 Maria Masnata1
Maria Masnata1 Francesca Cicchetti1,2*
Francesca Cicchetti1,2*- 1Axe Neurosciences, Centre de Recherche du CHU de Québec-Université Laval, Quebec, QC, Canada
- 2Département de Psychiatrie & Neurosciences, Université Laval, Quebec, QC, Canada
Neurodegenerative disorders are not only characterized by specific patterns of cell loss but the presence and accumulation of various pathological proteins—both of which correlate with disease evolution. There is now mounting evidence to suggest that these pathological proteins present with toxic, at times prion-like, properties and can therefore seed pathology in neighboring as well remotely connected healthy neurons as they spread across the brain. What is less clear, at this stage, is how much this actually contributes to, and drives, the core pathogenic events. In this review, we present a comprehensive, up-to-date summary of the reported in vitro studies that support the spreading and seeding capacities of pathological proteins, with an emphasis on mutant huntingtin protein in the context of Huntington's disease, although in vivo work remains to be performed to validate this theory in this particular disease. We have further reviewed these findings in light of their potential implications for the development of novel therapeutic approaches.
Are “Prionic” Proteins Pathogenic and Found in All Neurodegenerative Diseases?
The discovery of Lewy-body pathology within fetal ventral mesencephalic cells grafted in patients with Parkinson's disease (PD) years earlier has radically changed our views on the potential pathogenic mechanisms underlying sporadic neurodegenerative disorders of the central nervous system. This observation, initially reported by two independent teams (Kordower et al., 2008; Li et al., 2008), has led to the theory that the pathogenic α-synuclein protein can spread from the diseased brain to healthy tissue and cause protein aggregation and cellular dysfunction in a prion-like fashion (Olanow and Prusiner, 2009; Brundin et al., 2010; Soto, 2012). Indeed, it has been demonstrated both in vitro and in vivo that α-synuclein, the main component of Lewy bodies, can be released into the extracellular space and then be internalized by neighboring neurons (Desplats et al., 2009; Hansen et al., 2011), acting as a toxic agent that could seed pathology in the process. Furthermore, intracerebral inoculation of brain homogenates derived from aged α-synuclein transgenic mice, or injections of synthetic α-synuclein preformed fibrils, accelerates the formation of protein aggregates and precipitates neurological dysfunction in small animals (Luk et al., 2012a,b). It is also now known that there is pathology remote from the injection sites in these types of studies, which further supports an intercellular transneuronal spread of protein, as has also been demonstrated in rodent allografts placed in animals expressing human α-synuclein (Angot et al., 2012). In the latter study, human α-synuclein was shown to co-localize with markers of endosomes and exosomes (Angot et al., 2012), which could represent one of the routes by which the protein is transferred (Goedert et al., 2010; Angot et al., 2012). This mode of protein spread and disease propagation has been shown experimentally with several other proteins including amyloid, tau, SOD1, TDP-43 and FUS (Soto, 2012; Jucker and Walker, 2013; Guo and Lee, 2014). However, these studies have proposed a number of other putative mechanisms for protein spread which include nanotubes (Costanzo et al., 2013; Abounit et al., 2016a,b), vesicular transport (Angot et al., 2012; Lee et al., 2012), endocytosis (Hansen et al., 2011; Wu et al., 2013; Ruiz-Arlandis et al., 2016) or even direct penetration of the plasma membrane (Ren et al., 2009).
What is emerging from all this work is that a number of these processes may be common to all neurodegenerative disorders, not just sporadic but also monogenic diseases such as Huntington's disease (HD) (Brundin et al., 2010; Soto, 2012; Cicchetti et al., 2014). In HD, recent evidence has added weight to the idea that the mutant huntingtin protein (mHTT)—the genetic product that defines the disease—can propagate from cell-to-cell. In vivo observations for the ability of mHTT to travel across synapses has been collected in embryonic human stem cells differentiated into neurons and implanted in R6/2 mice, a transgenic mouse model of HD (Pecho-Vrieseling et al., 2014). It has also been demonstrated by expressing the human HTT gene (138 CAG) within olfactory receptor neurons which was subsequently found in the synaptically connected large posterior neurons in the brain of drosophila (Babcock and Ganetzky, 2015). Various cell types (HEK), including neuron-like cells (Cos-7 and PC-12), have been shown to be capable of internalizing synthetic mHTT aggregates from the extracellular milieu (Yang et al., 2002; Ren et al., 2009). The aggregates can be either translocated to the nucleus where their pathogenic effects on transcription can be exerted leading to cell dysfunction and death (Yang et al., 2002) or act as seeds for further protein aggregation within the cell itself (Herrera et al., 2011). Prion-like spread has also been suggested to take place following the phagocytosis of mHTT aggregates by glial cells in a Drosophila model of HD. The engulfed aggregates gained access to the cytoplasm of microglia where they interacted with soluble huntigtin (HTT), initiating a prion-like dissemination of pathology (Pearce et al., 2015). Although there are no other reports on the role of immune cells in mHTT propagation (Weiss et al., 2012), this is a likely scenario that may, at least partly, contribute to HD pathology and that certainly cannot be dismissed at this stage.
The notion that neurodegenerative disorders, including HD, share several features with classical prion diseases is gaining momentum—although this concept remains more debated for this disease and more in vivo work is clearly needed to substantiate the initial in vitro claims. Notwithstanding, at least two fundamental questions remain to be answered: What are the routes by which the pathological proteins, such as mHTT, propagate and therefore spread and seed their pathogenic effects? And can any of these routes be blocked to prevent disease dissemination? The aim of this manuscript is to review the in vitro evidence for the spreading and seeding capacities of mHTT and what implications this has for the development of novel therapeutic approaches.
Is mHTT Equally Toxic to Striatal and Cortical Neurons?
One of the most striking pathological features of HD is the massive loss of striatal projection neurons. However, the degeneration is not confined to this structure and cortical areas as well as a number of other sites all show cell loss early on in the disease process. However, cells in both of the cortical and striatal areas are tightly connected synaptically. They both express mHTT but their vulnerability to early degeneration in HD remains elusive, as does the chronology in which they degenerate with respect to one another. In vitro models in which mHTT expression is induced by high capacity adenoviral (Dong et al., 2012) or lentiviral vectors (Zala et al., 2005), have revealed that cortical neurons accumulate a significant number of inclusion bodies, but with no clear toxic consequences. In contrast, striatal neurons develop major morphological changes that are accompanied by the loss of neurofilaments and ultimately cell death, despite the fact that mHTT aggregates are rarely seen within these cells (Zala et al., 2005; Dong et al., 2012). The reasons for this differential vulnerability have been further investigated in the BACHD mouse model by genetically inhibiting mHTT production in the striatum and cortex simultaneously, or in either structure alone. Inhibition of mHTT synthesis in both the striatum and cortex combined had the most significant beneficial impact on motor and psychiatric HD-related behavioral phenotypes (Wang N. et al., 2014) with an abrogation of striatal degeneration. This may imply that the content of mHTT within cortical cells changes the cell properties; i.e. by producing more glutamate which creates excitotoxicity within their target structures such as the striatum. However, it may also indicate that preventing the propagation of mHTT via the cortico-striatal circuit can have a considerable impact on the disease.
Transneuronal/Transynaptic Propagation: How Neuronal Circuits Enable mHTT Spread
The validity of the transynaptic propagation theory (Figure 1) has gained additional support with the work of Pecho-Vrieseling et al. (2014). In this study, three distinct models were used to investigate this concept. In one set of experiments, the authors generated mixed-genotype (R6/2-wild-type) cortico-striatal cultures, more specifically combining R6/2 striatal neurons with wild-type cortical neurons or wild-type striatal neurons with R6/2 cortical neurons. Functional R6/2 cortical-wild-type striatal networks were created and used to study long-distance mHTT propagation from the cortex to wild-type DARPP-32+ medium spiny neurons. However, this was not seen in the R6/2 striatum-wild-type cortical circuit as no significant amounts of mHTT deposits were detected in the wild-type cortex.
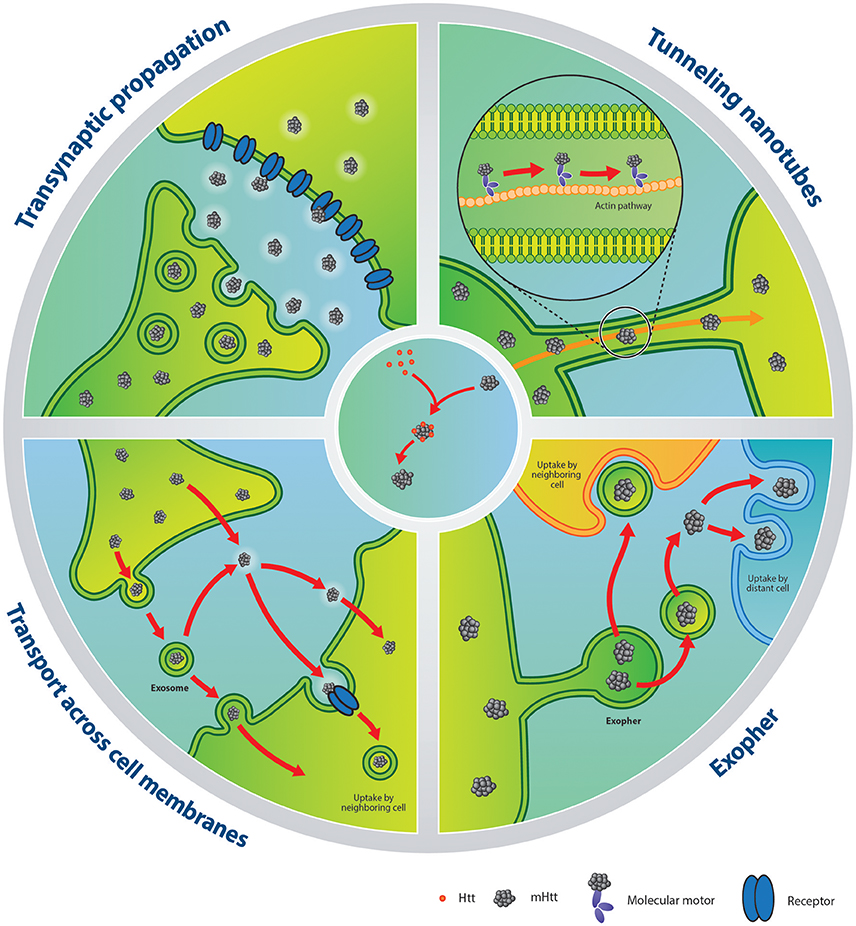
Figure 1. Putative mechanisms of mHTT spreading and seeding capacities. Left upper panel: illustration of transynaptic propagation of mHTT. Right upper panel: transport mechanism of mHTT via tunneling nanotubes. Left lower panel: mHTT can be released within exosomes or in a free form. After extrusion, exosomes carrying mHTT can fuse with the plasma membrane of a neighboring cell. Alternatively, mHTT can escape from the exosomal-vesicle into the extracellular compartment, with the same fate as the counterpart released directly as a free form. Finally, mHTT can be internalized by a recipient cell via receptor-mediated endocytosis or directly penetrate the plasma membrane. Right lower panel: In neurons of C. elegans, mHTT has been shown to be contained within exophers, an entity which resembles mammalian exosomes. Released exophers may be incorporated by adjacent or distant cells or secrete their contents into the milieu. Central panel: schematic of the seeding process of mHTT. The misfolded protein recruits HTT in an elongation process creating toxic aggregates. HTT, huntingtin; mHTT, mutant huntingtin.
In a second set of experiments, the authors pursued the transynaptic propagation theory using embryonic human stem cells differentiated into neurons and tagged with GFP (hGFP) which they then transplanted into organotypic brain slices derived from R6/2 mice. Cell inoculations were performed within the cortex and striatum and the identity of the transplanted cells was confirmed by immunostainings of mature and structure-specific cellular markers such as Tbr1 (cortex) and DARPP-32 (striatum). Two major observations were made relating to the propagation of mHTT: from endogenous neurons of the R6/2 mice or to the freshly transplanted human cells. Firstly, two waves of mHTT accumulation were seen between the mouse neurons: one wave occurred at 3–4 weeks and a second took place between 6 to 8 weeks following the initiation of cultures. In the human transplanted cells, mHTT accumulation progressively increased within the striatum between 4 and 8 weeks at which point it plateaued, while in the cortex, the pattern of propagation was identical to that seen in mouse neurons. The impact of mHTT propagation was seen primarily on neurites, which became atrophied. This was accompanied by a concomitant loss of DARPP-32+ neurons, one of the main features of HD pathology. Finally, the location of mHTT aggregates was first identified in the cytoplasm and subsequently in the cell nucleus. It should be noted that the authors repeated the experiment using human pluripotent stem cells differentiated into neurons and also showed the transynaptic spread of mHTT from R6/2 host tissue to human grafts, indicating that this was common to different cell types.
A final set of experiments was performed in vivo where wild-type mice were injected with Q72-HTT-Exon1 and synaptophysin-GFP viral vectors into cerebral cortical layers. In this case, mHTT aggregates were detected predominantly in medium spiny neurons expressing GFP which were innervated by cortical projection neurons previously transduced with the viruses, suggesting an active role for cortical projections in mHTT propagation to striatal neurons. Post-mortem analyses of grafted hGFP-neurons in cortical areas of 4-week-old R6/2 mice further confirmed these observations.
The seminal work of Pecho-Vrieseling et al. (2014) confirmed the hypothesis brought forward by Cicchetti et al. (2014) that mHTT could propagate transynaptically between disease and healthy tissue. But exactly how mHTT is transported whithin neurons remains unanswered. To understand this, microfluidic culture devices have been employed in which neuronal somata could be isolated from their processes and other cell types. With this system, it was shown that synthetic α-synuclein fibrils can be transported both anterogradely and retrogradely (Volpicelli-Daley et al., 2011; Freundt et al., 2012; Brahic et al., 2016), as well as to be released and taken up by second order neurons (Freundt et al., 2012). Similar observations have been made with Aβ42 (Freundt et al., 2012; Brahic et al., 2016), tau (Wu et al., 2013; Brahic et al., 2016) and HTTExon1 fibrils (Brahic et al., 2016). However, HTTExon1 fibrils showed limited anterograde transport, although retrograde transport efficiency was similar to that seen with α-synuclein fibrils (Brahic et al., 2016; Table 1).
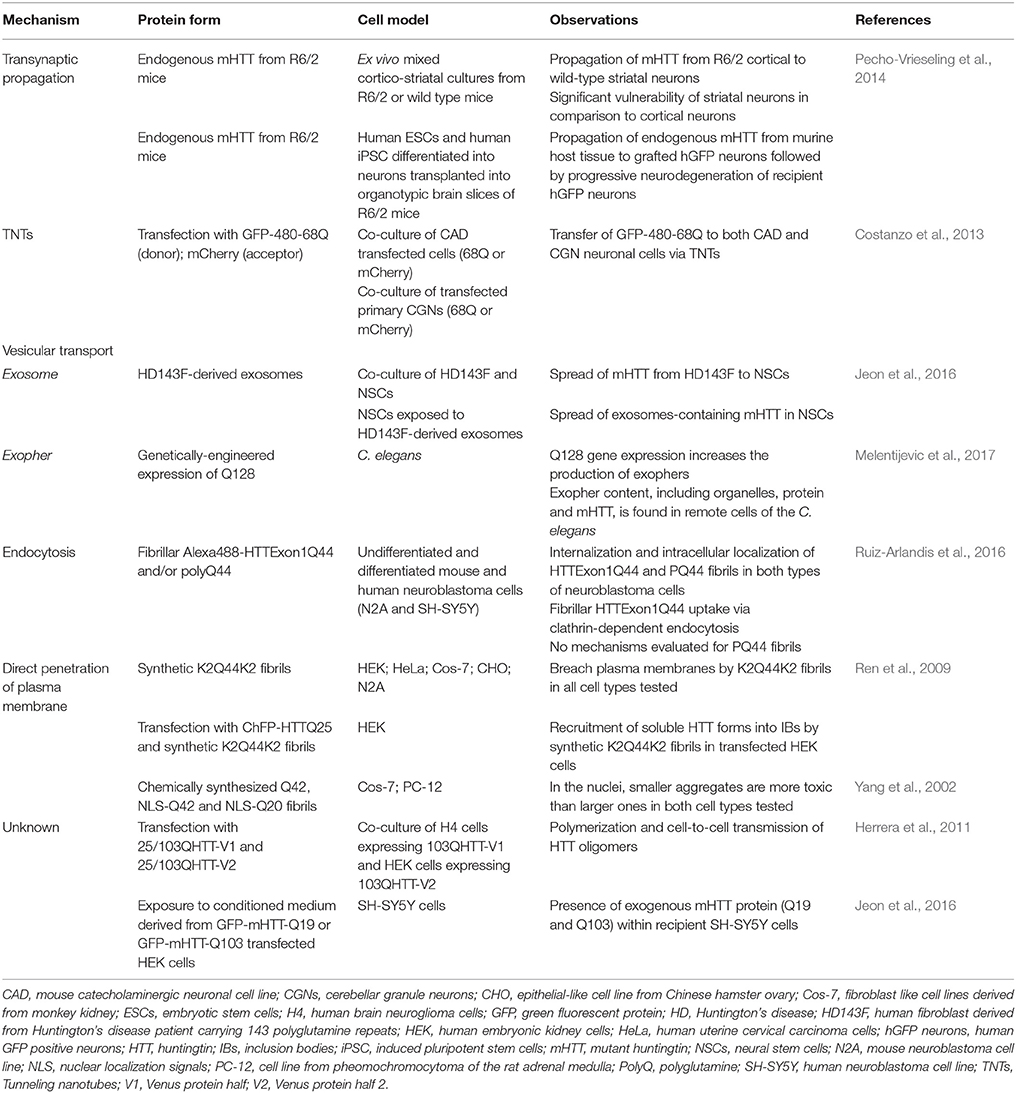
Table 1. In vitro evidence of mHTT spreading capacities.
It has now been demonstrated, in vitro, in vivo and in post-mortem human samples, that mHTT is able to disrupt vesicular and mitochondrial trafficking by recruiting and sequestering key elements of the axonal trafficking machinery, such as normal HTT, within aggregates (Trushina et al., 2004). This could explain the differences observed in the axonal transport between mHTT and other types of protein fibrils, in particular the fact that, after retrograde transport, HTTExon1 fibrils accumulate intracellularly, while more than half of the α-synuclein and Aβ42 fibrils are released into the media (Brahic et al., 2016). It is clear that the properties of mHTT do not simply allow for it to be transferred between cells but that it is also capable of triggering aggregate formation using endogenous proteins within the recipient cell - and by so doing display prionic characteristics.
Tunneling Nanotubes: A Channel for Disease Dissemination
Tunneling nanotubes (TNTs) are small entities that serve as a communication bridge between cells (Abounit and Zurzolo, 2012). Using adhesion proteins and fusion molecules (SNARE or viral fusion proteins), TNTs can change their configuration to merge to other cellular surfaces (Marzo et al., 2012). Neurons and various other cell types have the ability to produce these temporary and retractable protrusions which are made of F-actin strains and lipid bilayers containing organelles, plasma membrane components and ions such Ca2+ which are key to cell signaling (Smith et al., 2011). In normal physiological conditions, they have been noted to participate in cell development (Gurke et al., 2008), contribute to immune responses (Watkins and Salter, 2005), engage in regeneration processes (Wang et al., 2011) as well as facilitating electrical conduction between cells (Smith et al., 2011).
Within the F-actin path forming the TNTs, molecular motors can be hijacked to transport pathogens and prion-like proteins (Gousset et al., 2009; Abounit and Zurzolo, 2012; Figure 1). This has been demonstrated in mouse catecholaminergic neuronal cells for α-synuclein (Abounit et al., 2016a) and in both mouse catecholaminergic neuronal cells and mouse cerebellar granule neurons for mHTT (Costanzo et al., 2013). Furthermore, HTT and mHTT have been shown to form strong interactions with phospholipid bilayers suggesting that they can drift on F-actin streams and lipid surfaces (Marzo et al., 2012). More specifically, α-synuclein fibrils taken up from the media are almost exclusively found embedded in endolysosomal vesicles (Abounit et al., 2016a) within TNTs. In contrast, mHTT fibrils are identified in a free form state within the cell cytoplasm (Ren et al., 2009; Table 1) and their colocalization with vimentin hints to potential transport within aggresome-like structures (Costanzo et al., 2013). Although TNT formation can offer a defense mechanism for expelling material that the cell cannot digest/degrade—for example fibrillar amyloids—this system is not sufficient to completely restore the cell's health. Additionally, cells that contain pathological proteins produce more TNTs (Costanzo et al., 2013; Abounit et al., 2016a,b), creating opportunities for pathological protein transfer and potentially facilitating the seeding process in naïve cells.
Exosomes and Exophers as mHTT Cargo Carriers
It has now been clearly shown that misfolded proteins can be found within extracellular vesicles and that they can be carried and delivered to a recipient cell using this means (Figure 1). The demonstration that this applies to mHTT as well has also been shown using exosomes extracted from fibroblasts derived from a severe juvenile HD case harboring 143 CAG repeats (HD143F) and which were exposed to differentiated neuronal stem cells derived from embryonic cortical mouse tissue. After 4 days of contact between the exosomes and the cultured cells, the internalization of mHTT was detectable. Similar results were achieved when HD143F-derived exosomes were incubated with SH-SY5Y cells, confirming the spreading capacity of mHTT via exosomes in various cell types (Jeon et al., 2016; Table 1). Most strikingly, HD143F exosomes injected intraventricullarly into new-born wild-type animals led to the development of motor and cognitive HD-related behavioral phenotypes (Jeon et al., 2016). This study provided evidence that exosomes carrying mHTT could participate in disease induction/manifestation both in vitro and in vivo.
More recently, the C. elegans model has served to identify a novel vesicular entity capable of incorporating and extruding dysfunctional organelles as well as protein aggregates. This transporter was named an “exopher” (Melentijevic et al., 2017). Despite their larger size and the fact that they are born from a different genesis process, exophers resemble, in many ways, exosomes found in mammalian cells. They are released in a multiple-step process which includes protrusion, elongation and separation (Figure 1), emerging from the soma of different neurons, independently of cell division or cell death (Melentijevic et al., 2017). Their production is triggered by cellular stress, disruption of physiological conditions or, importantly, by the presence of inclusion bodies such as mHTT aggregates (Melentijevic et al., 2017). For example, the expression of aggregable HTT-Q128 in the genome of the nematode causes impairments of the normal functions of chaperones, autophagy pathways, ubiquitin-proteasome systems and provokes the collapse of the integrity maintenance machinery in cellular elements. The accumulated stress increases the production of exophers in touch-responsive ALMR neurons expressing Q128 in this animal which in fact serves as defense mechanisms to restore the cell's equilibrium (Melentijevic et al., 2017). However, the downside of exopher production is that they may, like exosomes, enable cell-to-cell delivery of insoluble pathogenic proteins (Melentijevic et al., 2017; Table 1).
Accessing the Membrane of Neighboring Cells by Endocytosis
Endocytosis is a cellular mechanism that controls various cellular functions such as internalization and recycling of plasma membrane components/ligands as well as the uptake and degradation of macromolecules and extracellular particles. The process begins with modifications of the plasma membrane, the generation of endocytic vesicles, which then matures into early and late endosomes, and finally leads to the degradation of the vesicle content through fusion with lysosomes (Miaczynska and Stenmark, 2008).
There is now compelling evidence that proteins, such as α-synuclein, can be incorporated into cells via clathrin-dependent endocytosis (Oh et al., 2016; Figure 1). Using undifferentiated and differentiated N2A cells exposed to HTTExon1Q44 and treated with various pharmacological compounds, clathrin-dependent endocytosis was identified to participate in mHTT uptake. In this particular study, HTTExon1Q44 fibrils were found in early endosomes of undifferentiated cells. This was observed as early as 6 h post-exposure to the toxic material or in both early endosomes and lysosomes 24 h post-exposure. At 48 h, concentrations of fibrils were unchanged within the lysosomes, but were found at lower concentrations in the early endosomes (Ruiz-Arlandis et al., 2016; Table 1). It was speculated that mHTT concentrations inside endosomes was lower because mHTT had been delivered to the lysosomes. In contrast, the stable concentrations of mHTT inside the lysosomes may have resulted from an equilibrium between the quantities of ingested vs. degraded fibrils. It is also feasible, according to the authors, that this observation reflects the saturation of the lysosome degradation machinery. It has been noted that the fate of the same fibrils differed in differentiated N2A, where co-localization within early endosomes could not be firmly established. Rather, mHTT fibrils were present within lysosomes, perhaps testifying to the cell's attempt to eliminate toxic mHTT through lysosomal degradation.
The idea that the turnover of HTT and mHTT aggregates is dependent on the endosomal–lysosomal system finds supports in another study in which it has been reported that following transfection, free HTT accumulated in the cytoplasm and inside autophagosome-like vacuoles, while mHTT localized both to the cytoplasm and nucleus. The presence of mHTT in vacuoles modified the morphology of the cells, which appeared atrophied, further implying that endosomal-lysosomal-vacuolar pathway activation may be responsible for this type of cell death (Kegel et al., 2000). HTT and mHTT have also be shown to co-localize with late endosome and lysosome markers and this has led to speculation that this was due to the lysosome's capacity to mediate the secretion of proteins fusing with the plasma membrane through an active calcium dependent process (Figure 1). Endosome ablation, calcium chelator or silencing synaptotagmin 7 (lysosome-specific calcium sensor) inhibited mHTT secretion, providing evidence for the involvement of late endosomal/lysosome pathway in secreting mHTT and, in smaller amounts, wild-type HTT (Trajkovic et al., 2017).
mHTT Transmission by Direct Penetration of Plasma Membranes
Release and uptake are the basic elements of a prion-like cell-to-cell protein propagation process. In order to dissect the mechanisms of transit between cells, artificially manufactured polyQ proteins were incorporated to the media of different mammalian cell cultures to verify and monitor their potential internalization, location after uptake and consequent toxic effects. Internalization of liposome-coated (4 h post-infection) and uncoated fibrils (24–48 h post-infection), characterized by 42 CAG repeats, were detected in both Cos-7 and PC-12 cells (Table 1). In this context, the presence of synthetic aggregates within the cytoplasm did not interfere with the physiological functions of the acceptor cells. To mimic the physiological expression of aggregates in the nucleus (Saudou et al., 1998)—which has been suggested to induce greater toxicity—the penetrance of the nuclear membrane was facilitated by the modification of the polyQ peptides by adding nuclear localization signals (NLS). While the F-Q42 was detected in proximity to the nuclear membrane, F-NLS-Q42 was easily delivered to the nucleus. Cell death was frequent in Cos-7 and PC-12 cells administered with F-LNS-Q42 or F-LNS-Q20 and was strictly associated with smaller sized fibrils (Yang et al., 2002), suggesting that the presence of polyQ into the nucleus is associated with high toxicity, independent of the polyQ length (Table 1).
A more elaborate study conducted in a wide variety of mammalian cells (Cos-7, HEK, N2A, CHO, and HeLa) revealed that only 1 h after contact, synthetic K2Q44K2 peptides were found within the cytoplasm, co-localizing with cytosolic quality control components in all cell types. Although their location was expected within the endosomal compartment (Lee et al., 2008), no co-localization with endosome, lysosome and autophagosome markers was revealed suggesting that fibrils were found free within the cytoplasm and were thus available to aggregate with other proteins including HTT. Transmission electron microscopy further uncovered that K2Q44K2 could reach the intracellular compartment by physically breaching plasma membranes (Figure 1; Table 1).
The hypothesis of free Q44 fibrils interacting with HTT was more specifically investigated by assessing the aggregation state of cyan fluorescent protein (CFP)-tagged HTTExon 1 Q25 (CFP–HTTQ25) in HEK cells after exposure with K2Q44K2. CFP fluorescence in cells not yet exposed to polyQ fibrils showed a diffuse nucleocytoplasmic distribution, as expected of the soluble HTTQ25 fragment. Following incubation with K2Q44K2 fibrils, CFP florescence co-localized with the fibrils in distinct puncta, suggesting the recruitment of the Q25 soluble fraction into the aggregates. Using other forms of fibrils (non-fibrillar HTTQ18 and fibrillar HTTQ51) in the same cell culture conditions, cytosolic nucleation was induced by fibrillar polyQ peptides (Ren et al., 2009; Figure 1; Table 2). This technique was used in several studies providing evidence for mHTT seeding capacities in different cell lines such as HeLa (Trevino et al., 2012) or human and murine neuroblastoma cells (Ruiz-Arlandis et al., 2016). Additionally, exposure of PC-12—that can inducibly express truncated exon1—with cerebrospinal fluid derived from postmortem samples of HD patients or living BACHD transgenic rats, showed that mHTT could trigger the aggregation process (Tan et al., 2015; Figure 1; Table 2).
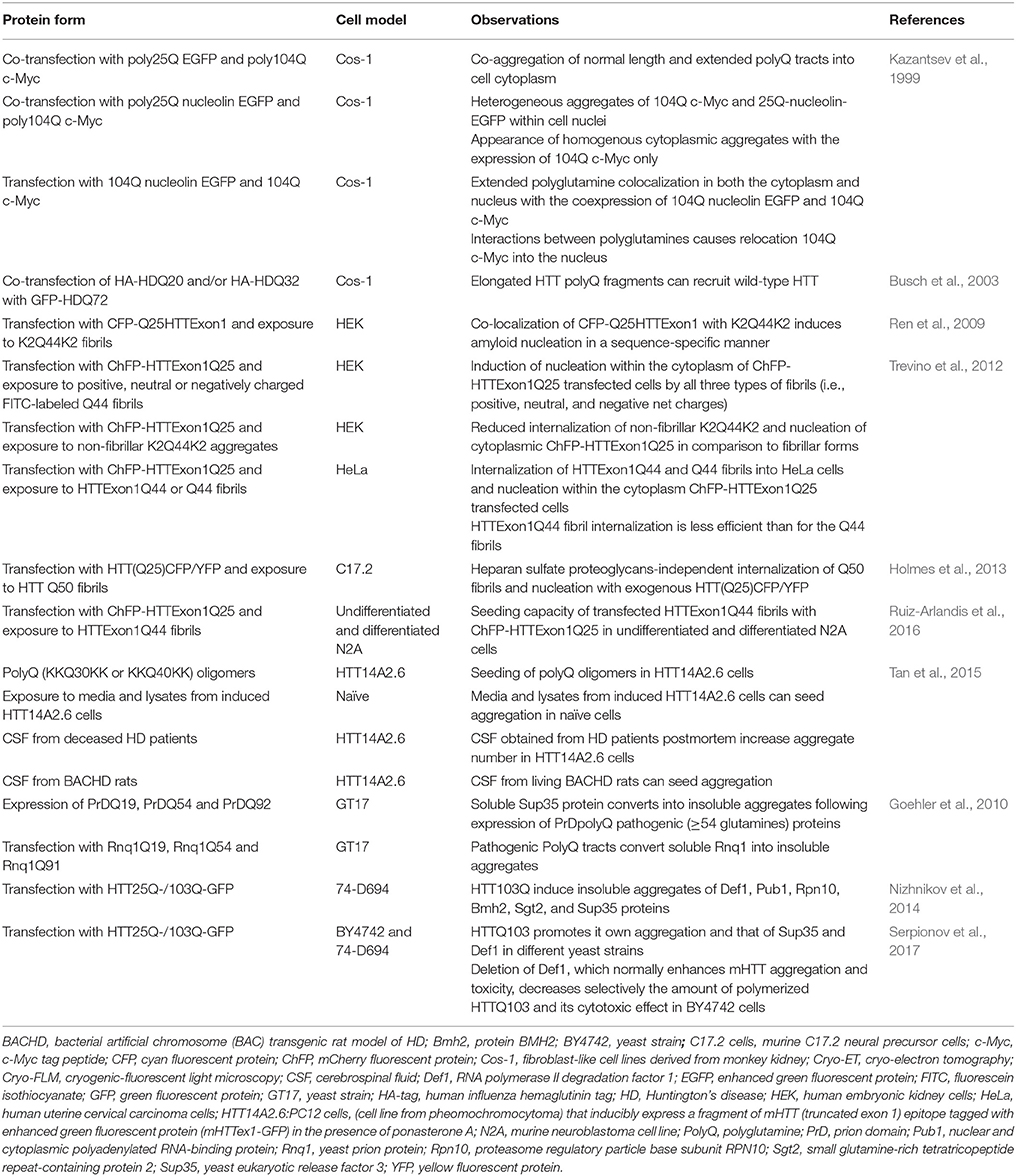
Table 2. In vitro evidence of mHTT seeding capacities.
mHTT Propagation Takes Place by Unknown Mechanisms
The seeding process of synthetic mHTT fibrils has been studied in greater detail with bimolecular fluorescence complementation assays (BiFC) alongside time-lapse microscopy, allowing for the visualization of HTTExon1 oligomer formation using halves of Venus fluorescent proteins (Herrera et al., 2011). With this approach, the reconstruction of a functional fluorophore testifies to the dimerization of mHTT fragments. A strong fluorescence signal was detected in Q103HTT-Venus transfected human glioma cells (H4), while much lower signals were measured in H4 cells expressing Q25HTT-Venus. Dimers of 103QHTT-Venus constructs appeared 30–45 min after exposure, and some cells began to show larger aggregates after only 1 h. Analyses at later time points, however, showed that all cells eventually died, indicating that both oligomers and inclusion bodies induced irreversible toxic effects. The cell-to-cell transmission potential of pathological mHTT exon1 fragment was further tested in H4 or HEK cells transfected with 103QHTT-V1 or 103QHTT-V2 plasmids and co-cultured. After 3 days, diffuse fluorescence revealed trafficking and, once again, the cell-to-cell transmission capacity of 103Q (Herrera et al., 2011; Table 1). One important point to take into consideration is that the mHTT propagation does not always require cell-to-cell contact. For example, the media of HEK cells overexpressing GFP-mHTT-Q19 or GFP-mHTT-Q103 triggers spreading of non-pathological (Q19) and pathological length (Q103) polyQ in SH-SY5Y cells after 5 days of incubation (Jeon et al., 2016; Table 2). This may indicate that, in in vivo contexts, mHTT could spread over extended distances and thereby exercise a toxic effect on remote cells.
Tau, α-synuclein and HTTQ50 fibrils can all be taken up by C17.2 cells and they are able to seed aggregation of intracellular tau RD-CFP/YFP, α-synuclein-CFP/YFP and HTT(Q25)CFP/YFP respectively (Table 2). In particular, internalization of tau and α-synuclein fibrils has been shown to be mediated by heparan sulfate proteoglycans (HSPGs)-binding, which stimulates cell uptake via macropinocytosis. This internalization can be inhibited by heparin, chlorate and heparinase III (Holmes et al., 2013). It should be noted, however, that such uptake has not been reported for HTTQ50 fibrils, which suggests it may use a different pathway (Holmes et al., 2013). Similarities and differences in uptake mechanisms between proteins will have to be carefully taken into account when designing treatment approaches.
Targeting mHTT Propagation Mechanisms as a Treatment Option
Taken together, there is now compelling evidence, with a large part emerging from in vitro studies, that mHTT, as for other proteins linked to neurodegenerative diseases, may behave in a prion-like fashion. However, given that HD is driven by a single gene that leads to the expression of mHTT in every cell of the body, is this of any relevance to the disease burden/onset? And can these propagation mechanisms be used for therapeutic benefits?
In many disorders, including HD, transynaptic/transneuronal spreading may be the favored route of protein transfer. Exocytosis, which occurs at the synaptic terminal, is regulated by a number of proteins, some of which interact pre- or post-synaptically with HTT. Consequently, mHTT can interfere with normal synaptic transmission by sequestering the wild-type protein, a phenomenon that is exacerbated as the polyQ length increases (Smith et al., 2005). The work of Pecho-Vrieseling and colleagues has provided evidence that mHTT itself can circulate between the pre- and post-synaptic membrane. Administration of BoNT—which works by cleaving and inactivating SNARE proteins (SNAP25 and VAMP-2)—blocked exocytosis and consequently mHTT transynaptic propagation (Pecho-Vrieseling et al., 2014).
mTOR inhibitors have also demonstrated to successfully rescue cortico-striatal degeneration in organotypic-striatal cultures derived from R6/2 and Hdh(CAG)150 HD mouse models. mTOR regulates a wide variety of cell functions—including autophagy downregulation—and the mTOR inhibitor AZD8055 has been shown to decrease the size of mHTT aggregates and the amount of insoluble mHTT in medium-spiny neurons in both the models cited above (Proenca et al., 2013). mTOR inhibitiors have also been found to reduce mHTT accumulation and alleviate toxicity in fly and mouse models of HD (Ravikumar et al., 2004), prevent levo-dopa induced dyskinesias in mouse models of PD (Santini et al., 2009) and ameliorate cognitive deficits in various AD animal models (Wang C. et al., 2014).
Transneuronal propagation of pathological proteins is not a unique characteristic of mHTT. It has been demonstrated for tau and β-amyloid in AD (de Calignon et al., 2012; Lee et al., 2012; Wu et al., 2013) and α-synuclein in PD (Desplats et al., 2009; Angot et al., 2012; Freundt et al., 2012; Luk et al., 2012a,b; Rey et al., 2016). Of great relevance is the fact that trans-neuronal spread of α-synuclein can be blocked by the monoclonal antibody 1H7 in vitro (Games et al., 2014) and in vivo (Spencer et al., 2017), ameliorating axonal transport and synaptic trafficking, raising the possibility of also treating HD through passive immunization. While the release of vesicles containing tau, α-synuclein and TDP-43 is increased by overexpression of the co-chaperone DnaJC5—possibly through non-canonical SNAP23 exocytosis—this is not the case for mHTT, indicating that not all pathological proteins may behave in the same manner under the same circumstances (Fontaine et al., 2016). We therefore may have to tailor therapies to specifically address the different mechanisms for propagation adopted by each protein, while other treatment options may be applicable to a range of proteins.
Previous reports have also indicated that tunneling nanotubes represent an efficient means for cells to communicate and share material. As we described above, F-actin is the principal component of the nanotube trafficking paths and its depolymerization by latrunculin or by cytochalasin B (Bukoreshtliev et al., 2009) abolishes the formation of TNTs, or at least reduces the number formed. If depolymerizing actin may be an attractive target by which to prevent bridge formation between cells—hence mHTT spreading—its implications for microfilaments and microtubules within the cytoskeleton organization would need to be known before applying such methodologies, given the detrimental consequences this may have on the cell integrity.
TNT formation is also promoted by oxidative stress, as shown with H2O2 administration and serum starvation (Wang et al., 2011). However, interventions designed to control oxidative stress are too non-specific to allow for the generation of a meaningful agent. Additionally, modulations of the M-Sec promoter protein (Hase et al., 2009), tumor suppressor p53, epidermal growth factor receptor (EGFR) and EGFR-regulated Akt, PI3K and mTOR (Wang et al., 2011) could be considered as targets given they all have a direct effect on nanotube growth. Unfortunately, reduction of physiological levels of any of these transcription factors could have devastating effects on the cell's vital functions.
If aiming to block the synthesis of F-actin, oxidative stress or various proteins involved in TNT formation is not a viable approach, the motor molecules that can carry mHTT could be a target option. For example, it has been reported that the transfer of endosome-related organelles is actomyosin dependent (Gurke et al., 2008) and therefore molecular motor myosin-X (Myo10) expression increases the number of TNTs and the transfer of vesicles between co-cultured cells (Gousset et al., 2013). In particular, a specific sequence of Myo10 is required for the formation and function of TNTs (Gousset et al., 2013), and its deletion does not affect filopodia involved in cell-to-cell communication (Bohil et al., 2006). However, it is important to note that TNT formation is beginning to be understood as a process that is cell-specific, in other words different cell lines can induce TNTs via different mechanisms (Gousset et al., 2013). This is a characteristic that is extremely important to take into consideration if we aim to inhibit TNT production in a specific cell population.
Endocytosis is also a very efficient pathway for mHTT internalization (Ruiz-Arlandis et al., 2016) and therefore could be considered as a process to target to halt propagation and disease dissemination. For example, chlorpromazine, monodansylcadaverine and dynasore can suppress mHTT uptake in N2A cells by a clathrin-dependent endocytosis mechanism (Ruiz-Arlandis et al., 2016). Although chlorpromazine has been used to treat psychotic disorders since the fifties, it is known to induce side-effects which include tardive dyskinesia, dystonia, motor restlessness and akathisia, which limits its use in a debilitating movement-disorder such as HD. Other pharmacological approaches could be employed to specifically inhibit clathrin-dependent endocytosis but a complete screening should be undertaken to identify such compounds, test their efficacy and monitor their potential side-effects as blocking this pathway could affect the well-being of the cells/neurons and only partially prevent mHTT propagation.
However, blocking mHTT uptake without targeting its release would not provide a fully efficient therapy and in order to address this issue, neutral sphingomyelinase and PI3-kinase inhibitors have been investigated and shown to be capable of reducing the secretion of mHTT–more specifically of 72Q and 25Q in transfected N2A cells, full length mHTT expressing lentiviruses were transduced in rat primary cortical neurons and endogenous mHTT of HdhQ111/Hdh+ and HdhQ111/HdhQ111 murine striatal cells. This event occurred simultaneously with the depletion of intracellular mHTT sequestered in vesicles, highlighting, once again, the role of late endosomes/lysosomes activity in mHTT secretion. Surprisingly, the knockdown of mHTT secretion by neutral sphingomyelinase inhibitors did not increase the amount of mHTT in the intracellular compartment, neither did it induce cell toxicity, raising the possibility to target release and propagation of free forms of mHTT. The side-effects of PI3-kinase inhibitors were not reported (Trajkovic et al., 2017). The absence of intracellular mHTT accumulation may indicate the existence of other release mechanisms (Jeon et al., 2016). Indeed, mHTT is very often detected outside the cell boundary, notably in the cerebrospinal fluid (Tan et al., 2015; Wild et al., 2015) within the extracellular matrix (Cicchetti et al., 2014), in blood vessels (Drouin-Ouellet et al., 2015) and possibly in plasma (Jeon et al., 2016). mHTT found in the extracellular milieu is also likely to emerge from cell death driven by mHTT toxicity (Cicchetti et al., 2014).
The plasma membrane constitutes a major barrier to external toxins. To preserve the homeostasis of its internal compartments, it selectivity permits compounds to cross or bind to the surface. Interactions between proteins and cell membranes can be physical, as determined by electrostatic attractions, or chemical, through specific or non-specific lipid, protein or carbohydrate bindings. In particular, binding of the HTT-N-terminus with the lipids of biological membrane has been shown to dependent on electrostatic interactions (Kegel et al., 2005). mHTT carries variable repetitions of polyglutamine residues at the N-terminus, and increasing the number of polyQ offers more insertion sites into the lipid bilayer. This has been demonstrated to occur via vesicles that simulate the biological membrane, corrupting its integrity (Kegel et al., 2009). Several other studies have demonstrated plasma membrane structural modifications following interactions with tau, Aβ or α-synuclein (Zhu et al., 2003; Flach et al., 2012). These interactions are well described in studies which compared the properties of α-synuclein and mHTT fibrils. They have revealed that soluble α-synuclein fibrils have differing binding capacities depending on their charge, while HTTExon1Q41 does not express this specificity, at least as demonstrated in synthetic vesicles (Pieri et al., 2012). It was also shown that HTTExon1 binding is dependent on the number of interaction sites, which is not the case for α-synuclein (Monsellier et al., 2016). Despite some dissimilarities, α-synuclein and mHTT fibrils do share common properties such as being able to modify the permeability of the lipid membrane by Ca2+ influxes (Monsellier et al., 2016). Furthermore, cholesterol concentration in the phospholipid bilayer impacts on its rigidity and stability, challenging the fibrils' capacity to alter the membrane's permeability (Pieri et al., 2012). In this regard, polyunsaturated fatty acid ethyl-eicosapentaenoic acid (ethyl-EPA) available in specific foods can dampen motor deficits in a HD mouse model (Clifford et al., 2002) probably due to its capacity to positively influence the plasma membrane properties, by preventing oxidative stress (Puri et al., 2005) or inhibiting apoptotic pathways (Murck and Manku, 2007). As its metabolite docosahexaenoic acid (DHA) (Wu et al., 2008), EPA was shown to modulate synaptic plasticity in ex vivo hippocampal slices from rats following the administration of ethyl-EPA for a period of 8 weeks (Kawashima et al., 2010). This compound has already completed a phase III clinical trial with some evidence of efficacy as shown by changes in the total motor scores in human patients with >45 CAG repeats (Puri et al., 2005).
Perspectives
In this review, we have presented the main findings from in vitro models, with a particular emphasis on mHTT propagation and seeding capacity, as it is becoming increasingly apparent that these mechanisms play a significant role in disease onset and manifestation and that targeting them may offer viable treatment options—keeping in mind that additional in vivo work must be conducted to go forward with the development of procedures that target this aspect of the disease. In the last few years, the focus of novel therapeutic approaches has been shifted and the aim is now to decrease the amounts of mHTT using approaches such as gene silencing (Lu and Yang, 2012) and editing (Yang et al., 2017). However, considerable challenges remain which relate to the capacity of mHTT to propagate between cells. Approaches designed to block propagation would not interfere with the primary aim of gene silencing/editing methodologies. This is particularly true in light of the fact that abnormalities in HD also lie outside the CNS (Sassone et al., 2009; Carroll et al., 2015). Future therapeutic perspectives will need to converge and tackle this genetic disorder from several fronts, including both pre-manifest and manifest patients, using combinations of agents that inhibit mHTT synthesis; restrict the damaging effects of the protein portion that is then secreted and by so doing prevent the accumulation and spread of this pathogenic protein.
Author Contributions
MM was responsible for conducting the literature search as well as designing tables and figures. FC conceptualized and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FC is a recipient of a Research Chair from the Fonds de Recherche du Québec en Santé (FRQS, 35059) providing salary support and operating funds, and receives funding from the Canadian Institutes of Health Research (CIHR, MOP136883) to conduct her HD-related research. The authors would like to thank Mr. Gilles Chabot for artwork.
References
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016a). Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Abounit, S., Wu, J. W., Duff, K., Victoria, G. S., and Zurzolo, C. (2016b). Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10, 344–351. doi: 10.1080/19336896.2016.1223003
Abounit, S., and Zurzolo, C. (2012). Wiring through tunneling nanotubes–from electrical signals to organelle transfer. J. Cell Sci. 125, 1089–1098. doi: 10.1242/jcs.083279
Angot, E., Steiner, J. A., Lema Tomé, C. M., Ekström, P., Mattsson, B., Björklund, A., et al. (2012). Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 7:e39465. doi: 10.1371/journal.pone.0039465
Babcock, D. T., and Ganetzky, B. (2015). Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc. Natl. Acad. Sci. U.S.A. 112, E5427–E5433. doi: 10.1073/pnas.1516217112
Bohil, A. B., Robertson, B. W., and Cheney, R. E. (2006). Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. U.S.A. 103, 12411–12416. doi: 10.1073/pnas.0602443103
Brahic, M., Bousset, L., Bieri, G., Melki, R., and Gitler, A. D. (2016). Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol. 131, 539–548. doi: 10.1007/s00401-016-1538-0
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Bukoreshtliev, N. V., Wang, X., Hodneland, E., Gurke, S., Barroso, J. F. V., and Gerdes, H.-H. (2009). Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett. 583, 1481–1488. doi: 10.1016/j.febslet.2009.03.065
Busch, A., Engemann, S., Lurz, R., Okazawa, H., Lehrach, H., and Wanker, E. E. (2003). Mutant Huntingtin Promotes the Fibrillogenesis of Wild-type Huntingtin a potential mechanism for loss of huntingtin function in huntington's disease. J. Biol. Chem. 278, 41452–41461. doi: 10.1074/jbc.M303354200
Carroll, J. B., Bates, G. P., Steffan, J., Saft, C., and Tabrizi, S. J. (2015). Treating the whole body in Huntington's disease. Lancet Neurol. 14, 1135–1142. doi: 10.1016/S1474-4422(15)00177-5
Cicchetti, F., Lacroix, S., Cisbani, G., Vallières, N., Saint-Pierre, M., St-Amour, I., et al. (2014). Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann. Neurol. 76, 31–42. doi: 10.1002/ana.24174
Clifford, J. J., Drago, J., Natoli, A. L., Wong, J. Y. F., Kinsella, A., Waddington, J. L., et al. (2002). Essential fatty acids given from conception prevent topographies of motor deficit in a transgenic model of Huntington's disease. Neuroscience 109, 81–88. doi: 10.1016/S0306-4522(01)00409-2
Costanzo, M., Abounit, S., Marzo, L., Danckaert, A., Chamoun, Z., Roux, P., et al. (2013). Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 126, 3678–3685. doi: 10.1242/jcs.126086
de Calignon, A., Polydoro, M., Suárez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Desplats, P., Lee, H.-J., Bae, E.-J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Dong, X., Zong, S., Witting, A., Lindenberg, K. S., Kochanek, S., and Huang, B. (2012). Adenovirus vector-based in vitro neuronal cell model for Huntington's disease with human disease-like differential aggregation and degeneration. J. Gene Med. 14, 468–481. doi: 10.1002/jgm.2641
Drouin-Ouellet, J., Sawiak, S. J., Cisbani, G., Lagacé, M., Kuan, W.-L., Saint-Pierre, M., et al. (2015). Cerebrovascular and blood-brain barrier impairments in Huntington's disease: potential implications for its pathophysiology. Ann. Neurol. 78, 160–177. doi: 10.1002/ana.24406
Flach, K., Hilbrich, I., Schiffmann, A., Gärtner, U., Krüger, M., Leonhardt, M., et al. (2012). Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 287, 43223–43233. doi: 10.1074/jbc.M112.396176
Fontaine, S. N., Zheng, D., Sabbagh, J. J., Martin, M. D., Chaput, D., Darling, A., et al. (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. doi: 10.15252/embj.201593489
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Games, D., Valera, E., Spencer, B., Rockenstein, E., Mante, M., Adame, A., et al. (2014). Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in parkinson's disease-like models. J. Neurosci. 34, 9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014
Goedert, M., Clavaguera, F., and Tolnay, M. (2010). The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325. doi: 10.1016/j.tins.2010.04.003
Goehler, H., Dröge, A., Lurz, R., Schnoegl, S., Chernoff, Y. O., and Wanker, E. E. (2010). Pathogenic polyglutamine tracts are potent inducers of spontaneous Sup35 and Rnq1 amyloidogenesis. PLoS ONE 5:e9642. doi: 10.1371/journal.pone.0009642
Gousset, K., Marzo, L., Commere, P.-H., and Zurzolo, C. (2013). Myo10 is a key regulator of TNT formation in neuronal cells. J. Cell Sci. 126, 4424–4435. doi: 10.1242/jcs.129239
Gousset, K., Schiff, E., Langevin, C., Marijanovic, Z., Caputo, A., Browman, D. T., et al. (2009). Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336. doi: 10.1038/ncb1841
Guo, J. L., and Lee, V. M. Y. (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. doi: 10.1038/nm.3457
Gurke, S., Barroso, J. F. V., and Gerdes, H.-H. (2008). The art of cellular communication: tunneling nanotubes bridge the divide. Histochem. Cell Biol. 129, 539–550. doi: 10.1007/s00418-008-0412-0
Hansen, C., Angot, E., Bergström, A.-L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hase, K., Kimura, S., Takatsu, H., Ohmae, M., Kawano, S., Kitamura, H., et al. (2009). M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 11, 1427–1432. doi: 10.1038/ncb1990
Herrera, F., Tenreiro, S., Miller-Fleming, L., and Outeiro, T. F. (2011). Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. 3:RRN1210. doi: 10.1371/currents.RRN1210
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Jeon, I., Cicchetti, F., Cisbani, G., Lee, S., Li, E., Bae, J., et al. (2016). Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 132, 577–592. doi: 10.1007/s00401-016-1582-9
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kawashima, A., Harada, T., Kami, H., Yano, T., Imada, K., and Mizuguchi, K. (2010). Effects of eicosapentaenoic acid on synaptic plasticity, fatty acid profile and phosphoinositide 3-kinase signaling in rat hippocampus and differentiated PC12 cells. J. Nutr. Biochem. 21, 268–277. doi: 10.1016/j.jnutbio.2008.12.015
Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. (1999). Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 96, 11404–11409. doi: 10.1073/pnas.96.20.11404
Kegel, K. B., Kim, M., Sapp, E., McIntyre, C., Castaño, J. G., Aronin, N., et al. (2000). Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. Off. J. Soc. Neurosci. 20, 7268–7278.
Kegel, K. B., Sapp, E., Yoder, J., Cuiffo, B., Sobin, L., Kim, Y. J., et al. (2005). Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 280, 36464–36473. doi: 10.1074/jbc.M503672200
Kegel, K. B., Schewkunow, V., Sapp, E., Masso, N., Wanker, E. E., DiFiglia, M., et al. (2009). Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem. Biophys. Res. Commun. 387, 472–475. doi: 10.1016/j.bbrc.2009.07.039
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506. doi: 10.1038/nm1747
Lee, H.-J., Suk, J.-E., Bae, E.-J., Lee, J.-H., Paik, S. R., and Lee, S.-J. (2008). Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. doi: 10.1016/j.biocel.2008.01.017
Lee, S., Kim, W., Li, Z., and Hall, G. F. (2012). Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int. J. Alzheimer's Dis. 2012:172837. doi: 10.1155/2012/172837
Li, J.-Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Lu, X.-H., and Yang, X. W. (2012). “Huntingtin holiday”: progress toward an Antisense Therapy for Huntington's Disease. Neuron 74, 964–966. doi: 10.1016/j.neuron.2012.06.001
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O'Brien, P., Trojanowski, J. Q., et al. (2012a). Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in non-transgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Kehm, V. M., Zhang, B., O'Brien, P., Trojanowski, J. Q., and Lee, V. M. Y. (2012b). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Marzo, L., Gousset, K., and Zurzolo, C. (2012). Multifaceted roles of tunneling nanotubes in intercellular communication. Front. Physiol. 3:72. doi: 10.3389/fphys.2012.00072
Melentijevic, I., Toth, M. L., Arnold, M. L., Guasp, R. J., Harinath, G., Nguyen, K. C., et al. (2017). C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542, 367–371. doi: 10.1038/nature21362
Miaczynska, M., and Stenmark, H. (2008). Mechanisms and functions of endocytosis. J. Cell Biol. 180, 7–11. doi: 10.1083/jcb.200711073
Monsellier, E., Bousset, L., and Melki, R. (2016). α-Synuclein and huntingtin exon 1 amyloid fibrils bind laterally to the cellular membrane. Sci. Rep. 6:19180. doi: 10.1038/srep19180
Murck, H., and Manku, M. (2007). Ethyl-EPA in Huntington disease: potentially relevant mechanism of action. Brain Res. Bull. 72, 159–164. doi: 10.1016/j.brainresbull.2006.10.020
Nizhnikov, A. A., Alexandrov, A. I., Ryzhova, T. A., Mitkevich, O. V., Dergalev, A. A., Ter-Avanesyan, M. D., et al. (2014). Proteomic screening for amyloid proteins. PLOS ONE 9:e116003. doi: 10.1371/journal.pone.0116003
Oh, S. H., Kim, H. N., Park, H. J., Shin, J. Y., Bae, E.-J., Sunwoo, M. K., et al. (2016). Mesenchymal stem cells inhibit transmission of α-synuclein by modulating clathrin-mediated endocytosis in a parkinsonian model. Cell Rep. 14, 835–849. doi: 10.1016/j.celrep.2015.12.075
Olanow, C. W., and Prusiner, S. B. (2009). Is Parkinson's disease a prion disorder? Proc. Natl. Acad. Sci. U.S.A. 106, 12571–12572. doi: 10.1073/pnas.0906759106
Pearce, M. M. P., Spartz, E. J., Hong, W., Luo, L., and Kopito, R. R. (2015). Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 6:6768. doi: 10.1038/ncomms7768
Pecho-Vrieseling, E., Rieker, C., Fuchs, S., Bleckmann, D., Esposito, M. S., Botta, P., et al. (2014). Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 17, 1064–1072. doi: 10.1038/nn.3761
Pieri, L., Madiona, K., Bousset, L., and Melki, R. (2012). Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905. doi: 10.1016/j.bpj.2012.04.050
Proenca, C. C., Stoehr, N., Bernhard, M., Seger, S., Genoud, C., Roscic, A., et al. (2013). Atg4b-dependent autophagic flux alleviates huntington's disease progression. PLoS ONE 8:e68357. doi: 10.1371/journal.pone.0068357
Puri, B. K., Leavitt, B. R., Hayden, M. R., Ross, C. A., Rosenblatt, A., Greenamyre, J. T., et al. (2005). Ethyl-EPA in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology 65, 286–292. doi: 10.1212/01.wnl.0000169025.09670.6d
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Ren, P.-H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225. doi: 10.1038/ncb1830
Rey, N. L., Steiner, J. A., Maroof, N., Luk, K. C., Madaj, Z., Trojanowski, J. Q., et al. (2016). Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J. Exp. Med. 213:1759. doi: 10.1084/jem.20160368
Ruiz-Arlandis, G., Pieri, L., Bousset, L., and Melki, R. (2016). Binding, internalization and fate of Huntingtin Exon1 fibrillar assemblies in mitotic and nonmitotic neuroblastoma cells. Neuropathol. Appl. Neurobiol. 42, 137–152. doi: 10.1111/nan.12258
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009). Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2:ra36. doi: 10.1126/scisignal.2000308
Sassone, J., Colciago, C., Cislaghi, G., Silani, V., and Ciammola, A. (2009). Huntington's disease: the current state of research with peripheral tissues. Exp. Neurol. 219, 385–397. doi: 10.1016/j.expneurol.2009.05.012
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/S0092-8674(00)81782-1
Serpionov, G. V., Alexandrov, A. I., and Ter-Avanesyan, M. D. (2017). Distinct mechanisms of mutant huntingtin toxicity in different yeast strains. FEMS Yeast Res. 17:fow102. doi: 10.1093/femsyr/fow102
Smith, I. F., Shuai, J., and Parker, I. (2011). Active generation and propagation of Ca2+ signals within tunneling membrane nanotubes. Biophys. J. 100, L37–L39. doi: 10.1016/j.bpj.2011.03.007
Smith, R., Brundin, P., and Li, J.-Y. (2005). Synaptic dysfunction in Huntington's disease: a new perspective. Cell. Mol. Life Sci. 62, 1901–1912. doi: 10.1007/s00018-005-5084-5
Soto, C. (2012). Transmissible proteins: expanding the prion heresy. Cell 149, 968–977. doi: 10.1016/j.cell.2012.05.007
Spencer, B., Valera, E., Rockenstein, E., Overk, C., Mante, M., Adame, A., et al. (2017). Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol. Commun. 5:7. doi: 10.1186/s40478-016-0410-8
Tan, Z., Dai, W., van Erp, T. G. M., Overman, J., Demuro, A., Digman, M. A., et al. (2015). Huntington's disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol. Psychiatry 20, 1286–1293. doi: 10.1038/mp.2015.81
Trajkovic, K., Jeong, H., and Krainc, D. (2017). Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci. 37, 9000–9012. doi: 10.1523/JNEUROSCI.0118-17.2017
Trevino, R. S., Lauckner, J. E., Sourigues, Y., Pearce, M. M., Bousset, L., Melki, R., et al. (2012). Fibrillar structure and charge determine the interaction of polyglutamine protein aggregates with the cell surface. J. Biol. Chem. 287, 29722–29728. doi: 10.1074/jbc.M112.372474
Trushina, E., Dyer, R. B., Badger, J. D., Ure, D., Eide, L., Tran, D. D., et al. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24, 8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wang, C., Yu, J.-T., Miao, D., Wu, Z.-C., Tan, M.-S., and Tan, L. (2014). Targeting the mTOR signaling network for Alzheimer's disease therapy. Mol. Neurobiol. 49, 120–135. doi: 10.1007/s12035-013-8505-8
Wang, N., Gray, M., Lu, X.-H., Cantle, J. P., Holley, S. M., Greiner, E., et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat. Med. 20, 536–541. doi: 10.1038/nm.3514
Wang, Y., Cui, J., Sun, X., and Zhang, Y. (2011). Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 18, 732–742. doi: 10.1038/cdd.2010.147
Watkins, S. C., and Salter, R. D. (2005). Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 23, 309–318. doi: 10.1016/j.immuni.2005.08.009
Weiss, A., Träger, U., Wild, E. J., Grueninger, S., Farmer, R., Landles, C., et al. (2012). Mutant huntingtin fragmentation in immune cells tracks Huntington's disease progression. J. Clin. Invest. 122, 3731–3736. doi: 10.1172/JCI64565
Wild, E. J., Boggio, R., Langbehn, D., Robertson, N., Haider, S., Miller, J. R. C., et al. (2015). Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J. Clin. Invest. 125, 1979–1986. doi: 10.1172/JCI80743
Wu, A., Ying, Z., and Gomez-Pinilla, F. (2008). DHA dietary supplementation enhances the effects of exercise on synaptic plasticity and cognition. Neuroscience 155, 751–759. doi: 10.1016/j.neuroscience.2008.05.061
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Yang, S., Chang, R., Yang, H., Zhao, T., Hong, Y., Kong, H. E., et al. (2017). CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J. Clin. Invest. 127, 2719–2724. doi: 10.1172/JCI92087
Yang, W., Dunlap, J. R., Andrews, R. B., and Wetzel, R. (2002). Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 11, 2905–2917. doi: 10.1093/hmg/11.23.2905
Zala, D., Benchoua, A., Brouillet, E., Perrin, V., Gaillard, M.-C., Zurn, A. D., et al. (2005). Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol. Dis. 20, 785–798. doi: 10.1016/j.nbd.2005.05.017
Keywords: Huntington's disease, Parkinson's disease, Alzheimer's disease, prions, tau, β-amyloid, α-synuclein, cell culture
Citation: Masnata M and Cicchetti F (2017) The Evidence for the Spread and Seeding Capacities of the Mutant Huntingtin Protein in in Vitro Systems and Their Therapeutic Implications. Front. Neurosci. 11:647. doi: 10.3389/fnins.2017.00647
Received: 19 September 2017; Accepted: 07 November 2017;
Published: 28 November 2017.
Edited by:
Dustin R. Wakeman, RxGen (United States), United StatesReviewed by:
Nolwen Lydiane Rey, Van Andel Institute, United StatesPatrícia Maciel, University of Minho, Portugal
Copyright © 2017 Masnata and Cicchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Cicchetti, ZnJhbmNlc2NhLmNpY2NoZXR0aUBjcmNodWwudWxhdmFsLmNh