 Michael R. Post
Michael R. Post Eugene V. Mosharov
Eugene V. Mosharov- Departments of Psychiatry and Neurology, New York State Psychiatric Institute, Columbia University Medical Center, New York, NY, United States
Several lines of evidence place alpha-synuclein (aSyn) at the center of Parkinson's disease (PD) etiology, but it is still unclear why overexpression or mutated forms of this protein affect some neuronal populations more than others. Susceptible neuronal populations in PD, dopaminergic neurons of the substantia nigra pars compacta (SNpc) and the locus coeruleus (LC), are distinguished by relatively high cytoplasmic concentrations of dopamine and calcium ions. Here we review the evidence for the multi-hit hypothesis of neurodegeneration, including recent papers that demonstrate synergistic interactions between aSyn, calcium ions and dopamine that may lead to imbalanced protein turnover and selective susceptibility of these neurons. We conclude that decreasing the levels of any one of these toxicity mediators can be beneficial for the survival of SNpc and LC neurons, providing multiple opportunities for targeted drug interventions aimed at modifying the course of PD.
Differential Susceptibility of Catecholaminergic Neurons in PD
Parkinson's disease (PD), the second most common neurodegenerative disorder (De Lau and Breteler, 2006), is marked by slowness (bradykinesia), resting tremor, muscular rigidity, and postural instability (Lang and Lozano, 1998). Although multiple brain regions are affected in late-stage PD (Braak et al., 1995), two catecholaminergic neuronal populations degenerate early, before the onset of the motor symptoms-dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc) and noradrenergic (NE) neurons of the locus coeruleus (LC). DA restoration through treatment with L-DOPA provides an effective symptomatic improvement, however, tolerance to treatment increases over time, accompanied by the development of severe side effects (L-DOPA-induced dyskinesia; Lewitt, 2015; Olanow, 2015). There is at the time no means available for delaying the progress of the disease, which is a critical goal in the field.
Rational design of disease-modifying therapies is complicated by the lack of a clear understanding of the pathophysiology of PD initiation and progression. The disease is predominantly sporadic, with an estimated 10% prevalence of familial cases (Eriksen et al., 2005). Alpha-synuclein (aSyn), encoded by the SNCA gene, plays a central role in both sporadic and familial PD. Mutations or multiplications of the SNCA cause autosomal dominant PD (Eriksen et al., 2005). Levels of phosphorylated aSyn are increased in post-mortem brains of PD patients and in patient-derived dopaminergic neurons (Fujiwara et al., 2002; Swirski et al., 2014). Genome-wide association studies (GWASs) have reported a correlation between the SNCA locus and the risk of developing sporadic PD (Simon-Sanchez et al., 2009; Chang et al., 2017). Importantly, post-mortem PD brains show proteinaceous aSyn-positive deposits called Lewy bodies (Spillantini et al., 1997; Baba et al., 1998). Conversely, deletion of aSyn is protective in mouse and cellular models of PD (Dauer et al., 2002; Alvarez-Fischer et al., 2008). Similarly, a recent study identified β2-adrenoreceptor (β2AR) agonists as negative regulators of the aSyn gene expression, and an association was found between the use of β2AR agonist salbutamol, a brain-penetrant asthma medication, and a reduced risk of developing PD (Mittal et al., 2017). Yet, Lewy body pathology occurs throughout the nervous system in PD patients and does not correlate well with cell death (Goedert et al., 2013; Surmeier et al., 2017a), suggesting that aSyn may be necessary but not sufficient for PD neurodegeneration.
Several cellular pathways are affected in PD, resulting in endoplasmic reticulum (ER) stress and activation of the unfolded protein response, disruption of lysosomal and proteasomal protein degradation, and impaired Ca2+ homeostasis and mitochondrial dysfunction (Rochet et al., 2004; Stefanis, 2012; Duda et al., 2016; Michel et al., 2016). Although there does not appear to be a unifying end-point toxicity pathway, inflammatory response and both necrotic and apoptotic degeneration are often observed in PD models (Perier et al., 2012). The central question in PD neuropathology, however, is why some neurons are highly susceptible to neurodegeneration while other, even closely related populations, are much less affected. Specifically, SNpc and LC catecholaminergic neurons degenerate in PD, whereas ventral tegmental area (VTA) and tuberoinfundibular DA neurons are relatively spared in both PD patients and laboratory models of the disorder (Hirsch et al., 1988; Braak et al., 1995). Two features of SNpc and LC neurons—the presence of elevated catecholamine and Ca2+ concentration in the cytosol—have consistently been suggested as modulators of their sensitivity to neurodegeneration.
Due to the ability of DA to produce oxidative stress and protein damage, it has long been speculated that a dysregulation of cytosolic DA homeostasis plays a role in PD (Edwards, 1993; Gainetdinov et al., 1998; Uhl, 1998; Schmitz et al., 2001; Lotharius and Brundin, 2002; Lohr et al., 2014; Pifl et al., 2014). Spontaneous DA oxidation at neutral pH of the cytosol yields DA-o-quinone and dopaminochrome (Graham, 1978; Sulzer and Zecca, 2000), which can then react with free cysteine and exposed cysteine residues of proteins and glutathione producing 5-S-cystenyl-DA. The latter can undergo further oxidation and is toxic to cultured cells (Spencer et al., 2002) or when injected into the mouse brain (Zhang and Dryhurst, 1994). 5-S-cystenyl adducts of DA and its metabolites are used as markers of excess cytosolic DA and oxidative stress in vivo (Hastings and Berman, 1999; Caudle et al., 2007) and are readily detected in human SNpc and LC, consistent with DA-induced protein damage in human PD (Fornstedt et al., 1989; Montine et al., 1995; Hastings and Berman, 1999). Other mechanisms of DA-mediated neurotoxicity include reactions of DA with nitric oxide (Daveu et al., 1997), peroxynitrite (Daveu et al., 1997; Vauzour et al., 2008) and aldehydes (Collins and Bigdeli, 1975; Deitrich and Erwin, 1980; Naoi et al., 1993; Marchitti et al., 2007). Accumulation of cytosolic DA is toxic to cells in vitro (Mytilineou et al., 1993; Pardo et al., 1995; Sulzer et al., 2000; Xu et al., 2002; Fuentes et al., 2007; Mosharov et al., 2009) and several reports confirm that a buildup of cytosolic DA is indeed sufficient to induce progressive nigrostriatal degeneration in rodents (Caudle et al., 2007; Chen et al., 2008), although clinical studies of L-DOPA toxicity produced controversial results (Fahn et al., 2004; Olanow et al., 2004; Holford et al., 2006).
Dysregulation of Ca2+ homeostasis is likewise frequently observed in models of both sporadic and familial PD (Goldberg et al., 2012; Hurley and Dexter, 2012; Surmeier et al., 2017b). This includes impairment of mitochondrial Ca2+ maintenance (Exner et al., 2012), disrupted communication between mitochondrial and ER Ca2+ stores (Ottolini et al., 2013; Guardia-Laguarta et al., 2014), decreased store-operated Ca2+ entry (Zhou et al., 2016), and additional mechanisms that may cause toxicity due to abnormally high or low Ca2+ levels (Duda et al., 2016; Michel et al., 2016; Surmeier et al., 2017b). SNpc and VTA neurons express drastically different levels of calbindin-D28K (Fu et al., 2012) and those expressing high levels of this Ca2+ buffering protein—the majority of VTA neurons and a small percentage of SNpc neurons—are spared from neurodegeneration in PD (Yamada et al., 1990; Rcom-H'cheo-Gauthier et al., 2014). Interestingly, at least some LC neurons appear to express Ca2+ buffering proteins calbindin-D28K, calretinin and parvalbumin (Bhagwandin et al., 2013), although no comparison was made with other brain areas, such as the VTA.
SNpc neurons have long axons that extend into the striatum and arborize extensively, with many DA release sites (Matsuda et al., 2009). Physiologically, these neurons display broad action potential spikes and an autonomous tonic firing pattern governed by the activity of the L-type Cav1.3 channels (LTCCs) (Hetzenauer et al., 2006; Surmeier et al., 2010). This drives a feed-forward stimulation of mitochondrial oxidative phosphorylation that maintains ATP production during increased neuronal activity (Chan et al., 2007; Surmeier et al., 2017b). Chronically increased cytoplasmic and mitochondrial Ca2+ levels may however drive the production of reactive oxygen and nitrogen species (ROS and RNS), leading to mitochondrial dysfunction. While Cav1.3 channels are expressed at similar levels in SNpc and neighboring VTA dopaminergic neurons (Dragicevic et al., 2014), they do not drive pacemaking in VTA neurons (Chan et al., 2007; Duda et al., 2016) (although, this remains controversial Liu et al., 2014), suggesting post-translational regulation of their activity. Pharmacological blockade of LTCCs with dihydropyridines alleviates mitochondrial oxidative stress in SNpc neurons in ex vivo mouse brain slices (Chan et al., 2007), and protects them in neurotoxin-based models of PD (Chan et al., 2007). Similarly, LC neurons display broad action potential spikes and autonomous pacemaking that is dependent on Cav1.2 and Cav1.3 L-type channels (Sanchez-Padilla et al., 2014) as well as the T-type channels (Matschke et al., 2015). Dihydropyridines also prevent mitochondrial oxidative stress in LC neurons in ex vivo brain slices (Sanchez-Padilla et al., 2014). Although LC neurons are selectively targeted by parkinsonian neurotoxins (Masilamoni et al., 2011), the effect of LTCC blockers on the survival of LC neurons in these models has not been studied. However, an LTCC inhibitor nimodipine was shown to protect both SNpc and LC neurons in a model of chronic neuroinflammation (Hopp et al., 2015).
Overall, SNpc and LC appear to share many of the same characteristics—a proteomic analysis identified similar changes in 61 PD-associated proteins in SNpc and LC neurons (Van Dijk et al., 2012)—and are uniquely situated with high levels of cytosolic catecholamines and Ca2+, which in the presence of aSyn may underlie their higher susceptibility to neurodegeneration. Below, we focus on the interactions between these three chemicals, highlighting recent developments in their role toward cell-selective PD pathogenesis.
aSyn AND Ca2+
aSyn is a protein widely expressed in the nervous system, with a subcellular localization at the presynaptic terminal. The protein is 140 amino acids in length (Figure 1), occurs as a helically folded tetramer under physiological conditions (Bartels et al., 2011) and is able to form oligomers, fibrils and more complex aggregates, eventually leading to Lewy bodies. The N-terminus is lysine-rich and is the site of the vesicle binding, with four lipid-binding KTK motif repeats in that region. Importantly, all known SNCA familial PD mutations to date—A30P, E46K, H50Q, G51D, A53E, and A53T—are found in this domain (Rcom-H'cheo-Gauthier et al., 2014). The central region of aSyn is known as the non-amyloid-β component (NAC) of amyloid plaques found in Alzheimer's disease patients and is responsible for aSyn aggregation and Lewy body formation (Li et al., 2002). The C-terminus is comprised of an EF-hand-like sequence that is capable of binding Ca2+; however, overexpression of truncated aSyn that lacks the C-terminus is sufficient to elicit a PD-like phenotype in mice (Tofaris et al., 2006). Normally, aSyn is involved in regulation of synaptic vesicles exocytosis, although its exact function is still debated (Imaizumi et al., 2005; Larsen et al., 2006; Burre et al., 2010; Nemani et al., 2010; Bendor et al., 2013). Although gain-of-function mechanisms of aSyn toxicity due to its post-translational modifications or oligomerization have been widely reported, recent data suggest that the loss-of-function mechanisms may also play a role (Collier et al., 2016).
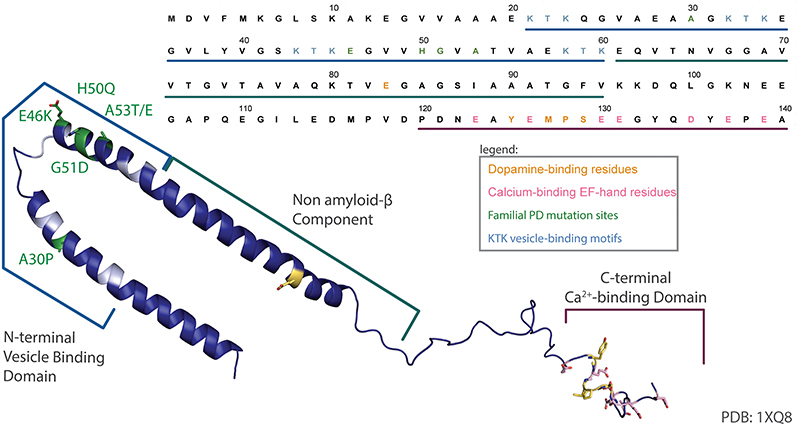
Figure 1. aSyn sequence map. An annotated structure of human micelle-bound aSyn solved by solution NMR (PDB 1XQ8) (Ulmer et al., 2005) and corresponding sequence have been color coded to highlight the lipid binding N-terminal domain and associated KTK motifs (cyan), common familial PD mutations (green), acidic residues in the C-terminal Ca2+-binding EF-hand-like motif (pink), and the dopamine-binding residues (yellow).
Effects of aSyn on Ca2+
Intracellular Ca2+ is a potent second-messenger that triggers many cellular events, and its concentration is tightly regulated by the activities of transporters and channels of the plasma, ER, and mitochondrial membranes, as well as calcium-binding proteins such as calbindin, parvalbumin, and calretinin (Zaichick et al., 2017). Various mechanisms by which aSyn is able to interfere with Ca2+ homeostasis in different cellular compartments have been reviewed in more detail elsewhere (Duda et al., 2016; Michel et al., 2016; Ottolini et al., 2017; Surmeier et al., 2017b), but are described here briefly.
First, aSyn localizes at the mitochondria-associated membranes of the ER (MAMs) where it can regulate IP3 receptor-mediated transfer of calcium (Cali et al., 2012; Guardia-Laguarta et al., 2014). Pathogenic PD mutations in aSyn result in reduced association with MAM accompanied by increased mitochondrial fragmentation and augmented autophagy (Guardia-Laguarta et al., 2014). Additionally, post-translationally modified aSyn interacts with TOM20, a translocase of the outer mitochondrial membrane, and impairs mitochondrial import of proteins required for oxidative phosphorylation both in vitro and in postmortem brain tissue from PD patients (Di Maio et al., 2016). Second, aSyn overexpression induces lysosomal permeability, allowing lysosomal calcium and protons to leak to the cytosol and induce cell death (Bourdenx et al., 2014). Third, aSyn can increase ion permeability of the plasma membrane or interfere with the activity of its channels resulting in dysregulated neuronal firing and Ca2+ dynamics patterns that precede neurodegeneration (Subramaniam et al., 2014; Angelova et al., 2016). Consistently, aSyn is required for cytosolic Ca2+ influx through the plasma membrane following exposure to the parkinsonian neurotoxin MPP+ via a putative interaction with LTCCs (Lieberman et al., 2017), although the exact mechanism of this interaction needs further investigation. Fourth, a recent study showed that extracellularly added aSyn increased the activity of the Cav2.2 channel, thus increasing cytoplasmic Ca2+ sufficiently to induce exocytotic DA release (Ronzitti et al., 2014). Interestingly, aSyn did not increase Cav2.2 expression, but rather caused a relocation of Cav2.2 from lipid rafts to cholesterol-poor domains, providing a novel mechanism by which aSyn may change the activity of Ca2+ channels via the reorganization of membrane microdomains indicating an indirect interaction between aSyn and Ca2+-channels. Finally, a study of the proximal aSyn intracellular partners using APEX2-based labeling found that aSyn might interact with calcineurin, a calmodulin dependent serine/threonine protein phosphatase that has ubiquitous intracellular substrates (Chung et al., 2017). This finding buttresses previous reports from the same group that demonstrated a functional relationship between aSyn levels and calcineurin activity (Caraveo et al., 2014).
Effects of Ca2+ on aSyn
Ca2+ binding seems to promote aSyn annular oligomer formation. These ring-like oligomers have been shown to insert in the membrane forming a pore, perhaps allowing more Ca2+ to enter the cell (Mironov, 2015). This oligomer formation is dependent on the C-terminus and is eliminated in truncated forms of aSyn (Lowe et al., 2004). Increasing internal Ca2+ concentration via thapsigargin or Ca2+ ionophore treatment causes an increase in aggregate formation, while Ca2+-chelators or Ca2+ channel inhibitors have the opposite effect (Danzer et al., 2007; Nath et al., 2011; Follett et al., 2013). The effect of Ca2+ on aSyn aggregation is mediated by the Ca2+-activated protease, calpain, which cleaves the C-terminus of aSyn (Dufty et al., 2007; Nath et al., 2011). This has been confirmed in vivo by the overexpression of the calpain inhibitor, calpastatin, which reduces PD-like symptoms and pathology in a mouse model of A30P aSyn overexpression (Diepenbroek et al., 2014). Additionally, an indirect effect of Ca2+ on aSyn aggregation can be mediated by disruptions in autophagy. As elevated Ca2+ leads to increased mitochondrial stress, it has been suggested that this adds demand to proteostasis systems by necessitating increased mitochondrial turnover. This process then reduces cell's capacity to degrade aSyn, leading to aSyn aggregation (Surmeier and Schumacker, 2013).
aSyn and DA
Due to the toxic potential of DA, it is not surprising that multiple cellular mechanisms exist to regulate its cytosolic concentration. It has been suggested that neuromelanin biosynthesis acts as one of the mechanisms for regulating toxic DA by-products by sequestering them into autophagic vacuoles (Sulzer and Zecca, 2000). Similarly, oxidized derivatives of both DA and NE are found in LC-derived neuromelanin (Wakamatsu et al., 2015). Other mechanisms include feedback inhibition of DA synthesis, catabolic DA cleavage and synaptic vesicle sequestration.
Catecholamines are synthesized from the non-essential amino acid tyrosine by a series of enzymatic reactions. In the first, rate-limiting step, tyrosine hydroxylase (TH) attaches a hydroxyl group to the aromatic ring of tyrosine, forming L-DOPA. TH activity is regulated on transcriptional, translational and post-translational levels (Goldstein and Lieberman, 1992; Kumer and Vrana, 1996; Fitzpatrick, 2000; Daubner et al., 2011), including phosphorylation-dependent activation of TH by various kinases and its inhibition by DA, which limits DA production when its cytosolic concentration increases. The second enzyme in DA biosynthesis, aromatic L-amino acid decarboxylaze (AADC), converts L-DOPA to DA. AADC activity can also be regulated by second messenger systems to decrease DA production when its extracellular concentration increases (Hadjiconstantinou and Neff, 2008). aSyn has been shown to co-immunoprecipitate with both TH (Perez et al., 2002) and AADC (Tehranian et al., 2006), and this interaction leads to decreased phosphorylation and activity of both enzymes. Decreased TH activity in the presence of aSyn overexpression depended on aSyn phosphorylation at Ser129 residue (Lou et al., 2010), which was modulated by the activity of protein phosphatase 2A (Peng et al., 2005). These data suggest that a loss of soluble aSyn due to reduced expression or aggregation may increase catecholamine synthesis.
Intracellular catecholamine catabolism starts with the cleavage by monoamine oxidase (MAO), which is localized at the outer mitochondrial membrane (Schnaitman et al., 1967), and produces two highly reactive compounds, hydrogen peroxide and 3,4-dihydroxyphenylacetaldehyde (DOPAL) (or 3,4-dihydroxyphenylglycolaldehyde for NE) (Richter, 1937). This is followed by the oxidation by aldehyde dehydrogenase (ALDH) to 3,4-dihydroxyphenylacetate (DOPAC) and 3,4-dihydroxyphenylglycol (DHPG), correspondingly. Although ALDH activity—there are both cytosolic and mitochondrial isoforms of this enzyme (Marchitti et al., 2007; Chen et al., 2014; Doorn et al., 2014) - is very high, possible neurotoxicity of the aldehyde metabolites of amines was predicted 60 years ago due to their extremely reactive nature (Blaschko, 1952). Indeed, the presence of DOPAL and its metabolites has been demonstrated both in vitro and in vivo (Burke et al., 2004; Goldstein et al., 2014). Additionally, a line of mice that are deficient for ALDH1a1 and ALDH2, the cytosolic and the mitochondrial isoforms expressed in SNpc DA neurons (Mccaffery and Drager, 1994; Galter et al., 2003), showed age-dependent, L-DOPA-responsive deficits in motor performance, significant increases in biogenic aldehydes and a loss of SNpc DA neurons (Wey et al., 2012), confirming that impaired detoxification of biogenic aldehydes may cause PD-like degeneration.
Finally, sequestration and compartmentalization of DA inside secretory vesicles is achieved via the activity of vesicular monoamine transporters (VMAT). The enzyme responsible for the conversion of DA to NE in noradrenergic neurons, dopamine beta-hydroxylase, is located in the lumen and the membrane of synaptic vesicles. Moreover, the acidic pH of vesicles prevents auto-oxidation of DA and NE, allowing high vesicular neurotransmitter concentrations without the formation of reactive species. Importantly, synaptic vesicle membrane is “leaky” and in vitro and in vivo studies have demonstrated that leakage of catecholamines from storage vesicles is the primary source of their catabolism in the cytosol (Goldstein et al., 1988; Halbrugge et al., 1989; Tyce et al., 1995).
High cytosolic DA levels following L-DOPA treatment have been shown to induce selective SNpc neuron degeneration and the formation of neuromelanin (Sulzer et al., 2000), whereas increased loading of DA from cytosol to vesicles following overexpression of vesicular monoamine transporter 2 (VMAT2) provides neuroprotection from L-DOPA (Mosharov et al., 2009). Consistent with this, higher levels of neuromelanin are found in SNpc neurons that degenerate in PD (Zucca et al., 2014). Striatal DA synaptic vesicles from PD patients were also found to have lower levels of VMAT2 (Pifl et al., 2014), although as these patients were almost certainly treated with L-DOPA, a decrease in VMAT expression could be a compensatory response rather than a cause of PD.
Oxidized DA and other catecholamines are able to interact with aSyn, producing DA-modified aSyn, which is less likely to fibrilize and instead forms soluble oligomers (Conway et al., 2001; Rochet et al., 2004). This interaction is non-covalent, reversible and occurs at the Y125EMPS129 pentapeptide in the C-terminal region of α-Syn with an additional long-range electrostatic interaction with E83 in the nAC region (Figure 1, in yellow) (Mazzulli et al., 2007; Herrera et al., 2008). Using fluorescence-lifetime imaging microscopy to monitor the relative position of the N- and C- terminals of aSyn, it was shown that DA induces a conformation where the termini are closer together, which may inhibit fibril formation (Outeiro et al., 2009). Additionally, DOPAL may cross-link aSyn lysine residues, also facilitating its aggregation (Werner-Allen et al., 2016). Intracellular aSyn oligomeric species can be cytotoxic by a variety of mechanisms, including permeabilization of vesicular and plasma membranes by pore-forming fibrils (Ding et al., 2002; Gosavi et al., 2002; Lashuel et al., 2002; Mosharov et al., 2006), disruption of proteasomal protein clearance, chronic ER stress, mitochondrial dysfunction and inhibition of SNARE complex formation and neurotransmitter release (Rochet et al., 2004; Ebrahimi-Fakhari et al., 2011; Choi et al., 2013; Kalia et al., 2013; Zaltieri et al., 2015).
Monomeric DA-aSyn, however, may also be toxic by interfering with protein degradation via a lysosomal pathway called chaperone-mediated autophagy (CMA) (Cuervo et al., 2004, 2010). CMA cytosolic substrates contain a KFERQ-like motif that can be recognized by the chaperone protein cyt-Hsc70 that delivers them to a lysosomal associated membrane protein (LAMP2A). LAMP2A forms a translocation complex once bound to a substrate and the unfolded protein crosses into the lysosomal lumen where it can be degraded. While aSyn, oxidized aSyn, and a phosphomimetic S129E aSyn mutant show similar LAMP2A binding levels, lysosomal uptake of the latter is significantly diminished. DA-aSyn demonstrates a similar CMA profile when compared to phosphorylated aSyn in that it binds to the lysosome without evidence of translocation. Furthermore, unlike phosphorylated aSyn, DA-aSyn blocks both the binding and uptake of a CMA substrate GAPDH, suggesting stronger binding to LAMP2A. A mutation in the DA-interacting region of aSyn (Y125EMPS129 to F125AAFA129) nullifies the effect, further demonstrating that the interaction of DA and oxidized forms of DA with aSyn leads to this change in CMA. In primary neuronal cultures, the same CMA blockade was demonstrated after exposure to a high dose of L-DOPA, but not in neurons derived from aSyn null animals (Martinez-Vicente et al., 2008).
A hypothesis that decreased uptake of DA into synaptic vesicles should lead to PD-like nigrostriatal neurodegeneration due to increased cytosolic transmitter levels was examined in mice that displayed a 95% reduction of VMAT2 expression due to a hypomorphic allele (Caudle et al., 2007). Surprisingly, the first generation of these mice (VMAT2-deficient KA1 line Mooslehner et al., 2001) did not show any PD phenotype, despite an ~85% reduction in brain levels of DA, NE and serotonin and their increased turnover. It was subsequently discovered, however, that this mouse line had a spontaneous deletion of the SNCA gene (Specht and Schoepfer, 2001; Colebrooke et al., 2006). After further breeding to reintroduce the wild-type aSyn gene, the resulting VMAT2-LO mice showed signs of PD-like progressive neurodegeneration, including L-DOPA-responsive motor deficits, oxidative stress and protein damage, decreased DA, DAT, and TH levels in the striatum, and pathological accumulations of aSyn and a reduced number of DA neurons in the SNpc (Caudle et al., 2007; Taylor et al., 2011). Overall, the VMAT2-LO mouse model not only demonstrated that a reduced capacity of cells to sequester cytosolic DA is sufficient to cause PD-like degeneration of neurons and their axonal projections, but also that this effect requires the presence of aSyn.
Another recent study investigated the toxic interaction between aSyn and DA in vivo by combining a common familial PD aSyn mutation with elevated cytosolic DA (Mor et al., 2017). Mice that overexpress PD mutant A53T aSyn were injected with a lentivirus containing TH with an R37R38 to E37E38 mutation. This mutation leads to a loss of feedback inhibition of TH by DA, resulting in increased neurotransmitter production in the cytosol (Nakashima et al., 2002). Elevation of cellular DA levels induced progressive motor impairment accompanied by nigrostriatal degeneration and increased formation of aSyn oligomers in A53T aSyn overexpressing mice but not in WT. Furthermore, in Caenorhabditis elegans overexpressing A53T aSyn, DA toxicity was prevented if DA-interacting residues of aSyn were mutated (Mor et al., 2017). Overall, both in vitro and in vivo data suggest that DA and aSyn have a synergetic effect on toxicity and that decreasing the levels of either of the compounds is neuroprotective.
aSyn, DA AND Ca2+
Ca2+ levels positively regulate the activity of both TH and AADC, providing a direct connection between synaptic activity and DA synthesis. However, because of Ca2+-driven pacemaking in SNpc and LC neurons, elevated levels of Ca2+ also lead to chronically increased cytosolic catecholamine levels. In agreement with this, L-DOPA treatment produces higher concentration of cytosolic catecholamines in cultured SNpc (Mosharov et al., 2009) and LC (unpublished data) compared to VTA neurons, which translated into higher susceptibility of these neurons to L-DOPA-induced degeneration. The difference between these cell types was normalized by pharmacological or genetic blockade of the LTCCs, confirming their role in selective PD-like neurodegeneration. Importantly, deletion of aSyn also protected SNpc neurons from L-DOPA-induced toxicity without changing cytosolic DA concentration, demonstrated that the levels of Ca2+, DA and aSyn are equally important for toxicity.
Using the same model system, we recently investigated metabolic changes in neurons exposed to the parkinsonian neurotoxin MPP+ (Lieberman et al., 2017). Similar to the difference observed in vivo described above, a significantly higher level of toxicity was observed in cultured SNpc than VTA neurons. In MPP+-treated SNpc, but not VTA neurons, neurotoxicity was caused by a transient increase in cytosolic Ca2+ that required the activity of LTCCs and ryanodine receptors. Combined with MPP+-mediated inhibition of DA cleavage by MAO (Choi et al., 2015), this caused upregulation of cytosolic DA and nitric oxide levels, mitochondria oxidation, and ER stress. As with L-DOPA toxicity, SNpc neurons from aSyn deficient mice were significantly more resistant to MPP+. Thus, in two different toxicity models we found that selective death of SNpc neurons results from a combination of “multiple hits,” including the activity of the LTCCs that create high basal cytoplasmic Ca2+ levels, an upregulation of DA synthesis and the presence of aSyn. Similar upregulation of Ca2+/NO with concomitant mitochondria oxidative stress was demonstrated in LC neurons (Sanchez-Padilla et al., 2014) and SN neurons exposed to preformed aSyn fibrils (Dryanovski et al., 2013), indicating that this pathway may be commonly activated under stress conditions.
A recent study of DA- and aSyn-mediated toxicity in human idiopathic and familial iPSC-derived DA neurons from patients with a DJ-1 mutation (PARK7) provided more evidence for the involvement of multiple factors in mediating PD-like neurotoxicity (Burbulla et al., 2017). The authors identified a DA- and Ca2+-dependent toxic cascade that started with mitochondrial oxidative stress leading to lysosomal dysfunction and aSyn accumulation. Interestingly, this toxicity pathway was not present in DJ-1 deficient mice or mouse iPSC-derived DA neurons generated from DJ-1 KO fibroblasts unless either DA production or aSyn expression was increased. Underlying species-specific differences may therefore explain the difficulties of creating an appropriate mouse model of PD.
Concluding Remarks and Future Directions
At the center of PD pathology is aSyn, which tends to form soluble oligomers and insoluble fibrils. Oligomerization is increased with increased Ca2+ or DA levels, while aSyn oligomers are able to increase internal Ca2+ and DA concentrations, forming a potential positive feedback cycle. Furthermore, DA-modified aSyn blocks CMA-mediated protein degradation, potentially causing a buildup of monomeric aSyn that then aggregates into more oligomers (Figure 2). These interactions demonstrate the precarious nature of SNpc and LC neuron health as, if one aspect of the homeostatic processes goes awry, the feedback loops activate and neurotoxicity ensues. Importantly, in this model it is possible to initiate the pathological sequence of events that lead to neurodegeneration by diverse insults, including elevation of Ca2+ levels, increased cytosolic DA unrelated to Ca2+-dependent regulation, mutation or overexpression of α-Syn, inhibition of CMA activity due to aging (Schneider et al., 2014, 2015), the presence of other parkinsonian mutations or other possible mechanisms.
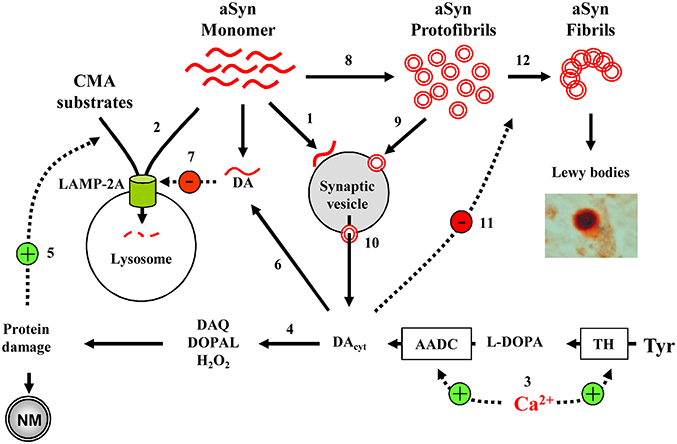
Figure 2. Model of cell-selective PD pathogenesis. (1) A physiological function of aSyn may be to bind synaptic vesicles in a reversible manner to inhibit exocytosis. (2) aSyn is degraded by CMA following LAMP-2A-mediated transport into the lysosomes. (3) In SN and LC neurons, high Ca2+ levels upregulate TH and AADC, leading to increased DA concentrations and (4) associated oxyradical stress, which induces various cell defense mechanisms, including (5) CMA. (6) DA-modified aSyn (7) blocks LAMP-2A-mediated uptake of CMA substrates, including aSyn itself. (8) aSyn may oligomerize to toxic protofibrils, which can (9) bind to and (10) permeabilize synaptic vesicles, leading to further increase in cytosolic DA. (11) DA stabilizes aSyn protofibrils, inhibiting the formation of larger polymers of aSyn (12). Overall, the presence of such interactions where DA and aSyn act as independent stressors that converge to produce neurotoxicity may explain why catecholaminergic neurons with Ca+2 channel- mediated pacemaking are more prone to produce neuromelanin and Lewy bodies, and are particularly vulnerable in PD and parkinsonian animal models. Only a few aSyn-DA-Ca2+ interactions discussed in the text are shown.
Therapeutically, this hypothesis provides several avenues to pursue the disease-modifying opportunities as decreasing the levels of any one of these key toxicity mediators should be beneficial for the survival of SNpc and LC neurons. Previous work has demonstrated the utility of immunotherapy to reduce aSyn levels in the CNS (Masliah et al., 2005) and prevent possible trans-synaptic spread of toxic aSyn species (Bae et al., 2012). A retrospective analysis demonstrated that the use of dihydropyridines correlates with decreased probability of developing PD (Pasternak et al., 2012), and an LTCC antagonist isradipine is currently in phase III clinical trials as a disease-modifying therapy for PD (Swart and Hurley, 2016). Combining these approaches with drugs that reduce toxic DA species might provide additional benefits. One important future focus will be the development of diagnostic tools to enable earlier disease-modifying treatments and stratification of patient populations to enhance beneficial outcomes. The level of aSyn peripherally and in the CNS (Malek et al., 2014) as well as the status of DA homeostasis (Niethammer et al., 2012) are currently the focus of studies aimed at developing bioassays and imaging approaches to identify pre-symptomatic PD cases with defined patho-physiologies to give “personalized” treatments.
Author Contributions
MP, OL, and EM gave their substantial contribution to conception and design of the manuscript, its drafting and revising it critically. All authors have approved the manuscript in its present form for publication and agree to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the National Institute of Neurological Disorders and Stroke (Grants NS075222 to EM) and the National Institute of Mental Health (F30MH114390 to OL and T32MH20004 to MP). We thank David Sulzer and Maha Subramaniam for their help during the preparation of the manuscript and Ellen Kanter for invaluable assistance.
References
Alvarez-Fischer, D., Henze, C., Strenzke, C., Westrich, J., Ferger, B., Hoglinger, G. U., et al. (2008). Characterization of the striatal 6-OHDA model of Parkinson's disease in wild type and alpha-synuclein-deleted mice. Exp. Neurol. 210, 182–193. doi: 10.1016/j.expneurol.2007.10.012
Angelova, P. R., Ludtmann, M. H., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bae, E. J., Lee, H. J., Rockenstein, E., Ho, D. H., Park, E. B., Yang, N. Y., et al. (2012). Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 32, 13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of alpha-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Bhagwandin, A., Gravett, N., Bennett, N. C., and Manger, P. R. (2013). Distribution of parvalbumin, calbindin and calretinin containing neurons and terminal networks in relation to sleep associated nuclei in the brain of the giant Zambian mole-rat (Fukomys mechowii). J. Chem. Neuroanat. 52, 69–79. doi: 10.1016/j.jchemneu.2013.06.002
Bourdenx, M., Bezard, E., and Dehay, B. (2014). Lysosomes and alpha-synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 8:83. doi: 10.3389/fnana.2014.00083
Braak, H., Braak, E., Yilmazer, D., Schultz, C., de Vos, R. A., and Jansen, E. N. (1995). Nigral and extranigral pathology in Parkinson's disease. J. Neural Transm. (Suppl. 46), 15–31.
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Burke, W. J., Li, S. W., Chung, H. D., Ruggiero, D. A., Kristal, B. S., Johnson, E. M., et al. (2004). Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology 25, 101–115. doi: 10.1016/S0161-813X(03)00090-1
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cali, T., Ottolini, D., Negro, A., and Brini, M. (2012). alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Caraveo, G., Auluck, P. K., Whitesell, L., Chung, C. Y., Baru, V., Mosharov, E. V., et al. (2014). Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 111, E3544–E3552. doi: 10.1073/pnas.1413201111
Caudle, W. M., Richardson, J. R., Wang, M. Z., Taylor, T. N., Guillot, T. S., Mccormack, A. L., et al. (2007). Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chang, D., Nalls, M. A., Hallgrimsdottir, I. B., Hunkapiller, J., Van Der Brug, M., Cai, F., et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet. 49, 1511–1516. doi: 10.1038/ng.3955
Chen, C. H., Ferreira, J. C., Gross, E. R., and Mochly-Rosen, D. (2014). Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol. Rev. 94, 1–34. doi: 10.1152/physrev.00017.2013
Chen, L., Ding, Y., Cagniard, B., Van Laar, A. D., Mortimer, A., Chi, W., et al. (2008). Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433. doi: 10.1523/JNEUROSCI.3602-07.2008
Choi, B. K., Choi, M. G., Kim, J. Y., Yang, Y., Lai, Y., Kweon, D. H., et al. (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U.S.A. 110, 4087–4092. doi: 10.1073/pnas.1218424110
Choi, S. J., Panhelainen, A., Schmitz, Y., Larsen, K. E., Kanter, E., Wu, M., et al. (2015). Changes in neuronal dopamine homeostasis following 1-methyl-4-phenylpyridinium (MPP+) exposure. J. Biol. Chem. 290, 6799–6809. doi: 10.1074/jbc.M114.631556
Chung, C. Y., Khurana, V., Yi, S., Sahni, N., Loh, K. H., Auluck, P. K., et al. (2017). In situ peroxidase labeling and mass-spectrometry connects alpha-synuclein directly to endocytic trafficking and mRNA metabolism in neurons. Cell Syst. 4, 242–250. doi: 10.1016/j.cels.2017.01.002
Colebrooke, R. E., Humby, T., Lynch, P. J., Mcgowan, D. P., Xia, J., and Emson, P. C. (2006). Age-related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson's disease. Eur. J. Neurosci. 24, 2622–2630. doi: 10.1111/j.1460-9568.2006.05143.x
Collier, T. J., Redmond, D. E. Jr., Steece-Collier, K., Lipton, J. W., and Manfredsson, F. P. (2016). Is alpha-synuclein loss-of-function a contributor to Parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 10:12. doi: 10.3389/fnins.2016.00012
Collins, M. A., and Bigdeli, M. G. (1975). Tetrahydroisoquinolines in vivo. I. Rat brain formation of salsolinol, a condensation product of dopamine and acetaldehyde, under certain conditions during ethanol intoxication. Life Sci. 16, 585–601.
Conway, K. A., Rochet, J. C., Bieganski, R. M., and Lansbury, P. T. Jr. (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294, 1346–1349. doi: 10.1126/science.1063522
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury Pt, J., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Cuervo, A. M., Wong, E. S., and Martinez-Vicente, M. (2010). Protein degradation, aggregation, and misfolding. Mov. Disord. 25(Suppl. 1), S49–S54. doi: 10.1002/mds.22718
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
Daubner, S. C., Le, T., and Wang, S. (2011). Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 508, 1–12. doi: 10.1016/j.abb.2010.12.017
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., et al. (2002). Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529. doi: 10.1073/pnas.172514599
Daveu, C., Servy, C., Dendane, M., Marin, P., and Ducrocq, C. (1997). Oxidation and nitration of catecholamines by nitrogen oxides derived from nitric oxide. Nitric Oxide 1, 234–243. doi: 10.1006/niox.1997.0123
Deitrich, R., and Erwin, V. (1980). Biogenic amine-aldehyde condensation products: tetrahydroisoquinolines and tryptolines (beta-carbolines). Annu. Rev. Pharmacol. Toxicol. 20, 55–80. doi: 10.1146/annurev.pa.20.040180.000415
De Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Diepenbroek, M., Casadei, N., Esmer, H., Saido, T. C., Takano, J., Kahle, P. J., et al. (2014). Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum. Mol. Genet. 23, 3975–3989. doi: 10.1093/hmg/ddu112
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci. Transl. Med. 8:342ra378. doi: 10.1126/scitranslmed.aaf3634
Ding, T. T., Lee, S. J., Rochet, J. C., and Lansbury, P. T. Jr. (2002). Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41, 10209–10217. doi: 10.1021/bi020139h
Doorn, J. A., Florang, V. R., Schamp, J. H., and Vanle, B. C. (2014). Aldehyde dehydrogenase inhibition generates a reactive dopamine metabolite autotoxic to dopamine neurons. Parkinsonism Relat Disord 20(Suppl. 1), S73–S75. doi: 10.1016/S1353-8020(13)70019-1
Dragicevic, E., Poetschke, C., Duda, J., Schlaudraff, F., Lammel, S., Schiemann, J., et al. (2014). Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 137, 2287–2302. doi: 10.1093/brain/awu131
Dryanovski, D. I., Guzman, J. N., Xie, Z., Galteri, D. J., Volpicelli-Daley, L. A., Lee, V. M., et al. (2013). Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 33, 10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013
Duda, J., Potschke, C., and Liss, B. (2016). Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson's disease. J. Neurochem. 139(Suppl. 1), 156–178. doi: 10.1111/jnc.13572
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., et al. (2007). Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 170, 1725–1738. doi: 10.2353/ajpath.2007.061232
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., et al. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 31, 14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011
Edwards, R. H. (1993). Neural degeneration and the transport of neurotransmitters. Ann. Neurol. 34, 638–645. doi: 10.1002/ana.410340504
Eriksen, J. L., Przedborski, S., and Petrucelli, L. (2005). Gene dosage and pathogenesis of Parkinson's disease. Trends Mol. Med. 11, 91–96. doi: 10.1016/j.molmed.2005.01.001
Exner, N., Lutz, A. K., Haass, C., and Winklhofer, K. F. (2012). Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062. doi: 10.1038/emboj.2012.170
Fahn, S., Oakes, D., Shoulson, I., Kieburtz, K., Rudolph, A., Lang, A., et al. (2004). Levodopa and the progression of Parkinson's disease. N. Engl. J. Med. 351, 2498–2508. doi: 10.1056/NEJMoa033447
Fitzpatrick, P. F. (2000). The aromatic amino acid hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 74, 235–294. doi: 10.1002/9780470123201.ch6
Follett, J., Darlow, B., Wong, M. B., Goodwin, J., and Pountney, D. L. (2013). Potassium depolarization and raised calcium induces alpha-synuclein aggregates. Neurotox. Res. 23, 378–392. doi: 10.1007/s12640-012-9366-z
Fornstedt, B., Brun, A., Rosengren, E., and Carlsson, A. (1989). The apparent autoxidation rate of catechols in dopamine-rich regions of human brains increases with the degree of depigmentation of substantia nigra. J. Neural Transm. Park. Dis. Dement. Sect. 1, 279–295. doi: 10.1007/BF02263482
Fu, Y., Yuan, Y., Halliday, G., Rusznak, Z., Watson, C., and Paxinos, G. (2012). A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct. 217, 591–612. doi: 10.1007/s00429-011-0349-2
Fuentes, P., Paris, I., Nassif, M., Caviedes, P., and Segura-Aguilar, J. (2007). Inhibition of VMAT-2 and DT-diaphorase induce cell death in a substantia nigra-derived cell line–an experimental cell model for dopamine toxicity studies. Chem. Res. Toxicol. 20, 776–783. doi: 10.1021/tx600325u
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gainetdinov, R. R., Fumagalli, F., Wang, Y. M., Jones, S. R., Levey, A. I., Miller, G. W., et al. (1998). Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J. Neurochem. 70, 1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x
Galter, D., Buervenich, S., Carmine, A., Anvret, M., and Olson, L. (2003). ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis. 14, 637–647. doi: 10.1016/j.nbd.2003.09.001
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Goldberg, J. A., Guzman, J. N., Estep, C. M., Ilijic, E., Kondapalli, J., Sanchez-Padilla, J., et al. (2012). Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat. Neurosci. 15, 1414–1421. doi: 10.1038/nn.3209
Goldstein, D. S., Eisenhofer, G., Stull, R., Folio, C. J., Keiser, H. R., and Kopin, I. J. (1988). Plasma dihydroxyphenylglycol and the intraneuronal disposition of norepinephrine in humans. J. Clin. Invest. 81, 213–220. doi: 10.1172/JCI113298
Goldstein, D. S., Kopin, I. J., and Sharabi, Y. (2014). Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol Ther. 144, 268–282. doi: 10.1016/j.pharmthera.2014.06.006
Goldstein, M., and Lieberman, A. (1992). The role of the regulatory enzymes of catecholamine synthesis in Parkinson's disease. Neurology 42, 8–12.
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Graham, D. G. (1978). Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 14, 633–643.
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Hadjiconstantinou, M., and Neff, N. H. (2008). Enhancing aromatic L-amino acid decarboxylase activity: implications for L-DOPA treatment in Parkinson's disease. CNS Neurosci. Ther. 14, 340–351. doi: 10.1111/j.1755-5949.2008.00058.x
Halbrugge, T., Wolfel, R., and Graefe, K. H. (1989). Plasma 3,4-dihydroxyphenylglycol as a tool to assess the role of neuronal uptake in the anaesthetized rabbit. Naunyn Schmiedebergs. Arch. Pharmacol. 340, 726–732. doi: 10.1007/BF00169681
Hastings, T. G., and Berman, S. B. (1999). “Dopamine-induced toxicity and quinone modification of proteins: implications for Parkinson's disease,” in Role of Catechol Quinone Species in Cellular Toxicity, ed C.R. Creveling (Johnson City, TN: F.P. Graham Publishing, Inc.), 69–89.
Herrera, F. E., Chesi, A., Paleologou, K. E., Schmid, A., Munoz, A., Vendruscolo, M., et al. (2008). Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE 3:e3394. doi: 10.1371/journal.pone.0003394
Hetzenauer, A., Sinnegger-Brauns, M. J., Striessnig, J., and Singewald, N. (2006). Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of Ca(V)1.3 and Ca(V)1.2 isoforms. Neuroscience 139, 1005–1015. doi: 10.1016/j.neuroscience.2006.01.059
Hirsch, E., Graybiel, A. M., and Agid, Y. A. (1988). Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334, 345–348. doi: 10.1038/334345a0
Holford, N. H., Chan, P. L., Nutt, J. G., Kieburtz, K., and Shoulson, I. (2006). Disease progression and pharmacodynamics in Parkinson disease - evidence for functional protection with levodopa and other treatments. J. Pharmacokinet. Pharmacodyn. 33, 281–311. doi: 10.1007/s10928-006-9012-6
Hopp, S. C., Royer, S. E., D'angelo, H. M., Kaercher, R. M., Fisher, D. A., and Wenk, G. L. (2015). Differential neuroprotective and anti-inflammatory effects of L-type voltage dependent calcium channel and ryanodine receptor antagonists in the substantia nigra and locus coeruleus. J. Neuroimmune Pharmacol. 10, 35–44. doi: 10.1007/s11481-014-9568-7
Hurley, M. J., and Dexter, D. T. (2012). Voltage-gated calcium channels and Parkinson's disease. Pharmacol. Ther. 133, 324–333. doi: 10.1016/j.pharmthera.2011.11.006
Imaizumi, T., Yamashita, K., Taima, K., Ishikawa, A., Yoshida, H., and Satoh, K. (2005). Effect of peroxisome proliferator-activated receptor-gamma ligands on the expression of retinoic acid-inducible gene-I in endothelial cells stimulated with lipopolysaccharide. Prostaglandins Other Lipid Mediat. 78, 46–54. doi: 10.1016/j.prostaglandins.2005.02.006
Kalia, L. V., Kalia, S. K., Mclean, P. J., Lozano, A. M., and Lang, A. E. (2013). alpha-Synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol. 73, 155–169. doi: 10.1002/ana.23746
Kumer, S. C., and Vrana, K. E. (1996). Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 67, 443–462. doi: 10.1046/j.1471-4159.1996.67020443.x
Lang, A. E., and Lozano, A. M. (1998). Parkinson's disease. Second of two parts. N. Engl. J. Med. 339, 1130–1143. doi: 10.1056/NEJM199810153391607
Larsen, K. E., Schmitz, Y., Troyer, M. D., Mosharov, E., Dietrich, P., Quazi, A. Z., et al. (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., et al. (2002). Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102. doi: 10.1016/S0022-2836(02)00735-0
Lewitt, P. A. (2015). Levodopa therapy for Parkinson's disease: pharmacokinetics and pharmacodynamics. Mov. Disord. 30, 64–72. doi: 10.1002/mds.26082
Li, H. T., Du, H. N., Tang, L., Hu, J., and Hu, H. Y. (2002). Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 64, 221–226. doi: 10.1002/bip.10179
Lieberman, O. J., Choi, S. J., Kanter, E., Saverchenko, A., Frier, M. D., Fiore, G. M., et al. (2017). Alpha-synuclein-dependent calcium entry underlies differential sensitivity of cultured, S. N., and VTA dopaminergic neurons to a Parkinsonian Neurotoxin. eNeuro 4:ENEURO.0167-17.2017. doi: 10.1523/ENEURO.0167-17.2017
Liu, Y., Harding, M., Pittman, A., Dore, J., Striessnig, J., Rajadhyaksha, A., et al. (2014). Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J. Neurophysiol. 112, 1119–1130. doi: 10.1152/jn.00757.2013
Lohr, K. M., Bernstein, A. I., Stout, K. A., Dunn, A. R., Lazo, C. R., Alter, S. P., et al. (2014). Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, 9977–9982. doi: 10.1073/pnas.1402134111
Lotharius, J., and Brundin, P. (2002). Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum. Mol. Genet. 11, 2395–2407. doi: 10.1093/hmg/11.20.2395
Lou, H., Montoya, S. E., Alerte, T. N., Wang, J., Wu, J., Peng, X., et al. (2010). Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 285, 17648–17661. doi: 10.1074/jbc.M110.100867
Lowe, R., Pountney, D. L., Jensen, P. H., Gai, W. P., and Voelcker, N. H. (2004). Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 13, 3245–3252. doi: 10.1110/ps.04879704
Malek, N., Swallow, D., Grosset, K. A., Anichtchik, O., Spillantini, M., and Grosset, D. G. (2014). Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson's disease - a systematic review. Acta Neurol. Scand. 130, 59–72. doi: 10.1111/ane.12247
Marchitti, S. A., Deitrich, R. A., and Vasiliou, V. (2007). Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol. Rev. 59, 125–150. doi: 10.1124/pr.59.2.1
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Masilamoni, G. J., Bogenpohl, J. W., Alagille, D., Delevich, K., Tamagnan, G., Votaw, J. R., et al. (2011). Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134, 2057–2073. doi: 10.1093/brain/awr137
Masliah, E., Rockenstein, E., Adame, A., Alford, M., Crews, L., Hashimoto, M., et al. (2005). Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron 46, 857–868. doi: 10.1016/j.neuron.2005.05.010
Matschke, L. A., Bertoune, M., Roeper, J., Snutch, T. P., Oertel, W. H., Rinne, S., et al. (2015). A concerted action of L- and T-type Ca(2+) channels regulates locus coeruleus pacemaking. Mol. Cell. Neurosci. 68, 293–302. doi: 10.1016/j.mcn.2015.08.012
Matsuda, W., Furuta, T., Nakamura, K. C., Hioki, H., Fujiyama, F., Arai, R., et al. (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453. doi: 10.1523/JNEUROSCI.4029-08.2009
Mazzulli, J. R., Armakola, M., Dumoulin, M., Parastatidis, I., and Ischiropoulos, H. (2007). Cellular oligomerization of alpha-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J. Biol. Chem. 282, 31621–31630. doi: 10.1074/jbc.M704737200
Mccaffery, P., and Drager, U. C. (1994). High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc. Natl. Acad. Sci. U.S.A. 91, 7772–7776. doi: 10.1073/pnas.91.16.7772
Michel, P. P., Hirsch, E. C., and Hunot, S. (2016). Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90, 675–691. doi: 10.1016/j.neuron.2016.03.038
Mironov, S. L. (2015). alpha-Synuclein forms non-selective cation channels and stimulates ATP-sensitive potassium channels in hippocampal neurons. J. Physiol. 593, 145–159. doi: 10.1113/jphysiol.2014.280974
Mittal, S., Bjornevik, K., Im, D. S., Flierl, A., Dong, X., Locascio, J. J., et al. (2017). beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science 357, 891–898. doi: 10.1126/science.aaf3934
Montine, T. J., Farris, D. B., and Graham, D. G. (1995). Covalent crosslinking of neurofilament proteins by oxidized catechols as a potential mechanism of Lewy body formation. J. Neuropathol. Exp. Neurol. 54, 311–319. doi: 10.1097/00005072-199505000-00004
Mooslehner, K. A., Chan, P. M., Xu, W., Liu, L., Smadja, C., Humby, T., et al. (2001). Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: potential mouse model for parkinsonism. Mol. Cell. Biol. 21, 5321–5331. doi: 10.1128/MCB.21.16.5321-5331.2001
Mor, D. E., Tsika, E., Mazzulli, J. R., Gould, N. S., Kim, H., Daniels, M. J., et al. (2017). Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 20, 1560–1568. doi: 10.1038/nn.4641
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Mosharov, E. V., Staal, R. G., Bove, J., Prou, D., Hananiya, A., Markov, D., et al. (2006). Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J. Neurosci. 26, 9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006
Mytilineou, C., Han, S. K., and Cohen, G. (1993). Toxic and protective effects of L-DOPA on mesencephalic cell cultures. J. Neurochem. 61, 1470–1478. doi: 10.1111/j.1471-4159.1993.tb13642.x
Nakashima, A., Kaneko, Y. S., Mori, K., Fujiwara, K., Tsugu, T., Suzuki, T., et al. (2002). The mutation of two amino acid residues in the N-terminus of tyrosine hydroxylase (TH) dramatically enhances the catalytic activity in neuroendocrine AtT-20 cells. J. Neurochem. 82, 202–206. doi: 10.1046/j.1471-4159.2002.00921.x
Naoi, M., Dostert, P., Yoshida, M., and Nagatsu, T. (1993). N-methylated tetrahydroisoquinolines as dopaminergic neurotoxins. Adv. Neurol. 60, 212–217.
Nath, S., Goodwin, J., Engelborghs, Y., and Pountney, D. L. (2011). Raised calcium promotes alpha-synuclein aggregate formation. Mol. Cell. Neurosci. 46, 516–526. doi: 10.1016/j.mcn.2010.12.004
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., et al. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. doi: 10.1016/j.neuron.2009.12.023
Niethammer, M., Feigin, A., and Eidelberg, D. (2012). Functional neuroimaging in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2:a009274. doi: 10.1101/cshperspect.a009274
Olanow, C. W. (2015). Levodopa: effect on cell death and the natural history of Parkinson's disease. Mov. Disord. 30, 37–44. doi: 10.1002/mds.26119
Olanow, C. W., Agid, Y., Mizuno, Y., Albanese, A., Bonuccelli, U., Damier, P., et al. (2004). Levodopa in the treatment of Parkinson's disease: current controversies. Mov. Disord. 19, 997–1005. doi: 10.1002/mds.20243
Ottolini, D., Cali, T., Negro, A., and Brini, M. (2013). The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 22, 2152–2168. doi: 10.1093/hmg/ddt068
Ottolini, D., Cali, T., Szabo, I., and Brini, M. (2017). Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications. Biol. Chem. 398, 77–100. doi: 10.1515/hsz-2016-0201
Outeiro, T. F., Klucken, J., Bercury, K., Tetzlaff, J., Putcha, P., Oliveira, L. M., et al. (2009). Dopamine-induced conformational changes in alpha-synuclein. PLoS ONE 4:e6906. doi: 10.1371/journal.pone.0006906
Pardo, B., Mena, M. A., Casarejos, M. J., Paino, C. L., and De Yebenes, J. G. (1995). Toxic effects of L-DOPA on mesencephalic cell cultures: protection with antioxidants. Brain Res. 682, 133–143. doi: 10.1016/0006-8993(95)00341-M
Pasternak, B., Svanstrom, H., Nielsen, N. M., Fugger, L., Melbye, M., and Hviid, A. (2012). Use of calcium channel blockers and Parkinson's disease. Am. J. Epidemiol. 175, 627–635. doi: 10.1093/aje/kwr362
Peng, X., Tehranian, R., Dietrich, P., Stefanis, L., and Perez, R. G. (2005). Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118, 3523–3530. doi: 10.1242/jcs.02481
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., and Zigmond, M. J. (2002). A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099.
Perier, C., Bove, J., and Vila, M. (2012). Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid. Redox Signal. 16, 883–895. doi: 10.1089/ars.2011.4074
Pifl, C., Rajput, A., Reither, H., Blesa, J., Cavada, C., Obeso, J. A., et al. (2014). Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 34, 8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014
Rcom-H'cheo-Gauthier, A., Goodwin, J., and Pountney, D. L. (2014). Interactions between calcium and alpha-synuclein in neurodegeneration. Biomolecules 4, 795–811. doi: 10.3390/biom4030795
Rochet, J. C., Outeiro, T. F., Conway, K. A., Ding, T. T., Volles, M. J., Lashuel, H. A., et al. (2004). Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson's disease. J. Mol. Neurosci. 23, 23–34. doi: 10.1385/JMN:23:1-2:023
Ronzitti, G., Bucci, G., Emanuele, M., Leo, D., Sotnikova, T. D., Mus, L. V., et al. (2014). Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 34, 10603–10615. doi: 10.1523/JNEUROSCI.0608-14.2014
Sanchez-Padilla, J., Guzman, J. N., Ilijic, E., Kondapalli, J., Galtieri, D. J., Yang, B., et al. (2014). Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 17, 832–840. doi: 10.1038/nn.3717
Schmitz, Y., Lee, C. J., Schmauss, C., Gonon, F., and Sulzer, D. (2001). Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J. Neurosci. 21, 5916–5924.
Schnaitman, C., Erwin, V. G., and Greenawalt, J. W. (1967). The submitochondrial localization of monoamine oxidase. An enzymatic marker for the outer membrane of rat liver mitochondria. J. Cell. Biol. 32, 719–735. doi: 10.1083/jcb.32.3.719
Schneider, J. L., Suh, Y., and Cuervo, A. M. (2014). Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 20, 417–432. doi: 10.1016/j.cmet.2014.06.009
Schneider, J. L., Villarroya, J., Diaz-Carretero, A., Patel, B., Urbanska, A. M., Thi, M. M., et al. (2015). Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249–264. doi: 10.1111/acel.12310
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Specht, C. G., and Schoepfer, R. (2001). Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2:11. doi: 10.1186/1471-2202-2-11
Spencer, J. P., Whiteman, M., Jenner, P., and Halliwell, B. (2002). 5-s-Cysteinyl-conjugates of catecholamines induce cell damage, extensive DNA base modification and increases in caspase-3 activity in neurons. J. Neurochem. 81, 122–129. doi: 10.1046/j.1471-4159.2002.00808.x
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stefanis, L. (2012). Alpha-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Subramaniam, M., Althof, D., Gispert, S., Schwenk, J., Auburger, G., Kulik, A., et al. (2014). Mutant alpha-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 34, 13586–13599. doi: 10.1523/JNEUROSCI.5069-13.2014
Sulzer, D., Bogulavsky, J., Larsen, K. E., Behr, G., Karatekin, E., Kleinman, M. H., et al. (2000). Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 97, 11869–11874. doi: 10.1073/pnas.97.22.11869
Sulzer, D., and Zecca, L. (2000). Intraneuronal dopamine-quinone synthesis: a review. Neurotox. Res. 1, 181–195. doi: 10.1007/BF03033289
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Goldberg, J. A. (2010). What causes the death of dopaminergic neurons in Parkinson's disease? Prog. Brain Res. 183, 59–77. doi: 10.1016/S0079-6123(10)83004-3
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017a). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Surmeier, D. J., and Schumacker, P. T. (2013). Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J. Biol. Chem. 288, 10736–10741. doi: 10.1074/jbc.R112.410530
Surmeier, D. J., Schumacker, P. T., Guzman, J. D., Ilijic, E., Yang, B., and Zampese, E. (2017b). Calcium and Parkinson's disease. Biochem. Biophys. Res. Commun. 483, 1013–1019. doi: 10.1016/j.bbrc.2016.08.168
Swart, T., and Hurley, M. J. (2016). Calcium channel antagonists as disease-modifying therapy for Parkinson's disease: therapeutic rationale and current status. CNS Drugs 30, 1127–1135. doi: 10.1007/s40263-016-0393-9
Swirski, M., Miners, J. S., De Silva, R., Lashley, T., Ling, H., Holton, J., et al. (2014). Evaluating the relationship between amyloid-beta and alpha-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson's disease. Alzheimers Res. Ther. 6:77. doi: 10.1186/s13195-014-0077-y
Taylor, T. N., Caudle, W. M., and Miller, G. W. (2011). VMAT2-deficient mice display nigral and extranigral pathology and motor and nonmotor symptoms of Parkinson's disease. Parkinsons Dis. 2011:124165. doi: 10.4061/2011/124165
Tehranian, R., Montoya, S. E., Van Laar, A. D., Hastings, T. G., and Perez, R. G. (2006). Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J. Neurochem. 99, 1188–1196. doi: 10.1111/j.1471-4159.2006.04146.x
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O'connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tyce, G. M., Hunter, L. W., Ward, L. E., and Rorie, D. K. (1995). Effluxes of 3,4-dihydroxyphenylalanine, 3,4-dihydroxyphenylglycol, and norepinephrine from four blood vessels during basal conditions and during nerve stimulation. J. Neurochem. 64, 833–841. doi: 10.1046/j.1471-4159.1995.64020833.x
Uhl, G. R. (1998). Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease. Ann. Neurol. 43, 555–560. doi: 10.1002/ana.410430503
Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005). Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 280, 9595–9603. doi: 10.1074/jbc.M411805200
Van Dijk, K. D., Berendse, H. W., Drukarch, B., Fratantoni, S. A., Pham, T. V., Piersma, S. R., et al. (2012). The proteome of the locus ceruleus in Parkinson's disease: relevance to pathogenesis. Brain Pathol. 22, 485–498. doi: 10.1111/j.1750-3639.2011.00540.x
Vauzour, D., Ravaioli, G., Vafeiadou, K., Rodriguez-Mateos, A., Angeloni, C., and Spencer, J. P. (2008). Peroxynitrite induced formation of the neurotoxins 5-S-cysteinyl-dopamine and DHBT-1: implications for Parkinson's disease and protection by polyphenols. Arch. Biochem. Biophys. 476, 145–151. doi: 10.1016/j.abb.2008.03.011
Wakamatsu, K., Tabuchi, K., Ojika, M., Zucca, F. A., Zecca, L., and Ito, S. (2015). Norepinephrine and its metabolites are involved in the synthesis of neuromelanin derived from the locus coeruleus. J. Neurochem. 135, 768–776. doi: 10.1111/jnc.13237
Werner-Allen, J. W., Dumond, J. F., Levine, R. L., and Bax, A. (2016). Toxic dopamine metabolite DOPAL forms an unexpected dicatechol pyrrole adduct with lysines of alpha-Synuclein. Angew. Chem. Int. Ed Engl. 55, 7374–7378. doi: 10.1002/anie.201600277
Wey, M. C., Fernandez, E., Martinez, P. A., Sullivan, P., Goldstein, D. S., and Strong, R. (2012). Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson's disease. PLoS ONE 7:e31522. doi: 10.1371/journal.pone.0031522
Xu, J., Kao, S. Y., Lee, F. J., Song, W., Jin, L. W., and Yankner, B. A. (2002). Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 8, 600–606. doi: 10.1038/nm0602-600
Yamada, T., Mcgeer, P. L., Baimbridge, K. G., and Mcgeer, E. G. (1990). Relative sparing in Parkinson's disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 526, 303–307. doi: 10.1016/0006-8993(90)91236-A
Zaichick, S. V., Mcgrath, K. M., and Caraveo, G. (2017). The role of Ca(2+) signaling in Parkinson's disease. Dis. Model. Mech. 10, 519–535. doi: 10.1242/dmm.028738
Zaltieri, M., Longhena, F., Pizzi, M., Missale, C., Spano, P., and Bellucci, A. (2015). Mitochondrial dysfunction and alpha-synuclein synaptic pathology in Parkinson's disease: who's on first? Parkinsons Dis. 2015:108029. doi: 10.1155/2015/108029
Zhang, F., and Dryhurst, G. (1994). Effects of L-cysteine on the oxidation chemistry of dopamine: new reaction pathways of potential relevance to idiopathic Parkinson's disease. J. Med. Chem. 37, 1084–1098. doi: 10.1021/jm00034a006
Zhou, Q., Yen, A., Rymarczyk, G., Asai, H., Trengrove, C., Aziz, N., et al. (2016). Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson's disease. Nat. Commun. 7:10332. doi: 10.1038/ncomms10332
Keywords: α-Synuclein, dopamine, calcium, Parkinson's disease, substantia nigra pars compacta, locus coeruleus, multiple hits
Citation: Post MR, Lieberman OJ and Mosharov EV (2018) Can Interactions Between α-Synuclein, Dopamine and Calcium Explain Selective Neurodegeneration in Parkinson's Disease? Front. Neurosci. 12:161. doi: 10.3389/fnins.2018.00161
Received: 28 December 2017; Accepted: 27 February 2018;
Published: 14 March 2018.
Edited by:
Ruth G. Perez, Texas Tech University Health Sciences Center, United StatesReviewed by:
Matthew John Benskey, College of Human Medicine, Michigan State University, United StatesElisa Greggio, Università degli Studi di Padova, Italy
Copyright © 2018 Post, Lieberman and Mosharov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugene V. Mosharov, ZW03MDZAY3VtYy5jb2x1bWJpYS5lZHU=