Abstract
Autistic Spectrum Disorder (ASD) is a complex neurodevelopmental brain disorder characterized by two core behavioral symptoms, namely impairments in social communication and restricted/repetitive behavior. The molecular mechanisms underlying ASD are not well understood. Recent genetic as well as non-genetic animal models contributed significantly in understanding the pathophysiology of ASD, as they establish autism-like behavior in mice and rats. Among the genetic causes, several chromosomal mutations including duplications or deletions could be possible causative factors of ASD. In addition, the biochemical basis suggests that several brain neurotransmitters, e.g., dopamine (DA), serotonin (5-HT), gamma-amino butyric acid (GABA), acetylcholine (ACh), glutamate (Glu) and histamine (HA) participate in the onset and progression of ASD. Despite of convincible understanding, risperidone and aripiprazole are the only two drugs available clinically for improving behavioral symptoms of ASD following approval by Food and Drug Administration (FDA). Till date, up to our knowledge there is no other drug approved for clinical usage specifically for ASD symptoms. However, many novel drug candidates and classes of compounds are underway for ASD at different phases of preclinical and clinical drug development. In this review, the diversity of numerous aetiological factors and the alterations in variety of neurotransmitter generation, release and function linked to ASD are discussed with focus on drugs currently used to manage neuropsychiatric symptoms related to ASD. The review also highlights the clinical development of drugs with emphasis on their pharmacological targets aiming at improving core symptoms in ASD.
Introduction
Autistic spectrum disorder (ASD) is a biologically based neurodevelopmental disorder affecting two major core behavioral symptoms, namely impairments in social skills and restricted /repetitive behavioral pattern or interest of ASD patients (Baronio et al., 2015). These core symptoms can be observed before the age of three years and are lasting for the whole lifetime (Andres, 2002). ASD has become high priority for scientists and health care providers, and has also attracted the public attention because of reported increase in its prevalence (Sheldrick and Carter, 2018; Xu et al., 2018). The worldwide estimated prevalence of individuals with ASD diagnosis is strikingly high with prevalence varying across numerous studies, but it is estimated that one in 160 children has an ASD worldwide, and it is expected to increase globally (Arvidsson et al., 2018). Despite its increasing prevalence, the pathophysiology of ASD is still understood incompletely, and this can be attributed to challenges in identifying suitable animal models and the complexity of the neurobiology in brain function (Nestler and Hyman, 2010). Several evidences suggest that strong genetic and environmental factors rise the occurrence of ASD in childhood (Baronio et al., 2015). To date, there are no efficient therapeutic interventions that target the core symptoms of ASD, namely social communication impairments and restricted/repetitive behavior (Sheldrick and Carter, 2018; Xu et al., 2018). However, pharmacological interventions may be used to provide symptomatic control of associated comorbidities but not to treat core deficits (Wong and Smith, 2006; Hanson et al., 2007). Notably, it is nowadays believed that ASD and schizophrenia (SCH) are conceptualized as two separate disorders, despite the fact that both ASD and SCH were found to share multiple aetiologies, phenotypic feature similarities, and risk factors, and were reported to co-occur at elevated rates (Chisholm et al., 2015).
Etiology of ASD
ASD is broadly considered to be a multi-factorial disorder that results from genetic as well as non-genetic risk factors. There is cumulative evidence for the involvement of genetic factors in the etiology of ASD, since siblings born in families with ASD are at 35–40% greater risk to develop ASD and with an increase in the current rate of approximately 1% from a rate of 0.05% in 1970s (Stubbs et al., 2016). Moreover, genetic studies revealed that alteration in the developmental pathways of neuronal and axonal structures that are strongly involved in synaptogenesis emerge from single gene mutations (Geschwind, 2011; Voineagu et al., 2011; Chang et al., 2015). It is likely that interactions between multiple genes, and variability in expression as a result of epigenetic factors and exposure to environmental factors are responsible for ASD (Muhle et al., 2004). In a previous clinical study involving a twin, it was appraised that the risk of developing ASD was 35–40% due to genetic variability, and the remaining 60% was contributed to by prenatal, perinatal, and postnatal environmental factors (Hallmayer et al., 2011). Accordingly, environmental factors implicated with ASD included prenatal and perinatal complications (Glasson et al., 2004; Maramara et al., 2014), birth and neonatal complications (Gardener et al., 2011; Guinchat et al., 2012), viral infection, autoimmune diseases, and exposure to teratogens and maternal anticonvulsants such as valproic acid (VPA) (Kern and Jones, 2006; Kolevzon et al., 2007). Therefore, an increased understanding of the interface between genetic and environmental factors in the pathogenesis of ASD may lead to an optimized therapeutic strategy.
Correlation of neurotransmitters dysfunction to ASD
Research has also focused on the study of neurotransmitters, in search of sensitive and specific markers of ASD as well as potential therapeutic interventions. The role of several central neurotransmitters (e.g., 5-HT, ACh, DA, GABA and Glu) in initial brain development may substantiate to be a significant area in studying the etiology of ASD. Certain disruption of brain neurotransmissions early during the development phase of the CNS may demonstrate early pharmacological intervention that helps to cure and maybe even preclude some of the severe behavioral symptoms of ASD. Ideally, the work in genetics may be able to explain these neurochemical defects at birth, providing possible appropriate medical treatment for infants who are at increased risk for ASD. This would completely exhibit new therapeutic tactic to the clinical control of ASD. Growing evidences suggest that a variety of several neurotransmitter systems such as ACh, 5-HT, DA, GABA, Glu, and HA are implicated in the onset and progression of ASD -along with genetic and environmental factors discussed below- (Shah and Wing, 2006; Bacchelli et al., 2015; Ellenbroek and Ghiabi, 2015; Wang et al., 2015; Chen et al., 2017; Hellings et al., 2017; Hellmer and Nystrom, 2017; Naaijen et al., 2017; Nakai et al., 2017; Paval, 2017; Paval et al., 2017; Figure 1).
Figure 1
Serotonin
Among all neurotransmitters investigated so far in ASD, 5-HT has motivated the most research efforts and investigations. 5-HT signaling facilitates several neural processes including neurogenesis, cell migration and survival, synaptogenesis, and synaptic plasticity. Interestingly, high 5-HT levels in the blood have been described for up to 45% of tested ASD subjects(Chen et al., 2015, 2017; Ellenbroek et al., 2016). Moreover, preclinical investigations using ASD-like animal models reported that hyperserotonemia significantly reduced the motivation for social interest through inhibition of separation distress, potentially accounting for the social impairments found in ASD individuals (Ellenbroek et al., 2016; Nakai et al., 2017). Furthermore, 5-HT was found to accumulate mainly in platelets utilizing the specific 5-HT transporter. In line with these findings, genetic studies of linkage stated that the 17q11.2 region containing the 5-HT transporter gene SLC6A4 polymorphisms appear to be associated with ASD, as Gly56 conversion to Ala56 in the transporter protein resulted in autistic phenotypic features combined with an amplified p38-mitogen-activated protein kinases (MAPK)-sensitive basal phosphorylation process. In addition and in a previous study, higher clearance rates of hippocampal 5-HT were observed and hence hyperserotonemia, which led to a significant hypersensitivity of brain 5-HT(1A) as well as 5-HT(2A) receptors, social impairment and repetitive behavior (Veenstra-VanderWeele et al., 2012; Figure 1).
Dopamine
Dopamine (DA) plays a fundamental role in brain functioning, and the pathophysiological role of dopaminergic system (DS) deficits in ASD is well recognized, with the wide clinical use of antipsychotics that mainly target the D2 receptors (D2Rs)(Seeman, 2010; Baronio et al., 2014) Interestingly, and in a very recent preclinical study, it has been shown that mice with increased dopaminergic neurotransmission in the dorsal striatum via the suppression of dopamine transporter expression in substantia nigra neurons or the optogenetic stimulation of the nigro-striatal circuitry exhibited significant deficits in sociability and repetitive behaviors relevant to ASD pathology in several rodent models, while these behavioral changes were blocked by using D1R antagonists (Lee et al., 2017). Therefore, D1R agonists produced typical autistic-like behaviors in normal mice or the genetic knockout (KO) of D2Rs (Lee et al., 2017). Furthermore, the siRNA-mediated inhibition of D2Rs in the dorsal striatum was shown to replicate ASD-like phenotypes in D2R KO mice (Lee et al., 2017). With regard to the DS, genetic studies have demonstrated that mutations of DS-associated genes such as the DA transporter (DAT) (Hamilton et al., 2013), DA receptors (Hettinger et al., 2008; Qian et al., 2013), and enzymes of DA biosynthesis (Nguyen et al., 2014) are implicated in ASD. These studies expanded the evidences of genetically linking between DA transporter and ASD (Hamilton et al., 2013; Bowton et al., 2014). ASD is strongly associated with a mutation in the DA transporter gene SLC6A3, which codes for a protein that contributes to regulation of DA levels in the brain. In fact, DAT is the crucial regulator of DA homeostasis and alteration of this homeostasis as a consequence of DAT dysfunction is associated with ASD and other neuropsychiatric conditions (Hamilton et al., 2013; Hellings et al., 2017; Paval, 2017; Paval et al., 2017). Moreover, it has been found that the dopaminergic fibers arising from the ventral tegmental area (VTA) project to the prefrontal cortex (PFC) and to regions of the limbic system, such as the nucleus accumbens (NAcc), forming the mesocorticolimbic (MCL) circuit. This circuit was found to be involved in high order brain functions, such as emotional social behavior, reward, motivation and cognition (Paval et al., 2017) (Hellings et al., 2017; Figure 1). Notably, the DS has been related to behavioral skills belonging to executive functioning, such as analyzing, planning and prioritizing (Seeman, 2010; Baronio et al., 2014). In line with that, children diagnosed with ASD were found to show deficits in tasks linked to executive functioning, including response/selection, planning/working memory and flexibility (Hellings et al., 2017; Paval et al., 2017). Moreover, the DS has strongly been linked to social behavior, attentional skills and perception, and motor activity, while development abnormalities in these areas have all been linked to ASD as well (Hellings et al., 2017; Paval et al., 2017).
GABA and glutamate
Gabaminergic as well as glutaminergic systems are also proposed as potential mechanisms for ASD. Consequently, mutations in the respective synaptic proteins would lead to defective neurotransmissions at excitatory and inhibitory synapses, leading to disruption of excitatory-inhibitory balance of neurotransmissions in postsynaptic neurons, a key mechanism which has strongly been associated with ASD (Jamain et al., 2002; Naaijen et al., 2017). In addition to numerous reports stating the duplication of 15q11-13 locus in ASD populations, a majority of the cases do not show mutations at this locus. However, they do exhibit abnormalities in the expression of protein encoded by 15q11-13 gene, as reported. These results demonstrated the involvement of 15q11-13 locus in ASD, either directly by mutation or indirectly by epigenetic factors (Hogart et al., 2007; Nakai et al., 2017). This site employs numerous genes coding for particular subunits of GABA receptors, namely GABRB3, GABRA5 and GABRG3. There are numerous signaling and scaffolding proteins which are implicated in normal development and GABA synapse function. Therefore, mutations in the genes encoding these proteins consequently result in GABAergic dysfunction, hence inhibitory signaling deficits. For example, deletions in gene encoding a protein contactin-associated protein 2 (CNTNAP2) has been linked to ASD (Stephan, 2008; Gregor et al., 2011; Nord et al., 2011). Moreover, deficiency in CNTNAP2 results in reduction in GAD1, parvalbumin and inhibitory interneurons along with impairments in inhibitory signaling (Peñagarikano et al., 2011; Soghomonian et al., 2017). This evidence suggests an association and the relation between mutations affecting the function of GABA and ASD. Furthermore, reduced expression of GABAergic genes and lower density of GABA related proteins have been found in individual brains of ASD patients. Consequently, alterations in GABA-A receptor genes and other genes expressed on GABA interneurons, as well as GABA biosynthesis enzymes (GAD 65 and GAD 67) in the cerebellum and parietal cortex, are strongly implicated in ASD (Fatemi et al., 2002; Coghlan et al., 2012). On the contrary, an increased level in the expression of excitatory Glu receptor AMPA, and of Glu transporter proteins were observed in individuals diagnosed with ASD, with highest expression abnormalities found in the cerebellum (Purcell et al., 2001; Figure 1). Based on the aforementioned, altered expression of genes related to GABA and/or Glu might be linked to several ASD phenotypic features including cognitive deficits and/or hyperactivity (Jamain et al., 2002; Naaijen et al., 2017). Moreover, abnormalities in both neurotransmitters including an increase in the excitatory Glu and/or decrease in the inhibitory GABA were found to lead to epileptic seizure, a clinical feature which is commonly observed in ASD patients (Ballaban-Gil and Tuchman, 2000; Levisohn, 2007; Gillberg et al., 2017).
Acetylcholine
The brain cholinergic neurotransmission system with ACh plays an essential role in regulating ASD-related behavioral symptoms including attention (Arnold et al., 2002), cognitive flexibility (Ragozzino et al., 1998), social interaction (Avale et al., 2011) and stereotypical behaviors (McConville et al., 1992; Bacchelli et al., 2015; Wang et al., 2015; Hellmer and Nystrom, 2017). Mounting evidences suggest the involvement of cholinergic system dysfunction in the phenotypic outcomes of ASD-related behavioral features, in both humans and animal models - (Karvat and Kimchi, 2014). In ASD patients, there are remarkable abnormalities in the cholinergic system. Anatomically there is irregularity in the number and structure of neurons in a basal forebrain cholinergic nucleus of patients diagnosed with ASD (Kemper and Bauman, 1998). Also, a decrease in the level of choline, the precursor of the neurotransmitter ACh and the agonist for nicotinic-cholinergic receptor (nAChR), was determined in individuals diagnosed with ASD (Friedman et al., 2006), with the severity of ASD related to the decreased level of cytosolic ACh (Sokol et al., 2002). Moreover, and by using immunohistochemical analyses, abnormalities in nAChR were observed in several brain regions (e.g., neocortex, cerebellum, thalamus and striatum) of ASD patients, specifically with the major abnormalities being reduction in nAChR subunits and muscarinic receptors (M1 type) (Mukaetova-Ladinska, 2017). Moreover, the brain ACh like several other brain neurotransmitters, e.g., DA, 5-HT and GABA, playing a key role in ASD are regulated by different central mechanisms including histamine H3 hetero-receptors (H3Rs), highlighting the attention to brain histaminergic system involvement in the ASD (Sadek and Stark, 2015; Sadek et al., 2016; Alachkar et al., 2017; Eissa et al., 2018). Genetically, studies reported duplications and mutations in CHRNA7, the gene encoding the α7nAChR subunit, in ASD patients (Mikhail et al., 2011; Leblond et al., 2012), whereas deletion of the CHRNB2 gene, encoding the β2-subunit of nAChR was observed in other cases (Granon et al., 2003). Furthermore, ASD related behavior may be linked to M1 type mAChR inhibition (McCool et al., 2008) and cholinergic cell damage (Walker et al., 2007; Figure 1). Consequently, social deficits and repetitive behaviors are the main phenotypic ASD features connected to disruption in cholinergic neurotransmission system (Wang et al., 2015). Also, reduced attention (Arnold et al., 2002), decreased cognitive flexibility (Ragozzino et al., 1998), reduced social communications (Avale et al., 2011) and conventional behaviors have been strongly linked to cholinergic neurotransmission dysfunction (McConville et al., 1992; Bacchelli et al., 2015; Wang et al., 2015; Hellmer and Nystrom, 2017).
Histamine
The brain histaminergic system was found to display a critical role in cognition, sleep and other neuropsychiatric disorders including schizophrenia (SCH) and Tourette syndrome that share comorbidity with ASD (Wright et al., 2017). Moreover, alteration in gene expression was found for histamine-N-methyltransferase enzyme (HNMT, enzyme responsible for metabolism of central histamine (HA) and for histamine receptor (HR) subtypes H1-, H2-, and H3R (Wright et al., 2017). Notably, there has in the last two decades been a rising interest in studying the role of brain HA on behaviors in both physiological conditions and psychiatric diseases, e.g., SCH (Sadek and Stark, 2015; Sadek et al., 2016). Interestingly and as revealed in several clinical reports, patients with SCH shared a variety of common symptoms and genetic factors with ASD (Konstantareas and Hewitt, 2001; Carroll and Owen, 2009). Moreover, a significant role for the brain HA has been projected, and a range of several H3R ligands has been developed so far for dual-targeting of both dopaminergic as well as histaminergic neurotransmissions (Bishara, 2010; Baronio et al., 2014). Furthermore and as discussed above, deficits in regulations of various other neurotransmitters including DA, 5-HT, GABA, and Glu are postulated (Witkin and Nelson, 2004). Therefore, the role of central HA that influences behavior in CNS disorder sheds light on the histaminergic system as pharmacological target for therapeutic purposes (Haas et al., 2008; Tiligada et al., 2011; Shan et al., 2012; Naddafi and Mirshafiey, 2013). Interestingly, brain H3Rs act as auto-receptors or hetero-receptors that regulates the biosynthesis and release of HA and several other neurotransmitters, which consequently play a role in cognitive and homeostatic processes, as shown in Figures 1, 2. Therefore, there is an indirect indication that histaminergic neurotransmission may have a significant role in SCH and that potent and selective H3R antagonists could lead to therapeutic improvements of cognitive symptoms associated with SCH and ASD (Witkin and Nelson, 2004; Esbenshade et al., 2008; von Coburg et al., 2009; Brown et al., 2013; Sadek et al., 2016). To date there are scarce literature studies concerning the association of H3R antagonists and treatment of ASD behavioral deficits. Accordingly and in a previous preclinical study, the imidazole-based H3R antagonists, namely thioperamide and ciproxifan, improved the decreased prepulse inhibition in an animal model of SCH (Brown et al., 2013; Figure 2, Table 4). Thioperamide and ciproxifan are selective and potent imidazole-based H3R antagonists that are widely used in preclinical animal experiments (Ligneau et al., 1998; Stark et al., 2000). However, the preclinical use of numerous non-imidazole-based H3R antagonists, e.g., ABT-239 and A-431404, ameliorated ketamine- and/or MK-801-induced cognitive impairments in experimental rats, demonstrating enhanced results when compared with reference antipsychotics like risperidone or olanzapine (Brown et al., 2013; Figure 2, Table 4). Interestingly, H3R antagonists possess antioxidant activity which could increase their potential clinical use, since oxidative stress is, also, considered as possible predictors of intensified symptoms of SCH and ASD (Mahmood et al., 2012). Moreover, a study considering the effectiveness of the non-imidazole H3R antagonist ABT-288 in the treatment of cognitive deficits linked with SCH revealed that schizophrenic features remained constant during the whole time period of the study, with acceptable safety, tolerability and pharmacokinetics profile of ABT-288 at a 15-fold higher dose and 12-fold higher exposures in subjects with SCH than previously observed in healthy volunteers (Coruzzi et al., 2012) (Hsieh et al., 2010). Moreover, the use of a H3R antagonist ameliorated behavioral impairments in an animal model of SCH, including spatial working memory deficit, an abnormality which also characterizes patients with ASD (Steele et al., 2007). Also, a recent preclinical study reported that an acute systemic administration of ciproxifan palliated some sociability impairments and stereotypic behavior in an animal model of VPA-induced ASD in mice (Baronio et al., 2015). However, further research efforts are still necessary to magnify these observed initial data and to achieve enhanced understanding of pathophysiology and therapeutic management of ASD. Based on the aforementioned observations, numerous brain neurotransmitters, e.g., 5-HT, DA, GABA, Glu, ACh, and HA appear to be implicated in the pathophysiology of ASD, since disruption of genes encoding different proteins (receptors and/or catalyzing biosynthesis enzymes for respective neurotransmitter) that affect the respective neurotransmission functionality leads to a variety of phenotypic features of ASD. Interestingly, the above discussed dysfunctions of neurotransmitters, namely 5-HT, DA, GABA, Glu, ACh, and HA were, also, found to be influentially implicated in the clinical outcome features of SCH, a disorders that is comorbid and shares multiple etiologies and risk factors with ASD (Chisholm et al., 2015; Devor et al., 2017).
Figure 2
Current pharmacological intervention
Based on the aforementioned abnormalities in genes as well as various neurotransmitter systems, studying the effects of a given drug on core symptoms in ASD is very challenging. Despite advances in early diagnosis and intervention, efficacious reversal of core autistic symptoms is still not accomplished to date. At present time there is no definite pharmacological treatment for ASD but treatments for ASD are based on behavioral therapies and the use of highly controlled learning environments. The recent approaches to treatment of ASD set behavioral therapy and atypical language development, as keystone for ASD therapy along with other treatments that tends to ameliorate associated symptoms and not the core deficits (Sathe et al., 2017; Weitlauf et al., 2017). The heterogeneity of clinical and behavioral features in children diagnosed with ASD contributes to the difficulty in understanding the pathophysiology of this disorder, and consequently, no specific treatment can be effective for all ASD children. Therefore, subgrouping of children based on responses to intervention is essential (James et al., 2009). As mentioned above, targeting ASD core symptoms for complete and effective treatment has been challenging and not yet achieved, however several pharmacological medications maybe effective in various associated symptoms that often cause significant impairments in ASD (Volkmar et al., 1999). These associated symptoms of ASD include inattention, hyperactivity, anxiety, sleep disturbances, irritability, repetitive behavior, aggression and self-injury. Antipsychotics are often used for therapeutic management of ASD symptoms in children (Findling et al., 2004). Currently, atypical antipsychotics risperidone and aripiprazole are the only two drugs which have so far been approved by FDA for improving behavioral symptoms associated with ASD (Matson et al., 2011), however, there are several other pharmacological interventions that show effective clinical management of ASD symptoms. The most promising drugs reported for managing behavioral and neurological symptoms of ASD are acting on different brain targets and are summarized in Table 1. Accordingly, therapeutic benefits have been observed and described with several classes of drugs including selective serotonin reuptake inhibitors (sertraline, citalopram, fluoxetine) for anxiety and repetitive behaviors, psychostimulant (methylphenidate) for hyperactivity, opioid antagonist (naltrexone) for hyperactivity, and atypical antipsychotics (risperidone, olanzapine, clozapine) for temper tantrums, aggression, or self-injurious behavior (Aman, 2004; Moore et al., 2004; Kumar et al., 2012). Notably, numerous candidates of the classes discussed below have progressed to several phases of clinical trials as shown in Figure 3.
Table 1
| Class | Medication | Structure | Pharmacological effects | Side effects | Reference |
|---|---|---|---|---|---|
| Atypical Antipsychotics | Clozapine | 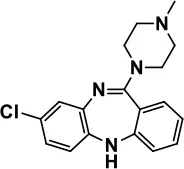 | Improves hyperactivity and aggression in ASD patients | Requires patient's hematological safety monitoring and it lowers seizure threshold | Zuddas et al., 1996; Chen et al., 2001; Gobbi and Pulvirenti, 2001; Sahoo et al., 2017 |
| Risperidone | 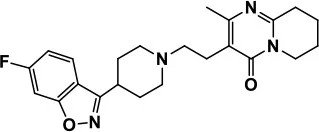 | Reduces irritability, repetitive behavior, aggression, anxiety, and depression & nervousness. It shows neuroprotective activity, modulates astroglia function, and increases the brain antioxidant activity. | Mild sedation, increased appetite, fatigue, dizziness, drowsiness, tremor, and constipation | McDougle et al., 1998b; McCracken et al., 2002; Hara et al., 2017 | |
| Aripiprazole |  | Reduces autistic symptoms in children such as irritability, stereotypy, and hyperactivity | Fatigue, vomiting, weight gain, tremor, and extrapyramidal symptoms | Marcus et al., 2009; Owen et al., 2009; Hirsch and Pringsheim, 2016 | |
| Selective Serotonin Reuptake inhibitor (SSRI) | Fluoxetine |  | Reduces stereotyped and repetitive behavior in children and adolescents with ASD. | Hypomania. agitation, and hyperactivity | Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005; Hendriksen et al., 2016 |
| Fluvoxamine | 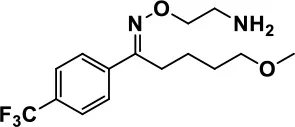 | Improves compulsive repetitive behaviors, aggression | Irritability, and increase risk of suicidal ideas | McDougle et al., 1996; Martin et al., 2003; Brown et al., 2017; Howes et al., 2018; Lee et al., 2018 | |
| Sertraline | 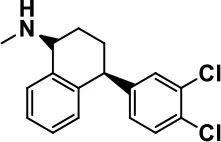 | All SSRIs possess similar effects to fluoxetine and fluvoxamine. Sertraline shows improvements in repetitive and disruptive behavior in adults with ASD, paroxetine reduced aggression, and escitalopram shows improvements in irritability, stereotypy, hyperactivity and inappropriate speech. | Hellings et al., 1996; McDougle et al., 1998a; AlOlaby et al., 2017 | ||
| Paroxetine | 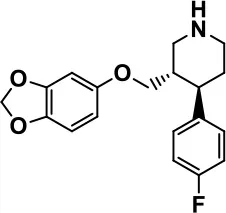 | Davanzo et al., 1998; Hellings et al., 2015 | |||
| Escitalopram | 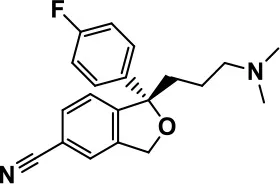 | Owley et al., 2005; Brown et al., 2017; Viktorin et al., 2017 | |||
| Venlafaxine |  | Improves restricted behavior and interest, social and communication deficits, and hyperactivity | Irritability, and increase risk of suicidal idea | Carminati et al., 2006, 2016 | |
| Tricyclic Antidepressant | Nortriptyline |  | Improves hyperactivity and aggressiveness in autistic children | Sedation, increase in aggression, irritability and hyperactivity | Kurtis, 1966; Campbell et al., 1971; Hong et al., 2017 |
| Clomipramine |  | Improves anger and compulsive and ritualized behavior | Gordon et al., 1993; Sanchez et al., 1996; Hong et al., 2017 | ||
| Anticonvulsants | Lamotrigine |  | Improves overall autistic symptoms | Life-threatening skin reactions, including Stevens-Johnson syndrome, drug reaction with eosinophilia and systemic symptoms, and toxic epidermal necrolysis | Uvebrant and Bauziene, 1994; Jobski et al., 2017 |
| Valproic acid | 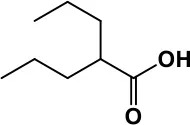 | Improves receptive language, affective instability, aggression, and social skills | Irritability, weight gain, anxiety | Anagnostou et al., 2006; Hollander et al., 2006, 2010; Jobski et al., 2017 | |
| Levetiracetam | 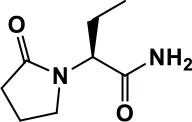 | Decreases symptoms such as hyperactivity, impulsivity, aggression, and affective lability | CNS effects such as somnolence, decreased energy, headache, dizziness, mood swings and coordination difficulties | Rugino and Samsock, 2002; Jobski et al., 2017 | |
| Glutamate antagonist | Amantadine |  | Improves hyperactivity and speech disturbance | Nervousness, anxiety, agitation, insomnia, difficulty in concentrating, and exacerbations of pre-existing seizure disorders and psychiatric symptoms in patients with schizophrenia or Parkinson's disease. | King et al., 2001; Naaijen et al., 2017 |
| Memantine |  | Improves memory, hyperactivity, irritability, social behavior and communication, and self-stimulatory behavior | Few autistic individuals experienced worsening of autistic symptoms | Owley et al., 2006; Chez et al., 2007; Naaijen et al., 2017; Vorstman et al., 2017 | |
| Acetylcholinesterase inhibitors | Rivastigmine | 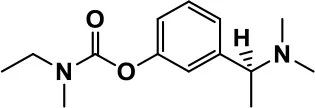 | Improves overall autistic behavior | Nausea, diarrhea, hyperactivity, and irritability | Vorstman et al., 2017 |
| Donepezil | 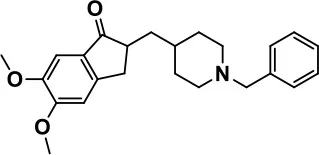 | Improves irritability and hyperactivity | Nausea and vomiting, decreased appetite and weight loss | Hardan and Handen, 2002; Vorstman et al., 2017 | |
| Galantamine | 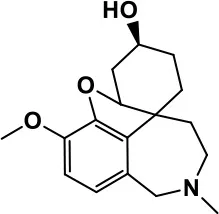 | Improves several autistic symptoms in children such as irritability, hyperactivity, social interaction deficits, inappropriate speech, loss of attention, and anger | Niederhofer et al., 2002; Nicolson et al., 2006; Vorstman et al., 2017 | ||
| Psycho-stimulant | Methylphenidate | 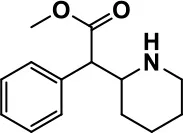 | Improves several autistic behavioral symptoms in children and adolescents such as hyperactivity, impulsivity, attention, social communication, and self-regulation | Anorexia, aggression, and insomnia | Handen et al., 2000; Di Martino et al., 2004; Jahromi et al., 2009; Kim et al., 2017 |
| Adrenergic α2receptor agonists | Clonidine | 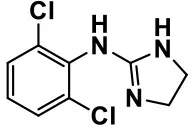 | Improves hyperactivity, mood fluctuation, aggressiveness and agitation, sleeping pattern and night time awakenings | Sedation, dry mouth, and hypotension | Fankhauser et al., 1992; Ming et al., 2008; Nash and Carter, 2016 |
| Guanfacine | 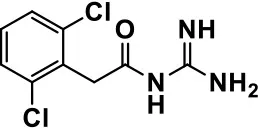 | Improves attention, hyperactivity, and tics | Insomnia, fatigue, blurred vision, mood instability, sedation, constipation, irritability, and aggression. | Posey et al., 2004; Scahill et al., 2006; Boellner et al., 2007; Nash and Carter, 2016 | |
| Opiate antagonist | Naltrexone | 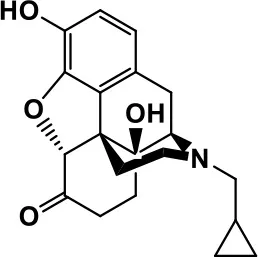 | Improves, hyperactivity, irritability, self- injuries. However, ineffective in social deficits. | Gastrointestinal complaints such as diarrhea and abdominal cramping. | Panksepp and Lensing, 1991; Bouvard et al., 1995; Kolmen et al., 1995; Elchaar et al., 2006; Clifford et al., 2007; Nash and Carter, 2016 |
Selected medications of different classes that are currently used to manage different ASD symptoms.
Data were obtained from https://clinicaltrials.gov/ last accessed 2017 August 20th.
Figure 3
Atypical antipsychotics
Clozapine belongs to the class of atypical antipsychotics because it shows capability of binding to 5-HT and DA receptors (Zuddas et al., 1996; Chen et al., 2001; Gobbi and Pulvirenti, 2001). It is an atypical antipsychotic medication mainly used for SCH that does not improve following the use of other antipsychotic medications. In patients with SCH and schizoaffective disorder it may decrease the rate of suicidal behavior. It is possibly more effective than typical antipsychotics and in patients who are resistant to other medications. Clozapine was found to improve hyperactivity and aggression in ASD children, adolescents and adults, but has a limited clinical use because of it's hematological safety profile a potential of lowering the seizure threshold in epilepsy patients, necessitating monitoring procedures of patients taking this medication (Zuddas et al., 1996; Chen et al., 2001; Gobbi and Pulvirenti, 2001). The antipsychotic medication with risperidone is mainly approached in ASD patients with SCH, bipolar disorder, and/or irritability symptoms, as this drug has revealed to be better than placebo in treating irritability, repetitive behavior, aggression, anxiety, depression and nervousness (McDougle et al., 1998b; McCracken et al., 2002). Moreover, risperidone has shown a neuroprotective effect and has enhanced the antioxidant and neuroprotective activity of astroglial cells in brain disorders such as ASD without clinical evidence of extrapyramidal side effects or seizures except mild sedative effects (McDougle et al., 1998b; McCracken et al., 2002). However, other side effects with use of risperidone were reported to include increased appetite, fatigue, dizziness and drowsiness (Table 1; McDougle et al., 1998b; McCracken et al., 2002). Aripiprazole is another atypical antipsychotic primarily recommended for the treatment of SCH and bipolar disorder. Other uses include an add-on treatment for major depressive disorder, tic disorders, and irritability associated with ASD, as it shows a different mechanism of action from those of the other atypical antipsychotics (e.g., clozapine, and risperidone) by acting rather as a partial agonist on the D2Rs and 5-HT1A receptors (Marcus et al., 2009; Owen et al., 2009; Hirsch and Pringsheim, 2016). However, it displays an antagonist profile at 5-HT2A and 5-HT7 receptors and acts as a partial agonist at the 5-HT2C receptor, both with high affinity (Marcus et al., 2009; Owen et al., 2009; Hirsch and Pringsheim, 2016). The latter action may be the reason for the minimal weight gain observed during the course of therapy with aripiprazole.
Neurotransmitter reuptake inhibitors
The class of neurotransmitter reuptake inhibitors, e.g., fluoxetine (selective serotonin reuptake inhibitor, SSRI) has shown numerous prospective therapeutic benefits, including decreases in rituals, stereotyped and repetitive monotonous behaviors in ASD children and adolescents (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Fluoxetine belongs to the class of SSRI which does not significantly inhibit norepinephrine and dopamine reuptake at therapeutic doses (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Fluoxetine was found to produce some adverse effects including disinhibition, hypomania, agitation, and hyperactivity (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Fluvoxamine is a drug which functions as a SSRI and σ1 receptor agonist (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Fluvoxamine is used mainly for the treatment of obsessive-compulsive disorder, and is also used to treat major depressive disorder and anxiety disorders such as panic disorder and post-traumatic stress disorder. Notably, fluvoxamine is approved to treat social anxiety disorder (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Fluvoxamine has also shown similar potential effects as fluoxetine against ASD (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). In a clinical trial, fluvoxamine was found to be well tolerated in ASD adults and it has improved compulsive as well as repetitive behaviors and aggression (Fatemi et al., 1998; DeLong et al., 2002; Hollander et al., 2005). Other SSRIs, including sertraline, paroxetine and escitalopram, showed almost the same potential benefits and adverse effects as compared to fluoxetine and fluvoxamine (Hellings et al., 1996; McDougle et al., 1998a). Venlafaxine is another SSRI which has, also, shown improvements of restricted behaviors, decreased interests, social deficits, hyperactivity and communication problems in individuals with ASD, (Table 1; Hellings et al., 1996; McDougle et al., 1998a).
Tricyclic antidepressants
The second-generation tricyclic antidepressant nortriptyline is used in the therapeutic management of major depression and childhood nocturnal enuresis (bedwetting), chronic fatigue syndrome, chronic pain and migraine, and labile affect in some neurological brain disorders (Kurtis, 1966; Campbell et al., 1971). Clomipramine is another tricyclic antidepressant used for the treatment of obsessive compulsive disorder, panic disorder, major depressive disorder, and chronic pain (Gordon et al., 1993; Sanchez et al., 1996). Interestingly, nortriptyline has been described to be effective in children with ASD as it improved the hyperactivity, aggressiveness, and ritualized behavior, while imipramine was not well tolerated in ASD children (Kurtis, 1966; Campbell et al., 1971). In a previous clinical trial, 58% of ASD subjects have found clomipramine to be superior to placebo and the antidepressant desipramine in improving ASD symptoms, anger, and compulsive and ritualized behaviors (Gordon et al., 1993). In another clinical study, clomipramine has caused several adverse effects such as sedation and worsening of behaviors like aggression, irritability, and hyperactivity (Sanchez et al., 1996; Table 1).
Anticonvulsants
Lamotrigine, a member of the sodium channel blocking class of anticonvulsants clinically used in the treatment of children diagnosed with epilepsy, decreased symptoms in approximately 62% of the ASD individuals, and no considerable change among placebo-treated and lamotrigine-treated patients was observed in a study comprising 35 patients diagnosed with ASD (Uvebrant and Bauziene, 1994). On the contrary, VPA with its anticonvulsant effect recognized based on its blockade of voltage-dependent sodium channels and increased GABA levels in the brain has shown -as an orphan drug- valuable effects in improving various symptoms and psychiatric comorbidities, e.g., receptive language, affective instability, and aggression, without appreciable clinical effects on core symptoms of ASD (Uvebrant and Bauziene, 1994; Jobski et al., 2017). Notably, VPA has been reported to be an inhibitor for histone deacetylase (HDAC), an enzyme which plays –together with other HDACs- an essential regulating role in gene transcription and phenotypic differentiation (Hsieh and Gage, 2004, 2005; Balasubramaniyan et al., 2006; Jessberger et al., 2007; Chomiak et al., 2013). Accordingly, numerous studies reported that specific expression forms of HDAC1 and HDAC2, which are categorized as class I of HDACs, in the murine brain are existent at various developmental ages with HDAC1 expressed in neural stem cells/progenitors and glia, and HDAC2 being upregulated in postmitotic neuroblasts and various but not in fully differentiated glia (Chomiak et al., 2013). Therefore, modulation of HDAC in diverse cell types and at several maturational time points may possibly lead to the observation of intensely diverse clinical outcomes and may explain why HDAC inhibition (for instance with VPA) in adulthood leads to improvement of ASD-like features in animals exposed prenatally to VPA (Chomiak et al., 2013). In previous studies, in utero exposure to VPA in mice induced ASD-like behavioral social interaction deficits, anxiety and spatial learning incapacity (Kataoka et al., 2013). However, all these behavioral impairments were ameliorated following chronic (5-week) treatment with VPA (30 mg/kg/d, i.p.) (Kataoka et al., 2013; Takuma et al., 2014), suggesting that dose (300-600/kg mg for acute induction of ASD or 30 mg/kg for chronic treatment goals) or time point (prenatal or 8-week old animals) of using VPA determines whether it is ASD-inducing or palliating cognitive dysfunction (Takuma et al., 2014). VPA-induced ASD in rodents will be discussed in more details under section Non-genetic Animal Models of ASD. Moreover, the anticonvulsant drug levetiracetam was found to be valuable in decreasing hyperactivity, impulsivity, aggression, and emotional lability (Rugino and Samsock, 2002) (Table 1). These clinical observations for several antiepileptic drugs demonstrate that the prevalence of psychopharmacotherapy and polypharmacy in ASD patients is considerable, which is probably due to the treatment of non-core ASD symptoms and psychiatric comorbidities, despite a lack of pharmacological treatment options for ASD core symptoms.
Glutamate antagonists
Levels of glutamate have been found to be excessively increased in post-mortem brain samples of some ASD individuals (Owley et al., 2006; Chez et al., 2007). Numerous studies have publicized the effectiveness of several glutamate antagonists, e.g., amantadine and memantine, in ASD patients (Owley et al., 2006; Chez et al., 2007). In a controlled clinical trial, amantadine showed improving effects on hyperactive behavior and inappropriate speech in ASD children (Owley et al., 2006; Chez et al., 2007). Also, the clinical use of memantine in the treatment of ASD individuals has shown therapeutic progresses in regard to memory, hyperactivity, irritability, language, social behavior and self-stimulatory behavior (Owley et al., 2006; Chez et al., 2007; Table 1).
Acetylcholinesterase inhibitors
Dysfunction of brain cholinergic neurotransmission has been described in several patients diagnosed with ASD (Perry et al., 2001; Hardan and Handen, 2002; Niederhofer et al., 2002; Nicolson et al., 2006). Therefore, acetylcholinesterase inhibitors, e.g., rivastigmine, donepezil, and galantamine, have in many studies been investigated for the use in ASD children (Hardan and Handen, 2002; Niederhofer et al., 2002; Nicolson et al., 2006). Interestingly, the clinical application of rivastigmine in ASD children significantly relieved overall ASD behaviors, however, several adverse effects including nausea, diarrhea, hyperactivity and irritability were reported (Hardan and Handen, 2002; Niederhofer et al., 2002; Nicolson et al., 2006). Among acetylcholinesterase inhibitors, donepezil has shown capability to improve irritability and hyperactivity of ASD children (Hardan and Handen, 2002; Niederhofer et al., 2002; Nicolson et al., 2006). Moreover, galantamine produced substantial improvements in hyperactivity, irritability, social withdrawal, inappropriate speech, attention deficiency, and reduction in anger in children diagnosed with ASD (Hardan and Handen, 2002; Niederhofer et al., 2002; Nicolson et al., 2006; Table 1). These improvements observed for several acetylcholine esterase inhibitors strongly support the hypothesis that enhancing the cholinergic neurotransmission in ASD results in positive therapeutic effects.
Psychostimulants
Methylphenidate, the most commonly known CNS stimulant widely used in the therapeutic management of attention deficit hyperactivity disorder (ADHD) and narcolepsy, is commonly indicated for ASD children and adolescents (Handen et al., 2000; Di Martino et al., 2004; Jahromi et al., 2009; Kim et al., 2017). Methylphenidate mainly acts as a norepinephrine-dopamine reuptake inhibitor (NDRI). In numerous controlled studies, methylphenidate palliated several behavioral ASD features including impulsivity, attention deficiency, and hyperactivity, but it correspondingly exhibited some initial adverse effects such as aggression, anorexia, and increased wakefulness (insomnia) (Handen et al., 2000; Di Martino et al., 2004; Jahromi et al., 2009; Kim et al., 2017; Table 1).
Adrenergic alpha (α)2 receptor agonists
Oral or transdermal administration of selective centrally acting α2 adrenergic agonist, e.g., clonidine, have revealed to improve mood instability, hyperactive behavior, aggressiveness and nervousness in ASD individuals (Fankhauser et al., 1992; Ming et al., 2008). Clonidine is a drug used to treat high blood pressure, ADHD, anxiety disorders, tic disorders, withdrawal (from either alcohol, opioids, or smoking), migraine, and certain pain conditions, with largely tolerable adverse effects (Fankhauser et al., 1992; Ming et al., 2008). Also, previous clinical trials carried out with clonidine in ASD subjects delivered evidences for the clinical effectiveness and safety profile in ASD and related brain disorders (Fankhauser et al., 1992; Ming et al., 2008). Moreover, a retrospective study revealed that the use of guanfacine, a selective α2 adrenergic receptor agonist used in the treatment of ADHD, anxiety, and hypertension, was accompanied with enhancements in insomnia, attention deficiency, hyperactivity, and tics (Posey et al., 2004; Scahill et al., 2006; Boellner et al., 2007). However, the most common adverse effects observed with guanfacine were mood alteration fatigue, blurred vision, and headache (Posey et al., 2004; Scahill et al., 2006; Boellner et al., 2007; Table 1).
Opiate antagonists
Based on the reputed role of endogenous opioids such as β-endorphin and encephalin in the regulation of social behavior, the opiate antagonist naltrexone has been assessed in ASD (Panksepp and Lensing, 1991; Bouvard et al., 1995; Kolmen et al., 1995; Elchaar et al., 2006; Clifford et al., 2007). The results observed in numerous studies for naltrexone showed that it might be able to treat behavioral aberrations perceived in ASD patients and induced by dysfunction of the brain opioid system (Panksepp and Lensing, 1991; Bouvard et al., 1995; Kolmen et al., 1995; Elchaar et al., 2006; Clifford et al., 2007). Moreover, numerous studies revealed the significant improvements of various behavioral symptoms obtained with the use of naltrexone in ASD children (Panksepp and Lensing, 1991; Bouvard et al., 1995; Kolmen et al., 1995; Elchaar et al., 2006; Clifford et al., 2007). Furthermore, these studies reported that naltrexone treatment provided substantial enhancements in self-injurious behavior, hyperactivity, social withdrawal, agitation and irritability in ASD patients (Panksepp and Lensing, 1991; Bouvard et al., 1995; Kolmen et al., 1995; Elchaar et al., 2006; Clifford et al., 2007; Table 1).
The aforementioned described drugs act on different targets to therapeutically manage the behavioral as well as psychiatric symptoms of ASD. However, several brain regions are altered in ASD individuals, resulting in loss of neuronal function, and behavioral and sensory impairments, including inattention, hyperactivity, mood fluctuations, aggressiveness, agitation, social deficits and repetitive and restricted behavior. In the brain, the plasticity of brain tissue, nerve tangling and imbalanced production of several neurotransmitters are all implicated on evolution in ASD individuals. Apart from environmental factors, other pathological conditions such as immunological problems, chronic neuroinflammation, oxidative stress, mitochondrial dysfunction are involved in etiopathogenesis of ASD (Kumar et al., 2012).
Up until now, there is no approved drug existing in the market which is specific for treating symptoms associated with ASD, however, preclinical and clinical research and development are in progress to find new therapeutic entities. Currently, there are several candidates that successfully passed different clinical phases of drug development, with different pharmacological targets to palliate ASD behavioral and neurological symptoms (Table 2, Figure 3). Interestingly, 10% of these clinical candidates are currently in phase 1, 46% have already progressed to phase 2, 27% advanced to phase 3, and 15% reached to phase 4 of clinical development, whereas 2% are currently of preclinical interest (Ruhela et al., 2015; Table 2, Figure 3).
Table 2
| Name | Class | Clinical indication/condition (No. of trials in phase 2) | Clinical indication/condition (No. of trials in phase 3) | Clinical indication/condition (No. of trials in phase 4) |
|---|---|---|---|---|
| Arbaclofen | Anticonvulsant | Autistic disorder (3) Neurobehavioral Manifestations (2) Neurodevelopmental Disorders (3) | Autistic disorder (2) Neurobehavioral Manifestations (3) Neurodevelopmental Disorders (1) | |
| Aripiprazole | Antipsychotic drug | Autistic Disorder (5) Neurobehavioral Manifestations (1) Neurodevelopmental Disorders (6) | Autistic Disorder (7) Neurobehavioral Manifestations (1) Neurocognitive Disorders (6) Neurodegenerative disease (11) Neurodevelopmental Disorders (13) | Autistic Disorder (5) Neurocognitive Disorders (3) Neurodegenerative disease (3) Neurodevelopmental Disorders (11) |
| Atomoxetine | Selective norepinephrine transporter | Neurobehavioral Manifestations (4) Neurocognitive Disorders (5) Neurodegenerative disease (4) Neurodevelopmental Disorders (14) | Autistic Disorder (1) Neurocognitive Disorders (1) Neurodegenerative disease (1) Neurodevelopmental Disorders (29) | Autistic Disorder (3) Neurobehavioral Manifestations (5) Neurocognitive Disorders (1) Neurodegenerative disease (2) Neurodevelopmental Disorders (48) |
| Baclofen | Central muscle relaxant | Autistic Disorder (2) Neurobehavioral Manifestations (1) Neurodegenerative Diseases (2) Neurologic Manifestations (9) | Neurobehavioral Manifestations (1) Neurodegenerative Diseases (1) | |
| Bumetanide | Diuretic action | Autistic Disorder (2) | Autistic Disorder (1) | Neurodevelopmental Disorders (1) |
| Buspirone | Anxiolytic | Autistic disorder (3) Neurodegenerative Diseases (1) Neurodevelopmental Disorders (4) | Autistic disorder (1) Neurocognitive Disorders (1) Neurodegenerative Diseases (1) | |
| Citalopram | Antidepressant | Autistic Disorder (1) Neurocognitive Disorders (4) Neurodegenerative Diseases (2) Neurodevelopmental Disorders (2) | Neurobehavioral Manifestations (3) Neurocognitive Disorders (7) Neurodegenerative Diseases (7) | Neurobehavioral Manifestations (3) Neurocognitive Disorders (4) Neurodegenerative Diseases (6) |
| CX516 | Autistic Disorder (1) Neurocognitive Disorders (2) Neurodegenerative Diseases (1) | |||
| D-Cyclocerine | NMDA modulator | |||
| Dimercaptosuccinic acid | Antidote for lead, mercury, and arsenic poisoning | Autistic Disorder (1) | ||
| Donepezil | Acetylcholinesterase inhibitor | Autistic Disorder (4) Neurobehavioral Manifestations (7) Neurocognitive Disorders (62) Neurodegenerative Diseases (57) | Neurobehavioral Manifestations (5) Neurocognitive Disorders (41) Neurodegenerative Diseases (33) | Autistic disorder (1) Neurobehavioral Manifestations (6) Neurocognitive Disorders (49) Neurodegenerative Diseases (37) Neurodevelopmental Disorders (1) |
| Fluoxetine | SSRI | Autistic disorder (3) Neurodegenerative disease (1) | Autistic disorder (3) Neurocognitive Disorders (1) | Neurodegenerative Diseases (1) Neurodevelopmental Disorders (1) |
| Fluvoxamine | SSRI | Autistic disorder (1) Neurocognitive Disorders (1) Neurodevelopmental Disorders (2) | Neurodegenerative Diseases (1) | |
| Galantamine | Cholinesterase inhibitor | Neurobehavioral Manifestations (2) Neurocognitive Disorders (12) Neurodegenerative disease (7) | Autistic Disorder (1) Neurocognitive Disorders (28) Neurodegenerative disease (21) Neurodevelopmental Disorders (1) | Neurobehavioral Manifestations (3) Neurocognitive Disorders (22) Neurodegenerative disease (21) Neurodevelopmental Disorders (2) |
| Lenalidomide | Anti-inflammatory | Autistic Disorder (1) Neurodegenerative Diseases (1) | ||
| Levetiracetam | Anticonvulsant | Neurobehavioral Manifestations (1) Neurocognitive Disorders (2) Neurodegenerative disease (5) | Neurocognitive Disorders (1) | Neurobehavioral Manifestations (2) Neurocognitive Disorders (1) Neurodegenerative disease (3) Neurodevelopmental Disorders (3) |
| Methyl cobalamin | Vitamin | Autistic disorder (2) Neurocognitive Disorders (1) Neurodegenerative Diseases (4) | Autistic disorder (2) Neurocognitive Disorders (1) Neurodegenerative Diseases (5) Neurodevelopmental Disorders (2) | Neurocognitive Disorders (4) Neurodegenerative Diseases (1) |
| Minocycline | Antibiotic | Autistic Disorder (1) Neurobehavioral Manifestations (3) Neurocognitive Disorders (5) Neurodegenerative disease (5) | Neurobehavioral Manifestations (3) Neurocognitive Disorders (2) Neurodegenerative disease (2) | Autistic Disorder (1) Neurobehavioral Manifestations (1) |
| N-Acetyl cysteine | Antioxidant | Autistic disorder (4) Neurobehavioral Manifestations (4) Neurocognitive Disorders (4) Neurodegenerative Diseases (7) Neurodevelopmental Disorders (5) | Neurobehavioral Manifestations (1) Neurodegenerative Diseases (2) | Neurocognitive Disorders (1) Neurodegenerative Diseases (1) |
| Naltrexone | Opioid receptor antagonist | Autistic Disorder (1) Neurobehavioral Manifestations (2) Neurocognitive Disorders (1) Neurodegenerative disease (1) | Neurobehavioral Manifestations (1) Neurodevelopmental Disorders (1) | Neurodegenerative disease (1) Neurodevelopmental Disorders (3) |
| Olanzapine | Atypical antipsychotic | Autistic Disorder (2) Neurocognitive Disorders (1) Neurodegenerative Diseases (1) Neurodevelopmental Disorders (2) | Autistic Disorder (1) Neurobehavioral Manifestations (2) Neurocognitive Disorders (3) Neurodegenerative Diseases (2) Neurodevelopmental Disorders (2) | Neurobehavioral Manifestations (2) Neurocognitive Disorders (6) Neurodegenerative Diseases (2) Neurodevelopmental Disorders (5) |
| Oralgam | human immunoglobulin | Autistic Disorder (1) Child Development Disorders, Pervasive (1) Neurodevelopmental Disorders (1) | ||
| Oxytocin hormone | Hormone | Autistic Disorder (5) | ||
| Paliperidone ER | Atypical antipsychotic | Autistic Disorder (1) Neurocognitive Disorders (1) Neurodevelopmental Disorders (1) | Neurobehavioral Manifestations (1) Neurocognitive Disorders (1) | |
| Pioglitazone | Antihyperglycemic | Autistic Disorder (1) Neurocognitive Disorders (2) Neurodegenerative Diseases (4) | Neurocognitive Disorders (2) Neurodegenerative Diseases (3) | |
| RG 1068 | Synthetic human secretin | Autistic Disorder (2) | ||
| Riluzole | Sodium channel blocker | Autistic Disorder (3) Neurocognitive Disorders (2) Neurodegenerative Disorders (34) | Autistic disorder (1) Neurocognitive Disorders (1) Neurodegenerative Diseases (11) | Neurobehavioral Manifestations (2) Neurodegenerative Diseases (3) Neurodevelopmental Disorders (1) |
| Risperidone | Atypical antipsychotic drug | Autistic Disorder (4) Neurocognitive Disorders (3) Neurodegenerative disease (1) Neurodevelopmental Disorders (7) | Autistic Disorder (6) Neurobehavioral Manifestations (4) Neurocognitive Disorders (6) Neurodegenerative disease (5) Neurodevelopmental Disorders (12) | Autistic Disorder (4) Neurobehavioral Manifestations (8) Neurocognitive Disorders (11) Neurodegenerative disease (5) Neurodevelopmental Disorders (10) |
| Saproterin | Enzymatic cofactor | |||
| Sertraline | SSRI | Autistic Disorder (2) Neurobehavioral Manifestations (2) Neurocognitive Disorders (1) Neurodegenerative disease (2) Neurodevelopmental Disorders (2) | Autistic Disorder (1) Neurobehavioral Manifestations (1) Neurocognitive Disorders (2) Neurodegenerative disease (1) Neurodevelopmental Disorders (2) | Neurocognitive Disorders (2) Neurodegenerative disease (1) Neurodevelopmental Disorders (2) |
| Valproic acid | Anticonvulsant | Autistic Disorder (3) Neurobehavioral Manifestations (2) Neurocognitive disorders (1) Neurodegenerative Diseases (7) | Autistic disorder (1) Neurobehavioral Manifestations (2) Neurocognitive Disorders (3) Neurodegenerative Diseases (3) Neurodevelopmental Disorders (1) | Autistic disorder (1) Neurobehavioral Manifestations (3) Neurocognitive Disorders (3) Neurodegenerative Diseases (1) Neurodevelopmental Disorders (6) |
| Valproic acid + sodium valproate | Anticonvulsant | Neurobehavioral Manifestations (1) Neurocognitive Disorders (1) | Autistic Disorder (1) Neurobehavioral Manifestations (1) Neurocognitive Disorders (1) | |
| Ziprasidone | Atypical antipsychotic | Autistic Disorder (1) Neurobehavioral Manifestations (1) Neurocognitive Disorders (2) Neurodevelopmental Disorders (2) | Autistic disorder (1) Neurobehavioral Manifestations (3) Neurocognitive Disorders (3) | Neurobehavioral Manifestations (2) Neurocognitive Disorders (2) Neurodevelopmental Disorders (2) |
Drugs currently in phase 1–4 of clinical development for neurobehavioral manifestations, neurodevelopmental, and autistic disorders.
Animal models of ASD
For the understanding of the etiology and pathogenesis of any human disease, e.g., ASD, experimental rodent models are of substantial meaning. Modelling of human neuropsychiatric diseases with animals is very challenging due to the multifaceted nature of these disorders, combined with the absence of effective diagnostic biomarkers and objective tests for accurate diagnosis (Nestler and Hyman, 2010). Moreover, neuropsychiatric diseases, such as ASD, are multifactorial with genetic, medical and neurodevelopmental conditions associated with the disease (Hoffman et al., 2017; Lyall et al., 2017; Xie et al., 2017). Generally, there are two types of rodent models for ASD, namely the genetic and non-genetic animal models. Accordingly, ASD genetic animal models are highly applicable when they reproduce the ASD features that are existent in an individual human genetic disease such as tuberous sclerosis and fragile X syndrome. Therefore, the ASD genetic models induced in mice should be established on an identified genetic cause of a disease, reflect key aspects of the human symptoms and respond to pharmacotherapies that are operational in the human diseases (Chadman et al., 2009).
Genetic animal models of ASD
A considerable advancement in detecting the genetic basis of several human diseases has been achieved by geneticists. These achievements include Alzheimer's disease, Huntington's disease, and some forms of breast cancer. However, more difficulties were associated with the detection of genetic factors for multifaceted diseases such as SCH, diabetes, bipolar disorder, and also ASD (Rao et al., 2013; Wei et al., 2015; Fregeac et al., 2016; Hu et al., 2017). In the case of ASD, extensive research efforts elucidated underlying mechanisms of the disorder and concluded that these mechanisms might include dysfunctions of neuronal microRNAs (miRNAs), which are regulators of gene expression, and which have gained much attention in pathophysiology of various psychiatric illnesses (Andolina et al., 2017). Accordingly, about half of all identified miRNAs in humans are expressed in the CNS and exhibit modulatory functions crucial for numerous biological processes associated to the development of the CNS (Hu et al., 2017). Consequently, disruptions in miRNA biogenesis and miRNA-target interaction have been linked with CNS diseases, including ASD (Hu et al., 2017). Moreover, three miRNA (miRNA-7, miRNA-9, and miRNA-106b) were found to be associated with neurodegenerative diseases and only one, namely miRNA-9, with intellectual disability (Doxakis, 2010; Wang et al., 2010; Xu et al., 2011; Hu et al., 2017). Furthermore, it was recognized that the intellectual disability in Down syndrome which is a neurodevelopmental disorder, was caused by an extra-copy of chromosome 21 (Lejeune et al., 1959). However, it is now being firmly established that deletions in the chromosome 21 are commonly discovered in some patients with intellectual disability including ASD (Hogart et al., 2010). Similarly, increased risk of delayed development, intellectual disability and neurological as well as psychiatric problems associated with ASD, SCH, epilepsy and hypotonia was found to be the result of micro deletions in the long (q) arm of the chromosome in a region designated q21.1 (Knight et al., 1999; Ravnan et al., 2006; Mefford et al., 2009). Moreover, chromosome 15 has been described to be as one of seven several chromosomes enriched in segmental low copy repeats or duplicons (Bailey et al., 2002). These duplicons were revealed to strongly contribute to a mechanism in which low copy repeats facilitated misalignment during meiosis I, leading to unequal recombination of nonallelic homologes and developing a sequence of common cut-off point along the 15q11.2-q13 region (Robinson et al., 1993a,b,c; Christian et al., 1999). Previous reports suggested, also, several cases of ASD with cytogenetic abnormalities in the 15q11–13 region (Cook et al., 1998), and the frequency of cytogenetic abnormalities in this region was found to be around 0-3% in ASD, and in fact, the most frequent cytogenetic abnormality in this population (Lord et al., 2000). Moreover, it has been reported that the paternal inheritance of a cytogenetical abnormality leads to a normal phenotype; maternal inheritance leads to autism or atypical autism (Tabet et al., 2015). Furthermore, previous efforts revealed that 16p13.1 micro duplication syndrome is caused by interstitial duplications encompassing 16p13.1, which is a risk factor for a range of neuropsychiatric disorders, including ASD, intellectual disability, SCH, and ADHD (Ullmann et al., 2007; Mefford et al., 2009; Ramalingam et al., 2011; Grozeva et al., 2012). There are, also, monogenic diseases that have been associated with ASD and display its characteristic features: tuberous sclerosis, fragile X syndrome, Rett syndrome and neurofibromatosis 1 (Wetmore and Garner, 2010; Hulbert and Jiang, 2016). Accordingly, single gene mutation gave rise to several human syndromes leading to an increase in the risk for developing ASD. Moreover, autistic traits are also connected to copy-number variants that lead to inheritance of maternal 15q11–13 duplication, resulting in Prader-Willi syndrome (Ogata et al., 2014). Furthermore, the developed strategies for the identification of genetic variants has led to the description of novel syndromic forms of ASD and facilitated the relationship between genetic traits and phenotype. Consequently, these genetic dissimilarities identified so far, along with the newly developed strategies for genetic engineering, improved the expansion of genetic animal models for ASD. Accordingly, mice are the major animal model for ASD due to their genetic manipulability and their talent to show behavioral deficits associated with ASD features (Chen et al., 2015). Moreover, construct as well as face validity of mice ASD animal models together with the availability of well-established techniques to manipulate their genome and study brain function at numerous levels of analysis constitute to the overall validity of mice model (Hulbert and Jiang, 2016). Also, mice as mammals are genetically and biologically similar to humans, however, their rapid reproduction and accelerated development allow for the testing of large numbers of animals at a relatively low cost (Hulbert and Jiang, 2016). Accordingly, animal models for genes of a syndromic disorder were commonly found to present autistic traits including Fragile X (FMR1) with social interaction deficits, hyperactivity, and cognitive impairments (Willem Verhoeven, 2011; Bhattacharya et al., 2012; Ronesi et al., 2012; Hulbert and Jiang, 2016; Hu et al., 2017), Rett syndrome and MECP2 mutations with the resulted repetitive and stereotypic/restricted behaviors, abnormal gait and reduced anxiety, decreased pain and normal olfactory discrimination (Chao et al., 2010; Samaco et al., 2013), tuberous sclerosis TSC1 or TSC2 with social interactions deficits and repetitive/restricted behavior or interest (Chao et al., 2010; Willem Verhoeven, 2011), Timothy syndrome (TS) CACNA1C with impairments in social interactions, repetitive/stereotypic behaviors, and increased fear conditioning (Ergaz et al., 2016), Phelan-McDermid syndrome with restricted interest as well as cognitive and motor deficits (Giza et al., 2010; Ergaz et al., 2016), PTEN mutations, cortical dysplasia focal epilepsy with deficits in regard to social interactions, restricted interest, sensory sensitivity, elevated anxiety, and seizures (Scott-Van Zeeland et al., 2010) (Cook and Scherer, 2008; LaSalle et al., 2015; Piochon et al., 2015) as behavioral features of the above described syndromes which are associated with the ASD (Table 3). Numerous inbred mouse strains show face validity as ASD models in addition to the genetically modified animal models of ASD, as such inbred strains display robust and well-replicated social deficits and repetitive behaviors (Kazdoba et al., 2016). These inbred strains are found to display idiopathic autism, as their ASD-relevant behaviors are not caused by known genetic mutations (Kazdoba et al., 2016). For instance and while assessing sociability, inbred strains like A/J, BALB/cByJ (BALB), BTBR T+Itpr3tf/J (BTBR), C58/J (C58), and 129S1/SvImJ mice showed lack of sociability, as compared to inbred mouse strains with high sociability, such as C57BL/6J (B6) and FVB/NJ mice (Brodkin, 2007; Moy et al., 2007, 2008; McFarlane et al., 2008; Kazdoba et al., 2016; Hu et al., 2017). Moreover, it has been shown that several mouse strains, e.g. BTBR and C58, also demonstrate explicit motoric stereotypies or repetitive behaviors, such as jumping, digging, and high levels of self-grooming and marble burying (Pasciuto et al., 2015; Kazdoba et al., 2016).
Table 3
| Mouse model | Associated syndrome | Targeted mutant gene (Chromosome) | Observed behavioral symptoms | References |
|---|---|---|---|---|
| Fmr1 KO mice | Fragile X syndrome | FMR1 (Xq27) | Social interaction deficits, hyperactivity, cognitive impairments. | Willem Verhoeven, 2011; Bhattacharya et al., 2012; Ronesi et al., 2012; Hulbert and Jiang, 2016; Hu et al., 2017 |
| Cntnap2−/− mice | Cortical dysplasia-focal epilepsy syndrome | CNTNAP2 (7q35) | Social interactions impairments, stereotypic behavior, sensory sensitivity, elevated motor activity, nest-building impairments. Normal olfactory discrimination and seizures. | Cook and Scherer, 2008; Scott-Van Zeeland et al., 2010; LaSalle et al., 2015; Piochon et al., 2015 |
| Tsc1+/− mice Tsc2+/− mice | Tuberous Sclerosis | TSC1 (9q34) TSC2 (16p13) | Social interactions deficits, normal olfaction. No difference in motor and sensory functions. Repetitive/restricted behavior or interest. | Chao et al., 2010; Willem Verhoeven, 2011; Ergaz et al., 2016 |
| Viaat-Mecp2 conditional mutant mice | Rett syndrome | MeCP2 (Xq28) | Repetitive and stereotypic/restricted behaviors, abnormal gait and reduced anxiety, irregular breathing, reduced pain and normal olfactory discrimination. | Amir et al., 1999; Chao et al., 2010; Katz et al., 2012; Kron et al., 2012; Samaco et al., 2013 |
| Shank3B mutant mice Shank3A mutant mice Shank3(e4-9) mutant | Phelan-McDermid syndrome | SHANK3 (22q13) | Restricted interest but no repetitive behavior, social interactions, learning, sensory, cognitive and motor deficits. | Giza et al., 2010; Phelan and McDermid, 2012; Ergaz et al., 2016 |
| Ube3A−/− mice (transgenic mice with triple dosage of Ube3A) | Angelman syndrome | UBE3A (15q11) | Impaired social interactions, repetitive behavior and restricted interest. | Kishino et al., 1997; Matsuura et al., 1997; Buiting et al., 2016 |
| TS2 mice | Timothy syndrome | CACNA1C (16p13) | Social interactions impairments, repetitive/stereotypic behaviors, and increased fear conditioning. | Chao et al., 2010; Willem Verhoeven, 2011; Ergaz et al., 2016 |
| PTEN mutant mice | Not defined | PTEN (10) | Social interactions deficits, restricted interest, sensory sensitivity, elevated anxiety, normal motor activity and seizures. | Cook and Scherer, 2008; LaSalle et al., 2015; Piochon et al., 2015 |
Most relevant ASD mouse models induced by targeting ASD-associated genes.
Table 4
| Ligand | Dose (mg/kg) | Species | Pharmacological effect observed | References |
|---|---|---|---|---|
| Thioperamide | 10 and 30 (i.p.) | Mice | Increase of prepulse inhibition | Browman et al., 2004; Sadek et al., 2016 |
| Ciproxifan | 1, 3, and 10 (i.p.) | Mice | Increase of prepulse inhibition | Browman et al., 2004; Sadek et al., 2016 |
| ABT-239 | 0.3-3.0 (i.p.) | Rats | Decrease of cognitive deficits induced by ketamine and MK-801 | Brown et al., 2013; Sadek et al., 2016 |
| A-431404 | 0.3-3.0 (i.p.) | Mice | Decrease of cognitive deficits induced by ketamine and MK-801 | Browman et al., 2004; Sadek et al., 2016 |
| SAR110894 | 0.3-3 (p.o.) | Mice | Decrease in Impaired social behaviors | Griebel et al., 2012; Sadek et al., 2016 |
| 0.3-1 (p.o.) | Rats | Decrease in Impaired social behaviors | Griebel et al., 2012; Sadek et al., 2016 |
Preclinical representation of several H3R antagonists in different animal models of SCH.
Non-genetic animal models of ASD
Environmental animal models are induced by prenatal exposure of pregnant animals to certain chemical compounds, infections, or inflammations. The environmental inducing factor is only of significance if the chemicals used in the animal model cause the same effects in humans. Accordingly, the antiepileptic drug, VPA was found to significantly increase the ASD degree in offspring of treated mothers (Christianson et al., 1994; Moore et al., 2000), and similarly showed to induce ASD-like behaviors in mice and rats (Rodier et al., 1997; Anshu et al., 2017; Nicolini and Fahnestock, 2017). Therefore, the ASD-like animal model induced by in utero exposure to the anticonvulsant drug VPA has been well established and recognized to study ASD features (Nicolini and Fahnestock, 2017). Moreover, the immunomodulatory drug thalidomide, which was used by pregnant women in the 1950s, was revealed to be associated with a marked escalation in the incidence of ASD in their offspring (Strömland et al., 1994). Furthermore, several reports revealed that methylmercury, which is formed from inorganic mercury by the action of microbes that live in aquatic systems including lakes, rivers, wetlands, sediments, and soils, exposure during childhood caused a variety of neuropsychological abnormalities, e.g., memory, attention, and language, (Grandjean et al., 1999; Falluel-Morel et al., 2007; Davidson et al., 2011). Also, a previous study has shown that acute systemic administration methylmercury during developmental phases elicited hippocampal cell death, reductions in neurogenesis, and severe learning impairments (Grandjean et al., 1999). Thimerosal was a widely used preservative in numerous biological and drug products since 1930s, including many vaccines. It is a mercury-containing organic compound which contains ethylmercury, a compound supposed to be the cause for several adverse neurodevelopmental deficits, including ASD (Hviid et al., 2003). Also, a previous study in which 10,000 cases have been examined showed that there is a significant association between maternal viral infection in the first trimester and ASD in the offspring (Atladóttir et al., 2010). Indeed, when animals were exposed to the maternal immune activation with polyinosine:cytosine (poly I:C) at embryonic day 9.5, the offspring displayed histological and behavioral abnormalities that resembled ASD (Shi et al., 2009; Hsiao et al., 2012; Garay et al., 2013). On behavioral level, these animals demonstrated impaired sociability, communication differences, and increased repetitive stereotyped behaviors and had smaller brain sizes at birth, followed by macroencephaly in adulthood. In line with these observations, it has been shown that viral infections in the mother can prompt substantial changes in the immune system in both mother and fetus, leading to long-term epigenetic alterations in the offspring (Kong et al., 2012). Consequently, the timing of immune insults during developmental phase may be one source of the heterogeneity in the phenotypic features observed in ASD (Kong et al., 2012). Accordingly, male rats treated with thalidomide/VPA demonstrated immunological changes such as decreased splenocyte proliferative response to mitogenic stimulation, lower thymus weight, and decreased interferon (IFN)-β/IL-10 ratio in peritoneal macrophages, whereas female rats in this study failed to show many of these behavioral and immunological changes, indicating that sex-specific responses to some environmental factors might play a significant role on the phenotypic outcomes of several ASD features (Schneider et al., 2008). Also and subsequent to viral infection, the immune response was found to produce various cytokines, such as interleukins (IL)-1,-2 and -6 which modulate the release of several monoamines such as 5-HT in the hippocampus and other brain regions (Libbey et al., 2005). Indeed, maternal infection was found to lead to elevated levels of cytokines and chemokines including interleukin-1β (IL-β), IL-6, IL-8, and IL-12p40 in the plasma of ASD children, and such increases were reported to be associated with more reduced communication skills and abnormal behaviors (Patterson, 2012). Also, the offspring of infected animal mothers given poly (I:C) exhibited both ASD-like behavioral and neuropathological deficits as described above (Shi et al., 2009; Hsiao et al., 2012; Patterson, 2012; Gadad et al., 2013; Garay et al., 2013). Based on the aforementioned results, environmental influences both during and after pregnancy can significantly influence the immune system and the developing nervous system to play a role in constructing several neurodevelopmental disorders including ASD.
It should be stressed that experimental animal models (genetically manipulated animal models, animal models obtained by destruction of certain CNS areas, and animal models obtained by using maternal factors) are indispensable for exploring the pathophysiologic causes of brain disorders, e.g., ASD, although they do not reflect the entire state of ASD disease. Moreover, animal models (mostly rodents) are widely used to study the development of cortical neurocircuit, genetic analysis and molecular mechanisms underlying ASD, and the palliative effects of newly developed drugs on core as well as associated symptoms of ASD. Notably, rat has become the most extensively used animals in the context of SCH and ASD neuroscience (Brudzynski, 2013). Accordingly, and as compared to mice, rats provide more experimental advantages including the fact that rats demonstrate richer social behavioral skills, show various types of social behaviors, and use a rich CNS communication system (Brudzynski, 2013). Furthermore, genetic as well as non-genetic rodent models have the capability of being the major objects of pharmaceutical industry in testing drug efficacy, dosage and acute as well as (sub)chronic toxicology.
Conclusion
The disciplines of neurochemistry, neuroanatomy, neuropsychopharmacology, genetics and molecular biology are encouraged to work simultaneously to further help uncover more details of the biological bases of ASD. It is motivating that numerous genes identified so far in human ASD genetic studies generate several valuable transgenic ASD rodent models, which display both ASD-like abnormalities in neuropathology as well as in behavioral phenotypic features. Moreover, several of the abnormalities involve the dysfunction of various neurotransmitter systems. Furthermore, a variety of multiple genetic and environmental factors induce alterations in brain development, leading to ASDs. Notably, there are associated behavioral abnormalities, e.g., anxiety-like behavior, seizure susceptibility, sensory processing, and motor abnormalities, which have been recognised in both the genetic and non-genetic rodent models of ASDs. Consequently, broader understanding of how the trajectory of patients affected with ASDs fits into several subcategories, with specific endophenotype concepts, will expedite both understanding of the causes of the disorders and can provide insight into specific possible future treatments. Preclinical experimental rodent models are supportive to determine the mechanistic pathways associated with the genetic and environmental targeted insults to the CNS and the pathogenesis of ASD. In recent times these animal models are also continuing to proliferate to assess the potential of different drugs and other related treatment modalities. Consequently, this is of valuable significance, as it may close a gap in our understanding of the effects of therapy and may suggest new and effective methods for ASD treatment. The experimental results observed so far clearly show that ASD involves cascade of complex gene-environmental interactions which requires experts of these research areas to work in collaboration together that may lead hopefully to improved pharmacological interventions, if not to cure but at least to reduce the impact of symptoms of this disorder. The majority of pharmacological interventions to date have focused on antipsychotics, antidepressants and psychostimulants that are the most commonly utilized medication classes for ASD. However, several other drug classes, e.g. SSRIs, glutamate antagonists, and antiepileptics, are currently progressing in clinical trials for their future potential use in ASD. In addition, future research in the ASD area needs to address individual differences within the ASD population in order to better account for etiological and phenotypic heterogeneity, and to emphasize on mechanistic processes as well as neurotransmitter dysfunctions and neurodevelopmental routes rather than outcomes or endpoints of the disorder. Moreover, understanding the high level of psychiatric comorbidities and overlapping features shared with other neurodevelopmental disorders together with integration of different research methodologies (e.g., neuropharmacological, behavioral, and brain imaging measures) could encourage further advances of the current level of knowledge in the field of therapeutic ASD intervention. Furthermore, it is becoming progressively obligatory to prospectively evaluate the co-occurrence of, and commonalities between SCH and ASD at the trait level applying the multifactorial data obtainable from large clinical samples, since undiagnosed co-occurring brain disorders in respective individuals may be the consequence of not receiving suitable facilities, assistances, or even pharmacological treatments. Given the similarities between several brain disorders, e.g., SCH and ASD, misdiagnosis is, also, possible. Therefore, it is imperative that future research efforts account for the heterogeneity of both disorders, and inspect evidence on several levels to categorize endophenotypic markers considering the dimensional nature of such brain disorders. In addition, future studies assessing new pharmacological treatment approaches combined with non-pharmacologic therapies are necessary to ensure that target core behaviors of ASD are properly managed.
Statements
Author contributions
NE and BS: Idea, design, writing, and submission. MA-H, AS, SO, and AS: Substantial contribution to the conception, formulation, and critical revision of the manuscript. All authors gave approval for the final submission of the review and agreed be accountable for all aspects of the work.
Acknowledgments
The Office of Graduate Studies and Research of UAE University as well as ADEK Award for Research Excellence (AARE) 2017 are highly thanked for the support provided to BS and MA-H in form of intramural College of Medicine and Health Sciences (Zayed Center for Health Sciences) as well as extramural funds from ADEK.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AlachkarA.LazewskaD.Kiec-KononowiczK.SadekB. (2017). The histamine H3 receptor antagonist E159 reverses memory deficits induced by dizocilpine in passive avoidance and novel object recognition paradigm in rats. Front. Pharmacol.8:709. 10.3389/fphar.2017.00709
2
AlOlabyR. R.SwehaS. R.SilvaM.Durbin-JohnsonB.YrigollenC. M.PrettoD.et al. (2017). Molecular biomarkers predictive of sertraline treatment response in young children with fragile X syndrome. Brain Dev.39, 483–492. 10.1016/j.braindev.2017.01.012
3
AmanM. G. (2004). Management of hyperactivity and other acting-out problems in patients with autism spectrum disorder. Semin. Pediatr. Neurol.11, 225–228. 10.1016/j.spen.2004.07.006
4
AmirR. E.Van den VeyverI. B.WanM.TranC. Q.FranckeU.ZoghbiH. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet.23, 185–188. 10.1038/13810
5
AnagnostouE.EspositoK.SooryaL.ChaplinW.WassermanS.HollanderE. (2006). Divalproex versus placebo for the prevention of irritability associated with fluoxetine treatment in autism spectrum disorder. J. Clin. Psychopharmacol. 26, 444–446. 10.1097/01.jcp.0000227703.72117.bc
6
AndolinaD.Di SegniM.VenturaR. (2017). MiRNA-34 and stress response. Oncotarget8, 5658–5659. 10.18632/oncotarget.13923
7
AndresC. (2002). Molecular genetics and animal models in autistic disorder. Brain Res. Bull.57, 109–119. 10.1016/S0361-9230(01)00642-6
8
AnshuK.NairA. K.KumaresanU. D.KuttyB. M.SrinathS.LaxmiT. R. (2017). Altered attentional processing in male and female rats in a prenatal valproic acid exposure model of autism spectrum disorder. Autism Res.10, 1929–1944. 10.1002/aur.1852
9
ArnoldH. M.BurkJ. A.HodgsonE. M.SarterM.BrunoJ. P. (2002). Differential cortical acetylcholine release in rats performing a sustained attention task versus behavioral control tasks that do not explicitly tax attention. Neuroscience114, 451–460. 10.1016/S0306-4522(02)00292-0
10
ArvidssonO.GillbergC.LichtensteinP.LundstromS. (2018). Secular changes in the symptom level of clinically diagnosed autism. J. Child. Psychol. Psychiatry. [Epub ahead of print]. 10.1111/jcpp.12864
11
AtladóttirH. O.ThorsenP.ØstergaardL.SchendelD. E.LemckeS.AbdallahM.et al. (2010). Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord.40, 1423–1430. 10.1007/s10803-010-1006-y
12
AvaleM. E.ChaboutJ.PonsS.SerreauP.De ChaumontF.Olivo-MarinJ. C.et al. (2011). Prefrontal nicotinic receptors control novel social interaction between mice. FASEB J.25, 2145–2155. 10.1096/fj.10-178558
13
BacchelliE.BattagliaA.CameliC.LomartireS.TancrediR.ThomsonS.et al. (2015). Analysis of CHRNA7 rare variants in autism spectrum disorder susceptibility. Am. J. Med. Genet. A167A, 715–723. 10.1002/ajmg.a.36847
14
BaileyJ. A.GuZ.ClarkR. A.ReinertK.SamonteR. V.SchwartzS.et al. (2002). Recent segmental duplications in the human genome. Science297, 1003–1007. 10.1126/science.1072047
15
BalasubramaniyanV.BoddekeE.BakelsR.KüstB.KooistraS.VenemanA.et al. (2006). Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience143, 939–951. 10.1016/j.neuroscience.2006.08.082
16
Ballaban-GilK.TuchmanR. (2000). Epilepsy and epileptiform EEG: association with autism and language disorders. Ment. Retard Dev. Disabil. Res. Rev.6, 300–308. 10.1002/1098-2779(2000)6:4<300::AID-MRDD9>3.0.CO;2-R
17
BaronioD.CastroK.GonchoroskiT.de MeloG. M.NunesG. D.Bambini-JuniorV.et al. (2015). Effects of an H3R antagonist on the animal model of autism induced by prenatal exposure to valproic acid. PLoS ONE10:e0116363. 10.1371/journal.pone.0116363
18
BaronioD.GonchoroskiT.CastroK.ZanattaG.GottfriedC.RiesgoR. (2014). Histaminergic system in brain disorders: lessons from the translational approach and future perspectives. Ann. Gen. Psychiatry13, 34. 10.1186/s12991-014-0034-y
19
BhattacharyaA.KaphzanH.Alvarez-DieppaA. C.MurphyJ. P.PierreP.KlannE. (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron76, 325–337. 10.1016/j.neuron.2012.07.022
20
BisharaD. (2010). Once-monthly paliperidone injection for the treatment of schizophrenia. Neuropsychiatr. Dis. Treat.6, 561–572. 10.2147/NDT.S8505
21
BoellnerS. W.PennickM.FiskeK.LyneA.ShojaeiA. (2007). Pharmacokinetics of a guanfacine extended-release formulation in children and adolescents with attention-deficit-hyperactivity disorder. Pharmacotherapy27, 1253–1262. 10.1592/phco.27.9.1253
22
BouvardM. P.LeboyerM.LaunayJ. M.RecasensC.PlumetM. H.Waller-PerotteD.et al. (1995). Low-dose naltrexone effects on plasma chemistries and clinical symptoms in autism: a double-blind, placebo-controlled study. Psychiatry Res.58, 191–201. 10.1016/0165-1781(95)02601-R
23
BowtonE.SaundersC.ReddyI. A.CampbellN. G.HamiltonP. J.HenryL. K.et al. (2014). SLC6A3 coding variant Ala559Val found in two autism probands alters dopamine transporter function and trafficking. Transl. Psychiatry4, e464. 10.1038/tp.2014.90
24
BrodkinE. S. (2007). BALB/c mice: low sociability and other phenotypes that may be relevant to autism. Behav. Brain Res.176, 53–65. 10.1016/j.bbr.2006.06.025
25
BrowmanK. E.KomaterV. A.CurzonP.RueterL. E.HancockA. A.DeckerM. W.et al. (2004). Enhancement of prepulse inhibition of startle in mice by the H3 receptor antagonists thioperamide and ciproxifan. Behav. Brain Res.153, 69–76. 10.1016/j.bbr.2003.11.001
26
BrownJ. T.EumS.CookE. H.BishopJ. R. (2017). Pharmacogenomics of autism spectrum disorder. Pharmacogenomics18, 403–414. 10.2217/pgs-2016-0167
27
BrownJ. W.WhiteheadC. A.BassoA. M.RueterL. E.ZhangM. (2013). Preclinical evaluation of non-imidazole histamine H3 receptor antagonists in comparison to atypical antipsychotics for the treatment of cognitive deficits associated with schizophrenia. Int. J. Neuropsychopharmacol.16, 889–904. 10.1017/S1461145712000739
28
BrudzynskiS. M. (2013). Ethotransmission: communication of emotional states through ultrasonic vocalization in rats. Curr. Opin. Neurobiol.23, 310–317. 10.1016/j.conb.2013.01.014
29
BuitingK.WilliamsC.HorsthemkeB. (2016). Angelman syndrome - insights into a rare neurogenetic disorder. Nat. Rev. Neurol.12, 584–593. 10.1038/nrneurol.2016.133
30
CampbellM.FishB.ShapiroT.FloydA.Jr. (1971). Imipramine in preschool autistic and schizophrenic children. J. Autism Child. Schizophr.1, 267–282. 10.1007/BF01557348
31
CarminatiG. G.DeriazN.BertschyG. (2006). Low-dose venlafaxine in three adolescents and young adults with autistic disorder improves self-injurious behavior and attention deficit/hyperactivity disorders (ADHD)-like symptoms. Prog. Neuropsychopharmacol. Biol. Psychiatry30, 312–315. 10.1016/j.pnpbp.2005.10.002
32
CarminatiG. G.GerberF.DarbellayB.KoselM. M.DeriazN.ChabertJ.et al. (2016). Using venlafaxine to treat behavioral disorders in patients with autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry65, 85–95. 10.1016/j.pnpbp.2015.09.002
33
CarrollL. S.OwenM. J. (2009). Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med.1, 102. 10.1186/gm102
34
ChadmanK. K.YangM.CrawleyJ. N. (2009). Criteria for validating mouse models of psychiatric diseases. Am. J. Med. Genet. B Neuropsychiatr. Genet.150B, 1–11. 10.1002/ajmg.b.30777
35
ChangJ.GilmanS. R.ChiangA. H.SandersS. J.VitkupD. (2015). Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci.18, 191–198. 10.1038/nn.3907
36
ChaoH. T.ChenH.SamacoR. C.XueM.ChahrourM.YooJ.et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature468, 263–269. 10.1038/nature09582
37
ChenJ. A.PeñagarikanoO.BelgardT. G.SwarupV.GeschwindD. H. (2015). The emerging picture of autism spectrum disorder: genetics and pathology. Annu. Rev. Pathol.10, 111–144. 10.1146/annurev-pathol-012414-040405
38
ChenN. C.BedairH. S.McKayB.BowersM. B.Jr.MazureC. (2001). Clozapine in the treatment of aggression in an adolescent with autistic disorder. J. Clin. Psychiatry62, 479–480. 10.4088/JCP.v62n0612h
39
ChenR.DavisL. K.GuterS.WeiQ.JacobS.PotterM. H.et al. (2017). Leveraging blood serotonin as an endophenotype to identify de novo and rare variants involved in autism. Mol. Autism8, 14. 10.1186/s13229-017-0130-3
40
ChezM. G.BurtonQ.DowlingT.ChangM.KhannaP.KramerC. (2007). Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol.22, 574–579. 10.1177/0883073807302611
41
ChisholmK.LinA.Abu-AkelA.WoodS. J. (2015). The association between autism and schizophrenia spectrum disorders: a review of eight alternate models of co-occurrence. Neurosci. Biobehav. Rev.55, 173–183. 10.1016/j.neubiorev.2015.04.012
42
ChomiakT.TurnerN.HuB. (2013). What we have learned about autism spectrum disorder from valproic acid. Patholog. Res. Int.2013, 712758. 10.1155/2013/712758
43
ChristianS. L.FantesJ. A.MewbornS. K.HuangB.LedbetterD. H. (1999). Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum. Mol. Genet.8, 1025–1037. 10.1093/hmg/8.6.1025
44
ChristiansonA. L.CheslerN.KrombergJ. G. (1994). Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev. Med. Child Neurol.36, 361–369. 10.1111/j.1469-8749.1994.tb11858.x
45
CliffordS.DissanayakeC.BuiQ. M.HugginsR.TaylorA. K.LoeschD. Z. (2007). Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord.37, 738–747. 10.1007/s10803-006-0205-z
46
CoghlanS.HorderJ.InksterB.MendezM. A.MurphyD. G.NuttD. J. (2012). GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci. Biobehav. Rev.36, 2044–2055. 10.1016/j.neubiorev.2012.07.005
47
CookE. H.Jr.CourchesneR. Y.CoxN. J.LordC.GonenD.GuterS. J.et al. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am. J. Hum. Genet.62, 1077–1083. 10.1086/301832
48
CookE. H.Jr.SchererS. W. (2008). Copy-number variations associated with neuropsychiatric conditions. Nature455, 919–923. 10.1038/nature07458
49
CoruzziG.PozzoliC.AdamiM.GrandiD.GuidoN.SmitsR.et al. (2012). Strain-dependent effects of the histamine H4 receptor antagonist JNJ7777120 in a murine model of acute skin inflammation. Exp. Dermatol.21, 32–37. 10.1111/j.1600-0625.2011.01396.x
50
DavanzoP. A.BelinT. R.WidawskiM. H.KingB. H. (1998). Paroxetine treatment of aggression and self-injury in persons with mental retardation. Am. J. Ment. Retard102, 427–437. 10.1352/0895-8017(1998)102<0427:PTOAAS>2.0.CO;2
51
DavidsonP. W.Cory-SlechtaD. A.ThurstonS. W.HuangL. S.ShamlayeC. F.GunzlerD.et al. (2011). Fish consumption and prenatal methylmercury exposure: cognitive and behavioral outcomes in the main cohort at 17 years from the Seychelles child development study. Neurotoxicology32, 711–717. 10.1016/j.neuro.2011.08.003
52
DeLongG. R.RitchC. R.BurchS. (2002). Fluoxetine response in children with autistic spectrum disorders: correlation with familial major affective disorder and intellectual achievement. Dev. Med. Child Neurol.44, 652–659. 10.1111/j.1469-8749.2002.tb00266.x
53
DevorA.AndreassenO. A.WangY.Mäki-MarttunenT.SmelandO. B.FanC. C.et al. (2017). Genetic evidence for role of integration of fast and slow neurotransmission in schizophrenia. Mol. Psychiatry22, 792–801. 10.1038/mp.2017.33
54
Di MartinoA.MelisG.CianchettiC.ZuddasA. (2004). Methylphenidate for pervasive developmental disorders: safety and efficacy of acute single dose test and ongoing therapy: an open-pilot study. J. Child Adolesc. Psychopharmacol.14, 207–218. 10.1089/1044546041649011
55
DoxakisE. (2010). Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem.285, 12726–12734. 10.1074/jbc.M109.086827
56
EissaN.KhanN.OjhaS. K.LazewskaD.Kiec-KononowiczK.SadekB. (2018). The histamine H3 receptor antagonist DL77 ameliorates MK801-induced memory deficits in rats. Front. Neurosci.12:42. 10.3389/fnins.2018.00042
57
ElchaarG. M.MaischN. M.AugustoL. M.WehringH. J. (2006). Efficacy and safety of naltrexone use in pediatric patients with autistic disorder. Ann. Pharmacother.40, 1086–1095. 10.1345/aph.1G499
58
EllenbroekB. A.AugustC.YounJ. (2016). Does prenatal valproate interact with a genetic reduction in the serotonin transporter? A rat study on anxiety and cognition. Front. Neurosci.10:424. 10.3389/fnins.2016.00424
59
EllenbroekB. A.GhiabiB. (2015). Do Histamine receptor 3 antagonists have a place in the therapy for schizophrenia?Curr. Pharm. Des.21, 3760–3770. 10.2174/1381612821666150605105325
60
ErgazZ.Weinstein-FudimL.OrnoyA. (2016). Genetic and non-genetic animal models for autism spectrum disorders (ASD). Reprod. Toxicol.64, 116–140. 10.1016/j.reprotox.2016.04.024
61
EsbenshadeT. A.BrowmanK. E.BitnerR. S.StrakhovaM.CowartM. D.BrioniJ. D. (2008). The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol.154, 1166–1181. 10.1038/bjp.2008.147
62
Falluel-MorelA.SokolowskiK.SistiH. M.ZhouX.ShorsT. J.Dicicco-BloomE. (2007). Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem.103, 1968–1981. 10.1111/j.1471-4159.2007.04882.x
63
FankhauserM. P.KarumanchiV. C.GermanM. L.YatesA.KarumanchiS. D. (1992). A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J. Clin. Psychiatry53, 77–82.
64
FatemiS. H.HaltA. R.StaryJ. M.KanodiaR.SchulzS. C.RealmutoG. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry52, 805–810. 10.1016/S0006-3223(02)01430-0
65
FatemiS. H.RealmutoG. M.KhanL.ThurasP. (1998). Fluoxetine in treatment of adolescent patients with autism: a longitudinal open trial. J. Autism Dev. Disord.28, 303–307. 10.1023/A:1026008602540
66
FindlingR. L.McNamaraN. K.GraciousB. L.O'RiordanM. A.ReedM. D.DemeterC.et al. (2004). Quetiapine in nine youths with autistic disorder. J. Child Adoles. Psychopharmacol.14, 287–294. 10.1089/1044546041649129
67
FregeacJ.ColleauxL.NguyenL. S. (2016). The emerging roles of MicroRNAs in autism spectrum disorders. Neurosci. Biobehav. Rev.71, 729–738. 10.1016/j.neubiorev.2016.10.018
68
FriedmanS. D.ShawD. W.ArtruA. A.DawsonG.PetropoulosH.DagerS. R. (2006). Gray and white matter brain chemistry in young children with autism. Arch. Gen. Psychiatry63, 786–794. 10.1001/archpsyc.63.7.786
69
GadadB. S.HewitsonL.YoungK. A.GermanD. C. (2013). Neuropathology and animal models of autism: genetic and environmental factors. Autism Res. Treat.2013:731935. 10.1155/2013/731935
70
GarayP. A.HsiaoE. Y.PattersonP. H.McAllisterA. K. (2013). Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav. Immun.31, 54–68. 10.1016/j.bbi.2012.07.008
71
GardenerH.SpiegelmanD.BukaS. L. (2011). Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics128, 344–355. 10.1542/peds.2010-1036
72
GeschwindD. H. (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci.15, 409–416. 10.1016/j.tics.2011.07.003
73
GillbergC.LundströmS.FernellE.NilssonG.NevilleB. (2017). Febrile seizures and epilepsy: association with autism and other neurodevelopmental disorders in the child and adolescent twin study in Sweden. Pediatr. Neurol.74, 80 e82–86 e82. 10.1016/j.pediatrneurol.2017.05.027
74
GizaJ.UrbanskiM. J.PrestoriF.BandyopadhyayB.YamA.FriedrichV.et al. (2010). Behavioral and cerebellar transmission deficits in mice lacking the autism-linked gene islet brain-2. J. Neurosci.30, 14805–14816. 10.1523/JNEUROSCI.1161-10.2010
75
GlassonE. J.BowerC.PettersonB.de KlerkN.ChaneyG.HallmayerJ. F. (2004). Perinatal factors and the development of autism: a population study. Arch. Gen. Psychiatry61, 618–627. 10.1001/archpsyc.61.6.618
76
GobbiG.PulvirentiL. (2001). Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J. Psychiatry Neurosci.26, 340–341.
77
GordonC. T.StateR. C.NelsonJ. E.HamburgerS. D.RapoportJ. L. (1993). A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Arch. Gen. Psychiatry50, 441–447. 10.1001/archpsyc.1993.01820180039004
78
GrandjeanP.Budtz-JørgensenE.WhiteR. F.JørgensenP. J.WeiheP.DebesF.et al. (1999). Methylmercury exposure biomarkers as indicators of neurotoxicity in children aged 7 years. Am. J. Epidemiol.150, 301–305. 10.1093/oxfordjournals.aje.a010002
79
GranonS.FaureP.ChangeuxJ. P. (2003). Executive and social behaviors under nicotinic receptor regulation. Proc. Natl. Acad. Sci. U.S.A.100, 9596–9601. 10.1073/pnas.1533498100
80
GregorA.AlbrechtB.BaderI.BijlsmaE. K.EkiciA. B.EngelsH.et al. (2011). Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med. Genet.12:106. 10.1186/1471-2350-12-106
81
GriebelG.PichatP.PruniauxM. P.BeeskeS.Lopez-GranchaM.GenetE.et al. (2012). SAR110894, a potent histamine H(3)-receptor antagonist, displays procognitive effects in rodents. Pharmacol. Biochem. Behav.102, 203–214. 10.1016/j.pbb.2012.04.004
82
GrozevaD.ConradD. F.BarnesC. P.HurlesM.OwenM. J.O'DonovanM. C.et al. (2012). Independent estimation of the frequency of rare CNVs in the UK population confirms their role in schizophrenia. Schizophr. Res.135, 1–7. 10.1016/j.schres.2011.11.004
83
GuinchatV.ThorsenP.LaurentC.CansC.BodeauN.CohenD. (2012). Pre-, peri- and neonatal risk factors for autism. Acta Obstet. Gynecol. Scand.91, 287–300. 10.1111/j.1600-0412.2011.01325.x
84
HaasH. L.SergeevaO. A.SelbachO. (2008). Histamine in the nervous system. Physiol. Rev.88, 1183–1241. 10.1152/physrev.00043.2007
85
HallmayerJ.ClevelandS.TorresA.PhillipsJ.CohenB.TorigoeT.et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry68, 1095–1102. 10.1001/archgenpsychiatry.2011.76
86
HamiltonP. J.CampbellN. G.SharmaS.ErregerK.Herborg HansenF.SaundersC.et al. (2013). De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry18, 1315–1323. 10.1038/mp.2013.102
87
HandenB. L.JohnsonC. R.LubetskyM. (2000). Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J. Autism Dev. Disord.30, 245–255. 10.1023/A:1005548619694
88
HansonE.KalishL. A.BunceE.CurtisC.McDanielS.WareJ.et al. (2007). Use of complementary and alternative medicine among children diagnosed with autism spectrum disorder. J. Autism Dev. Disord.37, 628–636. 10.1007/s10803-006-0192-0
89
HaraY.AgoY.TarutaA.HasebeS.KawaseH.TanabeW.et al. (2017). Risperidone and aripiprazole alleviate prenatal valproic acid-induced abnormalities in behaviors and dendritic spine density in mice. Psychopharmacology (Berl).234, 3217–3228. 10.1007/s00213-017-4703-9
90
HardanA. Y.HandenB. L. (2002). A retrospective open trial of adjunctive donepezil in children and adolescents with autistic disorder. J. Child Adolesc. Psychopharmacol.12, 237–241. 10.1089/104454602760386923
91
HellingsJ. A.ArnoldL. E.HanJ. C. (2017). Dopamine antagonists for treatment resistance in autism spectrum disorders: review and focus on BDNF stimulators loxapine and amitriptyline. Expert Opin. Pharmacother.18, 581–588. 10.1080/14656566.2017.1308483
92
HellingsJ. A.KelleyL. A.GabrielliW. F.KilgoreE.ShahP. (1996). Sertraline response in adults with mental retardation and autistic disorder. J. Clin. Psychiatry57, 333–336.
93
HellingsJ. A.ReedG.CainS. E.ZhouX.BarthF. X.AmanM. G.et al. (2015). Loxapine add-on for adolescents and adults with autism spectrum disorders and irritability. J. Child Adolesc. Psychopharmacol.25, 150–159. 10.1089/cap.2014.0003
94
HellmerK.NyströmP. (2017). Infant acetylcholine, dopamine, and melatonin dysregulation: neonatal biomarkers and causal factors for ASD and ADHD phenotypes. Med. Hypotheses100, 64–66. 10.1016/j.mehy.2017.01.015
95
HendriksenJ. G.KlinkenbergS.CollinP.WongB.NiksE. H.VlesJ. S. (2016). Diagnosis and treatment of obsessive compulsive behavior in a boy with Duchenne muscular dystrophy and autism spectrum disorder: a case report. Neuromuscul. Disord.26, 659–661. 10.1016/j.nmd.2016.08.003
96
HettingerJ. A.LiuX.SchwartzC. E.MichaelisR. C.HoldenJ. J. (2008). A DRD1 haplotype is associated with risk for autism spectrum disorders in male-only affected sib-pair families. Am. J. Med. Genet. B Neuropsychiatr. Genet.147b, 628–636. 10.1002/ajmg.b.30655
97
HirschL. E.PringsheimT. (2016). Aripiprazole for autism spectrum disorders (ASD). Cochrane Database Syst Rev,Cd009043. 10.1002/14651858.CD009043.pub3
98
HoffmanK.WeisskopfM. G.RobertsA. L.RazR.HartJ. E.LyallK.et al. (2017). Geographic patterns of autism spectrum disorder among children of participants in nurses' health study II. Am. J. Epidemiol.186, 834–842. 10.1093/aje/kwx158
99
HogartA.NagarajanR. P.PatzelK. A.YasuiD. H.LasalleJ. M. (2007). 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet.16, 691–703. 10.1093/hmg/ddm014
100
HogartA.WuD.LaSalleJ. M.SchanenN. C. (2010). The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis.38, 181–191. 10.1016/j.nbd.2008.08.011
101
HollanderE.ChaplinW.SooryaL.WassermanS.NovotnyS.RusoffJ.et al. (2010). Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology35, 990–998. 10.1038/npp.2009.202
102
HollanderE.PhillipsA.ChaplinW.ZagurskyK.NovotnyS.WassermanS.et al. (2005). A placebo controlled crossover trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology30, 582–589. 10.1038/sj.npp.1300627
103
HollanderE.SooryaL.WassermanS.EspositoK.ChaplinW.AnagnostouE. (2006). Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int. J. Neuropsychopharmacol.9, 209–213. 10.1017/S1461145705005791
104
HongM.LeeS. Y.HanJ.ParkJ. C.LeeY. J.HwangboR.et al. (2017). Prescription trends of psychotropics in children and adolescents with autism based on nationwide health insurance data. J. Korean Med. Sci.32, 1687–1693. 10.3346/jkms.2017.32.10.1687
105
HowesO. D.RogdakiM.FindonJ. L.WichersR. H.CharmanT.KingB. H.et al. (2018). Autism spectrum disorder: consensus guidelines on assessment, treatment and research from the British Association for psychopharmacology. J. Psychopharmacol.32, 3–29. 10.1177/0269881117741766
106
HsiaoE. Y.McBrideS. W.ChowJ.MazmanianS. K.PattersonP. H. (2012). Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. U.S.A.109, 12776–12781. 10.1073/pnas.1202556109
107
HsiehG. C.ChandranP.SalyersA. K.PaiM.ZhuC. Z.WensinkE. J.et al. (2010). H4 receptor antagonism exhibits anti-nociceptive effects in inflammatory and neuropathic pain models in rats. Pharmacol. Biochem. Behav.95, 41–50. 10.1016/j.pbb.2009.12.004
108
HsiehJ.GageF. H. (2004). Epigenetic control of neural stem cell fate. Curr. Opin. Genet. Dev.14, 461–469. 10.1016/j.gde.2004.07.006
109
HsiehJ.GageF. H. (2005). Chromatin remodeling in neural development and plasticity. Curr. Opin. Cell Biol.17, 664–671. 10.1016/j.ceb.2005.09.002
110
HuY.EhliE. A.BoomsmaD. I. (2017). MicroRNAs as biomarkers for psychiatric disorders with a focus on autism spectrum disorder: current progress in genetic association studies, expression profiling, and translational research. Autism Res.10, 1184–1203. 10.1002/aur.1789
111
HulbertS. W.JiangY. H. (2016). Monogenic mouse models of autism spectrum disorders: common mechanisms and missing links. Neuroscience321, 3–23. 10.1016/j.neuroscience.2015.12.040
112
HviidA.StellfeldM.WohlfahrtJ.MelbyeM. (2003). Association between thimerosal-containing vaccine and autism. JAMA290, 1763–1766. 10.1001/jama.290.13.1763
113
JahromiL. B.KasariC. L.McCrackenJ. T.LeeL. S.AmanM. G.McDougleC. J.et al. (2009). Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J. Autism Dev. Disord.39, 395–404. 10.1007/s10803-008-0636-9
114
JamainS.BetancurC.QuachH.PhilippeA.FellousM.GirosB.et al. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry7, 302–310. 10.1038/sj.mp.4000979
115
JamesS. J.MelnykS.FuchsG.ReidT.JerniganS.PavlivO.et al. (2009). Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am. J. Clin. Nutr.89, 425–430. 10.3945/ajcn.2008.26615
116
JessbergerS.NakashimaK.ClemensonG. D.Jr.MejiaE.MathewsE.UreK.et al. (2007). Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci.27, 5967–5975. 10.1523/JNEUROSCI.0110-07.2007
117
JobskiK.HöferJ.HoffmannF.BachmannC. (2017). Use of psychotropic drugs in patients with autism spectrum disorders: a systematic review. Acta Psychiatr. Scand.135, 8–28. 10.1111/acps.12644
118
KarvatG.KimchiT. (2014). Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology39, 831–840. 10.1038/npp.2013.274
119
KataokaS.TakumaK.HaraY.MaedaY.AgoY.MatsudaT. (2013). Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol.16, 91–103. 10.1017/S1461145711001714
120
KatzD. M.Berger-SweeneyJ. E.EubanksJ. H.JusticeM. J.NeulJ. L.Pozzo-MillerL.et al. (2012). Preclinical research in Rett syndrome: setting the foundation for translational success. Dis. Model. Mech.5, 733–745. 10.1242/dmm.011007
121
KazdobaT. M.LeachP. T.YangM.SilvermanJ. L.SolomonM.CrawleyJ. N. (2016). Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr. Top. Behav. Neurosci.28, 1–52. 10.1007/7854_2015_5003
122
KemperT. L.BaumanM. (1998). Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol.57, 645–652. 10.1097/00005072-199807000-00001
123
KernJ. K.JonesA. M. (2006). Evidence of toxicity, oxidative stress, and neuronal insult in autism. J. Toxicol. Environ. Health B Crit. Rev.9, 485–499. 10.1080/10937400600882079
124
KimS. J.ShonkaS.FrenchW. P.StricklandJ.MillerL.SteinM. A. (2017). Dose-response effects of long-acting liquid methylphenidate in children with Attention Deficit/Hyperactivity Disorder (ADHD) and Autism Spectrum Disorder (ASD): a pilot study. J. Autism Dev. Disord.47, 2307–2313. 10.1007/s10803-017-3125-1
125
KingB. H.WrightD. M.HandenB. L.SikichL.ZimmermanA. W.McMahonW.et al. (2001). Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry40, 658–665. 10.1097/00004583-200106000-00010
126
KishinoT.LalandeM.WagstaffJ. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet.15, 70–73. 10.1038/ng0197-70
127
KnightS. J.ReganR.NicodA.HorsleyS. W.KearneyL.HomfrayT.et al. (1999). Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet354, 1676–1681. 10.1016/S0140-6736(99)03070-6
128
KolevzonA.GrossR.ReichenbergA. (2007). Prenatal and perinatal risk factors for autism: a review and integration of findings. Arch. Pediatr. Adolesc. Med.161, 326–333. 10.1001/archpedi.161.4.326
129
KolmenB. K.FeldmanH. M.HandenB. L.JanoskyJ. E. (1995). Naltrexone in young autistic children: a double-blind, placebo-controlled crossover study. J. Am. Acad. Child Adolesc. Psychiatry34, 223–231. 10.1097/00004583-199502000-00018
130
KongA.FriggeM. L.MassonG.BesenbacherS.SulemP.MagnussonG.et al. (2012). Rate of de novo mutations and the importance of father's age to disease risk. Nature488, 471–475. 10.1038/nature11396
131
KonstantareasM. M.HewittT. (2001). Autistic disorder and schizophrenia: diagnostic overlaps. J. Autism Dev. Disord.31, 19–28. 10.1023/A:1005605528309
132
KronM.HowellC. J.AdamsI. T.RansbottomM.ChristianD.OgierM.et al. (2012). Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J. Neurosci.32, 13860–13872. 10.1523/JNEUROSCI.2159-12.2012
133
KumarB.PrakashA.SewalR. K.MedhiB.ModiM. (2012). Drug therapy in autism: a present and future perspective. Pharmacol. Rep.64, 1291–1304. 10.1016/S1734-1140(12)70927-1
134
KurtisL. B. (1966). Clinical study of the response to nortriptyline on autistic children. Int. J. Neuropsychiatry2, 298–301.
135
LaSalleJ. M.ReiterL. T.ChamberlainS. J. (2015). Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics7, 1213–1228. 10.2217/epi.15.70
136
LeblondC. S.HeinrichJ.DelormeR.ProepperC.BetancurC.HuguetG.et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet.8:e1002521. 10.1371/journal.pgen.1002521
137
LeeJ.KimB. H.KimE.HowesO. D.ChoK. I. K.YoonY. B.et al. (2018). Higher serotonin transporter availability in early-onset obsessive-compulsive disorder patients undergoing escitalopram treatment: a [(11) C]DASB PET study. Hum. Psychopharmacol.33:e2642. 10.1002/hup.2642
138
LeeY.KimH.KimJ. E.ParkJ. Y.ChoiJ.LeeJ. E.et al. (2017). Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol. Neurobiol.12, 1–14. 10.1007/s12035-017-0770-5
139
LejeuneJ.GautierM.TurpinR. (1959). [Study of somatic chromosomes from 9 mongoloid children]. C. R. Hebd. Seances Acad. Sci.248, 1721–1722.
140
LevisohnP. M. (2007). The autism-epilepsy connection. Epilepsia48(Suppl. 9), 33–35. 10.1111/j.1528-1167.2007.01399.x
141
LibbeyJ. E.SweetenT. L.McMahonW. M.FujinamiR. S. (2005). Autistic disorder and viral infections. J. Neurovirol.11, 1–10. 10.1080/13550280590900553
142
LigneauX.LinJ.Vanni-MercierG.JouvetM.MuirJ. L.GanellinC. R.et al. (1998). Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. J. Pharmacol. Exp. Ther.287, 658–666.
143
LordC.CookE. H.LeventhalB. L.AmaralD. G. (2000). Autism spectrum disorders. Neuron28, 355–363. 10.1016/S0896-6273(00)00115-X
144
LyallK.CroenL.DanielsJ.FallinM. D.Ladd-AcostaC.LeeB. K.et al. (2017). The changing epidemiology of autism spectrum disorders. Annu. Rev. Public Health38, 81–102. 10.1146/annurev-publhealth-031816-044318
145
MahmoodD.KhanamR.PillaiK. K.AkhtarM. (2012). Reversal of oxidative stress by histamine H3 receptor-ligands in experimental models of schizophrenia. Arzneimittelforschung.62, 222–229. 10.1055/s-0031-1301326
146
MaramaraL. A.HeW.MingX. (2014). Pre- and perinatal risk factors for autism spectrum disorder in a New Jersey cohort. J. Child Neurol.29, 1645–1651. 10.1177/0883073813512899
147
MarcusR. N.OwenR.KamenL.ManosG.McQuadeR. D.CarsonW. H.et al. (2009). A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry48, 1110–1119. 10.1097/CHI.0b013e3181b76658
148
MartinA.KoenigK.AndersonG. M.ScahillL. (2003). Low-dose fluvoxamine treatment of children and adolescents with pervasive developmental disorders: a prospective, open-label study. J. Autism Dev. Disord.33, 77–85. 10.1023/A:1022234605695
149
MatsonJ. L.SipesM.FodstadJ. C.FitzgeraldM. E. (2011). Issues in the management of challenging behaviours of adults with autism spectrum disorder. CNS Drugs25, 597–606. 10.2165/11591700-000000000-00000
150
MatsuuraT.SutcliffeJ. S.FangP.GaljaardR. J.JiangY. H.BentonC. S.et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet.15, 74–77. 10.1038/ng0197-74
151
McConvilleB. J.SanbergP. R.FogelsonM. H.KingJ.CirinoP.ParkerK. W.et al. (1992). The effects of nicotine plus haloperidol compared to nicotine only and placebo nicotine only in reducing tic severity and frequency in Tourette's disorder. Biol. Psychiatry31, 832–840. 10.1016/0006-3223(92)90315-Q
152
McCoolM. F.PatelS.TalatiR.RagozzinoM. E. (2008). Differential involvement of M1-type and M4-type muscarinic cholinergic receptors in the dorsomedial striatum in task switching. Neurobiol. Learn. Mem.89, 114–124. 10.1016/j.nlm.2007.06.005
153
McCrackenJ. T.McGoughJ.ShahB.CroninP.HongD.AmanM. G.et al. (2002). Risperidone in children with autism and serious behavioral problems. N. Engl. J. Med.347, 314–321. 10.1056/NEJMoa013171
154
McDougleC. J.BrodkinE. S.NaylorS. T.CarlsonD. C.CohenD. J.PriceL. H. (1998a). Sertraline in adults with pervasive developmental disorders: a prospective open-label investigation. J. Clin. Psychopharmacol.18, 62–66. 10.1097/00004714-199802000-00010
155
McDougleC. J.HolmesJ. P.CarlsonD. C.PeltonG. H.CohenD. J.PriceL. H. (1998b). A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch. Gen. Psychiatry55, 633–641. 10.1001/archpsyc.55.7.633
156
McDougleC. J.NaylorS. T.CohenD. J.VolkmarF. R.HeningerG. R.PriceL. H. (1996). A double-blind, placebo-controlled study of fluvoxamine in adults with autistic disorder. Arch. Gen. Psychiatry53, 1001–1008. 10.1001/archpsyc.1996.01830110037005
157
McFarlaneH. G.KusekG. K.YangM.PhoenixJ. L.BolivarV. J.CrawleyJ. N. (2008). Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav.7, 152–163. 10.1111/j.1601-183X.2007.00330.x
158
MeffordH. C.CooperG. M.ZerrT.SmithJ. D.BakerC.ShaferN.et al. (2009). A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res.19, 1579–1585. 10.1101/gr.094987.109
159
MikhailF. M.LoseE. J.RobinN. H.DescartesM. D.RutledgeK. D.RutledgeS. L.et al. (2011). Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med. Genet. A155a, 2386–2396. 10.1002/ajmg.a.34177
160
MingX.GordonE.KangN.WagnerG. C. (2008). Use of clonidine in children with autism spectrum disorders. Brain Dev.30, 454–460. 10.1016/j.braindev.2007.12.007
161
MooreM. L.EichnerS. F.JonesJ. R. (2004). Treating functional impairment of autism with selective serotonin-reuptake inhibitors. Ann. Pharmacother.38, 1515–1519. 10.1345/aph.1D543
162
MooreS. J.TurnpennyP.QuinnA.GloverS.LloydD. J.MontgomeryT.et al. (2000). A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet.37, 489–497. 10.1136/jmg.37.7.489
163
MoyS. S.NadlerJ. J.PoeM. D.NonnemanR. J.YoungN. B.KollerB. H.et al. (2008). Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav. Brain Res.188, 178–194. 10.1016/j.bbr.2007.10.029
164
MoyS. S.NadlerJ. J.YoungN. B.PerezA.HollowayL. P.BarbaroR. P.et al. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res.176, 4–20. 10.1016/j.bbr.2006.07.030
165
MuhleR.TrentacosteS. V.RapinI. (2004). The genetics of autism. Pediatrics113, e472–486. 10.1542/peds.113.5.e472
166
Mukaetova-LadinskaE. B. (2017). Silent lives: why do we fail community-dwelling people with dementia?Age Ageing46, 341–343. 10.1093/ageing/afx028
167
NaaijenJ.BraltenJ.PoelmansG.consortiumI.GlennonJ. C.FrankeB.et al. (2017). Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: association to overlapping traits in ADHD and autism. Transl. Psychiatry7, e999. 10.1038/tp.2016.273
168
NaddafiF.MirshafieyA. (2013). The neglected role of histamine in Alzheimer's disease. Am. J. Alzheimers. Dis. Other Demen.28, 327–336. 10.1177/1533317513488925
169
NakaiN.NaganoM.SaitowF.WatanabeY.KawamuraY.KawamotoA.et al. (2017). Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci. Adv.3:e1603001. 10.1126/sciadv.1603001
170
NashK.CarterK. J. (2016). Treatment options for the management of pervasive developmental disorders. Int. J. Psychiatry Med.51, 201–210. 10.1177/0091217416636600
171
NestlerE. J.HymanS. E. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci.13, 1161–1169. 10.1038/nn.2647
172
NguyenM.RothA.KyzarE. J.PoudelM. K.WongK.StewartA. M.et al. (2014). Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD). Neurochem. Int.66, 15–26. 10.1016/j.neuint.2014.01.002
173
NicoliniC.FahnestockM. (2017). The valproic acid-induced rodent model of autism. Exp. Neurol. 299(Pt A):217–227. 10.1016/j.expneurol.2017.04.017
174
NicolsonR.Craven-ThussB.SmithJ. (2006). A prospective, open-label trial of galantamine in autistic disorder. J. Child Adolesc. Psychopharmacol.16, 621–629. 10.1089/cap.2006.16.621
175
NiederhoferH.StaffenW.MairA. (2002). Galantamine may be effective in treating autistic disorder. BMJ325:1422. 10.1136/bmj.325.7377.1422/a
176
NordA. S.RoebW.DickelD. E.WalshT.KusendaM.O'ConnorK. L.et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet.19, 727–731. 10.1038/ejhg.2011.24
177
OgataH.IharaH.MurakamiN.GitoM.KidoY.NagaiT. (2014). Autism spectrum disorders and hyperactive/impulsive behaviors in Japanese patients with Prader-Willi syndrome: a comparison between maternal uniparental disomy and deletion cases. Am. J. Med. Genet. A164A, 2180–2186. 10.1002/ajmg.a.36615
178
OwenR.SikichL.MarcusR. N.Corey-LisleP.ManosG.McQuadeR. D.et al. (2009). Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics124, 1533–1540. 10.1542/peds.2008-3782
179
OwleyT.SaltJ.GuterS.GrieveA.WaltonL.AyuyaoN.et al. (2006). A prospective, open-label trial of memantine in the treatment of cognitive, behavioral, and memory dysfunction in pervasive developmental disorders. J. Child Adolesc. Psychopharmacol.16, 517–524. 10.1089/cap.2006.16.517
180
OwleyT.WaltonL.SaltJ.GuterS. J.Jr.WinnegaM.LeventhalB. L.et al. (2005). An open-label trial of escitalopram in pervasive developmental disorders. J. Am. Acad. Child Adolesc. Psychiatry44, 343–348. 10.1097/01.chi.0000153229.80215.a0
181
PankseppJ.LensingP. (1991). Brief report: a synopsis of an open-trial of naltrexone treatment of autism with four children. J. Autism Dev. Disord.21, 243–249. 10.1007/BF02284764
182
PasciutoE.BorrieS. C.KanellopoulosA. K.SantosA. R.CappuynsE.D'AndreaL.et al. (2015). Autism spectrum disorders: translating human deficits into mouse behavior. Neurobiol. Learn. Mem.124, 71–87. 10.1016/j.nlm.2015.07.013
183
PattersonP. H. (2012). Maternal infection and autism. Brain Behav. Immun.26, 393. 10.1016/j.bbi.2011.09.008
184
PavalD. (2017). A dopamine hypothesis of autism spectrum disorder. Dev. Neurosci.39, 355–360. 10.1159/000478725
185
PavalD.RadF.RusuR.NiculaeA. S.ColosiH. A.DobrescuI.et al. (2017). Low retinal dehydrogenase 1 (RALDH1) level in prepubertal boys with autism spectrum disorder: a possible link to dopamine dysfunction?Clin. Psychopharmacol. Neurosci.15, 229–236. 10.9758/cpn.2017.15.3.229
186
PeñagarikanoO.AbrahamsB. S.HermanE. I.WindenK. D.GdalyahuA.DongH.et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell147, 235–246. 10.1016/j.cell.2011.08.040
187
PerryE. K.LeeM. L.Martin-RuizC. M.CourtJ. A.VolsenS. G.MerritJ.et al. (2001). Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am. J. Psychiatry158, 1058–1066. 10.1176/appi.ajp.158.7.1058
188
PhelanK.McDermidH. E. (2012). The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol.2, 186–201. 10.1159/000334260
189
PiochonC.KlothA. D.GrasselliG.TitleyH. K.NakayamaH.HashimotoK.et al. (2015). Corrigendum: cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun. 6, 6014. 10.1038/ncomms7014
190
PoseyD. J.PuntneyJ. I.SasherT. M.KemD. L.McDougleC. J. (2004). Guanfacine treatment of hyperactivity and inattention in pervasive developmental disorders: a retrospective analysis of 80 cases. J. Child Adolesc. Psychopharmacol.14, 233–241. 10.1089/1044546041649084
191
PurcellA. E.JeonO. H.ZimmermanA. W.BlueM. E.PevsnerJ. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology57, 1618–1628. 10.1212/WNL.57.9.1618
192
QianY.ChenM.ForssbergH.Diaz HeijtzR. (2013). Genetic variation in dopamine-related gene expression influences motor skill learning in mice. Genes Brain Behav.12, 604–614. 10.1111/gbb.12062
193
RagozzinoM. E.PalS. N.UnickK.StefaniM. R.GoldP. E. (1998). Modulation of hippocampal acetylcholine release and spontaneous alternation scores by intrahippocampal glucose injections. J. Neurosci.18, 1595–1601. 10.1523/JNEUROSCI.18-04-01595.1998
194
RamalingamA.ZhouX. G.FiedlerS. D.BrawnerS. J.JoyceJ. M.LiuH. Y.et al. (2011). 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J. Hum. Genet.56, 541–544. 10.1038/jhg.2011.42
195
RaoP.BenitoE.FischerA. (2013). MicroRNAs as biomarkers for CNS disease. Front. Mol. Neurosci.6:39. 10.3389/fnmol.2013.00039
196
RavnanJ. B.TepperbergJ. H.PapenhausenP.LambA. N.HedrickJ.EashD.et al. (2006). Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet.43, 478–489. 10.1136/jmg.2005.036350
197
RobinsonW. P.BernasconiF.MutiranguraA.LedbetterD. H.LangloisS.MalcolmS.et al. (1993a). Nondisjunction of chromosome 15: origin and recombination. Am. J. Hum. Genet.53, 740–751.
198
RobinsonW. P.BinkertF.GinéR.VazquezC.MüllerW.RosenkranzW.et al. (1993b). Clinical and molecular analysis of five inv dup patients. Eur. J. Hum. Genet.1, 37–50. 10.1159/000472386
199
RobinsonW. P.SpiegelR.SchinzelA. A. (1993c). Deletion breakpoints associated with the Prader-Willi and Angelman syndromes (15q11-q13) are not sites of high homologous recombination. Hum. Genet.91, 181–184. 10.1007/BF00222722
200
RodierP. M.IngramJ. L.TisdaleB.CroogV. J. (1997). Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol.11, 417–422. 10.1016/S0890-6238(97)80001-U
201
RonesiJ. A.CollinsK. A.HaysS. A.TsaiN. P.GuoW.BirnbaumS. G.et al. (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci.15, 431. s431–440. s431. 10.1038/nn.3033
202
RuginoT. A.SamsockT. C. (2002). Levetiracetam in autistic children: an open-label study. J. Dev. Behav. Pediatr.23, 225–230. 10.1097/00004703-200208000-00006
203
RuhelaR. K.PrakashA.MedhiB. (2015). An urgent need for experimental animal model of autism in drug development. Ann Neurosci22, 44–49. 10.5214/ans.0972.7531.220210
204
SadekB.StarkH. (2015). Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology106, 56–73. 10.1016/j.neuropharm.2015.11.005
205
SadekB.SaadA.SadeqA.JalalF.StarkH. (2016). Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav. Brain Res.312, 415–430. 10.1016/j.bbr.2016.06.051
206
SahooS.PadhyS. K.SinglaN.SinghA. (2017). Effectiveness of clozapine for the treatment of psychosis and disruptive behaviour in a child with Atypical Autism: a case report and a brief review of the evidence. Asian J. Psychiatr.29, 194–195. 10.1016/j.ajp.2017.07.012
207
SamacoR. C.McGrawC. M.WardC. S.SunY.NeulJ. L.ZoghbiH. Y. (2013). Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet.22, 96–109. 10.1093/hmg/dds406
208
SanchezL. E.CampbellM.SmallA. M.CuevaJ. E.ArmenterosJ. L.AdamsP. B. (1996). A pilot study of clomipramine in young autistic children. J. Am. Acad. Child Adolesc. Psychiatry35, 537–544. 10.1097/00004583-199604000-00021
209
SatheN.AndrewsJ. C.McPheetersM. L.WarrenZ. E. (2017). Nutritional and dietary interventions for autism spectrum disorder: a systematic review. Pediatrics139:e20170346. 10.1542/peds.2017-0346
210
ScahillL.AmanM. G.McDougleC. J.McCrackenJ. T.TierneyE.DziuraJ.et al. (2006). A prospective open trial of guanfacine in children with pervasive developmental disorders. J. Child Adolesc. Psychopharmacol.16, 589–598. 10.1089/cap.2006.16.589
211
SchneiderT.RomanA.Basta-KaimA.KuberaM.BudziszewskaB.SchneiderK.et al. (2008). Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology33, 728–740. 10.1016/j.psyneuen.2008.02.011
212
Scott-Van ZeelandA. A.AbrahamsB. S.Alvarez-RetuertoA. I.SonnenblickL. I.RudieJ. D.GhahremaniD.et al. (2010). Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci. Transl. Med.2, 56ra80. 10.1126/scitranslmed.3001344
213
SeemanP. (2010). Dopamine D2 receptors as treatment targets in schizophrenia. Clin. Schizophr. Relat. Psychoses4, 56–73. 10.3371/CSRP.4.1.5
214
ShahA.WingL. (2006). Psychological approaches to chronic catatonia-like deterioration in autism spectrum disorders. Int. Rev. Neurobiol.72, 245–264. 10.1016/S0074-7742(05)72015-8
215
ShanL.BossersK.LuchettiS.BalesarR.LethbridgeN.ChazotP. L.et al. (2012). Alterations in the histaminergic system in the substantia nigra and striatum of Parkinson's patients: a postmortem study. Neurobiol. Aging33, 1488.e1481–1488.e1413. 10.1016/j.neurobiolaging.2011.10.016
216
SheldrickR. C.CarterA. S. (2018). State-level trends in the prevalence of Autism Spectrum Disorder (ASD) from 2000 to 2012: A reanalysis of findings from the autism and developmental disabilities network. J. Autism Dev. Disord. [Epub ahead of print]. 10.1007/s10803-018-3568-z
217
ShiL.SmithS. E.MalkovaN.TseD.SuY.PattersonP. H. (2009). Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun.23, 116–123. 10.1016/j.bbi.2008.07.012
218
SoghomonianJ. J.ZhangK.ReprakashS.BlattG. J. (2017). Decreased parvalbumin mRNA levels in cerebellar Purkinje cells in autism. Autism Res. 10, 1787–1796. 10.1002/aur.1835
219
SokolD. K.DunnD. W.Edwards-BrownM.FeinbergJ. (2002). Hydrogen proton magnetic resonance spectroscopy in autism: preliminary evidence of elevated choline/creatine ratio. J. Child Neurol.17, 245–249. 10.1177/088307380201700401
220
StarkH.SadekB.KrauseM.HülsA.LigneauX.GanellinC. R.et al. (2000). Novel histamine H(3)-receptor antagonists with carbonyl-substituted 4-(3-(phenoxy)propyl)-1H-imidazole structures like ciproxifan and related compounds. J. Med. Chem.43, 3987–3994. 10.1021/jm000966l
221
SteeleS. D.MinshewN. J.LunaB.SweeneyJ. A. (2007). Spatial working memory deficits in autism. J. Autism Dev. Disord.37, 605–612. 10.1007/s10803-006-0202-2
222
StephanD. A. (2008). Unraveling autism. Am. J. Hum. Genet.82, 7–9. 10.1016/j.ajhg.2007.12.003
223
StrömlandK.NordinV.MillerM.AkerströmB.GillbergC. (1994). Autism in thalidomide embryopathy: a population study. Dev. Med. Child Neurol.36, 351–356. 10.1111/j.1469-8749.1994.tb11856.x
224
StubbsG.HenleyK.GreenJ. (2016). Autism: will vitamin D supplementation during pregnancy and early childhood reduce the recurrence rate of autism in newborn siblings?Med. Hypotheses88, 74–78. 10.1016/j.mehy.2016.01.015
225
TabetA. C.VerloesA.PilorgeM.DelabyE.DelormeR.NygrenG.et al. (2015). Complex nature of apparently balanced chromosomal rearrangements in patients with autism spectrum disorder. Mol. Autism6, 19. 10.1186/s13229-015-0015-2
226
TakumaK.HaraY.KataokaS.KawanaiT.MaedaY.WatanabeR.et al. (2014). Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacol. Biochem. Behav.126, 43–49. 10.1016/j.pbb.2014.08.013
227
TiligadaE.KyriakidisK.ChazotP. L.PassaniM. B. (2011). Histamine pharmacology and new CNS drug targets. CNS Neurosci. Ther.17, 620–628. 10.1111/j.1755-5949.2010.00212.x
228
UllmannR.TurnerG.KirchhoffM.ChenW.TongeB.RosenbergC.et al. (2007). Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum. Mutat.28, 674–682. 10.1002/humu.20546
229
UvebrantP.BauzienèR. (1994). Intractable epilepsy in children. The efficacy of lamotrigine treatment, including non-seizure-related benefits. Neuropediatrics25, 284–289. 10.1055/s-2008-1073041
230
Veenstra-VanderWeeleJ.MullerC. L.IwamotoH.SauerJ. E.OwensW. A.ShahC. R.et al. (2012). Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. U.S.A.109, 5469–5474. 10.1073/pnas.1112345109
231
ViktorinA.UherR.ReichenbergA.LevineS. Z.SandinS. (2017). Autism risk following antidepressant medication during pregnancy. Psychol. Med.47, 2787–2796. 10.1017/S0033291717001301
232
VoineaguI.WangX.JohnstonP.LoweJ. K.TianY.HorvathS.et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature474, 380–384. 10.1038/nature10110
233
VolkmarF.CookE. H.Jr.PomeroyJ.RealmutoG.TanguayP. (1999). Practice parameters for the assessment and treatment of children, adolescents, and adults with autism and other pervasive developmental disorders. American Academy of child and adolescent psychiatry working group on quality issues. J. Am. Acad. Child Adolesc. Psychiatry38(12 Suppl.), 32s–54s. 10.1016/S0890-8567(99)80003-3
234
von CoburgY.KottkeT.WeizelL.LigneauX.StarkH. (2009). Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett.19, 538–542. 10.1016/j.bmcl.2008.09.012
235
VorstmanJ. A. S.ParrJ. R.Moreno-De-LucaD.AnneyR. J. L.NurnbergerJ. I.Jr.HallmayerJ. F. (2017). Autism genetics: opportunities and challenges for clinical translation. Nat. Rev. Genet.18, 362–376. 10.1038/nrg.2017.4
236
WalkerB. R.DiefenbachK. S.ParikhT. N. (2007). Inhibition within the nucleus tractus solitarius (NTS) ameliorates environmental exploration deficits due to cerebellum lesions in an animal model for autism. Behav. Brain Res.176, 109–120. 10.1016/j.bbr.2006.08.008
237
WangH.LiuJ.ZongY.XuY.DengW.ZhuH.et al. (2010). miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer's disease targets TGF-beta type II receptor. Brain Res.1357, 166–174. 10.1016/j.brainres.2010.08.023
238
WangL.AlmeidaL. E.SpornickN. A.KenyonN.KamimuraS.KhaibullinaA.et al. (2015). Modulation of social deficits and repetitive behaviors in a mouse model of autism: the role of the nicotinic cholinergic system. Psychopharmacology (Berl)232, 4303–4316. 10.1007/s00213-015-4058-z
239
WeiH.YuanY.LiuS.WangC.YangF.LuZ.et al. (2015). Detection of circulating miRNA levels in schizophrenia. Am. J. Psychiatry172, 1141–1147. 10.1176/appi.ajp.2015.14030273
240
WeitlaufA. S.SatheN.McPheetersM. L.WarrenZ. E. (2017). Interventions targeting sensory challenges in autism spectrum disorder: a systematic review. Pediatrics139:e20170347. 10.1542/peds.2017-0347
241
WetmoreD. Z.GarnerC. C. (2010). Emerging pharmacotherapies for neurodevelopmental disorders. J. Dev. Behav. Pediatr.31, 564–581. 10.1097/DBP.0b013e3181ee3833
242
Willem VerhoevenJ. E. A. I. F. (2011). Autism Spectrum Disorders: The Role of Genetics in Diagnosis and Treatment. Rijeka: InTech Janeza Trdine.
243
WitkinJ. M.NelsonD. L. (2004). Selective histamine H3 receptor antagonists for treatment of cognitive deficiencies and other disorders of the central nervous system. Pharmacol. Ther.103, 1–20. 10.1016/j.pharmthera.2004.05.001
244
WongH. H.SmithR. G. (2006). Patterns of complementary and alternative medical therapy use in children diagnosed with autism spectrum disorders. J. Autism Dev. Disord.36, 901–909. 10.1007/s10803-006-0131-0
245
WrightC.ShinJ. H.RajpurohitA.Deep-SoboslayA.Collado-TorresL.BrandonN. J.et al. (2017). Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry7:e1126. 10.1038/tp.2017.87
246
XieS.HeuvelmanH.MagnussonC.RaiD.LyallK.NewschafferC. J.et al. (2017). Prevalence of autism spectrum disorders with and without intellectual disability by gestational age at birth in the Stockholm youth cohort: a register linkage study. Paediatr. Perinat. Epidemiol.31, 586–594. 10.1111/ppe.12413
247
XuG.StrathearnL.LiuB.BaoW. (2018). Prevalence of Autism spectrum disorder among US children and adolescents, 2014–2016. JAMA319, 81–82. 10.1001/jama.2017.17812
248
XuX. L.ZongR.LiZ.BiswasM. H.FangZ.NelsonD. L.et al. (2011). FXR1P but not FMRP regulates the levels of mammalian brain-specific microRNA-9 and microRNA-124. J. Neurosci.31, 13705–13709. 10.1523/JNEUROSCI.2827-11.2011
249
ZuddasA.LeddaM. G.FrattaA.MugliaP.CianchettiC. (1996). Clinical effects of clozapine on autistic disorder. Am. J. Psychiatry153, 738. 10.1176/ajp.153.5.738a
Summary
Keywords
autistic spectrum disorder, genetic factors, environmental factors, neurotransmitter dysfunctions, neurodegeneration, neuroprotection, cognitive deficits, pharmacological intervention
Citation
Eissa N, Al-Houqani M, Sadeq A, Ojha SK, Sasse A and Sadek B (2018) Current Enlightenment About Etiology and Pharmacological Treatment of Autism Spectrum Disorder. Front. Neurosci. 12:304. doi: 10.3389/fnins.2018.00304
Received
30 October 2017
Accepted
19 April 2018
Published
16 May 2018
Volume
12 - 2018
Edited by
Francisco Lopez-Munoz, Universidad Camilo José Cela, Spain
Reviewed by
Dr. Girish Kumar Gupta, Maharishi Markandeshwar University, Mullana, India; Mohamed M. Abdel-Daim, Suez Canal University, Egypt
Updates
Copyright
© 2018 Eissa, Al-Houqani, Sadeq, Ojha, Sasse and Sadek.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bassem Sadek bassem.sadek@uaeu.ac.ae
This article was submitted to Neuropharmacology, a section of the journal Frontiers in Neuroscience
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.