 Alexander J. Moszczynski
Alexander J. Moszczynski Matthew A. Hintermayer
Matthew A. Hintermayer Michael J. Strong
Michael J. Strong- 1Molecular Medicine Research Group, Schulich School of Medicine & Dentistry, Robarts Research Institute, Western University, London, ON, Canada
- 2Department of Clinical Neurological Sciences, Schulich School of Medicine & Dentistry, Western University, London, ON, Canada
Approximately 50–60% of all patients with amyotrophic lateral sclerosis (ALS) will develop a deficit of frontotemporal function, ranging from frontotemporal dementia (FTD) to one or more deficits of neuropsychological, speech or language function which are collectively known as the frontotemporal spectrum disorders of ALS (ALS-FTSD). While the neuropathology underlying these disorders is most consistent with a widespread alteration in the metabolism of transactive response DNA-binding protein 43 (TDP-43), in both ALS with cognitive impairment (ALSci) and ALS with FTD (ALS-FTD; also known as MND-FTD) there is evidence for alterations in the metabolism of the microtubule associated protein tau. This alteration in tau metabolism is characterized by pathological phosphorylation at residue Thr175 (pThr175 tau) which in vitro is associated with activation of GSK3β (pTyr216GSK3β), phosphorylation of Thr231tau, and the formation of cytoplasmic inclusions with increased rates of cell death. This putative pathway of pThr175 induction of pThr231 and the formation of pathogenic tau inclusions has been recently shown to span a broad range of tauopathies, including chronic traumatic encephalopathy (CTE) and CTE in association with ALS (CTE-ALS). This pathway can be experimentally triggered through a moderate traumatic brain injury, suggesting that it is a primary neuropathological event and not secondary to a more widespread neuronal dysfunction. In this review, we discuss the neuropathological underpinnings of the postulate that ALS is associated with a tauopathy which manifests as a FTSD, and examine possible mechanisms by which phosphorylation at Thr175tau is induced. We hypothesize that this might lead to an unfolding of the hairpin structure of tau, activation of GSK3β and pathological tau fibril formation through the induction of cis-Thr231 tau conformers. A potential role of TDP-43 acting synergistically with pathological tau metabolism is proposed.
Introduction
Amyotrophic lateral sclerosis (ALS, Lou Gehrig's Disease) has classically been considered to be a disorder purely of the descending supraspinal and lower motor neurons, the net result of which is an insidiously progressive degenerative process culminating in death within 3–5 years following symptom onset for the majority of patients (Charcot and Joffroy, 1869; Strong, 2003). However, the concept that ALS is a pure motor disorder has been gradually replaced by one in which it is recognized that between 50 and 60% of all ALS patients will have a concurrent disorder of frontotemporal function (Strong, 2008; Elamin et al., 2011; Phukan et al., 2012; Oh et al., 2014; Montuschi et al., 2015; Strong et al., 2017). Although the literature historically contained a number of individual ALS case reports in whom a concurrent neuropsychological or behavioral disorder had been identified, the recognition that a significant proportion of ALS patients could also have a frank dementia is more recent (Hudson, 1981; Bak and Hodges, 2001). It is now recognized that ALS can include a broad range of neuropsychological, speech, language, or behavioral pathologies that have recently been termed “amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD)” (Strong et al., 2017). ALS-FTSD includes impairments in cognition (ALSci), behavioral dysfunction (ALSbi), a dysexecutive syndrome (ALScbi), or a frontotemporal dementia (ALS-FTD) meeting the Neary or Hodges criteria (Neary et al., 1998; Hodges and Miller, 2001; Rascovsky et al., 2007).
As our understanding of the clinical phenomenology of ALS-FTSD has evolved, so too has our understanding of its pathobiology. In contemporary nomenclature, the frontotemporal lobar degenerations (FTLD) are categorized into three neuropathological subtypes depending on the pattern of pathological protein deposition that is observed: FTLD-tau for those in which a tauopathy [pathological inclusions consisting of the microtubule associated protein tau (tau)] is clearly evident; FTLD-TDP for those in which transactive response DNA-binding protein of 43 kDa (TDP-43) deposition as neuronal or glial inclusions is the hallmark; and, FTLD-FUS for a small proportion of cases in which the DNA/RNA binding protein, fused in sarcoma (FUS), is deposited. For a minority of cases, none of these markers are evident, but there is evidence of widespread protein ubiquitin conjugation, suggesting a disorder of the ubiquitin-proteasome system (UPS). In this latter subgroup, the terminology FTLD-UPS is applied. Finally, an exceptionally rare subgroup will demonstrate a FTLD in which the molecular pathology (i.e., tau, TDP-43, FUS or ubiquitin) is not known, in which case the terminology of FTLD-NOS (not otherwise specified) is applied (Mackenzie et al., 2011; Irwin et al., 2015).
In its most florid form, ALS-FTSD bears all of the neuropathological features of a FTLD in which there is a strong positive correlation between the degree of cognitive dysfunction and the extent of pathological intraneuronal deposition of TDP-43 (Cykowski et al., 2017). Typically observed as a predominantly nuclear protein, in ALS and under conditions of neuronal stress or injury, TDP-43 undergoes a marked upregulation in its expression and adopts a predominantly cytosolic localization (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2011). This applies to both motor neurons and cortical neurons where the latter forms the basis for characterizing the FTLD of ALS as FTLD-TDP. This has been taken therefore to imply that the frontotemporal dysfunction of ALS cannot be grounded in alterations in the metabolism of tau, or potentially as an overlap syndrome. In this review, we will examine the evidence for alterations in tau metabolism in ALSci, and provide the basis for the consideration of this disorder as a tauopathy that is tightly integrated with the dysmetabolism of TDP-43.
ALS-FTSD: Clinical & Neuroimaging Characterization
The presence of frontotemporal dysfunction in an ALS patient is a negative prognostic indicator, heralding a significant reduction in survivorship. This is particularly true for those individuals in which the primary manifestation is either a dysexecutive syndrome or behavioral impairment (Olney et al., 2005; Elamin et al., 2011; Hu et al., 2013). While current epidemiological studies consider a range of prognostic factors that can have an impact on survivorship such as age of onset, rate of deterioration as measured by the ALSFRS–R Total Score, diagnostic delay, age at diagnosis, and metabolic markers, it is noteworthy that few account for the presence or absence of neuropsychological, speech or language dysfunction as a significant prognostic variable (Chiò et al., 2009; Lunetta et al., 2015; Couratier et al., 2016). In part, this may represent a bias introduced because of the complexity of detecting neuropsychological, speech, language, or behavioral impairments (Farhan et al., 2017). Because of this, the Strong criteria (2009) have been recently revised to increase the simplicity and applicability of these international consensus criteria (Strong et al., 2009, 2017).
Revision of the Strong criteria was also driven by the need to broaden the definition of frontotemporal spectrum disorder in ALS to include deficits in social cognition, including deficits in Theory of Mind (ToM). ToM refers specifically to the capacity to attribute independent mental states to others, and can be divided into an affective and cognitive component. The affective component of ToM refers to one's ability to make inferences regarding the emotions and feelings of others, whereas the cognitive component refers to the ability to infer others' intentions and beliefs (Shamay-Tsoory et al., 2009; Adenzato et al., 2010; Poletti et al., 2012). Deficits in both social cognition and ToM, localizing to the mesial frontal/anterior cingulate, are present in a significant proportion of ALS patients, even when there is no clear evidence to suggest more widespread higher-order cortical dysfunction (Meier et al., 2010; Girardi et al., 2011; Poletti et al., 2012; Cerami et al., 2014; van der Hulst et al., 2015). This suggests that deficits in social cognition and ToM might be the earliest harbinger of frontotemporal dysfunction in ALS.
The concept of non-motor dysfunction in ALS being reflective of a widespread higher cortical impairment—with a degree of preponderance to the frontal lobes, particularly the mesial frontal cortex—is supported by a broad array of neuroimaging studies (Abrahams et al., 2004; Lillo et al., 2010; Agosta et al., 2011, 2013; Goldstein et al., 2011; Prudlo et al., 2012; Ambikairajah et al., 2014). Such studies need to be interpreted in the light of underlying genetic traits of the individuals being studied, as it is increasingly clear that ALS is also syndromic with the clinical and molecular phenotype being driven by an ever-increasing array of known genetic variants (Strong, 2001, 2017; Turner and Verstraete, 2015). The contributions of neuroimaging have extended beyond identifying the general degree of cerebral atrophy accompanying frontotemporal syndromes, as visualized with either computed tomography (CT) or magnetic resonance imaging (MRI). Neuroimaging platforms such as single positron emission computerized tomography (SPECT) (Neary et al., 1990), MRI-based measures of functional connectivity (Douaud et al., 2011), and advanced structural MRI sequences that define subcortical frontotemporal white matter tract projection pathology (Agosta et al., 2016) can now provide an evaluation of the extent of loss of functional integrity. Positron emission tomography (PET) has proven invaluable in our understanding of the anatomic and cellular extent of the pathobiology of ALS-FTSD, including the involvement of non-neuronal cells in the disease process (Cistaro et al., 2012, 2014). Increasingly, these neuroimaging modalities are being linked to understanding the molecular pathology of ALS, such as the degree of deeper cortical structures and cerebellar pathology evident in those ALS patients carrying pathological hexanucleotide expansions in C9orf72, even in the presymptomatic stages (Mahoney et al., 2012; Bede et al., 2013; Rohrer et al., 2015; Walhout et al., 2015).
Our interest in the pathology of the anterior cingulate gyrus in ALS, and ultimately the importance of alterations in metabolism of the microtubule-associated protein tau, was driven by a broad array of evidence suggesting that alterations in verbal praxis and fluency were key harbingers of higher cortical dysfunction in the disease (Strong et al., 1996). In a prospective study designed to assess the degree of neuronal loss in the anterior cingulate gyrus using 1H-magnetic resonance spectroscopy (1H-MRS), we observed a significant reduction in the NAA/Cr ratio (indicative of neuronal loss) at baseline. This corresponded to early features of cognitive impairment at a time when no changes in the NAA/Cr ratio were observed in the motor cortex in the region of hand representation (Strong et al., 1999). In hindsight, when we returned to these studies to correlate the placement of the STEAM (Stimulated Echo Acquisition Mode) voxel for mesial frontal localization, the localization was predominantly area 24, showing that the region of neuronal loss correlated with those regions described earlier as encoding social cognition and ToM. As will be reviewed below, it was the neuropathological study of these cases that has driven our conceptualization of a tauopathy in association with ALS-FTSD.
ALS-FTSD: Neuropathological Characterization
Alterations in RNA Metabolism
Pathological intracellular protein inclusions are amongst the neuropathological hallmarks of ALS and include proteins such as copper/zinc superoxide dismutase 1 (SOD1) (Chou et al., 1996), TDP-43 (Arai et al., 2006; Neumann et al., 2006), fused in sarcoma/translocated in liposarcoma (FUS/TLS) (Kwiatkowski et al., 2009; Vance et al., 2009), TATA-binding protein-associated factor 15 (TAF-15) (Couthouis et al., 2011), Ewing sarcoma breakpoint region 1 (EWSR1) (Couthouis et al., 2011), RNA-binding motif 45 (RBM45) (Collins et al., 2012), Rho guanine nucleotide exchange factor (RGNEF) (Volkening et al., 2010; Keller et al., 2012), intermediate filament proteins (neurofilament Strong et al., 2005 or peripherin Migheli et al., 1993), 14-3-3 proteins (Malaspina et al., 2000; Kawamoto et al., 2004), and abnormal dipeptide repeat (DPR) proteins arising from C9orf72 G4C2 repeat expansions through repeat associated non-AUG (RAN) translation (Ash et al., 2013; Mori et al., 2013).
Amongst these proteins, it is the presence of TDP-43 immunoreactive neuronal cytoplasmic inclusions (NCIs) within degenerating motor neurons that is now recognized as being a ubiquitous neuropathological feature of all variants of ALS, with the exception of cases in which mutations in SOD1 are present. TDP-43 is a dual DNA/RNA binding protein whose metabolism is fundamentally altered in ALS and which forms the core protein of the vast majority of neuronal and glial inclusions in ALS, FTD, and ALS-FTSD (Arai et al., 2006; Neumann et al., 2006). Typically, TDP-43 is predominantly nuclear in its localization, but in response to cellular injury, its expression is markedly upregulated in a process that is accompanied by a nuclear to cytosolic shift in its localization (Moisse et al., 2009a,b). There is a high degree of correlation between the severity of the cognitive deficit that occurs in ALS and the distribution and extent of pathological TDP-43 deposition (Mackenzie, 2008; Mackenzie et al., 2011; Cykowski et al., 2017). Because of this prominence of TDP-43 pathology, and a general lack of deposition of the microtubule associated tau protein, the FTLD of ALS-FTD is currently classified as a “TDP-43 proteinopathy” and not a “tau proteinopathy.”
pThr175 Tau and Alterations in Tau Metabolism
Notwithstanding this, the evidence in support of alterations in tau metabolism in ALS is robust, including that observed in the hyperendemic focus of ALS in the western Pacific in which the deposition of tau protein is prominent throughout the neuroaxis (Hirano, 1966). The presence of tau pathology has also been described in both sporadic and familial ALS case reports (Orrell et al., 1995; Soma et al., 2012; Dobson-Stone et al., 2013; Nakamura et al., 2014; Takeuchi et al., 2016). Additionally, several more recent studies have reported an elevation in phosphorylated and truncated tau protein in both hippocampal and spinal cord neurons of various ALS populations (Gómez-Pinedo et al., 2016; Vintilescu et al., 2016).
Our interest in the pathological processing of tau protein in ALS was initially aimed at explaining the basis of the superficial linear spongiosus evident in the anterior cingulate gyri of those ALSci patients in whom we observed a loss of neurons in this region through 1H-MRS (Strong et al., 1999; Wilson et al., 2001). Using antibodies against tau protein as a marker of axonal projections which we postulated would be lost, we instead observed both neuronal and glial cytoplasmic inclusions which were tau immunoreactive throughout the affected anterior cingulate gyrus (Yang et al., 2003). This included a broad range of pathologies, including neurofibrillary tangle-like structures, dystrophic neurites, neuritic granules, and tau-immunoreactive tufted astrocytes. We demonstrated that this was not simply a function of aging as might be expected based on the subsequently described progressive age-related tauopathy (Yang et al., 2005; Crary et al., 2014; Jellinger et al., 2015). Following fractionation of tau protein into sarkosyl-soluble or insoluble fractions from either the cortical or subcortical white matter of the anterior cingulate gyrus of ALSci patients, we observed that in contrast to tau protein isolated from Alzheimer's disease in which the insoluble fraction contained the triplet tau isoforms characteristic of paired helical filaments (PHFs), all six isoforms of tau protein were expressed in both the soluble and insoluble fractions (Strong et al., 2006). We subsequently confirmed this finding independent of the presence or absence of pathological hexanucleotide expansions of C9orf 72 (Volkening et al., 2017). In the former study, we also observed that sarkosyl-insoluble tau protein from ALSci was resistant to enzymatic dephosphorylation, and, when examined using the Thioflavin S assay, had a significant increase in the tendency to form polymers ex vivo (Strong et al., 2006).
Given this suggestion of pathological tau protein phosphorylation in ALSci, we conducted a phospho-epitope analysis and identified a unique tau phosphorylation site at Threonine 175 (pThr175 tau) in ALS and ALSci that was not present in either control or AD (both soluble and insoluble tau protein isolates) (Strong et al., 2006). Using a novel polyclonal antibody against pThr175 tau, we subsequently confirmed the presence of prominent tau protein glial and neuronal cytoplasmic inclusions in ALSci, and to a lesser degree in ALS without cognitive deficits, in association with an increase in the expression of active GSK3β (Yang et al., 2008; Yang and Strong, 2012) (Figure 1). This deposition was accompanied by a diffuse increase in TDP-43 immunoreactivity, suggesting the co-occurrence of these pathological processes (Yang and Strong, 2012). The prominent deposition of tau protein, including pThr175 tau, has been subsequently validated in motor neuron disease (MND) and MND in the presence of a behavioral variant of FTD (bvFTD-MND), including the appearance of tau protein deposition in 10% of MND cases that did not conform to traditional morphological criteria (unclassified frontal tau protein) (Behrouzi et al., 2016). While the latter study suggested a morphological appearance reminiscent of AD, as discussed earlier, the characteristic sarkosyl-insoluble tau protein triplet protein molecular signature of AD has not been observed in ALS or ALSci. More recently, we have observed that the pathological phospho-epitope pThr175 tau protein is also observed across a broad range of tauopathies, strongly suggesting that its presence in ALSci is not simply incidental, but rather a marker of a fundamental alteration in tau protein processing across a range of tauopathies (Moszczynski et al., 2017).
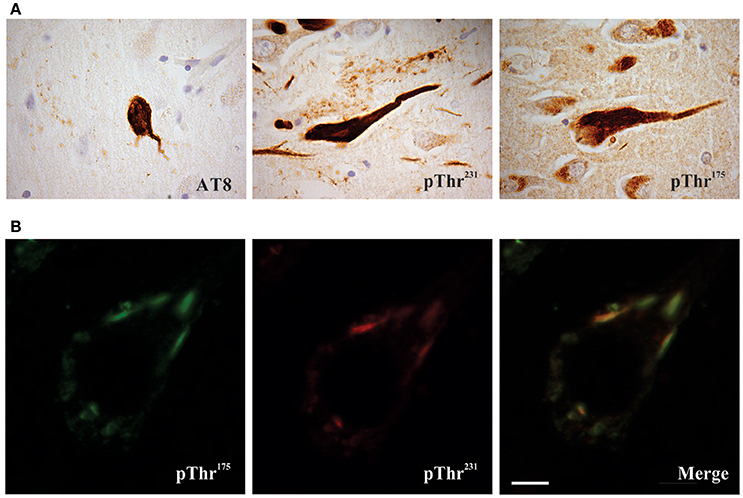
Figure 1. Phosphorylated tau pathology in amyotrophic latera sclerosis with cognitive impairment (ALSci) hippocampal neurons. (A) Immunohistochemical probing with AT8 (pSer202, pThr205), pThr175, pThr231 all reveal positive inclusions in ALS hippocampal neurons. Images taken with a 100x objective. (B) Representative image of pThr175 and pThr231 co-localization in ALSci hippocampal neuron. Scale bar = 5 μm.
Pathogenicity of pThr175 Tau
To confirm the pathogenicity of pThr175 tau, we transfected either HEK293T or Neuro2a cells with a pseudo-phosphorylated tau protein construct (Thr175Asp-tau), wild type human tau protein (WT-tau) or human tau protein in which phosphorylation at Thr175 was irreversibly inhibited (Thr175Ala-tau) (Gohar et al., 2009). We observed a significant induction of pathological tau protein cytoplasmic inclusions and enhanced rates of cell death in the presence of Thr175Asp-tau, regardless of which of the six human tau protein constructs was transfected. Consistent with the observed up-regulation of activated GSK3β (pTyr216GSK3β) that co-localized with tau protein pathology in ALSci, we demonstrated a significant increase in pTyr216 GSK3β expression in only the Thr175Asp-tau transfected cells (Moszczynski et al., 2015). Inhibition of GSK3β activity, either pharmacologically or by shRNA against GSK3β, abolished inclusion formation. We next demonstrated that the development of pathological inclusions in response to the presence of Thr175Asp-tau was dependent on the unprimed phosphorylation of the Thr231 residue of tau protein and that the presence of pThr231 was sufficient to induce tau protein inclusions. These observations suggest that a pathway of pThr175 induction of GSK3β activation and a consequent phosphorylation of Thr231 in tau protein fibril formation is a pathogenic mechanism contributing to neuronal death in ALSci. While investigating evidence for this pathway in human disease, we observed the co-localization of pThr175 and pThr231 in tau protein NCI-containing hippocampal neurons in ALSci and a broad range of tauopathies including Alzheimer's disease, diffuse Lewy body dementia, and FTLD among others (Moszczynski et al., 2017). What remains to be determined across both sets of experiments however is the ultrastructural characteristics of the cytoplasmic inclusions driven in vitro by pThr175 tau or as observed in vivo.
We then examined the tauopathy associated with chronic traumatic encephalopathy (CTE) and CTE with concurrent ALS (CTE-ALS) (Moszczynski et al., 2018). In both disease states, the neuropathological hallmark is a disseminated tauopathy including not only cortical neuronal and glial cytoplasmic inclusions, but also spinal cord tau protein pathology (McKee et al., 2009, 2010; Mez et al., 2017). Consistent with a role for pathological Thr175 tau phosphorylation, we observed pThr175, pThr231, and T22 immunoreactivity in both hippocampal and spinal motor neurons. Fractionation of the hippocampal tau protein into sarkosyl-soluble and insoluble fractions yielded all six tau protein isoforms partitioning into both fractions, similar to that observed in ALSci. In support of the hypothesis that this pathway is directly related to the induction of tau protein cytoplasmic inclusions, we were able to replicate this pathology following a single cortical impact (moderate brain trauma model) in young adult Sprague Dawley rats (Moszczynski et al., 2018).
While we have suggested that pThr175 tau is critical to the induction of a tauopathy, this evidence does not prove it to be sufficient for the development of tauopathy in vivo. To test this, we utilized the somatic gene transfer of a pseudophosphorylated human tau protein construct mimicking pThr175 tau. Young adult Sprague Dawley rats received bilateral stereotactic inoculums of a rAAV9 adenoviral construct containing one of either GFP tagged Thr175Asp-tau (pThr175 tau mimic), GFP-tagged WT-tau, GFP-tagged construct alone, or GFP-tagged Thr175Ala-tau (Moszcyznski et al., 2017). At 1 year following inoculation, expression of the viral vector remained prominent as assessed using anti eGFP immunohistochemistry. However, the brains of the Thr175Asp-tau inoculated rats demonstrated prominent neuronal tau protein pathology, including corkscrew neurites, and tau immunoreactive neuronal cytoplasmic inclusions.
Postulated Pathogenic Pathway
In support of the pathogenicity of pThr175 tau, phosphorylation at this site is not observed in fetal human tissue (which is normally hyperphosphorylated) (Kenessey and Yen, 1993) or tissue from healthy-aged individuals (Moszczynski et al., 2017). We have shown in vitro that a pseudophosphorylated human tau construct (Thr175Asp tau) induces pathological tau protein inclusions accompanied by the activation of GSK3β along with phosphorylation of tau at Thr231 (Moszczynski et al., 2015). The in vivo observation of co-expression of pThr175 tau, pTyr216GSK3β, pThr231 tau, and immunoreactivity to a tau protein antibody recognizing oligomeric tau (T22) strongly suggests that this pathway is critical to the induction of pathological tauopathies, including ALSci, CTE and CTE-ALS (Moszczynski et al., 2017, 2018).
The mechanism of Thr175 phosphorylation is not known. Candidate kinases include mitogen activated protein kinases (MAPK), GSK3β, p38, and leucine-rich repeat kinase 2 (LRRK2) (Hanger et al., 1998; Reynolds et al., 2000; Atzori et al., 2001) (see also https://docs.google.com/spreadsheets/d/1hGYs1ZcupmTnbB7n6qs1r_WVTXHt1O7NBLyKBN7EOUQ/edit#gid=0). Amongst these, a member of the MAPK family of kinases, c-Jun N-terminal kinase [JNK; also known as stress activated protein kinase (SAPK)], is of specific interest given its increased activity in response to various cellular stresses in vitro (Namgung and Xia, 2000) and in the acute phases following traumatic brain injury (TBI) in vivo (Tran et al., 2012). Additionally, the inhibition of JNK can reduce pathological tau protein phosphorylation following TBI in a rodent model (Tran et al., 2012).
Although the mechanism(s) by which the phosphorylation of tau at Thr175 leads to the activation of GSK3β are not yet known, it is likely that such mechanism(s) will involve altering tau's global hairpin structure given that Thr175 lies within the Pro-rich domain/hinge region of the hairpin as it has been shown that the phosphorylation of other residues within this domain can open the hairpin structure (Jeganathan et al., 2006, 2008). There are at least two potential mechanisms of GSK3β activation which may result from tau hairpin structure opening (Figure 2). Firstly, opening of the hairpin may expose the N-terminus proline-rich domain, which has been shown to interact with the SH3 domain of Src-family tyrosine kinase Fyn. In doing so, activated Fyn kinase may then phosphorylate GSK3β at the Tyr216 residue thereby enhancing its activity (Lee et al., 1998; Lesort et al., 1999; Klein et al., 2002). Secondly, opening of the hairpin conformation has been shown to expose the N-terminal phosphatase activating domain (PAD), consisting of amino acids 2–18. Exposure of this domain leads to activation of protein phosphatase-1 (PP1) which then dephosphorylates GSK3β at the Ser9 residue, enhancing GSK3β activity (Kanaan et al., 2011). In keeping with this mechanism, exposure of the PAD has been suggested to be important in tau protein mediated neuronal toxicity through interference with fast axonal transport, and may be an early event in the development of a number of tauopathies (Kanaan et al., 2011, 2016; Ward et al., 2012; Combs et al., 2016).
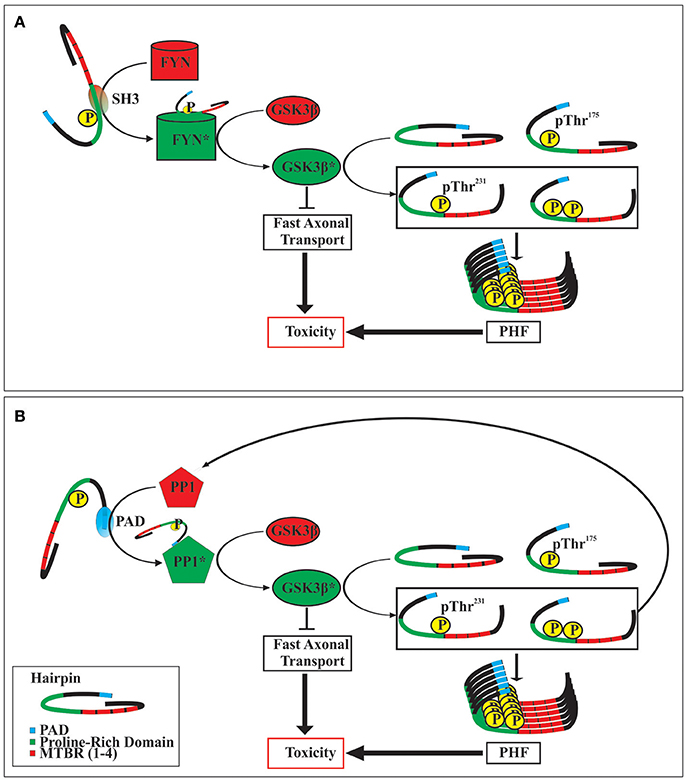
Figure 2. Two postulated mechanisms of toxicity following tau protein phosphorylation at Thr175. Opening of the tau hairpin conformation by phosphorylation at Thr175 exposes SH3 homology domains within the proline-rich region (A). This interacts with and activates FYN kinase, which in turn activates GSK3β by phosphorylating at Tyr216. Alternatively, opening of the tau hairpin can expose the N-terminal phosphatase activating domain (PAD) (B), which then activates protein phosphatase 1 (PP1). Activated PP1 dephosphorylates GSK3β at Ser9 increasing its activity. The activation of GSK3β may exert toxic effects in the cell by the inhibition of fast axonal transport, and by the phosphorylation of tau protein at Thr231, which promotes the formation of neurofibrillary tangles (paired helical filaments; PHF). Additionally, increased activity of GSK3β can increase the formation of pThr231 tau which also contains an exposed PAD, thereby initiating a dysregulated positive feedback loop. Activated enzymes are indicated by an asterisk (*).
In the studies cited above, tau phosphorylation at Thr231 is a critical step in the induction of pathological fibrils. Several mechanisms have been proposed by which this may occur, with alterations in tau degradation being key in a process that is dependent on tau conformation. Of note, cis-pThr231 tau demonstrates specificity in the development of pathology over trans-pThr231 tau, in which the cis conformer is pathological and cannot be degraded, whereas the trans conformer is physiological and can be degraded (Nakamura et al., 2012). The isomerase, peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1), converts the pathologic cis conformer to the physiologic trans conformer, allowing for dephosphorylation at this site by protein phosphatase 1 (Lu et al., 1996, 1999). It is of note, therefore, that cis-pThr231 tau is closely associated with a number of tauopathies including, AD, CTE and experimental CTE (Lu et al., 1996, 1999; Liou et al., 2003; Pastorino et al., 2006; Sultana et al., 2006; Lu and Zhou, 2007; Lim et al., 2008; Lee et al., 2011; Chen et al., 2015; Kondo et al., 2015; Albayram et al., 2017). Given that both cis-pThr231 tau and pThr175 tau are observed exclusively in disease or stressed states, it is attractive to postulate that the phosphorylation of tau protein at Thr175 increases the presence of cis-pThr231. This may occur through either enhancing the probability of cis phosphorylation by GSK3β or by inhibiting the ability of PIN1 to isomerize the epitope from cis to trans. This is the subject of current studies.
The Potential Role of Comorbid Pathologies
The fact that other cell stressors may induce Thr175 tau protein phosphorylation is of great significance to the mechanisms of neurodegenerative disease. Pathologies are rarely, if ever, observed in isolation, and it is common to identify co-morbid pathologies in the same brain (Amador-Ortiz et al., 2007; Josephs et al., 2014a,b; Smith, 2017). While this high incidence of comorbidity has traditionally been attributed to the aging process, it is increasingly evident that multiple pathologies are present even in young cohorts of patients afflicted with neurodegenerative disease. Therefore, there is likely an interplay between pathological processes that drives these co-morbid pathologies (Spires-Jones et al., 2017; Tan et al., 2017). As such, the possibility of synergistic toxicity must be considered when trying to understand the mechanisms that drive neurodegenerative disease, and when attempting to treat such diseases.
As discussed earlier, the FTLD of ALSci is classified as a FTLD-TDP based on the prominence of pathological TDP-43 deposition. The association of TDP-43 pathology with ALS and ALS-FTSD is well described (Arai et al., 2006; Neumann et al., 2006). However, there is also an increasing literature describing comorbid TDP-43 and tau pathologies, such as that observed in AD and CTE (McKee et al., 2010; Josephs et al., 2014a,b). The relationship between tau protein and TDP-43 pathology is strengthened in the observation that the effective elimination of aberrantly phosphorylated tau protein in a rodent model of CTE can also lead to the prevention of TDP-43 pathology (Albayram et al., 2017). It is possible that one pathological process primes the other. In this case, the toxicity of pseudophosphorylated Thr175Asp tau protein expression may be enhanced by the co-expression of TDP-43, serving as a second hit to the CNS. The potential of this synergistic toxicity is the focus of ongoing studies in our lab.
Summary
The presence of pathological tau processing is clearly evident across a broad range of ALS patients—including those found within the hyperendemic foci such as that seen in the western Pacific, a smattering of case reports, and ALSci as well as FTD-MND. This suggests that this is not a simple matter of incidental co-occurrence. Moreover, there is solid evidence to support the pathogenic role of pThr175 in the induction of a tauopathy, both in vitro and in vivo, and the pathological cascade induced by pThr175 which culminates in the generation of oligomeric tau has been observed in a broad range of tauopathies. The issue is therefore not whether a tauopathy exists in ALS-FTSD, but rather the extent to which it is the driving pathology—either alone, or in combination with the hallmark pathology of TDP-43—of the clinical and neuropathological phenotype.
Such a proposal however leaves many unanswered questions, including: the mechanism by which the Thr175 residue is initially phosphorylated; how pThr175 tau leads to the activation of GSK3β; whether pThr231 tau is necessary and sufficient to induce neuronal dysfunction and death, or whether additional tau protein pathological phosphorylation is needed; and what (if any) role for the co-existent TDP-43 pathology of ALS-FTSD is also necessary to drive the phenomenological aspects of pThr175 tau-mediated tauopathy.
Author Contributions
MS conceived and wrote the article. AM conceived and wrote the article. MH wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research funded by the Ontario Neurodegenerative Disorders Research Initiative (ONDRI) and the National Hockey League Players Association (NHLPA) Challenge Fund.
References
Abrahams, S., Goldstein, L. H., Simmons, A., Brammer, M., Williams, S. C., Giampietro, V., et al. (2004). Word retrieval in amyotrophic lateral sclerosis: a functional magnetic resonance imaging study. Brain 127, 1507–1517. doi: 10.1093/brain/awh170
Adenzato, M., Cavallo, M., and Enrici, I. (2010). Theory of mind ability in the behavioural variant of frontotemporal dementia: an analysis of the neural, cognitive, and social levels. Neuropsychologia 48, 2–12. doi: 10.1016/j.neuropsychologia.2009.08.001
Agosta, F., Canu, E., Valsasina, P., Riva, N., Prelle, A., Comi, G., et al. (2013). Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol. Aging 34, 419–427. doi: 10.1016/j.neurobiolaging.2012.04.015
Agosta, F., Ferraro, P. M., Riva, N., Spinelli, E. G., Chiò, A., Canu, E., et al. (2016). Structural brain correlates of cognitive and behavioral impairment in MND. Hum. Brain Mapp. 37, 1614–1626. doi: 10.1002/hbm.23124
Agosta, F., Valsasina, P., Absinta, M., Riva, N., Sala, S., Prelle, A., et al. (2011). Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cereb. Cortex 21, 2291–2298. doi: 10.1093/cercor/bhr002
Albayram, O., Kondo, A., Mannix, R., Smith, C., Tsai, C. Y., Li, C., et al. (2017). Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 8:1000. doi: 10.1038/s41467-017-01068-4
Amador-Ortiz, C., Lin, W. L., Ahmed, Z., Personett, D., Davies, P., Duara, R., et al. (2007). TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann. Neurol. 61, 435–445. doi: 10.1002/ana.21154
Ambikairajah, A., Devenney, E., Flanagan, E., Yew, B., Mioshi, E., Kiernan, M. C., et al. (2014). A visual MRI atrophy rating scale for the amyotrophic lateral sclerosis-frontotemporal dementia continuum. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 226–234. doi: 10.3109/21678421.2014.880180
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atzori, C., Ghetti, B., Piva, R., Srinivasan, A. N., Zolo, P., Delisle, M. B., et al. (2001). Activation of the JNK/p38 pathway occurs in diseases charcterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 60, 1190–1197. doi: 10.1093/jnen/60.12.1190
Bak, T. H., and Hodges, J. R. (2001). Motor neurone disease, dementia and aphasia; coincidence, co-occurrence or continuum? J. Neurol. 248, 260–270. doi: 10.1007/s004150170199
Bede, P., Bokde, A. L., Byrne, S., Elamin, M., McLaughlin, R. L., Kenna, K., et al. (2013). Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology 81, 361–369. doi: 10.1212/WNL.0b013e31829c5eee
Behrouzi, R., Liu, X., Wu, D., Robinson, A. C., Tanaguchi-Watanabe, S., Rollinson, S., et al. (2016). Pathological tau deposition in motor neurone disease and frontotemporal lobar degeneration associated with TDP-43 proteinopathy. Acta Neuropathol. Commun. 4:33. doi: 10.1186/s40478-016-0301-z
Cerami, C., Dodich, A., Canessa, N., Crespi, C., Iannaccone, S., Corbo, M., et al. (2014). Emotional empathy in amyotrophic lateral sclerosis: a behavioural and voxel-based morphometry study. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 21–29. doi: 10.3109/21678421.2013.785568
Charcot, J. M., and Joffroy, A. (1869). Deux cas d'atrophie musculaire progressive avec lésions de la substance grise et des faisceaux antérolatéraux de la moelle épinière. Arch. Physiol. Norm. Pathol. 2, 354–744.
Chen, C. H., Li, W., Sultana, R., You, M. H., Kondo, A., Shahpasand, K., et al. (2015). Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer's disease. Neurobiol. Dis. 76, 13–23. doi: 10.1016/j.nbd.2014.12.027
Chiò, A., Logroscino, G., Hardiman, O., Swingler, R., Mitchell, D., Beghi, E., et al. (2009). Prognostic factors in ALS: a critical review. Amyotroph. Lateral Scler. 10, 310–323. doi: 10.3109/17482960802566824
Chou, S. M., Wang, H. S., and Komai, K. (1996). Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J. Chem. Neuroanat. 10, 249–258. doi: 10.1016/0891-0618(96)00137-8
Cistaro, A., Cuccurullo, V., Quartuccio, N., Pagani, M., Valentini, M. C., and Mansi, L. (2014). Role of PET and SPECT in the study of amyotrophic lateral sclerosis. Biomed Res. Int. 2014:237437. doi: 10.1155/2014/237437
Cistaro, A., Valentini, M. C., Chiò, A., Nobili, F., Calvo, A., Moglia, C., et al. (2012). Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur. J. Nucl. Med. Mol. Imaging 39, 251–259. doi: 10.1007/s00259-011-1979-6
Collins, M., Riascos, D., Kovalik, T., An, J., Krupa, K., Krupa, K., et al. (2012). The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 124, 717–732. doi: 10.1007/s00401-012-1045-x
Combs, B., Hamel, C., and Kanaan, N. M. (2016). Pathological conformations involving the amino terminus of tau occur early in Alzheimer's disease and are differentially detected by monoclonal antibodies. Neurobiol. Dis. 94, 18–31. doi: 10.1016/j.nbd.2016.05.016
Couratier, P., Corcia, P., Lautrette, G., Nicol, M., Preux, P. M., and Marin, B. (2016). Epidemiology of amyotrophic lateral sclerosis: a review of literature. Rev. Neurol. 172, 37–45. doi: 10.1016/j.neurol.2015.11.002
Couthouis, J., Hart, M. P., Shorter, J., Dejesus-Hernandez, M., Erion, R., Oristano, R., et al. (2011). A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. U.S.A. 108, 20881–20890. doi: 10.1073/pnas.1109434108
Crary, J. F., Trojanowski, J. Q., Schneider, J. A., Abisambra, J. F., Abner, E. L., Alafuzoff, I., et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. doi: 10.1007/s00401-014-1349-0
Cykowski, M. D., Powell, S. Z., Peterson, L. E., Appel, J. W., Rivera, A. L., Takei, H., et al. (2017). Clinical significance of TDP-43 neuropathology in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 76, 402–413. doi: 10.1093/jnen/nlx025
Dobson-Stone, C., Luty, A. A., Thompson, E. M., Blumbergs, P., Brooks, W. S., Short, C. L., et al. (2013). Frontotemporal dementia-amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1-q12.2: genetic, clinical and neuropathological analysis. Acta Neuropathol. 125, 523–533. doi: 10.1007/s00401-013-1078-9
Douaud, G., Filippini, N., Knight, S., Talbot, K., and Turner, M. R. (2011). Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain 134, 3470–3479. doi: 10.1093/brain/awr279
Elamin, M., Phukan, J., Bede, P., Jordan, N., Byrne, S., Pender, N., et al. (2011). Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 76, 1263–1269. doi: 10.1212/WNL.0b013e318214359f
Farhan, S. M., Bartha, R., Black, S. E., Corbett, D., Finger, E., Freedman, M., et al. (2017). The Ontario Neurodegenerative Disease Research Initiative (ONDRI). Can. J. Neurol. Sci. 44, 196–202. doi: 10.1017/cjn.2016.415
Girardi, A., Macpherson, S. E., and Abrahams, S. (2011). Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology 25, 53–65. doi: 10.1037/a0020357
Gohar, M., Yang, W., Strong, W. L., Volkening, K., Leystra-Lantz, C., and Strong, M. J. (2009). Tau phosphorylation at 175Thr leads to fibril formation. Implications for the tauopathy of amyotrophic lateral sclerosis. J. Neurochem. 108, 634–643. doi: 10.1111/j.1471-4159.2008.05791.x
Goldstein, L. H., Newsom-Davis, I. C., Bryant, V., Brammer, M., Leigh, P. N., and Simmons, A. (2011). Altered patterns of cortical activation in ALS patients during attention and cognitive response inhibition tasks. J. Neurol. 258, 2186–2198. doi: 10.1007/s00415-011-6088-8
Gómez-Pinedo, U., Villar-Quiles, R. N., Galán, L., Matías-Guiu, J. A., Benito-Martin, M. S., Guerrero-Sola, A., et al. (2016). Immununochemical markers of the amyloid cascade in the hippocampus in motor neuron diseases. Front. Neurol. 7:195. doi: 10.3389/fneur.2016.00195
Hanger, D. P., Betts, J. C., Loviny, T. L. F., Blackstock, W. P., and Anderton, B. H. (1998). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament tau) from Alzheimer's disease brain using nanoelectrospray mass spectroscopy. J. Neurochem. 71, 2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x
Hirano, A. (1966). “Neuropathology of amyotrophic lateral sclerosis and parkinsonism-dementia complex on Guam,” in Proceedings of the Fifth International Congress of Neuropathology, eds L. Luthy and A. Bischoff (Amsterdam: Excerpta Medica), 190–194.
Hodges, J. R., and Miller, B. (2001). The classification, genetics and neuropathology of frontotemporal dementia. Introduction to the special topic papers: part 1. Neurocase 7, 31–35. doi: 10.1093/neucas/7.1.31
Hu, W. T., Shelnutt, M., Wilson, A., Yarab, N., Kelly, C., Grossman, M., et al. (2013). Behavior matters–cognitive predictors of survival in amyotrophic lateral sclerosis. PLoS ONE 8:e57584. doi: 10.1371/journal.pone.0057584
Hudson, A. J. (1981). Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain 194, 217–247. doi: 10.1093/brain/104.2.217
Irwin, D. J., Cairns, N. J., Grossman, M., McMillan, C. T., Lee, E. B., Van Deerlin, V. M., et al. (2015). Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 129, 469–491. doi: 10.1007/s00401-014-1380-1
Jeganathan, S., Hascher, A., Chinnathambi, S., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (2008). Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076. doi: 10.1074/jbc.M805300200
Jeganathan, S., von Bergen, M., Brutlach, H., Steinhoff, H. J., and Mandelkow, E. (2006). Global hairpin folding of tau in solution. Biochemistry 45, 2283–2293. doi: 10.1021/bi0521543
Jellinger, K. A., Alafuzoff, I., Attems, J., Beach, T. G., Cairns, N. J., Crary, J. F., et al. (2015). PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 129, 757–762. doi: 10.1007/s00401-015-1407-2
Josephs, K. A., Murray, M. E., Whitwell, J. L., Parisi, J. E., Petrucelli, L., Jack, C. R., et al. (2014a). Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol. 127, 441–450. doi: 10.1007/s00401-013-1211-9
Josephs, K. A., Whitwell, J. L., Weigand, S. D., Murray, M. E., Tosakulwong, N., Liesinger, A. M., et al. (2014b). TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 127, 811–824. doi: 10.1007/s00401-014-1269-z
Kanaan, N. M., Cox, K., Alvarez, V. E., Stein, T. D., Poncil, S., and McKee, A. C. (2016). Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 19–34. doi: 10.1093/jnen/nlv001
Kanaan, N. M., Morfini, G. A., LaPointe, N. E., Pigino, G. F., Patterson, K. R., Song, Y., et al. (2011). Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 31, 9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011
Kawamoto, Y., Akiguchi, I., Nakamura, A., and Budka, H. (2004). 14-3-3 proteins in Lewy body-like hyaline inclusion in patients with sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 108, 531–537. doi: 10.1007/s00401-004-0923-2
Keller, B. A., Volkening, K., Droppelmann, C. A., Ang, L. C., Rademakers, R., and Strong, M. J. (2012). Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. 124, 733–747. doi: 10.1007/s00401-012-1035-z
Kenessey, A., and Yen, S. H. (1993). The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 629, 40–46. doi: 10.1016/0006-8993(93)90478-6
Klein, C., Kramer, E. M., Cardine, A. M., Schraven, B., Brandt, R., and Trotter, J. (2002). Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 22, 698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002
Kondo, A., Shahpasand, K., Mannix, R., Qiu, J., Moncaster, J., Chen, C. H., et al. (2015). Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436. doi: 10.1038/nature14658
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lee, G., Newman, S. T., Gard, D. L., Band, H., and Panchamoorthy, G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111 (Pt 21), 3167–3177.
Lee, T. H., Chen, C. H., Suizu, F., Huang, P., Schiene-Fischer, C., Daum, S., et al. (2011). Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol. Cell 42, 147–159. doi: 10.1016/j.molcel.2011.03.005
Lesort, M., Jope, R. S., and Johnson, G. V. (1999). Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3β and Fyn tyrosine kinase. J. Neurochem. 72, 576–584. doi: 10.1046/j.1471-4159.1999.0720576.x
Lillo, P., Garcin, B., Hornberger, M., Bak, T. H., and Hodges, J. R. (2010). Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch. Neurol. 67, 826–830. doi: 10.1001/archneurol.2010.146
Lim, J., Balastik, M., Lee, T. H., Nakamura, K., Liou, Y. C., Sun, A., et al. (2008). Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Invest. 118, 1877–1889. doi: 10.1172/JCI34308
Liou, Y. C., Sun, A., Ryo, A., Zhou, X. Z., Yu, Z. X., Huang, H. K., et al. (2003). Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424, 556–561. doi: 10.1038/nature01832
Lu, K. P., Hanes, S. D., and Hunter, T. (1996). A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547. doi: 10.1038/380544a0
Lu, K. P., and Zhou, X. Z. (2007). The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916. doi: 10.1038/nrm2261
Lu, P. J., Wulf, G., Zhou, X. Z., Davies, P., and Lu, K. P. (1999). The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788. doi: 10.1038/21650
Lunetta, C., Lizio, A., Melazzini, M. G., Maestri, E., and Sansone, V. A. (2015). Amyotrophic Lateral Sclerosis Survival Score (ALS-SS): a simple scoring system for early prediction of patient survival. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 93–100. doi: 10.3109/21678421.2015.1083585
Mackenzie, I. R. (2008). The neuropathology of FTD associated with ALS. Alzheimer Dis. Assoc. Disord. 21, S44–S49. doi: 10.1097/WAD.0b013e31815c3486
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Mahoney, C. J., Downey, L. E., Ridgway, G. R., Beck, J., Clegg, S., Blair, M., et al. (2012). Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers. Res. Ther. 4:41. doi: 10.1186/alzrt144
Malaspina, A., Kaushik, N., and de Belleroche, J. (2000). A 14-3-3 mRNA is up-regulated in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 75, 2511–2520. doi: 10.1046/j.1471-4159.2000.0752511.x
McKee, A. C., Cantu, R. C., Nowinski, C. J., Hedley-Whyte, E. T., Gavett, B. E., Budson, A. E., et al. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735. doi: 10.1097/NEN.0b013e3181a9d503
McKee, A. C., Gavett, B. E., Stern, R. A., Nowinski, C. J., Cantu, R. C., Kowall, N. W., et al. (2010). TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 69, 918–929. doi: 10.1097/NEN.0b013e3181ee7d85
Meier, S. L., Charleston, A. J., and Tippett, L. J. (2010). Cognitive and behavioural deficits associated with the orbitomedial prefrontal cortex in amyotrophic lateral sclerosis. Brain 133, 3444–3457. doi: 10.1093/brain/awq254
Mez, J., Daneshvar, D. H., Kiernan, P. T., Abdolmohammadi, B., Alvarez, V. E., Huber, B. R., et al. (2017). Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318, 360–370. doi: 10.1001/jama.2017.8334
Migheli, A., Pezzulo, T., Attanasio, A., and Schiffer, D. (1993). Peripherin immunoreactive structures in amyotrophic lateral sclerosis. Lab. Invest. 68, 185–191.
Moisse, K., Mepham, J., Volkening, K., Welch, I., Hill, T., and Strong, M. J. (2009a). Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL (-/-) mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 1296, 176–186. doi: 10.1016/j.brainres.2009.07.023
Moisse, K., Volkening, K., Leystra-Lantz, C., Welch, I., Hill, T., and Strong, M. J. (2009b). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy. Brain Res. 1249, 202–211. doi: 10.1016/j.brainres.2008.10.021
Montuschi, A., Iazzolino, B., Calvo, A., Moglia, C., Lopiano, L., Restagno, G., et al. (2015). Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J. Neurol. Neurosurg. Psychiatry 86, 168–173. doi: 10.1136/jnnp-2013-307223
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Moszcyznski, A., Gopaul, J., McCunn, P., Volkening, K., Harvey, M., Bartha, R., et al. (2017). ALSci associated tau pathological phosphorylation of Thr175 induces a tau proteinopathy in vivo. Amyotroph. Lateral Scler. Frontotemporal Degener. 18:143.
Moszczynski, A. J., Gohar, M., Volkening, K., Leystra-Lantz, C., Strong, W., and Strong, M. J. (2015). Thr175-phosphorylated tau induces pathologic fibril formation via GSK3beta-mediated phosphorylation of Thr231 in vitro. Neurobiol. Aging 36, 1590–1599. doi: 10.1016/j.neurobiolaging.2014.12.001
Moszczynski, A. J., Strong, W., Xu, K., McKee, A., Brown, A., and Strong, M. J. (2018). Pathological Thr175 tau phosphorylationin CTE and CTE with ALS. Neurology 90, 1–8. doi: 10.1212/WNL.0000000000004899
Moszczynski, A. J., Yang, W., Hammond, R., Ang, L. C., and Strong, M. J. (2017). Threonine175, a novel pathological phosphorylation site on tau protein linked to multiple tauopathies. Acta Neuropathol. Commun. 5:6. doi: 10.1186/s40478-016-0406-4
Nakamura, K., Greenwood, A., Binder, L., Bigio, E. H., Denial, S., Nicholson, L., et al. (2012). Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149, 232–244. doi: 10.1016/j.cell.2012.02.016
Nakamura, S., Wate, R., Kaneko, S., Ito, H., Oki, M., Tsuge, A., et al. (2014). An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation. Neuropathology 34, 58–63. doi: 10.1111/neup.12049
Namgung, U., and Xia, Z. (2000). Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J. Neurosci. 20, 6442–6451. doi: 10.1523/JNEUROSCI.20-17-06442.2000
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/WNL.51.6.1546
Neary, D., Snowden, J. S., Mann, D. M., Northern, B., Goulding, P. J., and MacDermott, N. (1990). Frontal lobe dementia and motor neuron disease. J. Neurol. Neurosurg. Psychiatry 53, 23–32. doi: 10.1136/jnnp.53.1.23
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Oh, S. I., Park, A., Kim, H. J., Oh, K. W., Choi, H., Kwon, M. J., et al. (2014). Spectrum of cognitive impairment in Korean ALS patients without known genetic mutations. PLoS ONE 9:e87163. doi: 10.1371/journal.pone.0087163
Olney, R. K., Murphy, J., Forshew, D., Garwood, E., Miller, B. L., Langmore, S., et al. (2005). The effects of executive and behavioral dysfunction on the course of ALS. Neurology 65, 1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b
Orrell, R. W., King, A. W., Hilton, D. A., Campbell, M. J., Lane, R. J., and de Belleroche, J. S. (1995). Familial amyotrophic lateral sclerosis with a point mutation of SOD-1: intrafamilial heterogeneity of disease duration associated with neurofibrillary tangles. J. Neurol. Neurosurg. Psychiatry 59, 266–270. doi: 10.1136/jnnp.59.3.266
Pastorino, L., Sun, A., Lu, P. J., Zhou, X. Z., Balastik, M., Finn, G., et al. (2006). The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440, 528–534. doi: 10.1038/nature04543
Phukan, J., Elamin, M., Bede, P., Jordan, N., Gallagher, L., Byrne, S., et al. (2012). The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J. Neurol. Neurosurg. Psychiatry 83, 102–108. doi: 10.1136/jnnp-2011-300188
Poletti, M., Enrici, I., and Adenzato, M. (2012). Cognitive and affective theory of mind in neurodegenerative diseases: neuropsychological, neuroanatomical and neurochemical levels. Neurosci. Biobehav. Rev. 36, 2147–2164. doi: 10.1016/j.neubiorev.2012.07.004
Prudlo, J., Bißbort, C., Glass, A., Grossmann, A., Hauenstein, K., Benecke, R., et al. (2012). White matter pathology in ALS and lower motor neuron ALS variants: a diffusion tensor imaging study using tract-based spatial statistics. J. Neurol. 259, 1848–1859. doi: 10.1007/s00415-012-6420-y
Rascovsky, K., Hodges, J. R., Kipps, C. M., Johnson, J. K., Seeley, W. W., Mendez, M. F., et al. (2007). Diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD): current limitations and future directions. Alzheimer Dis. Assoc. Disord. 21, S14–S18. doi: 10.1097/WAD.0b013e31815c3445
Reynolds, C. H., Betts, J. C., Blackstock, W. P., Nebreda, A. R., and Anderton, B. H. (2000). Phosphorylation sites on tau identified by nanoelectrospray mass spectroscopy: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3β. J. Neurochem. 74, 1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x
Rohrer, J. D., Nicholas, J. M., Cash, D. M., van Swieten, J., Dopper, E., Jiskoot, L., et al. (2015). Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 14, 253–262. doi: 10.1016/S1474-4422(14)70324-2
Shamay-Tsoory, S. G., Aharon-Peretz, J., and Perry, D. (2009). Two systems for empathy: a double dissociation between emotional and cognitive empathy in inferior frontal gyrus versus ventromedial prefrontal lesions. Brain 132, 617–627. doi: 10.1093/brain/awn279
Smith, E. E. (2017). Clinical presentations and epidemiology of vascular dementia. Clin. Sci. 131, 1059–1068. doi: 10.1042/CS20160607
Soma, K., Fu, Y. J., Wakabayashi, K., Onodera, O., Kakita, A., and Takahashi, H. (2012). Co-occurrence of argyrophilic grain disease in sporadic amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 38, 54–60. doi: 10.1111/j.1365-2990.2011.01175.x
Spires-Jones, T. L., Attems, J., and Thal, D. R. (2017). Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134, 187–205. doi: 10.1007/s00401-017-1709-7
Strong, M. J. (2001). The evidence for ALS as a multisystems disorder of limited phenotypic expression. Can. J. Neurol. Sci. 28, 283–298. doi: 10.1017/S0317167100001505
Strong, M. J. (2003). The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol. Ther. 98, 379–414. doi: 10.1016/S0163-7258(03)00040-8
Strong, M. J. (2008). The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9, 323–338. doi: 10.1080/17482960802372371
Strong, M. J. (2017). Revisiting the concept of amyotrophic lateral sclerosis as a multisystems disorder of limited phenotypic expression. Curr. Opin. Neurol. 30, 599–607. doi: 10.1097/WCO.0000000000000488
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Strong, M. J., Grace, G. M., Freedman, M., Lomen-Hoerth, C., Woolley, S., Goldstein, L. H., et al. (2009). Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 131–146. doi: 10.1080/17482960802654364
Strong, M. J., Grace, G. M., Orange, J. B., and Leeper, H. A. (1996). Cognition, language and speech in amyotrophic lateral sclerosis: a review. J. Clin. Exp. Neuropsychol. 18, 291–303. doi: 10.1080/01688639608408283
Strong, M. J., Grace, G. M., Orange, J. B., Leeper, H. A., Menon, R. S., and Aere, C. (1999). A prospective study of cognitive impairment in ALS. Neurology 53, 1665–1670. doi: 10.1212/WNL.53.8.1665
Strong, M. J., Kesavapany, S., and Pant, H. C. (2005). The pathobiology of amyotrophic lateral sclerosis: a proteinopathy? J. Neuropathol. Exp. Neurol. 64, 649–664. doi: 10.1097/01.jnen.0000173889.71434.ea
Strong, M. J., Yang, W., Strong, W. L., Leystra-Lantz, C., Jaffe, H., and Pant, H. C. (2006). Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology 66, 1770–1771. doi: 10.1212/01.wnl.0000218161.15834.db
Sultana, R., Boyd-Kimball, D., Poon, H. F., Cai, J., Pierce, W. M., Klein, J. B., et al. (2006). Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol. Aging 27, 918–925. doi: 10.1016/j.neurobiolaging.2005.05.005
Takeuchi, R., Toyoshima, Y., Tada, M., Tanaka, H., Shimizu, H., Shiga, A., et al. (2016). Globular glial mixed four repeat tau and TDP-43 proteinopathy with motor neuron disease and frontotemporal dementia. Brain Pathol. 26, 82–94. doi: 10.1111/bpa.12262
Tan, R. H., Yang, Y., and Halliday, G. M. (2017). Multiple neuronal pathologies are common in young patients with pathologically proven Frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. doi: 10.1111/nan.12455. [Epub ahead of print].
Tran, H. T., Sanchez, L., and Brody, D. L. (2012). Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J. Neuropathol. Exp. Neurol. 71, 116–129. doi: 10.1097/NEN.0b013e3182456aed
Turner, M. R., and Verstraete, E. (2015). What does imaging reveal about the pathology of amyotrophic lateral sclerosis? Curr. Neurol. Neurosci. Rep. 15:45. doi: 10.1007/s11910-015-0569-6
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
van der Hulst, E. J., Bak, T. H., and Abrahams, S. (2015). Impaired affective and cognitive theory of mind and behavioural change in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 86, 1208–1215. doi: 10.1136/jnnp-2014-309290
Vintilescu, C. R., Afreen, S., Rubino, A. E., and Ferreira, A. (2016). The Neurotoxic TAU45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol. Med. 22. doi: 10.2119/molmed.2016.00095. [Epub ahead of print].
Volkening, K., Leystra-Lantz, C., and Strong, M. J. (2010). Human low molecular weight neurofilament (NFL) mRNA interacts with a predicted p190RhoGEF homologue (RGNEF). Amyotroph. Lateral Scler. 11, 97–103. doi: 10.3109/17482960902995584
Volkening, K., Strong, W. L., Seaton, S., Yang, W., and Strong, M. J. (2017). C9orf72 mutations do not influence the tau signature of amyotrophic lateral sclerosis with cognitive impairment (ALSci). Amyotroph. Lateral Scler. Frontotemporal. Degener. 18, 549–554. doi: 10.1080/21678421.2017.1332075
Walhout, R., Schmidt, R., Westeneng, H. J., Verstraete, E., Seelen, M., van Rheenen, W., et al. (2015). Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology 85, 1780–1788. doi: 10.1212/WNL.0000000000002135
Ward, S. M., Himmelstein, D. S., Lancia, J. K., and Binder, L. I. (2012). Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 40, 667–671. doi: 10.1042/BST20120134
Wilson, C. M., Grace, G. M., Munoz, D. G., He, B. P., and Strong, M. J. (2001). Cognitive impairment in sporadic ALS. A pathological continuum underlying a multisystem disorder. Neurology 57, 651–657. doi: 10.1212/WNL.57.4.651
Yang, W., Ang, L. C., and Strong, M. J. (2005). Tau protein aggregation in the frontal and entorhinal cortices as a function of aging. Dev. Brain Res. 156, 127–138. doi: 10.1016/j.devbrainres.2005.02.004
Yang, W., Leystra-Lantz, C., and Strong, M. J. (2008). Upregulation of GSK3β expression in frontal and temporal cortex of ALS with cognitive impairment (ALSci). Brain Res. 1196, 131–139. doi: 10.1016/j.brainres.2007.12.031
Yang, W., Sopper, M. M., Leystra-Lantz, C., and Strong, M. J. (2003). Microtubule-associated tau protein positive neuronal and glial inclusions in amyotrophic lateral sclerosis. Neurology 61, 1766–1773. doi: 10.1212/01.WNL.0000099372.75786.F8
Keywords: amyotrophic lateral sclerosis, chronic traumatic encephalopathy, frontotemporal dementia, TDP-43, microtubule associated tau protein
Citation: Moszczynski AJ, Hintermayer MA and Strong MJ (2018) Phosphorylation of Threonine 175 Tau in the Induction of Tau Pathology in Amyotrophic Lateral Sclerosis—Frontotemporal Spectrum Disorder (ALS-FTSD). A Review. Front. Neurosci. 12:259. doi: 10.3389/fnins.2018.00259
Received: 14 January 2018; Accepted: 04 April 2018;
Published: 20 April 2018.
Edited by:
Gen Sobue, Nagoya University, JapanReviewed by:
Jürgen Götz, The University of Queensland, AustraliaRyuichi Harada, Tohoku University, Japan
Copyright © 2018 Moszczynski, Hintermayer and Strong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Strong, bXN0cm9uZ0B1d28uY2E=
†These authors have contributed equally to this work.