 Serhiy Forostyak1,2*
Serhiy Forostyak1,2* Eva Sykova2,3
Eva Sykova2,3- 1Centre of Reconstructive Neuroscience, Institute of Experimental Medicine (ASCR), Czech Academy of Sciences, Prague, Czechia
- 2Department of Neuroscience, 2nd Faculty of Medicine, Charles University, Prague, Czechia
- 3Institute of Neuroimmunology, Slovak Academy of Sciences, Bratislava, Slovakia
Motor neurons (MN) degeneration is a main feature of amyotrophic lateral sclerosis (ALS), a neurological disorder with a progressive course. The diagnosis of ALS is essentially a clinical one. Most common symptoms include a gradual neurological deterioration that reflect the impairment and subsequent loss of muscle functions. Up-to-date ALS has no therapy that would prevent or cure a disease. Modern therapeutic strategies comprise of neuroprotective treatment focused on antiglutamatergic, antioxidant, antiapoptotic, and anti-inflammatory molecules. Stem cells application and gene therapy has provided researchers with a powerful tool for discovery of new mechanisms and therapeutic agents, as well as opened new perspectives for patients and family members. Here, we review latest progress made in basic, translational and clinical stem cell research related to the ALS. We overviewed results of preclinical and clinical studies employing cell-based therapy to treat neurodegenerative disorders. A special focus has been made on the neuroprotective properties of adult mesenchymal stromal cells (MSC) application into ALS patients. Finally, we overviewed latest progress in the field of embryonic and induced pluripotent stem cells used for the modeling and application during neurodegeneration in general and in ALS in particular.
History, Genetics, and Clinics of ALS
In 1848, Aran described for the first time a malady later become known as an amyotrophic lateral sclerosis (ALS). He reported 11 cases of the disease featuring a focal wasting and paresis, weakness and cramps in the upper extremities, and fatal end within 2 years (Aran, 1848). Aran proposed that the disease had been inherited from the parents. In 1873, Jean-Marie Charcot reported that ALS was never inherited, and that was the main reason for delineating ALS from muscular atrophy (Charcot, 1881). The view that ALS is rarely connected with family history persisted for almost 100 years. A new era in the field has started when several genes were linked with familial (FALS) and sporadic ALS (SALS) cases. It is thought that around 5–10 percent of all ALS incidents have a family history, whereas the rest are sporadic (Bento-Abreu et al., 2010; van Es et al., 2010; Andersen and Al-Chalabi, 2011). The mutations in the following genes have been found to result in FALS: superoxide dismutase 1 (SOD1), TARDBP, Ubiquilin 2, Alsin, Senataxin, FUS, Angiogenin, SIGMAR1 (Rosen et al., 1993; Hadano et al., 2001; Hand et al., 2002; Sapp et al., 2003; Chen et al., 2004; Nishimura et al., 2004; Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Vance et al., 2009; Elden et al., 2010). ALS has been recently associated with frontotemporal dementia, (FTD, ALS/FTD). A GGGGCC hexanucleotide repeat in the intron of protein C9ORF72 has been demonstrated to cause an alternative splicing of this protein that is leading to similar pathological events in two diseases (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Another pathological features of ALS and FTD are TDP-43 and p62 positive cytoplasmic depositions in the hippocampus and cerebellum (Achi and Rudnicki, 2012; Mahoney et al., 2012). The main differences between ALS/FTD patients and classical FTD cases are presences of psychiatric signs and the distribution of TDP-43 inclusions: SALS primarily features TDP-43 pathology in the spinal cord, patients with FTD primarily affect the cortex, while FTD-ALS patients have TDP-43 pathology in both areas (Geser et al., 2009; Neumann et al., 2009). The genetic screening of US population demonstrated that TDP-43 expansion occurs in 12% of familial FTD and 22.5% of FALS (DeJesus-Hernandez et al., 2011), while European population screening showed higher prevalence in FALS (46%), followed by familial FTD (29%) and SALS (21%) (Renton et al., 2011).
Despite diverse genetic backgrounds, SALS and FALS are clinically indistinguishable, 95% of all ALS cases are sporadic, and the other five percent have a genetic background. The clinical hallmark of both types of ALS is a progressive deterioration of neurological functions correlated (clinically and pathologically) with loss of primary and secondary MN, sparing of the oculomotor and the spinal Onuf's nuclei, coexistent neurogenic atrophy, weakness, and fasciculations caused by secondary MN degeneration, together with hyperactive deep tendon reflexes, pyramidal tract signs, and increased muscle tone (Borasio and Appel, 2003). Patients present a wide range of diverse clinical outcomes regarding disease onset, rate of progression and survival (Burkhardt et al., 2013). Disease's symptoms are typically asymmetrical. Some 20–30% of all cases have bulbar onset, with more than 50% of bulbar symptoms in older women. In FALS minor pathological changes could be diagnosed in the spinocerebellar tracts, typically without accompanying symptoms. Most commonly, the disease strikes people between the ages of 40 and 70, although the early onset is not exceptional. Unlike other neurodegenerative maladies, ALS is not age-related disease. However, aging is one of a many risk factors. Incidence of ALS is fairly uniform 1–2 per 100,000 individuals, except for an elevated incidence in Kii peninsula of Honshu island and Guam (Kuzuhara and Kokubo, 2005; Steele, 2005). A lifetime risk of ALS development approaches 1/400-1/700 with a somewhat more frequent occurrence in males than in females (ratio is ≈ 1.5; Johnston et al., 2006; Bento-Abreu et al., 2010).
Current Treatment and Management of ALS
Considering a great diversity of genetic and clinical forms of ALS, every therapeutic attempt could be regarded as experimental. Nevertheless, as for now a standard therapy for ALS includes an antiglutamatergic agent Riluzole that, however, does not alter the natural history of the disease (Lacomblez et al., 1996). Riluzole (100 mg/day), which reduces the presynaptic release of glutamate, remains the only effective drug that slows disease progression and extends the patients lifespan by 2–3 months (Lacomblez et al., 1996; Sykova et al., 2017). Additionally, all patients receive palliative or symptomatic therapy such as a non-invasive positive pressure ventilation (Hardiman, 2011), prescription of anticholinergic drugs (such as trihexyphenidyl, amitriptyline, or atropine) or the use of a portable suction machine if drooling is troublesome. Baclofen or diazepam might be used to deal with spasticity (Mustfa et al., 2006). Dysphagia could be managed by modifying food and fluid consistencies, postural advice, and in extreme cases of bulbar involvement, by gastrostomy or cricopharyngomyotomy. The results of such therapy are unsatisfactory, current clinical management is still extremely limited and novel therapeutic approaches are in an active search. The presymptomatic or at least the early diagnosis of ALS could offer wider possibilities for prevention and for the treatment of this devastating disease. Therefore, in the future screening of patients with FALS for mutations in the SOD1, TARDBP, FUS, and a several other genes might offer benefits in the diagnosis and treatment of ALS. Sadly, over 60 per cent of ALS patients die within 2–5 years of presentation mostly from pulmonary insufficiency with infections, with < 10 per cent survive rate longer than 8 years (Kiernan et al., 2018). Considering that the above therapies just improve patient's quality of life, but do not extend his/her survival the main task of treatment in the terminal stages is to keep patients as comfortable as possible (McGeer and McGeer, 2005).
Neuroprotective Strategies in ALS
Considering that the disease affects MN at different levels of the central nervous system (CNS), a neuroprotective strategy should aim to restore affected tissue homeostasis throughout the entire nervous system. Numerous attempts have focused on antiglutamatergic, antioxidant, antiapoptotic, anti-inflammatory, and neurotrophic molecules, as well as on gene therapy and stem cell application. These and other molecules are able to reach the MN after systemic (intravenous, intraarterial), local (intrathecal, intraspinal, intracerebral, etc.) or combined application, as it has been shown that the blood brain barrier (BBB) in ALS is also compromised (Garbuzova-Davis et al., 2007).
Antiglutamatergic Therapy
Antiglutamatergic Therapy has currently shown the best results in clinical trials. As already mentioned, the only anti-ALS medicine approved for the treatment of patients is Riluzole. The kynurenine pathway (KP), a major route for the metabolism of tryptophan, has been shown to play role in ALS. The KP excitotoxic catabolites such as N-methyl-D-aspartate receptor agonist quinolinic acid and the neuroprotective NMDA receptor antagonist kynurenic acid are involved into crosstalk between the CNS and immune systems, by modulating cell proliferation. Few companies such as Sanofi-Aventis or Teva Neuroscience developed KP inhibitors (Teriflunomide and Laquinimod, respectively), have entered clinical trials (Chen et al., 2009). Memantine, novel anti-excitatory drug is a non-competitive excitotoxic N-methyl-D-aspartate (NMDA)-receptor antagonist, has been shown to delay the loss of hind limb motor activity and extend the survival of SOD1G93A mice (Wang and Zhang, 2005).
Antioxidant Therapy
Antioxidant Therapy aimed at ameliorating oxidative stress, could provide a possible healing effect in ALS patients. However, clinical trials examining the application of vitamin E, acetylcysteine, methylcobalamine, glutathione, or coenzyme Q10 (CoQ10) indicate that these drugs are ineffective in ALS patients (Levy et al., 2006; Kaufmann et al., 2009). An antioxidant peptide called SS-31 has been shown to improve mitochondrial dynamics, resulting in a significant extension of survival, better motor activity, decreased MN loss and reduced immunostaining for oxidative stress markers in G93A mice (Petri et al., 2006).
Immunotherapeutic Strategies
Immunotherapeutic Strategies to combat ALS also could be an attractive therapeutic approach. Active vaccination with misfolded mSOD1 in the G37R SOD1 mouse model of FALS has been tried, resulting in the reduced loss of spinal cord neurons and a modest but statistically significant increase in life expectancy (Urushitani et al., 2007; Brody and Holtzman, 2008). However, much work remains to be done before clinical trials could be started.
The discovery of neurotrophic factors (NTF), their anti-apoptotic effect and the ability to promote the MN survival during development, made these molecules attractive candidates for the treatment of neurodegenerative disorders affecting motor system (Appel, 1981; Gould and Oppenheim, 2011). Increasing number of studies implicate an impaired production of vascular endothelial growth factor (VEGF) by MN, rather than a lack of functional receptors, is associated with ALS. The exogenous VEGF has been shown to cause a direct neuroprotective effect via the expression of VEGF-receptors in MN (Van Den Bosch et al., 2004). Various routes of insulin-like growth factor-1 (IGF-1) or VEGF delivery, was reported to slow disease progression and improved lifespan in animals studies (Kaspar et al., 2003; Nagano et al., 2005; Wang et al., 2007). Interestingly, clinical trials utilizing the subcutaneous delivery of IGF-1 failed to show a beneficial effect, mainly due to a reduced bioavailability of IGF-1 (Sorenson et al., 2008; Howe et al., 2009). The combined usage of IGF with IGF-binding protein 3 (IGFBP), also called IPLEX, significantly increased the serum half-life time of IGF-1 and proceeded toward an early-phase clinical trial. Despite a great deal of debate surrounding the effectiveness of IPLEX (Bedlack et al., 2009; Gould and Oppenheim, 2011), clinical trial employing a delivery of VEGF protein into ALS patients CSF is currently in progress (http://clinicaltrials.gov, identifier # NCT01384162 and NCT008005501).
One should also consider that MN exhibit trophic heterogeneity, that is they respond to a distinct type/s of NTFs during their development (Kanning et al., 2010). Studies on glial cell line-derived neurotrophic factor (GDNF) knockout mice showed a dramatic and restricted loss of small ventral lumbar myelinated axons (γ-MN) and spared large myelinated axons (α-MN) (Gould et al., 2008). Hence, a monotherapeutic strategy might bring some improvements in motor activity or even extend survival, but this demands distinct NTFs that will target specific types of MN. This concern might be resolved by using cocktails of NTFs delivered in such a way so that they would be able to pass through the BBB in an efficient and controlled way. Alternatively, application of stem cells, which are well-known to have paracrine properties, could serve as a vehicle for the delivery of NTFs. Stem cells research has reached a stage when unlimited number of non-engineered and engineered neuroglia could be produced and used for therapeutic purposes aiming at protection, repair, or replacement of affected cells in disorders affecting the brain and spinal cord, thus bringing a new hope for patients.
Cell-based Therapies
A new avenue has been opened for basic research and regenerative medicine with the discovery of stem cells (SC) and their regenerative capacities. By using a SC terminology one would expect to deal with a cell with a pluri- or multipotent features and unlimited self-renewal capacities. Pluripotency means that the cell is capable to differentiate into any mature cell type in the organism originating from all three germ layers (Nistor et al., 2005; Lee et al., 2007). SC classification considers the tissue of origin, the developmental stage when cells appear and could be isolated in the organism. Current research is dealing with the following types of SC: (1) embryonic (ESC), a pluripotent cells that could give rise to any cell in the body; (2) fetal (FSC), a multipotent cells that could give rise to any cell of certain germ layer; (3) somatic (adult-derived), multi- or oligopotent cells found in different tissues of the fully developed organisms; and 4) induced pluripotent (iPS), similar to ESC, but generated from mature somatic cells after the artificial introduction of transcriptional factors (Takahashi and Yamanaka, 2006).
Mesenchymal Stromal Cells (MSC)
MSCs are attractive and accepted target for use in cell-based therapies (autologous application). MSC are oligopotent cells that could be isolated by a relatively simple procedure (Forostyak et al., 2016a) from bone marrow (BMSC), umbilical cord (UMSC), fat tissue (AMSC), Wharton's jelly and other fetal tissues. To meet the “mesenchymness” criteria every cell should meet following criteria: ability for extensive in vitro growth on plastic; differentiate in vitro into chondrocytes, osteocytes and adipocytes; express “mesenchymal” surface markers (Mezey et al., 2000; Krause, 2002; Dominici et al., 2006; Forostyak et al., 2016a). It is interesting to note that despite great similarities in features and properties, MSCs of different origin display certain differences in growth rate, surface markers expression and even physiological activities (Forostyak et al., 2016b). Several groups also reported MSC differentiation toward functional neural phenotype (Tropel et al., 2006).
A therapeutic potential of MSC is very broad and still hides many unknown features to be exlored. However, it is generally accepted that despite the MSC's origin, they primarily act via the growth factors (GF) secreted either into the growth medium or within the recipient's tissue (paracrine function). A GF misbalance in the organism triggers cascades of intracellular changes that may result into various pathological states. Therefore, MSC ability to secrete a cocktail of growth factors brought attention for their use in regenerative medicine in general, and is very promising to use for the purpose of neuroprotection and neuroregeneration. We earlier reviewed available information about growth factors and corresponding genes that have been reported to be secreted by MSC (Forostyak et al., 2013b). Here, we will just name GFs that are known to contribute to ALS and were successfully tested in vivo for the purpose of neuroprotection or disease modification, those are: glia cell-line derived neurotrophic factor (GDNF), insulin growth factor type-1 (IGF-1), brain-derived neurotrophic factor (BDNF), neural growth factor (NGF), VEGF and others that play less significant role in ALS pathology. Apart from a paracrine effect, we have recently reported that after an intrathecal or combined application of MSC a level of apoptosis and inflammation decreases leading a better survival of host MN in host tissue (Forostyak et al., 2014). Moreover, MSC has been shown to repair a defective extracellular matrix (ECM) structure called perineuronal net (PNN) surrounding these MN in SOD1 rat transgenic model of ALS, by up-regulation of some chondroitin sulfate proteoglycan levels (CSPG) typically up-regulated at the end of a critical period of PNN development (Carulli et al., 2010; Forostyak et al., 2014). We could speculate that this finding could be indicating a reactivation of adult neural plasticity within a recipient's organism after MSC application. Notably, MSC do not stimulate alloreactivity, are able to pass a MHC barrier, and thus enabling appliocation between HLA-mismatched individuals (Le Blanc, 2003; Aggarwal and Pittenger, 2005; Rice and Scolding, 2008).
The above properties make these cells a very attractive target for neuroprotective and neuroreparative therapy. Increasing number of reports has led to the preclinical trials. These studies demonstrated positive effects of MSC on motor activity and survival after being delivered via various routes (mostly intrathecal, but also intravenous, intraspinal, intramuscular, or combined) using rodents' models of ALS (Mazzini et al., 2004; Garbuzova-Davis et al., 2008; Kim et al., 2010). Our preclinical study employing intrathecal (cisterna magna) injection of human MSC into symptomatic SOD1G93A transgenic rats, demonstrated prolonged survival of MSC-treated animals by more than 2 weeks compared with the vehicle-treated group, and also demonstrated decreased markers of inflammation in host CSF (Forostyak et al., 2014). Another studies using a combined (intraspinal and intravenous) BMSC transplantation, a part of an increased motor activity and survival and attenuated proliferation of microglial cells (Boucherie et al., 2009; Forostyak et al., 2011). Interestingly, we have noticed that despite a broad application of MSC in animal studies and in clinical trials involving patients, little has been known about MSC's physiology. The stem cells physiology may enable a better control over the graft and even further improvement of regenerative potential of cells prepared for human application. This knowledge may change and further improve effect of cell-based therapies of neurodegenerative and other types of diseases. These questions have been raised in several of our studies, where we have studied stem cells physiology with a help of Ca2+ signaling, ICC, electrophysiological methods and other methods of molecular biology. Ca2+ signals play a crucial role in the differentiation, proliferation and survival of stem cells starting from the early stages and later on mature cells (Forostyak et al., 2013a, 2016b). Dysregulation of calcium ([Ca2+]i) homeostasis is also crucial in the pathophysiology of ALS, and impaired [Ca2+]i in the cytoplasm of neurons is a potential mechanism of decreased cell survival (Dafinca et al., 2016). MSCs effects are dose- and passage-dependent, and we showed that MSC from earlier passages (up to the fifth) are more suitable therapeutic application due to their stability, anti-inflammatory and neuroprotective effects (Choi et al., 2010; Forostyak et al., 2016a). Similar outcomes were achieved by the administration of 106 cells in asymptomatic SOD1 animals, while 105 cells failed to change the prognosis of the disease (Habisch et al., 2007; Kim et al., 2010).
Embryonic and Induced Pluripotent Stem Cells
The advances in stem cell research demonstrated a great potential of cell-based approach in the treatment of currently incurable diseases and this brings hope for patients and their families, especially in the case of neurodegenerative disorders or neurotrauma. The Nobel Prize in Physiology or Medicine for “The discovery that mature cells can be reprogrammed to become pluripotent” highlights the importance of SC research in general and particularly iPS technology (Takahashi and Yamanaka, 2006).
At the moment several groups are working on substitution of defective neurons with donor stem cell-derived neuronal progenitors. The idea behind is that the grafted cell will integrate after transplantation into existing neural circuits and take over the functions of defective cells (Kallur et al., 2006; Lindvall and Kokaia, 2006). It has been reported that MN generated from ESC are not just able to maintain typical motor neuronal phenotype in vitro, but also to functionally incorporate into host CNS (Wichterle et al., 2002; Papadeas and Maragakis, 2009). A glial restricted precursors (GRP) or human neural stem/progenitor cells (hNSC) seem to have a greatest potential in ALS research. These cell types were described to modify ALS prognosis, to reduce MN death and to establish functional synapses with structural integration into the motor circuitry (Lepore et al., 2008; Xu et al., 2009). In ALS functional integration of grafted cells is crucial. However, MN replacement would also expect an axonal outgrowth and neuromuscular junction (NMJ) formation to innervate muscle fibers that otherwise are dedicated to be atrophied. We found only few studies that have demonstrated formation of new functional connections between grafted SC and the recipients' muscles after the grafting (Deshpande et al., 2006; Yohn et al., 2008; Gowing and Svendsen, 2011).
Taking into the consideration above limitations, generation and grafting of protective cells that will support the remaining MN might be more effective. It is known that in ALS astrocytic reactivity with proinflammatory transcriptional and translational profiles exacerbates motor neurons (MNs) dysfunction (Sun et al., 2015). These specific pathological events could be targeted by the application of either ES or iPS cells that were directed either toward progenitor or mature astrocytic phenotypes prior to transplantation (Popescu et al., 2013; Kondo et al., 2014).
The possibility of hyperproliferation and the formation of teratomas is always a risk related with ESC-/iPSC-derived neural progenitors (Seminatore et al., 2010). Latest preclinical studies demonstrate that this issue could be controlled, thus Food and Drug Administration (FDA) has permitted a phase I clinical study testing the safety of the intraspinal grafting of neural stem cells, into patients with confirmed ALS (http://clinicaltrials.gov, identifier #NCT01348451) (Gowing and Svendsen, 2011; Lunn et al., 2011). A transplantation of glial precursor cells or mature astroglia is another quit realistic approach to promote neuroprotection and trophic effects, that will support the failing motor neurons (Robberecht and Philips, 2013). These cells generated from somatic tissues, even from elderly patients, could be further reprogrammed to reach a pluripotent stage, and later differentiated toward neural/astroglial morphology of interest ready for delivery into the patients (Dimos et al., 2008; Hall et al., 2017). This alternative to embryonic/fetal cells, if isolated from affected individuals, could be used either as an in vitro model of neurodegenerative diseases, helping us to understand the mechanisms underlying the pathological processes, or could be used as therapeutic agents after avoiding the risk of graft rejection or the opposite, graft-vs.-host-disease if the donor and recipient is a same individual. The cutting-edge of modern cell biology is the generation of functional induced motor neurons or astrocytes from the patient's own fibroblasts, which after transplantation could protect the dying MN (Son et al., 2011; Hall et al., 2017).
Considering that neurodegenerative diseases often develop in elderly patients, there are some concerns related to the “quality” of stem cells if generated from an aged and sick organism and whether these cells might be used for cell replacement, neuroprotection and neuroregeneration. So far reports addressing this question are quit controversial. Dimos et al. by using the example of iPS cells generated from an 82-year-old woman with a FALS, showed that these cells possess the same properties as do ESC and that they could differentiate into MN (Dimos et al., 2008). However, in the case of iPS cells, which are generated using a cocktail of overexpressed transcriptional factors transferred to skin fibroblasts (or other somatic cells) by transfection of viral vector infection, it is necessary to note that we do not yet know whether the human use of these cells will not increase a risk of genetic modification of both donor and recipient cells (Wichterle and Przedborski, 2010). Concerns are also about the route of transplantation and the developmental stage of the pluripotent cells. It is necessary to keep in mind that the manipulation with the cells that are fully differentiated to the neural or neuronal phenotype is extremely difficult. Therefore, the cells at the precursor stage are potentially better candidates for the transplantation. According to the publicly available information, several clinical trials that are engaging human neural stem cells application to evaluate the safety and efficiency of such a therapy of ALS are currently recruiting patients or are on-going (Glass et al., 2012).
Clinical Trials of Stem Cell Therapy in ALS
In vivo experiments using rodent models of FALS formed a platform for clinical trials involving patients (Vercelli et al., 2008). We have analyzed supported past, current and future clinical trials from the clinicaltrial.gov website and summarized them into the overview table (Table 1). It is obvious that the number of phase 1/2 clinical trials is increasing annually. Mostly, these are safety studies involving small amounts of patients. Unfortunately, great majority of the trials does not describe details about the dosage, type of the cells, criteria for patients monitoring and does not report a feedback about the study outcomes. This complicates interpretation of the data and creates obstacles for further clinical application of various cell types. The majority of approved/available clinical trials employ mesenchymal stromal cells (MSCs) of different origin (mostly bone marrow) for the therapy of ALS. It could be explained mainly by the easiness of derivation and manipulation with autologous cells from patients, legal issues and long history of clinical application of bone marrow derived cells. On the other hand, this cells are quite unique, especially for their paracrine properties for detailed overview see (Forostyak et al., 2013b).
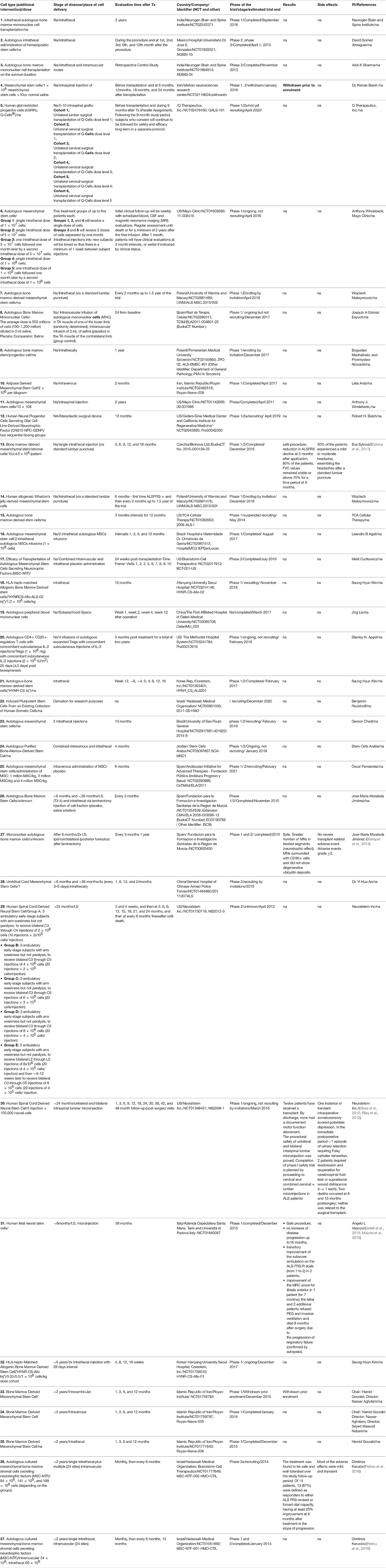
Table 1. An overview of clinical trials involving cell-based therapy to treat ALS (modified from www.clinicaltrials.gov).
Unfortunately, majority of the granted trials did not perform patients' follow-up longer than 24 months. The first long-term follow-up after nearly 9 years of patients monitoring, showed that dorsal application of MSC is safe procedure with no clear clinical benefits, but also without any structural changes or deterioration in psychosocial status (Mazzini et al., 2003, 2010, 2011). Trial with the mononuclear CD133(+) fraction isolated from the peripheral blood after the frontal motor cortex grafting were reported to significantly extend life of ALS patients and to improve their lifestyle compared with patients that were not treated with the cells (Martinez et al., 2009). Deda et al. reported that one year after the implantation of hematopoietic progenitor stem cells significantly improved tested parameters in 70 percent of the treated patients, compared with their pre-operative status (Deda et al., 2009). A latest prospective, non-randomized, open label clinical trial has been completed in 2016 in Prague, Czech Republic (Sykova et al., 2017). Study evaluated the safety and the efficacy of autologous multipotent MSC in the patients with confirmed diagnosis of ALS (http://www.sukl.eu). The trial involved 26 patients with sporadic ALS, who received a single intrathecal (via a lumbar puncture) dose of autologous MSCs applied via a lumbar puncture into the cerebrospinal fluid. As compared to previous trials, this study included the largest group of ALS patients, had a longer pre-and post-treatment assessment period, and a relatively small dose of stem cells was used. A potential adverse reactions were assessed by clinical, laboratory and MR examination for 18 months. Patients underwent clinical evaluation using ALS functional rating scale (ALSFRS), Norris spinal and bulbar scale (NSS and NSB), forced vital capacity (FVC) and weakness scale (WS). This study showed that 30 percent of the patients experienced mild/moderate headache after MSCs application, not connected with the actual cell application. No suspected serious adverse reactions or new cerebrospinal pathology on MR examinations were observed. Eighty percent of patients preserved FVC values above 60 percent for 12 months. Fourteen patients (out of 26) with a remarkable pretreatment decline in functional scales, had significant reduction/stabilization in their total functional score decline at 3 months after application, which was less pronounced at 6 and 9 months.
Despite an increasing number of clinical trials proving the safety of the procedure there is a great need for bigger multicentre trials. Even though some small series of experiments involving patients showed an improvement of motor and sensory functions after the administration of stem cells, there is a need for bigger multicentre studies with placebo group of patients. The above trials that resulted with a neuroprotective effect after cell-based therapy have employed various routes of application, different type of cells, and not same ways of clinical evaluation, therefore there is a need for unification of future clinical trials design. One could also speculate that the combination of different routes of cell delivery might bring even better results related to survival and motor functions. We would like to stress once again, that current research should also employ physiological characteristics of all cells types that are supposed to be delivered into patients. This could give us more homogenous data between the trials as well as develop an algorithm that will enable prognosis of cell-based therapy in the future (Forostyak et al., 2016b). Finally, specific markers, which will enable early disease diagnosis, are of a great importance for the successful cell-based therapy, mainly because at the beginning of neurodegeneration stem cells might bring more benefits in rescuing neurones from inevitable death, if compared with the therapy at the terminal-stage of ALS.
Author Contributions
SF and ES - data collection and literature overview, manuscript writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been funded by the Grant Agency of the Czech Republic (GACR 10504P, GACR 17-21146S and GACR 15-06958S). We would like to acknowledge the Ministry of Education, Youth and Sports (MEYS, MŠMT) of the Czech Republic (Centre of Reconstruction Neuroscience—NEURORECON, CZ.02.1.01/0.0/0.0/15_003/0000419).
References
Achi, E. Y., and Rudnicki, S. A. (2012). ALS and Frontotemporal Dysfunction: a review. Neurol. Res. Int. 2012:806306. doi: 10.1155/2012/806306
Aggarwal, S., and Pittenger, M. F. (2005). Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105, 1815–1822. doi: 10.1182/blood-2004-04-1559
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Appel, S. H. (1981). A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Ann. Neurol. 10, 499–505. doi: 10.1002/ana.410100602
Aran, F. A. (1848). Research on an as yet undescribed disease of the muscular system (progressive muscular atrophy). Arch. Gén. Méd. 24, 15–35.
Bedlack, R. S., Silani, V., and Cudkowicz, M. E. (2009). IPLEX and the telephone game: the difficulty in separating myth from reality on the internet. Amyotroph. Lateral. Scler. 10, 182–184. doi: 10.1080/17482960802673059
Bento-Abreu, A., Van Damme, P., Van Den Bosch, L., and Robberecht, W. (2010). The neurobiology of amyotrophic lateral sclerosis. Eur. J. Neurosci. 31, 2247–2265. doi: 10.1111/j.1460-9568.2010.07260.x
Blanquer, M., Moraleda, J. M., Iniesta, F., Gomez-Espuch, J., Meca-Lallana, J., Villaverde, R., et al. (2012). Neurotrophic bone marrow cellular nests prevent spinal motoneuron degeneration in amyotrophic lateral sclerosis patients: a pilot safety study. Stem Cells 30, 1277–1285. doi: 10.1002/stem.1080
Borasio, G. D., and Appel, S. H. (2003). “Upper and lower motor neuron disorders,” in Neurological Disorders: Course and Treatment 2nd edn., Chapter 81, eds T. Brandt, L. R. Caplan, J. Dichgans, H. C. Diener, and C. Kennard (San Diego, CA: Academic Press), 1165–1177.
Boucherie, C., Schafer, S., Lavand'homme, P., Maloteaux, J. M., and Hermans, E. (2009). Chimerization of astroglial population in the lumbar spinal cord after mesenchymal stem cell transplantation prolongs survival in a rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 87, 2034–2046. doi: 10.1002/jnr.22038
Brody, D. L., and Holtzman, D. M. (2008). Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev. Neurosci. 31, 175–193. doi: 10.1146/annurev.neuro.31.060407.125529
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi: 10.1016/j.mcn.2013.07.007
Carulli, D., Pizzorusso, T., Kwok, J. C., Putignano, E., Poli, A., Forostyak, S., et al. (2010). Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 133(Pt 8), 2331–2347. doi: 10.1093/brain/awq145
Charcot, J. M. (1881). Lectures on the Diseases of the Nervous System. (London: New Sydenham Society).
Chen, Y., Meininger, V., and Guillemin, G. J. (2009). Recent advances in the treatment of amyotrophic lateral sclerosis. emphasis on kynurenine pathway inhibitors. Cent. Nerv. Syst. Agents Med. Chem. 9, 32–39. doi: 10.2174/187152409787601941
Chen, Y. Z., Bennett, C. L., Huynh, H. M., Blair, I. P., Puls, I., Irobi, J., et al. (2004). DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135. doi: 10.1086/421054
Choi, M. R., Kim, H. Y., Park, J. Y., Lee, T. Y., Baik, C. S., Chai, Y. G., et al. (2010). Selection of optimal passage of bone marrow-derived mesenchymal stem cells for stem cell therapy in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 472, 94–98. doi: 10.1016/j.neulet.2010.01.054
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 Hexanucleotide expansions are associated with altered ER calcium homeostasis and stress granule formation in iPSC-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal Dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Deda, H., Inci, M. C., Kurekci, A. E., Sav, A., Kayihan, K., Ozgun, E., et al. (2009). Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cytotherapy 11, 18–25. doi: 10.1080/14653240802549470
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deshpande, D. M., Kim, Y. S., Martinez, T., Carmen, J., Dike, S., and Kerr, D. A. (2006). Recovery from paralysis in adult rats using embryonic stem cells. Ann. Neurol. 60, 32–44. doi: 10.1002/ana.20901
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., et al. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. the international society for cellular therapy position statement. Cytotherapy 8, 315–317. doi: 10.1080/14653240600855905
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Forostyak, O., Butenko, O., Anderova, M., Forostyak, S., Sykova, E., Verkhratsky, A., et al. (2016a). Specific profiles of ion channels and ionotropic receptors define adipose- and bone marrow derived stromal cells. Stem Cell Res. 16, 622–634. doi: 10.1016/j.scr.2016.03.010
Forostyak, O., Forostyak, S., Kortus, S., Sykova, E., Verkhratsky, A., and Dayanithi, G. (2016b). Physiology of Ca(2+) signalling in stem cells of different origins and differentiation stages. Cell Calcium 59, 57–66. doi: 10.1016/j.ceca.2016.02.001
Forostyak, O., Romanyuk, N., Verkhratsky, A., Sykova, E., and Dayanithi, G. (2013a). Plasticity of calcium signaling cascades in human embryonic stem cell-derived neural precursors. Stem Cells Dev. 22, 1506–1521. doi: 10.1089/scd.2012.0624
Forostyak, S., Homola, A., Turnovcova, K., Svitil, P., Jendelova, P., and Sykova, E. (2014). Intrathecal delivery of mesenchymal stromal cells protects the structure of altered perineuronal nets in SOD1 rats and amends the course of ALS. Stem Cells 32, 3163–3172. doi: 10.1002/stem.1812
Forostyak, S., Jendelova, P., Kapcalova, M., Arboleda, D., and Sykova, E. (2011). Mesenchymal stromal cells prolong the lifespan in a rat model of amyotrophic lateral sclerosis. Cytotherapy 13, 1036–1046. doi: 10.3109/14653249.2011.592521
Forostyak, S., Jendelova, P., and Sykova, E. (2013b). The role of mesenchymal stromal cells in spinal cord injury, regenerative medicine and possible clinical applications. Biochimie 95, 2257–2270. doi: 10.1016/j.biochi.2013.08.004
Garbuzova-Davis, S., Sanberg, C. D., Kuzmin-Nichols, N., Willing, A. E., Gemma, C., Bickford, P. C., et al. (2008). Human umbilical cord blood treatment in a mouse model of ALS: optimization of cell dose. PLoS ONE 3:e2494. doi: 10.1371/journal.pone.0002494
Garbuzova-Davis, S., Saporta, S., Haller, E., Kolomey, I., Bennett, S. P., Potter, H., et al. (2007). Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2:e1205. doi: 10.1371/journal.pone.0001205
Gelati, M., Profico, D., Projetti-Pensi, M., Muzi, G., Sgaravizzi, G., and Vescovi, A. L. (2013). Culturing and expansion of “clinical grade” precursors cells from the fetal human central nervous system. Methods Mol. Biol. 1059, 65–77. doi: 10.1007/978-1-62703-574-3_6
Geser, F., Martinez-Lage, M., Kwong, L. K., Lee, V. M., and Trojanowski, J. Q. (2009). Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J. Neurol. 256, 1205–1214. doi: 10.1007/s00415-009-5069-7
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Glass, J. D., Boulis, N. M., Johe, K., Rutkove, S. B., Federici, T., and Feldman, E. L. (2012). Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells 30, 1144–1151. doi: 10.1002/stem.1079
Gould, T. W., and Oppenheim, R. W. (2011). Motor neuron trophic factors: therapeutic use in ALS? Brain Res. Rev. 67, 1–39. doi: 10.1016/j.brainresrev.2010.10.003
Gould, T. W., Yonemura, S., Oppenheim, R. W., Ohmori, S., and Enomoto, H. (2008). The neurotrophic effects of glial cell line-derived neurotrophic factor on spinal motoneurons are restricted to fusimotor subtypes. J. Neurosci. 28, 2131–2146. doi: 10.1523/JNEUROSCI.5185-07.2008
Gowing, G., and Svendsen, C. N. (2011). Stem cell transplantation for motor neuron disease: current approaches and future perspectives. Neurotherapeutics 8, 591–606. doi: 10.1007/s13311-011-0068-7
Habisch, H. J., Janowski, M., Binder, D., Kuzma-Kozakiewicz, M., Widmann, A., Habich, A., et al. (2007). Intrathecal application of neuroectodermally converted stem cells into a mouse model of ALS: limited intraparenchymal migration and survival narrows therapeutic effects. J. Neural Transm. 114, 1395–1406. doi: 10.1007/s00702-007-0748-y
Hadano, S., Hand, C. K., Osuga, H., Yanagisawa, Y., Otomo, A., Devon, R. S., and Ikeda, J. E. (2001). A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173. doi: 10.1038/ng1001-166
Hall, C. E., Yao, Z., Choi, M., Tyzack, G. E., Serio, A., Luisier, R., et al. (2017). Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. 19, 1739–1749. doi: 10.1016/j.celrep.2017.05.024
Hand, C. K., Khoris, J., Salachas, F., Gros-Louis, F., Lopes, A. A., Mayeux-Portas, V., et al. (2002). A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q. Am. J. Hum. Genet. 70, 251–256. doi: 10.1086/337945
Hardiman, O. (2011). Management of respiratory symptoms in ALS. J. Neurol. 258, 359–365. doi: 10.1007/s00415-010-5830-y
Howe, C. L., Bergstrom, R. A., and Horazdovsky, B. F. (2009). Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 73, 1247; author reply 1247–1248. doi: 10.1212/WNL.0b013e3181b26ae6
Johnston, C. A., Stanton, B. R., Turner, M. R., Gray, R., Blunt, A. H., Butt, D., et al. (2006). Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J. Neurol. 253, 1642–1643. doi: 10.1007/s00415-006-0195-y
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., and Rouleau, G. A. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kallur, T., Darsalia, V., Lindvall, O., and Kokaia, Z. (2006). Human fetal cortical and striatal neural stem cells generate region-specific neurons in vitro and differentiate extensively to neurons after intrastriatal transplantation in neonatal rats. J. Neurosci. Res. 84, 1630–1644. doi: 10.1002/jnr.21066
Kanning, K. C., Kaplan, A., and Henderson, C. E. (2010). Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 33, 409–440. doi: 10.1146/annurev.neuro.051508.135722
Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842. doi: 10.1126/science.1086137
Kaufmann, P., Thompson, J. L., Levy, G., Buchsbaum, R., Shefner, J., Krivickas, L. S., et al. (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 66, 235–244. doi: 10.1002/ana.21743
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2018). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, H., Kim, H. Y., Choi, M. R., Hwang, S., Nam, K. H., Kim, H. C., et al. (2010). Dose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS mice. Neurosci. Lett. 468, 190–194. doi: 10.1016/j.neulet.2009.10.074
Kondo, T., Funayama, M., Tsukita, K., Hotta, A., Yasuda, A., Nori, S., et al. (2014). Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Rep. 3, 242–249. doi: 10.1016/j.stemcr.2014.05.017
Krause, D. S. (2002). Plasticity of marrow-derived stem cells. Gene Ther. 9, 754–758. doi: 10.1038/sj.gt.3301760
Kuzuhara, S., and Kokubo, Y. (2005). Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update. Mov. Disord. 20 (Suppl. 12,) S108–S113. doi: 10.1002/mds.20548
Lacomblez, L., Bensimon, G., Leigh, P. N., Guillet, P., and Meininger, V. (1996). Dose-ranging study of riluzole in amyotrophic lateral sclerosis. amyotrophic lateral sclerosis/riluzole study group II. Lancet 347, 1425–1431. doi: 10.1016/S0140-6736(96)91680-3
Le Blanc, K. (2003). Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy 5, 485–489. doi: 10.1080/14653240310003611
Lee, H., Shamy, G. A., Elkabetz, Y., Schofield, C. M., Harrsion, N. L., Panagiotakos, G., et al. (2007). Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells 25, 1931–1939. doi: 10.1634/stemcells.2007-0097
Lepore, A. C., Rauck, B., Dejea, C., Pardo, A. C., Rao, M. S., Rothstein, J. D., et al. (2008). Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 11, 1294–1301. doi: 10.1038/nn.2210
Levy, G., Kaufmann, P., Buchsbaum, R., Montes, J., Barsdorf, A., Arbing, R., et al. (2006). A two-stage design for a phase II clinical trial of coenzyme Q10 in ALS. Neurology 66, 660–663. doi: 10.1212/01.wnl.0000201182.60750.66
Lindvall, O., and Kokaia, Z. (2006). Stem cells for the treatment of neurological disorders. Nature 441, 1094–1096. doi: 10.1038/nature04960
Lunn, J. S., Sakowski, S. A., Federici, T., Glass, J. D., Boulis, N. M., and Feldman, E. L. (2011). Stem cell technology for the study and treatment of motor neuron diseases. Regen. Med. 6, 201–213. doi: 10.2217/rme.11.6
Mahoney, C. J., Beck, J., Rohrer, J. D., Lashley, T., Mok, K., Shakespeare, T., et al. (2012). Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135(Pt 3), 736–750. doi: 10.1093/brain/awr361
Martinez, H. R., Gonzalez-Garza, M. T., Moreno-Cuevas, J. E., Caro, E., Gutierrez-Jimenez, E., and Segura, J. J. (2009). Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 11, 26–34. doi: 10.1080/14653240802644651
Mazzini, L., Fagioli, F., and Boccaletti, R. (2004). Stem-cell therapy in amyotrophic lateral sclerosis. Lancet 364, 1936–1937. doi: 10.1016/S0140-6736(04)17470-9
Mazzini, L., Fagioli, F., Boccaletti, R., Mareschi, K., Oliveri, G., Olivieri, C., et al. (2003). Stem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humans. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 158–161. doi: 10.1080/14660820310014653
Mazzini, L., Ferrero, I., Luparello, V., Rustichelli, D., Gunetti, M., Mareschi, K., et al. (2010). Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: a Phase I clinical trial. Exp. Neurol. 223, 229–237. doi: 10.1016/j.expneurol.2009.08.007
Mazzini, L., Gelati, M., Profico, D. C., Sgaravizzi, G., Projetti Pensi, M., and Vescovi, A. L. (2015). Human neural stem cell transplantation in ALS: initial results from a phase I trial. J. Transl. Med. 13, 17. doi: 10.1186/s12967-014-0371-2
Mazzini, L., Mareschi, K., Ferrero, I., Miglioretti, M., Stecco, A., Servo, S., et al. (2011). Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: a long-term safety study. Cytotherapy 4, 56–60. doi: 10.3109/14653249.2011.613929
McGeer, E. G., and McGeer, P. L. (2005). Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. Biodrugs 19, 31–37. doi: 10.2165/00063030-200519010-00004
Mezey, E., Chandross, K. J., Harta, G., Maki, R. A., and McKercher, S. R. (2000). Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science 290, 1779–1782. doi: 10.1126/science.290.5497.1779
Mustfa, N., Walsh, E., Bryant, V., Lyall, R. A., Addington-Hall, J., Goldstein, L. H., et al. (2006). The effect of noninvasive ventilation on ALS patients and their caregivers. Neurology 66, 1211–1217. doi: 10.1212/01.wnl.0000208957.88534.11
Nagano, I., Ilieva, H., Shiote, M., Murakami, T., Yokoyama, M., Shoji, M., et al. (2005). Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 235, 61–68. doi: 10.1016/j.jns.2005.04.011
Neumann, M., Galushko, M., Karbach, U., Goldblatt, H., Visser, A., Wirtz, M., et al. (2009). Barriers to using psycho-oncology services: a qualitative research into the perspectives of users, their relatives, non-users, physicians, and nurses. Support. Care Cancer 18, 1147–1156. doi: 10.1007/s00520-009-0731-2
Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Nistor, G. I., Totoiu, M. O., Haque, N., Carpenter, M. K., and Keirstead, H. S. (2005). Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49, 85–396. doi: 10.1002/glia.20127
Papadeas, S. T., and Maragakis, N. J. (2009). Advances in stem cell research for Amyotrophic Lateral Sclerosis. Curr. Opin. Biotechnol. 20, 545–551. doi: 10.1016/j.copbio.2009.09.003
Petri, S., Kiaei, M., Damiano, M., Hiller, A., Wille, E., Manfredi, G., et al. (2006). Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 98, 1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x
Petrou, P., Gothelf, Y., Argov, Z., Gotkine, M., Levy, Y. S., Karussis, D., et al. (2016). Safety and Clinical Effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 73, 337–344. doi: 10.1001/jamaneurol.2015.4321
Popescu, I. R., Nicaise, C., Liu, S., Bisch, G., Knippenberg, S., Daubie, V., et al. (2013). Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl. Med. 2, 167–174. doi: 10.5966/sctm.2012-0042
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rice, C. M., and Scolding, N. J. (2008). Autologous bone marrow stem cells–properties and advantages. J. Neurol. Sci. 265, 59–62. doi: 10.1016/j.jns.2007.06.011
Riley, J., Federici, T., Polak, M., Kelly, C., Glass, J., Raore, B., et al. (2012). Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery 71, 405–416; discussion 416. doi: 10.1227/NEU.0b013e31825ca05f
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Sapp, P. C., Hosler, B. A. D., McKenna-Yasek Chin, W., Gann, A., Genise, H., et al. (2003). Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am. J. Hum. Genet. 73, 397–403. doi: 10.1086/377158
Seminatore, C., Polentes, J., Ellman, D., Kozubenko, N., Itier, V., Tine, S., et al. (2010). The postischemic environment differentially impacts teratoma or tumor formation after transplantation of human embryonic stem cell-derived neural progenitors. Stroke 41, 153–159. doi: 10.1161/STROKEAHA.109.563015
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Sorenson, E. J., Windbank, A. J., Mandrekar, J. N., Bamlet, W. R., Appel, S. H., Armon, C., et al. (2008). Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 71, 1770–1775. doi: 10.1212/01.wnl.0000335970.78664.36
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Steele, J. C. (2005). Parkinsonism-dementia complex of Guam. Mov. Disord. 20 (Suppl. 12), S99–S107. doi: 10.1002/mds.20547
Sun, S., Sun, Y., Ling, S. C., Ferraiuolo, L. M., and McAlonis-Downes Zou, Y. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U.S.A. 112, E6993–E7002. doi: 10.1073/pnas.1520639112
Sykova, E., Rychmach, P., Drahoradova, I., Konradova, S., Ruzickova, K., Vorisek, I., et al. (2017). Transplantation of mesenchymal stromal cells in patients with amyotrophic lateral sclerosis: results of phase I/IIa clinical trial. Cell Transplant. 26, 647–658. doi: 10.3727/096368916X693716
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tropel, P., Platet, N., Platel, J. C., Noel, D., Albrieux, M., Benabid, A. L., et al. (2006). Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells 24, 2868–2876. doi: 10.1634/stemcells.2005-0636
Urushitani, M., Ezzi, S. A., and Julien, J. P. (2007). Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 104, 2495–2500. doi: 10.1073/pnas.0606201104
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Van Den Bosch, L., Storkebaum, E., Vleminckx, V., Moons, L., Vanopdenbosch, L., Scheveneels, W., et al. (2004). Effects of vascular endothelial growth factor (VEGF) on motor neuron degeneration. Neurobiol. Dis. 17, 21–28. doi: 10.1016/j.nbd.2004.06.004
van Es, M. A., Dahlberg, C., Birve, A., Veldink, J. H., van den Berg, L. H., and Andersen, P. M. (2010). Large-scale SOD1 mutation screening provides evidence for genetic heterogeneity in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatr. 81, 562–566. doi: 10.1136/jnnp.2009.181453
Vercelli, A., Mereuta, O. M., Garbossa, D., Muraca, G., Mareschi, K., Rustichelli, D., et al. (2008). Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 31, 395–405. doi: 10.1016/j.nbd.2008.05.016
Wang, R., and Zhang, D. (2005). Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur. J. Neurosci. 22, 2376–2380. doi: 10.1111/j.1460-9568.2005.04431.x
Wang, Y., Mao, X. O., Xie, L., Banwait, S., Marti, H. H., Greenberg, D. A., et al. (2007). Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J. Neurosci. 27, 304–307. doi: 10.1523/JNEUROSCI.4433-06.2007
Wichterle, H., Lieberam, I., Porter, J. A., and Jessell, T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. doi: 10.1016/S0092-8674(02)00835-8
Wichterle, H., and Przedborski, S. (2010). What can pluripotent stem cells teach us about neurodegenerative diseases? Nat. Neurosci. 13, 800–804. doi: 10.1038/nn.2577
Xu, L., Ryugo, D. K., Pongstaporn, T., Johe, K., and Koliatsos, V. E. (2009). Human neural stem cell grafts in the spinal cord of SOD1 transgenic rats: differentiation and structural integration into the segmental motor circuitry. J. Comp. Neurol. 514, 297–309. doi: 10.1002/cne.22022
Keywords: stem cells, neurodegeneration, neuroprotection, clinical trials
Citation: Forostyak S and Sykova E (2017) Neuroprotective Potential of Cell-Based Therapies in ALS: From Bench to Bedside. Front. Neurosci. 11:591. doi: 10.3389/fnins.2017.00591
Received: 01 September 2017; Accepted: 09 October 2017;
Published: 24 October 2017.
Edited by:
Kim A. Staats, University of Southern California, United StatesReviewed by:
Alba Di Pardo, Centre for Neurogenetics and Rare Diseases, ItalyCorinne Lasmezas, Scripps Florida, United States
Copyright © 2017 Forostyak and Sykova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serhiy Forostyak, Zm9yb3N0eWFrc0BnbWFpbC5jb20=