 Amanda L. Lumsden1,2†
Amanda L. Lumsden1,2† Jack T. Rogers3†
Jack T. Rogers3† Shohreh Majd4†
Shohreh Majd4† Morgan Newman5
Morgan Newman5 Greg T. Sutherland6
Greg T. Sutherland6 Giuseppe Verdile7
Giuseppe Verdile7 Michael Lardelli5*
Michael Lardelli5*- 1College of Medicine and Public Health, Flinders University, Adelaide, SA, Australia
- 2South Australian Health and Medical Research Institute, Adelaide, SA, Australia
- 3Neurochemistry Laboratory, Department of Psychiatry-Neuroscience, Massachusetts General Hospital (East), Harvard Medical School, Harvard University, Charlestown, MA, United States
- 4Neuronal Injury and Repair Laboratory, Centre for Neuroscience, College of Medicine and Public Health, Flinders University, Adelaide, SA, Australia
- 5Centre for Molecular Pathology, School of Biological Sciences, University of Adelaide, Adelaide, SA, Australia
- 6Discipline of Pathology, Sydney Medical School, University of Sydney, Sydney, NSW, Australia
- 7School of Pharmacy and Biomedical Sciences, Faculty of Health Sciences, Curtin Health Innovation Research Institute, Curtin University, Bentley, WA, Australia
The overwhelming majority of dominant mutations causing early onset familial Alzheimer’s disease (EOfAD) occur in only three genes, PSEN1, PSEN2, and APP. An effect-in-common of these mutations is alteration of production of the APP-derived peptide, amyloid β (Aβ). It is this key fact that underlies the authority of the Amyloid Hypothesis that has informed Alzheimer’s disease research for over two decades. Any challenge to this authority must offer an alternative explanation for the relationship between the PSEN genes and APP. In this paper, we explore one possible alternative relationship – the dysregulation of cellular iron homeostasis as a common effect of EOfAD mutations in these genes. This idea is attractive since it provides clear connections between EOfAD mutations and major characteristics of Alzheimer’s disease such as dysfunctional mitochondria, vascular risk factors/hypoxia, energy metabolism, and inflammation. We combine our ideas with observations by others to describe a “Stress Threshold Change of State” model of Alzheimer’s disease that may begin to explain the existence of both EOfAD and late onset sporadic (LOsAD) forms of the disease. Directing research to investigate the role of dysregulation of iron homeostasis in EOfAD may be a profitable way forward in our struggle to understand this form of dementia.
Iron, Inflammation, and Neurodegeneration
Iron is the most abundant element in our planet and the most abundant transition metal in the human body (Emsley, 1998). It has been a central component of energy metabolism since the dawn of life, over one billion years before oxygenation of the Earth’s atmosphere drove the evolution of energy production by oxidative phosphorylation (Sheftel et al., 2012). Iron has been used medicinally for thousands of years before elucidation of its role in biology began with its discovery as a component of our blood [announced in 1746 (Busacchi, 1958; Sheftel et al., 2012)].
The most reactive form of iron, ferrous iron, Fe2+, was abundant in the biosphere before oxygen formation by cyanobacteria depleted it to form less soluble ferric (Fe3+) oxide. It is Fe2+ that is required to form the iron–sulfur complexes that are essential for aerobic respiration by bacteria and, consequently, for the function of mitochondria in our eukaryotic cells. However, the exploitation of Fe2+ in redox reactions involves the unavoidable generation of oxidative and nitrosative stress that must be properly managed lest it damage mitochondria, damage their antioxidant controls, and generate additional stress in a positive feedback loop (Leandro et al., 2015). Levels of bioavailable iron are tightly regulated by balancing of iron storage and iron acquisition through mechanisms that are exquisitely sensitive to iron requirements. Iron is normally acquired from the diet by absorption across enterocytes, transported around the body chaperoned by TRANSFERRIN, internalized by iron-requiring cells via TRANSFERRIN RECEPTOR (TFRC)-mediated endocytosis [and other pathways (Mills et al., 2010; Prus and Fibach, 2011)], and released from the endolysosomal compartment into the cytosol for cellular use. Excess iron is stored in numerous ways: in the cytosolic iron storage protein complex, FERRITIN; within mitochondria in MITOCHONDRIAL FERRITIN (FTMT, Levi et al., 2001); and in protein/chemical complexes such as hemosiderin (Iancu, 2011) and neuromelanin (Double et al., 2003) apparently in the lysosomal pathway (Iancu, 2011; Plum et al., 2016). Some cell types such as enterocytes, macrophages and hepatocytes have evolved increased iron storage capacity (Linder, 2013). Iron is also exported from cells via the protein SOLUTE CARRIER FAMILY 40 (IRON-REGULATED TRANSPORTER), MEMBER 1 (SLC40A1, better known as FERROPORTIN 1 or FPN1) (Donovan et al., 2000). FPN1 acts in conjunction with an iron oxidase (to convert Fe2+ to Fe3+) such as HEPHAESTIN (in intestinal epithelial cells absorbing iron in the gut) (Vulpe et al., 1999) or the homologous protein CERULOPLASMIN (in most other cell types) (Takahashi et al., 1984; Wang and Wang, 2018). The intracellular trafficking of iron within neurons will be discussed in more detail later.
Iron, in particular Fe2+, is so important for bacterial growth that denying its availability to bacteria is central to our innate immune defense against infection. In response to infection, an antibacterial peptide hormone, HEPCIDIN ANTIMICROBIAL PEPTIDE (HAMP), is produced (Pigeon et al., 2001). HAMP signaling drives internalization and degradation of FPN1 (Wang S.M. et al., 2010; Wessling-Resnick, 2010). This prevents dietary iron from exiting enterocytes into the circulation, and stored iron from exiting macrophages and other cells that have evolved to store it. This underlies the anemia associated with acute and chronic disease (Roy and Andrews, 2005). The iron storage capacity in mammalian systems is increased by upregulation of FERRITIN (Konijn and Hershko, 1977; Rogers, 1996), and FTMT (Yang et al., 2015). Another important protein that is a part of the mammalian innate immune system is the iron-binding protein LACTOTRANSFERRIN (LTF) that shows structural homology to TRANSFERRIN and has anti-microbial and anti-inflammatory activities (reviewed by Bonaccorsi di Patti et al., 2018). Of course, in defending against bacteria, the sequestration of iron also affects the functionality of our own mitochondria so that, during inflammation, cells may rely more heavily on energy generation by glycolysis (Gaber et al., 2017).
Inflammation is recognized as a key, unifying characteristic of all forms of neurodegeneration (Richards et al., 2016) so it is unsurprising that iron appears to play key roles in many neurodegenerative diseases. Some examples of this are to be provided but the main aim of this paper is to examine the role iron may play in the most common form of dementia, Alzheimer’s disease.
Alzheimer’s Disease
The current criteria for the pathological diagnosis of Alzheimer’s disease are used to compose an “ABC” score. The “A” describes the extent of Aβ peptide immunostaining (Thal et al., 2002) while “B” is derived from argyrophilic neurofibrillary tangles (NFTs, composed largely of hyperphosphorylated MICROTUBULE-ASSOCIATED PROTEIN TAU, MAPT) (Braak and Braak, 1991) and “C” from argyrophilic neuritic plaques (Mirra et al., 1991). Intermediate or high ABC scores are regarded as consistent with Alzheimer’s disease (Montine et al., 2012). Aβ is first seen throughout the ventral neocortex before spreading to the medial temporal lobe structures. In contrast, NFTs are first observed in the transentorhinal cortex before spreading throughout the hippocampal formation and neocortex with relative sparing of primary cortices (Braak and Braak, 1991). Unlike the linear course of events of Aβ aggregation followed by effects on MAPT that is described by the “Amyloid Hypothesis” (Hardy and Selkoe, 2002) there tends not to be a temporal or physical overlap of NFTs and Aβ in the cortex (Nelson et al., 2009). Indeed, it is the spread of NFTs rather than of Aβ plaques that is correlated with the severity and duration of Alzheimer’s disease (Arriagada et al., 1992). However, most neuritic plaques and some diffuse plaques are tau positive (Dickson and Vickers, 2001) while neuropil threads (composed of MAPT) are commonly seen among plaques suggesting that the overlap of Aβ and MAPT pathologies is more extensive that commonly appreciated when all types of MAPT pathology are considered.
Past and more recent research suggests that changes in iron trafficking and accumulation may be important in LOsAD. Two decades ago, Drs. Mark Smith and George Perry showed iron accumulation associated with the histological hallmarks of Alzheimer’s disease; Aβ plaques and NFTs (Smith et al., 1997) while Kennard et al. (1996) found that serum levels of the iron binding protein MELANOMA-ASSOCIATED ANTIGEN p97 (MFI2) were distinctly higher in individuals with Alzheimer’s disease compared to cognitively normal controls. More recently, the degree of iron accumulation in the brain has been seen to correlate with the degree of plaque and NFT pathology (van Duijn et al., 2017). While intracellular aggregation of Aβ may well contribute to plaque formation (Friedrich et al., 2010) and the rate of formation of an individual plaque is debated (Burgold et al., 2011; Hefendehl et al., 2011), the likelihood that microhemorrhages contribute to plaque formation is supported by their heme and fibrinogen content and their spatial correlation with blood vessels (Cullen et al., 2005, 2006). Since heme is rich in iron, an increasing brain Aβ plaque load would be expected to increase overall brain iron content.
Analysis of the iron storage complex FERRITIN shows great diagnostic and predictive potential for Alzheimer’s disease. In particular, FERRITIN levels in cerebrospinal fluid (an indicator of iron storage capacity) are very significantly higher in carriers of the major LOsAD risk allele, the 𝜀4 allele of the gene APOE (APOE4), and predict conversion from mild cognitive impairment (MCI) to Alzheimer’s disease (Ayton et al., 2015). Also, levels of FERRITIN in plasma correlate strongly with Aβ levels in the neocortex (as assessed by positron emission tomography) so that analyzing plasma FERRITIN may assist in identifying people at high risk for Alzheimer’s disease (Goozee et al., 2017).
Uncertainty Regarding the Mechanistic Role of Aβ in Alzheimer’s Disease
As of June 2018, a search in PubMed for the term “Alzheimer’s disease” finds over 105,000 papers (excluding reviews). Nevertheless, there is still no consensus on the pathological mechanism underlying this disease. Indeed, the very definition of the disease itself is disputed (Herrup, 2015; De La Torre, 2016; Iturria-Medina et al., 2016; Whitehouse and George, 2016).
The past two decades have seen great enthusiasm for the Amyloid Hypothesis of Alzheimer’s disease that posits a pathological role for the Aβ peptide cleaved from the AMYLOID BETA A4 PRECURSOR PROTEIN (APP). The Amyloid Hypothesis is an irrefutable tenet for many in the Alzheimer’s disease research community since changes in Aβ production appear to be the only common mechanism linking the few loci where mutations causing early onset familial Alzheimer’s disease (EOfAD) are found. However, the mode of Aβ’s purported toxicity remains to be clearly defined and recent prominent review papers have admitted to serious incongruities between observations of Aβ deposition in the brain and the progress of the disease (Herrup, 2015; De Strooper and Karran, 2016). It is thus uncertain whether Aβ, accumulation of which is currently a required element in the definition of Alzheimer’s disease (McKhann et al., 2011) is a causative agent, a protective mechanism, or an ‘innocent bystander’ in the pathological process. [It is also worth reflecting on the fact that a naïve, most parsimonious interpretation of the presence of iron in the plaques and NFTs of Alzheimer’s disease brains (Smith et al., 1997) – that resembles so clearly the silver staining used by Alois Alzheimer himself to identify these histological landmarks (Alzheimer et al., 1995) – is that iron is the common causative agent rather than the Aβ peptide somehow driving both plaque and NFT formation and then these coincidentally both binding iron.]
The dominant mutations causing EOfAD are now recognized to occur in four genes, APP, PRESENILIN 1 (PSEN1), PRESENILIN 2 (PSEN2), and (more recently) SORTILIN-RELATED RECEPTOR 1 (SORL1). Readers can refer to a number of excellent reviews and research papers describing these mutations (Tanzi, 2012; Guerreiro and Hardy, 2014; Verheijen et al., 2016). Mutations in SORL1 are thought to influence the production and secretion of Aβ by mechanisms that are not yet clearly defined (reviewed by, Yin et al., 2015). However, the involvement of APP and the PSENs in production of Aβ is considered clear: the PSENs form the catalytic core of γ-secretase complexes that cleave a transmembrane fragment of APP to release Aβ (Figure 1). [Intriguingly, SORL1 is also a substrate of γ-secretase (Bohm et al., 2006)]. EOfAD mutations in APP are thought to affect either the production rate and/or form of Aβ while EOfAD mutations in the PSENs have been thought to enhance the production of the longer forms of Aβ, in particular the 42 amino acid residue form, Aβ42, relative to the predominant, 40 amino acid residue form, Aβ40 (Chavez-Gutierrez et al., 2012; Szaruga et al., 2015). However, this idea remains controversial since many of these mutations may decrease total γ-secretase activity and reduce overall Aβ production (Jayne et al., 2016; Sun et al., 2017), casting doubt on a causative role for Aβ in Alzheimer’s disease pathology.
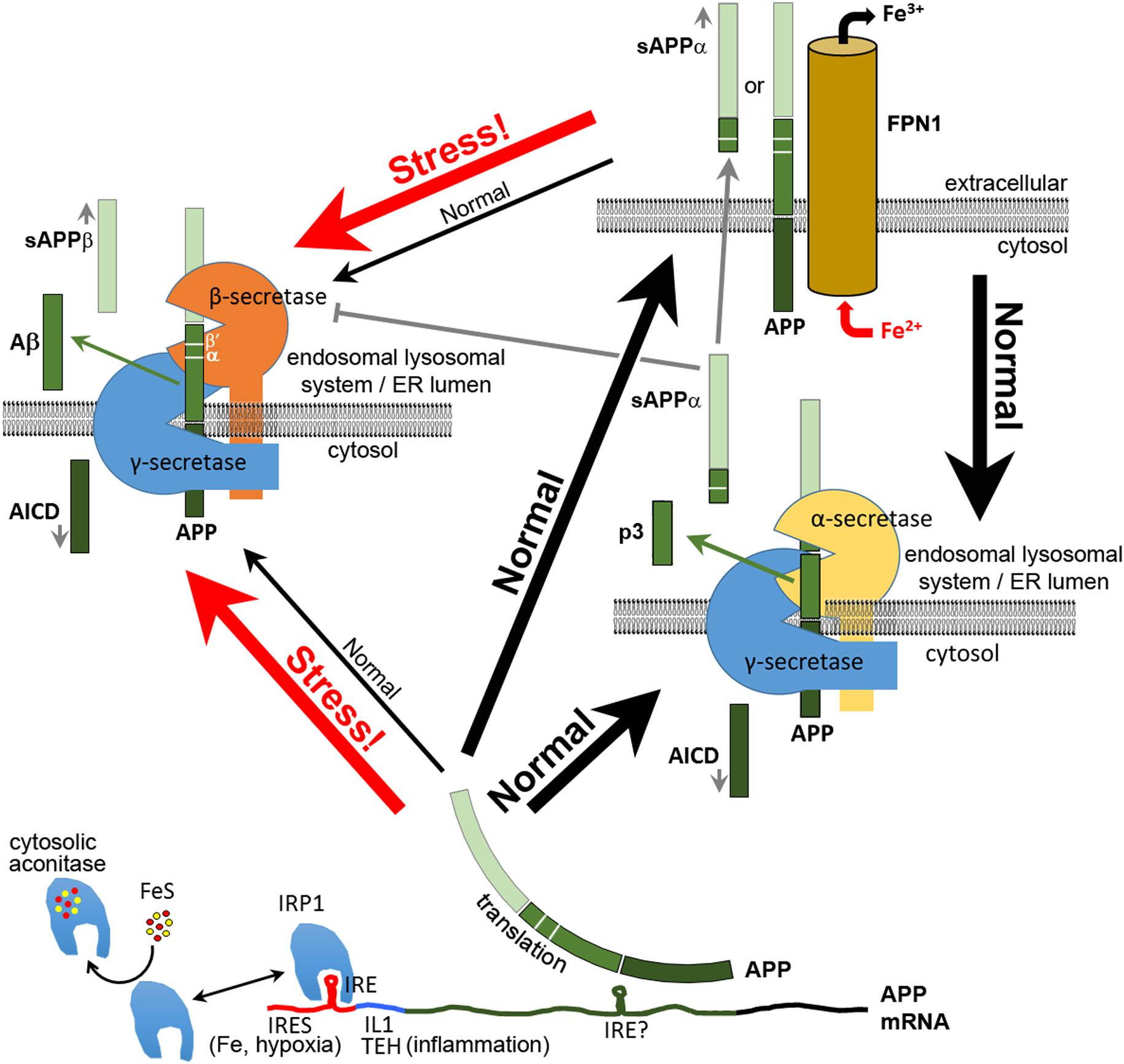
FIGURE 1. Multiple functions of APP in neurons. After its translation, APP can function in neurons to bind FPN1 and enhance the export of excess Fe2+. Some APP is cleaved by α-secretase or (mainly under stress) by β-secretase complexes, the latter producing the antimicrobial and antioxidant peptide Aβ. The secreted extracellular domains cleaved from APP by α–secretase (sAPPα) or β-secretase (sAPPβ) adopt different structures and activities. Like APP, sAPPα is able to increase the expression of FPN1 at the cell surface. sAPPα can also inhibit the activity of β-secretase. Translation of APP mRNA is suppressed by binding of IRP1 to an iron responsive element (IRE) (Rogers et al., 2002) when synthesis of Fe–S clusters is deficient. Under inflammatory stress and/or hypoxia, APP transcription is upregulated by oxidative stress, and APP translation is stimulated by binding of IL1 at a translation enhancer site (TEH) (Rogers et al., 1999) and by an internal ribosome entry site (IRES, Beaudoin et al., 2008) that permits translation in defiance of the unfolded protein response. The increased APP protein either increases iron export for sequestration by other cells or is diverted intracellularly to cleavage for production of Aβ and other APP proteolytic products. Plasma membrane APP may also be internalized for cleavage, probably due to interactions with SORTILIN and NGFR (p75) (Yang et al., 2013; Saadipour et al., 2017). β′ and α cleavage sites are indicated. It is not known what domains of α- and β-secretase interact with γ-secretase. Indeed, the functional existence of such complexes appears inconsistent with other data on the subcellular localization of these enzyme activities (e.g., see Pera et al., 2017). Note that the BACE1 protein that provides β-secretase activity was recently shown to be involved in intracellular Cu2+ homeostasis (Liebsch et al., 2017).
The PSENs are polyfunctional proteins (reviewed by, Duggan and McCarthy, 2016; Jayne et al., 2016; Otto et al., 2016). When cleaved within their “cytosolic loop” domain they apparently become catalytically active in γ-secretase complexes. However, before cleavage, PSEN holoproteins are involved in other cellular activities. The best defined of these is the role of PSEN1 as a chaperone for presentation of the protein ATPase, H+ TRANSPORTING, LYSOSOMAL, V0 SUBUNIT A1 (ATP6V0A1) for N-glycosylation by the Sec61alpha/oligosaccharyltransferase complex (Lee J.H. et al., 2010; Lee et al., 2015). Correct N-glycosylation of ATP6V0A1 is required for its targeting to lysosomes (Lee J.H. et al., 2010) where it functions as a subunit of the vacuolar ATPase (v-ATPase) complex required for lysosomal acidification. Thus, EOfAD mutations in PSEN1 appear to decrease the acidity, and reduce the activity, of lysosomes (Lee J.H. et al., 2010). This effect on lysosomes is specific to the holoprotein function since γ-secretase inhibition, or loss of the essential γ-secretase complex component NICASTRIN, do not affect lysosomal acidification (Lee J.H. et al., 2010). Furthermore, EOfAD mutations have never been discovered in the genes encoding the other components of γ-secretase complexes, while mutations in three of the four genes encoding components of γ-secretase complexes (including a frame-shift mutation in PSEN1) cause the skin disease acne inversa and not EOfAD (Wang B. et al., 2010; Li et al., 2011; Liu et al., 2011). Also, intriguingly, another Alzheimer’s disease-related phenomenon that appears to be relatively insensitive to changes in γ-secretase activity is the increased apposition between the endoplasmic reticulum (ER) and mitochondria seen in fibroblasts from both PSEN EOfAD and LOsAD patients (Area-Gomez et al., 2012) [but the apposition is sensitive to β-secretase inhibition (Pera et al., 2017)]. This is important since the PSEN proteins and γ-secretase activity are highly concentrated in this apposition structure known as the mitochondria-associated membranes (MAM; Area-Gomez et al., 2009, 2012).
In a review paper published in 2016, some of us examined the evidence for the involvement of γ-secretase activity in Alzheimer’s disease (Jayne et al., 2016). We found that the genetic data from disease-causing mutations in the PSENs and other components of γ-secretase complexes supported an alternative idea. We proposed that EOfAD mutations in the PSENs promote Alzheimer’s disease through their effect on holoprotein function. Indeed, their dominant action may be due to the formation of holoprotein multimers whereby mutant holoproteins bind to, and interfere with, the action of wild type PSENs. In that paper, we conceded that this idea could not explain some reported EOfAD-related phenomena. In particular, the role of APP in this alternative view was not obvious and we had no alternative explanation for the remarkable reported correlation between the concentration ratio of Aβ40 relative to Aβ42 and the mean age of onset of EOfAD for different mutations in PSEN1 (Duering et al., 2005).
In late 2016, Sun et al. (2017) published their comprehensive analysis of γ-secretase activity and Aβ formation for 138 different EOfAD mutations of PSEN1. They found that different EOfAD mutations can either increase or decrease γ-secretase activity and that the correlation of the Aβ40/Aβ42 ratio with the mean ages of onset of EOfAD mutations of PSEN1 is illusory. Furthermore, Arimon et al. (2015) showed that changes in the ratio of Aβ40 relative to Aβ42 can occur due to oxidative stress, which is a common phenomenon in Alzheimer’s disease brains (Martins et al., 1986; Sanabria-Castro et al., 2017). Thus, in reality, there is currently little genetic data to support a role for γ-secretase (and hence Aβ) in EOfAD (other than the existence of EOfAD mutations in the γ-secretase-cleavage site of APP).
An Alternative Link Between EOfAD-Associated Mutations in App and the PSENs
Since APP and the PSENs are linked in their common involvement in Aβ production, an alternative hypothesis for Alzheimer’s disease pathogenesis requires that a convincing alternative explanation is given for the relationship between the functions of APP and PSENs, and EOfAD pathology. While considerable effort has been devoted to understanding the relationship between EOfAD mutations and γ-secretase activity, relatively little is known about the effects of these mutations on PSEN holoprotein function or the normal functions of APP, and whether there is any commonality in function that links them. The genes PSEN1 and PSEN2 encode proteins with closely related structures and similar functions so it is perhaps unsurprising that EOfAD mutations should be found in both. However, APP is also part of a larger protein family. It shares structural and redundant functional activity with two other proteins, the AMYLOID BETA A4 PRECURSOR-LIKE PROTEINS 1 and 2 (APLP1, APLP2; structurally, APP is more similar to APLP1, Shariati and De Strooper, 2013). Why have EOfAD mutations never been found in the genes encoding these other proteins? What is unique about APP that is not shared with other members of its family?
Of course, of the three APP-related proteins, only APP itself can produce the Aβ peptide. Despite its close similarity to APP, the protein APLP1 apparently does not require cleavage by α- or β-secretase in order to be cleaved by γ-secretase (Schauenburg et al., 2018). However, both APLP1 (Li and Sudhof, 2004) and APLP2 (Pastorino et al., 2004) can be cleaved by β-secretase (Pastorino et al., 2004), and Aβ-equivalent peptides have been detected for these proteins (Eggert et al., 2004; Yanagida et al., 2009) although there is little to suggest that these peptides have pathological activity.
Another characteristic that APP does not share with the APLP proteins is its role in neuronal iron homeostasis. APP (and the α-secretase-cleaved secreted form of APP, sAPPα, but not APLP2) associates with FPN1 to facilitate the iron export function of FPN1 in the plasma membrane of neurons (Duce et al., 2010; Wong et al., 2014). APP’s role in iron export is so significant that it is amongst a list of other iron-related proteins (including FPN1 and others described later such as ACO2, FTH1, FTL, and SDHB (Kuhn, 2015)) whose expression is regulated post-transcriptionally by the presence of ‘iron responsive elements’ (IREs) in the untranslated regions (UTRs) of their respective mRNAs. When cellular iron levels are depleted, “IRON-RESPONSIVE ELEMENT-BINDING PROTEIN 1” (IRP1) (and related protein IRP2) are activated to bind to the stem-loop structure formed by the iron response element (IRE) motif. Whether IREs lie in the 5′ or 3′ UTR of a particular mRNA determines whether IRP binding results in translational silencing, or transcript stabilization, respectively. By regulating expression of key players in iron homeostasis, IRPs act to increase the bioavailability of iron under conditions of iron deficiency.
In the case of APP, IRP1 binds to an IRE motif in the 5′ UTR of APP mRNA to inhibit translation (Cho et al., 2010) and thus inhibit iron export (Rogers et al., 2002) in iron deficiency. This motif is not present in the 5′ UTR of APLP1 (Rogers et al., 2002) or APLP2 (Maloney et al., 2004).
In iron replete conditions, IRP1 incorporates an iron–sulfur cluster (ISC) to become enzymatically active as cytosolic protein ACONITASE1 (ACO1; related to the mitochondrial ACONITASE2 protein required by the tricarboxylic acid cycle, TCA, for energy production) (Hentze and Argos, 1991; Rouault et al., 1991). In this form, it is unable to bind IRE sequences. Notably, ACO1 may possibly also be converted to IRP1 under conditions of oxidative stress (Gehring et al., 1999), e.g., due to either hypoxia or hyperoxia (Terraneo and Samaja, 2017). Analysis of the subcellular localization of IRP1 shows it to be concentrated in the ER (Patton et al., 2005), which is consistent with the importance in Alzheimer’s disease of the MAM (see later).
The importance of APP in iron homeostasis is demonstrated by the structural similarity of its 5′ UTR sequence to those of FERRITIN LIGHT CHAIN (FTL) and FERRITIN HEAVY CHAIN 1 (FTH1) (Rogers et al., 1999), two proteins that together form the FERRITIN molecular cages in which iron is stored as Fe3+. Like APP, translation of the ferritin subunits is inhibited by IRP-IRE binding (to decrease iron storage when iron levels are depleted). Note that, unlike the 5′ UTR of FTH1 mRNA, APP mRNA’s 5′ UTR may not bind IRP2 (Cho et al., 2010). IRP2 is structurally similar to IRP1 (Rouault et al., 1990) but lacks aconitase activity (Guo et al., 1994; Samaniego et al., 1994) and becomes unstable when cellular iron levels are high (Iwai et al., 1995) (rather than becoming, like IRP1, unable to bind IREs due to possession of an iron–sulfur cluster).
Translation of APP, FTL, and FTH1 mRNAs are also all apparently stimulated by the binding of the pro-inflammatory cytokines INTERLEUKIN 1 ALPHA (IL1A) or INTERLEUKIN 1 BETA (IL1B) to INTERLEUKIN translation enhancer elements in their 5′ UTRs. In fact, in the case of FTH1, iron and IL1B act synergistically to increase translation of FTH1 mRNA (Thomson et al., 2005). Thus, APP, FTL, and FTH1 proteins all participate in the acute phase inflammatory response. FTL and FTH1 upregulation acts to sequester iron in FERRITIN making it unavailable to invading bacteria (Wessling-Resnick, 2010; Soares and Weiss, 2015). The upregulation of APP by inflammatory cytokines suggests that, in inflammation, there may be a need to export iron from the cytosol of cells that express APP, which in the brain is specifically neurons (and not glia or astrocytes) (Guo et al., 2012). APP’s role in the acute phase inflammatory response is discussed later. The regulation of APP mRNA translation has been reviewed by Ruberti et al. (2010) and some aspects of this are illustrated in Figure 1. The translation of APP mRNA is also regulated by FRAGILE X MENTAL RETARDATION PROTEIN (FMRP, encoded by gene FMR1) through its binding to a G-quartet-like RNA motif within the APP coding sequence (Westmark and Malter, 2007; Lee E.K. et al., 2010). Binding of other proteins, particularly in the 3′ UTR, regulates APP mRNA stability (e.g., Zaidi et al., 1994; Zaidi and Malter, 1995 and see reviews Ruberti et al., 2010; Westmark and Malter, 2012).
Interestingly, the trafficking of APP to the plasma membrane where it can interact with FPN1 is dependent on the activity of MAPT (Lei et al., 2012, 2017). Consequently, mice lacking MAPT expression show accumulation of iron in the brain (Lei et al., 2012). Apparently, iron can both induce MAPT hyperphosphorylation (Xie et al., 2012; Guo et al., 2013) and encourage aggregation of hyperphosphorylated MAPT (Yamamoto et al., 2002). Since MAPT becomes hyperphosphorylated and aggregates in the neurons of Alzheimer’s disease brains, we would expect this to reduce the available APP on the neuronal plasma membrane and so reduce iron export in aged neurons showing Alzheimer’s disease pathology. (Note the potential here for a positive feedback loop driving iron accumulation.)
Gene knockout studies in mice also point to the impact of age on APP’s effect on iron homeostasis. While mice lacking APP apparently show reduced body weight when young (Zheng et al., 1995), they do not show excess brain iron accumulation (compared to wild type) by 3 months of age. However, brain iron accumulation is significantly increased by 12 months of age (Needham et al., 2014). In contrast, mice lacking APLP2 show no difference in brain iron accumulation compared to wild type mice at any age (Needham et al., 2014). The age-dependence of iron accumulation due to APP dysfunction in mice is consistent with both familial and sporadic Alzheimer’s disease as adult onset diseases in humans. Apparently, neurons (and any other cells in which APP may function in iron export) are not critically dependent on this function during early development. In fact, iron accumulation is important for early brain development and insufficient early brain iron accumulation may determine an aberrant iron “set point” that is refractory to alteration by later iron supplementation (Hare et al., 2013).
Like APP (and as described later in this paper), the PSENs can be expected to play a role in iron homeostasis in neurons (and other cells) due to their role in acidification of the endo-lysosomal pathway (Lee J.H. et al., 2010; Lee et al., 2015). We propose that a shared function of EOfAD-associated proteins may be normal roles in regulation of neuronal iron homeostasis, particularly as animals age.
Cellular Iron Homeostasis and the EOfAD Mutations of App
Do the characteristics of APP’s EOfAD mutations support that APP’s role in iron homeostasis is critical for Alzheimer’s disease pathogenesis? There are two broad categories of EOfAD mutations affecting APP: (1) whole gene duplications and, (2) missense mutations (see the review by Tcw and Goate, 2017). Both categories of APP EOfAD mutations preserve the ability to produce a full-length APP protein (similar to EOfAD mutations in the PSEN genes). Individuals with trisomy of chromosome 21 possess three copies of the APP gene and show early onset Alzheimer’s disease (Prasher et al., 1998). A number of the missense mutations in APP might also be expected to increase the stability (and effective dosage) of APP expression. For example, mutations that inhibit cleavage of APP by α-secretase are suggested to be pathogenic by their enhancement of β-secretase cleavage to form Aβ rather than the shorter, non-pathogenic p3 peptide formed by α-secretase cleavage (Haass et al., 1994; Kaden et al., 2012). However, these mutations should also increase the stability of full-length APP, particularly when one remembers that cleavage of APP by β-secretase occurs mainly at the β′ site (Figure 1) rather than the β-site cleavage that produces Aβ during cellular stress (Deng et al., 2013). Similarly, even though γ-secretase cleavage of APP is thought to occur subsequent to α-secretase or β-secretase cleavage, there is evidence that these latter two enzymes act in complexes together with γ-secretase (Teng et al., 2010; Chen et al., 2015; Cui et al., 2015) suggesting that inhibition of γ-secretase might also lead to inhibition of initial cleavage by α- or β-secretases. Xu et al. (2016) recently showed that most EOfAD mutations in APP decrease the efficiency of its cleavage by γ-secretase. An effective over-expression of full-length APP would be expected to result in excessive Fe2+ export from neurons leading to a deficiency of cytosolic Fe2+. On the other hand, mutations in APP that increase β-secretase cleavage (such as the “Swedish” mutation, KM670/671NL, Citron et al., 1992) could lead to deficient Fe2+ export and neuronal iron accumulation.
The α-secretase-cleaved secreted form of APP, sAPPα, can facilitate FPN1 activity in a manner similar to full-length APP (Wong et al., 2014). This raises the important question of the activity of the β-secretase-cleaved secreted form, sAPPβ. An inability of sAPPβ to facilitate FPN1 activity would support the idea that iron homeostasis is the critical function affected by EOfAD mutations in APP. sAPPα and sAPPβ differ by only 16 amino acid residues in length and have largely similar functions although distinct differences in activity have also been observed (reviewed by Ludewig and Korte, 2016; Mockett et al., 2017). For example, expression of sAPPα, but not sAPPβ, can rescue the viability of mice that simultaneously lack both APP and APLP2 (Ring et al., 2007; Li et al., 2010; Weyer et al., 2011). [This shows that much of the activity of full-length APP can be attributed to sAPPα (Ring et al., 2007; Weyer et al., 2011)]. sAPPα can also facilitate long term potentiation in the hippocampus (Taylor et al., 2008; Hick et al., 2015) in contrast to sAPPβ (Hick et al., 2015). Chen et al. (2017) and Peters-Libeu et al. (2015) showed that sAPPα can inhibit the activity of β-secretase suggesting the existence of a “molecular switch” where one secretase pathway inhibits the other. Importantly, the latter study used small angle X-ray scattering to model the tertiary structures of sAPPα and sAPPβ and showed them to be very different. This was supported by fluorescence spectroscopy data (Peters-Libeu et al., 2015). However, the relative abilities of sAPPα and sAPPβ to stabilize FPN1 are yet to be tested. Further complicating this issue is the fact that, in the absence of cellular stress, most β-secretase cleavage of APP occurs at the β′ site 10 amino acid residues downstream of the β site (Deng et al., 2013) (Figure 2). Nevertheless, the activity of sAPPβ′ has been largely overlooked by researchers.
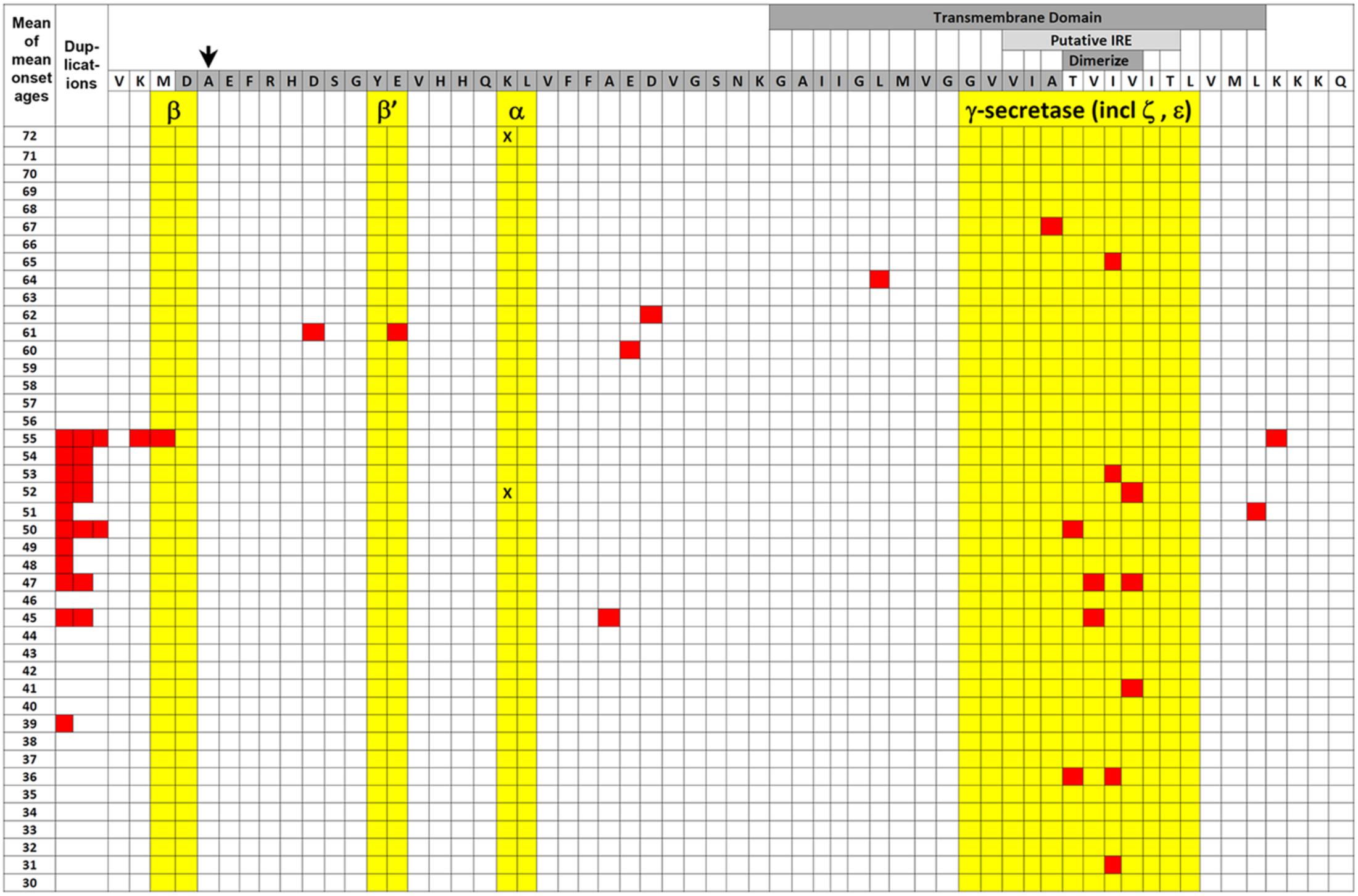
FIGURE 2. EOfAD mutations of APP. The transmembrane domain (labeled darker gray box) and Aβ region (darker gray-shaded amino acid residues) of APP protein are illustrated relative to the mean of mean onset ages (MMOA) of mutations (red squares) from the Alzheimer Disease & Frontotemporal Dementia Mutation Database (Cruts et al., 2012). MMOAs for duplications of the APP gene are shown on the left hand side. There is no MMOA for the K687N mutation in the α-secretase cleavage site but the two onset ages published (Kaden et al., 2012) are indicated by black “X”s. An arrow indicates the position of the protective A673T mutation that reduces Aβ production (Jonsson et al., 2012). Cleavages by various secretases occur between amino acid residues colored yellow. Positions of the possible downstream IRE (see also Figure 3) and the amino acid residues required for dimerization are also shown by labeled lighter and darker gray boxes, respectively. A two-tailed t-test assuming unequal variances for differences between the means of all the MMOAs of whole gene duplications versus non-γ-secretase-affecting missense mutations gives p = 0.05 suggesting that the mechanisms by which these mutation classes generate pathology may differ. We note that this data is also consistent with the suggestion by Area-Gomez et al. (2018) that increased formation of the β-secretase-cleaved transmembrane fragment of APP (the C99 fragment) is the critical effect of EOfAD mutations in APP. C99 accumulates in the MAM and appears to mediate the effects of PSEN activity on MAM formation with consequences for energy metabolism (Pera et al., 2017). EOfAD mutations of APP also alter the tendency of Aβ to aggregate (Hatami et al., 2017) but we know of no correlation between Aβ aggregation dynamics and MMOA.
An important possible implication of the ability of sAPPα to facilitate FPN1 activity is that increased gene dosage of APP (e.g., due to duplication mutations or in Down syndrome) could potentially cause excess sAPPα secretion from neurons that might act to stabilize FPN1 on the plasma membranes of proximal cells. Indeed, an elegant gene-trap experiment described by Liao et al. (2012) showed that only neurons in the developing brains of zebrafish embryos express APP but that APP’s extracellular domain accumulates in brain vasculature. Therefore, the potential may exist for non-autonomous cellular dyshomeostasis of iron due to changes in the expression and cleavage of APP in neurons. Interestingly, a common pathology of APP duplications (Cabrejo et al., 2006) and Down syndrome (Donahue et al., 1998; Buss et al., 2016) is intracerebral hemorrhage suggesting that increased APP expression disrupts the integrity of brain vasculature. Accumulation of Aβ in brain vasculature is also very common in Alzheimer’s disease [i.e., cerebral amyloid angiopathy (CAA; Vinters and Gilbert, 1983; Attems, 2005)] and probably contributes to this disruption (Samuraki et al., 2015) with the consequent release of heme contributing to higher iron levels in Alzheimer’s disease brains.
A recent paper by Lopez Sanchez et al. (2017) illustrated how different EOfAD mutations of APP might have differing, but still detrimental, effects on iron homeostasis. Forced over-expression of full-length wild type APP in human SH-SY5Y neuroblastoma cells led to decreased mitochondrial respiration and increased glycolysis (lactate production). This would be expected since the predicted cytosolic Fe2+ deficiency caused by APP overexpression would deprive the mitochondrial TCA and electron transport chain of iron–sulfur clusters while stabilizing the HIF1 transcription factor that regulates the hypoxia response (see later). In contrast, forced over-expression of the EOfAD “Swedish mutation” form of APP that shows enhanced β-secretase cleavage did not decrease mitochondrial respiration (despite increased production of Aβ42) unless β-secretase was inhibited, in which case it had the same effect as wild type APP. This supports that different EOfAD mutations of APP have different effects on energy metabolism, possibly as a result of differential effects on cytosolic Fe2+ availability. [The situation is further complicated by the observation of Chen et al. (2017) that iron can inhibit β-secretase activity while Bodovitz et al. (1995) noted that high iron levels can increase α-secretase activity.] In fact, examining the “mean of mean onset ages” (MMOA) of different EOfAD mutations in APP from the Alzheimer Disease & Frontotemporal Dementia Mutation Database (Cruts et al., 2012) shows that missense mutations (excluding those thought to affect γ-secretase cleavage) such as the Swedish mutation tend to cause later onset ages than duplications of APP (i.e., they are less pathogenic, Figure 2).
And how should we interpret the “Alzheimer’s disease-protective” mutation of APP, A673T? Icelandic carriers of this mutation show decreased risk of Alzheimer’s disease and increased cognitive performance when elderly (Jonsson et al., 2012). The allele is rare outside of Iceland (Kero et al., 2013; Ting et al., 2013; Bamne et al., 2014; Mengel-From et al., 2015; Wang L.S. et al., 2015). A673T is claimed to reduce cleavage at the β-site to reduce Aβ synthesis (Jonsson et al., 2012). Since cleavage at the β-site of APP is a stress response (particularly to infection – see later) and suppression of inflammatory stress is protective of aging vasculature (Ganjehei and Becker, 2015; Wang J. et al., 2015), it may be that A673T acts to reduce chronic inflammatory stress levels and preserve vascular health. Intriguingly, the Icelandic population (particularly the rural population living under conditions more similar to those prevailing for most of Iceland’s millennium of settlement), shows unusually high levels of serum FERRITIN (Jonsson et al., 1991). It is possible that an increased rate of iron export from neural cells is protective (i.e., advantageous in natural selection) under such conditions.
An additional complicating factor in considering how EOfAD mutations might affect iron homeostasis is our current limited knowledge regarding how FPN1 and APP interact. For example, APP can dimerize and transmembrane domain amino acid residues critical for this (and affecting γ-secretase cleavage) are mutated in EOfAD (Yan et al., 2017) (Figure 2). The dimerization of APP affects its interaction with the protein SORL1 and this influences the trafficking of APP from the ER to the plasma membrane (Eggert et al., 2017) where FPN1 acts to export Fe2+. The most severe EOfAD mutations in APP (those with the earliest Alzheimer’s disease onset ages) affect the transmembrane domain amino acid residues critical for dimerization (Figure 2). Intriguingly, the region of APP RNA that codes for these transmembrane amino acid residues is also predicted to fold into a possible second, downstream IRE (Figure 3).
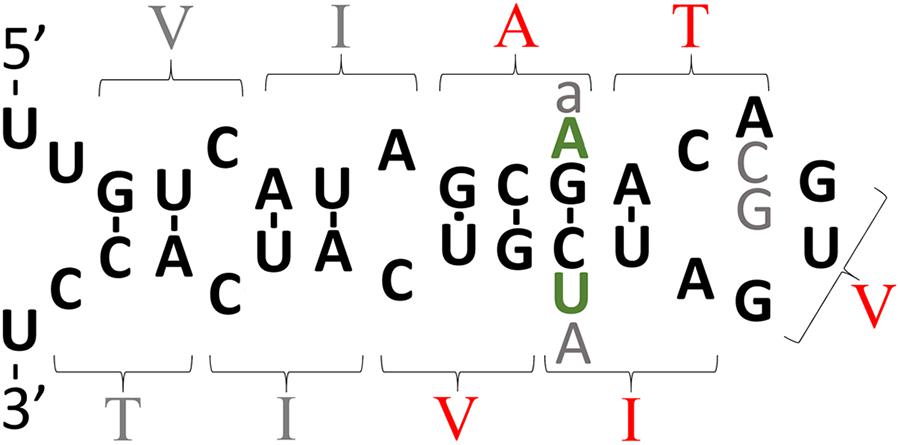
FIGURE 3. The intriguing story of APP’s possible downstream IRE. The theoretical secondary structure of the possible downstream IRE originally published by Tanzi and Hyman (1991) shortly before the Alzheimer’s disease field became focused on the idea that production of Aβ42 is critical to Alzheimer’s disease pathogenesis. The amino acid residues specified by codons are indicated by parentheses. Those amino acid residues shown in red are mutated in EOfAD. In 1991, it was suggested that transmembrane domain EOfAD mutations in APP might exert their pathological effect by disrupting regulation of APP by iron (and this was before APP’s role in iron homeostasis was known). However, the discovery in the following year of an apparently non-pathological (but theoretically IRE-disruptive) polymorphism in this region (Zubenko et al., 1992) (gray “A”) collapsed support for this suggestion. Most IREs are found either in the 5′- or 3′-UTRs of mRNAs where binding of IRPs inhibits or enhances, respectively, mRNA translation (Casey et al., 1988; Anderson et al., 2012). The position of APP’s putative, second IRE within transmembrane domain-coding sequences is unexpected and there is no experimental data supporting its functional reality. Rather, binding studies suggest that, unlike the IRE in APP’s 5′ UTR, this second, IRE-like structure does not bind IRP1 (Cho et al., 2010). However, it is intriguing that in the most diverged (from humans) animal known to possess an Aβ sequence, the Coelacanth, and in most placental mammals, there are two nucleotide differences in the area of the putative IRE and these are conservative since they would preserve base pairing in a double helical region of this structure (green “A” and “U,” found by inspection of genome sequences available at ensembl.org). Other variations found are: gray “a,” an alternative nucleotide in olive baboon and macaque; gray “C” and “G,” alternative nucleotides observed in mouse and kangaroo rat, respectively. If real, one possibility might be that this IRE-like structure only binds an IRP in co-operation with other protein(s) and that it controls mRNA stability. Alternatively, the two IRE’s of APP may interact via co-operative binding of IRPs, an idea alluded to by E. C. Theil (Silver and Walden, 1998) and consistent with formation of an IRP1 complex that can bind two IREs (Hu and Connor, 1996) but never tested for APP’s mRNA.
Cellular Iron Homoeostasis and the PSEN Proteins
The probable dysregulation of cytosolic Fe2+ homeostasis due to EOfAD mutations in APP is significant because EOfAD mutations in the PSENs are predicted to produce similar effects. The major route of iron entry into cells is via endosomes/lysosomes. TRANSFERRIN-Fe3+ complexes bind to TRANSFERRIN RECEPTOR on the surface of cells for endocytosis into endosomes (reviewed in Mayle et al., 2012). In the endosomal-lysosomal system, acidification by v-ATPase causes release of Fe3+ from TRANSFERRIN (Pantopoulos, 2004) and then the Fe3+ is reduced to the biologically active form, Fe2+, by SIX-TRANSMEMBRANE EPITHELIAL ANTIGEN OF PROSTATE 3 (STEAP3) (Passer et al., 2003; Grunewald et al., 2012). As stated earlier, PSEN1 is required for acidification of lysosomes (and, presumably, endosomes) by facilitating N-glycosylation of ATP6V0A1 (Figure 4).
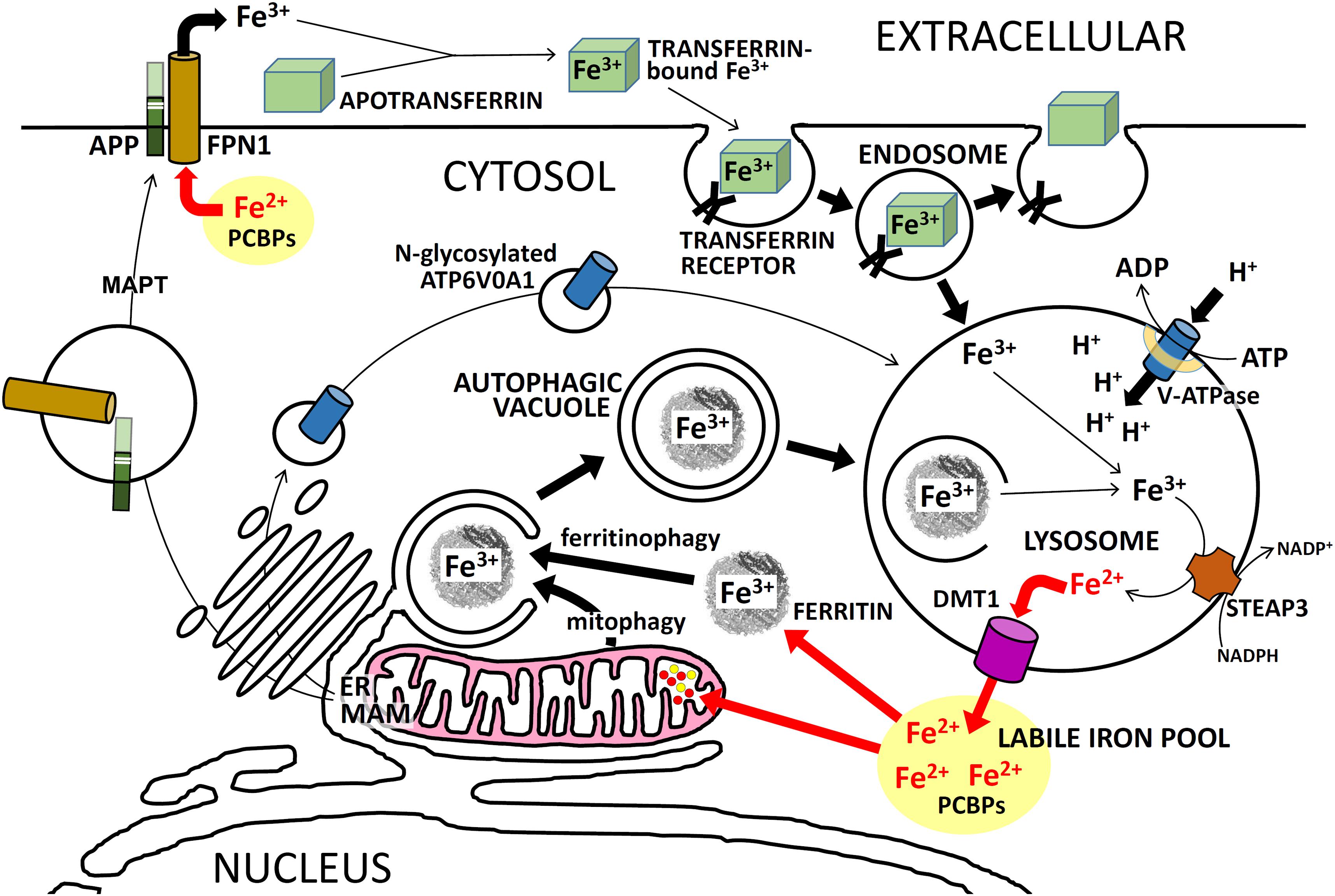
FIGURE 4. Intracellular iron trafficking in neurons. A simplified description of the trafficking of iron ions within neurons, showing the importance of APP and PSEN for these functions. PSENs (in the MAM of the ER) are required for normal N-glycosylation of ATP6V0A1 which is an essential component of the vacuolar ATPase (v-ATPase). Failure to sufficiently acidify endosomes/lysosomes retards Fe3+ release from TRANSFERRIN/TFRC complexes and should retard recycling of Fe3+ to Fe2+ via autophagy. This can cause iron to accumulate in cells as Fe3+. Excessive full-length APP may cause excessive FPN1 activity which would lower Fe2+ levels in the cytosol. Activity of MAPT (tau) is required for transport of APP to the plasma membrane. See the text for further explanation.
After formation, Fe2+ is transported out of endocytic vacuoles and into the cytosol by the protein SOLUTE CARRIER FAMILY 11 [PROTON-COUPLED DIVALENT METAL ION TRANSPORTER], MEMBER 2 (SLC11A2, more commonly known as DIVALENT METAL TRANSPORTER 1, DMT1) (Moos and Morgan, 2004). Other proteins that can transport Fe2+ out of lysosomes are MUCOLIPIN 1 (MCOLN1; Dong et al., 2008, previously known as TRPML1) and SLC11A1 (Evans et al., 2001) (previously named NRAMP1).
The Cytosolic Labile Iron Pool, Ferritin, and Autophagy
The nature of the cytosolic “labile iron pool” (simple iron ions and iron–sulfur clusters) has not been well characterized. Due to its highly reactive nature, free Fe2+ most likely does not occur in cells under normal conditions. Instead, this ion may always be chaperoned as it is distributed to the many proteins that require it for activity. Cytosolic Fe2+ can be chaperoned by POLY(rC)-BINDING PROTEINs (PCBPs) to, either, FERRITIN cages (Shi et al., 2008) (where it is re-oxidized to Fe3+ for storage), or to mitochondria [where it is imported via MITOFERRIN and other proteins (Richardson et al., 2010)], or to cytosolic target proteins (see recent reviews by Lane et al., 2015 and Philpott et al., 2017 and see Figure 4 and below). [2Fe-2S] iron–sulfur clusters appear to be chaperoned in the cytosol by complexes of the proteins GLUTAREDOXIN 3 (GLRX3) and BolA, E. COLI, HOMOLOG OF, 2 (BOLA2, reviewed by Philpott et al., 2017).
POLY(rC)-BINDING PROTEINs not only chaperone cytosolic Fe2+ to FERRITIN but also regulate FERRITIN synthesis through control of FTH1 mRNA translation. PCBP1 and PCBP2 have been observed to bind to the same stem-loop structure in the 5′ UTR of FTH1 mRNA as does IL1 (Thomson et al., 2005). The similarities between the 5′ UTRs of FERRITIN and APP mRNAs lead us to speculate that PCBPs may also regulate translation of APP although this is yet to be demonstrated experimentally.
In order to utilize the cytosolic Fe3+ stored in FERRITIN cages it must be reduced once again to Fe2+ within lysosomes after delivery by autophagy (Mancias et al., 2014). Autophagy of FERRITIN, or “ferritinophagy,” requires the specific cargo receptor protein NUCLEAR RECEPTOR COACTIVATOR 4 (NCOA4) (Dowdle et al., 2014; Mancias et al., 2014), expression of which is regulated by iron. High iron levels destabilize NCOA4 to slow the recycling of Fe3+ to Fe2+ via ferritinophagy (Mancias et al., 2015). Notably, autophagy was recently shown to initiate at the MAM (Hamasaki et al., 2013), a structure that also appears to regulate mitochondrial activity [by Ca2+ release (Carreras-Sureda et al., 2017)]. The MAM also coordinates EOfAD protein function (Area-Gomez et al., 2009), and is involved in Aβ production (discussed in Jayne et al., 2016), and in the initiation of inflammation (Misawa et al., 2013). The MAM of yeast was recently shown to be necessary for iron homeostasis (Xue et al., 2017) although whether this is conserved in humans is unknown. The autophagy of mitochondria, or “mitophagy,” is also an important part of cellular iron homeostasis since these organelles contain so much of the element. The necessity of autophagy for recycling of inactive, stored Fe3+ into active Fe2+ means that a cell with defective autophagy or deficient lysosomal acidification might accumulate iron to high levels while suffering an effective deficiency of cytosolic Fe2+. A similar situation occurs in the recessively inherited disorder mucolipidosis IV (ML4) where loss of MCOLN1 activity results in accumulation of Fe2+ in lysosomes with simultaneous cytosolic Fe2+ deficiency (Dong et al., 2008). This results in degeneration of retinas, motor impairment and intellectual disability (Dong et al., 2008).
The APOE gene that is the major locus for variation affecting risk of LOsAD (Tanzi, 2012) was recently shown to play a role in the transcriptional regulation of autophagy (Parcon et al., 2017; Theendakara et al., 2017). The Alzheimer’s disease-pathogenic 𝜀4 allele of APOE (APOE4) caused significantly lower levels of particular autophagy-critical gene transcripts compared to the non-pathogenic 𝜀3 allele (APOE3) both in Alzheimer’s disease brains and in a transfected astroglioma cell line (Parcon et al., 2017).
Finally, Aβ has been shown to bind Fe2+ (Bousejra-ElGarah et al., 2011; Boopathi and Kolandaivel, 2016). This means that accumulation of Aβ might sequester Fe2+ and contribute to a cytosolic deficiency, and is consistent with Aβ’s speculated antibacterial role (see later). Indeed, this might explain why one candidate anti-Aβ therapy has been seen to have ameliorative effects (even if somewhat minor) on Alzheimer’s disease progression in a phase 1b clinical trial (Sevigny et al., 2016, 2017).
The Intimate Relationship Between Iron Homeostasis, Energy Metabolism, and Responses to Hypoxia
Many proteins require Fe2+ as a cofactor, especially mitochondrial proteins involved in the TCA (Mailloux et al., 2007) (mitochondrial ACONITASE [ACO2], and SUCCINATE DEHYDROGENASE COMPLEX, SUBUNIT B, IRON SULFUR PROTEIN [SDHB]) and in the protein complexes of the electron transport chain (reviewed by Stiban et al., 2016). Therefore, a deficiency of cytosolic Fe2+ is expected to interfere with normal mitochondrial function and increase oxidative stress (Walter et al., 2002; Xu et al., 2013) while excessive cytosolic iron accumulation is thought to cause oxidative stress via Fenton chemistry (Winterbourn, 1995) (below). This is consistent with the fact that aberrant mitochondrial activity is a common phenomenon observed in Alzheimer’s disease studies (reviewed in Silva et al., 2012).
Electron transport chains causing reduction of oxygen (O2) exist both in mitochondria (as part of oxidative phosphorylation) and in the MAM of the ER [involved in the oxidative protein folding that forms disulfide bonds in proteins (Riemer et al., 2009)]. Both processes produce considerable quantities of H2O2 that can interact, in Fenton chemistry, with either Fe2+ or Fe3+ to produce highly reactive hydroxyl (HO•) or hydroxyperoxyl (HOO•) radicals, respectively (Wardman and Candeias, 1996). Oxidative stress that exceeds mitochondria’s antioxidant capacity will damage iron-containing proteins leading to additional iron release in a potential positive feedback loop. This further decreases mitochondrial function and cells’ capacity for production of energy by oxidative phosphorylation (Mandelker, 2008).
The critical dependence of mitochondria on both iron and oxygen for energy production is demonstrated by the intimate coupling of cellular responses to iron or oxygen deficiency. The concentration of Fe2+ is central to controlling the stability of the protein HYPOXIA-INDUCIBLE FACTOR 1, ALPHA SUBUNIT (HIF1α) that, together with its partner ARYL HYDROCARBON RECEPTOR NUCLEAR TRANSLOCATOR (ARNT, also known as HIF1β) forms the transcription factor HIF1, a master regulator of cellular responses to hypoxia. Low cytosolic Fe2+ concentrations lead to stabilization of HIF1α by inhibiting the activity of EGLN proteins (“EGL9, C. ELEGANS, HOMOLOG OF,” previously designated as the PHD proteins). EGLNs are Fe2+-dependent prolyl hydroxylases that act on HIF1α to stimulate its degradation (Bishop and Ratcliffe, 2015). Significantly, because HIF1 activity is sensitive to changes in cytosolic Fe2+ availability, this transcription factor also plays a role in cellular iron homeostasis by regulating the expression of numerous genes/proteins involved in iron trafficking including TRANSFERRIN, TRANSFERRIN RECEPTOR, SLC11A2, and FPN1 (see review by Lane et al., 2015).
The involvement of HIF1 in Fe2+ homeostasis is additionally significant since the EOfAD genes all show strong upregulation by hypoxia (Moussavi Nik et al., 2012 and references therein) and there is a great deal of circumstantial evidence implicating hypoxia as an important component in LOsAD (Raz et al., 2016). However, the PSENs are not simply targets of HIF1 regulation. Instead, they seem to be central to the activity of HIF1. Using immortalized mouse fibroblasts, De Gasperi et al. (2010) showed that Psen1 binds Hif1α and is required for normal Hif1α stabilization. Intriguingly, Hif1α stability can also be regulated by insulin (De Gasperi et al., 2010) providing a possible link between cytosolic Fe2+ and type II diabetes, a recognized risk factor for Alzheimer’s disease (Jayaraman and Pike, 2014). Dramatic, age-dependent changes in the ability to stabilize Hif1α under hypoxia have been seen in rat brains (Ndubuizu et al., 2009) while Liu et al. (2008) observed low levels of HIF1α in LOsAD brains compared to aged controls.
HIF1 plays a central role in regulation of energy metabolism since this transcription factor directly induces transcription of PYRUVATE DEHYDROGENASE KINASE, ISOENZYME 1 (PDK1) (Kim et al., 2006). The PDK1 protein phosphorylates the PYRUVATE DEHYDROGENASE COMPLEX, ALPHA-1 subunit (PDHA1) of the PYRUVATE DEHYDROGENASE COMPLEX thereby inhibiting the conversion of pyruvate to acetyl-CoA. This means that, under hypoxia, more of a cell’s glucose budget is directed away from the TCA and oxidative phosphorylation and into ATP production by the less efficient (but more rapid) process of anaerobic glycolysis. This maintains cellular ATP levels while protecting cells from the high levels of reactive oxygen species (ROS) that are generated by oxidative phosphorylation performed under low oxygen (Zhang et al., 2008). Deficient cytosolic Fe2+ levels would also be expected to stabilize HIF1α and skew cellular energy metabolism toward glycolysis, in essence imitating hypoxic stress. Here, we see a possible overlap with LOsAD since changes in vasculature are observed earlier than Aβ accumulation in development of LOsAD (Iturria-Medina et al., 2016) and it is well established that blood flow in Alzheimer’s disease brains is decreased (Farkas and Luiten, 2001; Marlatt et al., 2008; Nicolakakis and Hamel, 2011; Daulatzai, 2017).
An Overload of Cellular Iron Can Disguise a Cytosolic Fe2+ Deficiency
Because of iron’s central cellular role in energy metabolism, it is unsurprising that many neurological and neurodegenerative conditions show iron dysregulation (see recent excellent reviews, Mills et al., 2010; Lane et al., 2015; Meyer et al., 2015; Gozzelino and Arosio, 2016). The accumulation of iron with age appears to be a universal phenomenon in animals (Massie et al., 1985) and most neurodegenerative disorders apparently involve excess iron in the brain. Excess iron can promote Fenton chemistry that causes oxidative stress by production of hydroxyl radicals (Greenough et al., 2013). Significantly, oxidative stress is thought to stabilize HIF1α by inhibiting EGLN activity (Page et al., 2008). Therefore, iron accumulation should disturb the balance between glycolysis and oxidative phosphorylation, especially if age/pre-existing metabolic stress inhibits the production of antioxidants (Zhu et al., 2006).
Because the subcellular location and form of iron is critical to its function, it is possible to have a cytosolic deficiency of Fe2+ while accumulating abnormally high levels of iron in the endolysosomal compartment. As mentioned previously, this occurs in the neurodegenerative condition Mucolipidosis IV when loss of function of MCOLN1 inhibits transport of Fe2+ into the cytosol from endosomes and lysosomes (Dong et al., 2008). Similarly, we can predict that a failure to sufficiently acidify endolysosomes and lysosomes (such as due to EOfAD mutations in the PSEN genes) should inhibit importation of Fe2+ into the cytosol as well as the recycling of cytosolic Fe3+ stored within FERRITIN cages to Fe2+ via autophagy (and the recycling of iron in mitochondria via mitophagy). Indeed, this may occur in a juvenile onset form of autosomal recessive hereditary parkinsonism, Kufor-Rakeb syndrome, where loss of function of ATPase, TYPE 13A2 (ATP13A2) results in iron accumulation and neurodegeneration (Dehay et al., 2012; Usenovic et al., 2012; Meyer et al., 2015). There are many parallels between the pathologies of Alzheimer’s disease and Parkinson disease (PD) including effects on mitochondrial function and energy metabolism (Calderone et al., 2016). Indeed, there are striking parallels between APP and the major locus for familial PD, SYNUCLEIN, ALPHA (SNCA). Both genes encode proteins that contribute to protein inclusions in neurodegenerative disease, both show localization in the MAM (Area-Gomez et al., 2009; Guardia-Laguarta et al., 2014), and both have transcripts with 5′ UTR IREs that inhibit translation when iron levels are low (Febbraro et al., 2012). In fact, the SNCA protein appears to act as a ferrireductase that can convert Fe3+ to Fe2+ in the presence of Cu2+ and NADH (Davies et al., 2011) and it may also be involved in the uptake of TRANSFERRIN-bound Fe3+ (Baksi et al., 2016).
Iron accumulates in the brain with age (Pirpamer et al., 2016) and this accumulation may be enhanced in Alzheimer’s disease brains (Schrag et al., 2011). However, this may disguise an effective Fe2+ deficiency in the cytosol and mitochondria. The failure of autophagic flux observed in both LOsAD brains (Nixon et al., 2005; Nixon, 2007) and in fibroblasts from individuals with EOfAD due to PSEN1 mutations (Lee J.H. et al., 2010) implies an inability to recycle iron held within defective mitochondria and/or stored in FERRITIN. Likewise, mutations causing overexpression of full-length APP or sAPPα could cause excessive Fe2+ export even as Fe3+ accumulates due to ineffective autophagy and this will degrade mitochondrial performance leading to oxidative stress and decreased energy production.
Energy production is the central and most important cellular activity that makes possible all other activities. Falling energy production probably explains the increased tendency for proteins to aggregate with age (Cohen et al., 2006) [since cellular ATP levels decrease with age and ATP was recently shown to act as a hydrotrope that increases the general solubility of proteins (Patel et al., 2017)]. Failing cellular energy production will also affect the production of the reducing agent NADPH required for the conversion of Fe3+ to Fe2+ in lysosomes. Failing cellular energy production also means decreased production of the ATP required for synthesis of glutathione and other antioxidants. The accumulation of iron in an environment subject to oxidative stress (such as the aging brain) is potentially hazardous since ROS may disrupt iron-bearing proteins to release more iron in a positive feedback loop (Mills et al., 2010).
Non-Neuronal Cells Play Important Roles in Brain Iron Homeostasis
Another important factor to consider in understanding the role of iron homeostasis in Alzheimer’s disease is the roles that non-neuronal cell types play in iron trafficking and storage (reviewed in Belaidi and Bush, 2016). The arguments presented in this paper are centered on iron homeostasis in neurons. However, the highest concentrations of iron in the adult brain appear to be in oligodendrocytes (Connor et al., 1990) and it is known that iron deficiency has marked effects on myelin formation (Beard et al., 2003). Also, microglia [that are markedly dystrophic in Alzheimer’s disease (Streit et al., 2009) and are regarded as macrophages of the brain] show many-fold higher iron content than neurons or astrocytes (Urrutia et al., 2013). Interestingly astrocytes have emerged as a source of HAMP, when stimulated by inflammatory mediators secreted from activated glial cells (You et al., 2017). Furthermore, inflammatory triggers (lipopolysaccharide, cytokines TNFα and IL-6), have differential effects on iron sequestration, and on the expression of genes encoding FPN1 and HAMP, in neurons, astrocytes, and microglia (Urrutia et al., 2013). Understanding how EOfAD mutations affect iron homeostasis in, and between, different neural cell types will be critical to understanding the role of iron homeostasis in Alzheimer’s disease and requires much additional investigation.
A “Stress Threshold Change of State” Into Alzheimer’s Disease?
Can the many functions and interactions of the EOfAD genes and iron be crystalized into an explanation of the development of Alzheimer’s disease? We suggest that one unifying mechanistic scheme may involve an interplay between aging vasculature, hypoxia, iron, and energy production. Increasing hypoxia is a characteristic of normal aging in brains as indicated by rising levels of transcription of HIF1α and HIF1-responsive genes with age (Lu et al., 2004). This increasing hypoxia should increasingly skew metabolism toward glycolysis. However, at some point the aging brain can undergo a shift into a hypometabolic, dysfunctional state as seen in functional MRI analyses (Ashraf et al., 2015) and as evidenced by transcriptome-level comparisons of tissue from normal aged brains, MCI brains and brains from individuals with Alzheimer’s disease (Berchtold et al., 2014). We suggest the possibility that EOfAD mutation-driven cytosolic Fe2+ dysregulation may cause premature utilization of a proportion of the brain’s glycolytic capacity that would otherwise serve to cope with the rising hypoxia caused by the natural decline in function of cerebral vasculature with age. Eventually, a trigger such as an infection (Mawanda and Wallace, 2013), heart failure (Cermakova et al., 2015) or acute hypoxia event (Chen et al., 2014) may force the brain over a stress threshold causing a change of state into the levels of inflammation and hypometabolism that characterize Alzheimer’s disease. This stress threshold is exceeded sooner in the pre-stressed brains of EOfAD mutation carriers than in non-carriers, leading to an earlier onset of Alzheimer’s disease. The idea of a “discontinuous cellular change of state” (Herrup, 2010) into overt Alzheimer’s disease (described here as a “stress threshold change of state”) has also been suggested by others (discussed in Herrup, 2010). We summarize this idea in Figure 5.
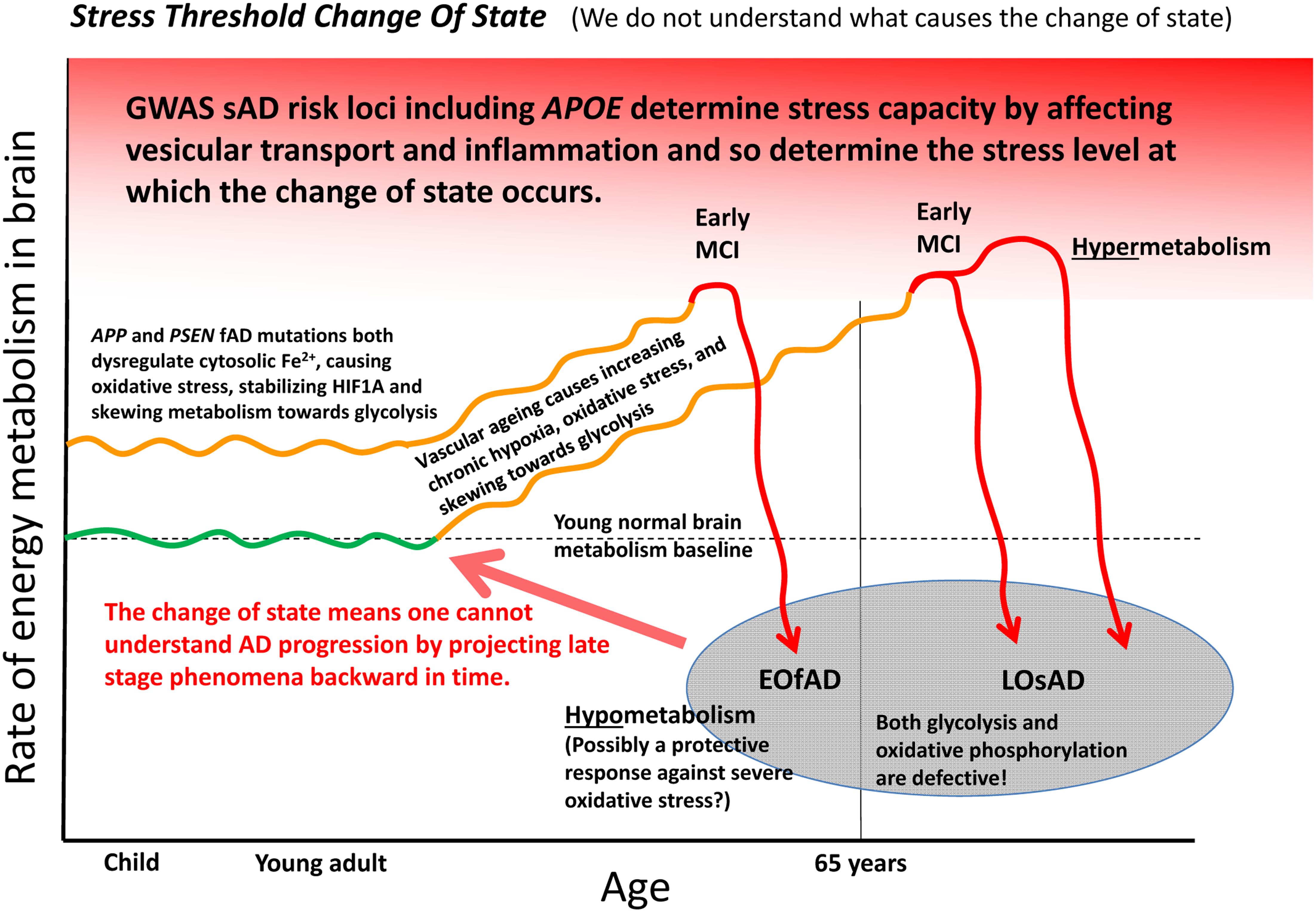
FIGURE 5. A model of Alzheimer’s disease development encompassing both EOfAD and LOsAD genetics. An early onset age of Alzheimer’s disease may occur due to metabolic stress on the brain, possibly due to iron dysregulation. This, in its effects, resembles hypoxic stress and supplements the gradually rising hypoxic stress normally driven by vascular aging so that a stress tolerance (homeostasis capacity) threshold is exceeded sooner, after which the brain shifts into a hypometabolic state, possibly in order to delay further damage due to rising oxidative stress. The timing and extent of this process will vary across different brain regions depending on basal metabolic load, glycolytic capacity, oxygen supply, etc.
An appealing aspect of the idea of iron dysregulation contributing to the stress that ultimately triggers a transition into Alzheimer’s disease is that it might explain the minimal overlap between the EOfAD mutant loci and the LOsAD risk loci. This lack of overlap has led some to question whether EOfAD and LOsAD represent the same disease (Jayne et al., 2016) although both forms of Alzheimer’s disease show similar disease progression once symptoms become overt (Masters et al., 2015). We see that the EOfAD mutations might initially create metabolic stress by disrupting iron homeostasis leaving EOfAD brains with less capacity to adjust to the rising hypoxia caused by age-related vascular changes. The LOsAD risk loci broadly appear to affect autophagy/endolysomal function or inflammation and so either contribute to a milder form of iron “dyshomeostasis” (and reduce cells’ ability to cope with the effects of energy deficiency such as protein aggregation) or reduce the threshold at which inflammation becomes chronic and self-destructive (Richards et al., 2016). Possibly, the change of state into hypometabolism in brains with profound Alzheimer’s disease is a last-ditch attempt by cells to avoid death in the face of critically high oxidative stress levels.
Notably, a metabolic inversion similar to the Alzheimer’s disease stress threshold change of state model above has been described for the monogenetic neurodegenerative condition, Huntington’s disease, in a review by Browne and Beal (2004). This hypothesis proposes that early increases in glucose utilization observed in presymptomatic mouse models and in some human studies, may be a widespread cellular response to perturbation of normal Huntingtin protein function, while decreased glucose utilization observed in the striatum in later stages of the disease may be due to specific cellular dysfunction (Browne and Beal, 2004). There is a long history of observations of cellular iron dysregulation when the HUNTINGTIN (HTT) gene is mutated (Muller and Leavitt, 2014), and wild-type huntingtin is required for normal distribution of iron, and/or expression of iron-related gene products, in the embryos of mice (Dragatsis et al., 1998) and zebrafish (Lumsden et al., 2007), in adult mouse brain (Dietrich et al., 2017) and in mouse embryonic stem cells (Hilditch-Maguire et al., 2000; Jacobsen et al., 2011).
App – An Expression Conundrum
An encouraging aspect of refocusing EOfAD analysis around changes in iron homeostasis is that it requires us to give greater attention to APP’s role in stress responses. Hypoxia causes oxidative stress that upregulates expression of APP, BACE1, and the PSENs (Sun et al., 2006; Wang et al., 2006; Zhang et al., 2007; Guglielmotto et al., 2009; Moussavi Nik et al., 2012, 2015; Villa et al., 2014) and can, thereby, greatly increase production of Aβ that can function as an antioxidant (Kontush, 2001; Kontush et al., 2001; Smith et al., 2002; Nadal et al., 2008; Baruch-Suchodolsky and Fischer, 2009) [although accumulated Aβ possibly generates ROS in the presence of Fe2+ via the Fenton reaction (Sutherland et al., 2013; Cheignon et al., 2017)]. Accumulating evidence also supports a role for Aβ as an antimicrobial peptide (Soscia et al., 2010; Kumar et al., 2016a,b). Aβ was reported to suppress the growth of Candida albicans and a variety of bacterial species at concentrations equal to, or lower than, another characterized human antimicrobial peptide, LL37, and depletion of Aβ from Alzheimer’s disease brain homogenates reduces the latter’s ability to inhibit growth of C. albicans (Soscia et al., 2010). An antimicrobial function for Aβ would explain its high structural conservation in most species of the tetrapod evolutionary lineage (Sharman et al., 2013; Moore et al., 2014).
That Aβ exhibits antioxidant and antimicrobial characteristics is consistent with the fact that inflammatory processes produce oxidative stress as a means of killing pathogens (Mittal et al., 2014). However, this also presents a conundrum: as mentioned previously, a rapid cellular and systemic (acute phase) response to bacterial infection is to sequester iron into cells and away from invading bacteria so as to inhibit their metabolism and growth (see, Urrutia et al., 2013 and reviewed in Soares and Weiss, 2015; Gozzelino and Arosio, 2016). While this explains why IL1 drives increased translation of the FERRITIN proteins to store iron as inactive Fe3+ (as described earlier), oxidative stress also increases APP transcription while IL1 increases APP translation. Increased full-length APP might be expected to stabilize FPN1 and promote iron export from, specifically, neurons in opposition to the action of HAMP (that drives FPN1 internalization and degradation (Nemeth et al., 2004; Ding et al., 2011)). Could it be that expulsion of “free” cytosolic iron from neurons via increased expression of APP promotes brain survival in the face of infection by allowing absorption of the iron by adjacent non-neuronal cells better equipped to sequester it? Simultaneously, increased cleavage of APP by β-secretase (encoded by BACE) may be important for diverting some of this increased APP protein into Aβ production for this peptide’s antioxidant and iron-sequestering capacity (Figure 1). Or does the increased β-secretase cleavage of APP due to inflammatory stress overwhelm the increase in APP transcription and translation to reduce levels of full-length APP and reduce iron export from neurons? Clearly, we need a much more detailed understanding of the relationship between iron homeostasis and the production of APP and Aβ at the cellular level during inflammatory responses in the brain.
A potential weakness in the idea of iron-dysregulation as unifying the effects of EOfAD mutations in APP and the PSEN genes, is that the various EOfAD mutations in APP are not all expected to produce the same effect on iron homeostasis. Some EOfAD mutations in APP (e.g., duplications and possibly those decreasing α-secretase cleavage) are expected to produce neuronal Fe2+ deficiency while others (e.g., those increasing β-secretase cleavage) might produce Fe2+ overload by decreasing normal APP function. However, the same criticism can be leveled at the Amyloid Hypothesis where the effects of γ-secretase site mutations are explained as causing changes in Aβ species length distribution while no explanation is clear for how APP duplication or mutations affecting the frequency of cleavage at the β-site can cause this. However, Arimon et al. (2015) showed that changes in Aβ length occur due to the effects of oxidative stress on the functioning of γ-secretase, while both cytosolic Fe2+ deficiency and overload would be expected to cause increased oxidative stress through interference with normal mitochondrial function. Thus, iron-dysregulation-driven oxidative stress potentially explains the common observation of changes in Aβ species length where the Amyloid Hypothesis cannot. We must note that most, if not all, of the EOfAD mutations in APP would appear to have the effect-in-common of increasing expression of APP’s transmembrane C99 fragment (that results from cleavage only at APP’s β-site) and Area-Gomez et al. (2018) have suggested that this may be critical for the increased MAM formation that is the basis of the MAM Hypothesis of Alzheimer’s disease (Schon and Area-Gomez, 2012). Interestingly, forced expression of C99 has metabolic effects since it appears to suppress ATP synthesis by oxidative phosphorylation in mitochondria while inhibition of C99 formation using an inhibitor of β-secretase increases oxidative phosphorylation (Pera et al., 2017).
Concluding Remarks
The concept described above for unifying EOfAD mutation function should, of course, only be regarded as an outline requiring a great deal of enhancement and testing. For example, the brain consists of many different cell types with specialized roles and greatly differing energy metabolisms and how APP and the PSENs act within these cells types may differ considerably. EOfAD mutations of the PSENs may affect metabolism and oxygen supply by a variety of independent routes such as direct effects on cells of the brain vasculature (Nakajima et al., 2006; Toussay et al., 2017) or via communication between neurons and brain microvasculature using unknown means (Gama Sosa et al., 2010). However, a conceptual focus on iron homeostasis coordinates many important aspects of Alzheimer’s disease including changes in metabolism/mitochondria, vascular function/blood flow/hypoxia, autophagy and inflammation and is not dependent on a central role for γ-secretase activity. We suggest that a concentration of effort in this area will give us valuable insights into the pathogenesis of Alzheimer’s disease.
Author Contributions
This project was initiated by ML and all co-authors contributed ideas, contributed text, and edited this work. Illustrations were drafted by ML and then adjusted after input from the co-authors.
Funding
JR was supported by grants from the Michael J. Fox Foundation (Grant ID: 15468), the United States National Institutes of Health (NIH 5R01MH102279-03, NIH 1R01AG056614-01, and R21NS077079) and a Zenith Grant from the Alzheimer’s Association. MN and ML are supported by a grant from Australia’s National Health and Medical Research Council (NHMRC), GNT1126422 and a donation from the Carthew family. GS received support from a Sydney Medical School Mid-career research Accelerator grant. GV was supported by NHMRC grants GNT1045507 and GNT1105698.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Louise O’Keefe for critical reading of the manuscript.
References
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An english translation of alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Anderson, C. P., Shen, M., Eisenstein, R. S., and Leibold, E. A. (2012). Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483. doi: 10.1016/j.bbamcr.2012.05.010
Area-Gomez, E., de Groof, A., Bonilla, E., Montesinos, J., Tanji, K., Boldogh, I., et al. (2018). A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 9:335. doi: 10.1038/s41419-017-0215-0
Area-Gomez, E., de Groof, A. J., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J. C., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Arimon, M., Takeda, S., Post, K. L., Svirsky, S., Hyman, B. T., and Berezovska, O. (2015). Oxidative stress and lipid peroxidation are upstream of amyloid pathology. Neurobiol. Dis. 84, 109–119. doi: 10.1016/j.nbd.2015.06.013
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42(3 Pt 1), 631–639. doi: 10.1212/WNL.42.3.631
Ashraf, A., Fan, Z., Brooks, D. J., and Edison, P. (2015). Cortical hypermetabolism in MCI subjects: a compensatory mechanism? Eur. J. Nucl. Med. Mol. Imaging 42, 447–458. doi: 10.1007/s00259-014-2919-z
Attems, J. (2005). Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 110, 345–359. doi: 10.1007/s00401-005-1074-9
Ayton, S., Faux, N. G., Bush, A. I., and Alzheimer’s Disease Neuroimaging Initiative. (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6:6760. doi: 10.1038/ncomms7760
Baksi, S., Tripathi, A. K., and Singh, N. (2016). Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: implications for visual manifestations of Parkinson’s disease. Free Radic. Biol. Med. 97, 292–306. doi: 10.1016/j.freeradbiomed.2016.06.025
Bamne, M. N., Demirci, F. Y., Berman, S., Snitz, B. E., Rosenthal, S. L., Wang, X., et al. (2014). Investigation of an amyloid precursor protein protective mutation (A673T) in a North American case-control sample of late-onset Alzheimer’s disease. Neurobiol. Aging 35, 1779.e15–1779.e16. doi: 10.1016/j.neurobiolaging.2014.01.020
Baruch-Suchodolsky, R., and Fischer, B. (2009). Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 48, 4354–4370. doi: 10.1021/bi802361k
Beard, J. L., Wiesinger, J. A., and Connor, J. R. (2003). Pre- and postweaning iron deficiency alters myelination in Sprague-Dawley rats. Dev. Neurosci. 25, 308–315. doi: 10.1159/000073507
Beaudoin, M. E., Poirel, V. J., and Krushel, L. A. (2008). Regulating amyloid precursor protein synthesis through an internal ribosomal entry site. Nucleic Acids Res. 36, 6835–6847. doi: 10.1093/nar/gkn792
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl. 1), 179–197. doi: 10.1111/jnc.13425
Berchtold, N. C., Sabbagh, M. N., Beach, T. G., Kim, R. C., Cribbs, D. H., and Cotman, C. W. (2014). Brain gene expression patterns differentiate mild cognitive impairment from normal aged and Alzheimer’s disease. Neurobiol. Aging 35, 1961–1972. doi: 10.1016/j.neurobiolaging.2014.03.031
Bishop, T., and Ratcliffe, P. J. (2015). HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 117, 65–79. doi: 10.1161/CIRCRESAHA.117.305109
Bodovitz, S., Falduto, M. T., Frail, D. E., and Klein, W. L. (1995). Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J. Neurochem. 64, 307–315. doi: 10.1046/j.1471-4159.1995.64010307.x
Bohm, C., Seibel, N. M., Henkel, B., Steiner, H., Haass, C., and Hampe, W. (2006). SorLA signaling by regulated intramembrane proteolysis. J. Biol. Chem. 281, 14547–14553. doi: 10.1074/jbc.M601660200
Bonaccorsi di Patti, M. C., Cutone, A., Polticelli, F., Rosa, L., Lepanto, M. S., Valenti, P., et al. (2018). The ferroportin-ceruloplasmin system and the mammalian iron homeostasis machine: regulatory pathways and the role of lactoferrin. Biometals 31, 399–414. doi: 10.1007/s10534-018-0087-5
Boopathi, S., and Kolandaivel, P. (2016). Fe(2+) binding on amyloid beta-peptide promotes aggregation. Proteins 84, 1257–1274. doi: 10.1002/prot.25075
Bousejra-ElGarah, F., Bijani, C., Coppel, Y., Faller, P., and Hureau, C. (2011). Iron(II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 50, 9024–9030. doi: 10.1021/ic201233b
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Browne, S. E., and Beal, M. F. (2004). The energetics of Huntington’s disease. Neurochem. Res. 29, 531–546. doi: 10.1023/B:NERE.0000014824.04728.dd
Burgold, S., Bittner, T., Dorostkar, M. M., Kieser, D., Fuhrmann, M., Mitteregger, G., et al. (2011). In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 121, 327–335. doi: 10.1007/s00401-010-0787-6
Busacchi, V. (1958). Vincenzo Menghini and the discovery of iron in the blood. Bull. Sci. Med. 130, 202–205.
Buss, L., Fisher, E., Hardy, J., Nizetic, D., Groet, J., Pulford, L., et al. (2016). Intracerebral haemorrhage in down syndrome: protected or predisposed? F1000Res. 5:876. doi: 10.12688/f1000research.7819.1
Cabrejo, L., Guyant-Marechal, L., Laquerriere, A., Vercelletto, M., De la Fourniere, F., Thomas-Anterion, C., et al. (2006). Phenotype associated with APP duplication in five families. Brain 129(Pt 11), 2966–2976. doi: 10.1093/brain/awl237
Calderone, A., Formenti, M., Aprea, F., Papa, M., Alberghina, L., Colangelo, A. M., et al. (2016). Comparing Alzheimer’s and Parkinson’s diseases networks using graph communities structure. BMC Syst. Biol. 10:25. doi: 10.1186/s12918-016-0270-7
Carreras-Sureda, A., Pihan, P., and Hetz, C. (2017). Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium 70, 24–31. doi: 10.1016/j.ceca.2017.08.004
Casey, J. L., Hentze, M. W., Koeller, D. M., Caughman, S. W., Rouault, T. A., Klausner, R. D., et al. (1988). Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science 240, 924–928. doi: 10.1126/science.2452485
Cermakova, P., Eriksdotter, M., Lund, L. H., Winblad, B., Religa, P., and Religa, D. (2015). Heart failure and Alzheimer’s disease. J. Intern. Med. 277, 406–425. doi: 10.1111/joim.12287
Chavez-Gutierrez, L., Bammens, L., Benilova, I., Vandersteen, A., Benurwar, M., Borgers, M., et al. (2012). The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274. doi: 10.1038/emboj.2012.79
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2017). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, A. C., Kim, S., Shepardson, N., Patel, S., Hong, S., and Selkoe, D. J. (2015). Physical and functional interaction between the alpha- and gamma-secretases: a new model of regulated intramembrane proteolysis. J. Cell Biol. 211, 1157–1176. doi: 10.1083/jcb.201502001
Chen, P. L., Yang, C. W., Tseng, Y. K., Sun, W. Z., Wang, J. L., Wang, S. J., et al. (2014). Risk of dementia after anaesthesia and surgery. Br. J. Psychiatry 204, 188–193. doi: 10.1192/bjp.bp.112.119610
Chen, Y. T., Chen, W. Y., Huang, X. T., Xu, Y. C., and Zhang, H. Y. (2017). Iron dysregulates APP processing accompanying with sAPPalpha cellular retention and beta-secretase inhibition in rat cortical neurons. Acta Pharmacol. Sin. 39, 177–183. doi: 10.1038/aps.2017.113
Cho, H. H., Cahill, C. M., Vanderburg, C. R., Scherzer, C. R., Wang, B., Huang, X., et al. (2010). Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 285, 31217–31232. doi: 10.1074/jbc.M110.149161
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Connor, J. R., Menzies, S. L., St Martin, S. M., and Mufson, E. J. (1990). Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J. Neurosci. Res. 27, 595–611. doi: 10.1002/jnr.490270421
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi: 10.1002/humu.22117
Cui, J., Wang, X., Li, X., Wang, X., Zhang, C., Li, W., et al. (2015). Targeting the gamma-/beta-secretase interaction reduces beta-amyloid generation and ameliorates Alzheimer’s disease-related pathogenesis. Cell Discov. 1:15021. doi: 10.1038/celldisc.2015.21
Cullen, K. M., Kocsi, Z., and Stone, J. (2005). Pericapillary haem-rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J. Cereb. Blood Flow Metab. 25, 1656–1667. doi: 10.1038/sj.jcbfm.9600155
Cullen, K. M., Kocsi, Z., and Stone, J. (2006). Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol. Aging 27, 1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016
Daulatzai, M. A. (2017). Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 95, 943–972. doi: 10.1002/jnr.23777
Davies, P., Moualla, D., and Brown, D. R. (2011). Alpha-synuclein is a cellular ferrireductase. PLoS One 6:e15814. doi: 10.1371/journal.pone.0015814
De Gasperi, R., Sosa, M. A., Dracheva, S., and Elder, G. A. (2010). Presenilin-1 regulates induction of hypoxia inducible factor-1alpha: altered activation by a mutation associated with familial Alzheimer’s disease. Mol. Neurodegener. 5:38. doi: 10.1186/1750-1326-5-38
De La Torre, J. C. (2016). “Alzheimer’s Turning Point : A Vascular Approach To Clinical Prevention”. Switzerland: Springer Publishing. doi: 10.1007/978-3-319-34057-9
De Strooper, B., and Karran, E. (2016). The cellular phase of alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Dehay, B., Ramirez, A., Martinez-Vicente, M., Perier, C., Canron, M. H., Doudnikoff, E., et al. (2012). Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 109, 9611–9616. doi: 10.1073/pnas.1112368109
Deng, Y., Wang, Z., Wang, R., Zhang, X., Zhang, S., Wu, Y., et al. (2013). Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur. J. Neurosci. 37, 1962–1969. doi: 10.1111/ejn.12235
Dickson, T. C., and Vickers, J. C. (2001). The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 105, 99–107. doi: 10.1016/S0306-4522(01)00169-5
Dietrich, P., Johnson, I. M., Alli, S., and Dragatsis, I. (2017). Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 13:e1006846. doi: 10.1371/journal.pgen.1006846
Ding, H., Yan, C. Z., Shi, H., Zhao, Y. S., Chang, S. Y., Yu, P., et al. (2011). Hepcidin is involved in iron regulation in the ischemic brain. PLoS One 6:e25324. doi: 10.1371/journal.pone.0025324
Donahue, J. E., Khurana, J. S., and Adelman, L. S. (1998). Intracerebral hemorrhage in two patients with Down’s syndrome and cerebral amyloid angiopathy. Acta Neuropathol. 95, 213–216. doi: 10.1007/s004010050789
Dong, X. P., Cheng, X., Mills, E., Delling, M., Wang, F., Kurz, T., et al. (2008). The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996. doi: 10.1038/nature07311
Donovan, A., Brownlie, A., Zhou, Y., Shepard, J., Pratt, S. J., Moynihan, J., et al. (2000). Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781. doi: 10.1038/35001596
Double, K. L., Gerlach, M., Schunemann, V., Trautwein, A. X., Zecca, L., Gallorini, M., et al. (2003). Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 66, 489–494. doi: 10.1016/S0006-2952(03)00293-4
Dowdle, W. E., Nyfeler, B., Nagel, J., Elling, R. A., Liu, S., Triantafellow, E., et al. (2014). Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 16, 1069–1079. doi: 10.1038/ncb3053
Dragatsis, I., Efstratiadis, A., and Zeitlin, S. (1998). Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development 125, 1529–1539.
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867. doi: 10.1016/j.cell.2010.08.014
Duering, M., Grimm, M. O., Grimm, H. S., Schroder, J., and Hartmann, T. (2005). Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol. Aging 26, 785–788. doi: 10.1016/j.neurobiolaging.2004.08.002
Duggan, S. P., and McCarthy, J. V. (2016). Beyond gamma-secretase activity: the multifunctional nature of presenilins in cell signalling pathways. Cell. Signal. 28, 1–11. doi: 10.1016/j.cellsig.2015.10.006
Eggert, S., Gonzalez, A. C., Thomas, C., Schilling, S., Schwarz, S. M., Tischer, C., et al. (2017). Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cell. Mol. Life Sci. 75, 301–322. doi: 10.1007/s00018-017-2625-7
Eggert, S., Paliga, K., Soba, P., Evin, G., Masters, C. L., Weidemann, A., et al. (2004). The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J. Biol. Chem. 279, 18146–18156. doi: 10.1074/jbc.M311601200
Evans, C. A., Harbuz, M. S., Ostenfeld, T., Norrish, A., and Blackwell, J. M. (2001). Nramp1 is expressed in neurons and is associated with behavioural and immune responses to stress. Neurogenetics 3, 69–78. doi: 10.1007/s100480100105
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 64, 575–611. doi: 10.1016/S0301-0082(00)00068-X
Febbraro, F., Giorgi, M., Caldarola, S., Loreni, F., and Romero-Ramos, M. (2012). alpha-Synuclein expression is modulated at the translational level by iron. Neuroreport 23, 576–580. doi: 10.1097/WNR.0b013e328354a1f0
Friedrich, R. P., Tepper, K., Ronicke, R., Soom, M., Westermann, M., Reymann, K., et al. (2010). Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947. doi: 10.1073/pnas.0904532106
Gaber, T., Strehl, C., and Buttgereit, F. (2017). Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 13, 267–279. doi: 10.1038/nrrheum.2017.37
Gama Sosa, M. A., Gasperi, R. D., Rocher, A. B., Wang, A. C., Janssen, W. G., Flores, T., et al. (2010). Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am. J. Pathol. 176, 353–368. doi: 10.2353/ajpath.2010.090482
Ganjehei, L., and Becker, R. C. (2015). Aspirin dosing in cardiovascular disease prevention and management: an update. J. Thromb. Thrombolysis 40, 499–511. doi: 10.1007/s11239-015-1267-6
Gehring, N. H., Hentze, M. W., and Pantopoulos, K. (1999). Inactivation of both RNA binding and aconitase activities of iron regulatory protein-1 by quinone-induced oxidative stress. J. Biol. Chem. 274, 6219–6225. doi: 10.1074/jbc.274.10.6219
Goozee, K., Chatterjee, P., James, I., Shen, K., Sohrabi, H. R., Asih, P. R., et al. (2017). Elevated plasma ferritin in elderly individuals with high neocortical amyloid-beta load. Mol. Psychiatry doi: 10.1038/mp.2017.146 [Epub ahead of print].
Gozzelino, R., and Arosio, P. (2016). Iron homeostasis in health and disease. Int. J. Mol. Sci. 17:E130. doi: 10.3390/ijms17010130
Greenough, M. A., Camakaris, J., and Bush, A. I. (2013). Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 62, 540–555. doi: 10.1016/j.neuint.2012.08.014
Grunewald, T. G., Bach, H., Cossarizza, A., and Matsumoto, I. (2012). The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol. Cell 104, 641–657. doi: 10.1111/boc.201200027
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Guerreiro, R., and Hardy, J. (2014). Genetics of Alzheimer’s disease. Neurotherapeutics 11, 732–737. doi: 10.1007/s13311-014-0295-9
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., et al. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J. Neurochem. 108, 1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x
Guo, B., Yu, Y., and Leibold, E. A. (1994). Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 269, 24252–24260.
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Guo, Q., Li, H., Gaddam, S. S., Justice, N. J., Robertson, C. S., and Zheng, H. (2012). Amyloid precursor protein revisited: neuron-specific expression and highly stable nature of soluble derivatives. J. Biol. Chem. 287, 2437–2445. doi: 10.1074/jbc.M111.315051
Haass, C., Hung, A. Y., Selkoe, D. J., and Teplow, D. B. (1994). Mutations associated with a locus for familial Alzheimer’s disease result in alternative processing of amyloid beta-protein precursor. J. Biol. Chem. 269, 17741–17748.
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hare, D., Ayton, S., Bush, A., and Lei, P. (2013). A delicate balance: iron metabolism and diseases of the brain. Front. Aging Neurosci. 5:34. doi: 10.3389/fnagi.2013.00034
Hatami, A., Monjazeb, S., Milton, S., and Glabe, C. G. (2017). Familial Alzheimer’s Disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-beta peptide. J. Biol. Chem. 292, 3172–3185. doi: 10.1074/jbc.M116.755264
Hefendehl, J. K., Wegenast-Braun, B. M., Liebig, C., Eicke, D., Milford, D., Calhoun, M. E., et al. (2011). Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J. Neurosci. 31, 624–629. doi: 10.1523/JNEUROSCI.5147-10.2011
Hentze, M. W., and Argos, P. (1991). Homology between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res. 19, 1739–1740. doi: 10.1093/nar/19.8.1739
Herrup, K. (2010). Reimagining Alzheimer’s disease–an age-based hypothesis. J. Neurosci. 30, 16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. doi: 10.1038/nn.4017
Hick, M., Herrmann, U., Weyer, S. W., Mallm, J. P., Tschape, J. A., Borgers, M., et al. (2015). Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta Neuropathol. 129, 21–37. doi: 10.1007/s00401-014-1368-x
Hilditch-Maguire, P., Trettel, F., Passani, L. A., Auerbach, A., Persichetti, F., and MacDonald, M. E. (2000). Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 9, 2789–2797. doi: 10.1093/hmg/9.19.2789
Hu, J., and Connor, J. R. (1996). Demonstration and characterization of the iron regulatory protein in human brain. J. Neurochem. 67, 838–844. doi: 10.1046/j.1471-4159.1996.67020838.x
Iancu, T. C. (2011). Ultrastructural aspects of iron storage, transport and metabolism. J. Neural. Transm. 118, 329–335. doi: 10.1007/s00702-011-0588-7
Iturria-Medina, Y., Sotero, R. C., Toussaint, P. J., Mateos-Perez, J. M., Evans, A. C., and Alzheimer’s Disease Neuroimaging Initiative (2016). Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 7:11934. doi: 10.1038/ncomms11934
Iwai, K., Klausner, R. D., and Rouault, T. A. (1995). Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J. 14, 5350–5357.
Jacobsen, J. C., Gregory, G. C., Woda, J. M., Thompson, M. N., Coser, K. R., Murthy, V., et al. (2011). HD CAG-correlated gene expression changes support a simple dominant gain of function. Hum. Mol. Genet. 20, 2846–2860. doi: 10.1093/hmg/ddr195
Jayaraman, A., and Pike, C. J. (2014). Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr. Diab. Rep. 14:476. doi: 10.1007/s11892-014-0476-2
Jayne, T., Newman, M., Verdile, G., Sutherland, G., Munch, G., Musgrave, I., et al. (2016). Evidence for and against a pathogenic role of reduced gamma-secretase activity in familial Alzheimer’s Disease. J. Alzheimers. Dis. 52, 781–799. doi: 10.3233/JAD-151186
Jonsson, J. J., Johannesson, G. M., Sigfusson, N., Magnusson, B., Thjodleifsson, B., and Magnusson, S. (1991). Prevalence of iron deficiency and iron overload in the adult Icelandic population. J. Clin. Epidemiol. 44, 1289–1297. doi: 10.1016/0895-4356(91)90090-V
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Kaden, D., Harmeier, A., Weise, C., Munter, L. M., Althoff, V., Rost, B. R., et al. (2012). Novel APP/Abeta mutation K16N produces highly toxic heteromeric Abeta oligomers. EMBO Mol. Med. 4, 647–659. doi: 10.1002/emmm.201200239
Kennard, M. L., Feldman, H., Yamada, T., and Jefferies, W. A. (1996). Serum levels of the iron binding protein p97 are elevated in Alzheimer’s disease. Nat. Med. 2, 1230–1235. doi: 10.1038/nm1196-1230
Kero, M., Paetau, A., Polvikoski, T., Tanskanen, M., Sulkava, R., Jansson, L., et al. (2013). Amyloid precursor protein (APP) A673T mutation in the elderly finnish population. Neurobiol. Aging 34, 1518.e1–1518.e3. doi: 10.1016/j.neurobiolaging.2012.09.017
Kim, J. W., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Konijn, A. M., and Hershko, C. (1977). Ferritin synthesis in inflammation. I. Pathogenesis of impaired iron release. Br. J. Haematol. 37, 7–16.
Kontush, A. (2001). Amyloid-beta: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radic. Biol. Med. 31, 1120–1131. doi: 10.1016/S0891-5849(01)00688-8
Kontush, A., Donarski, N., and Beisiegel, U. (2001). Resistance of human cerebrospinal fluid to in vitro oxidation is directly related to its amyloid-beta content. Free Radic. Res. 35, 507–517. doi: 10.1080/10715760100301521
Kuhn, L. C. (2015). Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 7, 232–243. doi: 10.1039/c4mt00164h
Kumar, D. K., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016a). Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8, 340ra372. doi: 10.1126/scitranslmed.aaf1059
Kumar, D. K., Eimer, W. A., Tanzi, R. E., and Moir, R. D. (2016b). Alzheimer’s disease: the potential therapeutic role of the natural antibiotic amyloid-beta peptide. Neurodegener. Dis. Manag. 6, 345–348. doi: 10.2217/nmt-2016-0035
Lane, D. J., Merlot, A. M., Huang, M. L., Bae, D. H., Jansson, P. J., Sahni, S., et al. (2015). Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 1853, 1130–1144. doi: 10.1016/j.bbamcr.2015.01.021
Leandro, G. S., Sykora, P., and Bohr, V. A. (2015). The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. 776, 31–39. doi: 10.1016/j.mrfmmm.2014.12.011
Lee, E. K., Kim, H. H., Kuwano, Y., Abdelmohsen, K., Srikantan, S., Subaran, S. S., et al. (2010). hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 17, 732–739. doi: 10.1038/nsmb.1815
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lee, J. H., McBrayer, M. K., Wolfe, D. M., Haslett, L. J., Kumar, A., Sato, Y., et al. (2015). Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444. doi: 10.1016/j.celrep.2015.07.050
Lei, P., Ayton, S., Appukuttan, A. T., Moon, S., Duce, J. A., Volitakis, I., et al. (2017). Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry 22, 396–406. doi: 10.1038/mp.2016.96
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Levi, S., Corsi, B., Bosisio, M., Invernizzi, R., Volz, A., Sanford, D., et al. (2001). A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 276, 24437–24440. doi: 10.1074/jbc.C100141200
Li, C. R., Jiang, M. J., Shen, D. B., Xu, H. X., Wang, H. S., Yao, X., et al. (2011). Two novel mutations of the nicastrin gene in Chinese patients with acne inversa. Br. J. Dermatol. 165, 415–418. doi: 10.1111/j.1365-2133.2011.10372.x
Li, H., Wang, B., Wang, Z., Guo, Q., Tabuchi, K., Hammer, R. E., et al. (2010). Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. U.S.A. 107, 17362–17367. doi: 10.1073/pnas.1012568107
Li, Q., and Sudhof, T. C. (2004). Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J. Biol. Chem. 279, 10542–10550. doi: 10.1074/jbc.M310001200
Liao, H. K., Wang, Y., Watt, K. E. N., Wen, Q., Breitbach, J., Kemmet, C. K., et al. (2012). Tol2 gene trap integrations in the zebrafish amyloid precursor protein genes appa and aplp2 reveal accumulation of secreted APP at the embryonic veins. Dev. Dyn. 241, 415–425. doi: 10.1002/Dvdy.23725
Liebsch, F., Aurousseau, M. R. P., Bethge, T., McGuire, H., Scolari, S., Herrmann, A., et al. (2017). Full-length cellular beta-secretase has a trimeric subunit stoichiometry, and its sulfur-rich transmembrane interaction site modulates cytosolic copper compartmentalization. J. Biol. Chem. 292, 13258–13270. doi: 10.1074/jbc.M117.779165
Linder, M. C. (2013). Mobilization of stored iron in mammals: a review. Nutrients 5, 4022–4050. doi: 10.3390/nu5104022
Liu, Y., Gao, M., Lv, Y. M., Yang, X., Ren, Y. Q., Jiang, T., et al. (2011). Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa). J. Invest. Dermatol. 131, 1570–1572. doi: 10.1038/jid.2011.62
Liu, Y., Liu, F., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2008). Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 582, 359–364. doi: 10.1016/j.febslet.2007.12.035
Lopez Sanchez, M. I. G., Waugh, H. S., Tsatsanis, A., Wong, B. X., Crowston, J. G., Duce, J. A., et al. (2017). Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci. Rep. 7:9835. doi: 10.1038/s41598-017-10233-0
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. doi: 10.1038/nature02661
Ludewig, S., and Korte, M. (2016). Novel insights into the physiological function of the APP (Gene) family and its proteolytic fragments in synaptic plasticity. Front. Mol. Neurosci. 9:161. doi: 10.3389/fnmol.2016.00161
Lumsden, A. L., Henshall, T. L., Dayan, S., Lardelli, M. T., and Richards, R. I. (2007). Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum. Mol. Genet. 16, 1905–1920. doi: 10.1093/hmg/ddm138
Mailloux, R. J., Beriault, R., Lemire, J., Singh, R., Chenier, D. R., Hamel, R. D., et al. (2007). The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One 2:e690. doi: 10.1371/journal.pone.0000690
Maloney, B., Ge, Y. W., Greig, N., and Lahiri, D. K. (2004). Presence of a “CAGA box” in the APP gene unique to amyloid plaque-forming species and absent in all APLP-1/2 genes: implications in Alzheimer’s disease. FASEB J. 18, 1288–1290. doi: 10.1096/fj.03-1703fje
Mancias, J. D., Pontano Vaites, L., Nissim, S., Biancur, D. E., Kim, A. J., Wang, X., et al. (2015). Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 4:e10308. doi: 10.7554/eLife.10308
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. doi: 10.1038/nature13148
Mandelker, L. (2008). Introduction to oxidative stress and mitochondrial dysfunction. Vet. Clin. North Am. Small Anim. Pract. 38, 1–30. doi: 10.1016/j.cvsm.2007.10.005
Marlatt, M. W., Lucassen, P. J., Perry, G., Smith, M. A., and Zhu, X. (2008). Alzheimer’s disease: cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J. Alzheimers Dis. 15, 199–210. doi: 10.3233/JAD-2008-15206
Martins, R. N., Harper, C. G., Stokes, G. B., and Masters, C. L. (1986). Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer’s disease may reflect oxidative stress. J. Neurochem. 46, 1042–1045. doi: 10.1111/j.1471-4159.1986.tb00615.x
Massie, H. R., Aiello, V. R., and Williams, T. R. (1985). Iron accumulation during development and ageing of Drosophila. Mech. Ageing Dev. 29, 215–220. doi: 10.1016/0047-6374(85)90020-X
Masters, C. L., Bateman, R. J., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Mawanda, F., and Wallace, R. (2013). Can infections cause Alzheimer’s disease? Epidemiol. Rev. 35, 161–180. doi: 10.1093/epirev/mxs007
Mayle, K. M., Le, A. M., and Kamei, D. T. (2012). The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 1820, 264–281. doi: 10.1016/j.bbagen.2011.09.009
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Mengel-From, J., Jeune, B., Pentti, T., McGue, M., Christensen, K., and Christiansen, L. (2015). The APP A673T frequency differs between Nordic countries. Neurobiol. Aging 36, 2909.e1–2909.e4. doi: 10.1016/j.neurobiolaging.2015.07.011
Meyer, E., Kurian, M. A., and Hayflick, S. J. (2015). Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu. Rev. Genomics Hum. Genet. 16, 257–279. doi: 10.1146/annurev-genom-090314-025011
Mills, E., Dong, X. P., Wang, F., and Xu, H. (2010). Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med. Chem. 2, 51–64. doi: 10.4155/fmc.09.140
Mirra, S. S., Heyman, A., McKeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., et al. (1991). The Consortium to establish a registry for Alzheimer’s Disease (CERAD). Part II. standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. doi: 10.1212/WNL.41.4.479
Misawa, T., Takahama, M., Kozaki, T., Lee, H., Zou, J., Saitoh, T., et al. (2013). Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460. doi: 10.1038/ni.2550
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P., and Malik, A. B. (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20, 1126–1167. doi: 10.1089/ars.2012.5149
Mockett, B. G., Richter, M., Abraham, W. C., and Muller, U. C. (2017). Therapeutic potential of secreted amyloid precursor protein APPsalpha. Front. Mol. Neurosci. 10:30. doi: 10.3389/fnmol.2017.00030
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Moore, D. B., Gillentine, M. A., Botezatu, N. M., Wilson, K. A., Benson, A. E., and Langeland, J. A. (2014). Asynchronous evolutionary origins of Abeta and BACE1. Mol. Biol. Evol. 31, 696–702. doi: 10.1093/molbev/mst262
Moos, T., and Morgan, E. H. (2004). The significance of the mutated divalent metal transporter (DMT1) on iron transport into the Belgrade rat brain. J. Neurochem. 88, 233–245. doi: 10.1046/j.1471-4159.2003.02142.x
Moussavi Nik, S. H., Newman, M., Wilson, L., Ebrahimie, E., Wells, S., Musgrave, I., et al. (2015). Alzheimer’s disease-related peptide PS2V plays ancient, conserved roles in suppression of the unfolded protein response under hypoxia and stimulation of gamma-secretase activity. Hum. Mol. Genet. 24, 3662–3678. doi: 10.1093/hmg/ddv110
Moussavi Nik, S. H., Wilson, L., Newman, M., Croft, K., Mori, T. A., Musgrave, I., et al. (2012). The BACE1-PSEN-AbetaPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. J. Alzheimers. Dis. 28, 515–530. doi: 10.3233/JAD-2011-110533
Muller, M., and Leavitt, B. R. (2014). Iron dysregulation in Huntington’s disease. J. Neurochem. 130, 328–350. doi: 10.1111/jnc.12739
Nadal, R. C., Rigby, S. E., and Viles, J. H. (2008). Amyloid beta-Cu2+ complexes in both monomeric and fibrillar forms do not generate H2O2 catalytically but quench hydroxyl radicals. Biochemistry 47, 11653–11664. doi: 10.1021/bi8011093
Nakajima, M., Ogawa, M., Shimoda, Y., Hiraoka, S., Iida, M., Koseki, H., et al. (2006). Presenilin-1 controls the growth and differentiation of endothelial progenitor cells through its beta-catenin-binding region. Cell Biol. Int. 30, 239–243. doi: 10.1016/j.cellbi.2005.11.003
Ndubuizu, O. I., Chavez, J. C., and LaManna, J. C. (2009). Increased prolyl 4-hydroxylase expression and differential regulation of hypoxia-inducible factors in the aged rat brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R158–R165. doi: 10.1152/ajpregu.90829.2008
Needham, B. E., Ciccotosto, G. D., and Cappai, R. (2014). Combined deletions of amyloid precursor protein and amyloid precursor-like protein 2 reveal different effects on mouse brain metal homeostasis. Metallomics 6, 598–603. doi: 10.1039/c3mt00358b
Nelson, P. T., Abner, E. L., Scheff, S. W., Schmitt, F. A., Kryscio, R. J., Jicha, G. A., et al. (2009). Alzheimer’s-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci. Lett. 450, 336–339. doi: 10.1016/j.neulet.2008.11.006
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093. doi: 10.1126/science.1104742
Nicolakakis, N., and Hamel, E. (2011). Neurovascular function in Alzheimer’s disease patients and experimental models. J. Cereb. Blood Flow Metab. 31, 1354–1370. doi: 10.1038/jcbfm.2011.43
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120(Pt 23), 4081–4091. doi: 10.1242/jcs.019265
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Otto, G. P., Sharma, D., and Williams, R. S. (2016). Non-catalytic roles of presenilin throughout evolution. J. Alzheimers Dis. 52, 1177–1187. doi: 10.3233/JAD-150940
Page, E. L., Chan, D. A., Giaccia, A. J., Levine, M., and Richard, D. E. (2008). Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell 19, 86–94. doi: 10.1091/mbc.E07-06-0612
Pantopoulos, K. (2004). Iron metabolism and the IRE/IRP regulatory system: an update. Ann. N. Y. Acad. Sci. 1012, 1–13. doi: 10.1196/annals.1306.001
Parcon, P. A., Balasubramaniam, M., Ayyadevara, S., Jones, R. A., Liu, L., Shmookler Reis, R. J., et al. (2017). Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 14, 230–242. doi: 10.1016/j.jalz.2017.07.754
Passer, B. J., Nancy-Portebois, V., Amzallag, N., Prieur, S., Cans, C., Roborel de Climens, A., et al. (2003). The p53-inducible TSAP6 gene product regulates apoptosis and the cell cycle and interacts with Nix and the Myt1 kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 2284–2289. doi: 10.1073/pnas.0530298100
Pastorino, L., Ikin, A. F., Lamprianou, S., Vacaresse, N., Revelli, J. P., Platt, K., et al. (2004). BACE (beta-secretase) modulates the processing of APLP2 in vivo. Mol. Cell. Neurosci. 25, 642–649. doi: 10.1016/j.mcn.2003.12.013
Patel, A., Malinovska, L., Saha, S., Wang, J., Alberti, S., Krishnan, Y., et al. (2017). ATP as a biological hydrotrope. Science 356, 753–756. doi: 10.1126/science.aaf6846
Patton, S. M., Pinero, D. J., Surguladze, N., Beard, J., and Connor, J. R. (2005). Subcellular localization of iron regulatory proteins to Golgi and ER membranes. J. Cell Sci. 118(Pt 19), 4365–4373. doi: 10.1242/jcs.02570
Pera, M., Larrea, D., Guardia-Laguarta, C., Montesinos, J., Velasco, K. R., Agrawal, R. R., et al. (2017). Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 36, 3356–3371. doi: 10.15252/embj.201796797
Peters-Libeu, C., Campagna, J., Mitsumori, M., Poksay, K. S., Spilman, P., Sabogal, A., et al. (2015). sAβPPα is a potent endogenous inhibitor of BACE1. J. Alzheimers Dis. 47, 545–555. doi: 10.3233/JAD-150282
Philpott, C. C., Ryu, M. S., Frey, A., and Patel, S. (2017). Cytosolic iron chaperones: proteins delivering iron cofactors in the cytosol of mammalian cells. J. Biol. Chem. 292, 12764–12771. doi: 10.1074/jbc.R117.791962
Pigeon, C., Ilyin, G., Courselaud, B., Leroyer, P., Turlin, B., Brissot, P., et al. (2001). A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819. doi: 10.1074/jbc.M008923200
Pirpamer, L., Hofer, E., Gesierich, B., De Guio, F., Freudenberger, P., Seiler, S., et al. (2016). Determinants of iron accumulation in the normal aging brain. Neurobiol. Aging 43, 149–155. doi: 10.1016/j.neurobiolaging.2016.04.002
Plum, S., Steinbach, S., Attems, J., Keers, S., Riederer, P., Gerlach, M., et al. (2016). Proteomic characterization of neuromelanin granules isolated from human Substantia nigra by laser-microdissection. Sci. Rep. 6:37139. doi: 10.1038/srep37139
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of Alzheimer-type dementia in down’s syndrome. Ann. Neurol. 43, 380–383. doi: 10.1002/ana.410430316
Prus, E., and Fibach, E. (2011). Uptake of non-transferrin iron by erythroid cells. Anemia 2011:945289. doi: 10.1155/2011/945289
Raz, L., Knoefel, J., and Bhaskar, K. (2016). The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. 36, 172–186. doi: 10.1038/jcbfm.2015.164
Richards, R. I., Robertson, S. A., O’Keefe, L. V., Fornarino, D., Scott, A., Lardelli, M., et al. (2016). The enemy within: innate surveillance-mediated cell death, the common mechanism of neurodegenerative disease. Front. Neurosci. 10:193. doi: 10.3389/fnins.2016.00193
Richardson, D. R., Lane, D. J., Becker, E. M., Huang, M. L., Whitnall, M., Suryo Rahmanto, Y., et al. (2010). Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U.S.A. 107, 10775–10782. doi: 10.1073/pnas.0912925107
Riemer, J., Bulleid, N., and Herrmann, J. M. (2009). Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324, 1284–1287. doi: 10.1126/science.1170653
Ring, S., Weyer, S. W., Kilian, S. B., Waldron, E., Pietrzik, C. U., Filippov, M. A., et al. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007
Rogers, J. T. (1996). Ferritin translation by interleukin-1and interleukin-6: the role of sequences upstream of the start codons of the heavy and light subunit genes. Blood 87, 2525–2537.
Rogers, J. T., Leiter, L. M., McPhee, J., Cahill, C. M., Zhan, S. S., Potter, H., et al. (1999). Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 274, 6421–6431. doi: 10.1074/jbc.274.10.6421
Rogers, J. T., Randall, J. D., Cahill, C. M., Eder, P. S., Huang, X., Gunshin, H., et al. (2002). An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 277, 45518–45528. doi: 10.1074/jbc.M207435200
Rouault, T. A., Stout, C. D., Kaptain, S., Harford, J. B., and Klausner, R. D. (1991). Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell 64, 881–883. doi: 10.1016/0092-8674(91)90312-M
Rouault, T. A., Tang, C. K., Kaptain, S., Burgess, W. H., Haile, D. J., Samaniego, F., et al. (1990). Cloning of the cDNA encoding an RNA regulatory protein–the human iron-responsive element-binding protein. Proc. Natl. Acad. Sci. U.S.A. 87, 7958–7962. doi: 10.1073/pnas.87.20.7958
Roy, C. N., and Andrews, N. C. (2005). Anemia of inflammation: the hepcidin link. Curr. Opin. Hematol. 12, 107–111. doi: 10.1097/00062752-200503000-00001
Ruberti, F., Barbato, C., and Cogoni, C. (2010). Post-transcriptional regulation of amyloid precursor protein by microRNAs and RNA binding proteins. Commun. Integr. Biol. 3, 499–503. doi: 10.4161/cib.3.6.13172
Saadipour, K., Manucat-Tan, N. B., Lim, Y., Keating, D. J., Smith, K. S., Zhong, J. H., et al. (2017). p75 neurotrophin receptor interacts with and promotes BACE1 localization in endosomes aggravating amyloidogenesis. J. Neurochem. 144, 302–317. doi: 10.1111/jnc.14206
Samaniego, F., Chin, J., Iwai, K., Rouault, T. A., and Klausner, R. D. (1994). Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J. Biol. Chem. 269, 30904–30910.
Samuraki, M., Matsunari, I., Yoshita, M., Shima, K., Noguchi-Shinohara, M., Hamaguchi, T., et al. (2015). Cerebral amyloid angiopathy-related microbleeds correlate with glucose metabolism and brain volume in Alzheimer’s Disease. J. Alzheimers Dis. 48, 517–528. doi: 10.3233/JAD-150274
Sanabria-Castro, A., Alvarado-Echeverria, I., and Monge-Bonilla, C. (2017). Molecular pathogenesis of Alzheimer’s Disease: an update. Ann. Neurosci. 24, 46–54. doi: 10.1159/000464422
Schauenburg, L., Liebsch, F., Eravci, M., Mayer, M. C., Weise, C., and Multhaup, G. (2018). APLP1 is endoproteolytically cleaved by gamma-secretase without previous ectodomain shedding. Sci. Rep. 8:1916. doi: 10.1038/s41598-018-19530-8
Schon, E. A., and Area-Gomez, E. (2012). Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci. 55, 26–36. doi: 10.1016/j.mcn.2012.07.011
Schrag, M., Mueller, C., Oyoyo, U., Smith, M. A., and Kirsch, W. M. (2011). Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 94, 296–306. doi: 10.1016/j.pneurobio.2011.05.001
Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces abeta plaques in Alzheimer’s disease. Nature 537, 50–56. doi: 10.1038/nature19323
Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2017). Addendum: the antibody aducanumab reduces abeta plaques in Alzheimer’s disease. Nature 546:564. doi: 10.1038/nature22809
Shariati, S. A., and De Strooper, B. (2013). Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 587, 2036–2045. doi: 10.1016/j.febslet.2013.05.026
Sharman, M. J., Moussavi Nik, S. H., Chen, M. M., Ong, D., Wijaya, L., Laws, S. M., et al. (2013). The guinea pig as a model for sporadic alzheimer’s disease (ad): the impact of cholesterol intake on expression of ad-related genes. PLoS One 8:e66235. doi: 10.1371/journal.pone.0066235
Sheftel, A. D., Mason, A. B., and Ponka, P. (2012). The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 1820, 161–187. doi: 10.1016/j.bbagen.2011.08.002
Shi, H., Bencze, K. Z., Stemmler, T. L., and Philpott, C. C. (2008). A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210. doi: 10.1126/science.1157643
Silva, D. F., Selfridge, J. E., Lu, J., E, L., Cardoso, S. M., and Swerdlow, R. H. (2012). Mitochondrial abnormalities in Alzheimer’s disease: possible targets for therapeutic intervention. Adv. Pharmacol. 64, 83–126. doi: 10.1016/B978-0-12-394816-8.00003-9
Silver, S., and Walden, W. (1998). Metal Ions in Gene Regulation. New York, NY: Chapman & Hall. doi: 10.1007/978-1-4615-5993-1
Smith, M. A., Casadesus, G., Joseph, J. A., and Perry, G. (2002). Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol. Med. 33, 1194–1199. doi: 10.1016/S0891-5849(02)01021-3
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A. 94, 9866–9868. doi: 10.1073/pnas.94.18.9866
Soares, M. P., and Weiss, G. (2015). The iron age of host-microbe interactions. EMBO Rep. 16, 1482–1500. doi: 10.15252/embr.201540558
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Stiban, J., So, M., and Kaguni, L. S. (2016). Iron-sulfur clusters in mitochondrial metabolism: multifaceted roles of a simple cofactor. Biochemistry 81, 1066–1080. doi: 10.1134/S0006297916100059
Streit, W. J., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Sun, L., Zhou, R., Yang, G., and Shi, Y. (2017). Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. U.S.A. 114, E476–E485. doi: 10.1073/pnas.1618657114
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18727–18732. doi: 10.1073/pnas.0606298103
Sutherland, G. T., Chami, B., Youssef, P., and Witting, P. K. (2013). Oxidative stress in Alzheimer’s disease: primary villain or physiological by-product? Redox Rep. 18, 134–141. doi: 10.1179/1351000213Y.0000000052
Szaruga, M., Veugelen, S., Benurwar, M., Lismont, S., Sepulveda-Falla, D., Lleo, A., et al. (2015). Qualitative changes in human gamma-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 212, 2003–2013. doi: 10.1084/jem.20150892
Takahashi, N., Ortel, T. L., and Putnam, F. W. (1984). Single-chain structure of human ceruloplasmin: the complete amino acid sequence of the whole molecule. Proc. Natl. Acad. Sci. U.S.A. 81, 390–394. doi: 10.1073/pnas.81.2.390
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Taylor, C. J., Ireland, D. R., Ballagh, I., Bourne, K., Marechal, N. M., Turner, P. R., et al. (2008). Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol. Dis. 31, 250–260. doi: 10.1016/j.nbd.2008.04.011
Tcw, J., and Goate, A. M. (2017). Genetics of beta-amyloid precursor protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 7:a024539. doi: 10.1101/cshperspect.a024539
Teng, L., Zhao, J., Wang, F., Ma, L., and Pei, G. (2010). A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res. 20, 138–153. doi: 10.1038/cr.2010.3
Terraneo, L., and Samaja, M. (2017). Comparative response of brain to chronic hypoxia and hyperoxia. Int. J. Mol. Sci. 18:1914. doi: 10.3390/ijms18091914
Thal, D. R., Rub, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Theendakara, V., Peters-Libeu, C. A., Bredesen, D. E., and Rao, R. V. (2017). Transcriptional effects of ApoE4: relevance to Alzheimer’s Disease. Mol. Neurobiol. 55, 5243–5254. doi: 10.1007/s12035-017-0757-2
Thomson, A. M., Cahill, C. M., Cho, H. H., Kassachau, K. D., Epis, M. R., Bridges, K. R., et al. (2005). The acute box cis-element in human heavy ferritin mRNA 5′-untranslated region is a unique translation enhancer that binds poly(C)-binding proteins. J. Biol. Chem. 280, 30032–30045. doi: 10.1074/jbc.M502951200
Ting, S. K., Chong, M. S., Kandiah, N., Hameed, S., Tan, L., Au, W. L., et al. (2013). Absence of A673T amyloid-beta precursor protein variant in Alzheimer’s disease and other neurological diseases. Neurobiol. Aging 34, 2441.e7–2441.e8. doi: 10.1016/j.neurobiolaging.2013.04.012
Toussay, X., Morel, J. L., Biendon, N., Rotureau, L., Legeron, F. P., Boutonnet, M. C., et al. (2017). Presenilin 1 mutation decreases both calcium and contractile responses in cerebral arteries. Neurobiol. Aging 58, 201–212. doi: 10.1016/j.neurobiolaging.2017.06.015
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Usenovic, M., Tresse, E., Mazzulli, J. R., Taylor, J. P., and Krainc, D. (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012
van Duijn, S., Bulk, M., van Duinen, S. G., Nabuurs, R. J. A., van Buchem, M. A., van der Weerd, L., et al. (2017). Cortical iron reflects severity of Alzheimer’s Disease. J. Alzheimers Dis. 60, 1533–1545. doi: 10.3233/JAD-161143
Verheijen, J., Van den Bossche, T., van der Zee, J., Engelborghs, S., Sanchez-Valle, R., Llado, A., et al. (2016). A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol. 132, 213–224. doi: 10.1007/s00401-016-1566-9
Villa, J. C., Chiu, D., Brandes, A. H., Escorcia, F. E., Villa, C. H., Maguire, W. F., et al. (2014). Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell Rep. 8, 1077–1092. doi: 10.1016/j.celrep.2014.07.028
Vinters, H. V., and Gilbert, J. J. (1983). Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14, 924–928. doi: 10.1161/01.STR.14.6.924
Vulpe, C. D., Kuo, Y. M., Murphy, T. L., Cowley, L., Askwith, C., Libina, N., et al. (1999). Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 21, 195–199. doi: 10.1038/5979
Walter, P. B., Knutson, M. D., Paler-Martinez, A., Lee, S., Xu, Y., Viteri, F. E., et al. (2002). Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. U.S.A. 99, 2264–2269. doi: 10.1073/pnas.261708798
Wang, B., and Wang, X. P. (2018). Does ceruloplasmin defend against neurodegenerative diseases? Curr. Neuropharmacol. doi: 10.2174/1570159X16666180508113025 [Epub ahead of print].
Wang, B., Yang, W., Wen, W., Sun, J., Su, B., Liu, B., et al. (2010). Gamma-secretase gene mutations in familial acne inversa. Science 330:1065. doi: 10.1126/science.1196284
Wang, S. M., Fu, L. J., Duan, X. L., Crooks, D. R., Yu, P., Qian, Z. M., et al. (2010). Role of hepcidin in murine brain iron metabolism. Cell Mol. Life Sci. 67, 123–133. doi: 10.1007/s00018-009-0167-3
Wang, J., Tan, L., Wang, H. F., Tan, C. C., Meng, X. F., Wang, C., et al. (2015). Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 44, 385–396. doi: 10.3233/JAD-141506
Wang, L. S., Naj, A. C., Graham, R. R., Crane, P. K., Kunkle, B. W., Cruchaga, C., et al. (2015). Rarity of the Alzheimer disease-protective APP A673T variant in the United States. JAMA Neurol. 72, 209–216. doi: 10.1001/jamaneurol.2014.2157
Wang, R., Zhang, Y. W., Zhang, X., Liu, R., Hong, S., Xia, K., et al. (2006). Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 20, 1275–1277. doi: 10.1096/fj.06-5839fje
Wardman, P., and Candeias, L. P. (1996). Fenton chemistry: an introduction. Radiat. Res. 145, 523–531. doi: 10.2307/3579270
Wessling-Resnick, M. (2010). Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 30, 105–122. doi: 10.1146/annurev.nutr.012809.104804
Westmark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5:e52. doi: 10.1371/journal.pbio.0050052
Westmark, C. J., and Malter, J. S. (2012). The regulation of AbetaPP expression by RNA-binding proteins. Ageing Res. Rev. 11, 450–459. doi: 10.1016/j.arr.2012.03.005
Weyer, S. W., Klevanski, M., Delekate, A., Voikar, V., Aydin, D., Hick, M., et al. (2011). APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280. doi: 10.1038/emboj.2011.119
Whitehouse, P. J., and George, D. R. (2016). A tale of two reports: what recent publications from the Alzheimer’s association and institute of medicine say about the state of the field. J. Alzheimers Dis. 49, 21–25. doi: 10.3233/JAD-150663
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 8, 969–974. doi: 10.1016/0378-4274(95)03532-X
Wong, B. X., Tsatsanis, A., Lim, L. Q., Adlard, P. A., Bush, A. I., and Duce, J. A. (2014). beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9:e114174. doi: 10.1371/journal.pone.0114174
Xie, L., Zheng, W., Xin, N., Xie, J. W., Wang, T., and Wang, Z. Y. (2012). Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 61, 334–340. doi: 10.1016/j.neuint.2012.05.016
Xu, T. H., Yan, Y., Kang, Y., Jiang, Y., Melcher, K., and Xu, H. E. (2016). Alzheimer’s disease-associated mutations increase amyloid precursor protein resistance to gamma-secretase cleavage and the Abeta42/Abeta40 ratio. Cell Discov. 2:16026. doi: 10.1038/celldisc.2016.26
Xu, W., Barrientos, T., and Andrews, N. C. (2013). Iron and copper in mitochondrial diseases. Cell Metab. 17, 319–328. doi: 10.1016/j.cmet.2013.02.004
Xue, Y., Schmollinger, S., Attar, N., Campos, O. A., Vogelauer, M., Carey, M. F., et al. (2017). Endoplasmic reticulum-mitochondria junction is required for iron homeostasis. J. Biol. Chem. 292, 13197–13204. doi: 10.1074/jbc.M117.784249
Yamamoto, A., Shin, R. W., Hasegawa, K., Naiki, H., Sato, H., Yoshimasu, F., et al. (2002). Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x
Yan, Y., Xu, T. H., Harikumar, K. G., Miller, L. J., Melcher, K., and Xu, H. E. (2017). Dimerization of the transmembrane domain of amyloid precursor protein is determined by residues around the gamma-secretase cleavage sites. J. Biol. Chem. 292, 15826–15837. doi: 10.1074/jbc.M117.789669
Yanagida, K., Okochi, M., Tagami, S., Nakayama, T., Kodama, T. S., Nishitomi, K., et al. (2009). The 28-amino acid form of an APLP1-derived Abeta-like peptide is a surrogate marker for Abeta42 production in the central nervous system. EMBO Mol. Med. 1, 223–235. doi: 10.1002/emmm.200900026
Yang, H., Guan, H., Yang, M., Liu, Z., Takeuchi, S., Yanagisawa, D., et al. (2015). Upregulation of mitochondrial ferritin by proinflammatory cytokines: implications for a role in Alzheimer’s disease. J. Alzheimers Dis. 45, 797–811. doi: 10.3233/JAD-142595
Yang, M., Virassamy, B., Vijayaraj, S. L., Lim, Y., Saadipour, K., Wang, Y. J., et al. (2013). The intracellular domain of sortilin interacts with amyloid precursor protein and regulates its lysosomal and lipid raft trafficking. PLoS One 8:e63049. doi: 10.1371/journal.pone.0063049
Yin, R. H., Yu, J. T., and Tan, L. (2015). The role of SORL1 in Alzheimer’s Disease. Mol. Neurobiol. 51, 909–918. doi: 10.1007/s12035-014-8742-5
You, L. H., Yan, C. Z., Zheng, B. J., Ci, Y. Z., Chang, S. Y., Yu, P., et al. (2017). Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 8:e2676. doi: 10.1038/cddis.2017.93
Zaidi, S. H., Denman, R., and Malter, J. S. (1994). Multiple proteins interact at a unique cis-element in the 3′-untranslated region of amyloid precursor protein mRNA. J. Biol. Chem. 269, 24000–24006.
Zaidi, S. H., and Malter, J. S. (1995). Nucleolin and heterogeneous nuclear ribonucleoprotein C proteins specifically interact with the 3′-untranslated region of amyloid protein precursor mRNA. J. Biol. Chem. 270, 17292–17298. doi: 10.1074/jbc.270.29.17292
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.M800102200
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., et al. (2007). Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 282, 10873–10880. doi: 10.1074/jbc.M608856200
Zheng, H., Jiang, M., Trumbauer, M. E., Sirinathsinghji, D. J., Hopkins, R., Smith, D. W., et al. (1995). beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531. doi: 10.1016/0092-8674(95)90073-X
Zhu, Y., Carvey, P. M., and Ling, Z. (2006). Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 1090, 35–44. doi: 10.1016/j.brainres.2006.03.063
Keywords: iron homeostasis, trafficking, familial Alzheimer’s disease, neurodegeneration, mutation, secretase, PRESENILIN, AMYLOID BETA A4 PRESCURSOR PROTEIN
Citation: Lumsden AL, Rogers JT, Majd S, Newman M, Sutherland GT, Verdile G and Lardelli M (2018) Dysregulation of Neuronal Iron Homeostasis as an Alternative Unifying Effect of Mutations Causing Familial Alzheimer’s Disease. Front. Neurosci. 12:533. doi: 10.3389/fnins.2018.00533
Received: 12 April 2018; Accepted: 16 July 2018;
Published: 13 August 2018.
Edited by:
Natalia N. Nalivaeva, University of Leeds, United KingdomReviewed by:
James Duce, University of Cambridge, United KingdomDaniel Pirici, University of Medicine and Pharmacy of Craiova, Romania
Copyright © 2018 Lumsden, Rogers, Majd, Newman, Sutherland, Verdile and Lardelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Lardelli, bWljaGFlbC5sYXJkZWxsaUBhZGVsYWlkZS5lZHUuYXU=
†These authors are first authors