 Rufus O. Akinyemi
Rufus O. Akinyemi Louise M. Allan2
Louise M. Allan2 Rajesh N. Kalaria
Rajesh N. Kalaria- 1Neuroscience and Ageing Research Unit, Institute for Advanced Medical Research and Training, College of Medicine, University of Ibadan, Oyo, Nigeria
- 2Neurovascular Research Group, Institute of Neuroscience, Newcastle University, Newcastle upon Tyne, United Kingdom
Neuroimaging evidence from older stroke survivors in Nigeria and Northeast England showed medial temporal lobe atrophy (MTLA) to be independently associated with post-stroke cognitive impairment and dementia. Given the hypothesis ascribing MTLA to neurodegenerative processes, we assessed Alzheimer pathology in the hippocampal formation and entorhinal cortex of autopsied brains from of post-stroke demented and non-demented subjects in comparison with controls and other dementias. We quantified markers of amyloid β (total Aβ, Aβ-40, Aβ-42, and soluble Aβ) and hyperphosphorylated tau in the hippocampal formation and entorhinal cortex of 94 subjects consisting of normal controls (n = 12), vascular dementia, VaD (17), post-stroke demented, PSD (n = 15), and post-stroke non-demented, PSND (n = 23), Alzheimer's disease, AD (n = 14), and mixed AD and vascular dementia, AD_VAD (n = 13) using immunohistochemical techniques. We found differential expression of amyloid and tau across the disease groups, and across hippocampal sub-regions. Among amyloid markers, the pattern of Aβ-42 immunoreactivity was similar to that of total Aβ. Tau immunoreactivity showed highest expression in the AD and mixed AD and vascular dementia, AD_VaD, which was higher than in control, post - stroke and VaD groups (p < 0.05). APOE ε4 allele positivity was associated with higher expression of amyloid and tau pathology in the subiculum and entorhinal cortex of post-stroke cases (p < 0.05). Comparison between PSND and PSD revealed higher total Aβ immunoreactivity in PSND compared to PSD in the CA1, subiculum and entorhinal cortex (p < 0.05) but no differences between PSND and PSD in Aβ-42, Aβ-40, soluble Aβ or tau immunoreactivities (p > 0.05). Correlation of MMSE and CAMCOG scores with AD pathological measures showed lack of correlation with amyloid species although tau immunoreactivity demonstrated correlation with memory scores (p < 0.05). Our findings suggest hippocampal AD pathology does not necessarily differ between demented and non-demented post-stroke subjects. The dissociation of cognitive performance with hippocampal AD pathological burden suggests more dominant roles for non-Alzheimer neurodegenerative and / or other non-neurodegenerative substrates for dementia following stroke.
Introduction
Stroke accounts for half of the global burden of neurological disorders while remaining the most common cause of acquired disability, and a common cause of cognitive impairment and dementia (Kalaria et al., 2016; Writing Group Members et al., 2016; GBD 2015 Neurological Disorders Collaborator Group, 2017). Magnetic resonance imaging studies in a cohort of older African stroke survivors participating in the Cognitive Function After STroke (CogFAST)—Nigeria Study showed that medial temporal lobe atrophy (MTLA) was an independent predictor of post-stroke cognitive impairment and dementia (Akinyemi et al., 2014, 2015). These findings were in concordance with previous findings of MTLA significantly predicting progression to cognitive impairment, dementia and death in the longitudinal CogFAST Newcastle Study (Firbank et al., 2007, 2012) while pathological studies unmasked hippocampal neuronal atrophy as an important neuropathological substrate of post-stroke dementia (PSD) in the Newcastle cohort (Gemmell et al., 2012).
The link between cerebrovascular disease, neurodegeneration and cognition has long been debated (de la Torre and Mussivand, 1993; Kalaria et al., 1993b). Evidence for this link has been provided by experimental models (Kalaria et al., 1993a; Kitaguchi et al., 2009) and from epidemiological studies (Schneider et al., 2004; Petrovitch et al., 2005). Accumulation of Alzheimer pathology in primary vascular brain disorders occurs largely as a result of shared mechanisms of neurovascular unit dysfunction (Iadecola, 2004; Kalaria et al., 2012). Experimental evidence from animal studies has shown that amyloid production may be exacerbated by cerebral hypoxia/ischaemia (Kalaria et al., 1993a; Whitehead et al., 2005).
In a large post-mortem study, Aho and colleagues using immunohistochemistry found no significant increase in amyloid load in subjects with cerebrovascular disease (CVD) compared to controls although there was a trend of increased deposition of Aβ-42 over Aβ-40 (Aho et al., 2006). In this context a post-mortem study of aging controls and subjects with vascular dementia (VaD), reported increased accumulation of total guanidine HCl extractable Aβ-42 peptides (over Aβ-40) by Enzyme Linked Immunosorbent Assay (ELISA) in the temporal cortex in the oldest VaD subjects as well as in older aged controls (Lewis et al., 2006) although these increases were not evident by immunohistochemistry.
Cross-sectional studies examining the relationship of Aβ with cognitive function have also yielded conflicting results. Whereas, some studies have reported significant correlation between metrics of AD pathology and cognitive performance (Bennett et al., 2006; Mormino et al., 2009) others have reported dissociation between Alzheimer pathological load and cognitive status especially in subjects with presumed high cognitive reserve, mixed pathologies or non-AD subjects (Mufson et al., 1999; Aizenstein et al., 2008; Stern, 2009). Similarly, the morphological variants and anatomical localization of the AD pathology may be important. Neuritic plaques and neurofibrillary tangles have stronger impact on cognition than diffuse plaques, and pathologies in the neocortical region exert more influence than those in the allocortical regions (Nelson et al., 2009, 2012).
The hippocampus plays a very strategic role in the neurobiology of memory, being involved in the hierarchical spread of amyloid and tau (neurofibrillary tangles) (Braak and Braak, 1991; Thal et al., 2002b). Whereas, amyloid is deposited in the hippocampus in late stages, tau deposition occurs quite early within the hippocampal formation during their natural histories, and both deposits appear to relate to the connections of the hippocampal circuitry (Lace et al., 2009).
Given the hypothesis ascribing MTLA to neurodegenerative processes (Henon et al., 1998; Cordoliani-Mackowiak et al., 2003; Firbank et al., 2007), we investigated Alzheimer pathology in the hippocampal formation and entorhinal cortex of brain tissues obtained from the CogFAST (Cognitive Function after Stroke) Newcastle post-stroke cohort. Our objective was to quantify the presence of markers of amyloid pathology (total Aβ, Aβ-40, Aβ-42, and soluble Aβ) and a marker of hyperphosphorylated tau pathology in post-stroke subjects with and without dementia compared to aging controls, Alzheimer's disease (AD) and mixed AD and vascular dementia (AD-VaD). We hypothesized that in tandem with MTLA, hippocampal Alzheimer pathology would be differentially expressed in demented and non-demented stroke survivors in comparison with other dementias and aging controls.
Methods
Subjects
Ninety-four human post-mortem brains were retrieved from the Newcastle Brain Tissue Resource (NBTR) at the Campus for Ageing and Vitality, Newcastle University, UK. Table 1 provides details of the demographic, cognitive and pathologic characteristics of the subjects. Post-stroke subjects in the longitudinal CogFAST-Newcastle Study were classified based on the performance at the last cognitive assessment before death. They were classified as post-stroke non-demented (PSND) if CAMCOG score was >80 and Clinical Dementia Rating (CDR) was less than 1, but as post-stroke demented (PSD) if CAMCOG score was <80 and CDR score was >1 (Allan et al., 2011). Control subjects were historical subjects that had no significant evidence of cognitive impairment upon scrutiny of their medical records and whose post-mortem brain tissue was considered devoid of sufficient vascular or degenerative pathologies beyond the threshold for assigning a specific pathologic diagnosis (Kalaria et al., 2004; Gemmell et al., 2012).
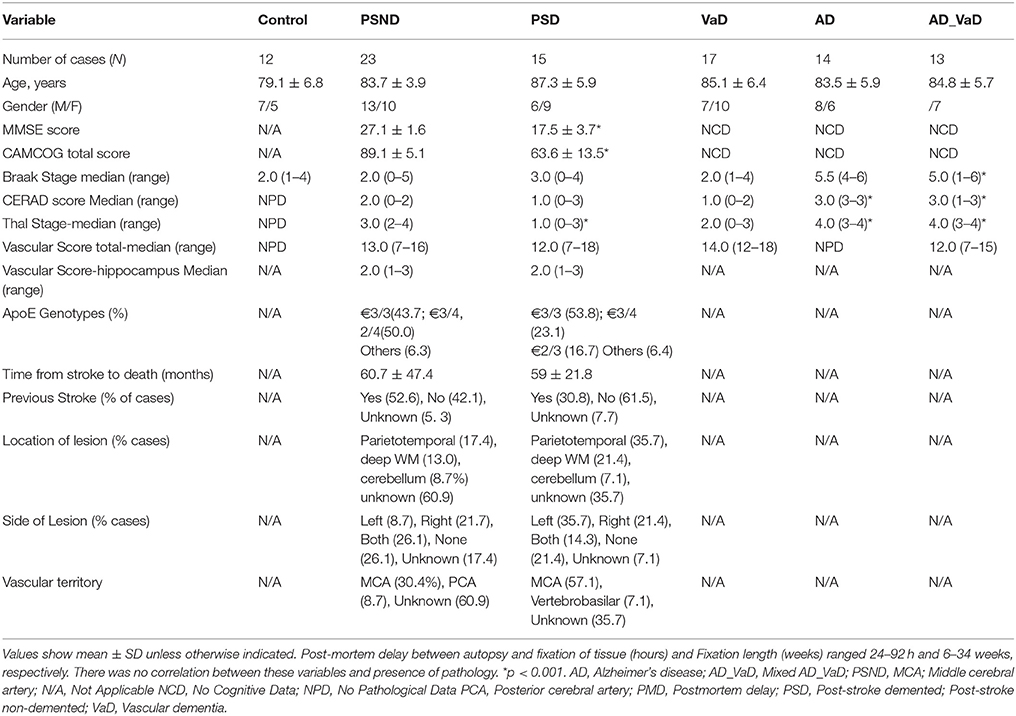
Table 1. Characteristics of study subjects.
Autopsies were performed between 24 and 92 h after death and brains were fixed for between 6 and 34 weeks. Cognitive scores on the Mini-Mental State Examination (MMSE) and Cambridge Cognitive Examination (CAMCOG) proximate to death as well as APOE genotypes were retrieved from clinical and research records of the subjects in the CogFAST-Newcastle cohort. The CogFAST-Newcastle Study and ancillary studies had ethical approval from the local Newcastle Ethical committees and participants gave written consent for brain tissue donation. Use of brain tissue was also approved by the local Ethical committees (Newcastle upon Tyne Hospitals National Health Service Trust, UK) and the committee of the NBTR.
Post-mortem reports were retrieved for all the cases used in this study. Primary neuropathological diagnoses were made from brain tissue sampled at several coronal levels (Perry and Oakley, 1993) to check for pathological changes consistent with AD, VaD and mixed AD_VaD in accordance with established pathologic diagnostic criteria (Hyman and Trojanowski, 1997; Kalaria et al., 2004), and following macroscopic and microscopic post-mortem examination of the brain tissue. Haematoxylin and Eosin was utilized as a standard stain for a general neuropathologic structural evaluation of the brain, and for the detection of infarcts and rarefactions. Gallyas and Bielschowsky's silver impregnation stains and AT8 immunohistochemistry were used to evaluate “CERAD” neuritic plaques and neurofibrillary tangles according to the methods of Braak (Braak and Braak, 1991; Mirra et al., 1991). In addition, vascular lesions (cortical and sub-cortical infarcts, border-zone infarcts, strategic infarcts, lacunar infarcts (<15 mm), microinfarcts (<5 mm) and mild, moderate and severe cerebral amyloid angiopathy were recorded (Kalaria et al., 2004). Final diagnoses were assigned during monthly clinicopathologic consensus meetings. A final diagnosis of VaD was made if there was clinical evidence of dementia (DSM IV) and pathologic evidence of multiple or cystic infarcts, lacunes, micro-infarcts, small vessel disease in the absence of severe degenerative pathology (Braak Stage <III) (Kalaria et al., 2004). Subjects were assigned mixed AD_VaD if there was pathologic evidence of cerebrovascular disease in the presence of significant AD pathology (Braak Stage V or VI) and moderate to severe CERAD scores. A diagnosis of AD was assigned when there was significant Alzheimer pathology-Braak V–VI, moderate to severe CERAD score and absence of significant vascular pathology.
APOE Genotyping
APOE genotyping was undertaken in the CogFAST cohort only (PSND and PSD) as shown in Table 1 using restriction enzyme isoform genotyping as previously described (Hixson and Vernier, 1990; Rowan et al., 2005). In brief, ApoE restriction isotyping used oligonucleotides to amplify apolipoprotein E gene sequences containing amino acid positions 112 and 158. The amplification products were digested with HhaI and subjected to electrophoresis on polyacrylamide gels. Each of the isoforms was distinguished by a unique combination of HhaI fragment sizes that enabled unambiguous typing of all homozygotic and heterozygotic combinations. HhaI cleaves at GCGC encoding 112arg (E4) and 158arg (E3, E4), but does not cut at GTGC encoding 112cys (E2, E3) and 158cys (E2).
Immunohistochemistry
Paraffin embedded brain tissue blocks taken from relevant coronal levels of the Newcastle Brain Map (Perry and Oakley, 1993) and containing the hippocampal formation and entorhinal cortex were cut into 10 μm serial sections using a rotary microtome. Sections were mounted on slides coated with 2% APES (3-aminopropyltrethoxysilane) solution in acetone, and dried in a pre-heated oven at 600C for 30 min. The cut sections were then serially immunostained in duplicates with primary antibodies to different amyloid-β species and tau (Table 2). The sections were first de-paraffinized in two sequential solutions of Xylene for 15 min and then rehydrated using decreasing concentrations of ethanol (100, 95, 70, and 50%) to deionized water. Antigen retrieval was performed for Aß-immunolabeling using formic acid-pretreatment for 4 h and for tau immunolabeling using heat in the form of microwaving sections for 10 min in a solution of 0.01M citrate buffer (PH 6.0). The buffer was brought to boil in the microwave, slides were added, buffer was microwaved at 450W for 10 min, and the solution was then allowed to cool for 20 min following which slides were transferred to deionized water. Non-specific reaction was quenched with 0.9% hydrogen peroxide (unless otherwise stated.) in 5 Mm Tris buffered Saline (TBS) solution (pH 7.6) for 15 min in order to remove endogenous peroxidise. Non-specific antigens were blocked using normal horse serum (anti-mouse antibody: 4G8, NU-1 and AT8) and normal goat serum (anti-rabbit antibody: T-40 and T-42 for 60 min. The slides were then incubated for 2 h at room temperature (AT8) or overnight at 40C (4G8, T-40, T-42, and NU-1) with the primary antibody diluted to specific concentration with buffer: total amyloid β (4G8, 1: 1000, Mouse monoclonal, Signet 9220-10), amyloid β-42 (T-42, 1: 5000, Rabbit polyclonal, gift; H. Mori, Japan), amyloid β-40 (T-40, 1: 5000, Rabbit polyclonal, gift; H. Mori, Japan), soluble amyloid oligomer (NU-1, Mouse monoclonal, gift: C. Klein, US) and antibody to hyperphosphorylated tau (AT8, 1: 1000, Mouse monoclonal Autogen Bioclear). After washes in buffer, biotinylated secondary antibody was applied to the sections with the blocking serum for 30 min, followed by the addition of the streptavidin/biotinyl-horseradish peroxidase (HRP) complex (SABC) for 30 min to remove excess secondary antibody. Finally, the slides were immersed in a 0.025% diaminobenzidine (DAB) solution for a variable short period of time to visualize the positive antibody reaction. Sections were then rinsed in water and counterstained in haematoxylin as indicated. Sections were then dehydrated back through graded alcohols, cleared in xylene and mounted with glass coverslips using DPX mounting medium (Sigma, UK). After each step, with the exception of the blocking stage, sections were rinsed in buffer (TBS) three times for 5 min each. All immuno-histochemical protocols included a positive control and a negative control for which the primary antibody is omitted. All the antibodies used in the analysis have been previously well characterized. We have published on use of these antibodies as described in our previous publications (Chang et al., 2003; Lewis et al., 2006; Ndung'u et al., 2012; Mukaetova-Ladinska et al., 2015).
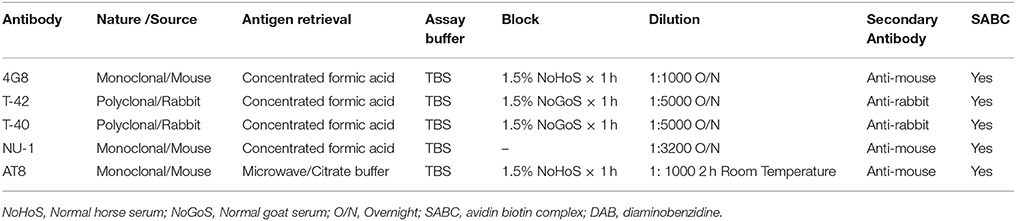
Table 2. Summary of immunoperoxidase staining with DAB as chromogen.
Microscopy and Image Analysis
Stained sections were examined and digitized using a Zeiss Axioplan 2 research grade microscope coupled to an Infinity 2 camera. Magnification was set at X10 for the hippocampal sub-regions CA1, CA2 and CA3, and X5 for the subiculum and entorhinal cortex (EC). Five images were taken at random from the CA1, CA3 and subiculum, 3 images from CA2 and 4 × 3 from the EC from the pial surface to the white matter. Approximately 2,820 images were taken.
Images were analyzed using Image Pro Plus 6.3 (Media Cybernetics, Silver Spring, MD, USA). The area of interest (AOI) was delineated on-screen using the wand tool and the size of the area selected was recorded. Histogram based analysis was used to measure the number of pixels stained (“per area,” PA) and intensity of stain (integrated optical density, IOD), as determined by the operator. Background was excluded by highlighting only the positive staining. To standardize the measurements, the range of stain intensity for each AOI was determined between 0 and 255 (where 0 = black and 255 = white, as set up for the Image Pro Plus Histogram based analysis). Using the sum of each of the three measures, further calculations were undertaken to generate the metrics of immunostaining: Percent of the area of interest positively stained, “Percent area (%PA)” = Per Area × 100; Mean intensity of stain per pixel, “Integrated Optical Density (IOD)” = 255 – (sum IOD/area). The mean % PA and IOD were then calculated for each subject by computing averages from the images taken from each hippocampal sub-region.
Statistics
Statistical analysis was carried out using the IBM SPSS software (version 19.0). The Kolmogorov-Smirnov Test was used to establish normality of data. Comparisons across groups of cases and across sub-regions were performed using parametric tests (ANOVA for group means and Tukey post-hoc analysis for between-group differences) and non-parametric tests Kruskal–Wallis and Mann–Whitney U-tests) for non-normally distributed dataset. The relationship among markers, and with demographic, cognitive and pathological variables were assessed using Pearson's correlation coefficient (r) or Spearman's correlation (rho) as necessary depending on the normality of the dataset. Appropriate power calculation was performed using the G*Power software (Faul et al., 2007) at significance level, α-level = 0.05 and assuming a moderate effect size Cohen's d = 0.4.
Results
Characteristics of Study Subjects
The demographic, cognitive and pathological characteristics of the study participants [non-demented (PSND), demented (PSD) subjects from the CogFAST-Newcastle study; Control, VaD, AD and mixed AD-VaD groups] are shown in Table 1. There were no significant differences in the age (p = 0.786), gender distribution (p = 0.493), post-mortem delay (p = 0.902) and length of fixation of tissues (p = 0.589) across the groups. However, the PSND group had significantly higher scores on the cognitive batteries MMSE and CAMCOG compared to the PSD group (p < 0.05). Similarly, Braak stage, CERAD score and Thal stage were significantly higher in the AD, AD-VaD groups compared to the VaD and post-stroke groups (PSND and PSD) (p < 0.05). Hippocampal vascular scores were similar among the PSND, PSD, VaD and AD-VaD groups. The distribution of the ε3 and ε4 alleles of APOE were not significantly different between the PSND and PSD groups (Fisher's exact test = 2.13; p = 0.249). Given a total sample size of 94, a significance level, α = 0.05 and assuming a moderate effect size Cohen's d = 0.4, 6 sub-groups and 5 degrees of freedom, the computed Power (1-β) = 0.8424 using the G*Power software (Faul et al., 2007).
Quantification of Amyloid (Aβ) Burden
In quantifying amyloid burden both parenchymal as well as vascular amyloid immunoreactivities were incorporated in order to capture the total quantity of the different species of amyloid detected within the defined area of interest as previously performed (Lewis et al., 2006). Figure 1 illustrates the immunostaining pattern with antibodies to various Aβ species in serial sections across the CA1 sub-region of the hippocampus.
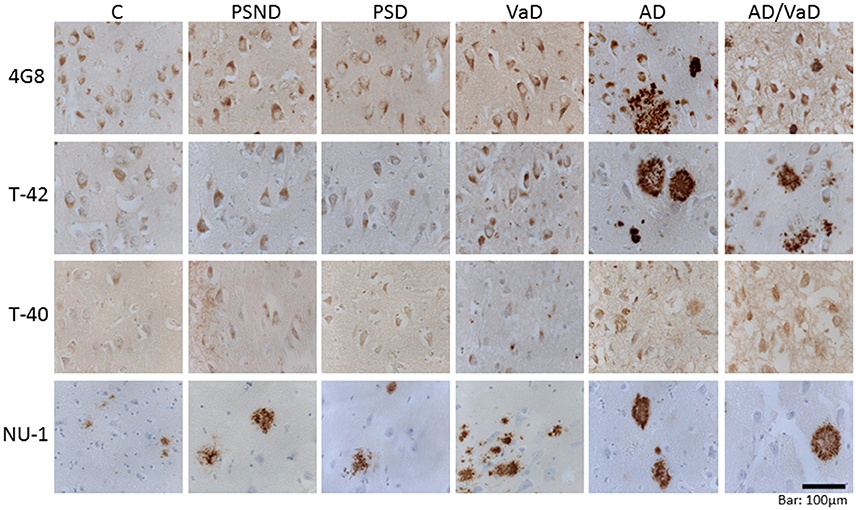
Figure 1. Illustrative images of amyloid pathology in the CA1 sub-region across different markers and across disease groups and aging controls. There is higher expression of amyloid in the AD and AD_VaD groups compared with the VaD, PSD, and PSND and control groups. The level of amyloid immunoreactivity is similar between 4G8 and T-42 but much lower in T-40 and NU-1.
Immunolabeling of Total Aβ with 4G8 Antibody
Distribution of 4G8 antibody immunostaining dataset assessed by Kolmogrov-Smirnov test showed non-normal distribution. Spearman's rank correlation analysis revealed no significant associations between post-mortem delay, length of fixation period and 4G8 immunoreactivity (IR) measures in the hippocampal sub-regions and entorhinal cortex. Furthermore, there was significant positive correlation between scores on the semi-quantitative amyloid rating scales of CERAD, Thal, Braak and tau stages with the metrics of 4G8 total IR in the entorhinal cortex, subiculum and CA1 sub-regions (Table 3) suggesting good agreement between semi-quantitative and quantitative measures of amyloid and tau quantification.
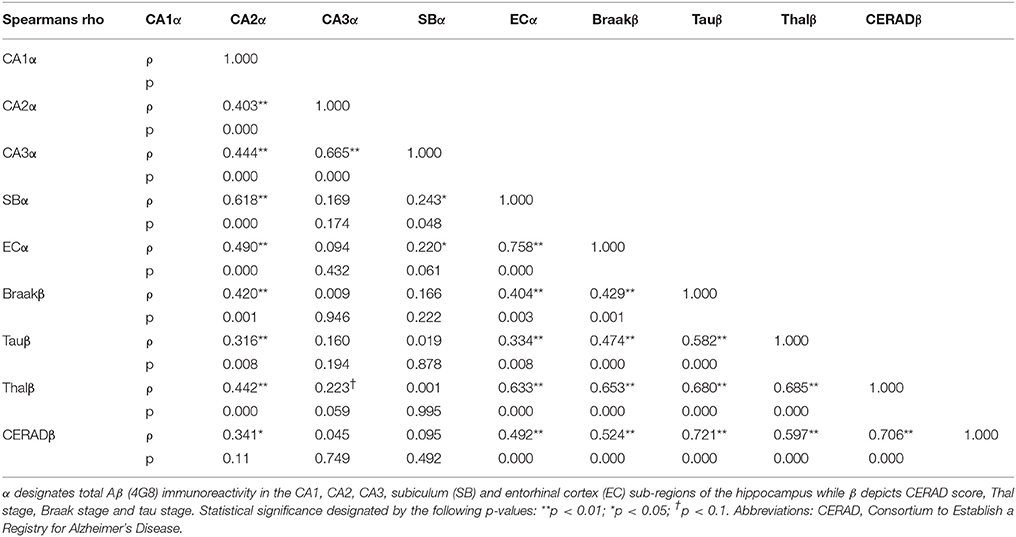
Table 3. Correlation Matrix of sub-regional total Aβ immunoreactivity, CERAD score, Thal stage, Braak stage and tau stage.
In the CA1 region, total Aβ IR varied significantly across groups (p = 0.008, Kruskal-Wallis Test) (Figure 2). Between group differences assessed with Mann–Whitney U-Test showed that compared to the control group, total AβIR was significantly higher in AD (p = 0.002), showed a trend with AD-VaD (p = 0.064) and PSND (p = 0.076) but not significantly different between PSD and VaD. In the CA2 and CA3 regions, there was no significant variation across disease groups. In the subiculum, there was significant variation across disease groups and controls (p < 0.001, Kruskal–Wallis Test) with total IR being significantly higher in the AD (p = 0.001), AD-VaD (p = 0.008) and PSND (p = 0.015) groups but not significantly different between PSD and VaD. In the entorhinal cortex, total Aβ IR similarly showed significant variation across the sub-region (p < 0.001, Kruskal–Wallis Test) with values significantly higher in AD (p = 0.022), AD-VaD (p = 0.016) than in controls while PSD was significantly lower than PSND (p = 0.019), AD-VaD (p = 0.002) and AD (p = 0.002) (Figure 2).
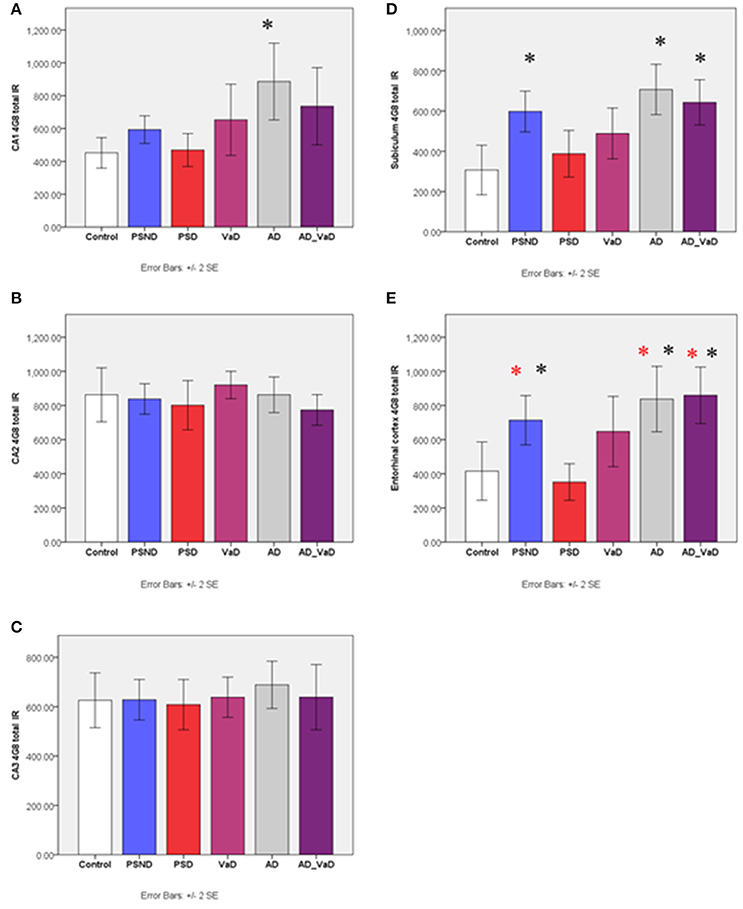
Figure 2. Bar graphs showing the distribution of total Aβ IR across hippocampal sub-regions (A) CA1, (B) CA2, (C) CA3, (D) Subiculum, (E) Entorhinal cortex in Controls, PSND, PSD, VaD, AD and AD_VaD. Bars show ± 2 SEM *Mann Whitney U-test was used to compare means of each group. *p < 0.05; + p < 0.1 (in comparison with the control group). (red) showed significant difference from PSD group. IR (immunoreactivity).
Aβ-42 Immunohistochemistry with T-42 Antibody
In the CA1 region, Aβ-42 immunoreactivity varied significantly across groups (p < 0.001, Kruskal-Wallis Test) (Figure 3). Between group differences assessed with Mann–Whitney U-Test showed that compared to the control group, T-42 immunoreactivity was significantly higher in AD (p = 0.002) and AD-VaD (p = 0.005), but not significantly different from VaD, PSND and PSD (Figure 3). In the CA2 region, immunoreactivity varied across disease groups (p = 0.001, Kruskal–Wallis Test) with the IR being significantly higher in AD-VaD (p = 0.046) and VaD (p = 0.014) groups compared to the control and PSD groups respectively (Figures 4B, 5). In the CA3 region, values revealed normal distribution by Kolmogorov–Smirnov test of normality and ANOVA showed significant variation of Aβ-42 immunoreactivity across the regions (p < 0.001). Compared to the PSND group, immunoreactivity was significantly higher in AD (p < 0.001) and AD-VaD (p = 0.002) groups but not significantly different from control PSD and VaD groups (Figure 3). In the subiculum, Aβ-42 immunoreactivity varied significantly across groups (p < 0.001) and was significantly higher in the AD (p = 0.001) and AD-VaD (p = 0.008) groups compared to the control and VaD groups respectively. However, the VaD groups did not differ significantly from the PSND and PSD groups respectively (Figures 4, 5). Similarly, Aβ-42 immunoreactivity varied significantly across the entorhinal cortex (p < 0.001) and was significantly higher in the AD (p = 0.001) and AD-VaD (p = 0.001) groups but not different from the VaD, PSND and PSD groups.
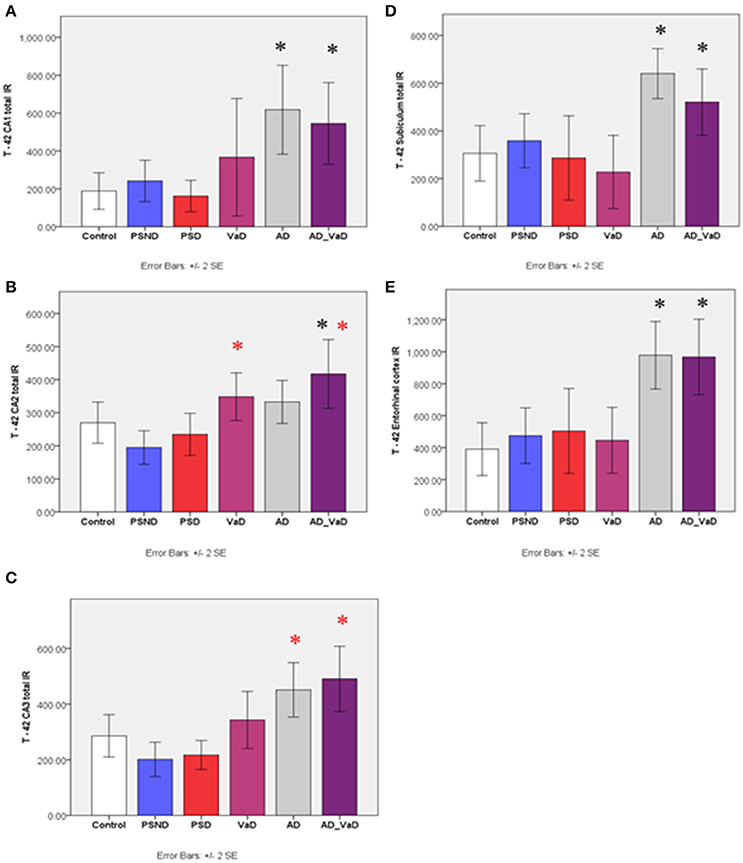
Figure 3. Bar graphs showing the distribution of Aβ-42 IR across hippocampal sub-regions (A) CA1, (B) CA2, (C) CA34, (D) Subiculum, (E) Entorhinal cortex in Controls, PSND, PSD, AD, VaD, and AD_VaD. Bars show ± 2 SEM *Mann Whitney U-test was used to compare means of each group. *p < 0.05; + p < 0.1 (in comparison with control group). (red) showed significant difference from PSND group. IR (immunoreactivity).
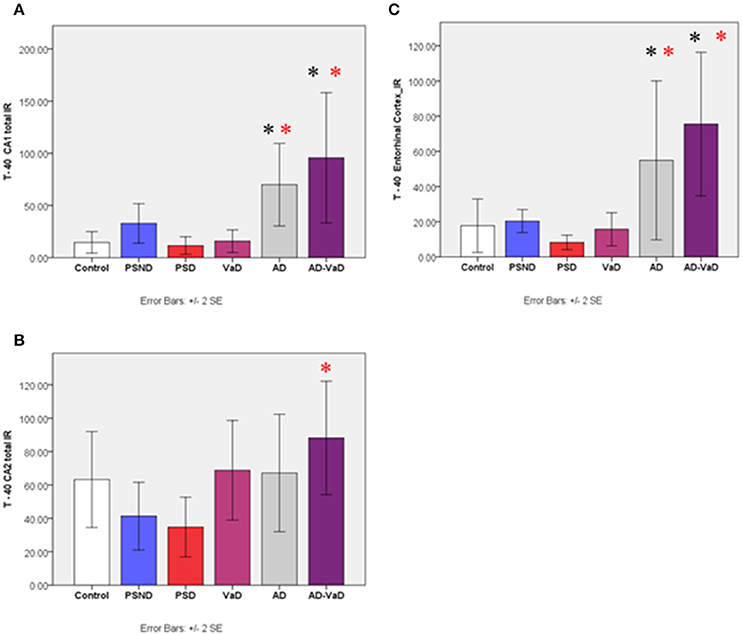
Figure 4. Bar graphs showing the distribution of Aβ-40 IR across hippocampal sub-regions (A) CA1, (B) CA2, (C) Entorbinal cortex in Controls, PSND, PSD, AD, VaD and AD_VaD. Bars show ± 2 SEM *Mann Whitney U–test was used to compare means of each group. *p < 0.05; + p < 0.1 (in comparison with the control group). (red) showed significant difference from PSD group. lR (immunoreactivity).
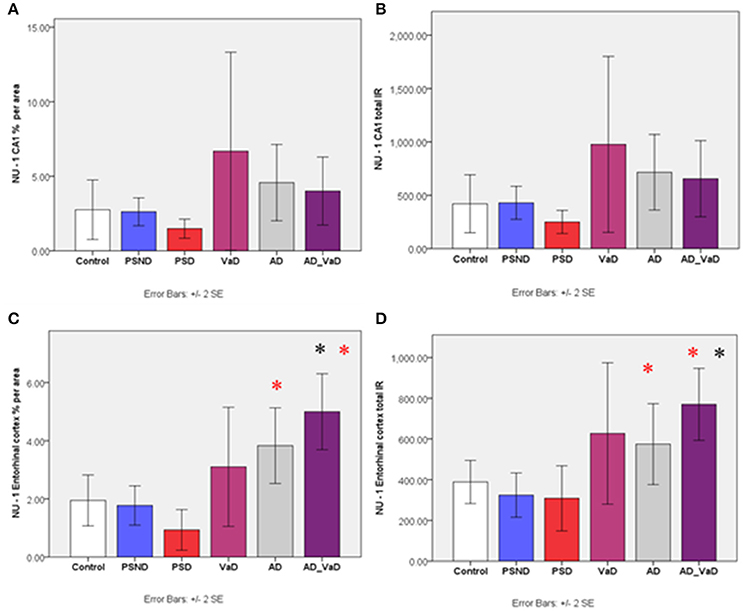
Figure 5. Bar graph shows percentage area and immunoreactivity of soluble Aβ (IR) across the CA1 region (A,B) and Entorhinal cortex (C,D) respectively in Control PSND, PSD, VaD, AD, and AD_VaD. Bars show ± 2 SEM *Mann Whitney U-test was used to compare means of each group. *p < 0.05 (in comparison with the *control group and PSD group). There were no significant differences across groups in the CA1, CA2, and CA3 regions (p > 0.05, Kruskal–Wallis Test).
Aβ-40 Immunoreactivity with T-40 Antibody
Immunostaining and distribution of T-40 antibody across disease groups varied significantly across the sub-regions (p = 0.001, Kruskal–Wallis Test). In CA1, the IR in the control group was not significantly different from that of PSND, PSD and, VaD but was significantly lower than AD (p = 0.004) and AD-VaD (p = 0.010). There was no difference between the PSND and PSD groups. In CA2, the control group IR was lower than AD, VaD and AD-VaD although this did not attain statistical significance (p > 0.05). However, VaD (p = 0.005) and AD-VaD (p = 0.006) groups were significantly higher than the PSD group. The PSND group did not differ from the PSD group (Figure 4). In the entorhinal cortex, Aβ-40 IR was significantly higher in the AD and AD-VaD groups compared to the control and PSD groups (p < 0.05, Kruska–Wallis Test). There was no significant variation in the CA3 region and subiculum.
We further explored the relationship of Aβ-42 and Aβ-40 based on the relative immuno-reactivities of T-42 and T-40 in the CA1 sub-region. The ratio varied between 1.17 and 748. 39 and the mean value was least in the control group and progressively increased in the PSND, PSD, VaD and AD groups to attain a maximum value in the AD-VaD group (Data not shown).
Soluble Aβ-Immunoreactivity with NU-1 Antibody
There were no significant differences in the percentage area and total immunoreactivity of NU-1 across the disease groups in the CA1, CA2, and CA3 regions (p > 0.05, Kruskal Wallis Test). However, in the subiculum and entorhinal cortex, the immunoreactivity varied significantly (p < 0.05 Kruskal–Wallis Test). Between group analysis showed significantly higher immunoreactivity in the AD-VaD (p = 0.007 Mann–Whitney U) group compared to the control group, and in the AD-VaD (p = 0.008) and AD (p = 0.040) compared to the PSD group (Figure 5). The PSND and PSD groups showed no significant differences.
Hyperphosphorylated tau Immunoreactivity with AT8 Antibody
Figure 6A shows tau (AT8) immunoreactivity across disease groups in the hippocampal subregion CA1 while Figure 6B shows the quantification across different subregions. Across disease groups, AT8 immunoreactivity was highest in AD and AD_ VaD groups in comparison to each of the other groups of PSND, PSD, VaD and Controls (p < 0.05). There was slightly higher AT8 immunoreactivity in PSD compared to the PSND group in the CA2 and CA3 sub-regions (Figure 6B) although the difference did not attain statistical significance.
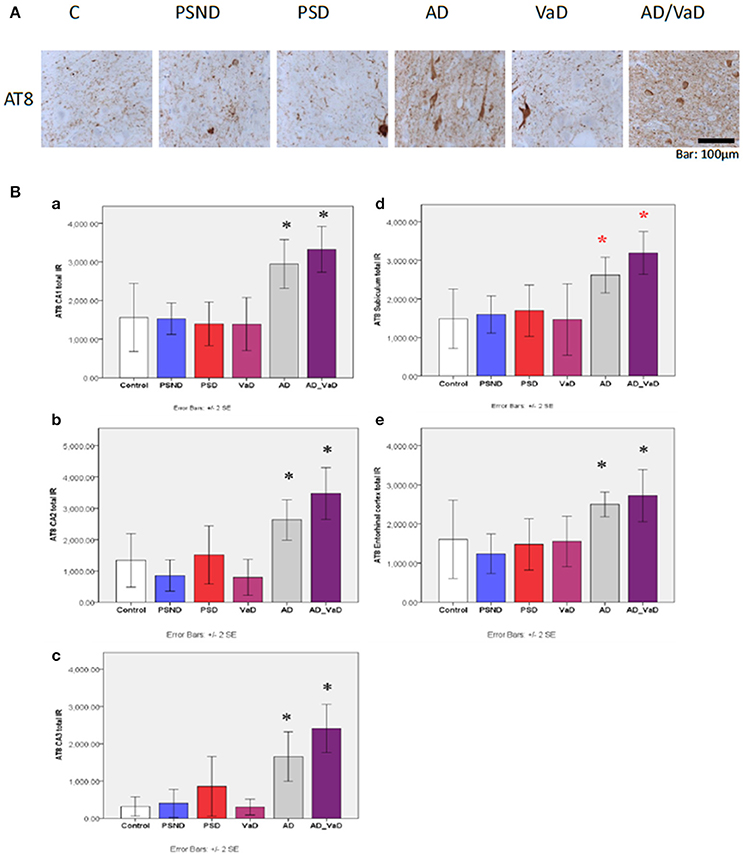
Figure 6. (A) Hyperphosphorylated tau (AT8) immunoreactivities showing neuropil threads, pretangles and tangles in hippocampal sub-region CAl. AT8 immunoreactivity is relatively higher in AD and AD_VaD compared to other groups. (B) Bar graph showing the distribution of AT8 IR across hippocampal sub-region. (a) CA1, (b) CA2, (c) CA3, (d) Subiculum (e) Entorhinal cortex in Controls. PSND. PSD. AD. VaD and AD_ VaD. Bars show ± 2 SEM *Mann Whitney U-test was used to compare means of each group. *p < 0.05; (in comparison with the control group). (red) showed significant difference from PSND group.
Influence of APOE ε4 Genotype on Amyloid and tau Accumulation in Post-Stroke Sub-Cohort
Eight out of 16 subjects with PSND (50%) were APO ε4 positive compared to 3 out of 13 PSD cases (23.1%). Overall, 11 out of 29 (37.9%) post-stroke subjects were APOE ε4 positive. Evaluation of the influence of APOE ε4 status on amyloid and tau deposition in the post-stroke sub-cohort revealed a statistically significant higher amyloid load in APOE ε4 positive post-stroke subjects compared to APOE ε4 negative subjects in the subiculum and entorhinal cortex respectively (p = 0.01) (Figure 7A). Presence of APOE ε4 also influenced tau deposition (AT8 immunoreactivity) in the post-stroke cohort, the immunoreactivity being significantly higher in APOE ε4 positive subjects in the subiculum (Mann–Whitney U-Test, p = 0.042) (Figure 7B).
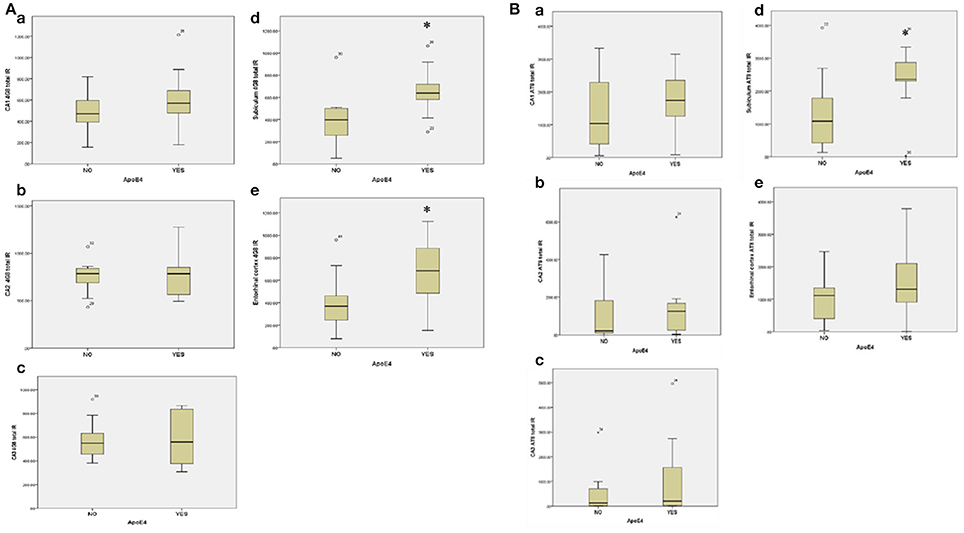
Figure 7. (A) Box Plots showing the influence of Apo E ε4 on total amyloid deposition across hippocampal sub-regions (a) CA1, (b) CA2, (c) CA3, (d) Subicuhun, and [e] Emorlrinal conex in the post-stroke sub-cohort Mann–Whitney U-test was used to compare the mean 4G8 total immunoreactivity between the Apo E ε4 positive and negative groups respectively. *p < 0.05. (B) Box Plot showing the influence of Apo E ε4 on total tau (AT8) deposition across hippocampal sub-regions (a) CA1 (b) CA2 (c) CA3 (d) Subiculum and (e) Entorhinol cortex in the post-stroke sub-cohort. Mann Whitney U-test was used to compare mean AT8 total immunoreactivity between the Apo E ε4 positive and negative groups respectively. *p < 0.05.
Clinico-Pathological Correlations
We explored relationships between the various markers of Alzheimer pathology used in this study and cognitive scores of the post-stroke group utilizing Spearman correlation analysis. Measures of general cognitive functioning (MMSE and CAMCOG total) and functioning in the memory domain (CAMCOG memory) were correlated with two measures of Alzheimer pathological burden: Aβ-42 immunoreactivity (being the predominant β-amyloid species deposited in the hippocampus) and tau immunoreactivity measures in the CA1, subiculum and entorhinal cortex: sub-regions which demonstrated the most consistent patterns of variation of immunoreactivity across the hippocampal formation. Table 4 demonstrates significant correlation of only AT8 immunoreactivity in the subiculum with CAMCOG memory (rho = −0.425, p = 0.024), whereas there was no significant correlation with Aβ-42 immunoreactivity.
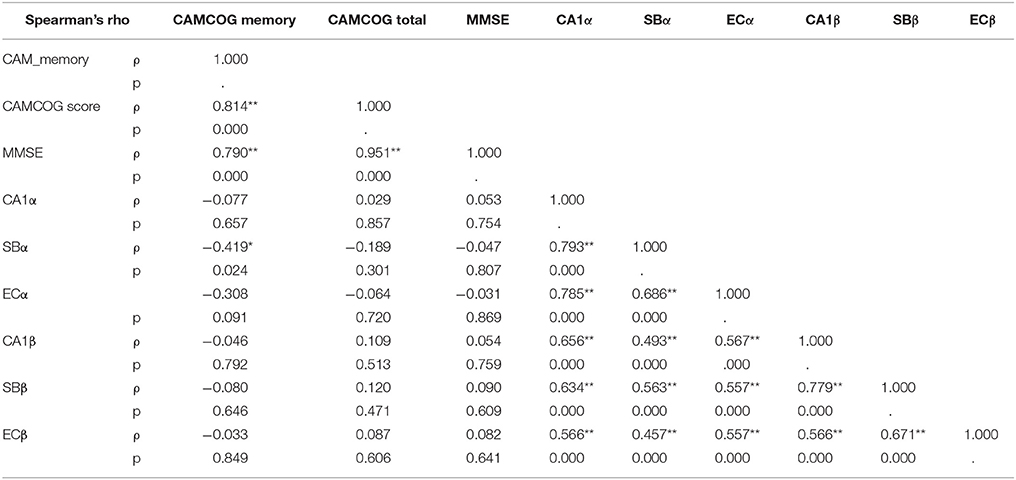
Table 4. Correlation matrix showing association of neuropathologic measures of amyloid and tau pathology with cognitive scores in selected hippocampal sub-regions and entorhinal cortex.
Discussion
We hypothesized that Alzheimer pathology would be differentially expressed in demented and non-demented post-stroke subjects in comparison to normal aging controls and other dementias. We found (1) amyloid beta deposition was not remarkably different between PSD and PSND groups despite the finding that MTLA was a significant feature in PSD subjects. (2) As expected, concentrations of total amyloid beta and amyloid β-42 were significantly greater in AD and mixed subjects but low in post-stroke subjects consistent with insufficient concentrations for a diagnosis of AD (3) an association between APOE ε4 allele positivity and higher load of amyloid and tau pathology in the subiculum and entorhinal cortex of post-stroke cases (4) hyperphosphorylated tau immunoreactivity did not differ significantly between PSND and PSD groups and (5) poor correlation of cognitive measures with the burden of amyloid and tau pathology in post-stroke subjects.
Amyloid Accumulation, Aging and Cerebrovascular Disease
Consistently across all the markers of amyloid pathology, we found evidence of increasing accumulation of amyloid in controls, post-stroke groups, AD and AD_VaD in that order. Our finding of amyloid accumulation in normal aging controls concurs with the biological phenomenon of aging-associated accumulation of amyloid that has been reported across species: in drosophilia (Rogers et al., 2012), mice (Yamada et al., 2011), non-human primates (Ndung'u et al., 2012) and man (Tomlinson et al., 1968; Katzman et al., 1988; Bennett et al., 2006; Lewis et al., 2006; Boyle et al., 2013b). This occurs as a result of aging-related compromise of the neurovascular unit resulting in increased production of amyloid and reduced clearance through the perivascular space (Iadecola, 2004; Kalaria, 2009; Kalaria et al., 2012). Amyloid accumulation in the post-stroke groups mirrored that in the VaD group and was less than in the AD and AD-VaD groups (Figures 2, 3) in consonance with the findings of Lewis et al. (2006) showing enhancement of amyloid accumulation in VaD possibly triggered by cerebral hypoxia consequent to cerebral vascular disease (Lewis et al., 2006; Kalaria et al., 2016). This is in tandem with previous findings of enhanced accumulation of amyloid in animal models of chronic cerebral hypoperfusion (Kalaria et al., 1993a; Yamada et al., 2011) as well as increased Pittsburgh Compound B (PIB) uptake in PSD subjects in a pilot study (Mok et al., 2010). In addition, this suggests that beyond age-associated accumulation of amyloid, cerebral vascular disorders including stroke do exacerbate brain amyloid deposition as previously demonstrated in brain tissue from hypertensive (Petrovitch et al., 2000) and diabetic subjects (Luchsinger, 2010). Although Marchant et al. failed to establish a direct relationship between CVD and Aβ using PIB-PET approach in a cohort of non-demented elderly subjects (Marchant et al., 2012), and further suggested that the PIB-PET amyloid measures did not influence cognition, the authors admitted that the limited statistical power of the study may have failed to detect any existent interaction. Utilizing a semi-quantitative approach, a neuropathological study of 484 post-mortem brains did not find a relationship between amyloid deposition and cerebrovascular lesions (Aho et al., 2006). The previous study by Lewis et al. (2006) and this current work have utilized sensitive quantitative approaches to detect amyloid load. Besides, findings from studies in non-demented elderly subjects may not necessarily simulate those in demented subjects with significant CVD as the mechanisms that produce cognitive decline and dementia may differ in different clinicopathological scenarios (Kalaria, 2012a,b).
Although the main Aβ variants detected in the human brain are Aβ1–40 and Aβ1–42, a significant proportion consists also of N-terminal truncated species (Aβn-40/42 where n = 2 to 11) of which pyroglutamate-modified Aβ peptides are predominant components. Most N-truncated Aβ are considered to be the degradation products of full-length Aβ although Aβ11–40/42 may be generated intracellularly directly from APP by BACE proteolysis occurring in trans-Golgi network (Perez-Garmendia et al., 2014). AβN3(pE), Aβ peptide bearing amino-terminal pyroglutamate at position 3, has been shown to be a major N-truncated/modified constituent of intracellular, extracellular and vascular Aβ deposits in AD brain tissue (Saido et al., 1995) which progressively accumulates in the brain and could predate the development of AD symptoms. These truncated species are believed to constitute a potential seeding specie in the formation of pathological amyloid aggregates (Saido et al., 1995; Perez-Garmendia et al., 2014) Although we did not specifically assay for these pyroglutamate-modified Aβ peptides, they may have contributed to the intraneuronal and vascular amyloid which was detected by the immunolabeling of total Aß (4G8) in the current study.
Sub-Regional Variation in Hippocampal Amyloid Accumulation
Across the sub-regions of the hippocampal formation and entorhinal cortex, amyloid deposition was significantly higher in the CA1, subiculum and entorhinal cortex compared to the CA2 and CA3 regions respectively. This differential pattern may be related to the spatial localization and role of these regions in the hippocampal circuitry (Lavenex and Banta Lavenex, 2013), differential susceptibility of these sub-regions to different pathologies (Small et al., 2011) or the temporal evolution and hierarchical progression of cerebral amyloidosis (Thal et al., 2006).
The entorhinal cortex has been described as the gateway into the hippocampal formation whereas the subiculum and CA1 regions constitute the outflow stations (Goldman-Rakic et al., 1984; Suzuki and Amaral, 2004). Alzheimer pathology tends to spread along the hippocampal circuitry (Thal et al., 2002a; Lace et al., 2009) and this may explain the differential susceptibility and high β-amyloid load in these sub-regions. Besides, in the hierarchical evolution and natural history of amyloid and tau pathologies, these sub-regions are affected earlier in the disease course, compared to other regions such as CA2 and CA3 (Thal et al., 2002b; Lace et al., 2009). The later effect in the CA2 region, in particular may reflect the natural course of disease or the existence of some underlying protective mechanisms operating in the early stages of disease and only giving way in the advanced stage of the disease (Caruana et al., 2012).
The finding of a relatively higher ratio of Aβ-42 compared to Aβ-40 which further increases with the degree of accumulation of AD pathology is in concordance with previous findings (Aho et al., 2006; Selkoe, 2008) demonstrating the predominance of Aβ-42 over Aβ-40 in brain parenchymal amyloid deposits. We have also previously demonstrated a preponderance of Aβ (42) over Aβ-40 in parenchymal and vascular amyloid deposits in non-human primates including squirrel, rhesus monkeys and aging baboons (Ndung'u et al., 2012).
Differential Amyloid and tau Deposition between PSND and PSD
Largely, there were no significant differences in the quantity of amyloid and tau deposited across hippocampal regions and markers in PSND compared to PSD groups. This suggests age-related deposition of amyloid and tau in post-stroke survivors. However, total Aβ immunoreactivity was unexpectedly higher in the entorhinal cortex of PSND than PSD. Though not statistically significant, a similar pattern was observed with Aβ-42 in the subiculum, Aβ-40 in the entorhinal cortex, and soluble Aβ in the CA1 sub-region. This suggests that diffuse early amyloid accumulation in post-stroke subjects alone does not explain why some stroke survivors become demented while others remain cognitively intact. Cognitively normal elderly subjects with huge quantities of amyloid pathology but preserved cognitive functioning have been described in the literature (Bennett et al., 2006; Chetelat et al., 2010). It may also imply that amyloid needs the synergy of other pathologies including tau pathology, vascular lesions, brain atrophy, white matter pathology, MTLA in order to produce significant cognitive decline and dementia (Mormino et al., 2009). In a PIB-PET study of elderly subjects-normal, mild cognitive impairment (MCI) and AD, the investigators found that whereas amyloid load (PIB index) and hippocampal atrophy both predicted loss of episodic memory, amyloid deposition alone in the absence of hippocampal atrophy failed to predict episodic memory loss (Mormino et al., 2009). In addition, a recent study by Wong et al. (2016), found that amyloid retention measured by 11C-PiB PET showed no association with cognitive impairment and clusters of neuropsychiatric symptoms suggesting that other plausible biological pathways could be advanced to explain the development and progression of cognitive impairment and neuropsychiatric symptoms following stroke (Wong et al., 2016). A complementary study of our current cohort (Gemmell et al., 2012) found that whereas pyramidal neuronal volume was preserved in the CA regions and entorhinal cortex of the control and PSND groups, subjects in all the demented groups (including PSD) had significant atrophy of these neurons. This would, therefore, suggest that high amyloid load in the PSND group was insufficient to produce dementia because of preserved neuronal volume. This, indeed, may be a signature of brain/cognitive reserve, preserved synaptic integrity or some other compensatory mechanisms (Stern, 2009; Boyle et al., 2013a).
The observation of similarity between amyloid load in the PSD compared to the control group could imply that given similar quantities of amyloid pathology with respect to the controls, the PSD group was demented possibly because of the presence of additional cerebrovascular lesions which lowered the pathological threshold (Snowdon et al., 1997; Esiri et al., 1999); the presence of neuronal atrophy (Gemmell et al., 2012) or the slightly higher and more advanced hyperphosphorylated tau pathology in the CA2 and CA3 sub-regions of the PSD group. A recent study in which total and phosphorylated tau proteins were quantified in the frontal and temporal cortices of subjects with vascular dementia found a selective loss of total tau protein in VaD compared with controls and AD, whereas phosphorylated tau levels were similar to controls in VaD in both regions, but they were increased in the temporal lobes of patients with AD (Mukaetova-Ladinska et al., 2015). These results demonstrated that breaches of microvascular or microstructural tissue integrity subsequent to ischemic injury in older age may modify tau protein metabolism or phosphorylation and have effects on the burden of neurofibrillary pathology (Mukaetova-Ladinska et al., 2015).
APOE ε4 Genotype and Accumulation of Amyloid and tau Pathologies
Further analysis of the post-stroke cohort suggested that the presence of the APOE ε4 allele was responsible for driving amyloid and tau accumulation in those who possessed the allele (which was present in 50%) of the PSND subjects. Despite the limited size of the sub-cohort, the amyloid load was significantly higher in the PSND group than the PSD group. APOE ε4 has been associated with accumulation of amyloid and/or tau pathology (Nagy et al., 1995; Saito et al., 2002) in AD but the relationship with post-stroke dementia has been conflicting and less well defined. Previous studies in the Newcastle cohort failed to establish a relationship between APOE ε4 with post-stroke cognitive impairment at 3 months after the stroke (Rowan et al., 2005) but predicted decline at 1-year follow up (Ballard et al., 2004). Furthermore, these results are in concordance with a previous study examining the genetic associations of vascular dementia subtypes in which an association was found between APOE-ε4 allele and mixed dementia, stroke-related dementia and subcortical ischemic vascular dementia (SIVD) as well as higher Aβ-(42) levels (Jones et al., 2011). Other studies had also reported both positive (Packard et al., 2007; Liu et al., 2012) and negative associations (Gdovinova et al., 2006) although these were largely clinical studies. It is plausible that the APOE ε4 allele might have contributed to the higher quantity of amyloid in subregions of the PSND group compared to the PSD group, but further research is required to explore this relationship further.
Correlation of Cognitive Scores with AD Pathology
In the post-stroke cohort with available cognitive scores, very limited correlation of tau pathology with CAMCOG memory score was established. In this cohort, there was dissociation of cognitive performance and hippocampal Alzheimer pathologic burden. The implication of this may be that AD pathology probably does not contribute very strongly to the substrates of dementia after a stroke event as previously hypothesized (Henon et al., 1997). And if there was any contribution at all, tau pathology probably contributed more than amyloid pathology. Recent reports also suggest that MTLA which had hitherto been widely ascribed to AD pathology, may have a vascular basis (Bastos-Leite et al., 2007; O'Sullivan et al., 2008). The lack of association may also be due to the presence of other pathologies such as synucleinopathies, (the determination of which is beyond the scope of the current study), presence of robust cognitive reserve and other lifestyle and psychosocial factors might also offer reasons for the dissociation between clinical and cognitive measures in the current study. Our study had limitations. We did not quantify specific isoforms of phosphorylated tau protein (R3 or R4) in our cohort as this could yield different amounts in the different subgroups. Alternative techniques including neurochemical approaches such as ELISA could have also be employed to further substantiate the findings from this current study and the potential existence of familywise Type 1 error associated with multiple pairwise comparisons is also acknowledged.
In conclusion, we did not find quantitative AD pathological markers sufficient to separate the non-demented post-stroke from demented post-stroke subjects. Thus, hippocampal AD-pathological mechanisms do not separate non-demented from demented stroke subjects. Neocortical Alzheimer pathology, other non-Alzheimer neurodegenerative pathologies as well as other non-neurodegenerative mechanisms such as vascular and inflammatory/immune mechanisms require further research in order to fully determine the precise pathological substrates of dementia following stroke.
Author Contributions
ROA performed experiments in the study and drafted the first draft of the manuscript. AO provided technical support. LA and AO contributed to critically revising the manuscript for important intellectual content. RK corrected drafts and obtained the funding. All co-authors approved the final version of the manuscript for submission.
Funding
Our work is supported by grants from the Dunhill Medical Trust (UK), Medical Research Council (MRC, G0500247), Newcastle Centre for Brain Ageing and Vitality (BBSRC, EPSRC, ESRC and MRC, LLHW), and Alzheimer's Research (ARUK). Our work is also supported by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK MRC (G0400074), by the Newcastle NIHR Biomedical Research Centre in Ageing and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust, and by a grant from the Alzheimer's Society and ARUK as part of the Brains for Dementia Research Project. ROA is also supported by a grant from the University of Ibadan College of Medicine (CTR16A012) for the IBADAN Brain Bank Project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
ROA was recipient of a research fellowship and a return home fellowship from the International Brain Research Organization (IBRO), and an Overseas Research Studentship from Newcastle University, UK. We are also grateful to the patients, families, and clinical house staff for supporting our longitudinal studies. We are grateful for the technical assistance of Janet Slade in performing experiments.
References
Aho, L., Jolkkonen, J., and Alafuzoff, I. (2006). Beta-amyloid aggregation in human brains with cerebrovascular lesions. Stroke 37, 2940–2945. doi: 10.1161/01.STR.0000248777.44128.93
Aizenstein, H. J., Nebes, R. D., Saxton, J. A., Price, J. C., Mathis, C. A., Tsopelas, N. D., et al. (2008). Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 65, 1509–1517. doi: 10.1001/archneur.65.11.1509
Akinyemi, R. O., Allan, L., Owolabi, M. O., Akinyemi, J. O., Ogbole, G., Ajani, A., et al. (2014). Profile and determinants of vascular cognitive impairment in African stroke survivors: the CogFAST Nigeria Study. J. Neurol. Sci. 346, 241–249. doi: 10.1016/j.jns.2014.08.042
Akinyemi, R. O., Firbank, M., Ogbole, G. I., Allan, L. M., Owolabi, M. O., Akinyemi, J. O., et al. (2015). Medial temporal lobe atrophy, white matter hyperintensities and cognitive impairment among Nigerian African stroke survivors. BMC Res. Notes 8, 625. doi: 10.1186/s13104-015-1552-7
Allan, L. M., Rowan, E. N., Firbank, M. J., Thomas, A. J., Parry, S. W., Polvikoski, T. M., et al. (2011). Long term incidence of dementia, predictors of mortality and pathological diagnosis in older stroke survivors. Brain 134(Pt 12), 3716–3727. doi: 10.1093/brain/awr273
Ballard, C. G., Morris, C. M., Rao, H., O'Brien, J. T., Barber, R., Stephens, S., et al. (2004). APOE epsilon4 and cognitive decline in older stroke patients with early cognitive impairment. Neurology 63, 1399–1402. doi: 10.1212/01.WNL.0000141851.93193.17
Bastos-Leite, A. J., van der Flier, W. M., van Straaten, E. C., Staekenborg, S. S., Scheltens, P., and Barkhof, F. (2007). The contribution of medial temporal lobe atrophy and vascular pathology to cognitive impairment in vascular dementia. Stroke 38, 3182–3185. doi: 10.1161/STROKEAHA.107.490102
Bennett, D. A., Schneider, J. A., Arvanitakis, Z., Kelly, J. F., Aggarwal, N. T., Shah, R. C., et al. (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6
Boyle, P. A., Wilson, R. S., Yu, L., Barr, A. M., Honer, W. G., Schneider, J. A., et al. (2013a). Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann. Neurol. 74, 478–489. doi: 10.1002/ana.23964
Boyle, P. A., Yu, L., Wilson, R. S., Schneider, J. A., and Bennett, D. A. (2013b). Relation of neuropathology with cognitive decline among older persons without dementia. Front. Aging Neurosci. 5:50. doi: 10.3389/fnagi.2013.00050
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Caruana, D. A., Alexander, G. M., and Dudek, S. M. (2012). New insights into the regulation of synaptic plasticity from an unexpected place: hippocampal area CA2. Learn. Mem. 19, 391–400. doi: 10.1101/lm.025304.111
Chang, L., Bakhos, L., Wang, Z., Venton, D. L., and Klein, W. L. (2003). Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer's disease drug candidate screening. J. Mol. Neurosci. 20, 305–313. doi: 10.1385/JMN:20:3:305
Chetelat, G., Villemagne, V. L., Pike, K. E., Baron, J. C., Bourgeat, P., Jones, G., et al. (2010). Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133, 3349–3358. doi: 10.1093/brain/awq187
Cordoliani-Mackowiak, M. A., Henon, H., Pruvo, J. P., Pasquier, F., and Leys, D. (2003). Poststroke dementia: influence of hippocampal atrophy. Arch. Neurol. 60, 585–590. doi: 10.1001/archneur.60.4.585
de la Torre, J. C., and Mussivand, T. (1993). Can disturbed brain microcirculation cause Alzheimer's disease? Neurol. Res. 15, 146–153. doi: 10.1080/01616412.1993.11740127
Esiri, M. M., Nagy, Z., Smith, M. Z., Barnetson, L., and Smith, A. D. (1999). Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer's disease. Lancet 354, 919–920. doi: 10.1016/S0140-6736(99)02355-7
Faul, F., Erdfelder, E., Lang, A. G., and Buchner, A. (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191. doi: 10.3758/BF03193146
Firbank, M. J., Allan, L. M., Burton, E. J., Barber, R., O'Brien, J. T., and Kalaria, R. N. (2012). Neuroimaging predictors of death and dementia in a cohort of older stroke survivors. J. Neurol. Neurosurg. Psychiatry 83, 263–267. doi: 10.1136/jnnp-2011-300873
Firbank, M. J., Burton, E. J., Barber, R., Stephens, S., Kenny, R. A., Ballard, C., et al. (2007). Medial temporal atrophy rather than white matter hyperintensities predict cognitive decline in stroke survivors. Neurobiol. Aging 28, 1664–1669. doi: 10.1016/j.neurobiolaging.2006.07.009
GBD 2015 Neurological Disorders Collaborator Group (2017). Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16, 877–897. doi: 10.1016/S1474-4422(17)30299-5
Gdovinova, Z., Habalova, V., and Novosadova, Z. (2006). Polymorphism of apolipoproteine E in relation with Alzheimer and vascular dementia. Cell Mol. Neurobiol. 26, 1219–1224. doi: 10.1007/s10571-006-9043-y
Gemmell, E., Bosomworth, H., Allan, L., Hall, R., Khundakar, A., Oakley, A. E., et al. (2012). Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 43, 808–814. doi: 10.1161/STROKEAHA.111.636498
Goldman-Rakic, P. S., Selemon, L. D., and Schwartz, M. L. (1984). Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience 12, 719–743. doi: 10.1016/0306-4522(84)90166-0
Henon, H., Pasquier, F., Durieu, I., Godefroy, O., Lucas, C., Lebert, F., et al. (1997). Preexisting dementia in stroke patients. baseline frequency, associated factors, and outcome. Stroke 28, 2429–2436. doi: 10.1161/01.STR.28.12.2429
Henon, H., Pasquier, F., Durieu, I., Pruvo, J. P., and Leys, D. (1998). Medial temporal lobe atrophy in stroke patients: relation to pre-existing dementia. J. Neurol. Neurosurg. Psychiatry 65, 641–647. doi: 10.1136/jnnp.65.5.641
Hixson, J. E., and Vernier, D. T. (1990). Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J. Lipid Res. 31, 545–548.
Hyman, B. T., and Trojanowski, J. Q. (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the national institute on aging and the reagan institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 1095–1097. doi: 10.1097/00005072-199710000-00002
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Jones, E. L., Kalaria, R. N., Sharp, S. I., O'Brien, J. T., Francis, P. T., and Ballard, C. G. (2011). Genetic associations of autopsy-confirmed vascular dementia subtypes. Dement Geriatr. Cogn. Disord. 31, 247–253. doi: 10.1159/000327171
Kalaria, R. N. (2009). Linking cerebrovascular defense mechanisms in brain ageing and Alzheimer's disease. Neurobiol. Aging 30, 1512–1514. doi: 10.1016/j.neurobiolaging.2007.10.020
Kalaria, R. N. (2012a). Cerebrovascular disease and mechanisms of cognitive impairment: evidence from clinicopathological studies in humans. Stroke 43, 2526–2534. doi: 10.1161/STROKEAHA.112.655803
Kalaria, R. N. (2012b). Risk factors and neurodegenerative mechanisms in stroke related dementia. Panminerva Med. 54, 139–148.
Kalaria, R. N., Akinyemi, R., and Ihara, M. (2012). Does vascular pathology contribute to Alzheimer changes? J. Neurol. Sci. 322, 141–147. doi: 10.1016/j.jns.2012.07.032
Kalaria, R. N., Akinyemi, R., and Ihara, M. (2016). Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 1862, 915–925. doi: 10.1016/j.bbadis.2016.01.015
Kalaria, R. N., Bhatti, S. U., Lust, W. D., and Perry, G. (1993a). The amyloid precursor protein in ischemic brain injury and chronic hypoperfusion. Ann. N. Y. Acad. Sci. 695, 190–193. doi: 10.1111/j.1749-6632.1993.tb23050.x
Kalaria, R. N., Bhatti, S. U., Palatinsky, E. A., Pennington, D. H., Shelton, E. R., Chan, H. W., et al. (1993b). Accumulation of the beta amyloid precursor protein at sites of ischemic injury in rat brain. Neuroreport 4, 211–214. doi: 10.1097/00001756-199302000-00025
Kalaria, R. N., Kenny, R. A., Ballard, C. G., Perry, R., Ince, P., and Polvikoski, T. (2004). Towards defining the neuropathological substrates of vascular dementia. J. Neurol. Sci. 226, 75–80. doi: 10.1016/j.jns.2004.09.019
Katzman, R., Terry, R., DeTeresa, R., Brown, T., Davies, P., Fuld, P., et al. (1988). Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 23, 138–144. doi: 10.1002/ana.410230206
Kitaguchi, H., Tomimoto, H., Ihara, M., Shibata, M., Uemura, K., Kalaria, R. N., et al. (2009). Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 1294, 202–210. doi: 10.1016/j.brainres.2009.07.078
Lace, G., Savva, G. M., Forster, G., de Silva, R., Brayne, C., Matthews, F. E., et al. (2009). Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain 132(Pt 5), 1324–1334. doi: 10.1093/brain/awp059
Lavenex, P., and Banta Lavenex, P. (2013). Building hippocampal circuits to learn and remember: insights into the development of human memory. Behav. Brain Res. 254, 8–21. doi: 10.1016/j.bbr.2013.02.007
Lewis, H., Beher, D., Cookson, N., Oakley, A., Piggott, M., Morris, C. M., et al. (2006). Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 32, 103–118. doi: 10.1111/j.1365-2990.2006.00696.x
Liu, X., Li, L., Liu, F., Deng, S., Zhu, R., Li, Q., et al. (2012). ApoE gene polymorphism and vascular dementia in Chinese population: a meta-analysis. J. Neural. Transm. 119, 387–394. doi: 10.1007/s00702-011-0714-6
Luchsinger, J. A. (2010). Diabetes, related conditions, and dementia. J. Neurol. Sci. 299, 35–38. doi: 10.1016/j.jns.2010.08.063
Marchant, N. L., Reed, B. R., DeCarli, C. S., Madison, C. M., Weiner, M. W., Chui, H. C., et al. (2012). Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol. Aging 33, 1006.e25-100636. doi: 10.1016/j.neurobiolaging.2011.10.001
Mirra, S. S., Heyman, A., McKeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., et al. (1991). The consortium to establish a registry for Alzheimer's Disease (CERAD). part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41, 479–486. doi: 10.1212/WNL.41.4.479
Mok, V., Leung, E. Y., Chu, W., Chen, S., Wong, A., Xiong, Y., et al. (2010). Pittsburgh compound B binding in poststroke dementia. J. Neurol. Sci. 290, 135–137. doi: 10.1016/j.jns.2009.12.014
Mormino, E. C., Kluth, J. T., Madison, C. M., Rabinovici, G. D., Baker, S. L., Miller, B. L., et al. (2009). Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 132(Pt 5), 1310–1323. doi: 10.1093/brain/awn320
Mufson, E. J., Chen, E. Y., Cochran, E. J., Beckett, L. A., Bennett, D. A., and Kordower, J. H. (1999). Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp. Neurol. 158, 469–490. doi: 10.1006/exnr.1999.7086
Mukaetova-Ladinska, E. B., Abdel-All, Z., Mugica, E. S., Li, M., Craggs, L. J., Oakley, A. E., et al. (2015). Tau proteins in the temporal and frontal cortices in patients with vascular dementia. J. Neuropathol. Exp. Neurol. 74, 148–157. doi: 10.1097/NEN.0000000000000157
Nagy, Z., Esiri, M. M., Jobst, K. A., Johnston, C., Litchfield, S., Sim, E., et al. (1995). Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience 69, 757–761. doi: 10.1016/0306-4522(95)00331-C
Ndung'u, M., Hartig, W., Wegner, F., Mwenda, J. M., Low, R. W., Akinyemi, R. O., et al. (2012). Cerebral amyloid beta(42) deposits and microvascular pathology in ageing baboons. Neuropathol. Appl. Neurobiol. 38, 487–499. doi: 10.1111/j.1365-2990.2011.01246.x
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nelson, P. T., Braak, H., and Markesbery, W. R. (2009). Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J. Neuropathol. Exp. Neurol. 68, 1–14. doi: 10.1097/NEN.0b013e3181919a48
O'Sullivan, M., Jouvent, E., Saemann, P. G., Mangin, J. F., Viswanathan, A., Gschwendtner, A., et al. (2008). Measurement of brain atrophy in subcortical vascular disease: a comparison of different approaches and the impact of ischaemic lesions. Neuroimage 43, 312–320. doi: 10.1016/j.neuroimage.2008.07.049
Packard, C. J., Westendorp, R. G., Stott, D. J., Caslake, M. J., Murray, H. M., Shepherd, J., et al. (2007). Association between apolipoprotein E4 and cognitive decline in elderly adults. J. Am. Geriatr. Soc. 55, 1777–1785. doi: 10.1111/j.1532-5415.2007.01415.x
Perez-Garmendia, R., Hernandez-Zimbron, L. F., Morales, M. A., Luna-Munoz, J., Mena, R., Nava-Catorce, M., et al. (2014). Identification of N-terminally truncated pyroglutamate amyloid-beta in cholesterol-enriched diet-fed rabbit and AD brain. J. Alzheimers Dis. 39, 441–455. doi: 10.3233/JAD-130590
Petrovitch, H., Ross, G. W., Steinhorn, S. C., Abbott, R. D., Markesbery, W., Davis, D., et al. (2005). AD lesions and infarcts in demented and non-demented Japanese-American men. Ann. Neurol. 57, 98–103. doi: 10.1002/ana.20318
Petrovitch, H., White, L. R., Izmirilian, G., Ross, G. W., Havlik, R. J., Markesbery, W., et al. (2000). Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu-Asia aging study. Neurobiol. Aging 21, 57–62. doi: 10.1016/S0197-4580(00)00106-8
Rogers, I., Kerr, F., Martinez, P., Hardy, J., Lovestone, S., and Partridge, L. (2012). Ageing increases vulnerability to abeta42 toxicity in Drosophila. PLoS ONE 7:e40569. doi: 10.1371/journal.pone.0040569
Rowan, E., Morris, C. M., Stephens, S., Ballard, C., Dickinson, H., Rao, H., et al. (2005). Impact of hypertension and apolipoprotein E4 on poststroke cognition in subjects >75 years of age. Stroke 36, 1864–1868. doi: 10.1161/01.STR.0000177524.17424.2a
Saido, T. C., Iwatsubo, T., Mann, D. M., Shimada, H., Ihara, Y., and Kawashima, S. (1995). Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 14, 457–466. doi: 10.1016/0896-6273(95)90301-1
Saito, Y., Suzuki, K., Nanba, E., Yamamoto, T., Ohno, K., and Murayama, S. (2002). Niemann-pick type C disease: accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann. Neurol. 52, 351–355. doi: 10.1002/ana.10266
Schneider, J. A., Wilson, R. S., Bienias, J. L., Evans, D. A., and Bennett, D. A. (2004). Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155. doi: 10.1212/01.WNL.0000118211.78503.F5
Selkoe, D. J. (2008). Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer's disease. Handb. Clin. Neurol. 89, 245–260. doi: 10.1016/S0072-9752(07)01223-7
Small, S. A., Schobel, S. A., Buxton, R. B., Witter, M. P., and Barnes, C. A. (2011). A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 12, 585–601. doi: 10.1038/nrn3085
Snowdon, D. A., Greiner, L. H., Mortimer, J. A., Riley, K. P., Greiner, P. A., and Markesbery, W. R. (1997). Brain infarction and the clinical expression of Alzheimer disease. the nun study. JAMA 277, 813–817. doi: 10.1001/jama.1997.03540340047031
Stern, Y. (2009). Cognitive reserve. Neuropsychologia 47, 2015–2028. doi: 10.1016/j.neuropsychologia.2009.03.004
Suzuki, W. A., and Amaral, D. G. (2004). Functional neuroanatomy of the medial temporal lobe memory system. Cortex 40, 220–222. doi: 10.1016/S0010-9452(08)70958-4
Thal, D. R., Capetillo-Zarate, E., Del Tredici, K., and Braak, H. (2006). The development of amyloid beta protein deposits in the aged brain. Sci. Aging Knowledge Environ. 2006:re1. doi: 10.1126/sageke.2006.6.re1
Thal, D. R., Ghebremedhin, E., Rub, U., Yamaguchi, H., Del Tredici, K., and Braak, H. (2002a). Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293. doi: 10.1093/jnen/61.3.282
Thal, D. R., Rub, U., Orantes, M., and Braak, H. (2002b). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Tomlinson, B. E., Blessed, G., and Roth, M. (1968). Observations on the brains of non-demented old people. J. Neurol. Sci. 7, 331–356. doi: 10.1016/0022-510X(68)90154-8
Whitehead, S. N., Hachinski, V. C., and Cechetto, D. F. (2005). Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity: inflammatory responses. Stroke 36, 107–112. doi: 10.1161/01.STR.0000149627.30763.f9
Wong, A., Lau, A. Y., Yang, J., Wang, Z., Liu, W., Lam, B. Y., et al. (2016). Neuropsychiatric symptom clusters in stroke and transient ischemic attack by cognitive status and stroke subtype: frequency and relationships with vascular lesions, brain atrophy and amyloid. PLoS ONE 11:e0162846. doi: 10.1371/journal.pone.0162846
Writing Group Members, Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., et al. (2016). Executive summary: heart disease and stroke statistics– update: a report from the American heart association. Circulation 133, 447–454. doi: 10.1161/CIR.0000000000000366
Keywords: Alzheimer's disease, cerebrovascular disease, mixed dementia, post-stroke dementia, stroke, vascular dementia
Citation: Akinyemi RO, Allan LM, Oakley A and Kalaria RN (2017) Hippocampal Neurodegenerative Pathology in Post-stroke Dementia Compared to Other Dementias and Aging Controls. Front. Neurosci. 11:717. doi: 10.3389/fnins.2017.00717
Received: 30 September 2017; Accepted: 08 December 2017;
Published: 19 December 2017.
Edited by:
Vivienne Ann Russell, University of Cape Town, South AfricaReviewed by:
Wolfgang Härtig, Leipzig University, GermanyScott Edward Counts, Michigan State University, United States
Copyright © 2017 Akinyemi, Allan, Oakley and Kalaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rufus O. Akinyemi, cm9ha2lueWVtaUBjb20udWkuZWR1Lm5n; cnVmdXNha2lueWVtaUB5YWhvby5jb20=
Rajesh N. Kalaria, cmFqLmthbGFyaWFAbmNsLmFjLnVr