 Andrew Kneynsberg
Andrew Kneynsberg Benjamin Combs
Benjamin Combs Kyle Christensen1,2
Kyle Christensen1,2 Gerardo Morfini
Gerardo Morfini Nicholas M. Kanaan
Nicholas M. Kanaan- 1Neuroscience Program, Michigan State University, East Lansing, MI, United States
- 2Department of Translational Science and Molecular Medicine, College of Human Medicine, Michigan State University, Grand Rapids, MI, United States
- 3Department of Anatomy and Cell Biology, University of Illinois at Chicago, Chicago, IL, United States
- 4Hauenstein Neuroscience Center, Mercy Health Saint Mary's, Grand Rapids, MI, United States
Tauopathies are a diverse group of diseases featuring progressive dying-back neurodegeneration of specific neuronal populations in association with accumulation of abnormal forms of the microtubule-associated protein tau. It is well-established that the clinical symptoms characteristic of tauopathies correlate with deficits in synaptic function and neuritic connectivity early in the course of disease, but mechanisms underlying these critical pathogenic events are not fully understood. Biochemical in vitro evidence fueled the widespread notion that microtubule stabilization represents tau's primary biological role and that the marked atrophy of neurites observed in tauopathies results from loss of microtubule stability. However, this notion contrasts with the mild phenotype associated with tau deletion. Instead, an analysis of cellular hallmarks common to different tauopathies, including aberrant patterns of protein phosphorylation and early degeneration of axons, suggests that alterations in kinase-based signaling pathways and deficits in axonal transport (AT) associated with such alterations contribute to the loss of neuronal connectivity triggered by pathogenic forms of tau. Here, we review a body of literature providing evidence that axonal pathology represents an early and common pathogenic event among human tauopathies. Observations of axonal degeneration in animal models of specific tauopathies are discussed and similarities to human disease highlighted. Finally, we discuss potential mechanistic pathways other than microtubule destabilization by which disease-related forms of tau may promote axonopathy.
Introduction
Proper brain function relies on appropriate connectivity between specific neuronal populations. An essential cellular process underlying such connectivity involves the generation and continued maintenance of molecular constituents within axons and dendrites (Conde and Caceres, 2009; Rasband, 2010). Maintenance of axons is particularly challenging in neurons because of their large size and complex biochemical heterogeneity of discrete functional compartments (i.e., nodes of Ranvier and synapses) within this major neuronal subdomain (Morfini et al., 2001). The axonal cytoskeleton features a polarized microtubule organization, a characteristic that allows for bidirectional transport of membrane-bounded organelles (MBOs) to and from the neuronal soma (Baas et al., 2016). Specialized intracellular transport events include the regulated delivery of MBOs from the soma to the pre- and post-synaptic compartments, the removal of old materials from these compartments, and the maintenance of neurotrophic support, among many others (Morfini et al., 2001). Long distance MBO trafficking events, collectively referred to as axonal transport (AT), are mainly powered by the kinesin and dynein superfamily of microtubule-based molecular motors (Black, 2016; Morfini et al., 2016). To date, a large body of genetic and experimental evidence indicates that maintenance of the unique cytoarchitecture and connectivity of neurons depends on appropriate functionality of major AT components, including microtubules, molecular motors, and protein kinases involved in their regulation (Gibbs et al., 2015; Morfini et al., 2016). Accordingly, genetic mutations in specific molecular motor protein subunits are implicated as potential causative factors for various neurodegenerative diseases (Morfini et al., 2009; Kanaan et al., 2013; Brady and Morfini, 2017).
Tauopathies comprise a heterogeneous group of diseases, including Alzheimer's disease (AD), frontal temporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), chronic traumatic encephalopathy (CTE), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick's disease (PiD) (Arendt et al., 2016). Being originally limited to observations derived from post-mortem brains at advanced disease stages, much of the past research on tauopathies focused on mechanisms linking aberrant accumulation of filamentous tau aggregates to cognitive decline and neuronal cell death. However, the recent development of animal models of tauopathies facilitated the identification of much earlier pathogenic events. Detailed pathological analysis of these models indicates that disease-specific symptoms coincide with reductions in synaptic function and neuronal connectivity that long precede the loss of neurons (Morfini et al., 2009). Collectively, the available evidence indicates that neurons affected in tauopathies follow a dying back pattern of degeneration (Higuchi et al., 2002; Kanaan et al., 2013; Brady and Morfini, 2017). Based on this knowledge, interventions focused on maintenance of neuronal connectivity, rather than prevention of cell death, may represent a more pressing therapeutic need for tauopathies (Cheng et al., 2010; Lingor et al., 2012). Identification of disease-relevant mechanisms linking tau to axonal and neuritic degeneration may provide specific molecular targets to improve neuronal connectivity in human tauopathies (Kanaan et al., 2013).
Evidence of Axonopathy in Human Tauopathies
Degenerating axons follow a stereotypical set of morphological changes, typically initiated by enlargement of areas known as swellings or spheroid bodies (Raff et al., 2002). Concomitantly, axons undergo thinning between spheroids, ultimately displaying a beaded appearance as dystrophy progresses. This thinning process continues until the axon becomes fragmented, and eventually degraded (Zhou et al., 1998). Accordingly, the extent of these progressive morphological changes can be used to evaluate axonal degeneration in vivo and in vitro (Gatto et al., 2015; Kneynsberg et al., 2016). Demyelination often accompanies this process, highlighting the interdependence among oligodendrocytes and their myelinated axon tract (Barres et al., 1993).
Distinctive clinical and neuropathological features of specific tauopathies including AD (reviewed in Masters et al., 2015); FTDP-17, (reviewed in Ghetti et al., 2015); PSP, (Williams and Lees, 2009); CBD, (reviewed in Kouri et al., 2011); PiD, (Mckhann et al., 2001); and CTE, (reviewed in Kiernan et al., 2015) are reviewed elsewhere. Below, we provide a concise review of independent studies in human brains, which collectively provide strong evidence supporting the contention that axonal pathology represents an early and critical pathogenic event common to multiple human tauopathies (Table 1).
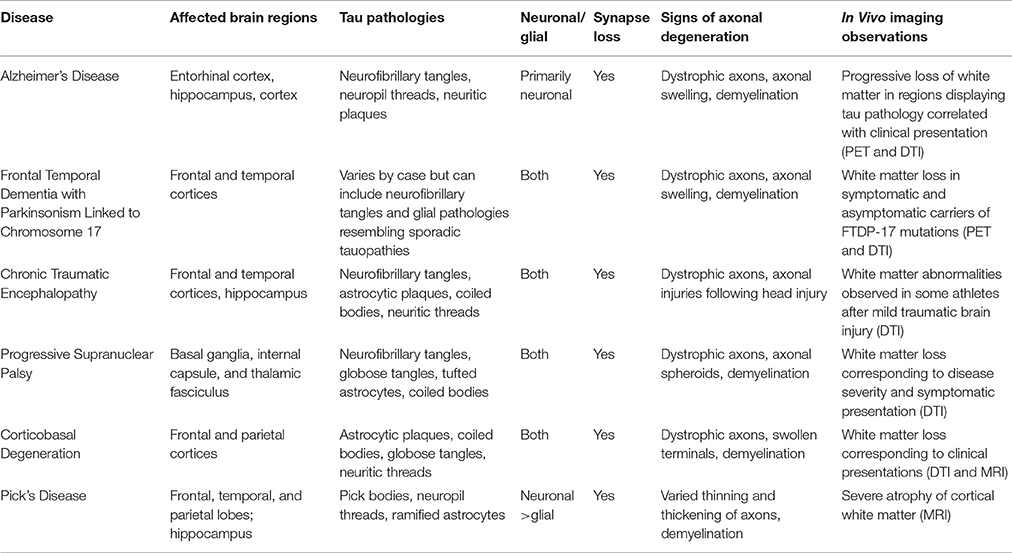
Table 1. Features of axonopathy in human tauopathies.
Alzheimer's Disease (AD)
The strong focus on tau lesions in AD yielded a large body of data establishing axonal degeneration as a prominent neuropathological hallmark of this disease (Kanaan et al., 2013). Tau inclusions within dystrophic neurites, known as neuropil threads, are a robust neuropathological feature of AD brains that appear before neurofibrillary tangles (NFTs) form in neuronal somata (Kowall and Kosik, 1987; Ghoshal et al., 2002). Immunohistochemical evidence demonstrates that pathological forms of tau known to inhibit AT accumulate in neuropil threads, suggesting localized disruption of cellular processes critical for axonal maintenance in AD, including AT (Kanaan et al., 2011; Combs et al., 2016a). Observations of abnormal vesicle accumulations within dystrophic neurites of AD brains provide further support for this notion (Praprotnik et al., 1996; Dessi et al., 1997).
Consistent with AT deficits in AD, results from ultrastructural and immunohistochemical analyses of AD brains reveal a clear correlation between loss of synapses and the manifestation of cognitive deficits (Dekosky and Scheff, 1990; Terry et al., 1991). Highlighting the pathological relevance of these findings, brain imaging studies in living AD patients document significant atrophy of axon-rich white matter structures, suggesting that axonal degeneration represents a prominent feature of the disease. Brain regions affected in patients with mild cognitive impairment, a well-established prodromal AD stage, mainly include axonal projections in the perforant pathway and cortical regions (Stoub et al., 2006; Huang and Auchus, 2007), where axon demyelination also occurs (Sjobeck and Englund, 2003; Sjobeck et al., 2005; Ihara et al., 2010). In more advanced AD cases, white matter atrophy extends to the corpus callosum, where the degree of atrophy directly correlates with cognitive decline (Vermersch et al., 1996; Hampel et al., 1998). Diffuse tensor imaging (DTI) techniques quantitatively measure water diffusion within cellular structures to identify microstructural changes in white matter that may not be evident with other imaging techniques (Meerschaert et al., 2016). Significantly, results from DTI studies show alterations in specific axonal tracts connecting association cortices, while others involved with motor or visual functions are largely spared. Again, the extent to which these tracts are affected displays a close correlation with the degree of cognitive decline (Bozzali et al., 2002).
Frontotemporal Dementia with Parkinsonism Linked to Chromosome 17 (FTDP-17)
FTDP-17 represents a subgroup of inherited early-onset tauopathies resulting from mutations in the gene encoding tau. The discovery of FTDP-17 tau mutations demonstrated that tau dysfunction alone suffices to cause neuronal dysfunction and degeneration (Foster et al., 1997; Hutton et al., 1998), and axonal pathology is seen in FTDP-17 brains. Specifically, positron emission tomographic (PET) imaging studies in FTDP-17 patients show the accumulation of tau aggregates in white matter areas (Wszolek et al., 1992). Interestingly, brain imaging studies found white matter alterations in pre-symptomatic disease stages (Rohrer et al., 2010; Dopper et al., 2014). Similar to neuropil threads in AD, mutant tau aggregates localize within dystrophic axons of FTDP-17 brains (Delisle et al., 1999; Murrell et al., 1999; Lippa et al., 2000; Kouri et al., 2014). As discussed below, animal models of specific FTDP-17 variants largely recapitulate the axonopathy phenotype observed in affected human brains.
Chronic Traumatic Encephalopathy (CTE)
CTE is a neurodegenerative disease associated with repetitive subconcussive and mild traumatic brain injuries (reviewed in Blennow et al., 2016). Focal areas of neuronal and glial tau inclusions at the depths of cortical sulci and in perivascular regions are pathognomonic CTE lesions found in the frontal and temporal cortices, as well as the hippocampus (Mckee et al., 2012). Symptoms of diffuse axonal injuries, including axonal swellings, unregulated calcium influx, and cytoskeletal abnormalities are evident within the first 24 h after concussion and may persist for weeks (Blumbergs et al., 1994; Maxwell et al., 1995; Giza and Hovda, 2001). Tau-positive neuropil threads also are a prominent neuropathological feature of CTE and cognitive decline correlates with axonal atrophy in subcortical white matter (Tokuda et al., 1991; Kraus et al., 2007; Mckee et al., 2009). Pathological forms of tau, identified by conformation-dependent tau antibodies, were recently found to localize within axons of cortical white matter and neuropil threads in the cholinergic basal forebrain (Kanaan et al., 2016; Mufson et al., 2016). Using a PET tracer, protein aggregates, likely made up of tau, were identified within white matter tracts of former professional football players suspected of having CTE (Barrio et al., 2015). Membrane-associated proteins, including amyloid precursor protein, accumulate in axons after traumatic brain injuries, suggesting AT disruption may play a role in CTE pathogenesis (Uryu et al., 2007). Additionally, a multitude of DTI studies have identified white matter changes in athletes at risk for concussions or repetitive subconcussive impacts from several sports, including boxing, football, soccer, ice hockey, as well as in veterans exposed to blast trauma (Zhang et al., 2003; Koerte et al., 2012a,b; McAllister et al., 2014; Petrie et al., 2014).
Progressive Supranuclear Palsy (PSP)
In PSP, tau aggregates are observed in both neurons (NFTs and globose tangles) and glial cells (tufted astrocytes and coiled bodies) (Pollock et al., 1986; Hauw et al., 1994). The presence of neuropil threads in PSP indicates the presence pathological tau in axons, particularly in the basal ganglia, internal capsule, and thalamic fasciculus (Hauw et al., 1990; Dickson, 1999). Additionally, axonal spheroids are found in subcortical white matter, the putamen, globus pallidus, and the subthalamic nucleus (Probst et al., 1988; Ahmed et al., 2008). Extending these findings, DTI studies in early PSP cases found evidence of white matter degeneration within the pons, substantia nigra, cerebellar peduncles, and corpus callosum and the degree of atrophy in some of these regions correlates to disease severity and onset of symptoms (Padovani et al., 2006; Knake et al., 2010; Whitwell et al., 2011; Zhang et al., 2016). Interestingly, PSP is characterized by abundant tau lesions termed coiled bodies within oligodendrocytes, the cells responsible for generating myelin sheaths in the central nervous system. Demyelination is particularly evident in white matter tracts of PSP-affected brains, directly correlating with tau burden in the superior cerebellar peduncle and red nucleus. These findings suggest that tau-induced oligodendrocyte dysfunction could indirectly contribute to the axonal degeneration phenotype observed in PSP (Ishizawa et al., 2000).
Corticobasal Degeneration (CBD)
The characteristic tau pathology of CBD includes astrocytic plaques, occasional coiled bodies, and neuronal globose inclusions that are primarily found in the frontal and parietal lobes of the cerebral cortex, the cerebellum, and substantia nigra (Rebeiz et al., 1968; Feany and Dickson, 1995). Neuropil thread pathology is prominent in cortical gray and white matter, the subthalamic nucleus, and the striatum of CBD brains (Ikeda et al., 1994; Dickson, 1999). MRI and DTI-based studies find significantly greater white matter abnormalities in CBD (Doi et al., 1999; Zhang et al., 2016). Additional studies identified pathological changes in specific hand sensorimotor fiber tracts in patients who manifested limb apraxia at early CBD stages (Borroni et al., 2008), furthering the linkage between axonal degeneration and specific symptomatic outcomes. The extensive astroglial lesions in CBD and other tauopathies suggest that pathological forms of tau may affect astrocyte-specific functions critical to neuronal health, including sustained trophic support (Figure 1C; Kahlson and Colodner, 2015).
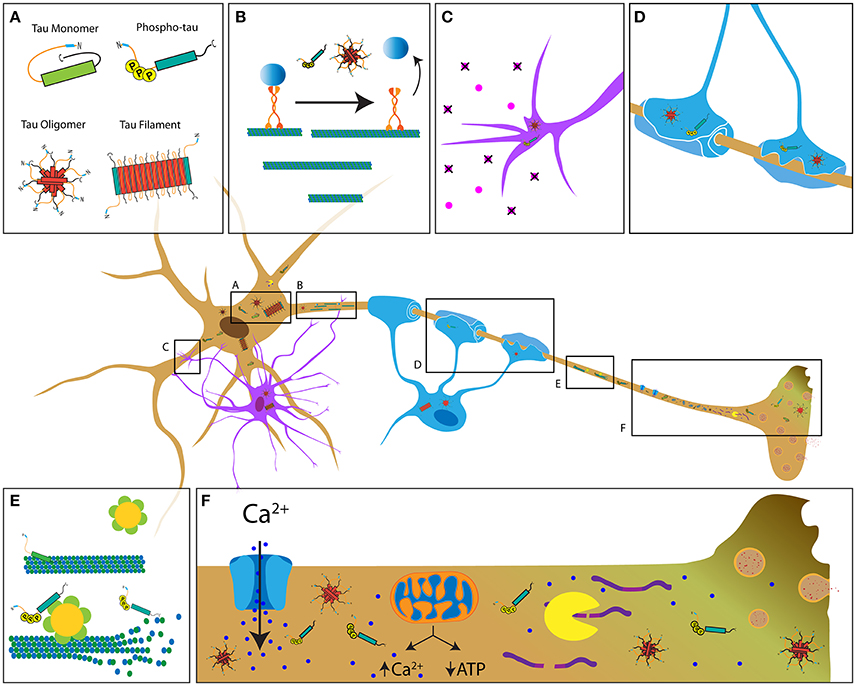
Figure 1. Potential mechanisms contributing to tau-induced axonal degeneration in tauopathies. (A) Pathological forms of tau include phosphorylated tau, tau oligomers, and tau filaments, all of which feature increased exposure of the PAD (blue region of tau) (Kanaan et al., 2011). In contrast, this domain is hidden in soluble tau monomers, which feature a paperclip conformation (Jeganathan et al., 2008). Depending on the specific tauopathy, pathological tau is present in neurons (brown cell), astrocytes (purple cell, C) and oligodendrocytes (blue cell, D). (B) Pathological conformations of tau impair axonal transport through activation of a phosphotransferase-based signaling pathway that promotes detachment of transported cargoes from the motor protein conventional kinesin. (C,D) Accumulation of pathological tau may also interfere with cellular processes involved in trophic support by astrocytes and oligodendrocytes, ultimately resulting in axonal dysfunction and demyelination. (E) Disease-related modifications of tau (e.g., phosphorylation) that reduce its binding to microtubules may promote aberrant katanin-mediated microtubule severing. (F) Disease-related forms of tau can alter Ca2+ homeostasis through various mechanisms, including abnormal modulation of ion channel activity, the endoplasmic reticulum and/or mitochondrial Ca2+ buffering. Enhanced Ca2+ levels may in turn increase calpain activity, leading to abnormal cleavage of cytoskeletal proteins. Together, these mechanisms represent the multifaceted pathways by which tau that likely contributes to axonopathy.
Pick's Disease (PiD)
Pick's disease (PiD) is characterized by severe atrophy of the frontal, temporal, and parietal lobes. Cytoplasmic neuronal tau inclusions known as Pick bodies represent the major neuropathological hallmark of PiD, and these are typically found in the dentate gyrus of the hippocampus and frontal and temporal cortices (Pollock et al., 1986; Probst et al., 1996). MRI studies of Pick's disease cases document severe atrophy of cortical white matter (Wang et al., 2006; Yamakawa et al., 2006). Microscopic evaluation of post-mortem brain tissue reveals that axons surrounding the dendrites of neurons in the affected brain regions (e.g., polymorphic layer of the dentate gyrus) often display pathological tau, in addition, glial tau pathology is observed (Cochran et al., 1994; Probst et al., 1996). Neuritic threads and spheroids are observed in mossy fibers projecting to the dentate nucleus along with abnormal tau in cerebellar white matter and other axons in PiD brains (Probst et al., 1996; Braak et al., 1999). A marked loss of myelinated axons is observed in subcortical white matter (Dickson, 1998) as well as perforant pathway synaptic loss (Lippa, 2004). Accumulation of AT-related molecules in Pick bodies (e.g., kinesin and synpatophysin) suggests AT is impaired in PiD (Nakamura et al., 1994).
Collectively, results from ultrastructural, immunohistochemical, and brain imaging-based studies strongly implicate axonopathy and the consequent neural disconnection as a pathogenic component common to all tauopathies. Localization of pathological forms of tau within dystrophic axons in each tauopathy further suggests alterations in one or more cellular processes critical for axonal maintenance, including AT (Morfini et al., 2002a; Kanaan et al., 2013).
Animal Models of Tauopathies
Models of tauopathy using the expression of specific tau variants include transgenic mice and viral vector-mediated gene delivery of tau-encoding cDNA constructs in rodent brains, among others (Ballatore et al., 2007; LaFerla and Green, 2012; Combs et al., 2016b). As discussed below, these animal models exhibit characteristics reminiscent of those seen in human tauopathies, further supporting a role of axonopathy in the disease process.
Several transgenic tauopathy models were produced over the years, which replicate selected cardinal features of human tauopathies (Gotz et al., 2007). The early animal models are based on expression of wild-type human 4R (Gotz et al., 1995) and 3R (Brion et al., 1999) tau variants and these mice show only mild phenotypes. However, tau transgenic mice with stronger gene promoters produce a clear neurodegenerative phenotype and many of these transgenic models (e.g., the T44 and ALZ17 lines) feature tau pathology mainly in the brainstem and spinal cord (Ishihara et al., 1999; Spittaels et al., 1999; Probst et al., 2000). Axonopathy in these models manifests in the form of axonal spheroids, axonal neurofilament- and tau-immunoreactive inclusions (Spittaels et al., 1999). Expanding on these early models, the 8c transgenic mouse line expresses a human tau gene cassette that produces all six human tau isoforms and these mice display signs of axonopathy including axonal swellings and spheroids, but do not develop NFTs (Duff et al., 2000). The 8c line was later crossed with a tau knockout line to produce mice that exclusively express human tau isoforms in the absence of endogenous mouse tau (Tucker et al., 2001). Interestingly, mice resulting from this cross show early accumulation of pathological tau within axons (e.g., CP13 positive phospho-tau), before such changes are detected in neuronal somata (Andorfer et al., 2003). Thus, overexpression of exogenous human tau alone suffices to cause axonopathy, which appears inconsistent with microtubule destabilization playing a role in tauopathies. Instead, these observations suggest that exogenous transgenic tau promotes a toxic gain of function that affects the maintenance of axons (Kanaan et al., 2011).
After the discovery of autosomal dominant tau mutations in FTDP-17 families (Hutton et al., 1998), several groups generated transgenic mice expressing FTDP-17-related mutant forms of tau. Unlike wild-type tau expressing models, these mice successfully recapitulate NFT-like inclusions typical of tauopathies (Lewis et al., 2000; Gotz et al., 2001; Allen et al., 2002). P301L tau remains the most commonly studied FTDP-17 mutation (Dumanchin et al., 1998; Hutton et al., 1998; Rizzu et al., 1999), and several transgenic mouse models are based on expression of this mutant form of tau. JNPL3 transgenic mice for example, feature expression of 4R/0N P301L tau under control of the mouse prion promoter (Lewis et al., 2000). Interestingly, these mice feature not only NFT-like and pre-tangle tau inclusions, but also axonal spheroids and degenerated myelinated axonal tracts, predominantly in the spinal cord (Lewis et al., 2000; Lin et al., 2003, 2005). In rTg4510 mice, the P301L tau transgene is downstream of a tetracycline operon-responsive element (TRE), and they express a tetracycline-controlled transactivator (tTA) that is under control of the Ca2+-calmodulin kinase II-α (CaMKIIα) promoter, which provides relatively selective expression of tau in forebrain neurons (Mayford et al., 1996; Santacruz et al., 2005; Spires et al., 2006). Accordingly, mutant tau expression can be suppressed through doxycycline treatment, thus providing spatial and temporal control of tau expression (Santacruz et al., 2005). Interestingly, signs of axonopathy in the rTg4510 model are comparable to those in JNPL3 mice, including swollen axons, NFT-like inclusions, myelin loss, and accumulation of mitochondria in swollen axons (Ludvigson et al., 2011). The rTgTauEC model uses a similar Tet-Off system as the rTg4510 model. However, tTA expression is controlled by the neuropsin promoter (de Calignon et al., 2012), which restricts mutant tau expression to a subset of entorhinal cortex (EC) neurons (Yasuda and Mayford, 2006). In this model, initial signs of axonopathy include strong Alz50 immunoreactivity [Alz50 is an antibody that recognizes a pathological tau conformation (Carmel et al., 1996)] within axonal terminals of the outer two-thirds molecular layer of the dentate gyrus, followed by age-dependent axonal degeneration and loss of hippocampal neurons (de Calignon et al., 2012). Microglia activation and astrogliosis is observed in rTgTauEC mice, suggesting that glial dysfunction might exacerbate axonopathy in this model. Additionally, the rTgTauEC model shows spread of pathological tau through interconnected neurons, suggesting transsynaptic release of misfolded or aggregated tau into the synaptic cleft and uptake by downstream neurons (de Calignon et al., 2012). Based on these observations, it is reasonable to posit that synaptic and axonal degeneration may not only be a primary source of neuronal disconnection, but also promote the release and spread of pathological tau species through synaptically connected networks (reviewed in Walsh and Selkoe, 2016).
Glial contributions to axonopathy are an active area of research, and models that selectively overexpress tau in glial cells are valuable models of tauopathies. One such model expresses 4R/1N P301L tau specifically in oligodendrocytes, achieved by controlling expression through the 2′,3′-cyclic nucleotide 3′ phosphodiesterase promoter. These mice displayed oligodendrocyte tau inclusions, AT deficits that preceded axonal degeneration, and deficits in myelination (Higuchi et al., 2005). This model highlights the importance of oligodendrocytes on axonal maintenance because tau inclusions appear to promote deficits in oligodendrocyte function that in turn indirectly contribute to axonopathy (Figure 1D; Chin and Goldman, 1996; Dickson et al., 1996).
Viral vector-mediated gene delivery facilitates the introduction of exogenous genes with extensive spatial and temporal control over its expression. In wild-type mice, injection of recombinant adeno-associated viral (rAAV) vectors encoding P301L tau into the EC (Siman et al., 2013, 2015) or the hippocampus (Jaworski et al., 2009, 2011) results in axonal degeneration. Injected mice display accumulation of tau phosphorylated at the AT8 epitope within synapses, degeneration of axons in the performant pathway and spread of P301L tau into the dentate gyrus (Siman et al., 2013). Similar to some transgenic tau models, rAAV-based tauopathy models also display reactive gliosis around degenerating axons, which may contribute to axonal pathology (Siman et al., 2013, 2015). Furthermore, injection of an rAAV vector encoding pseudophosphorylated AT8 tau in the EC led to early signs of axonopathy, including the development of spheroids and a swollen morphology (Combs et al., 2016b). Similarly, lentiviral vectors encoding wild-type or P301L tau induce markers of axonopathy when injected in the rat hippocampus, that includes a time-dependent increase in swollen, fragmented, and dystrophic axons, as well as microglial activation (Caillierez et al., 2013; Hebron et al., 2014).
Collectively, data from various animal models of tauopathy reveal axonal degeneration as a prominent pathological feature, and further suggest there is a contribution to this phenotype from glial cells. Based on their resemblance to this specific aspect of human tauopathies, these animal models may help define mechanisms and specific molecular components mediating the toxic effect of pathogenic tau on axonal maintenance and function.
Pathogenic Mechanisms Linking Abnormal Tau to Axonopathy
The initial discovery of tau protein was based on biochemical procedures that revealed its ability to associate with purified microtubules and modulate their dynamic growth behavior in vitro (Weingarten et al., 1975; Cleveland et al., 1977a,b). These biochemical properties of tau quickly led to its designation as a microtubule-associated protein, and the suggestion that microtubule stabilization represents tau's primary functional role in neurons (Weingarten et al., 1975; Cleveland et al., 1977a,b). Several subsequent reports document various effects of tau on microtubule dynamics in cultured cell lines (reviewed in Feinstein and Wilson, 2005). By extension, microtubule destabilization resulting from loss of tau function is widely believed to represent a major mechanism underlying neuronal dysfunction and degeneration in human tauopathies (Guo et al., 2017). However, direct experimental evidence that tau is required for the maintenance of microtubule stability in cultured primary neurons or in neurons in vivo has yet to be provided.
Collectively, data from the available four transgenic tau knockout mouse lines (reviewed in Ke et al., 2012) reveal little to no effect on behavior, cognition, and neuropathology associated with germline removal of the tau gene (Harada et al., 1994; Dawson et al., 2001; Tucker et al., 2001; Fujio et al., 2007; Morris et al., 2011; van Hummel et al., 2016). These findings are inconsistent with a critical role for tau in sustaining microtubule stabilization in vivo. Furthermore, axonal development is normal in cultured primary neurons obtained from some tau knockout mice (Harada et al., 1994; Takei et al., 2000). Deleterious effects of removing tau documented so far are limited to mild behavioral and parkinsonian-like deficiencies with advancing age in a couple of mouse lines (Ikegami et al., 2000; Lei et al., 2014), alterations in microtubule density limited to small caliber cerebellar parallel fibers in one model (Harada et al., 1994), and delayed neurite development in cultured neurons from another (Dawson et al., 2001). Additional experiments using primary cultured neurons show that immunodepletion of axonal tau does not lead to appreciable alterations in axonal microtubule content or dynamics (Tint et al., 1998); thus, extending findings from tau knockout mice. Tau-independent mechanisms for microtubule stabilization, including post-translational modifications of tubulin and the existence of additional microtubule-associated proteins, could explain the marginal impact of tau deletion on microtubule stability (Song and Brady, 2015; Matamoros and Baas, 2016).
Available experimental evidence suggests that tau performs functions other than microtubule stabilization in neurons. Numerous studies suggest a role for tau in multiple cellular processes and compartments, some of which may not include microtubule interactions. The functional repertoire of tau includes microtubule-actin cytoskeleton interactions (Selden and Pollard, 1986; Liu et al., 1999), regulation of AT (LaPointe et al., 2009; Kanaan et al., 2011), nuclear and nucleolar functions (Papasozomenos and Binder, 1987; Loomis et al., 1990; Sultan et al., 2011), end-binding protein regulation (Ramirez-Rios et al., 2016), cytoskeleton-plasma membrane interactions (Brandt et al., 1995), RNA-binding protein and stress granule regulation (Vanderweyde et al., 2016), as well as scaffolding of phosphotransferases (Lee et al., 1998; Liao et al., 1998; Ittner et al., 2010; Kanaan et al., 2013). Supporting this later role, a number of protein kinases and phosphatases reportedly interact with tau including PP1 (Liao et al., 1998), GSK3β (Sun et al., 2002), Cdk5 (Li et al., 2006), Fyn (Lee et al., 1998), and others (Liu et al., 2016). Together, these reports support a role for tau as a scaffolding protein that targets phosphotransferases to microtubules; thus, facilitating their interaction with specific microtubule-associated protein substrates (e.g., motor proteins) (reviewed in Morfini et al., 2002b; Kanaan et al., 2013). Experimental evidence also suggests such a role may extend to the dendritic compartment, where tau appears to regulate localization and activity of the tyrosine protein kinase Fyn (Xia et al., 2015; Lau et al., 2016). Collectively, the available evidence supports a broader functional repertoire for tau unrelated to microtubule stabilization, providing clues to the potential mechanisms by which pathogenic forms of tau may promote axonopathy (Figure 1).
The dependence of axons on proper anterograde and retrograde AT is evident because mutations in selected conventional kinesins or cytoplasmic dynein subunits suffice to promote dying-back degeneration of neurons (Morfini et al., 2009). Providing an explanation for the axonal degeneration phenotype observed in tauopathies, multiple independent studies documented deficits in AT triggered by pathogenic forms of tau, including oligomeric tau (Higuchi et al., 2005; Gotz et al., 2006; Cox et al., 2016; Swanson et al., 2017). Several mechanisms are proposed to mediate this toxic effect. The early observation that tau overexpression leads to alterations in intracellular trafficking of cellular organelles in cultured cell lines suggested that tau inhibits AT (Stamer et al., 2002). In vitro studies using purified components of the AT system further supported a model where tau would elicit this effect by competing with conventional kinesin heavy chain subunits for microtubule binding (Seitz et al., 2002; Mandelkow et al., 2003). However, studies in the squid axoplasm preparation reveal that even supraphysiological (i.e., ~20-fold higher) levels of human tau monomers do not negatively impact AT (Morfini et al., 2007). Pulse chase studies in vivo extend the conclusions from squid axoplasm studies to mammalian neurons, showing normal AT rates in the optic nerve of tau-overexpressing mice (Yuan et al., 2008).
The highly dynamic nature of the interaction between tau and microtubules in vitro and in situ also appears inconsistent with the notion that tau physically blocks motor proteins (Samsonov et al., 2004; Janning et al., 2014; Stern et al., 2017). Recent work suggests that disease-related phosphorylation does not cause tau to fall off microtubules as the off-rate is unaffected by pseudophosphorylation at the PHF1 site (i.e., S396 and S404), but may reduce microtubule binding by decreasing the on-rate (Niewidok et al., 2016). The highly dynamic conformational flexibility of tau likely facilitates its interaction with a large repertoire of binding partners (Jeganathan et al., 2006; Uversky, 2015; Stern et al., 2017) (reviewed in Bakota et al., 2017). Further, tau's role as a scaffolding protein and the recruitment of phosphotransferases to the microtubule cystoskeleton associated with this function could in turn be subject to phosphoregulation.
The major motor proteins responsible for AT, conventional kinesin and cytoplasmic dynein, are regulated by specific protein kinases providing a novel mechanism linking pathological forms of tau to deficits in AT (Kanaan et al., 2013; Gibbs et al., 2015; Brady and Morfini, 2017). Unlike soluble tau monomers, tau aggregates inhibit anterograde AT in the isolated squid axoplasm when perfused at physiological levels (LaPointe et al., 2009). Remarkably, this effect is dependent upon amino acids 2–18 in the extreme amino terminus of tau, a motif termed the phosphatase-activating domain (PAD) (Kanaan et al., 2011). PAD is sufficient to activate protein phosphatase 1 (PP1) and GSK3 in a manner independent of microtubule binding (Kanaan et al., 2011). PP1 reportedly activates GSK3β by dephosphorylating the autoinhibitory residue (Ser 9), causing its activation (Morfini et al., 2004). Active GSK3 in turn phosphorylates kinesin light chain subunits of conventional kinesin, promoting release of transported cargoes from this motor protein (Morfini et al., 2002b). Systematic evaluation of the effects that numerous tau construct variants elicit on AT reveals that different pathological changes (e.g., mutations, aggregation, oligomerization and phosphorylation) promote increased PAD exposure (Figure 1A; Kanaan et al., 2012). Given the unique dependence of axons on sustained AT, these studies highlight a specific kinase-dependent mechanism by which pathological forms of tau may cause axonal dysfunction, further supporting a role of tau as a modulator of protein kinases involved in AT regulation (Figure 1B). More recent studies provide further support for this mechanism in mammalian neurons by showing that amyloid-β oligomer-mediated impairments in AT require both tau and specific phosphotransferases (Decker et al., 2010; Vossel et al., 2015). Additionally, phosphorylation of tau at specific epitopes modulates both its dynamic behavior and its effect on AT (Kanaan et al., 2012; Tiernan et al., 2016; Stern et al., 2017). For example, tyrosine 18 phosphorylation within PAD by non-receptor kinases blocks the inhibitory effect of pathogenic tau on anterograde AT (Kanaan et al., 2012). In contrast, phospho-mimicking residues within the proline-rich region and the C-terminus confer a toxic effect on anterograde AT upon wild type tau monomers (Kanaan et al., 2011; Tiernan et al., 2016; Stern et al., 2017). Interestingly, tau-induced toxicity in cultured cortical astrocytes is associated with kinesin-dependent transport deficits (Yoshiyama et al., 2003), suggesting that tau may impair microtubule-dependent transport in glial cells as well.
In addition to the mechanisms above, mechanisms linking pathological tau to Ca2+ dysregulation may negatively impact axonal connectivity (Figure 1F). For example, injection of wild-type tau in the squid giant synapse promotes release of Ca2+ from the endoplasmic reticulum, leading to inhibition of synaptic vesicle exocytosis and synaptic transmission through a mechanism involving PAD exposure and GSK3 activation (Figure 1F; Moreno et al., 2016). Oligomeric tau is particularly toxic to synaptic function, causing impaired long-term potentiation (Fa et al., 2016) and long-term depression (Decker et al., 2015) in cultured neurons and mouse brain slice cultures, as well as memory deficits in living mice (Fa et al., 2016). Studies also show that tau oligomers can cause Ca2+ imbalance and cell death in induced pluripotent stem cells (Imamura et al., 2016), and tau may further increase Ca2+ levels by directly inhibiting plasma membrane Ca2+ ATPase (Berrocal et al., 2016). Besides the endoplasmic reticulum, mitochondria are a main source of Ca2+ buffering in cells (Werth and Thayer, 1994). Interestingly, P301L mutant tau reduces the number of axonal mitochondria (Rodriguez-Martin et al., 2016) and the N-terminal domain of tau may stimulate autophagic turnover of mitochondria in neurons (Amadoro et al., 2014). Reduced numbers and/or dysfunction of axonal mitochondria could in turn promote abnormal Ca2+ levels (reviewed in Eckert et al., 2014). Ultimately, pathogenic tau-mediated increases in Ca2+ levels may promote abnormal activation of calcium-activated proteases and proteolysis of critical cytoskeletal protein components (Figure 1F; Yin et al., 2016). Consistent with this notion, increased calpain activation occurs in AD and other tauopathy brains (Adamec et al., 2002), and inhibition of calcium activated protease reduces axonal degeneration in the P301L transgenic mouse model (Rao et al., 2014). Interestingly, studies suggest that microtubules devoid of tau are more vulnerable to degradation by the microtubule-severing protein katanin, providing another route by which abnormal tau modifications promote cytoskeletal degradation (Figure 1E; Qiang et al., 2006; Sudo and Baas, 2011). Collectively, these studies suggest various mechanism linking pathogenic forms of tau to axonal degeneration. A better understanding of these mechanisms may provide a broader set of tau-based therapeutic targets.
Conclusions
The landscape of tauopathies is marked by significant heterogeneity in clinical presentation, cellular topography, and neuropathological features. Despite this diversity, an examinations of human brains and animal models of tauopathies reveal axonopathy as a common feature of these diverse diseases (Hyman et al., 1990; Ahmed et al., 2008; Kovacs et al., 2008; Ling et al., 2015). A comprehensive analysis demonstrates that tau likely plays a central role in axonal dysfunction and degeneration and that disease-related modifications of tau contribute to axonopathy and synaptic dysfunction through multiple pathways including misregulation of phosphotransferase activities, AT deficits, disruption of Ca2+ homeostasis, altered glial function, and others (Figure 1). Developing and implementing therapeutic strategies based on preserving neuronal connectivity will require a continued deepening of our understanding of these mechanisms and the identification of specific molecular components linking pathological tau to axonopathy in the context of each tauopathy.
Author Contributions
AK, BC, KC, GM, and NK all contributed equally with writing, editing and figure preparation for the manuscript.
Funding
This work was supported by the National Institute of Health grants AG044372 and NS082730 (NK), NS066942 and NS096642 (GM), the BrightFocus Foundation grant A2013364S (NK), the Jean P. Schultz Biomedical Research Endowment (NK), and the Secchia Family Foundation Research Fund (NK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
PET, Positron emission tomography; DTI, Diffuse tensor imaging; MRI, Magnetic resonance imaging; AD, Alzheimer's disease; FTDP-17, Frontal temporal dementia with parkinsonism linked to chromosome 17; CTE, Chronic traumatic encephalopathy; PSP, Progressive supranuclear palsy; CBD, Corticobasal degeneration; PiD, Pick's disease.
References
Adamec, E., Mohan, P., Vonsattel, J. P., and Nixon, R. A. (2002). Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol. 104, 92–104. doi: 10.1007/s00401-002-0528-6
Ahmed, Z., Josephs, K. A., Gonzalez, J., Delledonne, A., and Dickson, D. W. (2008). Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain 131, 460–472. doi: 10.1093/brain/awm301
Allen, B., Ingram, E., Takao, M., Smith, M. J., Jakes, R., Virdee, K., et al. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 22, 9340–9351.
Amadoro, G., Corsetti, V., Florenzano, F., Atlante, A., Ciotti, M. T., Mongiardi, M. P., et al. (2014). AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol. Dis. 62, 489–507. doi: 10.1016/j.nbd.2013.10.018
Andorfer, C., Kress, Y., Espinoza, M., De Silva, R., Tucker, K. L., Barde, Y. A., et al. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. doi: 10.1046/j.1471-4159.2003.01879.x
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126, 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Baas, P. W., Rao, A. N., Matamoros, A. J., and Leo, L. (2016). Stability properties of neuronal microtubules. Cytoskeleton (Hoboken) 73, 442–460. doi: 10.1002/cm.21286
Bakota, L., Ussif, A., Jeserich, G., and Brandt, R. (2017). Systemic and network functions of the microtubule-associated protein tau: implications for tau-based therapies. Mol. Cell. Neurosci. 16, 30257–30263. doi: 10.1016/j.mcn.2017.03.003
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Barres, B. A., Jacobson, M. D., Schmid, R., Sendtner, M., and Raff, M. C. (1993). Does oligodendrocyte survival depend on axons? Curr. Biol. 3, 489–497. doi: 10.1016/0960-9822(93)90039-Q
Barrio, J. R., Small, G. W., Wong, K. P., Huang, S. C., Liu, J., Merrill, D. A., et al. (2015). In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc. Natl. Acad. Sci. U.S.A. 112, E2039–E2047. doi: 10.1073/pnas.1409952112
Berrocal, M., Corbacho, I., Sepulveda, M. R., Gutierrez-Merino, C., and Mata, A. M. (2016). Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta 1864, 1028–1035. doi: 10.1016/j.bbamcr.2016.10.023
Black, M. M. (2016). Axonal transport: the orderly motion of axonal structures. Methods Cell Biol. 131, 1–19. doi: 10.1016/bs.mcb.2015.06.001
Blennow, K., Brody, D. L., Kochanek, P. M., Levin, H., Mckee, A., Ribbers, G. M., et al. (2016). Traumatic brain injuries. Nat. Rev. Dis. Primers 2:16084. doi: 10.1038/nrdp.2016.84
Blumbergs, P. C., Scott, G., Manavis, J., Wainwright, H., Simpson, D. A., and Mclean, A. J. (1994). Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet 344, 1055–1056. doi: 10.1016/S0140-6736(94)91712-4
Borroni, B., Garibotto, V., Agosti, C., Brambati, S. M., Bellelli, G., Gasparotti, R., et al. (2008). White matter changes in corticobasal degeneration syndrome and correlation with limb apraxia. Arch. Neurol. 65, 796–801. doi: 10.1001/archneur.65.6.796
Bozzali, M., Falini, A., Franceschi, M., Cercignani, M., Zuffi, M., Scotti, G., et al. (2002). White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatr. 72, 742–746. doi: 10.1136/jnnp.72.6.742
Braak, E., Arai, K., and Braak, H. (1999). Cerebellar involvement in Pick's disease: affliction of mossy fibers, monodendritic brush cells, and dentate projection neurons. Exp. Neurol. 159, 153–163. doi: 10.1006/exnr.1999.7131
Brady, S. T., and Morfini, G. A. (2017). Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol. Dis. 105, 273–282. doi: 10.1016/j.nbd.2017.04.010
Brandt, R., Leger, J., and Lee, G. (1995). Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J. Cell Biol. 131, 1327–1340. doi: 10.1083/jcb.131.5.1327
Brion, J. P., Tremp, G., and Octave, J. N. (1999). Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer's disease. Am. J. Pathol. 154, 255–270. doi: 10.1016/S0002-9440(10)65272-8
Caillierez, R., Begard, S., Lecolle, K., Deramecourt, V., Zommer, N., Dujardin, S., et al. (2013). Lentiviral delivery of the human wild-type tau protein mediates a slow and progressive neurodegenerative tau pathology in the rat brain. Mol. Ther. 21, 1358–1368. doi: 10.1038/mt.2013.66
Carmel, G., Mager, E. M., Binder, L. I., and Kuret, J. (1996). The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J. Biol. Chem. 271, 32789–32795. doi: 10.1074/jbc.271.51.32789
Cheng, H. C., Ulane, C. M., and Burke, R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725. doi: 10.1002/ana.21995
Chin, S. S., and Goldman, J. E. (1996). Glial inclusions in CNS degenerative diseases. J. Neuropathol. Exp. Neurol. 55, 499–508. doi: 10.1097/00005072-199605000-00001
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977a). Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 116, 227–247. doi: 10.1016/0022-2836(77)90214-5
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977b). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225. doi: 10.1016/0022-2836(77)90213-3
Cochran, E. J., Fox, J. H., and Mufson, E. J. (1994). Severe panencephalic Pick's disease with Alzheimer's disease-like neuropil threads and synaptophysin immunoreactivity. Acta Neuropathol. 88, 479–484. doi: 10.1007/BF00389503
Combs, B., Hamel, C., and Kanaan, N. M. (2016a). Pathological conformations involving the amino terminus of tau occur early in Alzheimer's disease and are differentially detected by monoclonal antibodies. Neurobiol. Dis. 94, 18–31. doi: 10.1016/j.nbd.2016.05.016
Combs, B., Kneynsberg, A., and Kanaan, N. M. (2016b). Gene therapy models of Alzheimer's disease and other dementias. Methods Mol. Biol. 1382, 339–366. doi: 10.1007/978-1-4939-3271-9_25
Conde, C., and Caceres, A. (2009). Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 10, 319–332. doi: 10.1038/nrn2631
Cox, K., Combs, B., Abdelmesih, B., Morfini, G., Brady, S. T., and Kanaan, N. M. (2016). Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol. Aging 47, 113–126. doi: 10.1016/j.neurobiolaging.2016.07.015
Dawson, H. N., Ferreira, A., Eyster, M. V., Ghoshal, N., Binder, L. I., and Vitek, M. P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187.
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Decker, H., Lo, K. Y., Unger, S. M., Ferreira, S. T., and Silverman, M. A. (2010). Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J. Neurosci. 30, 9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010
Decker, J. M., Kruger, L., Sydow, A., Zhao, S., Frotscher, M., Mandelkow, E., et al. (2015). Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++) dysregulation. Acta Neuropathol. Commun. 3, 23. doi: 10.1186/s40478-015-0193-3
Dekosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Delisle, M. B., Murrell, J. R., Richardson, R., Trofatter, J. A., Rascol, O., Soulages, X., et al. (1999). A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 98, 62–77. doi: 10.1007/s004010051052
Dessi, F., Colle, M. A., Hauw, J. J., and Duyckaerts, C. (1997). Accumulation of SNAP-25 immunoreactive material in axons of Alzheimer's disease. Neuroreport 8, 3685–3689. doi: 10.1097/00001756-199712010-00006
Dickson, D. W. (1998). Pick's disease: a modern approach. Brain Pathol. 8, 339–354. doi: 10.1111/j.1750-3639.1998.tb00158.x
Dickson, D. W. (1999). Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J. Neurol. 246(Suppl. 2), II6–II15. doi: 10.1007/BF03161076
Dickson, D. W., Feany, M. B., Yen, S. H., Mattiace, L. A., and Davies, P. (1996). Cytoskeletal pathology in non-Alzheimer degenerative dementia: new lesions in diffuse Lewy body disease, Pick's disease, and corticobasal degeneration. J. Neural Transm. Suppl. 47, 31–46. doi: 10.1007/978-3-7091-6892-9_2
Doi, T., Iwasa, K., Makifuchi, T., and Takamori, M. (1999). White matter hyperintensities on MRI in a patient with corticobasal degeneration. Acta Neurol. Scand. 99, 199–201. doi: 10.1111/j.1600-0404.1999.tb07345.x
Dopper, E. G., Rombouts, S. A., Jiskoot, L. C., Den Heijer, T., De Graaf, J. R., De Koning, I., et al. (2014). Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology 83, e19–e26. doi: 10.1212/WNL.0000000000000583
Duff, K., Knight, H., Refolo, L. M., Sanders, S., Yu, X., Picciano, M., et al. (2000). Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 7, 87–98. doi: 10.1006/nbdi.1999.0279
Dumanchin, C., Camuzat, A., Campion, D., Verpillat, P., Hannequin, D., Dubois, B., et al. (1998). Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum. Mol. Genet. 7, 1825–1829. doi: 10.1093/hmg/7.11.1825
Eckert, A., Nisbet, R., Grimm, A., and Gotz, J. (2014). March separate, strike together–role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer's disease. Biochim. Biophys. Acta 1842, 1258–1266. doi: 10.1016/j.bbadis.2013.08.013
Fa, M., Puzzo, D., Piacentini, R., Staniszewski, A., Zhang, H., Baltrons, M. A., et al. (2016). Extracellular Tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep. 6:19393. doi: 10.1038/srep19393
Feany, M. B., and Dickson, D. W. (1995). Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am. J. Pathol. 146, 1388–1396.
Feinstein, S. C., and Wilson, L. (2005). Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim. Biophys. Acta 1739, 268–279. doi: 10.1016/j.bbadis.2004.07.002
Foster, N. L., Wilhelmsen, K., Sima, A. A., Jones, M. Z., D'amato, C. J., and Gilman, S. (1997). Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference participants. Ann. Neurol 41, 706–715. doi: 10.1002/ana.410410606
Fujio, J., Hosono, H., Ishiguro, K., Ikegami, S., and Fujita, S. C. (2007). Tau phosphorylation in the mouse brain during aversive conditioning. Neurochem. Int. 51, 200–208. doi: 10.1016/j.neuint.2007.04.024
Gatto, R. G., Chu, Y., Ye, A. Q., Price, S. D., Tavassoli, E., Buenaventura, A., et al. (2015). Analysis of YFP(J16)-R6/2 reporter mice and postmortem brains reveals early pathology and increased vulnerability of callosal axons in Huntington's disease. Hum. Mol. Genet. 24, 5285–5298. doi: 10.1093/hmg/ddv248
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. doi: 10.1111/nan.12213
Ghoshal, N., Garcia-Sierra, F., Wuu, J., Leurgans, S., Bennett, D. A., Berry, R. W., et al. (2002). Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp. Neurol. 177, 475–493. doi: 10.1006/exnr.2002.8014
Gibbs, K. L., Greensmith, L., and Schiavo, G. (2015). Regulation of axonal transport by protein kinases. Trends Biochem. Sci. 40, 597–610. doi: 10.1016/j.tibs.2015.08.003
Giza, C. C., and Hovda, D. A. (2001). The Neurometabolic Cascade of Concussion. J. Athl. Train. 36, 228–235.
Gotz, J., Chen, F., Van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Gotz, J., Deters, N., Doldissen, A., Bokhari, L., Ke, Y., Wiesner, A., et al. (2007). A decade of tau transgenic animal models and beyond. Brain Pathol. 17, 91–103. doi: 10.1111/j.1750-3639.2007.00051.x
Gotz, J., Ittner, L. M., and Kins, S. (2006). Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer's disease? J. Neurochem. 98, 993–1006. doi: 10.1111/j.1471-4159.2006.03955.x
Gotz, J., Probst, A., Spillantini, M. G., Schafer, T., Jakes, R., Burki, K., et al. (1995). Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 14, 1304–1313.
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Hampel, H., Teipel, S. J., Alexander, G. E., Horwitz, B., Teichberg, D., Schapiro, M. B., et al. (1998). Corpus callosum atrophy is a possible indicator of region- and cell type-specific neuronal degeneration in Alzheimer disease: a magnetic resonance imaging analysis. Arch. Neurol. 55, 193–198. doi: 10.1001/archneur.55.2.193
Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T., et al. (1994). Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491. doi: 10.1038/369488a0
Hauw, J. J., Daniel, S. E., Dickson, D., Horoupian, D. S., Jellinger, K., Lantos, P. L., et al. (1994). Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44, 2015–2019. doi: 10.1212/WNL.44.11.2015
Hauw, J. J., Verny, M., Delaere, P., Cervera, P., He, Y., and Duyckaerts, C. (1990). Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer's disease and aging. Neurosci. Lett. 119, 182–186. doi: 10.1016/0304-3940(90)90829-X
Hebron, M. L., Algarzae, N. K., Lonskaya, I., and Moussa, C. (2014). Fractalkine signaling and Tau hyper-phosphorylation are associated with autophagic alterations in lentiviral Tau and Abeta1-42 gene transfer models. Exp. Neurol. 251, 127–138. doi: 10.1016/j.expneurol.2013.01.009
Higuchi, M., Lee, V. M., and Trojanowski, J. Q. (2002). Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med. 2, 131–150. doi: 10.1385/NMM:2:2:131
Higuchi, M., Zhang, B., Forman, M. S., Yoshiyama, Y., Trojanowski, J. Q., and Lee, V. M. (2005). Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J. Neurosci. 25, 9434–9443. doi: 10.1523/JNEUROSCI.2691-05.2005
Huang, J., and Auchus, A. P. (2007). Diffusion tensor imaging of normal appearing white matter and its correlation with cognitive functioning in mild cognitive impairment and Alzheimer's disease. Ann. N. Y. Acad. Sci. 1097, 259–264. doi: 10.1196/annals.1379.021
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Hyman, B. T., Van Hoesen, G. W., and Damasio, A. R. (1990). Memory-related neural systems in Alzheimer's disease: an anatomic study. Neurology 40, 1721–1730. doi: 10.1212/WNL.40.11.1721
Ihara, M., Polvikoski, T. M., Hall, R., Slade, J. Y., Perry, R. H., Oakley, A. E., et al. (2010). Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer's disease, and dementia with Lewy bodies. Acta Neuropathol. 119, 579–589. doi: 10.1007/s00401-009-0635-8
Ikeda, K., Akiyama, H., Haga, C., Kondo, H., Arima, K., and Oda, T. (1994). Argyrophilic thread-like structure in corticobasal degeneration and supranuclear palsy. Neurosci. Lett. 174, 157–159. doi: 10.1016/0304-3940(94)90010-8
Ikegami, S., Harada, A., and Hirokawa, N. (2000). Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 279, 129–132. doi: 10.1016/S0304-3940(99)00964-7
Imamura, K., Sahara, N., Kanaan, N. M., Tsukita, K., Kondo, T., Kutoku, Y., et al. (2016). Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 6:34904. doi: 10.1038/srep34904
Ishihara, T., Hong, M., Zhang, B., Nakagawa, Y., Lee, M. K., Trojanowski, J. Q., et al. (1999). Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24, 751–762. doi: 10.1016/S0896-6273(00)81127-7
Ishizawa, K., Lin, W. L., Tiseo, P., Honer, W. G., Davies, P., and Dickson, D. W. (2000). A qualitative and quantitative study of grumose degeneration in progressive supranuclear palsy. J. Neuropathol. Exp. Neurol. 59, 513–524. doi: 10.1093/jnen/59.6.513
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., Van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Janning, D., Igaev, M., Sundermann, F., Bruhmann, J., Beutel, O., Heinisch, J. J., et al. (2014). Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell 25, 3541–3551. doi: 10.1091/mbc.E14-06-1099
Jaworski, T., Dewachter, I., Lechat, B., Croes, S., Termont, A., Demedts, D., et al. (2009). AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE 4:e7280. doi: 10.1371/journal.pone.0007280
Jaworski, T., Lechat, B., Demedts, D., Gielis, L., Devijver, H., Borghgraef, P., et al. (2011). Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. 179, 2001–2015. doi: 10.1016/j.ajpath.2011.06.025
Jeganathan, S., Hascher, A., Chinnathambi, S., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (2008). Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076. doi: 10.1074/jbc.M805300200
Jeganathan, S., Von Bergen, M., Brutlach, H., Steinhoff, H. J., and Mandelkow, E. (2006). Global hairpin folding of tau in solution. Biochemistry 45, 2283–2293. doi: 10.1021/bi0521543
Kahlson, M. A., and Colodner, K. J. (2015). Glial Tau pathology in tauopathies: functional consequences. J. Exp. Neurosci. 9, 43–50. doi: 10.4137/JEN.S25515
Kanaan, N. M., Cox, K., Alvarez, V. E., Stein, T. D., Poncil, S., and Mckee, A. C. (2016). Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 19–34. doi: 10.1093/jnen/nlv001
Kanaan, N. M., Morfini, G. A., Lapointe, N. E., Pigino, G. F., Patterson, K. R., Song, Y., et al. (2011). Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 31, 9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011
Kanaan, N. M., Morfini, G., Pigino, G., Lapointe, N. E., Andreadis, A., Song, Y., et al. (2012). Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 33, 826 e815–830. doi: 10.1016/j.neurobiolaging.2011.06.006
Kanaan, N. M., Pigino, G. F., Brady, S. T., Lazarov, O., Binder, L. I., and Morfini, G. A. (2013). Axonal degeneration in Alzheimer's disease: when signaling abnormalities meet the axonal transport system. Exp. Neurol. 246, 44–53. doi: 10.1016/j.expneurol.2012.06.003
Ke, Y. D., Suchowerska, A. K., Van Der Hoven, J., De Silva, D. M., Wu, C. W., Van Eersel, J., et al. (2012). Lessons from tau-deficient mice. Int. J. Alzheimers. Dis. 2012:873270. doi: 10.1155/2012/873270
Kiernan, P. T., Montenigro, P. H., Solomon, T. M., and Mckee, A. C. (2015). Chronic traumatic encephalopathy: a neurodegenerative consequence of repetitive traumatic brain injury. Semin. Neurol. 35, 20–28. doi: 10.1055/s-0035-1545080
Knake, S., Belke, M., Menzler, K., Pilatus, U., Eggert, K. M., Oertel, W. H., et al. (2010). In vivo demonstration of microstructural brain pathology in progressive supranuclear palsy: a DTI study using TBSS. Mov. Disord. 25, 1232–1238. doi: 10.1002/mds.23054
Kneynsberg, A., Collier, T. J., Manfredsson, F. P., and Kanaan, N. M. (2016). Quantitative and semi-quantitative measurements of axonal degeneration in tissue and primary neuron cultures. J. Neurosci. Methods 266, 32–41. doi: 10.1016/j.jneumeth.2016.03.004
Koerte, I. K., Ertl-Wagner, B., Reiser, M., Zafonte, R., and Shenton, M. E. (2012a). White matter integrity in the brains of professional soccer players without a symptomatic concussion. JAMA 308, 1859–1861. doi: 10.1001/jama.2012.13735
Koerte, I. K., Kaufmann, D., Hartl, E., Bouix, S., Pasternak, O., Kubicki, M., et al. (2012b). A prospective study of physician-observed concussion during a varsity university hockey season: white matter integrity in ice hockey players. Part 3 of 4. Neurosurg. Focus 33, E3:1–7. doi: 10.3171/2012.10.FOCUS12303
Kouri, N., Carlomagno, Y., Baker, M., Liesinger, A. M., Caselli, R. J., Wszolek, Z. K., et al. (2014). Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 127, 271–282. doi: 10.1007/s00401-013-1193-7
Kouri, N., Whitwell, J. L., Josephs, K. A., Rademakers, R., and Dickson, D. W. (2011). Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat. Rev. Neurol. 7, 263–272. doi: 10.1038/nrneurol.2011.43
Kovacs, G. G., Majtenyi, K., Spina, S., Murrell, J. R., Gelpi, E., Hoftberger, R., et al. (2008). White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J. Neuropathol. Exp. Neurol. 67, 963–975. doi: 10.1097/NEN.0b013e318187a80f
Kowall, N. W., and Kosik, K. S. (1987). Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann. Neurol. 22, 639–643. doi: 10.1002/ana.410220514
Kraus, M. F., Susmaras, T., Caughlin, B. P., Walker, C. J., Sweeney, J. A., and Little, D. M. (2007). White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain 130, 2508–2519. doi: 10.1093/brain/awm216
LaFerla, F. M., and Green, K. N. (2012). Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006320. doi: 10.1101/cshperspect.a006320
LaPointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Kozikowski, A. P., Binder, L. I., et al. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. doi: 10.1002/jnr.21850
Lau, D. H., Hogseth, M., Phillips, E. C., O'neill, M. J., Pooler, A. M., Noble, W., et al. (2016). Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer's disease. Acta Neuropathol. Commun. 4, 49. doi: 10.1186/s40478-016-0317-4
Lee, G., Newman, S. T., Gard, D. L., Band, H., and Panchamoorthy, G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 111(Pt 21), 3167–3177.
Lei, P., Ayton, S., Moon, S., Zhang, Q., Volitakis, I., Finkelstein, D. I., et al. (2014). Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 9:29. doi: 10.1186/1750-1326-9-29
Lewis, J., Mcgowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., et al. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405. doi: 10.1038/78078
Li, T., Hawkes, C., Qureshi, H. Y., Kar, S., and Paudel, H. K. (2006). Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 45, 3134–3145. doi: 10.1021/bi051635j
Liao, H., Li, Y., Brautigan, D. L., and Gundersen, G. G. (1998). Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J. Biol. Chem. 273, 21901–21908. doi: 10.1074/jbc.273.34.21901
Lin, W. L., Lewis, J., Yen, S. H., Hutton, M., and Dickson, D. W. (2003). Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 32, 1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95
Lin, W. L., Zehr, C., Lewis, J., Hutton, M., Yen, S. H., and Dickson, D. W. (2005). Progressive white matter pathology in the spinal cord of transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 34, 397–410. doi: 10.1007/s11068-006-8726-0
Ling, H., Hardy, J., and Zetterberg, H. (2015). Neurological consequences of traumatic brain injuries in sports. Mol. Cell. Neurosci. 66, 114–122. doi: 10.1016/j.mcn.2015.03.012
Lingor, P., Koch, J. C., Tonges, L., and Bahr, M. (2012). Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 349, 289–311. doi: 10.1007/s00441-012-1362-3
Lippa, C. F. (2004). Synaptophysin immunoreactivity in Pick's disease: comparison with Alzheimer's disease and dementia with Lewy bodies. Am. J. Alzheimers. Dis. Other Demen. 19, 341–344. doi: 10.1177/153331750401900606
Lippa, C. F., Zhukareva, V., Kawarai, T., Uryu, K., Shafiq, M., Nee, L. E., et al. (2000). Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann. Neurol. 48, 850–858. doi: 10.1002/1531-8249(200012)48:6<850::AID-ANA5>3.0.CO;2-V
Liu, C., Song, X., Nisbet, R., and Gotz, J. (2016). Co-immunoprecipitation with Tau Isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188. doi: 10.1074/jbc.M115.641902
Liu, C. W., Lee, G., and Jay, D. G. (1999). Tau is required for neurite outgrowth and growth cone motility of chick sensory neurons. Cell Motil. Cytoskeleton 43, 232–242. doi: 10.1002/(SICI)1097-0169(1999)43:3<232::AID-CM6>3.0.CO;2-7
Loomis, P. A., Howard, T. H., Castleberry, R. P., and Binder, L. I. (1990). Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 87, 8422–8426. doi: 10.1073/pnas.87.21.8422
Ludvigson, A. E., Luebke, J. I., Lewis, J., and Peters, A. (2011). Structural abnormalities in the cortex of the rTg4510 mouse model of tauopathy: a light and electron microscopy study. Brain Struct. Funct. 216, 31–42. doi: 10.1007/s00429-010-0295-4
Mandelkow, E. M., Stamer, K., Vogel, R., Thies, E., and Mandelkow, E. (2003). Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer's disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Matamoros, A. J., and Baas, P. W. (2016). Microtubules in health and degenerative disease of the nervous system. Brain Res. Bull. 126, 217–225. doi: 10.1016/j.brainresbull.2016.06.016
Maxwell, W. L., Mccreath, B. J., Graham, D. I., and Gennarelli, T. A. (1995). Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J. Neurocytol. 24, 925–942. doi: 10.1007/BF01215643
Mayford, M., Bach, M. E., Huang, Y. Y., Wang, L., Hawkins, R. D., and Kandel, E. R. (1996). Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683. doi: 10.1126/science.274.5293.1678
McAllister, T. W., Ford, J. C., Flashman, L. A., Maerlender, A., Greenwald, R. M., Beckwith, J. G., et al. (2014). Effect of head impacts on diffusivity measures in a cohort of collegiate contact sport athletes. Neurology 82, 63–69. doi: 10.1212/01.wnl.0000438220.16190.42
Mckee, A. C., Cantu, R. C., Nowinski, C. J., Hedley-Whyte, E. T., Gavett, B. E., Budson, A. E., et al. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735. doi: 10.1097/NEN.0b013e3181a9d503
Mckee, A. C., Stein, T. D., Nowinski, C. J., Stern, R. A., Daneshvar, D. H., Alvarez, V. E., et al. (2012). The spectrum of disease in chronic traumatic encephalopathy. Brain 136(Pt 1), 43–64. doi: 10.1093/brain/aws307
Mckhann, G. M., Albert, M. S., Grossman, M., Miller, B., Dickson, D., Trojanowski, J. Q., et al. (2001). Clinical and pathological diagnosis of frontotemporal dementia: report of the work group on frontotemporal dementia and pick's disease. Arch. Neurol. 58, 1803–1809. doi: 10.1001/archneur.58.11.1803
Meerschaert, M. M., Magin, R. L., and Ye, A. Q. (2016). Anisotropic fractional diffusion tensor imaging. J. Vib. Control 22, 2211–2221. doi: 10.1177/1077546314568696
Moreno, H., Morfini, G., Buitrago, L., Ujlaki, G., Choi, S., Yu, E., et al. (2016). Tau pathology-mediated presynaptic dysfunction. Neuroscience 325, 30–38. doi: 10.1016/j.neuroscience.2016.03.044
Morfini, G. A., Burns, M., Binder, L. I., Kanaan, N. M., Lapointe, N., Bosco, D. A., et al. (2009). Axonal transport defects in neurodegenerative diseases. J. Neurosci. 29, 12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009
Morfini, G., Pigino, G., Beffert, U., Busciglio, J., and Brady, S. T. (2002a). Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med. 2, 89–99. doi: 10.1385/NMM:2:2:089
Morfini, G., Pigino, G., Mizuno, N., Kikkawa, M., and Brady, S. T. (2007). Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J. Neurosci. Res. 85, 2620–2630. doi: 10.1002/jnr.21154
Morfini, G., Schmidt, N., Weissmann, C., Pigino, G., and Kins, S. (2016). Conventional kinesin: biochemical heterogeneity and functional implications in health and disease. Brain Res. Bull. 126, 347–353. doi: 10.1016/j.brainresbull.2016.06.009
Morfini, G., Szebenyi, G., Brown, H., Pant, H. C., Pigino, G., Deboer, S., et al. (2004). A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 23, 2235–2245. doi: 10.1038/sj.emboj.7600237
Morfini, G., Szebenyi, G., Elluru, R., Ratner, N., and Brady, S. T. (2002b). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293. doi: 10.1093/emboj/21.3.281
Morfini, G., Szebenyi, G., Richards, B., and Brady, S. T. (2001). Regulation of kinesin: implications for neuronal development. Dev. Neurosci. 23, 364–376. doi: 10.1159/000048720
Morris, M., Koyama, A., Masliah, E., and Mucke, L. (2011). Tau reduction does not prevent motor deficits in two mouse models of Parkinson's disease. PLoS ONE 6:e29257. doi: 10.1371/journal.pone.0029257
Mufson, E. J., Perez, S. E., Nadeem, M., Mahady, L., Kanaan, N. M., Abrahamson, E. E., et al. (2016). Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain Inj. 30, 1399–1413. doi: 10.1080/02699052.2016.1219058
Murrell, J. R., Spillantini, M. G., Zolo, P., Guazzelli, M., Smith, M. J., Hasegawa, M., et al. (1999). Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J. Neuropathol. Exp. Neurol. 58, 1207–1226. doi: 10.1097/00005072-199912000-00002
Nakamura, Y., Takeda, M., Yoshimi, K., Hattori, H., Hariguchi, S., Hashimoto, S., et al. (1994). Involvement of clathrin light chains in the pathology of Pick's disease; implication for impairment of axonal transport. Neurosci. Lett. 180, 25–28. doi: 10.1016/0304-3940(94)90905-9
Niewidok, B., Igaev, M., Sundermann, F., Janning, D., Bakota, L., and Brandt, R. (2016). Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau's interaction with microtubules in axon-like processes. Mol. Biol. Cell 27, 3537–3549. doi: 10.1091/mbc.E16-06-0402
Padovani, A., Borroni, B., Brambati, S. M., Agosti, C., Broli, M., Alonso, R., et al. (2006). Diffusion tensor imaging and voxel based morphometry study in early progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatr. 77, 457–463. doi: 10.1136/jnnp.2005.075713
Papasozomenos, S. C., and Binder, L. I. (1987). Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil. Cytoskeleton 8, 210–226. doi: 10.1002/cm.970080303
Petrie, E. C., Cross, D. J., Yarnykh, V. L., Richards, T., Martin, N. M., Pagulayan, K., et al. (2014). Neuroimaging, behavioral, and psychological sequelae of repetitive combined blast/impact mild traumatic brain injury in Iraq and Afghanistan war veterans. J. Neurotrauma 31, 425–436. doi: 10.1089/neu.2013.2952
Pollock, N. J., Mirra, S. S., Binder, L. I., Hansen, L. A., and Wood, J. G. (1986). Filamentous aggregates in Pick's disease, progressive supranuclear palsy, and Alzheimer's disease share antigenic determinants with microtubule-associated protein, tau. Lancet 2:1211. doi: 10.1016/S0140-6736(86)92212-9
Praprotnik, D., Smith, M. A., Richey, P. L., Vinters, H. V., and Perry, G. (1996). Filament heterogeneity within the dystrophic neurites of senile plaques suggests blockage of fast axonal transport in Alzheimer's disease. Acta Neuropathol. 91, 226–235. doi: 10.1007/s004010050420
Probst, A., Gotz, J., Wiederhold, K. H., Tolnay, M., Mistl, C., Jaton, A. L., et al. (2000). Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 99, 469–481. doi: 10.1007/s004010051148
Probst, A., Langui, D., Lautenschlager, C., Ulrich, J., Brion, J. P., and Anderton, B. H. (1988). Progressive supranuclear palsy: extensive neuropil threads in addition to neurofibrillary tangles. Very similar antigenicity of subcortical neuronal pathology in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol. 77, 61–68. doi: 10.1007/BF00688244
Probst, A., Tolnay, M., Langui, D., Goedert, M., and Spillantini, M. G. (1996). Pick's disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 92, 588–596. doi: 10.1007/s004010050565
Qiang, L., Yu, W., Andreadis, A., Luo, M., and Baas, P. W. (2006). Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 26, 3120–3129. doi: 10.1523/JNEUROSCI.5392-05.2006
Raff, M. C., Whitmore, A. V., and Finn, J. T. (2002). Axonal self-destruction and neurodegeneration. Science 296, 868–871. doi: 10.1126/science.1068613
Ramirez-Rios, S., Denarier, E., Prezel, E., Vinit, A., Stoppin-Mellet, V., Devred, F., et al. (2016). Tau antagonizes end-binding protein tracking at microtubule ends through a phosphorylation-dependent mechanism. Mol. Biol. Cell 27, 2924–2934. doi: 10.1091/mbc.E16-01-0029
Rao, M. V., Mcbrayer, M. K., Campbell, J., Kumar, A., Hashim, A., Sershen, H., et al. (2014). Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J. Neurosci. 34, 9222–9234. doi: 10.1523/JNEUROSCI.1132-14.2014
Rasband, M. N. (2010). The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562. doi: 10.1038/nrn2852
Rebeiz, J. J., Kolodny, E. H., and Richardson, E. P. Jr. (1968). Corticodentatonigral degeneration with neuronal achromasia. Arch. Neurol. 18, 20–33. doi: 10.1001/archneur.1968.00470310034003
Rizzu, P., Van Swieten, J. C., Joosse, M., Hasegawa, M., Stevens, M., Tibben, A., et al. (1999). High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 64, 414–421. doi: 10.1086/302256
Rodriguez-Martin, T., Pooler, A. M., Lau, D. H., Morotz, G. M., De Vos, K. J., Gilley, J., et al. (2016). Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol. Dis. 85, 1–10. doi: 10.1016/j.nbd.2015.10.007
Rohrer, J. D., Ridgway, G. R., Modat, M., Ourselin, S., Mead, S., Fox, N. C., et al. (2010). Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53, 1070–1076. doi: 10.1016/j.neuroimage.2009.12.088
Samsonov, A., Yu, J. Z., Rasenick, M., and Popov, S. V. (2004). Tau interaction with microtubules in vivo. J. Cell Sci. 117, 6129–6141. doi: 10.1242/jcs.01531
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Seitz, A., Kojima, H., Oiwa, K., Mandelkow, E. M., Song, Y. H., and Mandelkow, E. (2002). Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 21, 4896–4905. doi: 10.1093/emboj/cdf503
Selden, S. C., and Pollard, T. D. (1986). Interaction of actin filaments with microtubules is mediated by microtubule-associated proteins and regulated by phosphorylation. Ann. N. Y. Acad. Sci. 466, 803–812. doi: 10.1111/j.1749-6632.1986.tb38464.x
Siman, R., Cocca, R., and Dong, Y. (2015). The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage alzheimer-type tauopathy. PLoS ONE 10:e0142340. doi: 10.1371/journal.pone.0142340
Siman, R., Lin, Y. G., Malthankar-Phatak, G., and Dong, Y. (2013). A rapid gene delivery-based mouse model for early-stage Alzheimer disease-type tauopathy. J. Neuropathol. Exp. Neurol. 72, 1062–1071. doi: 10.1097/NEN.0000000000000006
Sjobeck, M., and Englund, E. (2003). Glial levels determine severity of white matter disease in Alzheimer's disease: a neuropathological study of glial changes. Neuropathol. Appl. Neurobiol. 29, 159–169. doi: 10.1046/j.1365-2990.2003.00456.x
Sjobeck, M., Haglund, M., and Englund, E. (2005). Decreasing myelin density reflected increasing white matter pathology in Alzheimer's disease–a neuropathological study. Int. J. Geriatr. Psychiatry 20, 919–926. doi: 10.1002/gps.1384
Song, Y., and Brady, S. T. (2015). Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 25, 125–136. doi: 10.1016/j.tcb.2014.10.004
Spires, T. L., Orne, J. D., Santacruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., et al. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607. doi: 10.2353/ajpath.2006.050840
Spittaels, K., Van Den Haute, C., Van Dorpe, J., Bruynseels, K., Vandezande, K., Laenen, I., et al. (1999). Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol. 155, 2153–2165. doi: 10.1016/S0002-9440(10)65533-2
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stern, J. L., Lessard, D. V., Hoeprich, G. J., Morfini, G. A., and Berger, C. L. (2017). Phospho-regulation of tau modulates inhibition of kinesin-1 motility. Mol. Biol. Cell. 28, 1079–1087. doi: 10.1091/mbc.E16-10-0728
Stoub, T. R., Detoledo-Morrell, L., Stebbins, G. T., Leurgans, S., Bennett, D. A., and Shah, R. C. (2006). Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 10041–10045. doi: 10.1073/pnas.0603414103
Sudo, H., and Baas, P. W. (2011). Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum. Mol. Genet. 20, 763–778. doi: 10.1093/hmg/ddq521
Sultan, A., Nesslany, F., Violet, M., Begard, S., Loyens, A., Talahari, S., et al. (2011). Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 286, 4566–4575. doi: 10.1074/jbc.M110.199976
Sun, W., Qureshi, H. Y., Cafferty, P. W., Sobue, K., Agarwal-Mawal, A., Neufield, K. D., et al. (2002). Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J. Biol. Chem. 277, 11933–11940. doi: 10.1074/jbc.M107182200
Swanson, E., Breckenridge, L., Mcmahon, L., Som, S., Mcconnell, I., and Bloom, G. S. (2017). Extracellular tau oligomers induce invasion of endogenous tau into the somatodendritic compartment and axonal transport dysfunction. J. Alzheimers. Dis. 58, 803–820. doi: 10.3233/JAD-170168
Takei, Y., Teng, J., Harada, A., and Hirokawa, N. (2000). Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 150, 989–1000. doi: 10.1083/jcb.150.5.989
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., Deteresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Tiernan, C. T., Combs, B., Cox, K., Morfini, G., Brady, S. T., Counts, S. E., et al. (2016). Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp. Neurol. 283, 318–329. doi: 10.1016/j.expneurol.2016.06.030
Tint, I., Slaughter, T., Fischer, I., and Black, M. M. (1998). Acute inactivation of tau has no effect on dynamics of microtubules in growing axons of cultured sympathetic neurons. J. Neurosci. 18, 8660–8673.
Tokuda, T., Ikeda, S., Yanagisawa, N., Ihara, Y., and Glenner, G. G. (1991). Re-examination of ex-boxers' brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol. 82, 280–285. doi: 10.1007/BF00308813
Tucker, K. L., Meyer, M., and Barde, Y. A. (2001). Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37. doi: 10.1038/82868
Uryu, K., Chen, X. H., Martinez, D., Browne, K. D., Johnson, V. E., Graham, D. I., et al. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192. doi: 10.1016/j.expneurol.2007.06.018
Uversky, V. N. (2015). Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7:18. doi: 10.3389/fnagi.2015.00018
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E., Cook, C., Lummertz Da Rocha, E., et al. (2016). Interaction of tau with the RNA-Binding Protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466. doi: 10.1016/j.celrep.2016.04.045
van Hummel, A., Bi, M., Ippati, S., Van Der Hoven, J., Volkerling, A., Lee, W. S., et al. (2016). No overt deficits in aged tau-deficient C57Bl/6.Mapttm1(EGFP)Kit GFP knockin mice. PLoS ONE 11:e0163236. doi: 10.1371/journal.pone.0163236
Vermersch, P., Roche, J., Hamon, M., Daems-Monpeurt, C., Pruvo, J. P., Dewailly, P., et al. (1996). White matter magnetic resonance imaging hyperintensity in Alzheimer's disease: correlations with corpus callosum atrophy. J. Neurol. 243, 231–234. doi: 10.1007/BF00868519
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Walsh, D. M., and Selkoe, D. J. (2016). A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 17, 251–260. doi: 10.1038/nrn.2016.13
Wang, L. N., Zhu, M. W., Feng, Y. Q., and Wang, J. H. (2006). Pick's disease with Pick bodies combined with progressive supranuclear palsy without tuft-shaped astrocytes: a clinical, neuroradiologic and pathological study of an autopsied case. Neuropathology 26, 222–230. doi: 10.1111/j.1440-1789.2006.00671.x
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Werth, J. L., and Thayer, S. A. (1994). Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 14, 348–356.
Whitwell, J. L., Master, A. V., Avula, R., Kantarci, K., Eggers, S. D., Edmonson, H. A., et al. (2011). Clinical correlates of white matter tract degeneration in progressive supranuclear palsy. Arch. Neurol. 68, 753–760. doi: 10.1001/archneurol.2011.107
Williams, D. R., and Lees, A. J. (2009). Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 8, 270–279. doi: 10.1016/S1474-4422(09)70042-0
Wszolek, Z. K., Pfeiffer, R. F., Bhatt, M. H., Schelper, R. L., Cordes, M., Snow, B. J., et al. (1992). Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann. Neurol. 32, 312–320. doi: 10.1002/ana.410320303
Xia, D., Li, C., and Gotz, J. (2015). Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 1852, 913–924. doi: 10.1016/j.bbadis.2014.12.017
Yamakawa, K., Takanashi, M., Watanabe, M., Nakamura, N., Kobayashi, T., Hasegawa, M., et al. (2006). Pathological and biochemical studies on a case of Pick disease with severe white matter atrophy. Neuropathology 26, 586–591. doi: 10.1111/j.1440-1789.2006.00738.x
Yasuda, M., and Mayford, M. R. (2006). CaMKII activation in the entorhinal cortex disrupts previously encoded spatial memory. Neuron 50, 309–318. doi: 10.1016/j.neuron.2006.03.035
Yin, Y., Wang, Y., Gao, D., Ye, J., Wang, X., Fang, L., et al. (2016). Accumulation of human full-length tau induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain-2. Sci. Rep. 6:27283. doi: 10.1038/srep27283
Yoshiyama, Y., Zhang, B., Bruce, J., Trojanowski, J. Q., and Lee, V. M. (2003). Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J. Neurosci. 23, 10662–10671.
Yuan, A., Kumar, A., Peterhoff, C., Duff, K., and Nixon, R. A. (2008). Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 28, 1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008
Zhang, L., Ravdin, L. D., Relkin, N., Zimmerman, R. D., Jordan, B., Lathan, W. E., et al. (2003). Increased diffusion in the brain of professional boxers: a preclinical sign of traumatic brain injury? AJNR Am. J. Neuroradiol. 24, 52–57.
Zhang, Y., Walter, R., Ng, P., Luong, P. N., Dutt, S., Heuer, H., et al. (2016). Progression of microstructural degeneration in progressive supranuclear palsy and corticobasal syndrome: a longitudinal diffusion tensor imaging study. PLoS ONE 11:e0157218. doi: 10.1371/journal.pone.0157218
Keywords: axonal transport, Alzheimer's disease (AD), chronic traumatic encephalopathy (CTE), pick's disease, progressive supranuclear palsy, corticobasal degeneration, neurodegeneration, animal models of tauopathies
Citation: Kneynsberg A, Combs B, Christensen K, Morfini G and Kanaan NM (2017) Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Front. Neurosci. 11:572. doi: 10.3389/fnins.2017.00572
Received: 31 July 2017; Accepted: 29 September 2017;
Published: 17 October 2017.
Edited by:
Samuel David Crish, Northeast Ohio Medical University, United StatesReviewed by:
Gloria Lee, University of Iowa, United StatesRoland Brandt, University of Osnabrück, Germany
Copyright © 2017 Kneynsberg, Combs, Christensen, Morfini and Kanaan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas M. Kanaan, bmljaG9sYXMua2FuYWFuQGhjLm1zdS5lZHU=
Gerardo Morfini, Z21vcmZpbmlAdWljLmVkdQ==
†Co-senior authors.