Rubi, B., Ljubicic, S., Pournourmohammadi, S., Carobbio, S., Armanet, M., Bartley, C., et al. (2005). Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 280, 36824–36832. doi: 10.1074/jbc.M505560200
Rubi, B., and Maechler, P. (2010). Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let's seek the balance. Endocrinology 151, 5570–5581. doi: 10.1210/en.2010-0745
Shams, T. A., and Muller, D. J. (2014). Antipsychotic induced weight gain: genetics, epigenetics, and biomarkers reviewed. Curr. Psychiatry Rep. 16:473. doi: 10.1007/s11920-014-0473-9
Sharpe, J. K., Byrne, N. M., Stedman, T. J., and Hills, A. P. (2005). Resting energy expenditure is lower than predicted in people taking atypical antipsychotic medication. J. Am. Diet. Assoc. 105, 612–615. doi: 10.1016/j.jada.2005.01.005
Sharpe, J. K., Stedman, T. J., Byrne, N. M., Wishart, C., and Hills, A. P. (2006). Energy expenditure and physical activity in clozapine use: implications for weight management. Aust. N. Z. J. Psychiatry 40, 810–814. doi: 10.1080/j.1440-1614.2006.01888.x
Simpson, N., Maffei, A., Freeby, M., Burroughs, S., Freyberg, Z., Javitch, J., et al. (2012). Dopamine-mediated autocrine inhibitory circuit regulating human insulin secretion in vitro. Mol. Endocrinol. 26, 1757–1772. doi: 10.1210/me.2012-1101
Song, X., Fan, X., Song, X., Zhang, J., Zhang, W., Li, X., et al. (2013). Elevated levels of adiponectin and other cytokines in drug naïve, first episode schizophrenia patients with normal weight. Schizophr. Res. 150, 269–273. doi: 10.1016/j.schres.2013.07.044
Spelman, L. M., Walsh, P. I., Sharifi, N., Collins, P., and Thakore, J. H. (2007). Impaired glucose tolerance in first-episode drug-naïve patients with schizophrenia. Diabetic Med. 24, 481–485. doi: 10.1111/j.1464-5491.2007.02092.x
Szmulewicz, A. G., Angriman, F., Pedroso, F. E., Vazquez, C., and Martino, D. J. (2017). Long-term antipsychotic use and major cardiovascular events: a retrospective cohort study. J. Clin. Psychiatry. doi: 10.4088/JCP.16m10976. [Epub ahead of print].
Tagami, K., Kashiwase, Y., Yokoyama, A., Nishimura, H., Miyano, K., Suzuki, M., et al. (2016). The atypical antipsychotic, olanzapine, potentiates ghrelin-induced receptor signaling: an in vitro study with cells expressing cloned human growth hormone secretagogue receptor. Neuropeptides 58, 93–101. doi: 10.1016/j.npep.2015.12.010
Tang, H., McGowan, O. O., and Reynolds, G. P. (2014). Polymorphisms of serotonin neurotransmission and their effects on antipsychotic drug action. Pharmacogenomics 15, 1599–1609. doi: 10.2217/pgs.14.111
Tanyanskiy, D. A., Martynikhin, I. A., Rotar, O. P., Konradi, A. O., Sokolian, N. A., Neznanov, N. G., et al. (2015). Association of adipokines with metabolic disorders in patients with schizophrenia: results of comparative study with mental healthy cohort. Diabetes Metab. Syndr. 9, 163–167. doi: 10.1016/j.dsx.2015.04.009
Thiselton, D. L., Vladimirov, V. I., Kuo, P. H., McClay, J., Wormley, B., Fanous, A., et al. (2008). AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol. Psychiatry 63, 449–457. doi: 10.1016/j.biopsych.2007.06.005
Tiwari, A. K., Zhang, D., Pouget, J. G., Zai, C. C., Chowdhury, N. I., Brandl, E. J., et al. (2016). Impact of histamine receptors H1 and H3 polymorphisms on antipsychotic-induced weight gain. World J. Biol. Psychiatry. doi: 10.1080/15622975.2016.1262061. [Epub ahead of print].
Valiquette, G. (2011). Bromocriptine for diabetes mellitus type II. Cardiol. Rev. 19, 272–275. doi: 10.1097/CRD.0b013e318229d2d2
Vancampfort, D., Stubbs, B., Mitchell, A. J., De Hert, M., Wampers, M., Ward, P. B., et al. (2015). Risk of metabolic syndrome and its components in people with schizophrenia and related psychotic disorders, bipolar disorder and major depressive disorder: a systematic review and meta-analysis. World Psychiatry 14, 339–347. doi: 10.1002/wps.20252
van der Zwaal, E. M., Janhunen, S. K., La Fleur, S. E., and Adan, R. A. (2014). Modelling olanzapine-induced weight gain in rats. Int. J. Neuropsychopharmacol. 17, 169–186. doi: 10.1017/S146114571300093X
van der Zwaal, E. M., Merkestein, M., Lam, Y. K., Brans, M. A., Luijendijk, M. C., Bok, L. I., et al. (2012). The acute effects of olanzapine on ghrelin secretion, CCK sensitivity, meal size, locomotor activity and body temperature. Int. J. Obes. 36, 254–261. doi: 10.1038/ijo.2011.97
Vehapoglu, A., Turkmen, S., and Terzioglu, S. (2016). Alpha-melanocyte-stimulating hormone and agouti-related protein: do they play a role in appetite regulation in childhood obesity? J. Clin. Res. Pediatr. Endocrinol. 8, 40–47. doi: 10.4274/jcrpe.2136
Vidarsdottir, S., Vlug, P., Roelfsema, F., Frolich, M., and Pijl, H. (2010). Orally disintegrating and oral standard olanzapine tablets similarly elevate the homeostasis model assessment of insulin resistance index and plasma triglyceride levels in 12 healthy men: a randomized crossover study. J. Clin. Psychiatry 71, 1205–1211. doi: 10.4088/JCP.08m04654yel
Wang, G. J., Volkow, N. D., and Fowler, J. S. (2002). The role of dopamine in motivation for food in humans: implications for obesity. Expert Opin. Ther. Targets 6, 601–609. doi: 10.1517/14728222.6.5.601
Wang, G. J., Volkow, N. D., Logan, J., Pappas, N. R., Wong, C. T., Zhu, W., et al. (2001). Brain dopamine and obesity. Lancet 357, 354–357. doi: 10.1016/S0140-6736(00)03643-6
Weston-Green, K., Huang, X. F., and Deng, C. (2011). Olanzapine treatment and metabolic dysfunction: a dose response study in female Sprague dawley rats. Behav. Brain Res. 217, 337–346. doi: 10.1016/j.bbr.2010.10.039
Wilson, J. M., Sanyal, S., and van Tol, H. H. (1998). Dopamine D2 and D4 receptor ligands: relation to antipsychotic action. Eur. J. Pharmacol. 351, 273–286. doi: 10.1016/S0014-2999(98)00312-4
Xu, M. Q., Xing, Q. H., Zheng, Y. L., Li, S., Gao, J. J., He, G., et al. (2007). Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J. Clin. Psychiatry 68, 1358–1367. doi: 10.4088/JCP.v68n0906
Xu, Y., Wu, Z., Sun, H., Zhu, Y., Kim, E. R., Lowell, B. B., et al. (2013). Glutamate mediates the function of melanocortin receptor 4 on Sim1 neurons in body weight regulation. Cell Metab. 18, 860–870. doi: 10.1016/j.cmet.2013.11.003
Yanik, T., Kursungoz, C., Sutcigil, L., and Ak, M. (2013). Weight gain in risperidone therapy: investigation of peripheral hypothalamic neurohormone levels in psychotic patients. J. Clin. Psychopharmacol. 33, 608–613. doi: 10.1097/JCP.0b013e318297980e
Yilmaz, Z., Davis, C., Loxton, N. J., Kaplan, A. S., Levitan, R. D., Carter, J. C., et al. (2015). Association between MC4R rs17782313 polymorphism and overeating behaviors. Int. J. Obes. 39, 114–120. doi: 10.1038/ijo.2014.79
Zai, C. C., Tiwari, A. K., Chowdhury, N. I., Brandl, E. J., Shaikh, S. A., Freeman, N., et al. (2016). Association study of Serotonin 3 receptor subunit gene variants in antipsychotic-induced weight gain. Neuropsychobiology 74, 169–175. doi: 10.1159/000457903
Zhang, J. P., Lencz, T., Zhang, R. X., Nitta, M., Maayan, L., John, M., et al. (2016). Pharmacogenetic associations of antipsychotic drug-related weight gain: a systematic review and meta-analysis. Schizophr. Bull. 42, 1418–1437. doi: 10.1093/schbul/sbw058
Keywords: schizophrenia, antipsychotic drugs, metabolism, insulin, diabetes, dyslipidemia, dopamine, monoamines
Citation: Freyberg Z, Aslanoglou D, Shah R and Ballon JS (2017) Intrinsic and Antipsychotic Drug-Induced Metabolic Dysfunction in Schizophrenia. Front. Neurosci. 11:432. doi: 10.3389/fnins.2017.00432
Received: 17 May 2017; Accepted: 13 July 2017;
Published: 28 July 2017.
Edited by:
Chao Deng, University of Wollongong, AustraliaCopyright © 2017 Freyberg, Aslanoglou, Shah and Ballon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zachary Freyberg, ZnJleWJlcmdAcGl0dC5lZHU=