 Lindsey B. Shelton
Lindsey B. Shelton Laura J. Blair
Laura J. Blair- Department of Molecular Medicine and USF Health Byrd Institute, University of South Florida, Tampa, FL, United States
The ATP-dependent 90 kDa heat shock protein, Hsp90, is a major regulator of protein triage, from assisting in nascent protein folding to refolding or degrading aberrant proteins. Tau, a microtubule associated protein, aberrantly accumulates in Alzheimer's disease (AD) and other neurodegenerative diseases, deemed tauopathies. Hsp90 binds to and regulates tau fate in coordination with a diverse group of co-chaperones. Imbalances in chaperone levels and activity, as found in the aging brain, can contribute to disease onset and progression. For example, the levels of the Hsp90 co-chaperone, FK506-binding protein 51 kDa (FKBP51), progressively increase with age. In vitro and in vivo tau models demonstrated that FKBP51 synergizes with Hsp90 to increase neurotoxic tau oligomer production. Inversely, protein phosphatase 5 (PP5), which dephosphorylates tau to restore microtubule-binding function, is repressed with aging and activity is further repressed in AD. Similarly, levels of cyclophilin 40 (CyP40) are reduced in the aged brain and further repressed in AD. Interestingly, CyP40 was shown to breakup tau aggregates in vitro and prevent tau-induced neurotoxicity in vivo. Moreover, the only known stimulator of Hsp90 ATPase activity, Aha1, increases tau aggregation and toxicity. While the levels of Aha1 are not significantly altered with aging, increased levels have been found in AD brains. Overall, these changes in the Hsp90 heterocomplex could drive tau deposition and neurotoxicity. While the relationship of tau and Hsp90 in coordination with these co-chaperones is still under investigation, it is clear that imbalances in these proteins with aging can contribute to disease onset and progression. This review highlights the current understanding of how the Hsp90 family of molecular chaperones regulates tau or other misfolded proteins in neurodegenerative diseases with a particular emphasis on the impact of aging.
Introduction
Aging is the biggest risk factor for developing a neurodegenerative disease, but the specific factors which cause these predominantly sporadic diseases are still under investigation (Reeve et al., 2014). As cells within the body age, the cellular homeostasis network must deal with an increasing amount of misfolded and aggregated proteins that can pathogenically accumulate leading to cell death. Aging is caused by compromised cellular homeostasis, fitness, and plasticity, leading to degeneration and cell death in vital organs. According to the “garbage catastrophe” hypothesis, aged differentiated cells lose the capacity to dispose of damaged and malfunctioning proteins (Terman, 2001). Such damaged proteins can assume cytotoxic properties, and their constant removal is thus essential for cell survival. Not only does aging lead to an increased likelihood of protein misfolding and aggregation, it is compounded by a decrease in the efficiency of the protein degradation machinery. The activity of both the proteasome, which is the main mechanism of protein degradation (Rock et al., 1994; Conconi et al., 1996), and chaperone-mediated autophagy (CMA; Cuervo and Dice, 2000b) is significantly impaired with aging and is especially pronounced in post-mitotic cells, such as neurons, potentially resulting in neurodegenerative disease (Terman, 2001). Fortunately, there is a system in place to help the body maintain proteostasis in times of stress and disease: the molecular chaperone network (Söti and Csermely, 2002). This network is comprised of a diverse family of proteins which contains members that are constitutively expressed to help in normal cell maintenance as well as members that become activated during times of stress. All of these chaperones assist in various ways to help fold, refold and degrade misfolded proteins.
The molecular chaperone network is comprised of diverse families of heat shock proteins (Hsps) that are divided based on their molecular mass. The small Hsps regulate general protein aggregation, Hsp40s regulate Hsp70 ATP hydrolysis, Hsp70 folds proteins during translation, and Hsp90 maintains and triages a subset of clients (Liberek et al., 2008; Miyata et al., 2011). While Hsp70 and Hsp90 perform many overlapping roles in the cell, Hsp90 shows more client selectivity. Hsp90 requires ATP to perform these functions including protein degradation, protein folding, prevention of protein aggregation, and protein modification (Echeverría et al., 2011). These regulatory processes are particularly important for intrinsically disordered proteins (IDPs) which have a high propensity to aggregate (Schopf et al., 2017).
Hsp90 binds to one of these IDPs, tau, in a broad region that includes aggregation prone areas (Karagöz et al., 2014). Tau normally functions to stabilize the microtubules and regulate axonal transport (Guo et al., 2017). The pathological accumulation of tau is a hallmark in several neurodegenerative disorders collectively termed tauopathies (Kovacs, 2015); a series of diseases including Alzheimer's disease (AD), progressive supranuclear palsy (PSP), Pick's disease, and chronic traumatic encephalopathy (CTE; Guo et al., 2017). Currently there are no treatment options available which regulate tau pathogenesis (Orr et al., 2017), therefore more work needs to be done to identify potential tau regulating therapeutic strategies.
A promising avenue to target tau is through Hsp90 inhibition. In fact, Hsp90 ATPase-inhibitors rapidly degrade tau aggregates in vivo (Dickey et al., 2007a; Luo et al., 2007), but these inhibitors have not yet been successful in clinical trials due to lack of efficacy and associated toxicities (Bhat et al., 2014; Renouf et al., 2016; Thakur et al., 2016). However, Hsp90 regulates tau and other aggregating proteins in coordination with a diverse group of co-chaperones (Schopf et al., 2017). In fact, the levels of many of these co-chaperones have been shown to change with aging, which can alter the fate of tau and potentially contribute to disease onset or severity (Blair et al., 2013; Brehme et al., 2014). It is possible that a more successful treatment strategy may be found by a therapeutic aimed toward regulating these co-chaperones or Hsp90/co-chaperone heterocomplexes (Kamal et al., 2003; Rodina et al., 2016). This review discusses the involvement of Hsp90 and its co-chaperones in disease and how alterations in levels and activity with aging can affect this process (Table 1). Current Hsp90 therapeutic interventions for neurodegenerative diseases will also be reviewed.
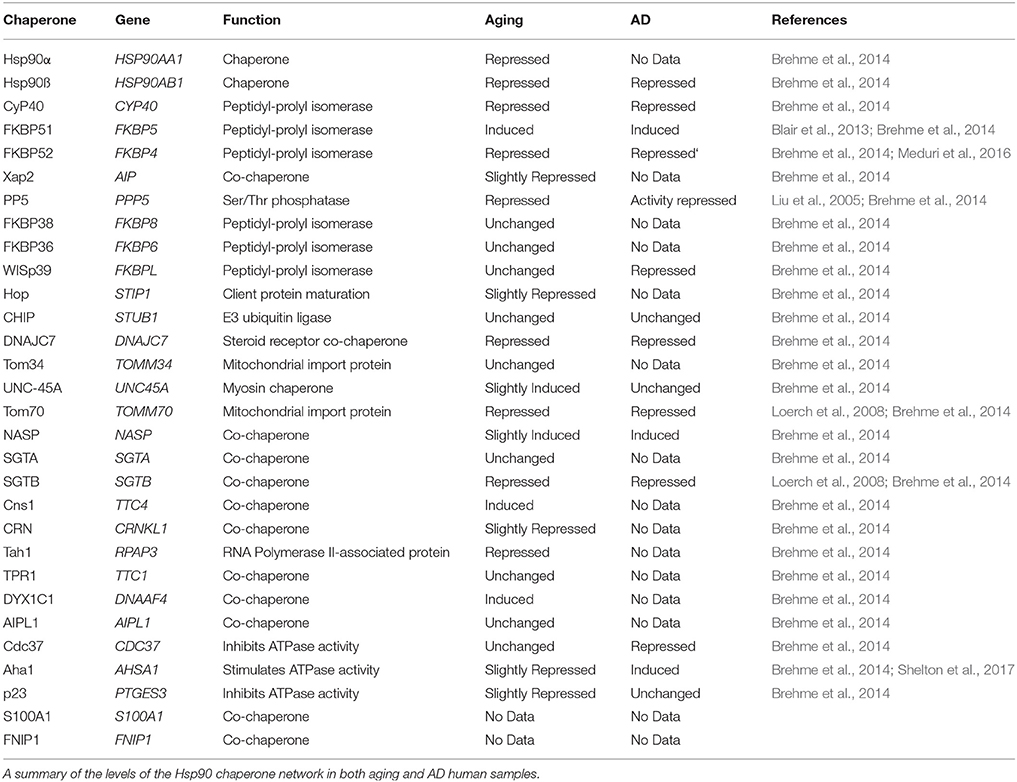
Table 1. Summary of Hsp90 and Hsp90 co-chaperone levels in aging and Alzheimer's disease (AD).
Hsp90
Hsp90 is critical to maintaining proteostasis (Brehme et al., 2014) and accounts for up to 6% of all protein within the cell during times of stress (Picard, 2002; Prodromou, 2016). Hsp90 consists of three domains: an N-terminal ATP-binding domain, a middle domain, and a C-terminal domain responsible for the inherent dimerization of the protein (Li and Buchner, 2013). Hsp90 requires ATP in order to dimerize and properly assist in protein folding. The Hsp90 ATPase cycle consists of four stages: the ATP-bound state, an initial intermediate state (I1), a second intermediate state (I2), and finally a closed state in which ATP hydrolysis occurs (Li and Buchner, 2013). There are several different isoforms of Hsp90, however this review will only focus on Hsp90 in the cytosol, which includes Hsp90α (stress-inducible) and Hsp90β (constitutively active) (Li et al., 2012).
The two different cytosolic forms of Hsp90 are 86% genetically identical and have 93% amino acid sequence homology, showing lots of similarities in structure and function. However, there are some differences that set these two isoforms apart. The first difference is the viability of Hsp90 knock-out mice. Mice lacking Hsp90β are embryonically lethal and do not survive past day 9, whereas mice lacking Hsp90α are viable but leads to sterility in male mouse (Table 2; Voss et al., 2000; Grad et al., 2010). There are also some differences in the cellular functions of Hsp90α and Hsp90β. Hsp90α is involved in growth promotion, cell cycle regulation, stress-induced cytoprotection, and cancer cell invasiveness; whereas Hsp90β is involved with early embryonic development, germ cell maturation, cytoskeletal stabilization, cellular transformation, signal transduction, and long-term cell adaptation (Eustace et al., 2004; Sreedhar et al., 2004). While there are some general functional differences between the two cytosolic isoforms more studies are needed to better understand the role of these different isoforms on tau pathology.
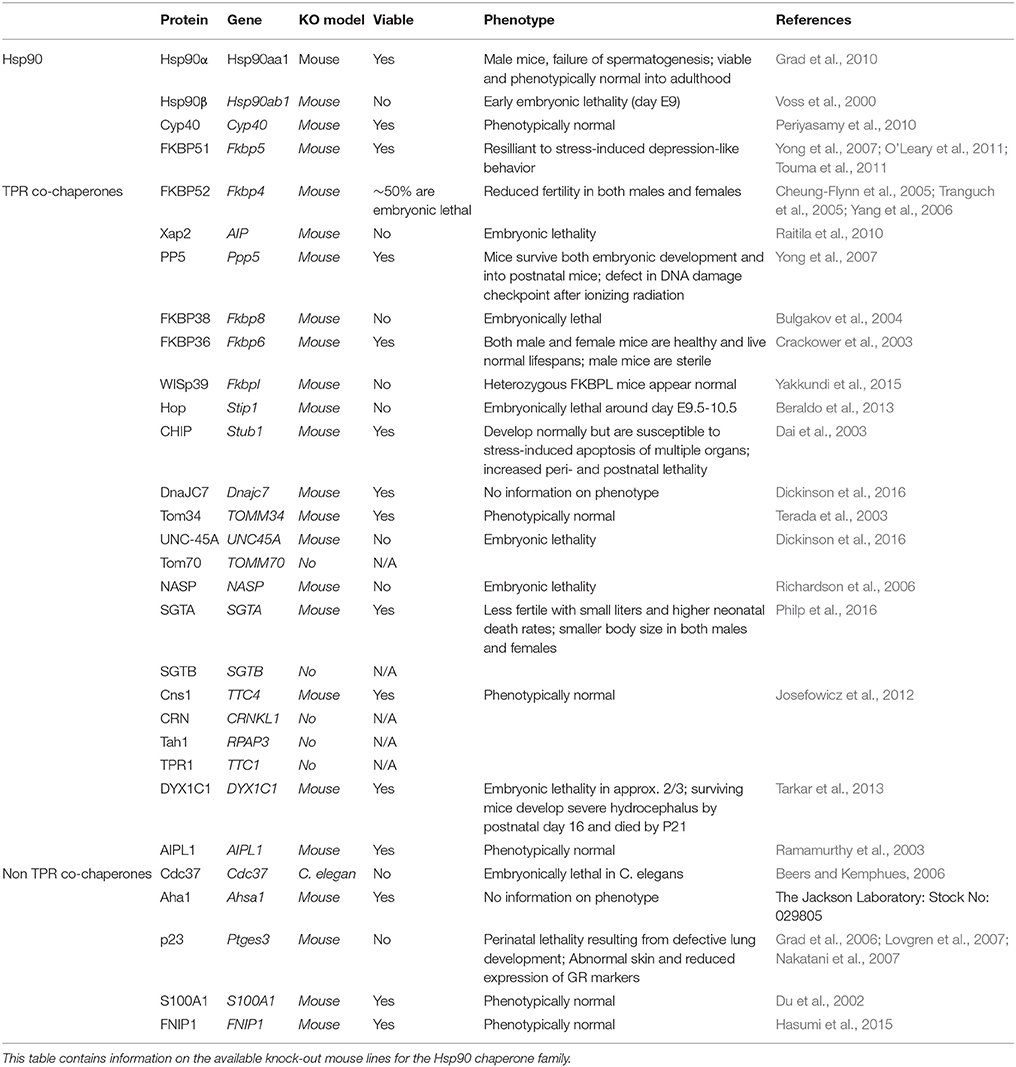
Table 2. Summary of Hsp90 and Hsp90 co-chaperone knockout mice.
Alterations in chaperone expression are commonly seen in aging, leading to complications within the Hsp90 chaperone network. In fact, recent work has shown the levels of many Hsp90 co-chaperones are also altered in the aged brain (Brehme et al., 2014). These co-chaperones are necessary for client selection and triage. There are two main categories of Hsp90 co-chaperones: tetratricopeptide repeat (TPR) and non-TPR containing (Prodromou et al., 2000). Therefore, changes in Hsp90 levels are part of a larger imbalance in the chaperone network which contribute to aging and age-related neurodegenerative disorders.
TPR Co-Chaperones
TPR-containing Hsp90 co-chaperones interact with the C-terminal MEEVD peptide motif on Hsp90 (Li et al., 2012). Since Hsp90 functions as a dimer, two TPR-containing co-chaperones could interact simultaneously. However, these interactions are dependent on the isoform of Hsp90 and the repertoire of expressed co-chaperones. In fact, co-chaperones do compete for binding to Hsp90 (Harst et al., 2005; Hildenbrand et al., 2011). This competition can have beneficial or detrimental effects on tau pathology. Known examples include co-chaperones which interact with Hsp90 to promote the degradation of aberrant tau or others which drive tau oligomerization and aggregation. Therefore, an imbalance in protein levels with aging and AD compound this already complex competition for binding Hsp90 to regulate tau fate (Figure 1). Here, we will describe these TPR-co-chaperones, and how their interaction with Hsp90 regulates tau.
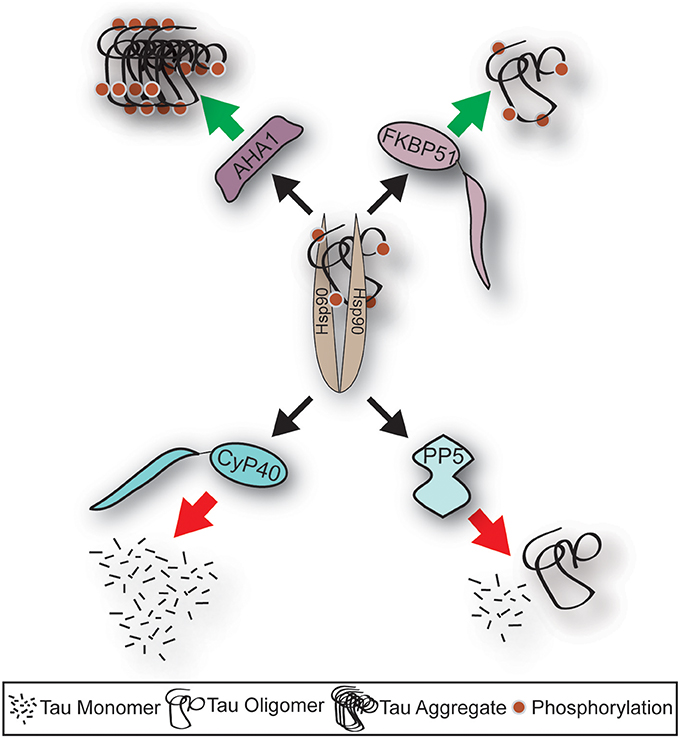
Figure 1. Schematic depicting fate of tau following Hsp90 interaction with distinct co-chaperone; the impact of Alzheimer's disease on the levels of co-chaperones. Aha1 and FKBP51 protein levels are induced in AD, and their association to tau leads to increased aggregation. Whereas, CyP40 and PP5 levels are repressed in AD, and their association to tau leads to reduced tau aggregation. This schematic highlights the important role of co-chaperones in AD.
Immunophilins and Immunophilin Homologs
Hsp90 interacts with six immunophilins that display peptidyl-prolyl isomerase (PPIase) activity through TPR domains including cyclophilin 40 (CyP40) and five FK506-binding proteins: FKBP51, FKBP52, FKBP36, FKBP38, FKBPL/WISp39 (Jascur et al., 2005; Jarczowski et al., 2009; Guy et al., 2015; Blundell et al., 2017). These PPIases regulate the twisting of proline bonds through stabilization of the cis-trans transition state and accelerate the isomerization process. This is particularly important for tau, which has 40 proline residues that regulate phosphorylation and aggregation propensity (Mandelkow and Mandelkow, 2012). Hsp90 also interacts with two immunophilin homologs: protein phosphatase 5 (PP5) and XAP2/FKBP37. Altered levels of many of these immunophilins and immunophilin-like proteins have been found in aging and AD (Table 1), which could skew the competition dynamics for Hsp90 binding (discussed later in this review) and may promote toxic tau accumulation.
CyP40
An interesting PPIase, CyP40, decreases in aging and is further repressed in AD (Table 1; Brehme et al., 2014). CyP40 was recently shown to disaggregate tau fibrils in vitro and prevents toxic tau accumulation in vivo preserving memory, demonstrating a neuroprotective role for CyP40 in the brain (Baker et al., 2017). The PPIase activity of CyP40 is slightly repressed when bound to Hsp90, but under cellular stress CyP40 can release from Hsp90 increasing its isomerase and chaperone activity (Blackburn et al., 2015). However, as CyP40 levels decrease with aging, it is possible that the pool of free CyP40 is not sufficient to help disentangle aggregating proteins, like tau.
FKBP51
Contrary to the neuroprotective effects of CyP40, two FK506-binding proteins (FKBPs) have been shown to stimulate toxic tau aggregation (Blair et al., 2013; Giustiniani et al., 2015; Kamah et al., 2016). One of these, FKBP51, coordinates with Hsp90 to preserve toxic tau oligomers in vivo (Blair et al., 2013). In fact, mice lacking FKBP51 have decreased tau levels in the brain (Jinwal et al., 2010; Blair et al., 2013). However, throughout aging, FKBP51 levels progressively increase and are further increased in AD brain samples (Table 1; Blair et al., 2013; Sabbagh et al., 2014). Previous studies have also shown that FKBP51 can form complexes with tau in both human AD brain samples and control samples (Jinwal et al., 2010). Additionally, this study showed that FKBP51 was able to stabilize microtubules, suggesting a novel and unique function for FKBP51 (Jinwal et al., 2010). Taken together, the increase in FKBP51 in aging and AD suggest that targeting FKBP51 could offer a potential therapeutic strategy for tauopathies such as AD.
FKBP52
FKBP52 interacts both physically and functionally with tau and promotes tau aggregation in vitro (Giustiniani et al., 2015; Meduri et al., 2016). FKBP52 induces oligomers from both P301L and truncated wild-type tau. Interestingly, this oligomerization is not due to the PPIase activity of FKBP52, instead the oligomerization of tau appears to occur via molecular interaction (Kamah et al., 2016). FKBP52 can also induce aggregation of a truncated form of tau that appears to have prion like behavior, suggesting a possible mechanism for the spread of tau pathology throughout the brain in diseases such as AD (Giustiniani et al., 2015). However, it is interesting to note that FKBP52 levels are lower in the cortex of AD patients' brains (Table 1; Brehme et al., 2014; Meduri et al., 2016).
FKBP36, FKBP38, and FKBPL
There are several other FKBPs that act as co-chaperones to Hsp90 including FKBP36, FKBP38, and FKBPL (WISp39), however their relationship to tau, if any, is still unknown at this point and so they will not be discussed in detail.
XAP2
XAP2, otherwise known as FKBP37 or Aryl hydrocarbon receptor interacting protein (AIP), contains a PPIase homologous domain. While a direct role of XAP2 in tau pathogenesis has not been described, studies have shown that XAP2 is activated by histone deacetylase (HDAC) 6, which has been linked to pathogenic tau (Kekatpure et al., 2009; Cook et al., 2012; Selenica et al., 2014). In addition, XAP2 coordinates with Hsp90 to regulate glucocorticoid receptor signaling (Laenger et al., 2009), which has also been implicated in the production of pathogenic tau (Pinheiro et al., 2016). Additional studies are needed to determine the direct or indirect effects of XAP2 on tau pathology.
PP5
Another member of this family, protein phosphatase 5 (PP5), is repressed in aging. PP5 is contains a PPIase homology domain, but does not display classical PPIase activity, since it can bind FK506 (Silverstein et al., 1997). Instead, PP5 acts as a Ser/Thr phosphatase, which activates when bound to Hsp90 (Conde et al., 2005). PP5 activity has been shown to be repressed in AD (Table 1; Liu et al., 2005). Studies have shown that PP5 is able to dephosphorylate tau at several phosphorylation sites connected to AD pathology (Gong et al., 2004). Further studies are needed to better understand if the upregulation of PP5 could be used to slow or prevent tau pathogenesis.
Hop
The Hsp70-Hsp90 organizing protein, otherwise known as Hop and sometimes STIP1 (stress inducible protein 1), is involved in helping transfer client proteins from the early stages of protein maturation involving Hsp70 and Hsp40 to the later stages of the cycle involving Hsp90 (Baindur-Hudson et al., 2015). As such, Hop plays a crucial role in the maturation of client proteins, like tau. A previous study found that when Hop was depleted using siRNA, there was an accumulation of tau (Jinwal et al., 2013). This suggests that Hop is necessary for tau clearance via Hsp70/Hsp90. In fact, loss-of-function mutations in Hop drive toxic tau accumulation in a fly model of tauopathy (Ambegaokar and Jackson, 2011). Together these studies demonstrate a protective role of Hop in a tauopathic brain.
CHIP
C-terminus of Hsc70-interacting protein (CHIP) is highly involved in the Hsp70-Hsp90 machinery acting not only as a co-chaperone, but also as an E3 ubiquitin ligase responsible for ubiquitin-dependent proteasomal degradation (Edkins, 2015). CHIP has many roles within the cell including stress-response activation, protein triage, and restitution of the stress response (Dickey et al., 2007b). CHIP has been linked to several neurodegenerative disorders including Huntington's disease, Parkinson's disease and AD as well as other diseases such as cystic fibrosis and cancer (Dickey et al., 2007b; Edkins, 2015). In tauopathic mice, CHIP regulates the removal of tau species that have undergone abnormal phosphorylation and folding (Dickey et al., 2007b). Additionally, silencing CHIP via siRNA, led to a massive increase in tau levels (Dickey et al., 2008). Similarly, CHIP/Stub1-knockout mice have increased accumulation of phospho- and total tau species (Table 2; Palubinsky et al., 2015). Overexpression of CHIP could represent a therapeutic strategy to prevent neuronal cell death and improve outcomes of neurodegenerative diseases by promoting the degradation of tau.
DnaJC7
DnaJC7, also known as Tpr2, simultaneously binds Hsp70 and Hsp90 via its two TPR domains (Brychzy et al., 2003). To date, a link between DnaJC7 and tau has not been investigated. However, it is known that DnaJC7 plays an important role in steroid receptor chaperoning, as well as recycling substrates from Hsp90 back to Hsp70 via this unique TPR interaction (Brychzy et al., 2003; Moffatt et al., 2008). Additional studies are needed to understand if DnaJC7 regulates tau pathogenesis.
Tom34
Tom34 is a co-chaperone involved in mitochondrial protein import. One study found that in Drosophila, impaired Tom34 gene function led to enhanced tau pathology (Ambegaokar and Jackson, 2011). Conversely, the same study demonstrated Tom34 overexpression was able to suppress tau toxicity elucidating a role for Tom34 in tau pathology in Drosophila. The mechanism by which Tom34 promotes tau pathology remains unclear. It is possible that mitochondrial dysfunction could lead to cellular stress which, in turn, could enhance tau pathology. Additional studies are needed to fully elucidate this interaction.
In addition, there are other TPR-containing Hsp90 co-chaperones such as UNC-45, Tom70, NASP, SGTA, SGTB, Cns1, CRN, Tah1, TPR1, DYX1C1, and AIPL1. However, very little is known about most of these co-chaperones in the brain and even less is known about their interactions with tau, therefore they will not be discussed in detail in this review.
Non-TPR Co-chaperones
Cdc37
Cell division cycle 37 (Cdc37) slows the ATPase activity of Hsp90 allowing a prolonged interaction between Hsp90 and its client proteins (Cox and Johnson, 2011). Cdc37 is also required for the stable folding of protein kinases in coordination with Hsp90 (Calderwood, 2015). Many of these kinases are known to phosphorylate tau at sites associated with AD, such as GSK3β and MAPK13 (Taipale et al., 2012; Jin et al., 2016). Interestingly, overexpression of Cdc37 preserves tau, and its suppression reduces tau (Jinwal et al., 2012). However, additional studies are needed to better understand the dynamics between Cdc37 and tau phosphorylation.
Aha1
The activator of Hsp90 ATPase homolog 1 (Aha1) works as a co-chaperone to stimulate the ATPase function of Hsp90 to regulate the folding and activation of client proteins. Aha1 interacts with Hsp90 independent of its nucleotide status and allows the Hsp90 ATPase cycle to skip the I1 phase, thus accelerating the progression of the ATPase cycle dramatically (Li and Buchner, 2013; Wolmarans et al., 2016). Aha1 levels have been shown to increase with AD. In fact, we have found that Aha1 levels in the medial temporal gyrus of human brain correlated with increased tau Braak staging (Shelton et al., 2017). In the same study, we found that high levels of Aha1 in a tau transgenic mouse model increased tau oligomers as well as neuronal loss concomitant with cognitive deficits (Shelton et al., 2017). Since Aha1 levels are repressed in aging, but are abnormally preserved in AD, tau aggregation could be accelerated in part by Aha1 in the AD brain. Previous studies have also implicated Aha1 for a role in cystic fibrosis. In fact, one study showed that knockdown of Aha1 promotes the translocation of the disease-related mutant of cystic fibrosis transmembrane conductance receptor, CFTR, to the plasma membrane, allowing it to properly function (Wang et al., 2006). Thus, treatments which reduce Aha1 may be beneficial for both AD and cystic fibrosis.
P23
p23 has an opposing effect on Hsp90 compared to Aha1. p23 works by inhibiting the ATPase activity of Hsp90. The interaction between Hsp90 and p23 is nucleotide-dependent meaning that p23 can only interact with Hsp90 when ATP is bound (Sullivan et al., 1997). p23 works in a unique way to inhibit ATPase activity, it can either inhibit the hydrolysis process or it can impede the release of ADP and Pi (Rehn and Buchner, 2015). As a co-chaperone, p23 works to suppress protein aggregation and exhibits chaperoning activity, although p23 is not able to refold proteins on its own (Freeman et al., 1996). Inhibition of p23 in an siRNA screen of Hsp90 co-chaperones showed that silencing p23 reduced both total and phospho-tau (Jinwal et al., 2012, 2013). p23 also plays an important role in preventing endoplasmic reticulum (ER) stress-induced cell death, which can be triggered by misfolded proteins, like tau (Rao et al., 2006; Abisambra et al., 2013). However, p23 can be cleaved during ER stress-induced cell death into a smaller p19 fragment which is then unable to exert its anti-apoptotic effects (Zhang et al., 2013). A mutant p23 (p23D142N) that is uncleavable was shown to ameliorate the ER stress-induced cell death in vitro and suggests that this mutant p23 could be a potential therapeutic target in neurodegenerative diseases (Zhang et al., 2013).
S100A1
S100 calcium-binding protein A1 (S100A1) interacts with Hsp90. One study used siRNA to screen several Hsp90 co-chaperones to investigate the effect on tau. This study found that reductions in S100A1 also led to massive reductions in both phospho- and total tau levels in cells (Jinwal et al., 2013). S100A1 could play a role in stabilizing tau, thus leading to a worsening of tau pathology. Therefore, silencing or knocking down S100A1 could offer a potential therapeutic strategy for tauopathies.
FNIP1
The folliculin-interacting protein 1 (FNIP1) is able to interact with Hsp90 as a co-chaperone in order to inhibit its ATPase activity. One study found that FNIP1, in complex with FNIP2 and Hsp90, was able to stabilize the tumor suppressor folliculin (FLCN; Woodford et al., 2016). FNIP1 was shown to interact directly with Hsp90, and it can also interact with other co-chaperones such as p23, Hop and Cdc37 (Woodford et al., 2016). FNIP1 was also shown to compete for binding with Aha1 suggesting an important role for FNIP1 in the Hsp90 chaperone network (Woodford et al., 2016). Additional studies are needed to determine if FNIP1 could regulate tau directly or potentially through competition with Aha1 to bind Hsp90 and alter its ATPase activity.
Aging in the Hsp90 Chaperone Network
All of the above mentioned co-chaperones interact with Hsp90 in order to form diverse heterocomplexes, however, changes in Hsp90 expression with aging can alter their composition. There is conflicting data on Hsp90 levels with age in both human and animal studies. One study focused on the basal levels of cytosolic Hsp90 in peripheral blood mononuclear cells (PBMC) and found that in aged human samples there was an increase in Hsp90 under normal physiological conditions when compared to young samples (Njemini et al., 2007). Another study had similar findings in the hippocampus of aged gerbils. This study demonstrated that cytosolic Hsp90 levels were significantly increased in the hippocampus of aged gerbils (24 months) compared to adult gerbils (6 months) (Lee et al., 2011). Conversely, there are also studies showing decreased levels of Hsp90 in aged human brain samples. For instance, two other studies investigated the levels of chaperone proteins in the human brain. One study found that cytosolic Hsp90 was repressed in the superior frontal gyrus, while another demonstrated a similar repression in the prefrontal cortex of aged patients compared to controls (Berchtold et al., 2008; Loerch et al., 2008; Brehme et al., 2014). Taken together, this data suggests that alterations in Hsp90 levels do not occur uniformly and that changes in the expression of Hsp90 with aging may vary between cell types and brain regions (Berchtold et al., 2008). While Hsp90 protein levels are an important factor with aging, co-chaperone expression levels could be equally important in heterocomplex formation.
In addition to the differences in expression levels of Hsp90, there are also changes in expression levels of co-chaperone proteins during the aging process. Almost all of the Hsp90 co-chaperones are repressed in aging, suggesting that these proteins could play important roles in maintaining homeostasis within the cell (Brehme et al., 2014). For instance, CyP40, FKBP52, PP5, Hop, p23, and Aha1 are all repressed in the aged brain. All of these proteins are integral to the Hsp90 chaperone system and when levels of these proteins go down the Hsp90 chaperone network can no longer function normally, which can lead to an increased risk of developing a neurodegenerative disease. Interestingly, one co-chaperone is significantly induced in the aged brain and that is FKBP51. FKBP51 has several important roles within the cell including immunoregulation as well as helping with protein folding and trafficking in complex with Hsp90. Because FKBP51 is induced in aging, while many other co-chaperones are reduced, this suggests that the imbalance seen in these proteins during aging could lead to completely different Hsp90 heterocomplexes resulting in the dysfunction of cellular homeostasis during aging.
Hsp90 is able to form many unique heterocomplexes with different co-chaperones in order to regulate protein triage. Hsp90 heterocomplexes are unique in that there is usually a specific progression of co-chaperones that interact with Hsp90 (Schopf et al., 2017). One interesting aspect to these heterocomplexes is the fact that Hsp90 can bind multiple co-chaperones simultaneously. One study found that Hsp90 could form stable complexes with Hsp90, FKBP52, Hop, and p23 (Hildenbrand et al., 2011). There does appear to be a hierarchy though, with some co-chaperones able to bind more strongly than others. For example, Aha1 has been shown to compete with Hop, FNIP1, and p23 for the ability to bind with Hsp90 (Harst et al., 2005; Woodford et al., 2016). These competition dynamics between Aha1 and p23/FNIP1 suggest that there is a constant battle for control of the ATPase activity of Hsp90. Additionally, FKBP51 and FKBP52 have been shown to have greater relative binding to Hsp90 compared to other TPR co-chaperones (Schülke et al., 2010). While not as strong as FKBP51 and FKBP52, PP5 forms more complexes with Hsp90 than most other TPR co-chaperones. Taken together, increased FKBP51 and decreased PP5 and CyP40 could contribute to an imbalance in Hsp90 heterocomplexes which may promote increased tau phosphorylation and aggregation causing neurotoxicity (Blair et al., 2013). This suggests an even more complex system in place because depending on the amount of certain co-chaperones and their relative ability to bind to Hsp90; certain maladaptive complexes could be more abundant than others with aging.
In addition to altering Hsp90 heterocomplex composition and client selection, altered Hsp90 co-chaperone expression can interfere with degradation of aberrant proteins via the proteasome or autophagy. As mentioned above, aged cells are often inundated with misfolded and aggregated proteins, which can overload the Hsp90 chaperone network causing a negative spiral where there are not enough healthy chaperone molecules to refold or degrade aberrant proteins. In addition to the problems faced with an overwhelmed chaperone network, the proteolytic activity of the proteasome also declines with aging, and in fact Hsp90 has been shown to protect the proteasome from age-related, oxidative-dependent decline (Conconi and Friguet, 1997). However, with advanced aging, the association between Hsp90 and the proteasome drastically decreases (Conconi et al., 1996). This suggests that because the Hsp90 chaperone system and the proteasome are so connected, when one starts to fail the other will fail as well leading to cytotoxicity and cell death. Proteins can also be degraded by CMA; however, CMA activity also decreases with age (Cuervo and Dice, 2000a). Hsp90 and Hop are both involved in the CMA system; helping to unfold the target substrate before it can translocate into the lysosome for degradation. As mentioned previously, both Hsp90 and Hop are repressed in aging and therefore may not be able to assist in the translocation of substrates, leading to a buildup of misfolded or aggregated proteins. Post-translational modifications (PTMs) of Hsp90 can also complicate the matter further.
There are many different PTMs that can affect Hsp90 including phosphorylation, acetylation, S-nitrosylation, oxidation, and ubiquitination; and all of these PTMs can impact the chaperoning function of Hsp90. Phosphorylation of Hsp90 leads to reduced chaperoning ability and phosphorylation of specific tyrosine residues can affect the ability of Hsp90 to interact with distinct client proteins (Zhao et al., 2001; Mollapour and Neckers, 2012). Acetylation of Hsp90 affects client protein interaction and also decreases binding of Hsp90 to ATP (Yu et al., 2002; Mollapour and Neckers, 2012). S-nitrosylation, oxidation and ubiquitination also inhibit Hsp90 chaperone activity (Blank et al., 2003; Martínez-Ruiz et al., 2005; Chen et al., 2008). These PTMs increase with aging and can alter the ability of Hsp90 to function properly as well as change the ability of different co-chaperones to bind. As the chaperone network declines with aging, so does the ability of the cell to recover from damaged proteins and stress, thus leading to an environment which promotes aberrant protein accumulation and neurotoxicity.
Targeting the Hsp90 Chaperone Network
Inhibition of the ATPase activity of Hsp90 has been shown to have positive outcomes in cell culture and animal models of tauopathy. Hsp90 ATPase inhibitors have been developed to target each of the three domains; with the majority of Hsp90 inhibitors targeting the N-terminal domain (Bhat et al., 2014). Inhibition of Hsp90 induces the expression of protective chaperones, Hsp70 and Hsp40, further promoting the degradation of aberrant proteins (Carman et al., 2013). Previous studies have shown that Hsp90 inhibition decreased the levels of hyperphosphorylated and/or mutated tau species both in cells and mice. The Hsp90 N-terminal domain inhibitor, EC102, was used to demonstrate degradation of hyperphosphorylated pathologically relevant tau in cells (Dickey et al., 2007a). Another N-terminal Hsp90 ATPase inhibitor, 17-AAG, was shown to decrease levels of phosphorylated tau in cells, and a related N-terminal Hsp90 ATPase inhibitor, PU-DZ8, reduced soluble and insoluble tau in tauP301L mice (Luo et al., 2007). Although Hsp90 inhibitors have been in clinical development since 1999, none have reached New Drug Application (NDA) status (Bhat et al., 2014). All of these clinical trials were focused on investigating Hsp90 inhibitors on various cancers. Hsp90 plays a similar role in both neurodegenerative disorders and cancer, however because of the complexity of the brain and the need for a blood-brain barrier (BBB) permeable drug, the clinical development of Hsp90 inhibitors for neurodegenerative diseases has been even less successful. While the development of Hsp90 inhibitors is still underway, it is possible that the development of therapeutics which target Hsp90 heterocomplexes or discrete Hsp90 co-chaperones could open up additional avenues for success in developing a BBB-permeable drug.
In addition to offering more potential therapeutic targets, small molecules which modify Hsp90/co-chaperone interactions may also show more specificity for a specific pool of Hsp90 which may reduce the number of on-target side effects. There are very few Hsp90 co-chaperone targeting small molecules, and of these, only a handful of these have been investigated for their role in effecting tau. There is a Hop/Hsp90 complex specific inhibitor, C9, however, there is no available data on how chemical inhibition of this complex affects tau accumulation (Pimienta et al., 2011). The Cdc37/Hsp90 inhibitors, Celasterol and Withaferin A (Zhang et al., 2008; Yu et al., 2010), reduce tau levels and a new compound, platycodin D has just been discovered (Li et al., 2017). Platycodin D does not affect the ATPase activity of Hsp90, but instead disrupts the interaction between Hsp90 and Cdc37 leading to client protein degradation without an increase in Hsp70 (Li et al., 2017). More work still needs to be done to better understand the role of Cdc37 in tau phosphorylation to determine if targeting this complex is of therapeutic benefit. Developing drugs to target discrete FKBPs has been challenging due to their homology, however, despite their structural similarity they do display differences in conformational flexibility which could be a way to potentially target specific FKBPs in the future (LeMaster and Hernandez, 2015). Interestingly, one study demonstrated that patients chronically treated with FK506, which inhibits the PPIase domain of many of the FKBPs, significantly reduced the incidence of AD (Taglialatela et al., 2015). The targeting FKBP51 is of great interest for the treatment of tauopathies as well as mood disorders (Zannas and Binder, 2014). Recently a PPIase antagonist has been developed which shows selectivity for FKBP51, but additional studies are needed to determine if targeting the PPIase domain of FKBP51 will be effective in regulating tau accumulation (Jinwal et al., 2010). There is one compound, MJC13, which targets the FKBP52-Hsp90-androgen receptor complex (De Leon et al., 2011). MJC13 results in a reduced stress response which has demonstrated therapeutic potential in cancer, but so far MJC13 has not been investigated for a role in tau pathology (De Leon et al., 2011). Additionally, Aha1-specific inhibitors have been recently developed (Hall et al., 2014). One of these inhibitors, KU-177, reduced insoluble tauP301L levels in cells (Shelton et al., 2017). While this is an exciting result, more studies are needed to determine if Aha1 inhibitors regulate tau similarly in vivo. There is still much to be done to develop compounds which target the Hsp90 chaperone network, but there are a lot of promising leads which can be targeted to develop disease-modifying therapeutics.
Conclusions
The Hsp90 chaperone machinery plays a huge role in both aging and neurodegenerative diseases. Hsp90 is one of the most highly expressed proteins in the cell and is involved in a myriad of cellular processes. Previous work has focused on inhibition of Hsp90 to triage misfolded proteins. There are also many co-chaperones that associate with Hsp90 and play their own roles in aging and neurodegeneration. As these Hsp90 co-chaperones change with age they can significantly impact the propensity for certain neurodegenerative diseases. FKBP51 steadily increases with age and both FKBP51 and Aha1 are induced in the AD brain suggesting that these two co-chaperones negatively affect tau pathology. On the other hand, both CyP40 and PP5 are repressed in aged and AD brains. CyP40 disaggregates tau fibrils in vitro and PP5 dephosphorylates tau restoring microtubule binding, suggesting that increasing the levels or activity of these co-chaperones could have a beneficial, neuroprotective role in diseases such as AD. These are just a few examples of how important maintaining the balance of Hsp90 co-chaperones is to homeostasis, and what can happen when they are altered in aging and disease. Hsp90 co-chaperones offer a unique target for potential therapeutics due to their specific roles within the Hsp90 machinery. Overall, more work needs to be done to develop BBB-permeable therapeutics to target discrete Hsp90 co-chaperones for the treatment of AD and other tauopathies.
Author Contributions
LS wrote the manuscript. JK created the figures and edited the manuscript. LB critiqued and edited the manuscript drafts.
Funding
This work was supported by NIH Grants NS073899 and MH103848.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O'Leary, J. C., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. Off. J. Soc. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Ambegaokar, S. S., and Jackson, G. R. (2011). Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 20, 4947–4977. doi: 10.1093/hmg/ddr432
Baindur-Hudson, S., Edkins, A. L., and Blatch, G. L. (2015). Hsp70/Hsp90 organising protein (hop): beyond interactions with chaperones and prion proteins. Subcell. Biochem. 78, 69–90. doi: 10.1007/978-3-319-11731-7_3
Baker, J. D., Shelton, L. B., Zheng, D., Favretto, F., Nordhues, B. A., Darling, A., et al. (2017). Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 15:e2001336. doi: 10.1371/journal.pbio.2001336
Beers, M., and Kemphues, K. (2006). Depletion of the co-chaperone CDC-37 reveals two modes of PAR-6 cortical association in C. elegans embryos. Dev. Camb. Engl. 133, 3745–3754. doi: 10.1242/dev.02544
Beraldo, F. H., Soares, I. N., Goncalves, D. F., Fan, J., Thomas, A. A., Santos, T. G., et al. (2013). Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 27, 3594–3607. doi: 10.1096/fj.13-232280
Berchtold, N. C., Cribbs, D. H., Coleman, P. D., Rogers, J., Head, E., Kim, R., et al. (2008). Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. U.S.A. 105, 15605–15610. doi: 10.1073/pnas.0806883105
Bhat, R., Tummalapalli, S. R., and Rotella, D. P. (2014). Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J. Med. Chem. 57, 8718–8728. doi: 10.1021/jm500823a
Blackburn, E. A., Wear, M. A., Landré, V., Narayan, V., Ning, J., Erman, B., et al. (2015). Cyclophilin40 isomerase activity is regulated by a temperature-dependent allosteric interaction with Hsp90. Biosci. Rep. 35:e00258. doi: 10.1042/BSR20150124
Blair, L. J., Nordhues, B. A., Hill, S. E., Scaglione, K. M., O'Leary, J. C., Fontaine, S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169. doi: 10.1172/JCI69003
Blank, M., Mandel, M., Keisari, Y., Meruelo, D., and Lavie, G. (2003). Enhanced ubiquitinylation of heat shock protein 90 as a potential mechanism for mitotic cell death in cancer cells induced with hypericin. Cancer Res. 63, 8241–8247.
Blundell, K. L., Pal, M., Roe, S. M., Pearl, L. H., and Prodromou, C. (2017). The structure of FKBP38 in complex with the MEEVD tetratricopeptide binding-motif of Hsp90. PLoS ONE 12:e0173543. doi: 10.1371/journal.pone.0173543
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. doi: 10.1016/j.celrep.2014.09.042
Brychzy, A., Rein, T., Winklhofer, K. F., Hartl, F. U., Young, J. C., and Obermann, W. M. J. (2003). Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. EMBO J. 22, 3613–3623. doi: 10.1093/emboj/cdg362
Bulgakov, O. V., Eggenschwiler, J. T., Hong, D.-H., Anderson, K. V., and Li, T. (2004). FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Dev. Camb. Engl. 131, 2149–2159. doi: 10.1242/dev.01122
Calderwood, S. K. (2015). Cdc37 as a co-chaperone to Hsp90. Subcell. Biochem. 78, 103–112. doi: 10.1007/978-3-319-11731-7_5
Carman, A., Kishinevsky, S., Koren, J., Lou, W., and Chiosis, G. (2013). Chaperone-dependent neurodegeneration: a molecular perspective on therapeutic intervention. J. Alzheimers Dis. Park. 2013:007. doi: 10.4172/2161-0460.S10-007
Chen, W.-Y., Chang, F.-R., Huang, Z.-Y., Chen, J.-H., Wu, Y.-C., and Wu, C.-C. (2008). Tubocapsenolide A, a novel withanolide, inhibits proliferation and induces apoptosis in MDA-MB-231 cells by thiol oxidation of heat shock proteins. J. Biol. Chem. 283, 17184–17193. doi: 10.1074/jbc.M709447200
Cheung-Flynn, J., Prapapanich, V., Cox, M. B., Riggs, D. L., Suarez-Quian, C., and Smith, D. F. (2005). Physiological role for the cochaperone FKBP52 in androgen receptor signaling. Mol. Endocrinol. Baltim. Md 19, 1654–1666. doi: 10.1210/me.2005-0071
Conconi, M., and Friguet, B. (1997). Proteasome inactivation upon aging and on oxidation-effect of HSP 90. Mol. Biol. Rep. 24, 45–50. doi: 10.1023/A:1006852506884
Conconi, M., Szweda, L. I., Levine, R. L., Stadtman, E. R., and Friguet, B. (1996). Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Arch. Biochem. Biophys. 331, 232–240. doi: 10.1006/abbi.1996.0303
Conde, R., Xavier, J., McLoughlin, C., Chinkers, M., and Ovsenek, N. (2005). Protein phosphatase 5 is a negative modulator of heat shock factor 1. J. Biol. Chem. 280, 28989–28996. doi: 10.1074/jbc.M503594200
Cook, C., Gendron, T. F., Scheffel, K., Carlomagno, Y., Dunmore, J., DeTure, M., et al. (2012). Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Hum. Mol. Genet. 21, 2936–2945. doi: 10.1093/hmg/dds125
Cox, M. B., and Johnson, J. L. (2011). The role of p23, Hop, immunophilins, and other co-chaperones in regulating Hsp90 function. Methods Mol. Biol. Clifton NJ 787, 45–66. doi: 10.1007/978-1-61779-295-3_4
Crackower, M. A., Kolas, N. K., Noguchi, J., Sarao, R., Kikuchi, K., Kaneko, H., et al. (2003). Essential role of Fkbp6 in male fertility and homologous chromosome pairing in meiosis. Science 300, 1291–1295. doi: 10.1126/science.1083022
Cuervo, A. M., and Dice, J. F. (2000a). Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513. doi: 10.1074/jbc.M002102200
Cuervo, A. M., and Dice, J. F. (2000b). When lysosomes get old. Exp. Gerontol. 35, 119–131. doi: 10.1016/S0531-5565(00)00075-9
Dai, Q., Zhang, C., Wu, Y., McDonough, H., Whaley, R. A., Godfrey, V., et al. (2003). CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 22, 5446–5458. doi: 10.1093/emboj/cdg529
De Leon, J. T., Iwai, A., Feau, C., Garcia, Y., Balsiger, H. A., Storer, C. L., et al. (2011). Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 11878–11883. doi: 10.1073/pnas.1105160108
Dickey, C. A., Kamal, A., Lundgren, K., Klosak, N., Bailey, R. M., Dunmore, J., et al. (2007a). The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658. doi: 10.1172/JCI29715
Dickey, C. A., Koren, J., Zhang, Y.-J., Xu, Y.-F., Jinwal, U. K., Birnbaum, M. J., et al. (2008). Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. U.S.A. 105, 3622–3627. doi: 10.1073/pnas.0709180105
Dickey, C. A., Patterson, C., Dickson, D., and Petrucelli, L. (2007b). Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol. Med. 13, 32–38. doi: 10.1016/j.molmed.2006.11.003
Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. doi: 10.1038/nature19356
Du, X.-J., Cole, T. J., Tenis, N., Gao, X.-M., Köntgen, F., Kemp, B. E., et al. (2002). Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice. Mol. Cell. Biol. 22, 2821–2829. doi: 10.1128/MCB.22.8.2821-2829.2002
Echeverría, P. C., Bernthaler, A., Dupuis, P., Mayer, B., and Picard, D. (2011). An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS ONE. 6:e26044. doi: 10.1371/journal.pone.0026044
Edkins, A. L. (2015). CHIP: a co-chaperone for degradation by the proteasome. Subcell. Biochem. 78, 219–242. doi: 10.1007/978-3-319-11731-7_11
Eustace, B. K., Sakurai, T., Stewart, J. K., Yimlamai, D., Unger, C., Zehetmeier, C., et al. (2004). Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat. Cell Biol. 6, 507–514. doi: 10.1038/ncb1131
Freeman, B. C., Toft, D. O., and Morimoto, R. I. (1996). Molecular chaperone machines: chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science 274, 1718–1720. doi: 10.1126/science.274.5293.1718
Giustiniani, J., Guillemeau, K., Dounane, O., Sardin, E., Huvent, I., Schmitt, A., et al. (2015). The FK506-binding protein FKBP52 in vitro induces aggregation of truncated Tau forms with prion-like behavior. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 29, 3171–3181. doi: 10.1096/fj.14-268243
Gong, C.-X., Liu, F., Wu, G., Rossie, S., Wegiel, J., Li, L., et al. (2004). Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5. J. Neurochem. 88, 298–310. doi: 10.1111/j.1471-4159.2004.02147.x
Grad, I., Cederroth, C. R., Walicki, J., Grey, C., Barluenga, S., Winssinger, N., et al. (2010). The molecular chaperone Hsp90α is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS ONE 5:e15770. doi: 10.1371/journal.pone.0015770
Grad, I., McKee, T. A., Ludwig, S. M., Hoyle, G. W., Ruiz, P., Wurst, W., et al. (2006). The Hsp90 cochaperone p23 is essential for perinatal survival. Mol. Cell. Biol. 26, 8976–8983. doi: 10.1128/MCB.00734-06
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. (Berl.) 133, 665–704. doi: 10.1007/s00401-017-1707-9
Guy, N. C., Garcia, Y. A., Sivils, J. C., Galigniana, M. D., and Cox, M. B. (2015). Functions of the Hsp90-binding FKBP immunophilins. Subcell. Biochem. 78, 35–68. doi: 10.1007/978-3-319-11731-7_2
Hall, J. A., Forsberg, L. K., and Blagg, B. S. J. (2014). Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med. Chem. 6, 1587–1605. doi: 10.4155/fmc.14.89
Harst, A., Lin, H., and Obermann, W. M. J. (2005). Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 387, 789–796. doi: 10.1042/BJ20041283
Hasumi, H., Baba, M., Hasumi, Y., Lang, M., Huang, Y., Oh, H. F., et al. (2015). Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc. Natl. Acad. Sci. U.S.A. 112, E1624–E1631. doi: 10.1073/pnas.1419502112
Hildenbrand, Z. L., Molugu, S. K., Herrera, N., Ramirez, C., Xiao, C., and Bernal, R. A. (2011). Hsp90 can accommodate the simultaneous binding of the FKBP52 and HOP proteins. Oncotarget 2, 43–58. doi: 10.18632/oncotarget.225
Jarczowski, F., Jahreis, G., Erdmann, F., Schierhorn, A., Fischer, G., and Edlich, F. (2009). FKBP36 is an inherent multifunctional glyceraldehyde-3-phosphate dehydrogenase inhibitor. J. Biol. Chem. 284, 766–773. doi: 10.1074/jbc.M709779200
Jascur, T., Brickner, H., Salles-Passador, I., Barbier, V., El Khissiin, A., Smith, B., et al. (2005). Regulation of p21(WAF1/CIP1) stability by WISp39, a Hsp90 binding TPR protein. Mol. Cell 17, 237–249. doi: 10.1016/j.molcel.2004.11.049
Jin, J., Tian, R., Pasculescu, A., Dai, A. Y., Williton, K., Taylor, L., et al. (2016). Mutational analysis of glycogen synthase kinase 3β protein kinase together with kinome-wide binding and stability studies suggests context-dependent recognition of kinases by the chaperone heat shock protein 90. Mol. Cell. Biol. 36, 1007–1018. doi: 10.1128/MCB.01045-15
Jinwal, U. K., Abisambra, J. F., Zhang, J., Dharia, S., O'Leary, J. C., Patel, T., et al. (2012). Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 287, 24814–24820. doi: 10.1074/jbc.M112.367268
Jinwal, U. K., Koren, J., Borysov, S. I., Schmid, A. B., Abisambra, J. F., Blair, L. J., et al. (2010). The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. Off. J. Soc. Neurosci. 30, 591–599. doi: 10.1523/JNEUROSCI.4815-09.2010
Jinwal, U. K., Koren, J., and Dickey, C. A. (2013). Reconstructing the Hsp90/Tau machine. Curr. Enzyme Inhib. 9, 41–45. doi: 10.2174/1573408011309010006
Josefowicz, S. Z., Niec, R. E., Kim, H. Y., Treuting, P., Chinen, T., Zheng, Y., et al. (2012). Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399. doi: 10.1038/nature10772
Kamah, A., Cantrelle, F. X., Huvent, I., Giustiniani, J., Guillemeau, K., Byrne, C., et al. (2016). Isomerization and oligomerization of truncated and mutated tau forms by FKBP52 are independent processes. J. Mol. Biol. 428, 1080–1090. doi: 10.1016/j.jmb.2016.02.015
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., et al. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410. doi: 10.1038/nature01913
Karagöz, G. E., Duarte, A. M. S., Akoury, E., Ippel, H., Biernat, J., Morán Luengo, T., et al. (2014). Hsp90-tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974. doi: 10.1016/j.cell.2014.01.037
Kekatpure, V. D., Dannenberg, A. J., and Subbaramaiah, K. (2009). HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 284, 7436–7445. doi: 10.1074/jbc.M808999200
Kovacs, G. G. (2015). Invited review: neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23. doi: 10.1111/nan.12208
Laenger, A., Lang-Rollin, I., Kozany, C., Zschocke, J., Zimmermann, N., Rüegg, J., et al. (2009). XAP2 inhibits glucocorticoid receptor activity in mammalian cells. FEBS Lett. 583, 1493–1498. doi: 10.1016/j.febslet.2009.03.072
Lee, C. H., Park, J. H., Choi, J. H., Yoo, K.-Y., Ryu, P. D., and Won, M.-H. (2011). Heat shock protein 90 and its cochaperone, p23, are markedly increased in the aged gerbil hippocampus. Exp. Gerontol. 46, 768–772. doi: 10.1016/j.exger.2011.05.002
LeMaster, D. M., and Hernandez, G. (2015). Conformational dynamics in FKBP domains: relevance to molecular signaling and drug design. Curr. Mol. Pharmacol. 9, 5–26. doi: 10.2174/1874467208666150519113146
Li, J., and Buchner, J. (2013). Structure, function and regulation of the hsp90 machinery. Biomed. J. 36, 106–117. doi: 10.4103/2319-4170.113230
Li, J., Soroka, J., and Buchner, J. (2012). The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635. doi: 10.1016/j.bbamcr.2011.09.003
Li, T., Chen, X., Dai, X.-Y., Wei, B., Weng, Q.-J., Chen, X., et al. (2017). Novel Hsp90 inhibitor platycodin D disrupts Hsp90/Cdc37 complex and enhances the anticancer effect of mTOR inhibitor. Toxicol. Appl. Pharmacol. 330, 65–73. doi: 10.1016/j.taap.2017.07.006
Liberek, K., Lewandowska, A., and Zietkiewicz, S. (2008). Chaperones in control of protein disaggregation. EMBO J. 27, 328–335. doi: 10.1038/sj.emboj.7601970
Liu, F., Iqbal, K., Grundke-Iqbal, I., Rossie, S., and Gong, C.-X. (2005). Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer's disease. J. Biol. Chem. 280, 1790–1796. doi: 10.1074/jbc.M410775200
Loerch, P. M., Lu, T., Dakin, K. A., Vann, J. M., Isaacs, A., Geula, C., et al. (2008). Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 3:e3329. doi: 10.1371/journal.pone.0003329
Lovgren, A. K., Kovarova, M., and Koller, B. H. (2007). cPGES/p23 is required for glucocorticoid receptor function and embryonic growth but not prostaglandin E2 synthesis. Mol. Cell. Biol. 27, 4416–4430. doi: 10.1128/MCB.02314-06
Luo, W., Dou, F., Rodina, A., Chip, S., Kim, J., Zhao, Q., et al. (2007). Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. U.S.A. 104, 9511–9516. doi: 10.1073/pnas.0701055104
Mandelkow, E.-M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Martínez-Ruiz, A., Villanueva, L., González de Orduña, C., López-Ferrer, D., Higueras, M. A., Tarín, C., et al. (2005). S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. U.S.A. 102, 8525–8530. doi: 10.1073/pnas.0407294102
Meduri, G., Guillemeau, K., Dounane, O., Sazdovitch, V., Duyckaerts, C., Chambraud, B., et al. (2016). Caspase-cleaved Tau-D(421) is colocalized with the immunophilin FKBP52 in the autophagy-endolysosomal system of Alzheimer's disease neurons. Neurobiol. Aging 46, 124–137. doi: 10.1016/j.neurobiolaging.2016.06.017
Miyata, Y., Koren, J., Kiray, J., Dickey, C. A., and Gestwicki, J. E. (2011). Molecular chaperones and regulation of tau quality control: strategies for drug discovery in tauopathies. Future Med. Chem. 3, 1523–1537. doi: 10.4155/fmc.11.88
Moffatt, N. S., Bruinsma, E., Uhl, C., Obermann, W. M., and Toft, D. (2008). Role of the cochaperone Tpr2 in Hsp90 chaperoning. Biochemistry (Mosc.) 47, 8203–8213. doi: 10.1021/bi800770g
Mollapour, M., and Neckers, L. (2012). Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823, 648–655. doi: 10.1016/j.bbamcr.2011.07.018
Nakatani, Y., Hokonohara, Y., Kakuta, S., Sudo, K., Iwakura, Y., and Kudo, I. (2007). Knockout mice lacking cPGES/p23, a constitutively expressed PGE2 synthetic enzyme, are peri-natally lethal. Biochem. Biophys. Res. Commun. 362, 387–392. doi: 10.1016/j.bbrc.2007.07.180
Njemini, R., Lambert, M., Demanet, C., Kooijman, R., and Mets, T. (2007). Basal and infection-induced levels of heat shock proteins in human aging. Biogerontology 8, 353–364. doi: 10.1007/s10522-006-9078-y
O'Leary, J. C., Dharia, S., Blair, L. J., Brady, S., Johnson, A. G., Peters, M., et al. (2011). A new anti-depressive strategy for the elderly: ablation of FKBP5/FKBP51. PLoS ONE 6:e24840. doi: 10.1371/journal.pone.0024840
Orr, M. E., Sullivan, A. C., and Frost, B. (2017). A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol. Sci. 38, 637–648. doi: 10.1016/j.tips.2017.03.011
Palubinsky, A. M., Stankowski, J. N., Kale, A. C., Codreanu, S. G., Singer, R. J., Liebler, D. C., et al. (2015). CHIP is an essential determinant of neuronal mitochondrial stress signaling. Antioxid. Redox Signal. 23, 535–549. doi: 10.1089/ars.2014.6102
Periyasamy, S., Hinds, T., Shemshedini, L., Shou, W., and Sanchez, E. R. (2010). FKBP51 and Cyp40 are positive regulators of androgen-dependent prostate cancer cell growth and the targets of FK506 and cyclosporin A. Oncogene 29, 1691–1701. doi: 10.1038/onc.2009.458
Philp, L. K., Day, T. K., Butler, M. S., Laven-Law, G., Jindal, S., Hickey, T. E., et al. (2016). Small glutamine-rich tetratricopeptide repeat-containing protein alpha (SGTA) ablation limits offspring viability and growth in mice. Sci. Rep. 6:28950. doi: 10.1038/srep28950
Picard, D. (2002). Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. CMLS 59, 1640–1648. doi: 10.1007/PL00012491
Pimienta, G., Herbert, K. M., and Regan, L. (2011). A compound that inhibits the HOP-Hsp90 complex formation and has unique killing effects in breast cancer cell lines. Mol. Pharm. 8, 2252–2261. doi: 10.1021/mp200346y
Pinheiro, S., Silva, J., Mota, C., Vaz-Silva, J., Veloso, A., Pinto, V., et al. (2016). Tau mislocation in glucocorticoid-triggered hippocampal pathology. Mol. Neurobiol. 53, 4745–4753. doi: 10.1007/s12035-015-9356-2
Prodromou, C. (2016). Mechanisms of Hsp90 regulation. Biochem. J. 473, 2439–2452. doi: 10.1042/BCJ20160005
Prodromou, C., Panaretou, B., Chohan, S., Siligardi, G., O'Brien, R., Ladbury, J. E., et al. (2000). The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 19, 4383–4392. doi: 10.1093/emboj/19.16.4383
Raitila, A., Lehtonen, H. J., Arola, J., Heliövaara, E., Ahlsten, M., Georgitsi, M., et al. (2010). Mice with inactivation of aryl hydrocarbon receptor-interacting protein (Aip) display complete penetrance of pituitary adenomas with aberrant ARNT expression. Am. J. Pathol. 177, 1969–1976. doi: 10.2353/ajpath.2010.100138
Ramamurthy, V., Roberts, M., van den Akker, F., Niemi, G., Reh, T. A., and Hurley, J. B. (2003). AIPL1, a protein implicated in Leber's congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A. 100, 12630–12635. doi: 10.1073/pnas.2134194100
Rao, R. V., Niazi, K., Mollahan, P., Mao, X., Crippen, D., Poksay, K. S., et al. (2006). Coupling endoplasmic reticulum stress to the cell-death program: a novel HSP90-independent role for the small chaperone protein p23. Cell Death Differ. 13, 415–425. doi: 10.1038/sj.cdd.4401761
Reeve, A., Simcox, E., and Turnbull, D. (2014). Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. doi: 10.1016/j.arr.2014.01.004
Rehn, A. B., and Buchner, J. (2015). p23 and Aha1. Subcell. Biochem. 78, 113–131. doi: 10.1007/978-3-319-11731-7_6
Renouf, D. J., Hedley, D., Krzyzanowska, M. K., Schmuck, M., Wang, L., and Moore, M. J. (2016). A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pancreatic cancer. Cancer Chemother. Pharmacol. 78, 541–545. doi: 10.1007/s00280-016-3102-y
Richardson, R. T., Alekseev, O. M., Grossman, G., Widgren, E. E., Thresher, R., Wagner, E. J., et al. (2006). Nuclear autoantigenic sperm protein (NASP), a linker histone chaperone that is required for cell proliferation. J. Biol. Chem. 281, 21526–21534. doi: 10.1074/jbc.M603816200
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. doi: 10.1016/S0092-8674(94)90462-6
Rodina, A., Wang, T., Yan, P., Gomes, E. D., Dunphy, M. P. S., Pillarsetty, N., et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. doi: 10.1038/nature19807
Sabbagh, J. J., O'Leary, J. C., Blair, L. J., Klengel, T., Nordhues, B. A., Fontaine, S. N., et al. (2014). Age-associated epigenetic upregulation of the FKBP5 gene selectively impairs stress resiliency. PLoS ONE 9:e107241. doi: 10.1371/journal.pone.0107241
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360. doi: 10.1038/nrm.2017.20
Schülke, J.-P., Wochnik, G. M., Lang-Rollin, I., Gassen, N. C., Knapp, R. T., Berning, B., et al. (2010). Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS ONE 5:e11717. doi: 10.1371/journal.pone.0011717
Selenica, M.-L., Benner, L., Housley, S. B., Manchec, B., Lee, D. C., Nash, K. R., et al. (2014). Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 6:12. doi: 10.1186/alzrt241
Shelton, L. B., Baker, J. D., Zheng, D., Sullivan, L. E., Solanki, P. K., Webster, J. M., et al. (2017). Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. U.S.A. 114, 9707–9712. doi: 10.1073/pnas.1707039114
Silverstein, A. M., Galigniana, M. D., Chen, M. S., Owens-Grillo, J. K., Chinkers, M., and Pratt, W. B. (1997). Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J. Biol. Chem. 272, 16224–16230. doi: 10.1074/jbc.272.26.16224
Söti, C., and Csermely, P. (2002). Chaperones and aging: role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 41, 383–389. doi: 10.1016/S0197-0186(02)00043-8
Sreedhar, A. S., Kalmár, E., Csermely, P., and Shen, Y.-F. (2004). Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 562, 11–15. doi: 10.1016/S0014-5793(04)00229-7
Sullivan, W., Stensgard, B., Caucutt, G., Bartha, B., McMahon, N., Alnemri, E. S., et al. (1997). Nucleotides and two functional states of hsp90. J. Biol. Chem. 272, 8007–8012. doi: 10.1074/jbc.272.12.8007
Taglialatela, G., Rastellini, C., and Cicalese, L. (2015). Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J. Alzheimers Dis. JAD 47, 329–333. doi: 10.3233/JAD-150065
Taipale, M., Krykbaeva, I., Koeva, M., Kayatekin, C., Westover, K. D., Karras, G. I., et al. (2012). Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001. doi: 10.1016/j.cell.2012.06.047
Tarkar, A., Loges, N. T., Slagle, C. E., Francis, R., Dougherty, G. W., Tamayo, J. V., et al. (2013). DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 45, 995–1003. doi: 10.1038/ng.2707
Terada, K., Ueno, S., Yomogida, K., Imai, T., Kiyonari, H., Takeda, N., et al. (2003). Expression of Tom34 splicing isoforms in mouse testis and knockout of Tom34 in mice. J. Biochem. 133, 625–631.
Terman, A. (2001). Garbage catastrophe theory of aging: imperfect removal of oxidative damage? Redox Rep. Commun. Free Radic. Res. 6, 15–26. doi: 10.1179/135100001101535996
Thakur, M. K., Heilbrun, L. K., Sheng, S., Stein, M., Liu, G., Antonarakis, E. S., et al. (2016). A phase II trial of ganetespib, a heat shock protein 90 Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trials consortium (PCCTC) study. Invest. New Drugs 34, 112–118. doi: 10.1007/s10637-015-0307-6
Touma, C., Gassen, N. C., Herrmann, L., Cheung-Flynn, J., Büll, D. R., Ionescu, I. A., et al. (2011). FK506 binding protein 5 shapes stress responsiveness: modulation of neuroendocrine reactivity and coping behavior. Biol. Psychiatry 70, 928–936. doi: 10.1016/j.biopsych.2011.07.023
Tranguch, S., Cheung-Flynn, J., Daikoku, T., Prapapanich, V., Cox, M. B., Xie, H., et al. (2005). Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc. Natl. Acad. Sci. U.S.A. 102, 14326–14331. doi: 10.1073/pnas.0505775102
Voss, A. K., Thomas, T., and Gruss, P. (2000). Mice lacking HSP90beta fail to develop a placental labyrinth. Dev. Camb. Engl. 127, 1–11.
Wang, X., Venable, J., LaPointe, P., Hutt, D. M., Koulov, A. V., Coppinger, J., et al. (2006). Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815. doi: 10.1016/j.cell.2006.09.043
Wolmarans, A., Lee, B., Spyracopoulos, L., and LaPointe, P. (2016). The mechanism of Hsp90 ATPase stimulation by Aha1. Sci. Rep. 6, 33179. doi: 10.1038/srep33179
Woodford, M. R., Dunn, D. M., Blanden, A. R., Capriotti, D., Loiselle, D., Prodromou, C., et al. (2016). The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat. Commun. 7:12037. doi: 10.1038/ncomms12037
Yakkundi, A., Bennett, R., Hernández-Negrete, I., Delalande, J.-M., Hanna, M., Lyubomska, O., et al. (2015). FKBPL is a critical antiangiogenic regulator of developmental and pathological angiogenesis. Arterioscler. Thromb. Vasc. Biol. 35, 845–854. doi: 10.1161/ATVBAHA.114.304539
Yang, Z., Wolf, I. M., Chen, H., Periyasamy, S., Chen, Z., Yong, W., et al. (2006). FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol. Endocrinol. Baltim. Md 20, 2682–2694. doi: 10.1210/me.2006-0024
Yong, W., Yang, Z., Periyasamy, S., Chen, H., Yucel, S., Li, W., et al. (2007). Essential role for co-chaperone FKBP52 but not FKBP51 in androgen receptor-mediated signaling and physiology. J. Biol. Chem. 282, 5026–5036. doi: 10.1074/jbc.M609360200
Yu, X., Guo, Z. S., Marcu, M. G., Neckers, L., Nguyen, D. M., Chen, G. A., et al. (2002). Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl. Cancer Inst. 94, 504–513. doi: 10.1093/jnci/94.7.504
Yu, Y., Hamza, A., Zhang, T., Gu, M., Zou, P., Newman, B., et al. (2010). Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 79, 542–551. doi: 10.1016/j.bcp.2009.09.017
Zannas, A. S., and Binder, E. B. (2014). Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav. 13, 25–37. doi: 10.1111/gbb.12104
Zhang, J., Spilman, P., Chen, S., Gorostiza, O., Matalis, A., Niazi, K., et al. (2013). The small co-chaperone p23 overexpressing transgenic mouse. J. Neurosci. Methods 212, 190–194. doi: 10.1016/j.jneumeth.2012.09.022
Zhang, T., Hamza, A., Cao, X., Wang, B., Yu, S., Zhan, C.-G., et al. (2008). A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170. doi: 10.1158/1535-7163.MCT-07-0484
Keywords: Hsp90, aging, tau, chaperones, co-chaperones, proteostasis
Citation: Shelton LB, Koren J III and Blair LJ (2017) Imbalances in the Hsp90 Chaperone Machinery: Implications for Tauopathies. Front. Neurosci. 11:724. doi: 10.3389/fnins.2017.00724
Received: 30 September 2017; Accepted: 12 December 2017;
Published: 22 December 2017.
Edited by:
Naruhiko Sahara, National Institute of Radiological Sciences (NIRS), JapanReviewed by:
Shin-ichi Hisanaga, Tokyo Metropolitan University, JapanJason Eriksen, University of Houston, United States
Copyright © 2017 Shelton, Koren and Blair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura J. Blair, bGJsYWlyQGhlYWx0aC51c2YuZWR1