 Thomas B. Stoker
Thomas B. Stoker Kelli M. Torsney1,4†
Kelli M. Torsney1,4† Roger A. Barker
Roger A. Barker- 1John van Geest Centre for Brain Repair, Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 2Wellcome Trust – Medical Research Council Stem Cell Institute, University of Cambridge, Cambridge, United Kingdom
- 3Department of Neurology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
- 4Department of Medicine for the Elderly, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
Parkinson’s disease (PD) is the second most common neurodegenerative disease, manifesting as a characteristic movement disorder with a number of additional non-motor features. The pathological hallmark of PD is the presence of intra-neuronal aggregates of α-synuclein (Lewy bodies). The movement disorder of PD occurs largely due to loss of dopaminergic neurons of the substantia nigra, resulting in striatal dopamine depletion. There are currently no proven disease modifying treatments for PD, with management options consisting mainly of dopaminergic drugs, and in a limited number of patients, deep brain stimulation. Long-term use of established dopaminergic therapies for PD results in significant adverse effects, and there is therefore a requirement to develop better means of restoring striatal dopamine, as well as treatments that are able to slow progression of the disease. A number of exciting treatments have yielded promising results in pre-clinical and early clinical trials, and it now seems likely that the landscape for the management of PD will change dramatically in the short to medium term future. Here, we discuss the promising regenerative cell-based and gene therapies, designed to treat the dopaminergic aspects of PD whilst limiting adverse effects, as well as novel approaches to reducing α-synuclein pathology.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease, typically manifesting with a characteristic movement disorder, consisting of bradykinesia, rigidity, rest tremor and postural instability (Kalia and Lang, 2015). Additional non-motor manifestations occur, including depression, anxiety, sleep abnormalities, constipation and cognitive decline with dementia, which can significantly impair the patient’s quality of life (Khoo et al., 2013).
Pathologically, PD is characterized by the presence of abnormal intra-neuronal aggregates of α-synuclein, termed Lewy bodies and Lewy neurites (Spillantini et al., 1997). Whilst the mechanistic role of Lewy bodies is not fully understood, α-synuclein is clearly central to the pathogenesis of PD, as is highlighted by the fact that rare SNCA mutations, duplications, or triplications cause autosomal dominant familial PD (Klein and Westenberger, 2012). The movement disorder of PD occurs in part due to the selective loss of dopaminergic neurons of the substantia nigra pars compacta, resulting in depletion of dopamine in the striatum, whilst non-motor manifestations predominantly occur due to more widespread neurodegeneration, affecting the cortex and a number of brainstem regions (Selikhova et al., 2009; Kalia and Lang, 2015). Dopaminergic loss also has wider effects, including on sleep and cognition (Williams-Gray et al., 2009).
Since the introduction of levodopa in the 1960s, there have been relatively few developments in the treatment of PD. There are no disease-modifying treatments, and the chronic use of levodopa results in significant adverse effects, which themselves constitute an important part of advanced PD (Jenner, 2003; Kalia and Lang, 2015). However, a number of exciting treatment approaches are either already in, or will soon begin in clinical trials, and the landscape of PD treatment is likely to change dramatically over the coming decades. In this review, we discuss the emerging treatment approaches, and the form that future PD management might take in the next few years.
Current Treatment Options for Parkinson’s Disease
There are currently no disease-modifying treatments for PD, and management predominantly consists of dopaminergic drugs. The most commonly used of these are preparations of levodopa, the precursor of dopamine, which is administered in combination with a dopa-decarboxylase inhibitor which acts to limit some of the side effects, such as nausea. Dopamine agonists, such as ropinirole or rotigotine, are also used. Monoamine oxidase B inhibitors, such as rasagiline and selegiline, and catechol-O-methyltransferase (COMT) inhibitors such as entacapone, can be used to reduce the metabolism of endogenous dopamine. These treatments can restore dopaminergic activity in the striatum, heralding improvements in the motor features of PD. However, they do not treat many of the non-motor features, which are particularly disabling for many patients. Indeed, in some cases treatments may exacerbate some of the non-motor symptoms, such as postural hypotension and neuropsychiatric problems (Young et al., 1997; Kujawa et al., 2000).
Whilst these treatments can cause dramatic improvements in the motor features of PD, especially in the early stages, prolonged use of levodopa in particular results in significant adverse effects, which form an important part of the clinical picture in advanced PD. The non-physiological continuous delivery of dopamine to the striatum is thought to underlie the problematic dyskinesias (abnormal involuntary jerky movements) (Jenner, 2003; Huot et al., 2013), and significant fluctuations in motor function can occur due to erratic absorption of the drug and variable transit of levodopa into the brain – giving the so called on-off phenomenon (Nutt et al., 1984). These medications also result in off-target effects, resulting from their delivery to areas of the brain other than the striatum, which is thought to be the basis for the neuropsychiatric adverse effects that can occur, including hallucinations and impulse control disorder (Ernst, 1969; Voon et al., 2009).
Other treatment options include deep brain stimulation (DBS), which can be very effective in controlling the movement disorder of PD, but like the dopaminergic medications it does not help with most of the non-motor manifestations (Kalia et al., 2013). Though DBS is a safe treatment approach, there are other potentially problematic adverse effects including speech dysfunction and psychiatric disturbance, as well as the general risks associated with a neurosurgical procedure, and this treatment is only suitable in a minority of PD cases (Benabid, 2003).
One approach to delivering dopamine in a more physiological manner is the use of levodopa-intestinal gel, which results in a more predictable release of dopamine than is possible with oral preparations. This may be useful in reducing the motor adverse effects of dopaminergic treatment, but is currently very expensive for widespread use, and is not without complications given the surgery that is necessary for its placement (Olanow et al., 2014). In addition, as with DBS and apomorphine pumps, patients are attached to a device which must be worn all of the time, which is undesirable for many individuals.
So even though there are effective treatment options for the motor features of PD, these come with significant problems, and none of them are able to slow the progression of disease, or improve the disabling non-motor features. Indeed some of these non-motor features are even partly driven by these drugs. There is therefore a need to identify novel methods of restoring striatal dopamine in a targeted and physiological manner, as well as a need to identify treatments that are able to prevent ongoing neurodegeneration and progression of disease. Several experimental approaches are currently being investigated in pre-clinical studies and clinical trials, and it seems likely that the treatment of PD will see dramatic changes in the coming decades. Here, we discuss some of the most promising prospective treatment approaches.
Restoration of Dopaminergic Deficits
Treatment of many of the motor symptoms of PD can be achieved through restoration of striatal dopaminergic tone. This may be accomplished through targeted delivery of dopamine-producing cells, or the use of viruses to deliver genes encoding the enzymes required for dopamine biosynthesis into the striatum. Targeting these regenerative treatments to the striatum, the site of greatest dopamine loss in PD, would minimize the off-target effects seen with oral dopamine-replacement.
Cell-Based Approaches
A variety of cell sources have now been investigated for transplantation in PD, with varying degrees of success (Table 1; Barker et al., 2015). The use of human fetal ventral mesencephalon (VM) tissue grafts in humans has provided proof-of-concept that such cell-based approaches can be effective in treating many of the critical features of PD, but ethical and logistical barriers (chiefly the unpredictable and inadequate supply of fetal tissue) means that this approach will never be a viable mainline therapy for this condition (Barker et al., 2015). What is required, is a renewable source of dopamine-producing cells, or their progenitors, that have the ability to integrate into the host brain, extend axons over adequate distances to innervate the whole striatum, and survive within the host for years. These criteria are most likely to be met by stem cell-derived neurons, which now offer the most likely approach to delivering a clinically useful and scalable cell-based therapy for PD (Barker et al., 2018).
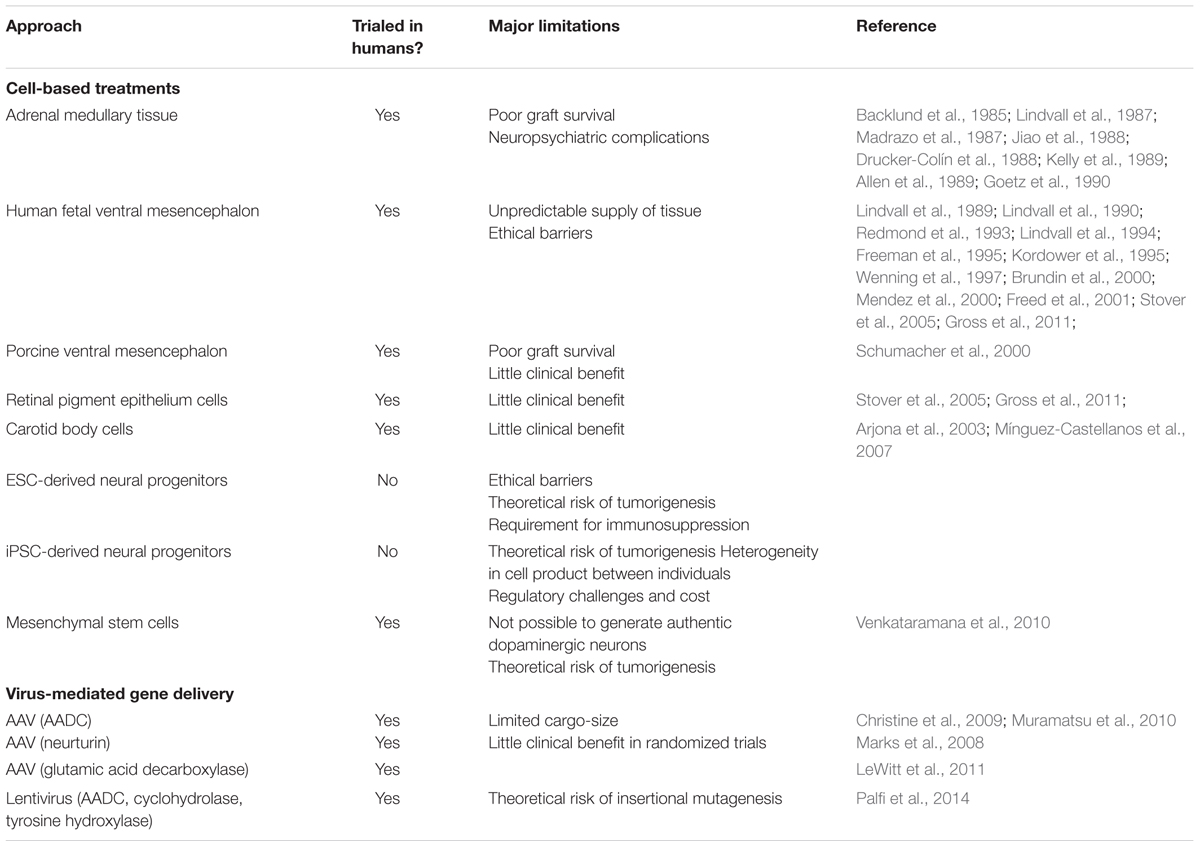
TABLE 1. Experimental regenerative approaches to treating Parkinson’s disease.
Trials of embryonic stem cell (ESC)-derived and induced pluripotent stem cell (iPSC)-derived dopaminergic neuron progenitors are due to commence over the next one or 2 years (Barker et al., 2017). Details on design of these trials, including cell-delivery method, patient-selection criteria, immunosuppression, and study end-points are discussed in detail elsewhere (Barker et al., 2017). ESC lines are isolated from the early blastocyst, and have been generated from surplus human embryos derived from in vitro fertilization procedures (Thomson et al., 1998). It is now possible for these to be reprogrammed to midbrain dopaminergic neuronal progenitors with high efficiency, which have been safely grafted into rodents, with restoration of motor deficits (Kriks et al., 2011; Grealish et al., 2014). As with the prior human fetal mesencephalon grafts, these cells would be allogenic, so would likely require a period of immunosuppression, which is associated with risks to the host, including infection and malignancy (Gutierrez-Dalmau and Campistol, 2007). There are of course ethical considerations with the use of embryonic tissue, but in most cultures this is probably considered acceptable in comparison to the use of fetal tissue (Barker and de Beaufort, 2013).
In contrast, iPSCs are derived through the reprogramming of somatic cells such as fibroblasts, into pluripotent cells, which can subsequently be converted into dopaminergic neuron progenitors (Takahashi et al., 2007; Kikuchi et al., 2017). These have been shown to integrate into the host striatum in non-human primates, yielding motor improvement with no tumor formation at 2 years (Kikuchi et al., 2017). This concept is appealing, as it could potentially provide autologous tissue grafts, which may circumvent the need for immunosuppressive agents. One disadvantage of using autologous cells, however, is that the grafted product will contain any genetic PD-susceptibility factors that contributed to the development of PD in the host. Acquisition of PD pathology has been seen in allogeneic fetal VM grafts, and it may be that this occurs more rapidly in cells with an inherent genetic predisposition to this type of pathology, meaning that the benefit of iPSC-derived grafts may be lost sooner in comparison to ESC-derived grafts (Li et al., 2016). Whilst ESC reprogramming can be highly reproducible, providing a consistent cell product for the use in a large population of patients, there is a high degree of variability between individuals in the reprogramming of adult fibroblasts into iPSCs, and subsequent differentiation to neural cells. This poses regulatory challenges, as each iPSC line derived would essentially be a different product and would potentially be subject to regulatory approval and extensive preclinical testing which brings with it substantial costs. The impact of this may be reduced by the use of haplobanks and accepting a degree of human leukocyte antigen (HLA) mismatch, but this would probably necessitate the use of immunosuppressant agents, meaning that the major advantage of iPSC-derived grafts is lost (Taylor et al., 2012).
Though developments in the field of cell-based therapies have taken decades, there is much hope surrounding forthcoming trials of ESC-derived and iPSC-derived neural progenitor grafts. Increasingly, there is consideration being given to the practicalities of how these treatments may be delivered, highlighting the progress that has been made toward a useful cell-based therapy.
An alternative stem cell source that has been considered as a potential means of treating PD are bone marrow-derived mesenchymal stem cells (MSCs). These are multipotent cells that can be differentiated into tyrosine hydroxylase positive cells, which have improved motor behavior in 6-hydroxydopamine lesioned rodents (Offen et al., 2007). However, differentiation into dopaminergic neurons has been challenging, with only few published reports of this (Trzaska et al., 2007), and the utility of MSCs is likely to depend on a period of optimisation, as has been seen with iPSCs and ESCs. It has also been suggested that MSCs themselves may have neuroprotective properties through anti-inflammatory and paracrine activity, and that they may be useful as a regenerative therapy even without differentiation into dopaminergic neurons (Kim Y.J. et al., 2009; Delcroix et al., 2011). MSC grafts have been delivered to PD patients in a small open-label trial, with short-term safety demonstrated (Venkataramana et al., 2010). This study was not able to comment on whether or not there was any clinical benefit, with short follow up time, and the role of MSCs going forward remains unclear.
The main concern regarding the use of stem cell-based therapies for PD, is the theoretical potential for tumor formation. This may occur due to overgrowth of the graft, or the presence of mutations in oncogenes or tumor suppressor genes in the grafted cells. Though tumors were seen in some of the early pre-clinical trials of ESC-derived transplants, these have not been observed with improved differentiation protocols (Sonntag et al., 2007; Kikuchi et al., 2017). However, the limited follow-up time that is possible in animal models should be acknowledged, and it may be that when present for many years, as would be the case in PD patients, that these grafts do convey a risk of tumorigenesis. Furthermore, interpretation of genetic abnormalities within a graft product is challenging, and because of this, forthcoming clinical trials have adopted different strategies for genetic testing (Barker et al., 2017). Robust investigation of safety, and thorough surveillance will be vital to ensure that the risk of tumor formation is negligible, if these treatments are to be successful.
Viral Gene-Delivery Approaches
An alternative approach toward a regenerative treatment for PD involves the use of viral vectors to achieve expression of specific genes in the striatum. These have included vectors delivering genes encoding the enzymes responsible for the production of dopamine – aromatic amine decarboxylase (AADC), tyrosine hydroxylase and cyclohydrolase-1.
Adeno-associated virus (AAV) vectors carrying AADC have been shown to result in persistent transgene expression, with consequent long-term conversion of levodopa to dopamine in non-human primates (Bankiewicz et al., 2006). Phase I trials have subsequently investigated the safety of AAV vectors delivering AADC to the putamina of PD patients (Christine et al., 2009; Muramatsu et al., 2010). Both trials reported an improvement in clinical and PET imaging parameters at 6 months, and the procedures were well tolerated. Two further phase I clinical trials conducted in PD patients using AAV vectors have been undertaken, but these have taken different therapeutic approaches. One has used a trophic factor for dopaminergic neurons (neurturin) and the other has sought to change the identity of the subthalamic output neurons from glutamatergic to glutamic acid-decarboxylase expressing ones. These trials showed initial promise, and supported the safety of gene-based therapy approaches (Kaplitt et al., 2007; Marks et al., 2008). However, randomized phase II trials were not so promising, showing no, or only mild benefit in comparison to placebo (Marks et al., 2010; LeWitt et al., 2011). AAV vectors are non-integrating vectors, so do not carry the risk of insertional mutagenesis that may occur with integrating viral vectors (Mukherjee and Thrasher, 2013). However, the main drawback of AAV vectors is that the potential size of the genetic cargo is relatively limited, meaning that the gene for only one of the enzymes in the dopamine synthesis pathway could be delivered in the aforementioned trials.
In contrast, lentivirus vectors are integrating viral vectors, with capacity for a much larger genetic cargo than AAVs – an attribute that has been exploited in viral gene-delivery trials (Palfi et al., 2014). ProSavin (Oxford Biomedica, United Kingdom) is a lentiviral product containing the genes for AADC, along with those for tyrosine hydroxylase and cyclohydrolase-1. A phase I/II trial showed that this product was well tolerated, though a significant number of patients did experience an increase in on-medication dyskinesias (Palfi et al., 2014). A further trial of a new version of ProSavin, OXB -102, is about to start in the United Kingdom and France.
α-Synuclein as a Therapeutic Target
Accumulation and aggregation of α-synuclein is the pathological hallmark of PD, and though its role is not completely understood, it appears pivotal in the pathogenesis of PD (and other α-synucleinopathies such as dementia with Lewy bodies and multiple system atrophy) (Spillantini et al., 1997). It follows therefore, that reducing the levels of pathological forms of α-synuclein may alter the course of PD. A number of experimental approaches have been investigated or are currently under investigation for their ability to potentially offer a disease-modifying effect in PD (see Figure 1) through targeting α-synuclein. Here, we provide an overview of some of the most promising approaches.
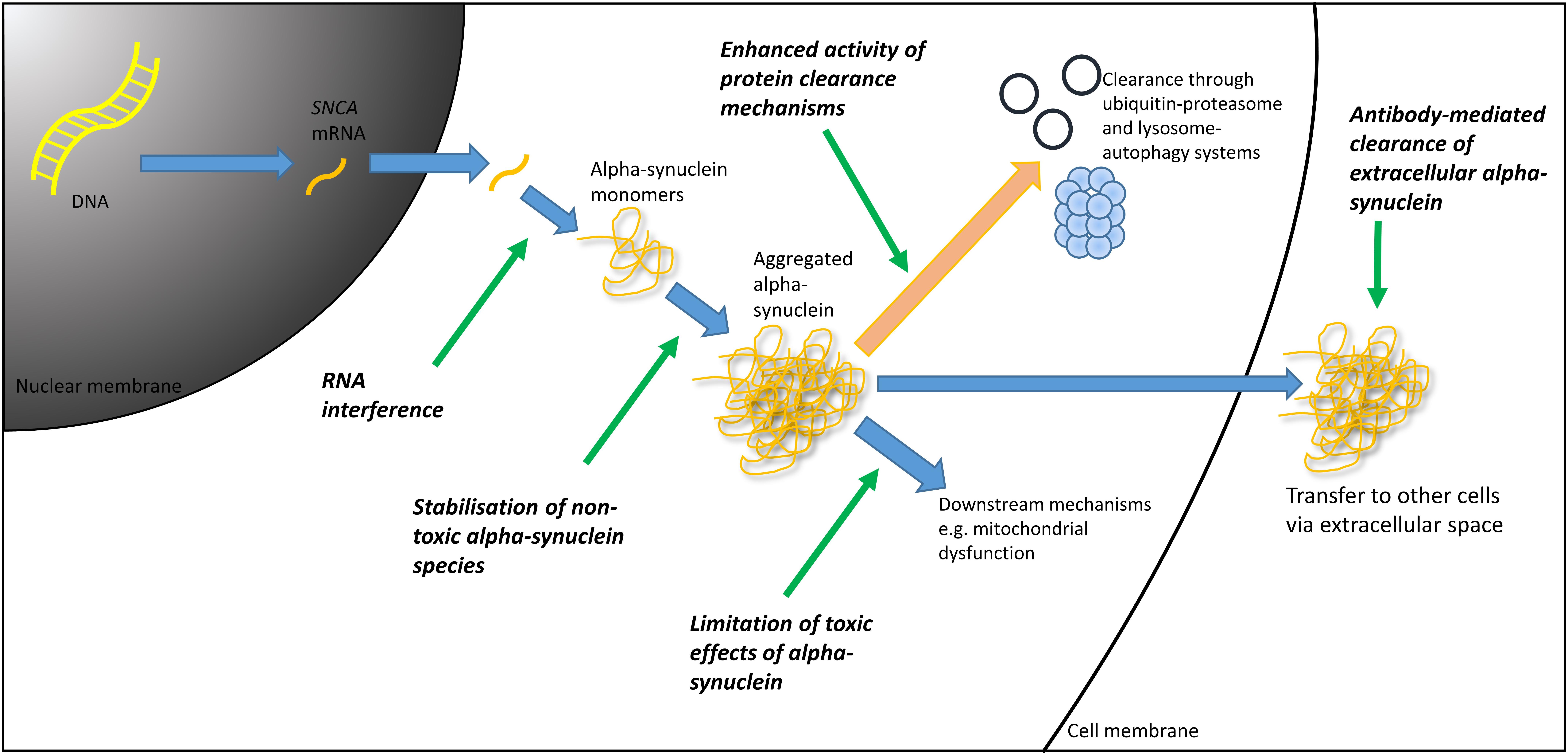
FIGURE 1. Therapeutic targets in α-synuclein mediated pathology. DNA, deoxyribonucleic acid; mRNA, messenger ribonucleic acid; RNA, ribonucleic acid.
Reduction of α-Synuclein Production
One mechanism by which the pathological effect of α-synuclein could be mitigated is to reduce its synthesis. This may be achieved through RNA interference technologies, in which exogenous introduction of synthetic ribonucleic acid (RNA) molecules are used to trigger selective post-transcriptional silencing of the α-synuclein gene, through messenger RNA (mRNA) degradation. Lentiviral delivery of a short-hairpin RNA (shRNA) targeting α-synuclein has been shown to silence ectopic α-synuclein expression in a rodent model and in SH-SY5Y cells, offering support to this concept (Sapru et al., 2006). Additionally, direct infusion of a small-interfering RNA (siRNA) directed against α-synuclein into the mouse hippocampus lead to a reduction in the expression of α-synuclein (Lewis et al., 2008). Following on from this, work conducted in non-human primates, in which an infusion of siRNA directed against α-synuclein was administered, demonstrated a reduction in α-synuclein mRNA, and a 40–50% reduction in α-synuclein protein levels (McCormack et al., 2010). It remains to be seen whether or not this reduction in α-synuclein levels will translate into a clinical benefit, but demonstration that these techniques have the ability to reduce α-synuclein levels is certainly promising.
Of course, one concern with suppression of α-synuclein production is that there may be negative implications due to silencing of normal α-synuclein and with this a loss of its normal physiological function. This function is not completely understood, but it appears to play a role in the docking of vacuoles to the cell membrane, so could potentially be important in neurotransmitter release (Burré et al., 2014). Reassuringly, despite a reduction in α-synuclein levels in the rodent models discussed, α-synuclein suppression did not result in any toxicity (Lewis et al., 2008). However, other studies have reported significant neurotoxicity in association with the suppression of α-synuclein including in the substantia nigra pars compacta (Robertson et al., 2004; Gorbatyuk et al., 2010; Kanaan and Manfredsson, 2012). In one of these studies, the neurodegeneration could be rescued by supplementing α-synuclein levels, suggesting that it was indeed the loss of α-synuclein that precipitated the pathology (Gorbatyuk et al., 2010). These reports highlight the need for adequate safety data if these techniques are to progress from pre-clinical to clinical studies.
Whilst these RNA-based approaches reduce translation of the α-synuclein gene, an alternative target would be to reduce transcription of the α-synuclein gene. Recently, β2-adrenergic receptor agonists (β2 agonists) including clenbuterol, have been associated with a reduction in α-synuclein gene expression (Mittal et al., 2017). Clenbuterol, a medication used in the management of asthma, was shown to reduce α-synuclein expression by 35% in a neuroblastoma cell line and in rat cortical neurons. Additionally it has the ability to cross the blood brain barrier – a necessary requirement for any of these proposed treatment approaches (Mittal et al., 2017). These preclinical trials have been supported by the results of two large Norwegian epidemiological cohorts, in which four million individuals were followed up. In these, β2 agonists were associated with a reduced risk of developing PD. Conversely, β2 antagonists (such as propranolol) were associated with an increased risk of PD (Mittal et al., 2017). Although this association requires further investigation, the prospect that β2 agonists may offer a disease-modifying effect in PD is an exciting one, particularly given the fact that there is already abundant experience with the use of these drugs for other conditions, meaning that their introduction to the clinic could be relatively quick.
Increasing α-Synuclein Clearance
An alternative approach to targeting α-synuclein, is to enhance its clearance. This may potentially be achieved through increasing the intracellular degradation of α-synuclein through autophagy pathways and the ubiquitin-proteasome system for example, or by utilizing immune-therapies to clear extra-cellular α-synuclein, particularly given that it has been hypothesized that α-synuclein pathology may spread between cells in a prion-like fashion.
Immunotherapies
Active and passive immunotherapies have been investigated as a means to target and degrade extracellular α-synuclein, and have been shown to reduce α-synuclein aggregation and prevent behavioral deficits in transgenic mice (George and Brundin, 2015). Some of these treatments have reached clinical trials, with promising safety data and early results (Brundin et al., 2017).
A phase I clinical trial of an active immunotherapy vaccine (AFFITOPE PD03A, AFFiRiS, Austria) has recently been completed1. This is a synthetically produced vaccine containing an α-synuclein mimicking peptide. In this trial, patients with early-stage PD received repeated subcutaneous injections of AFFITOPE PD03A. Thirty-six patients were randomized to high dose or low dose AFFITOPE PD03A or placebo. A dose-dependent immune response against the peptide, as well as cross-reactivity against α-synuclein, was reported, with increasing antibody titres over time1. Encouragingly both doses were well tolerated and no serious drug related side effects were reported. Another one of the company’s active immunotherapies, PD01A, has been reported to be safe in early PD, over 4 years2.
A passive immunotherapy approach using humanized monoclonal antibodies against α-synuclein (PRX001) has also been tested. Prothena conducted a phase Ia clinical trial demonstrating a reduction in α-synuclein levels by up to 96.5% in healthy volunteers, and a subsequent phase 1b trial in PD patients in which a reduction in α-synuclein was also seen (Schenk et al., 2017). PRX001 was well tolerated and a phase II trial in patients with early stage PD is currently under way3.
Additional immunotherapies targeting α-synuclein are on the horizon, with the AstraZeneca and Takeda Pharmaceutical companies announcing the development of an α-synuclein antibody MEDI1341 for PD4. The press release regarding MEDI1341 claimed that this drug has a high affinity for the target and a reduced effector function, potentially offering a safer and more efficacious treatment, in comparison to other α-synuclein immunotherapies.
Enhancement of Autophagy
The proposed mechanism for the immunotherapies that have been discussed is clearance of extracellular α-synuclein, but it may also be possible to clear intra-cellular α-synuclein through other means. The lysosome-autophagy system plays an important role in α-synuclein clearance, through both chaperone-mediated autophagy and macroautophagy (Decressac et al., 2013). Enhancement of activity within this system could therefore reduce α-synuclein levels, and its propensity to accumulate and aggregate. A number of drugs have been investigated as potential means by which autophagy can be activated, including novel agents as well as the repurposing of drugs with other established indications.
Rapamycin for example, has historically been used as an immunosuppressant agent, and is a well-established inducer of macroautophagy (Moors et al., 2017), and it has been shown to reduce α-synuclein accumulation in an in vitro model of GBA1 mutation-associated PD (Cullen et al., 2011). Although the unfavorable adverse effect profile of rapamycin means that is unlikely to be useful as a chronic treatment for PD, results such as this suggest that enhancing autophagy can indeed reduce total α-synuclein levels (Cullen et al., 2011; Moors et al., 2017). Several compounds have been suggested to increase autophagy activity, including for example the disaccharide trehalose (Sarkar et al., 2007; Rodríguez-Navarro et al., 2010) and the tricyclic antidepressant nortriptyline (Gassen et al., 2014), which are under consideration for entry into clinical trials.
Trehalose is of particular interest having been shown to reduce protein aggregates in models of other neurodegenerative diseases (Tanaka et al., 2004; Davies et al., 2006). Trehalose is a naturally occurring disaccharide, which appears to play an important role in stress responses in yeast (Singer and Lindquist, 1998). Whilst it has been suggested that its ability to reduce protein aggregation occurs due to a chaperone activity, or through binding and stabilization of abnormal proteins (Tanaka et al., 2004; Davies et al., 2006), it has also been shown to act via an mTOR-independent pathway to increase autophagy (Sarkar et al., 2007; Rodríguez-Navarro et al., 2010). These studies have clearly prompted interest in this compound, though a recent study of mouse primary cortical neurons found that trehalose did not prevent toxicity from exposure to α-synuclein pre-formed fibrils (Redmann et al., 2017).
With an increased understanding of the genetic basis of PD it is becoming increasingly clear that different pathogenic mechanisms may be more pronounced in certain groups of patients. It thus follows that, as our knowledge of pathological subtypes of PD expands, then different targeted therapeutic approaches may emerge. GBA1 mutations are the most common genetic risk factor for PD (Sidransky et al., 2009). The GBA1 gene encodes for the enzyme β-glucocerebrosidase (GCase), and homozygous mutations in this gene result in the lysosomal storage disorder, Gaucher disease (Migdalska-Richards and Schapira, 2016). Though the mechanism by which GBA1 mutations increase the risk of PD are not fully understood, there is an established literature suggesting that dysfunction of the lysosome-autophagy system is impaired in this setting (Schöndorf et al., 2014; Bae et al., 2015; Fernandes et al., 2016). Therefore, in this group of patients, targeting autophagy may be particularly relevant. There are two clinical trials ongoing in which PD patients with GBA1 mutations are receiving treatments aiming to correct abnormalities in the lysosomal environment. In the first, a phase II trial (MOVES-PD, Genzyme/Sanofi), patients are receiving a compound called GZ/SAR4026715. This inhibits the production of glycosphingolipids, which are normally catabolized by GCase, and build up in cells in PD patients with GBA1 mutations. In the other, ambroxol, a Food and Drug Administration (FDA)-approved mucolytic, is being investigated as a potential treatment for GBA1 mutation-associated PD (ClinicalTrials.gov Identifier: NCT02941822 and NCT02914366). Ambroxol has chaperone properties, so potentially facilitates the transit of misfolded GCase to the lysosome (Maegawa et al., 2009). In in vitro studies, it improves lysosomal function in dermal fibroblasts with GBA1 mutations (Bendikov-Bar et al., 2013; McNeill et al., 2014) and increases GCase activity in non-human primates in vivo (Migdalska-Richards et al., 2017).
Repurposing of Other Drugs
Two other drugs have recently been repurposed and trialed in PD patients – the glucagon-like peptide-1 (GLP-1) agonist, exenatide, and the tyrosine kinase inhibitor, nilotinib. Exenatide is an established treatment for type two diabetes mellitus (Lovshin and Drucker, 2009), whilst nilotinib is used in the treatment of chronic myelogenous leukemia, so data already exists on the safety and tolerability of these agents in patient populations, which helped expedite their progress through clinical trials, which have shown promising results.
Exenatide has been shown to provide neuroprotective, and neurorestorative effects in toxin-based rodent models of nigrostriatal degeneration, with improvements in motor function, behavior, learning, and memory (Bertilsson et al., 2008; Kim S. et al., 2009). A recent double blinded placebo-controlled trial studied the effects of subcutaneous exenatide in patients with moderate PD (Athauda et al., 2017). The treatment was associated with positive and lasting effects on off-medication motor scores at 60 weeks. In the context of PD this follow up period is relatively short, and it remains unclear as to whether this drug reduces the progression of neurodegeneration in PD, but these early results appear promising (Athauda et al., 2017).
Nilotinib has been shown to enhance amyloid clearance, and hence has been purported as a potential means of reducing α-synuclein levels (Lonskaya et al., 2014). It acts as an inhibitor of c-abl, an oncogene involved in the regulation of cell growth, differentiation, proliferation and survival of cells. Increased levels of c-abl have been associated with PD, which is thought to result in an increase in phosphorylation and aggregation of α-synuclein (Brahmachari et al., 2016; Lindholm et al., 2016). In addition, an increase in c-abl activity leads to a reduction in the function of parkin, which is a key protein in mitochondrial biogenesis, and in which mutations result in familial PD (Lonskaya et al., 2014). Nilotinib has been shown to attenuate exogenously expressed α-synuclein levels in mice, and to reduce α-synuclein induced nigral degeneration (Hebron et al., 2013). A recent open-label study in which patients with PD with dementia or Lewy body dementia received nilotinib, demonstrated safety and tolerability of the drug in these groups, at doses of 150 and 300 mg (lower than what is used in its established indications) (Pagan et al., 2016). This study, however, involved no placebo group, and there were significant baseline differences between the two small groups, so it was not possible to comment on any clinical benefits that the drug may offer. Though the pre-clinical studies raise interesting prospects for nilotinib, there is as yet no convincing evidence of its efficacy in PD patients (Wyse et al., 2016), although another trial (NILO-PD) with this agent is currently ongoing in the United States (ClinicalTrials.gov identifier: NCT03205488).
Conclusion
Current treatment options for PD are limited to symptomatic measures, predominantly in the form of dopaminergic medications and DBS. Though they can confer significant symptomatic benefit, they also result in troubling adverse effects, which can impair the quality of life of patients. Furthermore, none of these alter the course of disease. There is therefore a need to develop treatments that are able to restore dopaminergic tone in the striatum, in a targeted, physiological manner, and to identify drugs, which can prevent or slow ongoing neurodegeneration. A number of exciting treatment approaches are beginning to enter clinical trials. These include regenerative treatments in the form of stem cell-derived grafts and viral gene therapies designed to replace the function of the neurons that have been lost, as well as novel and repurposed drugs targeting the pathogenic mechanisms of PD, with potentially disease-modifying properties. Expanding knowledge of the pathophysiology and genetics of PD, has allowed for the development of specific treatment approaches, and it is probable that the standard treatment of PD will be dramatically altered over the coming decades, as these new treatment options emerge and are used in combination.
Author Contributions
TS and KT contributed equally to the writing of the manuscript. RB reviewed and edited the manuscript.
Funding
The authors acknowledge financial support from the following organizations: Wellcome Trust Stem Cell Institute (Cambridge), Medical Research Council, National Institute for Health Research Biomedical Research Centre, and the Cure Parkinson’s Trust.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://www.affiris.com/news/affiris-announces-top-line-results-of-first-in-human-clinical-study-using-affitope/
- ^ http://www.affiris.com/news/affiris-announces-encouraging-long-term-data-from-a-series-of-first-in-human-studies-using-affitope-pd01a-targeting-oligomeric-alpha-synuclein-in-early-parkinsons-disease-patients/
- ^ http://ir.prothena.com/news-releases/news-release-details/prothena-announces-initiation-phase-2-pasadena-study?ReleaseID=1032188
- ^ https://www.astrazeneca.com/media-centre/press-releases/2017/astrazeneca-and-takeda-establish-collaboration-to-develop-and-commercialise-medi1341-for-parkinsons-disease-25082017.html
- ^ http://news.genzyme.com/press-release/sanofi-initiates-phase-2-clinical-trial-evaluate-therapy-genetic-form-parkinsons-disea
References
Allen, G. S., Burns, R. S., Tulipan, N. B., and Parker, R. A. (1989). Adrenal medullary transplantation to the caudate nucleus in Parkinson’s disease. Initial clinical results in 18 patients. Arch. Neurol. 46, 487–491. doi: 10.1001/archneur.1989.00520410021016
Arjona, V., Mínguez-Castellanos, A., Montoro, R. J., Ortega, A., Escamilla, F., Toledo-Aral, J. J., et al. (2003). Autotransplantation of human carotid body cell aggregates for treatment of Parkinson’s disease. Neurosurgery 53, 321-328; discussion 328–330.
Athauda, D., Maclagan, K., Skene, S. S., Bajwa-Joseph, M., Letchford, D., Chowdhury, K., et al. (2017). Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. doi: 10.1016/S0140-6736(17)31585-4
Backlund, E. O., Granberg, P. O., Hamberger, B., Knutsson, E., Mårtensson, A., Sedvall, G., et al. (1985). Transplantation of adrenal medullary tissue to striatum in parkinsonism. First clinical trials. J. Neurosurg. 62, 169–173. doi: 10.3171/jns.1985.62.2.0169
Bae, E. J., Yang, N. Y., Lee, C., Lee, H. J., Kim, S., Sardi, S. P., et al. (2015). Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 47:e153. doi: 10.1038/emm.2014.128
Bankiewicz, K. S., Forsayeth, J., Eberling, J. L., Sanchez-Pernaute, R., Pivirotto, P., Bringas, J., et al. (2006). Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol. Ther. 14, 564–570. doi: 10.1016/j.ymthe.2006.05.005
Barker, R. A., and de Beaufort, I. (2013). Scientific and ethical issues related to stem cell research and interventions in neurodegenerative disorders of the brain. Prog. Neurobiol. 110, 63–73. doi: 10.1016/j.pneurobio.2013.04.003
Barker, R. A., Drouin-Ouellet, J., and Parmar, M. (2015). Cell-based therapies for Parkinson disease—past insights and future potential. Nat. Rev. Neurol. 11, 492–503. doi: 10.1038/nrneurol.2015.123
Barker, R. A., Götz, M., and Parmar, M. (2018). New approaches for brain repair-from rescue to reprogramming. Nature 557, 329–334. doi: 10.1038/s41586-018-0087-1
Barker, R. A., Parmar, M., Studer, L., and Takahashi, J. (2017). Human trials of stem cell-derived dopamine neurons for Parkinson’s disease: dawn of a new Era. Cell Stem Cell 21, 569–573. doi: 10.1016/j.stem.2017.09.014
Benabid, A. L. (2003). Deep brain stimulation for Parkinson’s disease. Curr. Opin. Neurobiol. 13, 696–706. doi: 10.1016/j.conb.2003.11.001
Bendikov-Bar, I., Maor, G., Filocamo, M., and Horowitz, M. (2013). Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. doi: 10.1016/j.bcmd.2012.10.007
Bertilsson, G., Patrone, C., Zachrisson, O., Andersson, A., Dannaeus, K., Heidrich, J., et al. (2008). Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 86, 326–338. doi: 10.1002/jnr.21483
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to α-synuclein-induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Brundin, P., Dave, K. D., and Kordower, J. H. (2017). Therapeutic approaches to target α-synuclein pathology. Exp. Neurol. 298, 225–235. doi: 10.1016/j.expneurol.2017.10.003
Brundin, P., Pogarell, O., Hagell, P., Piccini, P., Widner, H., Schrag, A., et al. (2000). Bilateral caudate and putamen grafts of embryonic mesencephalic tissue treated with lazaroids in Parkinson’s disease. Brain 123(Pt 7), 1380–1390. doi: 10.1093/brain/123.7.1380
Burré, J., Sharma, M., and Südhof, T. C. (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Christine, C. W., Starr, P. A., Larson, P. S., Eberling, J. L., Jagust, W. J., Hawkins, R. A., et al. (2009). Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669. doi: 10.1212/WNL.0b013e3181c29356
Cullen, V., Sardi, S. P., Ng, J., Xu, Y. H., Sun, Y., Tomlinson, J. J., et al. (2011). Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann. Neurol. 69, 940–953. doi: 10.1002/ana.22400
Davies, J. E., Sarkar, S., and Rubinsztein, D. C. (2006). Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 15, 23–31. doi: 10.1093/hmg/ddi422
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Björklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U.S.A. 110, E1817–E1826. doi: 10.1073/pnas.1305623110
Delcroix, G. J., Garbayo, E., Sindji, L., Thomas, O., Vanpouille-Box, C., Schiller, P. C., et al. (2011). The therapeutic potential of human multipotent mesenchymal stromal cells combined with pharmacologically active microcarriers transplanted in hemi-parkinsonian rats. Biomaterials 32, 1560–1573. doi: 10.1016/j.biomaterials.2010.10.041
Drucker-Colín, R., Madrazo, I., Ostrosky-Solís, F., Shkurovich, M., Franco, R., and Torres, C. (1988). Adrenal medullary tissue transplants in the caudate nucleus of Parkinson’s patients. Prog. Brain Res. 78, 567–574. doi: 10.1016/S0079-6123(08)60332-5
Ernst, A. M. (1969). The role of biogenic amines in the extra-pyramidal system. Acta Physiol. Pharmacol. Neerl. 15, 141–154.
Fernandes, H. J., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Freed, C. R., Greene, P. E., Breeze, R. E., Tsai, W. Y., DuMouchel, W., Kao, R., et al. (2001). Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med. 344, 710–719. doi: 10.1056/NEJM200103083441002
Freeman, T. B., Olanow, C. W., Hauser, R. A., Nauert, G. M., Smith, D. A., Borlongan, C. V., et al. (1995). Bilateral fetal nigral transplantation into the postcommissural putamen in Parkinson’s disease. Ann. Neurol. 38, 379–388. doi: 10.1002/ana.410380307
Gassen, N. C., Hartmann, J., Zschocke, J., Stepan, J., Hafner, K., Zellner, A., et al. (2014). Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: evidence in cells, mice, and humans. PLoS Med. 11:e1001755. doi: 10.1371/journal.pmed.1001755
George, S., and Brundin, P. (2015). Immunotherapy in Parkinson’s disease: micromanaging alpha-synuclein aggregation. J. Parkinsons Dis. 5, 413–424. doi: 10.3233/JPD-150630
Goetz, C. G., Tanner, C. M., Penn, R. D., Stebbins, G. T., Gilley, D. W., Shannon, K. M., et al. (1990). Adrenal medullary transplant to the striatum of patients with advanced Parkinson’s disease: 1-year motor and psychomotor data. Neurology 40, 273–276. doi: 10.1212/WNL.40.2.273
Gorbatyuk, O. S., Li, S., Nash, K., Gorbatyuk, M., Lewin, A. S., Sullivan, L. F., et al. (2010). In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 18, 1450–1457. doi: 10.1038/mt.2010.115
Grealish, S., Diguet, E., Kirkeby, A., Mattsson, B., Heuer, A., Bramoulle, Y., et al. (2014). Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 15, 653–665. doi: 10.1016/j.stem.2014.09.017
Gross, R. E., Watts, R. L., Hauser, R. A., Bakay, R. A., Reichmann, H., von Kummer, R., et al. (2011). Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 10, 509–519. doi: 10.1016/S1474-4422(11)70097-7
Gutierrez-Dalmau, A., and Campistol, J. M. (2007). Immunosuppressive therapy and malignancy in organ transplant recipients: a systematic review. Drugs 67, 1167–1198. doi: 10.2165/00003495-200767080-00006
Hebron, M. L., Lonskaya, I., and Moussa, C. E. (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 22, 3315–3328. doi: 10.1093/hmg/ddt192
Huot, P., Johnston, T. H., Koprich, J. B., Fox, S. H., and Brotchie, J. M. (2013). The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 65, 171–222. doi: 10.1124/pr.111.005678
Jenner, P. (2003). Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson’s disease. Curr. Opin. Neurol. 16(Suppl. 1), S3–S7. doi: 10.1097/00019052-200312001-00002
Jiao, S. S., Zhang, W. C., Cao, J. K., Zhang, Z. M., Wang, H., Ding, M. C., et al. (1988). Study of adrenal medullary tissue transplantation to striatum in parkinsonism. Prog. Brain Res. 78, 575–580. doi: 10.1016/S0079-6123(08)60333-7
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kalia, S. K., Sankar, T., and Lozano, A. M. (2013). Deep brain stimulation for Parkinson’s disease and other movement disorders. Curr. Opin. Neurol. 26, 374–380. doi: 10.1097/WCO.0b013e3283632d08
Kanaan, N. M., and Manfredsson, F. P. (2012). Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? J. Parkinsons Dis. 2, 249–267. doi: 10.3233/JPD-012138
Kaplitt, M. G., Feigin, A., Tang, C., Fitzsimons, H. L., Mattis, P., Lawlor, P. A., et al. (2007). Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369, 2097–2105. doi: 10.1016/S0140-6736(07)60982-9
Kelly, P. J., Ahlskog, J. E., van Heerden, J. A., Carmichael, S. W., Stoddard, S. L., and Bell, G. N. (1989). Adrenal medullary autograft transplantation into the striatum of patients with Parkinson’s disease. Mayo Clin. Proc. 64, 282–290. doi: 10.1016/S0025-6196(12)65248-3
Khoo, T. K., Yarnall, A. J., Duncan, G. W., Coleman, S., O’Brien, J. T., Brooks, D. J., et al. (2013). The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 80, 276–281. doi: 10.1212/WNL.0b013e31827deb74
Kikuchi, T., Morizane, A., Doi, D., Magotani, H., Onoe, H., Hayashi, T., et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596. doi: 10.1038/nature23664
Kim, S., Moon, M., and Park, S. (2009). Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 202, 431–439. doi: 10.1677/JOE-09-0132
Kim, Y. J., Park, H. J., Lee, G., Bang, O. Y., Ahn, Y. H., Joe, E., et al. (2009). Neuroprotective effects of human mesenchymal stem cells on dopaminergic neurons through anti-inflammatory action. Glia 57, 13–23. doi: 10.1002/glia.20731
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Kordower, J. H., Freeman, T. B., Snow, B. J., Vingerhoets, F. J., Mufson, E. J., Sanberg, P. R., et al. (1995). Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson’s disease. N. Engl. J. Med. 332, 1118–1124. doi: 10.1056/NEJM199504273321702
Kriks, S., Shim, J. W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. doi: 10.1038/nature10648
Kujawa, K., Leurgans, S., Raman, R., Blasucci, L., and Goetz, C. G. (2000). Acute orthostatic hypotension when starting dopamine agonists in Parkinson’s disease. Arch. Neurol. 57, 1461–1463. doi: 10.1001/archneur.57.10.1461
Lewis, J., Melrose, H., Bumcrot, D., Hope, A., Zehr, C., Lincoln, S., et al. (2008). In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 3:19. doi: 10.1186/1750-1326-3-19
LeWitt, P. A., Rezai, A. R., Leehey, M. A., Ojemann, S. G., Flaherty, A. W., Eskandar, E. N., et al. (2011). AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 10, 309–319. doi: 10.1016/S1474-4422(11)70039-4
Li, W., Englund, E., Widner, H., Mattsson, B., van Westen, D., Lätt, J., et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. U.S.A. 113, 6544–6549. doi: 10.1073/pnas.1605245113
Lindholm, D., Pham, D. D., Cascone, A., Eriksson, O., Wennerberg, K., and Saarma, M. (2016). c-Abl Inhibitors Enable Insights into the pathophysiology and neuroprotection in Parkinson’s disease. Front. Aging Neurosci. 8:254. doi: 10.3389/fnagi.2016.00254
Lindvall, O., Backlund, E. O., Farde, L., Sedvall, G., Freedman, R., Hoffer, B., et al. (1987). Transplantation in Parkinson’s disease: two cases of adrenal medullary grafts to the putamen. Ann. Neurol. 22, 457–468. doi: 10.1002/ana.410220403
Lindvall, O., Brundin, P., Widner, H., Rehncrona, S., Gustavii, B., Frackowiak, R., et al. (1990). Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science 247, 574–577. doi: 10.1126/science.2105529
Lindvall, O., Rehncrona, S., Brundin, P., Gustavii, B., Astedt, B., Widner, H., et al. (1989). Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 46, 615–631. doi: 10.1001/archneur.1989.00520420033021
Lindvall, O., Sawle, G., Widner, H., Rothwell, J. C., Björklund, A., Brooks, D., et al. (1994). Evidence for long-term survival and function of dopaminergic grafts in progressive Parkinson’s disease. Ann. Neurol. 35, 172–180. doi: 10.1002/ana.410350208
Lonskaya, I., Hebron, M. L., Desforges, N. M., Schachter, J. B., and Moussa, C. E. (2014). Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 92, 373–386. doi: 10.1007/s00109-013-1112-3
Lovshin, J. A., and Drucker, D. J. (2009). Incretin-based therapies for type 2 diabetes mellitus. Nat. Rev. Endocrinol. 5, 262–269. doi: 10.1038/nrendo.2009.48
Madrazo, I., Drucker-Colín, R., Díaz, V., Martínez-Mata, J., Torres, C., and Becerril, J. J. (1987). Open microsurgical autograft of adrenal medulla to the right caudate nucleus in two patients with intractable Parkinson’s disease. N. Engl. J. Med. 316, 831–834. doi: 10.1056/NEJM198704023161402
Maegawa, G. H., Tropak, M. B., Buttner, J. D., Rigat, B. A., Fuller, M., Pandit, D., et al. (2009). Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. doi: 10.1074/jbc.M109.012393
Marks, W. J., Bartus, R. T., Siffert, J., Davis, C. S., Lozano, A., Boulis, N., et al. (2010). Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 9, 1164–1172. doi: 10.1016/S1474-4422(10)70254-4
Marks, W. J., Ostrem, J. L., Verhagen, L., Starr, P. A., Larson, P. S., Bakay, R. A., et al. (2008). Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 7, 400–408. doi: 10.1016/S1474-4422(08)70065-6
McCormack, A. L., Mak, S. K., Henderson, J. M., Bumcrot, D., Farrer, M. J., and Di Monte, D. A. (2010). Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS One 5:e12122. doi: 10.1371/journal.pone.0012122
McNeill, A., Magalhaes, J., Shen, C., Chau, K. Y., Hughes, D., Mehta, A., et al. (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495. doi: 10.1093/brain/awu020
Mendez, I., Dagher, A., Hong, M., Hebb, A., Gaudet, P., Law, A., et al. (2000). Enhancement of survival of stored dopaminergic cells and promotion of graft survival by exposure of human fetal nigral tissue to glial cell line–derived neurotrophic factor in patients with Parkinson’s disease. Report of two cases and technical considerations. J. Neurosurg. 92, 863–869. doi: 10.3171/jns.2000.92.5.0863
Migdalska-Richards, A., Ko, W. K. D., Li, Q., Bezard, E., and Schapira, A. H. V. (2017). Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 71:e21967. doi: 10.1002/syn.21967
Migdalska-Richards, A., and Schapira, A. H. (2016). The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem.
Mínguez-Castellanos, A., Escamilla-Sevilla, F., Hotton, G. R., Toledo-Aral, J. J., Ortega-Moreno, A., Méndez-Ferrer, S., et al. (2007). Carotid body autotransplantation in Parkinson disease: a clinical and positron emission tomography study. J. Neurol. Neurosurg. Psychiatry 78, 825–831. doi: 10.1136/jnnp.2006.106021
Mittal, S., Bjørnevik, K., Im, D. S., Flierl, A., Dong, X., Locascio, J. J., et al. (2017). β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 357, 891–898. doi: 10.1126/science.aaf3934
Moors, T. E., Hoozemans, J. J., Ingrassia, A., Beccari, T., Parnetti, L., Chartier-Harlin, M. C., et al. (2017). Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 12:11. doi: 10.1186/s13024-017-0154-3
Mukherjee, S., and Thrasher, A. J. (2013). Gene therapy for PIDs: progress, pitfalls and prospects. Gene 525, 174–181. doi: 10.1016/j.gene.2013.03.098
Muramatsu, S., Fujimoto, K., Kato, S., Mizukami, H., Asari, S., Ikeguchi, K., et al. (2010). A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol. Ther. 18, 1731–1735. doi: 10.1038/mt.2010.135
Nutt, J. G., Woodward, W. R., Hammerstad, J. P., Carter, J. H., and Anderson, J. L. (1984). The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N. Engl. J. Med. 310, 483–488. doi: 10.1056/NEJM198402233100802
Offen, D., Barhum, Y., Levy, Y. S., Burshtein, A., Panet, H., Cherlow, T., et al. (2007). Intrastriatal transplantation of mouse bone marrow-derived stem cells improves motor behavior in a mouse model of Parkinson’s disease. J. Neural Transm. Suppl. 72, 133–143. doi: 10.1007/978-3-211-73574-9_16
Olanow, C. W., Goetz, C. G., Kordower, J. H., Stoessl, A. J., Sossi, V., Brin, M. F., et al. (2003). A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 54, 403–414. doi: 10.1002/ana.10720
Olanow, C. W., Kieburtz, K., Odin, P., Espay, A. J., Standaert, D. G., Fernandez, H. H., et al. (2014). Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 13, 141–149. doi: 10.1016/S1474-4422(13)70293-X
Pagan, F., Hebron, M., Valadez, E. H., Torres-Yaghi, Y., Huang, X., Mills, R. R., et al. (2016). Nilotinib effects in Parkinson’s disease and dementia with lewy bodies. J. Parkinsons Dis. 6, 503–517. doi: 10.3233/JPD-160867
Palfi, S., Gurruchaga, J. M., Ralph, G. S., Lepetit, H., Lavisse, S., Buttery, P. C., et al. (2014). Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146. doi: 10.1016/S0140-6736(13)61939-X
Redmann, M., Wani, W. Y., Volpicelli-Daley, L., Darley-Usmar, V., and Zhang, J. (2017). Trehalose does not improve neuronal survival on exposure to alpha-synuclein pre-formed fibrils. Redox Biol. 11, 429–437. doi: 10.1016/j.redox.2016.12.032
Redmond, D. E., Robbins, R. J., Naftolin, F., Marek, K. L., Vollmer, T. L., Leranth, C., et al. (1993). Cellular replacement of dopamine deficit in Parkinson’s disease using human fetal mesencephalic tissue: preliminary results in four patients. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 71, 325–359.
Robertson, D. C., Schmidt, O., Ninkina, N., Jones, P. A., Sharkey, J., and Buchman, V. L. (2004). Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem. 89, 1126–1136. doi: 10.1111/j.1471-4159.2004.02378.x
Rodríguez-Navarro, J. A., Rodríguez, L., Casarejos, M. J., Solano, R. M., Gómez, A., Perucho, J., et al. (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 39, 423–438. doi: 10.1016/j.nbd.2010.05.014
Sapru, M. K., Yates, J. W., Hogan, S., Jiang, L., Halter, J., and Bohn, M. C. (2006). Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 198, 382–390. doi: 10.1016/j.expneurol.2005.12.024
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D. C. (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 282, 5641–5652. doi: 10.1074/jbc.M609532200
Schenk, D. B., Koller, M., Ness, D. K., Griffith, S. G., Grundman, M., Zago, W., et al. (2017). First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 32, 211–218. doi: 10.1002/mds.26878
Schöndorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028
Schumacher, J. M., Ellias, S. A., Palmer, E. P., Kott, H. S., Dinsmore, J., Dempsey, P. K., et al. (2000). Transplantation of embryonic porcine mesencephalic tissue in patients with PD. Neurology 54, 1042–1050. doi: 10.1212/WNL.54.5.1042
Selikhova, M., Williams, D. R., Kempster, P. A., Holton, J. L., Revesz, T., and Lees, A. J. (2009). A clinico-pathological study of subtypes in Parkinson’s disease. Brain 132, 2947–2957. doi: 10.1093/brain/awp234
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Singer, M. A., and Lindquist, S. (1998). Multiple effects of trehalose on protein folding in vitro and in vivo. Mol. Cell 1, 639–648. doi: 10.1016/S1097-2765(00)80064-7
Sonntag, K. C., Pruszak, J., Yoshizaki, T., van Arensbergen, J., Sanchez-Pernaute, R., and Isacson, O. (2007). Enhanced yield of neuroepithelial precursors and midbrain-like dopaminergic neurons from human embryonic stem cells using the bone morphogenic protein antagonist noggin. Stem Cells 25, 411–418. doi: 10.1634/stemcells.2006-0380
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stover, N. P., Bakay, R. A., Subramanian, T., Raiser, C. D., Cornfeldt, M. L., Schweikert, A. W., et al. (2005). Intrastriatal implantation of human retinal pigment epithelial cells attached to microcarriers in advanced Parkinson disease. Arch. Neurol. 62, 1833–1837. doi: 10.1001/archneur.62.12.1833
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H., et al. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 10, 148–154. doi: 10.1038/nm985
Taylor, C. J., Peacock, S., Chaudhry, A. N., Bradley, J. A., and Bolton, E. M. (2012). Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 11, 147–152. doi: 10.1016/j.stem.2012.07.014
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Trzaska, K. A., Kuzhikandathil, E. V., and Rameshwar, P. (2007). Specification of a dopaminergic phenotype from adult human mesenchymal stem cells. Stem Cells 25, 2797–2808. doi: 10.1634/stemcells.2007-0212
Venkataramana, N. K., Kumar, S. K., Balaraju, S., Radhakrishnan, R. C., Bansal, A., Dixit, A., et al. (2010). Open-labeled study of unilateral autologous bone-marrow-derived mesenchymal stem cell transplantation in Parkinson’s disease. Transl. Res. 155, 62–70. doi: 10.1016/j.trsl.2009.07.006
Voon, V., Fernagut, P. O., Wickens, J., Baunez, C., Rodriguez, M., Pavon, N., et al. (2009). Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol. 8, 1140–1149. doi: 10.1016/S1474-4422(09)70287-X
Wenning, G. K., Odin, P., Morrish, P., Rehncrona, S., Widner, H., Brundin, P., et al. (1997). Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson’s disease. Ann. Neurol. 42, 95–107. doi: 10.1002/ana.410420115
Williams-Gray, C. H., Evans, J. R., Goris, A., Foltynie, T., Ban, M., Robbins, T. W., et al. (2009). The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 132, 2958–2969. doi: 10.1093/brain/awp245
Wyse, R. K., Brundin, P., and Sherer, T. B. (2016). Nilotinib - differentiating the hope from the hype. J. Parkinsons Dis. 6, 519–522. doi: 10.3233/JPD-160904
Keywords: α-synuclein, novel therapies, Parkinson’s disease, regenerative therapies, stem cells
Citation: Stoker TB, Torsney KM and Barker RA (2018) Emerging Treatment Approaches for Parkinson’s Disease. Front. Neurosci. 12:693. doi: 10.3389/fnins.2018.00693
Received: 13 August 2018; Accepted: 18 September 2018;
Published: 08 October 2018.
Edited by:
Shane V. Hegarty, Harvard University, United StatesReviewed by:
Yvonne Nolan, University College Cork, IrelandAnelyssa D’Abreu, Brown University, United States
Copyright © 2018 Stoker, Torsney and Barker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas B. Stoker, dGJzMjZAY2FtLmFjLnVr
†Joint first authors