 Matthew J. Fogarty
Matthew J. Fogarty Erica W. H. Mu
Erica W. H. Mu Nickolas A. Lavidis
Nickolas A. Lavidis Peter G. Noakes
Peter G. Noakes Mark C. Bellingham
Mark C. Bellingham- 1Faculty of Medicine, School of Biomedical Sciences, University of Queensland, St Lucia, QLD, Australia
- 2Queensland Brain Institute, University of Queensland, St Lucia, QLD, Australia
Objective: Motor neurons (MNs) die in amyotrophic lateral sclerosis (ALS), a clinically heterogeneous neurodegenerative disease of unknown etiology. In human or rodent studies, MN loss is preceded by increased excitability. As increased neuronal excitability correlates with structural changes in dendritic arbors and spines, we have examined longitudinal changes in dendritic structure in vulnerable neuron populations in a mouse model of familial ALS.
Methods: We used a modified Golgi-Cox staining method to determine the progressive changes in dendritic structure of hippocampal CA1 pyramidal neurons, striatal medium spiny neurons, and resistant (trochlear, IV) or susceptible (hypoglossal, XII; lumbar) MNs from brainstem and spinal cord of mice over-expressing the human SOD1G93A (SOD1) mutation, in comparison to wild-type (WT) mice, at four postnatal (P) ages of 8–15, 28–35, 65–75, and 120 days.
Results: In SOD1 mice, dendritic changes occur at pre-symptomatic ages in both XII and spinal cord lumbar MNs. Spine loss without dendritic changes was present in striatal neurons from disease onset. Spine density increases were present at all ages studied in SOD1 XII MNs. Spine density increased in neonatal lumbar MNs, before decreasing to control levels by P28-35 and was decreased by P120. SOD1 XII MNs and lumbar MNs, but not trochlear MNs showed vacuolization from the same time-points. Trochlear MN dendrites were unchanged.
Interpretation: Dendritic structure and spine alterations correlate with the neuro-motor phenotype in ALS and with cognitive and extra-motor symptoms seen in patients. Prominent early changes in dendritic arbors and spines occur in susceptible cranial and spinal cord MNs, but are absent in MNs resistant to loss in ALS.
Introduction
In amyotrophic lateral sclerosis (ALS) both upper and lower motor neurons (MNs), as well as the corticospinal tract, undergo progressive degeneration (Charcot and Joffroy, 1869; Cleveland and Rothstein, 2001). This loss of MNs in the brainstem and spinal cord inevitably causes worsening muscular atrophy and weakness, typically causing death 3–5 years following diagnosis (Cleveland and Rothstein, 2001; Hardiman et al., 2011), usually from respiratory complication or failure (Connolly et al., 2015). The Golgi-Cox technique has demonstrated significant structural changes, including dendritic shortening and decreased spine density, in upper and lower motor neurons of ALS patients and animal models of familial ALS (Hammer et al., 1979; Horoupian et al., 1984; Udaka et al., 1986; Kato et al., 1987; Fogarty et al., 2016c). These structural abnormalities are correlated with cortical hyper-excitability (Eisen et al., 1993; Mills and Nithi, 1997; Zanette et al., 2002; Turner et al., 2005; Vucic and Kiernan, 2006; Fogarty et al., 2015, 2016b), which can be observed before diagnosis in certain ALS patients (Vucic et al., 2008), suggesting that protracted preclinical structure/function alterations play a key role in disease pathogenesis (van Zundert et al., 2012; Eisen et al., 2014).
The dendritic structure of the neuron integrates the diverse stream of excitatory, inhibitory and neuromodulatory synaptic inputs, which are the key determinants of whether an action potential is generated (Luebke et al., 2010). Regulation of dendrite length and the density of dendritic spines normally occurs during brain development and aging, in addition to many psychiatric or neurodegenerative pathologies, and these changes are correlated with alteration in neurotransmitter activity (Luebke et al., 2010; Sala and Segal, 2014). In particular, glutamatergic neurotransmission is thought to be a significant regulator of dendritic structure, and glutamatergic excitotoxicity is one proposed mechanism in ALS pathogenesis (Eisen et al., 1993; Mills and Nithi, 1997; van Zundert et al., 2008, 2012; Bogaert et al., 2010; Bellingham, 2011; Turner et al., 2013; Devlin et al., 2015; Fogarty et al., 2015, 2016b; Saba et al., 2015).
The hSOD1G93A transgenic mouse (SOD1) is the most prevalent animal model of ALS, overexpressing the human SOD1G93A mutation (Gurney et al., 1994; Turner and Talbot, 2008). SOD1 mutations are found in 10–20% of familial ALS and in 1–2% of sporadic ALS cases, thus accounting for about 2% of all ALS cases (Taylor et al., 2016). This SOD1 mouse model of familial ALS shows significant alterations in dendritic structure, including the density of dendritic spines, of primary motor cortex upper MNs (Spalloni et al., 2011; Jara et al., 2012; Fogarty et al., 2015, 2016b; Saba et al., 2015), pyramidal neurons of medial pre-frontal cortex (MPFC) (Sgobio et al., 2008; Fogarty et al., 2016c), and lower MNs in the brainstem and spinal cord (van Zundert et al., 2008; Martin et al., 2013). Although these studies have provided insights into individual components of the neuro-motor network at specific time points in disease pathogenesis, there remains a need to define the onset and progression of neuronal pathology at successive stages of disease in ALS in key subcortical structures receiving cortical outputs: the striatum, hippocampus (Spalloni and Longone, 2015), brainstem, and spinal cord.
Our results show that dendritic atrophy and spine loss in striatal medium spiny neurons (MSNs) and lower MNs of lumbar spinal cord contrast starkly with our observations of increased dendritic arbor and spine density in brainstem hypoglossal (XII) MNs and unchanged morphology of trochlear (IV) MNs, a brainstem lower MN population which is resistance to degeneration in the SOD1 mouse (Ferrucci et al., 2010). Dendritic vacuolisation of SOD1 XII MNs and lumbar MNs occurred from the same time-point, while IV MNs did not show significant vacuolization or dendritic changes at any time point studied. These data may hint at independent mechanisms of degeneration between these MN populations and other upper and lower MNs. In addition to dendritic changes in the lower MN populations severely affected by ALS (Hammer et al., 1979; Horoupian et al., 1984; Udaka et al., 1986; Kato et al., 1987; Cleveland and Rothstein, 2001; Aggarwal and Nicholson, 2002; Hardiman et al., 2011; Turner et al., 2013; Eisen et al., 2014), we also observed hitherto unreported degenerative changes in neurons within the dorsal striatum.
Materials and Methods
Ethics Statement
Fifty two age- and litter-matched wild-type (WT) and heterozygous transgenic mice overexpressing the hSOD1G93A mutation were utilized. This study was conducted following the guidelines of the Queensland Government Animal Research Act 2001, associated Animal Care and Protection Regulations (2002 and 2008), as well as the Australian Code for the Care and Use of Animals for Scientific Purposes, 8th Edition (National Health and Medical Research Council, 2013). The University of Queensland Anatomical Biosciences Animal Ethics Committee approved all protocols involving animal use.
Golgi-Cox Impregnation and Processing
Mice from four previously characterized phenotypic stages were used—(1) postnatal (P) days P8-15 (7 WT and 5 SOD1 mice), when lower MNs show early signs of intrinsic and synaptic hyper-excitability (van Zundert et al., 2008; Fogarty et al., 2015); (2) P28-25 (5 WT and 5 SOD1 mice), pre-symptomatic stage when mice do not show discernable symptoms but do show synaptic and structural changes in neurons (Sgobio et al., 2008; van Zundert et al., 2008; Ozdinler et al., 2011; Fogarty et al., 2015; Saba et al., 2015); (3) P65-75 (5 WT and 5 SOD1 mice), representing disease onset, with the earliest period of MN loss, muscle weakness, and appearance of overt symptoms (Ozdinler et al., 2011; Jara et al., 2012; Lee et al., 2013; Steyn et al., 2013) and; (4) P110-130 (8 WT and 12 SOD1 mice, a mid-disease stage where mice show marked muscle mass and loss of muscle strength, respectively (Lee et al., 2013). In our SOD1 colony, end stage disease (loss of righting reflex) occurs between 150 and 175 days (Ngo et al., 2012).
Mice were anesthetized with sodium pentobarbitone (60–80 mg/kg intraperitoneally, Vetcare) and then a heparinized needle (Sigma-Aldrich) was used to intracardially exsanguinate the animal. Whole brain, brainstem, and lumbar spinal cord tissues were incubated for 5 days at 37°C in the dark in a modified rapid Golgi-Cox solution that contained 5% potassium dichromate, 5% potassium chromate, 5% mercuric chloride, as described previously (Rutledge et al., 1969) (all chemicals Sigma-Aldrich).
Brain slices (300 μm thickness) were cut with a vibrating Zeiss Hyrax V50 microtome (Carl Zeiss) to produce off-coronal (15° rostral rotation) forebrain sections, transverse brainstem sections, and parasagittal lumbar spinal cord sections (embedded in 10% agarose block in 0.1 M phosphate buffered saline [PBS]). Serial sections in 24-well plates were sequentially incubated in 30% sucrose in 0.1 M PBS (30 min), 50% ethanol (dehydration, 5 min), 0.1 M ammonium hydroxide solution (30 min, rinsed twice in distilled water (5 min), and then immersed in aluminum sulfate-based Fujihunt photo fixer (Fuijfilm, Singapore, 30 min) while protected from light exposure. After rinsing in distilled water twice (5 min each), sections were dehydrated in ethanol (70, 90, 95, and three rinses of 100%, 5 min each) and transferred to chloroform: xylene: alcohol (CXA) solution (1:1:1 ratio, 10 min). Sections were cleared in xylene (2 × 5 min each) and mounted using DPX (Sigma-Aldrich) on Superfrost Plus (Lomb Menzel Glaser) slides. Slides were air dried and stored in the dark. We note that aged mice brain tissue required longer xylene (extra 5 min) clearing due to a higher fat content.
Region Selection
Dorsal striatum neurons for tracing were selected from sections between bregma ~1.9 and 0.6 mm, within the striatum, which was located lateral to the lateral ventricle and ventral and medial to the corpus callosum. Hippocampal CA1 pyramidal neurons for tracing were selected from sections between bregma ~−1.1 and −2.6 mm. A mouse brain atlas (Franklin and Paxinos, 2008) was used to help define the boundaries of these areas.
Neuronal Tracing
Morphological properties (dendritic branching, length and dendritic spines) of Golgi-impregnated neurons were traced with a 63x objective (NA 1.4) of an Axioskop 2 microscope (Carl Zeiss) with an automated z-stage controlled by Neurolucida™ software (MBF Bioscience Inc.) as for previous reports (Fogarty et al., 2015, 2016c; Kanjhan et al., 2015). We classified small processes as spines only if they met criteria of <3 μm length and <0.8 μm cross-sectional diameter (Harris, 1999; Fogarty et al., 2015). These tracings yielded: (i) soma (irregular ellipsoid) volume (based on cross-sectional areas as previously; Kanjhan et al., 2015); (ii) total arbor length (the summed dendritic arbor length); (iii) apical or basal length (summed single apical tree length or summed length of all basal trees); (iv) mean basal tree or mean dendritic length (the mean length of each basal or dendritic tree from the soma); (v) apical and basal reach (farthest linear distance between apical or basal dendrite ending and soma); (vi) apical and basal ramifications (number of bifurcations before dendritic termination, a measure of neuronal complexity); and (vii) apical or basal dendritic spine density (number of dendritic spines per 100 μm of dendrite), as defined in our previous study (Fogarty et al., 2015; Kanjhan et al., 2015). In total, 1.34 m of dendrite length were traced in the 404 neurons that were used in this study.
All analyzed pyramidal neurons had a minimum of three intact basal dendritic trees and an apical dendrite that reached dendritic termination without exiting the brain section (Klenowski et al., 2015). Dendritic spines were assessed along the entire apical or basal dendritic arbor, as for previous studies (Fogarty et al., 2016c). Striatal MSNs were identified as for previous studies (Rafols and Fox, 1979), with spine densities of >10 spines/μm (Klenowski et al., 2016) required for inclusion, as aspiny neurons with similar shaped dendritic trees occur in rodent striatum (Rafols and Fox, 1979). The IV and XII motor nucleus were identified with a mouse brain atlas, relative to major landmarks including the aqueduct, medial longitudinal fasciculus and central canal (Franklin and Paxinos, 2008), with MNs selected by their large soma and multipolar dendrites. Lumbar MNs were identified as large neurons with longitudinal soma lengths >25 μm and dendrites arranged rostro-caudally, as for past studies (Bellinger and Anderson, 1987).
Imaging
Photomicrographs of dendritic arbors were generated from minimum intensity z-stack projections (z-step size of 2 μm between images; stack depth between 20 and 100 images) that were joined digitally to form mosaics of complete cells. The imaged area was estimated from the overlaid tracing file. The mosaics appear “tessellated” because of different individual z-stack thicknesses. Representative images of spine density were taken with a 100x objective (Carl Zeiss) image at a single focal plane.
Statistical Methods
Prism 6 (Graphpad) was used for all statistical analysis. Data are given as mean ± SEM and represent the mean of all single neurons from an individual brain, brainstem or spinal cord region per mouse i.e., the n used for statistical purposes was an individual animal as the independent variable, as consistent with previous reports using the Golgi technique (Fogarty et al., 2016c). For the striatum, CA1, and brainstem regions, a minimum of two thick sections was used. The number of individual neuronal observations that contributed to these summary data is reported in the relevant tables, with a priori exclusion criteria, namely, individual data points from neurons that were beyond two standard deviations from the mean. Data was assessed for normality using a D'Agostino and Pearson omnibus test. Two-way ANOVAs with Bonferroni's post-test were applied to data with significant differences due to genotype were accepted as statistically significant with adjusted P (adj. P) values *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Percentage changes are reported in relation to the WT mean for significantly different age groups. The researcher performing tracings (MJF) was blind to genotype.
Results
Spine Density Loss Occurs from P65-75 Onward in Striatum MSNs from SOD1 Mice
The soma volume, total arbor length, mean tree length, dendritic reach or dendritic ramifications of striatum MSNs in SOD1 mice did not differ from WT MSNs at any age (Table 1, Figure 1). By contrast, SOD1 MSNs showed significantly decreased dendritic spine density from P65-75 onward (Figures 1C–F,H). While dendritic spine density of SOD1 and WT striatal MSNs was not significantly different at P8-16 or P28-35 (Table 1, Figure 1H), the dendritic spine density of SOD1 MSNs was reduced by 40% at P65-75 (adj. P = 0.0082**, Table 1, Figure 1H), and by 33% at P120 (adj. P = 0.043*, Table 1, Figure 1H).
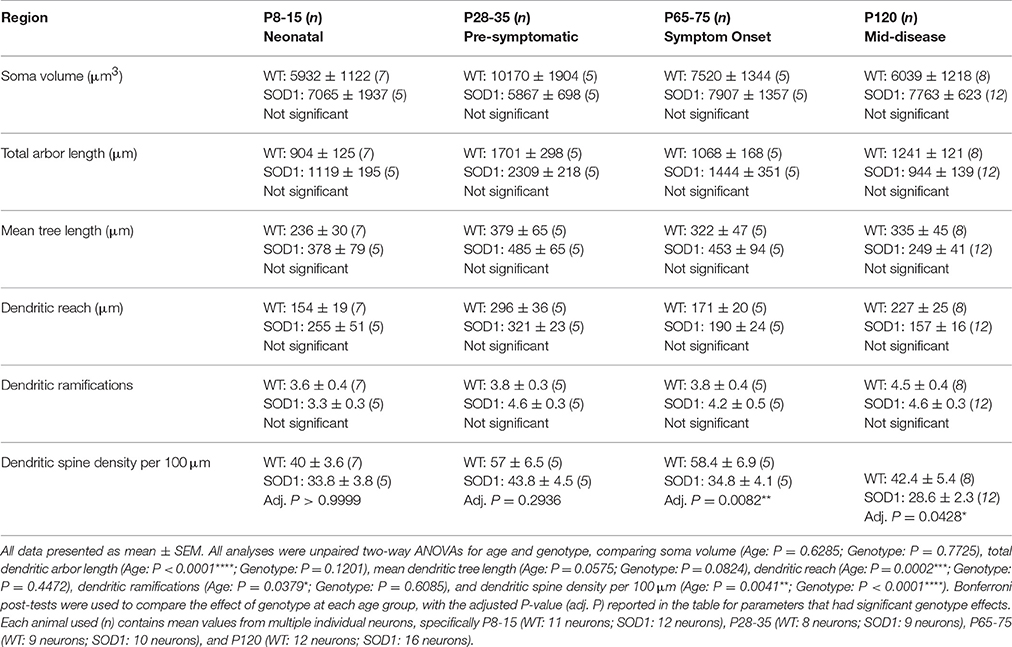
Table 1. Morphometric dendritic and dendritic spine parameters of MSNs within the striatum.
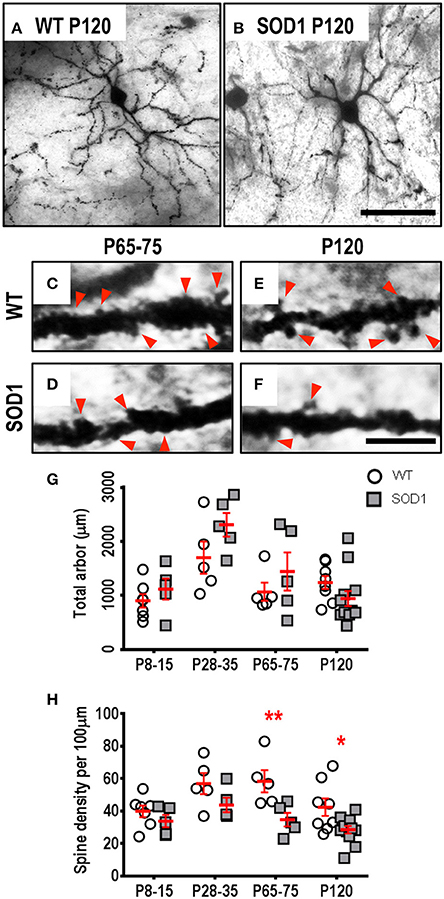
Figure 1. Decreased dendritic spine density of MSNs within the striatum commencing from P65-75 in SOD1 mice, compared to WT controls. Images show mosaics of striatum MSNs from P120 WT (A) and SOD1 (B) mice, note our representative image represents the mean MSN dendritic length in 7 of 12 P120 SOD1 mice was decreased compared to the lowest WT value at mid-disease ages. High-magnification images of dendrites from P65-75 WT (C), SOD1 (D), and P120 WT (E), and SOD1 (F) mice MSNs are also shown, with examples of some of the dendritic spines identified using red arrows, representative of the spiny structures and not an exhaustive identification of all quantified spines on these images. (G) shows a scatterplot quantifying unchanged total dendritic arbor length (μm) of SOD1 MSNs (gray squared), compared to WT controls (white circles). (H) shows a scatterplot quantifying significantly decreased dendritic spine density per 100 μm dendrite of SOD1 MSNs (gray squared) compared to WT controls (white circles) at P65-75 and P120. All data mean ± SEM, with two-way ANOVAs followed by Bonferroni post-tests, *P < 0.05 and **P < 0.01. n = 7, 5, 5, and 8 for WT P8-15, P28-35, P65-75, and P120 respectively. n = 6, 5, 5, and 12 for SOD1 P8-15, P28-35, P65-75, and P120 respectively. Scale bar: (A,B), 75 μm. Dendritic frame width (C–F), 20 μm.
Spine loss in MSNs has not been reported in any ALS rodent model. Corticostriatal inputs from motor and prefrontal cortex are critical for striatal regulation of goal-directed movements (Anderson et al., 2010; Rothwell et al., 2015). TDP-43 positive-inclusions occur in the striatum of ALS patients (Zhang et al., 2008), and spine density loss and synaptic hyper-activity in the SOD1 mouse motor cortex precedes dendritic shrinkage (Jara et al., 2012; Yasvoina et al., 2013; Fogarty et al., 2015, 2016c). Thus, increased corticostriatal output may contribute to MSN synaptic dysfunction, which may be involved in extrapyramidal symptoms of ALS.
SOD1 Hippocampal CA1 Pyramidal Neurons Show No Morphological Differences to WT Neurons at Any Age
While morphological changes in motor and prefrontal cortex of SOD1 mice occurs well before any disease phenotype (Jara et al., 2012; Yasvoina et al., 2013; Fogarty et al., 2015, 2016c), entorhinal cortex pyramidal neurons of SOD1 mice are unaltered (Fogarty et al., 2016c). The hippocampal CA1 region is a major output target of the entorhinal cortex, which also processes inputs from multiple cortical areas (van Groen et al., 2003) and is associated with cognitive deficits in clinical ALS (Strong et al., 2009). We therefore measured the dendritic arbors and dendritic spine densities of hippocampal CA1 pyramidal neurons, and found that there were no differences in any morphological measurement of hippocampal CA1 pyramidal neurons of SOD1 mice compared to WT controls at any age (Table 2). This lack of hippocampal pathology in SOD1 mice accords with the rarity of cognitive dysfunction in ALS patients with SOD1 mutations (Wicks et al., 2009).
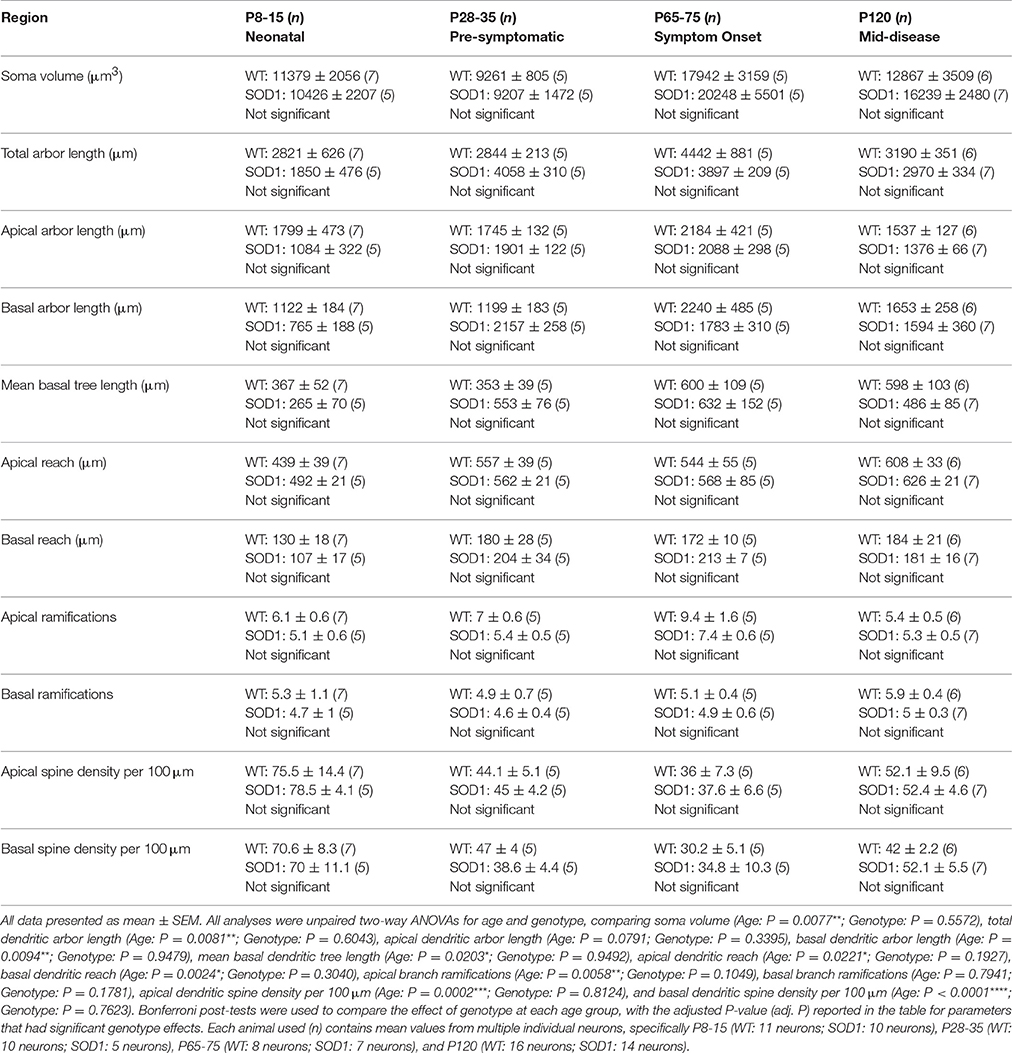
Table 2. Morphometric dendritic and dendritic spine parameters of pyramidal neurons within the hippocampus CA1.
Brainstem SOD1 XII MNs Show an Early Increase in Dendritic Arbor Length, Followed by a Later Decrease in Dendritic Arbor Length
Approximately one third of ALS patients show bulbar deficits as their first symptom, evident as impaired speech or swallowing that can lead to malnutrition, choking, and death (Fujimura-Kiyono et al., 2011). Synaptic and intrinsic hyper-excitability of XII MNs occurs in neonatal SOD1 mice (van Zundert et al., 2008), and oral motor deficits also occur in ALS rodent models (Smittkamp et al., 2008). At P8-15 in SOD1 XII MNs, the total arbor length was increased by 78%, compared to WT MNs (P = 0.015*, Table 3, Figure 2Q). This increase persisted at a lower level of 20% at P28-35 in SOD1 XII MNs, compared to WT MNs (P = 0.021*, Table 3, Figure 2Q). By P65-75, the total arbor length of SOD1 XII MNs was the same as WT controls (P = 0.48, Table 3, Figure 2Q). By P120, total arbor length of SOD1 XII MNs was reduced by 54%, compared to WT XII MNs (P < 0.0001****, Table 3, Figure 2Q). This pattern of changes was repeated for the mean dendritic tree length. Compared to WT XII MNs, the mean tree length of SOD1 XII MNs SOD1 XII MNs was increased by 51% at P8-15 increased (P < 0.009**, Table 3, Figure 2R), increased by 60% at P28-35 (P < 0.0005***, Table 3, Figure 2R), not different at P65-75 (Table 3, Fig. 2 Q), and decreased by 46% decrease at P120 (P < 0.0001****, Table 3, Figure 2R). The soma volume of XII MNs was unchanged between genotypes at all ages studied (Table 3). There was also no significant difference in the dendritic reach or dendritic ramifications of XII MNs of SOD1 and WT mice for any age studied (Table 3).
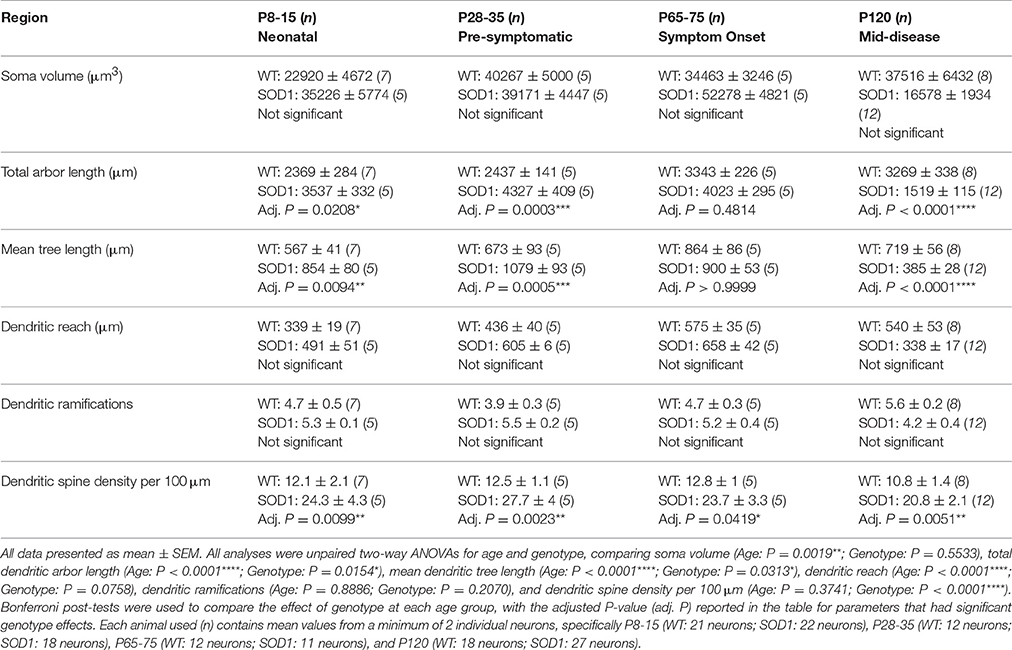
Table 3. Morphometric dendritic and dendritic spine parameters of XII MNs within the brainstem.
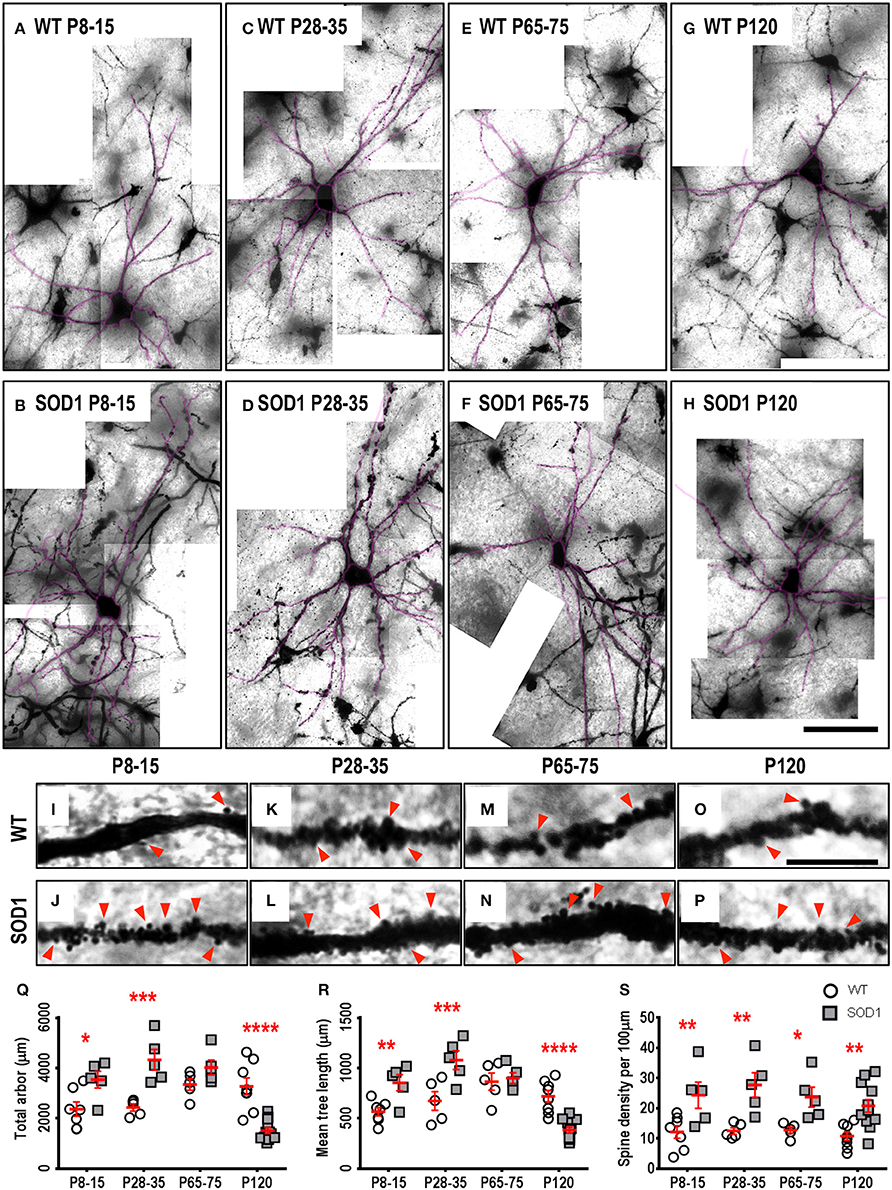
Figure 2. Altered dendritic arbors and increased spine density of SOD1 XII MNs commencing from P8-15 compared to WT controls. Images show mosaics of XII MNs from P8-15, P28-35, P65-75, and P120 WT (A,C,E,G) and SOD1 (B,D,F,H) mice brainstem. Superimposed in purple are dendritic tracings from the XII MN in order to illustrate more clearly the dendritic arbor size differences. High-magnification images of dendrites from P8-15, P28-35, P65-75, and P120 WT (I,K,M,O) and SOD1 (J,L,N,P) XII MNs are also shown, with examples of some of the dendritic spines identified using red arrows. Note that this is representative of the spiny structures and not an exhaustive identification of all quantified spines on these images. (Q) shows a scatterplot quantifying significantly increased total dendritic length (μm) of SOD1 XII MNs (gray squared) at P8-15 and 28-35 and significant decreases at P120 compared to WT controls (white circles). (R) shows a scatterplot quantifying significantly increased mean tree dendritic length (μm) of SOD1 XII MNs (gray squared) at P8-15 and 28-35 and significant decreases at P120 compared to WT controls (white circles). (S) shows a scatterplot quantifying significantly increased dendritic spine density per 100 μm of SOD1 XII MNs (gray squared) compared to WT controls (white circles) at P8-15, P28-35, P65-75, and P120. All data mean ± SEM, with two-way ANOVAs followed by Bonferroni post-tests, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. n = 7, 5, 5, and 8 for WT P8-15, P28-35, P65-75, and P120 respectively. n = 6, 5, 5, and 12 for SOD1 P8-15, P28-35, P65-75, and P120 respectively. Scale bar: (A–H), 100 μm. Dendritic frame width (I–P), 15 μm.
Brainstem SOD1 XII MNs Show Increased Dendritic Spine Density from P8-15 Until P120
The dendritic spine is the postsynaptic compartment for excitatory synapses (Harris, 1999), and is present on the dendrites of XII MNs in neonatal WT mice (Kanjhan et al., 2015). Compared to WT littermates, the dendritic spine density of XII MNs from SOD1 mice was increased at all ages (Table 3, Figure 2). At P8-15 in SOD1 XII MNs, SOD1 XII MN spine density was increased by 101% (P = 0.0099**, Table 3, Figures 2I,J,S); at P28-35, spine density was increased by 122% (P = 0.0023**, Table 3, Figures 2K,L,S); at P65-75, spine density was increased by 85% (P = 0.042*, Table 3, Figures 2M,N,S); and at P120 (P = 0.0051**, Table 3, Figures 2O,P,S), spine density was increased by 93%, compared to WT controls at the same ages (Table 3, Figure 2S).
Lumbar Spinal Cord MNs of SOD1 Mice Show Reductions in Dendritic Length from P28-35 Onward
As respiratory MNs (which include XII MNs) tend to degenerate later than spinal cord MNs innervating limb muscles (Haenggeli and Kato, 2002; Ngo et al., 2012; Lee et al., 2013), we determined whether the dendritic changes in XII MNs were reiterated in hindlimb MNs of the lumbar spinal cord MNs of SOD1, compared to WT mice. By contrast to the early increase in dendritic length seen in XII MNs from neonatal or pre-symptomatic SOD1 mice, the dendritic arbors of SOD1 lumbar MNs did not increase, but were decreased from P28-35 onward, compared to WT controls (Figure 3). Compared to WT controls, the total arbor length of SOD1 lumbar MNs was unchanged at P8-15 (Table 4, Figure 3R), decreased by 43% at P28-35 (P = 0.023*, Table 4, Figure 3R), decreased by 51% at P65-75 (P = 0.0001***, Table 4, Figure 3R), and decreased by 64% at P120 (P < 0.0001****, Table 4, Figure 3R). This pattern was repeated for mean dendritic tree lengths of SOD1 lumbar MNs, compared to WT controls, being unchanged at P8-15 (Table 4, Figure 3S), decreased by 37% at P28-35 (P = 0.0073**, Table 4, Figure 3S), decreased by 44% at P65-75 (P = 0.0009***, Table 4, Figure 3S), and decreased by 65% at P120 (P < 0.0001****, Table 4, Figure 3S). A similar pattern was seen for changes in dendritic reach, which was unchanged for P8-15 SOD1 lumbar MNs (P > 0.9999, Table 4), decreased at P28-35 by 31% (P = 0.033*, Table 4), decreased at P65-75 by 32% (P = 0.0032**, Table 4), and further decreased at P120 by 46% (P < 0.0001****, Table 4), compared to WT controls at the same ages.
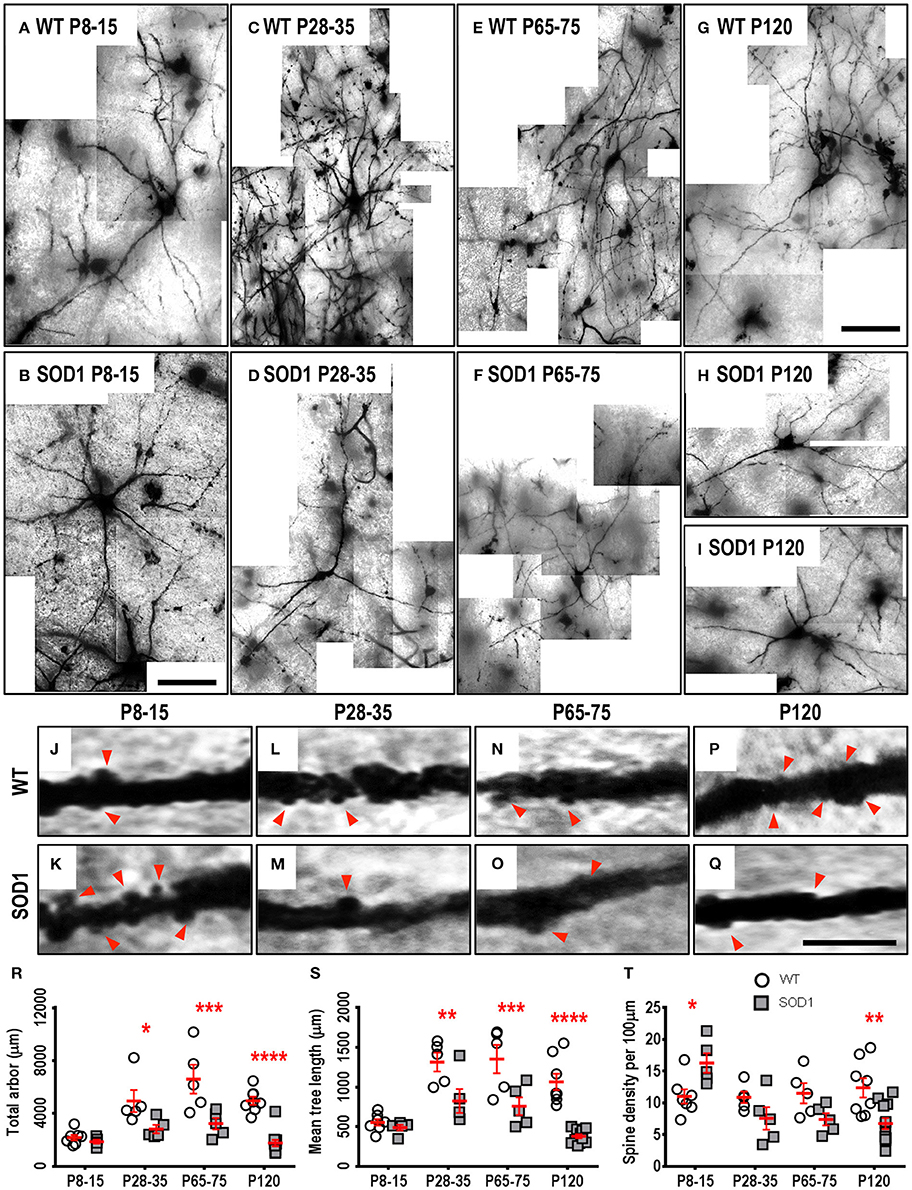
Figure 3. Decreased dendritic arbors of SOD1 lumbar MNs from P28-35 onward, and altered spine density from P8-15 onward, compared to WT controls. Images show mosaics of lumbar MNs from P8-15, P28-35, P65-75, and P120 WT (A,C,E,G) and SOD1 (B,D,F,H,I) spinal cords. High-magnification images of dendrites from P8-15, P28-35, P65-75, and P120 WT (J,L,N,P) and SOD1 (K,M,O,Q) lumbar MNs are also shown, with examples of some of the dendritic spines identified using red arrows. Note that this is representative of the spiny structures and not an exhaustive identification of all quantified spines on these images. (R) shows a scatterplot quantifying significantly decreased total dendritic length (μm) of SOD1 lumbar MNs (gray squared) compared to WT controls (white circles) at P28-35, P65-75, and P120. (S) shows a scatterplot quantifying significantly decreased mean tree dendritic length (μm) of SOD1 lumbar MNs (gray squared) compared to WT controls (white circles) at P28-35, P65-75, and P120. (T) shows a scatterplot quantifying significantly increased dendritic spine density per 100 μm of SOD1 lumbar MNs (gray squared) at P8-16 and significant decreases at P120 compared to WT controls (white circles). All data mean ± SEM, with two-way ANOVAs followed by Bonferroni post-tests, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. n = 7, 5, 5, and 8 for WT P8-15, P28-35, P65-75, and P120 respectively. n = 6, 5, 5, and 12 for SOD1 P8-15, P28-35, P65-75, and P120 respectively. Scale bar: (A,B), 75 μm. (C–I), 150 μm. Dendritic frame width, 20 μm (J–Q).
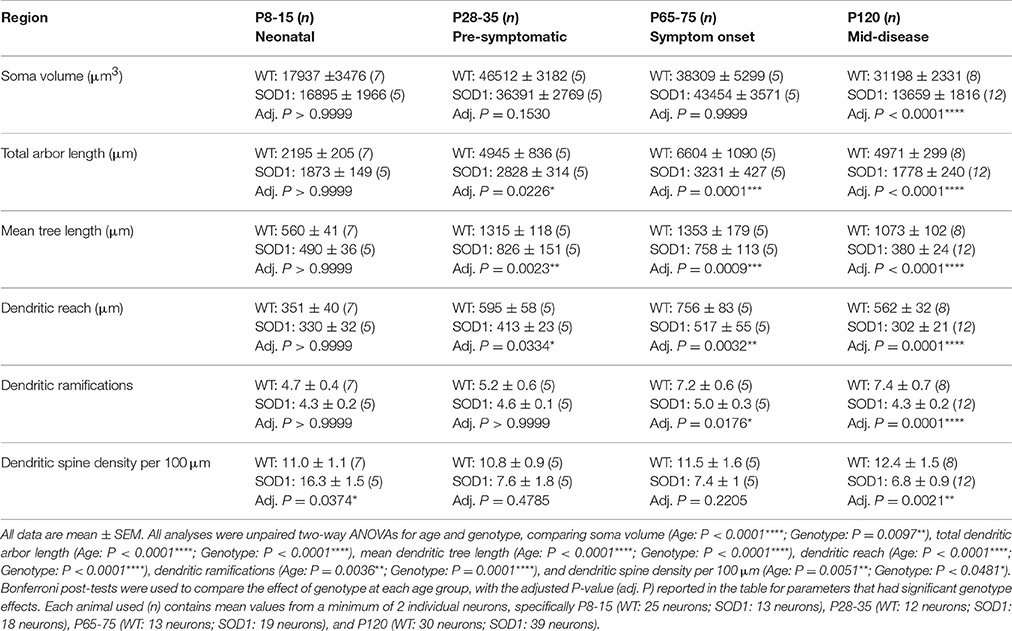
Table 4. Morphometric dendritic and dendritic spine parameters of MNs in the lumbar spinal cord.
The branching of SOD1 lumbar MN dendrites was also reduced at later disease stages. At P8-15 and P28-35, the dendritic ramifications of SOD1 lumbar MNs were unchanged, compared to WT controls (Table 4), while dendritic ramifications decreased by 31% at P65-75 (P = 0.018*, Table 4), and then declined further to 42% at P120 in SOD1 lumbar MNs (P < 0.0001****, Table 4), compared to WT controls at the same ages.
Finally, SOD1 lumbar MN soma volume was unchanged for the three earliest age groups (Table 4). However, by P120, the soma volume of SOD1 lumbar MNs, was reduced by 56%, compared to WT controls (P < 0.0001****, Table 4).
Dendritic Spine Density of SOD1 Lumbar MNs Is Increased at P8-15, Followed by a Decrease in Dendritic Spine Density by P120
We next determined whether reduced dendritic arbors of SOD1 lumbar MNs were associated with decreases in dendritic spine density, similar to changes in motor cortex and medial prefrontal cortex pyramidal neurons of SOD1 mice (Sgobio et al., 2008; Jara et al., 2012; Fogarty et al., 2015, 2016c) or whether dendritic spines were increased, as for XII MNs. Compared to WT controls, dendritic spine density of SOD1 lumbar MNs increased by 48% at P8-15 (P = 0.037*, Table 4, Figures 3J,K,T), then returned to control levels at P28-35 and P65-75 (Table 4, Figures 3L–O,T), and decreased by 45% at P120 compared to WT controls by (P = 0.0021**, Table 4, Figures 3P,Q,T). Thus, both XII MNs and lumbar MNs had increased dendritic spine densities at P8-16, compared to WT controls. However, while spine density in lumbar MNs then returned to control levels at P28-25 and P65-75, and then decreased at P120, spine density remained elevated in XII MNs at all ages studied. Elevated spine density could be thus interpreted as a neuroprotective response, as lumbar MNs are beginning to die by P30-35 in SOD1 mice (Ngo et al., 2012; Vinsant et al., 2013), while XII MN loss occurs later, at P90-100 (Haenggeli and Kato, 2002).
Brainstem IV SOD1 MNs Show No Dendritic or Spine Differences Compared to WT Controls at Any Age Studied
Some motor neuron pools are relatively resistant to death in ALS, such as those innervating the external muscles of the eye (Ferrucci et al., 2010). We therefore assessed the dendritic arbor and dendritic spine densities of trochlear (IV) MNs in the brainstem. There was no morphological changes in soma volume, total arbor length, mean tree length, dendritic reach, dendritic ramifications, or dendritic spine density of IV MNs from SOD1 mice, compared to WT IV MNS, for any age studied (Table 5). This lack of morphological change in IV MNs of SOD1 mice fits with the relative sparing of IV MNs in this model (Ferrucci et al., 2010).
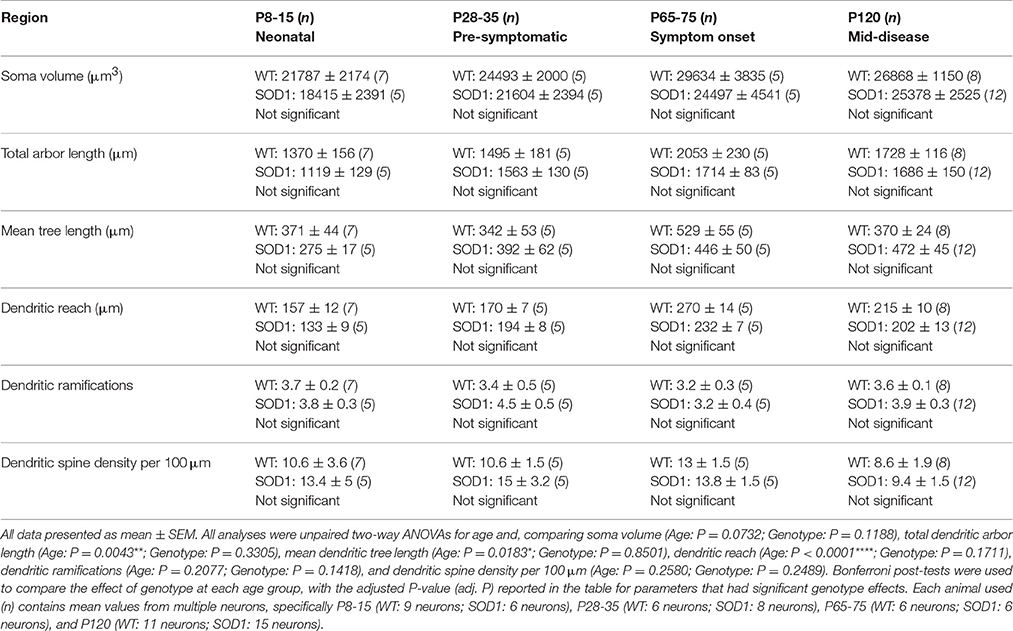
Table 5. Morphometric dendritic and dendritic spine parameters of IV MNs within the brainstem.
Vacuolation Occurs in XII MNs and Lumbar MNs from P65-76 Onward, But Is Not Present in IV MNs
Dendritic vacuoles, derived from degenerating mitochondria, are prevalent in lumbar MNs from P60 onward in SOD1 mice (Kong and Xu, 1998; Vinsant et al., 2013) and in the dendrites of human Betz cells in ALS post mortem tissue (Genç et al., 2017). Similar to other previous studies of vacuolization (Rafols and Fox, 1979; Goldstein et al., 1983), we found abundant dendritic vacuolation in XII MNs and lumbar MNs, at both P65-75 and P120, compared to WT MNs, quantified on a presence/absence basis (Figure 4). For the two earlier age groups, there were no differences in the percentage of XII MNs with vacuolization (Figure 4D); however, by P65-75, the percentage of MNs with vacuolization increased by ~6-fold, compared to WT controls (P = 0.028*, Figure 4D), and this increase persisted at P120 (P < 0.0001**** Figure 4D). Similarly, at P8-15 and P28-35, lumbar MNs in SOD1 mice did not show an increase in vacuolization, compared to WT controls (Figure 4E), but, at P65-75, there was a ~6-fold increase in the percentage of MNs with vacuolization (P = 0.034*, Figure 4E) and by P120, this became a 10-fold increase in the percentage of lumbar MNs with vacuolization, (P < 0.0001**** Figure 4E). By contrast, IV MNs from SOD1 mice did not exhibit significant differences in vacuolization at any age studied, compared to WT controls (P = 0.37, Figure 4C).
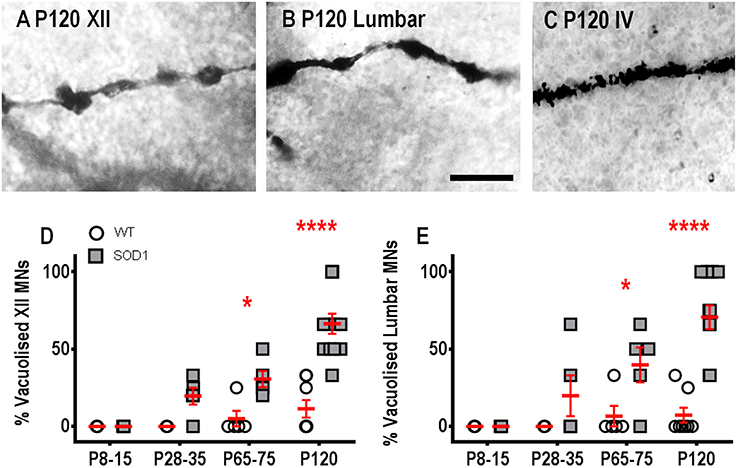
Figure 4. Increased proportion of vacuolated dendrites from SOD1 XII and lumbar MNs from P65-75 onward, compared to WT controls. Images show “beads on a string” appearance of dendritic vacuoles from P120 SOD1 XII MNs (A) and lumbar MNs (B). High-magnification images of dendrites from P120 SOD1 IV MNs (C) shows lack of hollowing and vacuoles in this neuronal population. (D) shows a scatterplot quantifying increased percentages of vacuolated SOD1 XII MNs (gray squared) compared to WT controls (white circles) at P65-75 and P120. (E) shows a scatterplot quantifying increased percentages of vacuolated SOD1 lumbar MNs (gray squared) compared to WT controls (white circles) at P65-75 and P120. All data mean ± SEM, with two-way ANOVAs followed by Bonferroni post-tests, *P < 0.05 and *****P < 0.0001. n = 7, 5, 5, and 8 for WT P8-15, P28-35, P65-75, and P120 respectively. n = 6, 5, 5, and 12 for SOD1 P8-15, P28-35, P65-75, and P120 respectively. Scale bar: (A,B), 25 μm.
Discussion
There are no longitudinal studies of dendritic morphology and spine density of several subcortical and motor neuron populations in any rodent model of ALS. We present a number of new insights, showing that morphological changes occur in MSNs from the dorsal striatum, brainstem XII MNs, and lumbar MNs, but not in hippocampal CA1 pyramidal neurons, and brainstem IV MNs. The structural changes in brainstem and spinal cord MNs susceptible to death in ALS exhibited differing degrees of dendritic and dendritic spine plasticity, as well as different rates of change, which may reflect differential susceptibility to dendritic degeneration. In total, our morphological assessment of dendritic changes strengthens the idea that changes in dendritic structure and synaptic inputs are drivers of both motor and non-motor phenotypes in ALS. As glutamate is the most widespread CNS neurotransmitter, our results provide additional support for the hypothesis that glutamate excitotoxicity as one of the pathological factors in ALS.
The striatum participates in control of mood, language and behavior, all of which can be abnormal in ALS (Josephs et al., 2009). Changes in striatum volume are present in ALS (Bede et al., 2013), and TDP-43 positive inclusions are found in striatal neurons in ALS patients (Josephs et al., 2009; Brettschneider et al., 2013; Riku et al., 2016). In the dorsal striatum, we found that there was a decrease in dendritic spine density of MSNs of SOD1 mice, without any changes in dendritic length or structure. Our dendritic length measurements agree with previous morphological studies of striatal MSNs; however, our spine densities are lower (for both genotypes) than those derived from fluorescent 3-dimensional rendering methodology (Judson et al., 2010), suggesting that the Golgi-Cox impregnation may be underestimating spine density differences between genotypes. Excitotoxic cell death in the striatum is associated with spine loss in MSNs (Garcia et al., 2010), and is also postulated to be a mechanism in neurodegenerative diseases of adulthood, including Parkinson's and Huntington's disease (Cepeda et al., 2003; Chase et al., 2003; Plotkin and Surmeier, 2015).
SOD1 mice show enhanced learning behavior and hippocampal long term synaptic plasticity (Spalloni et al., 2006). Unexpectedly, hippocampal CA1 pyramidal neurons from SOD1 mice displayed no morphological differences to controls at any age studied. The two major sources of excitatory input to CA1 pyramidal neurons are the Schaeffer collaterals of CA3 neurons, which synapse on the proximal apical dendrite, and layer II/III pyramidal neurons of the entorhinal cortex, which synapse on distal apical dendrites (van Groen et al., 2003). We have previously reported that pyramidal neurons in layer II/III of the entorhinal cortex, which also are the main efferent target of CA1 pyramidal neurons (van Groen et al., 2003), show no significant morphological changes in SOD1 mice (Fogarty et al., 2016c). Further functional studies of synaptic transmission within this hippocampal-entorhinal circuit are needed to test whether this circuitry undergoes significant modification in the SOD1 mouse.
In ALS, loss of brainstem XII MNs innervating the tongue contribute to disease morbidity and mortality, through impairment or loss of swallowing and speech, malnutrition, choking, and decreased ability to maintain upper airway patency for ventilation (Connolly et al., 2015). In SOD1 ALS models, XII MNs appear to undergo cell death later and at lower levels in comparison to spinal MNs (Haenggeli and Kato, 2002; Ngo et al., 2012; Lee et al., 2013). Thus, our observations of significant increases in dendritic arbor length and dendritic spine density suggest that these changes may underpin an enhanced capacity of SOD1 XII MNs to stave off degeneration. Alternatively, increased arborization and increased dendritic spines in SOD1 XII MNs may be maladaptive, increasing the substrate for later glutamate-induced excitotoxicity.
We found that XII MNs from SOD1 mice showed increased dendritic arbor length, from a week after birth until disease onset, followed by dendritic retraction by mid-disease age. Dendritic spine density was also elevated, compared to WT controls, at all ages studied. These findings in a vulnerable lower motor neuron population stand in contrast to the early dendritic shrinkage and spine loss of upper MNs and other vulnerable lower MNs reported here and elsewhere (Hammer et al., 1979; Horoupian et al., 1984; Udaka et al., 1986; Kato et al., 1987; Jara et al., 2012; Fogarty et al., 2015, 2016c). Although loss of dendritic arbor and dendritic spines are seen as hallmarks of neurodegeneration (Hammer et al., 1979; Horoupian et al., 1984; Udaka et al., 1986; Kato et al., 1987), other conditions, such as autism, exhibit increased spine density (Hutsler and Zhang, 2010), thereby suggesting that changes in circuit activity may not always be associated with spine loss, particularly in XII MNs (Fogarty et al., 2016a, 2017). Alternatively, since XII MNs have reduced calcium-buffering capacity (Rothstein et al., 1990; von Lewinski et al., 2008; Jaiswal and Keller, 2009), increased spine density may help to protect XII MNs, as spines can serve to confine calcium transients to the spine head (Koester and Sakmann, 1998; Yuste et al., 1999; Burette et al., 2010; Kenyon et al., 2010; Sala and Segal, 2014). This is consistent with reports of increased glutamatergic neurotransmission (van Zundert et al., 2008; Fogarty et al., 2016a, 2017) and intracellular calcium concentration (Bogaert et al., 2010; Luebke et al., 2010; Bellingham, 2011; van Zundert et al., 2012; Sala and Segal, 2014) in animal models showing increased MN excitability and of ALS.
We also found reduction in the dendritic arbor length of lumbar spinal cord MNs, compared to WT controls, commencing from P28-35. Similar to XII MNs, SOD1 lumbar MNs showed higher spine density compared to WT lumbar MNS at neonatal ages (P8-15), however, by disease onset (P65-75) and later, there was spine loss compared to WT controls. It is notable that hindlimb lumbar MNs are lost as early as P35 in SOD1 mice (Ngo et al., 2012), when both dendritic arbor length and spine density significantly decrease below WT controls. Previous reports have identified dendritic length recession in SOD1 mice at late-embryonic ages and at P6-10 (Martin et al., 2013; Leroy et al., 2014). The dendritic lengths reported here are lower than those obtained by single neuron dye-filling methods (Martin et al., 2013; Leroy et al., 2014); however, the dendritic length and spine density quantification were comparable to past studies using Golgi staining methods (Ma and Vacca-Galloway, 1991; Bou-Flores et al., 2000). Neonatal spinal cord MN morphology has been studied intensely in an alternate SOD1 model, the G83R mutation, which also shows increased dendritic length, compared to WT controls, at P3-4, P4-P9, and P8-9 (Amendola et al., 2007; Amendola and Durand, 2008; Filipchuk and Durand, 2012); a caveat is that dendritic lengthening is associated with MN hypoexcitability in this model (Pambo-Pambo et al., 2009; Hadzipasic et al., 2014). By contrast, lumbar MNs in SOD1 mice are hyper-excitable at postnatal ages (Quinlan et al., 2011), but begin to show hypo-excitability by P40 (Delestree et al., 2014). Further study of the relationship between functional MN excitability and MN morphology (Fogarty et al., 2013) will be needed to determine whether dendritic changes are driven by a shift from hyper-activity to hypo-activity.
Brainstem MNs controlling the extrinsic eye muscles, and sacral MNs controlling bladder and bowel function are relatively resistant to death in in clinical ALS and SOD1 models (Ferrucci et al., 2010; Spalloni and Longone, 2015). We found that there were no significant changes in dendritic structure or spine density at all ages studied in IV MNs, which are one of these resilient MN populations. Post mortem studies of ALS spinal cord show that another resilient MN population, Onuf's nucleus MNs, have shorter dendritic arbors (Takeda et al., 2014). Again, functional studies of IV MNs and MNs from Onuf's nucleus are needed, to determine whether these MNs show disease-related alteration in their activity.
There has been some preliminary evidence supporting disease-related changes in dendritic spine subtypes, classified conventionally as stubby, mushroom, and thin (Jones and Powell, 1969) in cortical neurons from a TDP-43 rodent model (Handley et al., 2017). Limitations of the light microscopic method (Harris et al., 1992; Arellano et al., 2007; Maiti et al., 2015) and evidence of a continuum of intermediate spine morphologies in past reports (Peters and Kaiserman-Abramof, 1970; Harris et al., 1992; Maiti et al., 2015) have precluded a robust and repeatable taxonomy of dendritic spines in material from the present study. Although there may be inherent limitations with differential impregnation of neurons with Golgi-Cox in case of neuro-degeneration, evidence from the literature demonstrates successful staining in animal models of neurodegenerative diseases (Ukabam, 1988; Spires et al., 2004) and human post-mortem tissue in cases of aging and Alzheimer's disease (Hanks and Flood, 1991).
In summary, we report the first longitudinal study of dendritic arbors, dendritic spines and dendritic vacuoles of several sub-cortical neuron populations, including multiple MN types, in a widely utilized mouse model of ALS. We describe dendritic reduction in susceptible neurons in SOD1 mice, compared to WT controls, commencing relatively early, at P8-15 or P28-35, ages prior to the emergence of symptoms, in spinal cord lumbar MNs, and at mid-disease stage in XII MNs. Spine density increases were present at all ages studied in SOD1 XII MNs, while spine density of lumbar MNs was increased neonatally, returning to control levels by P28-35 and decreasing compared to WT control by P120. Spine loss occurred in the absence of dendritic alteration in the MSNs of the striatum from P65-75, the age of disease symptom onset. Establishing whether dendritic spine loss in in ALS is a protective change, or a maladaptive change, (Kawashima et al., 1998) is an important avenue of investigation. The morphologic evidence we present here, along with past studies (Fogarty et al., 2015), suggests that spine loss largely precedes dendritic pathology in SOD1 mice. It will also be important to determine whether similar morphological changes occur in susceptible neurons in the more recently developed animal models of ALS-related fronto-temporal dementia that show inclusion bodies (Leigh et al., 1991; Katsuse and Dickson, 2005).
Characterizing changes in dendritic arbor and dendritic spines is useful to identify areas where neuronal alterations occur in ALS. Our results indicate early and persistent changes in the dendritic network of neuro-motor and extra-motor regions in the SOD1 model. The alterations in glutamatergic excitatory synaptic transmission previously reported at early ages in this mouse model (van Zundert et al., 2008; Fogarty et al., 2015; Saba et al., 2015) and which also occur in human induced pluripotent stem cells derived from ALS patients (Devlin et al., 2015) are consistent with our morphological results, lending support to the hypothesis of glutamate excitotoxicity. Our study provides a useful reference point for more detailed characterization of other models of ALS or fronto-temporal dementia that continue to emerge, in addition to being an important baseline for the study of possible therapies using functional or behavioral outcomes.
Author Contributions
EM, MF, NL, PN, and MB contributed to study design. EM and MF performed all experiments. MF, EM, NL, PN, and MB contributed to data analysis, writing, and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Maryam Shayegh and Mary White for mouse genotyping. This work was supported by grants from the National Health & Medical Research Council to MB and PN (#1065884) and by grants to MB and to PN from the Motor Neurone Disease Research Institute of Australia (GA1403 and GA1719). MF is supported by an NHMRC early career fellowship (CJ Martin).
References
Aggarwal, A., and Nicholson, G. (2002). Detection of preclinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation. J. Neurol. Neurosurg. Psychiatry 73, 199–201. doi: 10.1136/jnnp.73.2.199
Amendola, J., and Durand, J. (2008). Morphological differences between wild-type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J. Comp. Neurol. 511, 329–341. doi: 10.1002/cne.21818
Amendola, J., Gueritaud, J. P., Lamotte d'Incamps, B., Bories, C., Liabeuf, S., Allene, C., et al. (2007). Postnatal electrical and morphological abnormalities in lumbar motoneurons from transgenic mouse models of amyotrophic lateral sclerosis. Arch. Ital. Biol. 145, 311–323.
Anderson, C. T., Sheets, P. L., Kiritani, T., and Shepherd, G. M. (2010). Sublayer-specific microcircuits of corticospinal and corticostriatal neurons in motor cortex. Nat. Neurosci. 13, 739–744. doi: 10.1038/nn.2538
Arellano, J. I., Benavides-Piccione, R., Defelipe, J., and Yuste, R. (2007). Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front. Neurosci. 1, 131–143. doi: 10.3389/neuro.01.1.1.010.2007
Bede, P., Elamin, M., Byrne, S., McLaughlin, R. L., Kenna, K., Vajda, A., et al. (2013). Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology 81, 2107–2115. doi: 10.1212/01.wnl.0000437313.80913.2c
Bellinger, D. L., and Anderson, W. J. (1987). Postnatal development of cell columns and their associated dendritic bundles in the lumbosacral spinal cord of the rat. I. The ventrolateral cell column. Brain Res. 432, 55–67.
Bellingham, M. C. (2011). A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci. Ther. 17, 4–31. doi: 10.1111/j.1755-5949.2009.00116.x
Bogaert, E., d'Ydewalle, C., and Van Den Bosch, L. (2010). Amyotrophic lateral sclerosis and excitotoxicity: from pathological mechanism to therapeutic target. CNS Neurol. Disord. Drug Targets 9, 297–304. doi: 10.2174/187152710791292576
Bou-Flores, C., Lajard, A. M., Monteau, R., De Maeyer, E., Seif, I., Lanoir, J., et al. (2000). Abnormal phrenic motoneuron activity and morphology in neonatal monoamine oxidase A-deficient transgenic mice: possible role of a serotonin excess. J. Neurosci. 20, 4646–4656.
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Burette, A. C., Strehler, E. E., and Weinberg, R. J. (2010). A plasma membrane Ca2+ ATPase isoform at the postsynaptic density. Neuroscience 169, 987–993. doi: 10.1016/j.neuroscience.2010.05.062
Cepeda, C., Hurst, R. S., Calvert, C. R., Hernandez-Echeagaray, E., Nguyen, O. K., Jocoy, E., et al. (2003). Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J. Neurosci. 23, 961–969.
Charcot, J.-M., and Joffroy, A. (1869). Deux cas d'atrophie musculaire progressive avec lésions de la substance grise et de faisceaux antérolatéraux de la moelle épiniàre. Arch. Physiol. Norm. Pathol. 1, 354–367; 628–649; 744–757.
Chase, T. N., Bibbiani, F., and Oh, J. D. (2003). Striatal glutamatergic mechanisms and extrapyramidal movement disorders. Neurotox. Res. 5, 139–146. doi: 10.1007/BF03033378
Cleveland, D. W., and Rothstein, J. D. (2001). From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819. doi: 10.1038/35097565
Connolly, S., Galvin, M., and Hardiman, O. (2015). End-of-life management in patients with amyotrophic lateral sclerosis. Lancet Neurol. 14, 435–442. doi: 10.1016/S1474-4422(14)70221-2
Delestree, N., Manuel, M., Iglesias, C., Elbasiouny, S. M., Heckman, C. J., and Zytnicki, D. (2014). Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J. Physiol. 592, 1687–1703. doi: 10.1113/jphysiol.2013.265843
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 5999. doi: 10.1038/ncomms6999
Eisen, A., Kiernan, M., Mitsumoto, H., and Swash, M. (2014). Amyotrophic lateral sclerosis: a long preclinical period? J. Neurol. Neurosurg. Psychiatry 85, 1232–1238. doi: 10.1136/jnnp-2013-307135
Eisen, A., Pant, B., and Stewart, H. (1993). Cortical excitability in amyotrophic lateral sclerosis: a clue to pathogenesis. Can. J. Neurol. Sci. 20, 11–16.
Ferrucci, M., Spalloni, A., Bartalucci, A., Cantafora, E., Fulceri, F., Nutini, M., et al. (2010). A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol. Dis. 37, 370–383. doi: 10.1016/j.nbd.2009.10.017
Filipchuk, A. A., and Durand, J. (2012). Postnatal dendritic development in lumbar motoneurons in mutant superoxide dismutase 1 mouse model of amyotrophic lateral sclerosis. Neuroscience 209, 144–154. doi: 10.1016/j.neuroscience.2012.01.046
Fogarty, M. J., Hammond, L. A., Kanjhan, R., Bellingham, M. C., and Noakes, P. G. (2013). A method for the three-dimensional reconstruction of Neurobiotin-filled neurons and the location of their synaptic inputs. Front. Neural Circuits 7:153. doi: 10.3389/fncir.2013.00153
Fogarty, M. J., Kanjhan, R., Bellingham, M. C., and Noakes, P. G. (2016a). Glycinergic neurotransmission: a potent regulator of embryonic motor neuron dendritic morphology and synaptic plasticity. J. Neurosci. 36, 80–87. doi: 10.1523/JNEUROSCI.1576-15.2016
Fogarty, M. J., Kanjhan, R., Yanagawa, Y., Noakes, P. G., and Bellingham, M. C. (2017). Alterations in hypoglossal motor neurons due to GAD67 and VGAT deficiency in mice. Exp. Neurol. 289, 117–127. doi: 10.1016/j.expneurol.2016.12.004
Fogarty, M. J., Klenowski, P. M., Lee, J. D., Drieberg-Thompson, J. R., Bartlett, S. E., Ngo, S. T., et al. (2016b). Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 6:37968. doi: 10.1038/srep37968
Fogarty, M. J., Mu, E. W., Noakes, P. G., Lavidis, N. A., and Bellingham, M. C. (2016c). Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 4, 77. doi: 10.1186/s40478-016-0347-y
Fogarty, M. J., Noakes, P. G., and Bellingham, M. C. (2015). Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neurosci. 35, 643–647. doi: 10.1523/JNEUROSCI.3483-14.2015
Franklin, K. B. J., and Paxinos, G. (2008). The Mouse Brain in Stereotaxic Coordinates, 3rd Edn. Sydney, NSW: Academic Press.
Fujimura-Kiyono, C., Kimura, F., Ishida, S., Nakajima, H., Hosokawa, T., Sugino, M., et al. (2011). Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 1244–1249. doi: 10.1136/jnnp-2011-300141
Garcia, B. G., Neely, M. D., and Deutch, A. Y. (2010). Cortical regulation of striatal medium spiny neuron dendritic remodeling in parkinsonism: modulation of glutamate release reverses dopamine depletion-induced dendritic spine loss. Cereb. Cortex 20, 2423–2432. doi: 10.1093/cercor/bhp317
Genç, B., Jara, J. H., Lagrimas, A. K., Pytel, P., Roos, R. P., Mesulam, M. M., et al. (2017). Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 7:41765. doi: 10.1038/srep41765
Goldstein, B., Maxwell, D. S., Ellison, G., and Hammer, R. P. (1983). Dendritic vacuolization in the central nervous system of rats after long-term voluntary consumption of ethanol. J. Neuropathol. Exp. Neurol. 42, 579–589.
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775.
Hadzipasic, M., Tahvildari, B., Nagy, M., Bian, M., Horwich, A. L., and McCormick, D. A. (2014). Selective degeneration of a physiological subtype of spinal motor neuron in mice with SOD1-linked ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 16883–16888. doi: 10.1073/pnas.1419497111
Haenggeli, C., and Kato, A. C. (2002). Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci. Lett. 335, 39–43. doi: 10.1016/S0304-3940(02)01140-0
Hammer, R. P. Jr., Tomiyasu, U., and Scheibel, A. B. (1979). Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp. Neurol. 63, 336–346.
Handley, E. E., Pitman, K. A., Dawkins, E., Young, K. M., Clark, R. M., Jiang, T. C., et al. (2017). Synapse dysfunction of layer V pyramidal neurons precedes neurodegeneration in a mouse model of TDP-43 proteinopathies. Cereb. Cortex 27, 3630–3647. doi: 10.1093/cercor/bhw185
Hanks, S. D., and Flood, D. G. (1991). Region-specific stability of dendritic extent in normal human aging and regression in Alzheimer's disease. I. CA1 of hippocampus. Brain Res. 540, 63–82.
Hardiman, O., van den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
Harris, K. M. (1999). Structure, development, and plasticity of dendritic spines. Curr. Opin. Neurobiol. 9, 343–348.
Harris, K. M., Jensen, F. E., and Tsao, B. (1992). Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 12, 2685–2705.
Horoupian, D. S., Thal, L., Katzman, R., Terry, R. D., Davies, P., Hirano, A., et al. (1984). Dementia and motor neuron disease: morphometric, biochemical, and Golgi studies. Ann. Neurol. 16, 305–313. doi: 10.1002/ana.410160306
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Jara, J. H., Villa, S. R., Khan, N. A., Bohn, M. C., and Ozdinler, P. H. (2012). AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 47, 174–183. doi: 10.1016/j.nbd.2012.03.036
Jones, E. G., and Powell, T. P. (1969). Morphological variations in the dendritic spines of the neocortex. J. Cell Sci. 5, 509–529.
Josephs, K. A., Stroh, A., Dugger, B., and Dickson, D. W. (2009). Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 118, 349–358. doi: 10.1007/s00401-009-0547-7
Judson, M. C., Eagleson, K. L., Wang, L., and Levitt, P. (2010). Evidence of cell-nonautonomous changes in dendrite and dendritic spine morphology in the met-signaling-deficient mouse forebrain. J. Comp. Neurol. 518, 4463–4478. doi: 10.1002/cne.22467
Kanjhan, R., Fogarty, M. J., Noakes, P. G., and Bellingham, M. C. (2015). Developmental changes in the morphology of mouse hypoglossal motor neurons. Brain Struct. Funct. 221, 3755–3786. doi: 10.1007/s00429-015-1130-8
Kato, T., Hirano, A., and Donnenfeld, H. (1987). A Golgi study of the large anterior horn cells of the lumbar cords in normal spinal cords and in amyotrophic lateral sclerosis. Acta Neuropathol. 75, 34–40.
Katsuse, O., and Dickson, D. W. (2005). Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis. Assoc. Disord. 19(Suppl. 1), S37–S43. doi: 10.1097/01.wad.0000183889.61421.a8
Kawashima, T., Kikuchi, H., Takita, M., Doh-ura, K., Ogomori, K., Oda, M., et al. (1998). Skein-like inclusions in the neostriatum from a case of amyotrophic lateral sclerosis with dementia. Acta Neuropathol. 96, 541–545.
Kenyon, K. A., Bushong, E. A., Mauer, A. S., Strehler, E. E., Weinberg, R. J., and Burette, A. C. (2010). Cellular and subcellular localization of the neuron-specific plasma membrane calcium ATPase PMCA1a in the rat brain. J. Comp. Neurol. 518, 3169–3183. doi: 10.1002/cne.22409
Klenowski, P. M., Fogarty, M. J., Belmer, A., Noakes, P. G., Bellingham, M. C., and Bartlett, S. E. (2015). Structural and functional characterization of dendritic arbors and GABAergic synaptic inputs on interneurons and principal cells in the rat basolateral amygdala. J. Neurophysiol. 114, 942–957. doi: 10.1152/jn.00824.2014
Klenowski, P. M., Shariff, M. R., Belmer, A., Fogarty, M. J., Mu, E. W., Bellingham, M. C., et al. (2016). Prolonged consumption of sucrose in a binge-like manner, alters the morphology of medium spiny neurons in the nucleus accumbens shell. Front. Behav. Neurosci. 10:54. doi: 10.3389/fnbeh.2016.00054
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U.S.A. 95, 9596–9601.
Kong, J., and Xu, Z. (1998). Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 18, 3241–3250.
Lee, J. D., Kamaruzaman, N. A., Fung, J. N., Taylor, S. M., Turner, B. J., Atkin, J. D., et al. (2013). Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 10:119. doi: 10.1186/1742-2094-10-119
Leigh, P. N., Whitwell, H., Garofalo, O., Buller, J., Swash, M., Martin, J. E., et al. (1991). Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114 (Pt 2), 775–788.
Leroy, F., Lamotte d'Incamps, B., Imhoff-Manuel, R. D., and Zytnicki, D. (2014). Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3:e04046. doi: 10.7554/eLife.04046
Luebke, J. I., Weaver, C. M., Rocher, A. B., Rodriguez, A., Crimins, J. L., Dickstein, D. L., et al. (2010). Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct. Funct. 214, 181–199. doi: 10.1007/s00429-010-0244-2
Ma, W. Y., and Vacca-Galloway, L. L. (1991). Reduced branching and length of dendrites detected in cervical spinal cord motoneurons of Wobbler mouse, a model for inherited motoneuron disease. J. Comp. Neurol. 311, 210–222. doi: 10.1002/cne.903110204
Maiti, P., Manna, J., and McDonald, M. P. (2015). Merging advanced technologies with classical methods to uncover dendritic spine dynamics: a hot spot of synaptic plasticity. Neurosci. Res. 96, 1–13. doi: 10.1016/j.neures.2015.02.007
Martin, E., Cazenave, W., Cattaert, D., and Branchereau, P. (2013). Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 54, 116–126. doi: 10.1016/j.nbd.2013.02.011
Mills, K. R., and Nithi, K. A. (1997). Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve 20, 1137–1141.
Ngo, S. T., Baumann, F., Ridall, P. G., Pettitt, A. N., Henderson, R. D., Bellingham, M. C., et al. (2012). The relationship between Bayesian motor unit number estimation and histological measurements of motor neurons in wild-type and SOD1(G93A) mice. Clin. Neurophysiol. 123, 2080–2091. doi: 10.1016/j.clinph.2012.01.028
Ozdinler, P. H., Benn, S., Yamamoto, T. H., Guzel, M., Brown, R. H. Jr., and Macklis, J. D. (2011). Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G(9)(3)A transgenic ALS mice. J. Neurosci. 31, 4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011
Pambo-Pambo, A., Durand, J., and Gueritaud, J. P. (2009). Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J. Neurophysiol. 102, 3627–3642. doi: 10.1152/jn.00482.2009
Peters, A., and Kaiserman-Abramof, I. R. (1970). The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am. J. Anat. 127, 321–355. doi: 10.1002/aja.1001270402
Plotkin, J. L., and Surmeier, D. J. (2015). Corticostriatal synaptic adaptations in Huntington's disease. Curr. Opin. Neurobiol. 33, 53–62. doi: 10.1016/j.conb.2015.01.020
Quinlan, K. A., Schuster, J. E., Fu, R., Siddique, T., and Heckman, C. J. (2011). Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 589(Pt 9), 2245–2260. doi: 10.1113/jphysiol.2010.200659
Rafols, J. A., and Fox, C. A. (1979). Fine structure of the primate striatum. Appl. Neurophysiol. 42, 13–16.
Riku, Y., Watanabe, H., Yoshida, M., Mimuro, M., Iwasaki, Y., Masuda, M., et al. (2016). Marked involvement of the striatal efferent system in TAR DNA-binding protein 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 75, 801–811. doi: 10.1093/jnen/nlw053
Rothstein, J. D., Tsai, G., Kuncl, R. W., Clawson, L., Cornblath, D. R., Drachman, D. B., et al. (1990). Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 28, 18–25. doi: 10.1002/ana.410280106
Rothwell, P. E., Hayton, S. J., Sun, G. L., Fuccillo, M. V., Lim, B. K., and Malenka, R. C. (2015). Input- and output-specific regulation of serial order performance by corticostriatal circuits. Neuron 88, 345–356. doi: 10.1016/j.neuron.2015.09.035
Rutledge, L. T., Duncan, J., and Beatty, N. (1969). A study of pyramidal cell axon collaterals in intact and partially isolated adult cerebral cortex. Brain Res. 16, 15–22.
Saba, L., Viscomi, M. T., Caioli, S., Pignataro, A., Bisicchia, E., Pieri, M., et al. (2015). Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb. Cortex 26, 1512–1528. doi: 10.1093/cercor/bhu317
Sala, C., and Segal, M. (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev. 94, 141–188. doi: 10.1152/physrev.00012.2013
Sgobio, C., Trabalza, A., Spalloni, A., Zona, C., Carunchio, I., Longone, P., et al. (2008). Abnormal medial prefrontal cortex connectivity and defective fear extinction in the presymptomatic G93A SOD1 mouse model of ALS. Genes Brain Behav. 7, 427–434. doi: 10.1111/j.1601-183X.2007.00367.x
Smittkamp, S. E., Brown, J. W., and Stanford, J. A. (2008). Time-course and characterization of orolingual motor deficits in B6SJL-Tg(SOD1-G93A)1Gur/J mice. Neuroscience 151, 613–621. doi: 10.1016/j.neuroscience.2007.10.017
Spalloni, A., and Longone, P. (2015). Cognitive impairment in amyotrophic lateral sclerosis, clues from the SOD1 mouse. Neurosci. Biobehav. Rev. 60, 12–25. doi: 10.1016/j.neubiorev.2015.11.006
Spalloni, A., Geracitano, R., Berretta, N., Sgobio, C., Bernardi, G., Mercuri, N. B., et al. (2006). Molecular and synaptic changes in the hippocampus underlying superior spatial abilities in pre-symptomatic G93A+/+ mice overexpressing the human Cu/Zn superoxide dismutase (Gly93 –> ALA) mutation. Exp. Neurol. 197, 505–514. doi: 10.1016/j.expneurol.2005.10.014
Spalloni, A., Origlia, N., Sgobio, C., Trabalza, A., Nutini, M., Berretta, N., et al. (2011). Postsynaptic alteration of NR2A subunit and defective autophosphorylation of alphaCaMKII at threonine-286 contribute to abnormal plasticity and morphology of upper motor neurons in presymptomatic SOD1G93A mice, a murine model for amyotrophic lateral sclerosis. Cereb. Cortex 21, 796–805. doi: 10.1093/cercor/bhq152
Spires, T. L., Grote, H. E., Garry, S., Cordery, P. M., Van Dellen, A., Blakemore, C., et al. (2004). Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur. J. Neurosci. 19, 2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x
Steyn, F. J., Lee, K., Fogarty, M. J., Veldhuis, J. D., McCombe, P. A., Bellingham, M. C., et al. (2013). Growth hormone secretion is correlated with neuromuscular innervation rather than motor neuron number in early-symptomatic male amyotrophic lateral sclerosis mice. Endocrinology 154, 4695–4706. doi: 10.1210/en.2013-1570
Strong, M. J., Grace, G. M., Freedman, M., Lomen-Hoerth, C., Woolley, S., Goldstein, L. H., et al. (2009). Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 131–146. doi: 10.1080/17482960802654364
Takeda, T., Uchihara, T., Nakayama, Y., Nakamura, A., Sasaki, S., Kakei, S., et al. (2014). Dendritic retraction, but not atrophy, is consistent in amyotrophic lateral sclerosis-comparison between Onuf's neurons and other sacral motor neurons. Acta Neuropathol. Commun. 2:11. doi: 10.1186/2051-5960-2-11
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Turner, B. J., and Talbot, K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. doi: 10.1016/j.pneurobio.2008.01.001
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., de Carvalho, M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322. doi: 10.1016/S1474-4422(13)70036-X
Turner, M. R., Osei-Lah, A. D., Hammers, A., Al-Chalabi, A., Shaw, C. E., Andersen, P. M., et al. (2005). Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J. Neurol. Neurosurg. Psychiatry 76, 1279–1285. doi: 10.1136/jnnp.2004.054429
Udaka, F., Kameyama, M., and Tomonaga, M. (1986). Degeneration of Betz cells in motor neuron disease. A Golgi study. Acta Neuropathol. 70, 289–295.
Ukabam, C. U. (1988). A Golgi-Cox study of the hypoglossal nucleus of “wobbler” and normal mice. Ric. Clin. Lab. 18, 313–318.
van Groen, T., Miettinen, P., and Kadish, I. (2003). The entorhinal cortex of the mouse: organization of the projection to the hippocampal formation. Hippocampus 13, 133–149. doi: 10.1002/hipo.10037
van Zundert, B., Izaurieta, P., Fritz, E., and Alvarez, F. J. (2012). Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell Biochem. 113, 3301–3312. doi: 10.1002/jcb.24234
van Zundert, B., Peuscher, M. H., Hynynen, M., Chen, A., Neve, R. L., Brown, R. H. Jr., et al. (2008). Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 28, 10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008
Vinsant, S., Mansfield, C., Jimenez-Moreno, R., Del Gaizo Moore, V., Yoshikawa, M., Hampton, T. G., et al. (2013). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 3, 431–457. doi: 10.1002/brb3.142
von Lewinski, F., Fuchs, J., Vanselow, B. K., and Keller, B. U. (2008). Low Ca2+ buffering in hypoglossal motoneurons of mutant SOD1 (G93A) mice. Neurosci. Lett. 445, 224–228. doi: 10.1016/j.neulet.2008.08.084
Vucic, S., and Kiernan, M. C. (2006). Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129(Pt 9), 2436–2446. doi: 10.1093/brain/awl172
Vucic, S., Nicholson, G. A., and Kiernan, M. C. (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131(Pt 6), 1540–1550. doi: 10.1093/brain/awn071
Wicks, P., Abrahams, S., Papps, B., Al-Chalabi, A., Shaw, C. E., Leigh, P. N., et al. (2009). SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J. Neurol. 256, 234–241. doi: 10.1007/s00415-009-0078-0
Yasvoina, M. V., Genç, B., Jara, J. H., Sheets, P. L., Quinlan, K. A., Milosevic, A., et al. (2013). eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci. 33, 7890–7904. doi: 10.1523/JNEUROSCI.2787-12.2013
Yuste, R., Majewska, A., Cash, S. S., and Denk, W. (1999). Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J. Neurosci. 19, 1976–1987.
Zanette, G., Tamburin, S., Manganotti, P., Refatti, N., Forgione, A., and Rizzuto, N. (2002). Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin. Neurophysiol. 113, 1688–1697.
Keywords: dendrite, spine density, striatum, hippocampus, hypoglossal motor neuron, trochlear motor neuron, lumbar motor neuron
Citation: Fogarty MJ, Mu EWH, Lavidis NA, Noakes PG and Bellingham MC (2017) Motor Areas Show Altered Dendritic Structure in an Amyotrophic Lateral Sclerosis Mouse Model. Front. Neurosci. 11:609. doi: 10.3389/fnins.2017.00609
Received: 24 August 2017; Accepted: 18 October 2017;
Published: 01 November 2017.
Edited by:
Stefano L. Sensi, Università degli Studi “G. d'Annunzio” Chieti - Pescara, ItalyReviewed by:
Patrizia Longone, Fondazione Santa Lucia (IRCCS), ItalyIan Paul Johnson, University of Adelaide, Australia
Copyright © 2017 Fogarty, Mu, Lavidis, Noakes and Bellingham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark C. Bellingham, bWFyay5iZWxsaW5naGFtQHVxLmVkdS5hdQ==
†These authors have contributed equally to this work.