 Driton Vela
Driton Vela- Department of Physiology, Faculty of Medicine, University of Pristina, Pristina, Kosovo
Hepcidin is the major regulator of systemic iron metabolism, while the role of this peptide in the brain has just recently been elucidated. Studies suggest a dual role of hepcidin in neuronal iron load and inflammation. This is important since neuronal iron load and inflammation are pathophysiological processes frequently associated with neurodegeneration. Furthermore, manipulation of hepcidin activity has recently been used to recover neuronal damage due to brain inflammation in animal models and cultured cells. Therefore, understanding the mechanistic insights of hepcidin action in the brain is important to uncover its role in treating neuronal damage in neurodegenerative diseases.
Introduction
Iron is a crucial element for maintaining brain homeostasis. It is used by brain cells to synthesize myelin, produce neurotransmitters, and realize a wide range of important cellular biochemical reactions (Ward et al., 2014). But, iron excess is toxic for brain cells. This is observed during brain hemorrhage, where inflammatory signaling is responsible for the increase in brain iron load, which is associated with oxidative damage and cognitive decline (Wu et al., 2003; Ward et al., 2014; Garton et al., 2016). Furthermore, iron dysregulation has been proposed as a pathogenic factor in neurodegenerative diseases as well (Ward et al., 2014). Recent research suggests that brain iron dysmetabolism can be tackled via hepcidin manipulation, since local hepcidin production in the brain has been shown to affect cellular iron transport (Urrutia et al., 2013; Du et al., 2015; Gong et al., 2016; Xiong et al., 2016; Zhou et al., 2017). But, it seems that the presence or absence of inflammation dictates the deleterious vs. beneficial effects of hepcidin in the brain. Therefore, unraveling mechanistic aspects of the dual nature of hepcidin in the brain is important to understand its therapeutic potential.
Regulation of Systemic Hepcidin and its Actions
Most of systemic hepcidin is produced in hepatocytes via paracrine signaling from liver sinusoidal endothelial cells (LSECs) (Canali et al., 2017). LSECs respond to iron-mediated pathways by producing bone morphogenetic protein 6 (BMP6), which then activates BMP receptor (BMPR) in hepatocytes (Steinbicker et al., 2011). Activated BMPR induces s-mothers against decapentaplegic (SMAD) protein phosphorylation. Phosphorylated SMADs migrate to nucleus where they cause hepcidin upregulation (Casanovas et al., 2009). Although iron-mediated signals act mainly through BMP/SMAD pathway, other pathways are also involved, like mediation through transferrin receptor 2 (TFR2) and hemochromatosis protein (HFE) (Fleming, 2009). The second most studied stimulator of hepcidin expression is inflammation. It induces hepcidin expression via interleukin-6/janus kinase 2/signal transducer and activator of transcription 3 (IL-6/JAK2/STAT3) pathway (Pietrangelo et al., 2007). This pathway does not dominate hepcidin expression in basal conditions, but during high levels of inflammation it can override hepcidin regulation by iron signaling. In addition to these pathways, hepcidin is controlled by negative regulation, which includes signaling via erythroferrone (ERFE), heparin, vitamin D (Poli et al., 2011; Bacchetta et al., 2014; Kautz et al., 2014). Tight regulation of hepcidin production ensures a direct control of iron metabolism. This is attributed to the ability of hepcidin to induce ferroportin (FPN) degradation in target cells (Ramey et al., 2010; Zhang et al., 2011). FPN is known as the major cellular iron exporter, which is the main target protein of hepcidin.
Regulation of Brain Iron Metabolism
Iron homeostasis in the brain must be finely regulated to serve the purposes of brain cells without damaging cell structures. There are different mechanisms that allow for this to occur; first, blood brain barrier (BBB) is a natural barrier that does not allow passing of iron with ease, which means that iron has to be transported transcellularly via TFR (Bien-Ly et al., 2014; McCarthy and Kosman, 2015a). The iron-TFR complex undergoes endocytosis, while iron gets released intracellularly through divalent metal transporter 1 (DMT1) (Ward et al., 2014). Iron exits from brain cells via FPN (Ward et al., 2014). Higher density of FPN in neurons compared to other brain cells shows why FPN downregulation causes more marked cellular iron load in neurons (Moos and Rosengren Nielsen, 2006; Boserup et al., 2011; Urrutia et al., 2013). Studies suggest that astrocytes encircling brain microvascular endothelial cells (BMVECs) influence their FPN activity by releasing local hepcidin, but also by secreting ferroxidases [including amyloid-β precursor protein (APP)] to stabilize FPN (McCarthy and Kosman, 2014, 2015a,b). In any case, iron released into brain parenchyma enters neuronal cells via TFR1, transient receptor potential canonical (TRPC) channel, DMT1 and newly discovered players, such as Zip8 and Steap2, with DMT1 role being more prominent during pathophysiological processes (Ji and Kosman, 2015; Skjørringe et al., 2015).
Role of Systemic Hepcidin in Brain Homeostasis
It is believed that systemic hepcidin can cross blood barriers such as BBB (Xiong et al., 2016). Hepcidin is a member of the big family of antimicrobial peptides (AMPs), which have been shown to cross BBB via different pathways (Welling et al., 2015). This argument is further enforced by the ability of a specific group of AMPs named defensins (which includes hepcidin) to cross intact BBB rather easily (Schluesener and Meyermann, 1995). On the other hand, the idea that hepcidin passes intact BBB has been proposed by Raha-Chowdhury et al. (2015). Authors base their conclusion on their observation that hepcidin expression was seen in nearly all cellular layers of BBB, and also due to the fact that protein expression of hepcidin is higher and more robust than its mRNA expression, which has also been corroborated by other studies (Zechel et al., 2006). The idea that hepcidin could pass through intact BBB has been shared by other authors as well (Myhre et al., 2013; Hofer and Perry, 2016). It seems that the amphipathic and cationic chemical structure of defensins in general and hepcidin in particular, might make them potential candidates for BBB pass via transcytosis (Kumagai et al., 1987; Rousselle et al., 2000; Hunter et al., 2002), though this property of hepcidin has still not been conclusively shown in intact BBB. Still, hepcidin passage through BBB is significantly increased with its disruption, and it is observed even with larger peptides than hepcidin (Ostergaard et al., 2009).
It has to be mentioned that models with liver hepcidin knockout do not result in significant brain iron dysmetabolism (Xiong et al., 2016). This would be the reason why we do not observe significant brain iron overload in hemochromatosis (Rutgers et al., 2007). This means that autonomous mechanisms in the brain are responsible for keeping iron metabolism in check. These mechanisms are mostly realized through the activity of glial cells, which are capable of buffering excess iron (Pelizzoni et al., 2013; Codazzi et al., 2015; McCarthy et al., 2018). Astrocytes are especially active during iron transport through BBB (McCarthy and Kosman, 2015b), while microglia are responsible for the control of parenchymal brain iron transport (McCarthy et al., 2018). Microglia are important “managers” of the response to brain damage due to their first-time reaction and commanding role in this respect (Shinozaki et al., 2017). On the other hand, astrocytes are able to transport the much needed iron for neuronal needs as well, which is why conditional deletion of FPN in astrocytes damages the process of myelination (Schulz et al., 2012). FPN is also important for the iron transport through BMVEC, which is why specific BMVEC FPN mutations cause transient brain iron deficiency. This process is reversible due to upregulation of iron import proteins in BMVEC (Iacovelli et al., 2009). This means that significant injury to brain cells from iron overload will occur if both, cellular iron import and export proteins are affected. Cellular iron overload seems to be more detrimental to neurons due to their lower iron buffering capacity (Bishop et al., 2011). But, studies suggest that glial cell beneficial phenotype can also be dysregulated in the presence of high ironload (Song et al., 2018).
Animal models suggest that liver hepcidin has important consequences in brain pathologies, such as brain hemorrhage/ischemia. During brain hemorrhage/ischemia liver hepcidin is upregulated, which is accompanied with increased global brain iron content after peak levels of hepcidin (Xiong et al., 2016). Importantly, hepcidin knockout decreases brain iron content in models with brain hemorrhage, but not in physiological conditions (Tan et al., 2016; Xiong et al., 2016). Xiong et al. (2016) study found a significantly lower brain iron content in hepcidin knockout rodents compared to normal rodents during intracerebral hemorrhage (ICH). In addition, brain markers of oxidative stress and brain water content were lower in hepcidin knockout rodents compared to the brain of normal rodents during ICH. Finally, hepcidin knockout rodents performed better than their normal counterparts during cognitive tests. Together these data suggest that the presence of hepcidin impacts brain iron content and function during ICH. Data from human studies seem to complement findings from animal studies because they suggest that systemic hepcidin could contribute to increased brain damage during ICH. This observation is based on the existence of the correlation between serum levels of hepcidin and clinical scores that predict poor outcome in these patients (Xiong et al., 2015). This is the reason why Xiong et al. (2016) conclude that hepcidin affects brain iron efflux during ICH, but how could this action occur is unknown. It is interesting to notice that hepcidin has opposite effects on brain iron content during brain iron-overload that occurs without inflammation (Du et al., 2015; Gong et al., 2016). Du et al. (2015) study showed that the use ad-hepcidin after injection of holotransferrin into left carotid artery results in reduced brain iron content compared to rodents not treated with ad-hepcidin (Du et al., 2015). In vitro results from this study showed that ad-hepcidin and hepcidin peptide reduced brain iron uptake by decreasing TFR1, DMT1, and FPN expression in brain endothelial cells. So, how could hepcidin presence during ICH cause opposite results? During ICH brain iron is increased even many weeks after the insult (Dietrich and Bradley, 1988; Wu et al., 2003; Hua et al., 2006; Liu et al., 2016). The increase in brain iron content in this setting might occur because during BBB disruption iron leakage to brain parenchyma cannot be accompanied with reactive iron efflux since hepcidin blocks excess iron from getting out of brain cells. In this respect, hepcidin further enhances the effect of inflammatory signals on cellular iron accumulation (Sansing et al., 2011; Urrutia et al., 2013; Zhou et al., 2014; Xiong et al., 2016; Goldstein et al., 2017). This occurs because during brain inflammation, cytokines, and hepcidin have agonistic effects in suppressing cellular iron efflux (Urrutia et al., 2013; Xiong et al., 2016; Zhao Y. et al., 2018). In addition, inflammatory signaling overrides the blocking effect of hepcidin on cellular iron uptake which creates an ideal environment that promotes brain iron overload by both, inflammatory signals and hepcidin (Urrutia et al., 2013; Du et al., 2015; Gong et al., 2016; Xiong et al., 2016; Zhou et al., 2017; Zhao Y. et al., 2018).
Brain Hepcidin Expression and Mechanisms of Regulation
Local hepcidin expression in the brain has been an object of investigation of different studies. In physiological conditions hepcidin expression in brain structures and cells (neurons, glial cells, endothelial cells, epithelial cells of choroid plexus) has been consistently observed by in vivo studies in humans and rodents, albeit in low levels (Krause et al., 2000; Pigeon et al., 2001; Zechel et al., 2006; Wang et al., 2008, 2010; Hänninen et al., 2009; Raha et al., 2013; Wei et al., 2014; Farajdokht et al., 2015; Raha-Chowdhury et al., 2015; Graf et al., 2016; Li Y. et al., 2016; Pan et al., 2016; Tan et al., 2016; Lu et al., 2017; You et al., 2017; Zhang et al., 2017). Data from human and animal studies suggest that local hepcidin is more robustly expressed in pathophysiological states (Sun et al., 2012; Urrutia et al., 2013; Tan et al., 2016; Xiong et al., 2016; You et al., 2017; Zhang et al., 2017). Similar to other cells, hepcidin main target protein in brain cells is FPN, but also iron import proteins (Sun et al., 2012; Urrutia et al., 2013; Tan et al., 2016; Xiong et al., 2016; You et al., 2017; Zhang et al., 2017).
Hepcidin upregulation can be elicited by acute iron load in astrocytes and microglia, while in neurons this response seems to become more evident with higher doses of iron supplementation (Sun et al., 2012; Urrutia et al., 2013; Simpson et al., 2015). On the other hand, according to Burkhart et al. (2016) hepcidin expression in the brain stem is scarce and probably has no effect during physiological conditions. It has to be mentioned that Burkhart et al. (2016) model of study has many differences compared to other authors. Burkhart et al. (2016) study did not include a bichamber model of BBB as compared to others, which imitates the cellular environment of brain endothelial cells and adjacent astrocytes (McCarthy and Kosman, 2014; Simpson et al., 2015). Still, even authors of this study acknowledge that hepcidin has an important role in brain iron homeostasis during iron-overload and conditions associated with BBB disruption (Burkhart et al., 2016). Unfortunately, compared to liver hepcidin, we still do not know the detailed mechanistic aspects of iron-induced regulation of hepcidin expression in brain cells. It is interesting to notice that results from studies with BMP6 pretreatment experiments are similar to those that have used pretreatment with hepcidin in terms of protection of brain cells from oxidative stress. But, whether BMP6 can control hepcidin expression in response to ironload in brain cells is still not known (Wang et al., 2001; Urrutia et al., 2017).
On the other hand, there is quite robust data concerning hepcidin regulation in the brain via an inflammatory cascade which involves lipopolysaccharide (LPS), toll-like receptor 4 (TLR4), IL-6, STAT3 molecules (Figure 1). The magnitude of cell-specific response to LPS is highest in glial cells and lowest in neurons (Urrutia et al., 2013; You et al., 2017). Furthermore, TLR4 as the main membrane receptor for LPS is more consistently present in microglia (Trotta et al., 2014). In addition, studies suggest that IL-6 (which is a downstream molecule induced by LPS activity) is the mediator of hepcidin upregulation in the brain during inflammation (Zhang et al., 2017). Knockdown of IL-6 during brain inflammation reduces significantly the extent of STAT3 phosphorylation and resultant hepcidin upregulation and FPN downregulation (Zhang et al., 2017). Furthermore, IL-6 activity is associated with overexpression of DMT1, though this effect is not as robust as the consequences in reduction of iron export through FPN (Zhang et al., 2017). Urrutia et al. (2013) and Zhang et al. (2017) have revealed that although the effect of IL-6 in brain hepcidin expression is robust, it is not the only cytokine through which LPS regulates hepcidin. Intriguingly, FPN downregulation during inflammation is not related only to STAT3 or hepcidin levels, but due to an unknown IL-6 dependent pathway (Zhang et al., 2017). This finding is in-line with the suggestion that inflammatory signaling independent of hepcidin expression is the main culprit behind downregulation of FPN in brain cells. Cellular analysis shows that in an inflammatory setting the most evident changes in cellular iron load are observed in neurons and microglia, but less in astrocytes (Urrutia et al., 2013). This is probably due to the ability of astrocytes to facilitate iron transport in and out of their cellular environment (Xu H. et al., 2017). This would make sense if we take into account the general role of astrocytes as supporting cells that mediate transport and metabolism of different molecules.
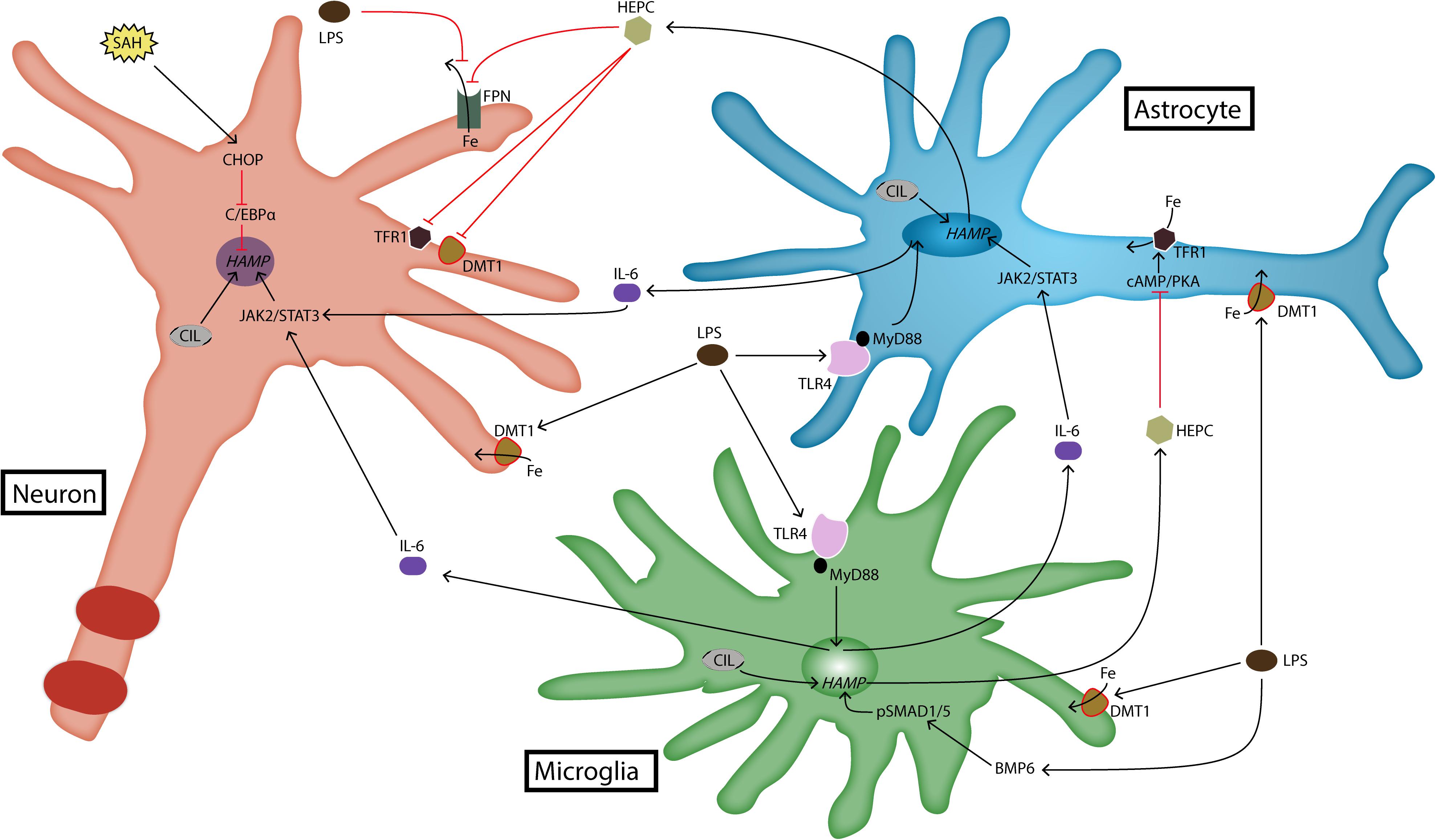
FIGURE 1. Expression and regulation of hepcidin in brain cells. Brain hepcidin expression is upregulated during inflammation. LPS is a product of inflammation that activates TLR4 receptor. Activated TLR4 induces the production of cytokines such as IL-6, which then goes on to upregulate hepcidin expression via JAK2/STAT3 pathway. Recently, it has been shown that LPS can induce hepcidin expression in microglia through BMP/SMAD pathway as well. LPS is a strong stimulus for iron import, because it induces upregulation of DMT1. Another pathway that can induce hepcidin expression in neurons is realized through CHOP and C/EBPα molecules. This occurs during SAH. In any case, most of hepcidin is produced by glial cells during inflammatory conditions. It acts on target cells by degrading FPN, which reduces iron export out of brain cells. Hepcidin also downregulates iron import proteins, DMT1 and TFR1. In astrocytes this action is realized through cAMP/PKA pathway. BMP6, bone morphogenetic protein 6; cAMP, cyclic adenosine monophosphate; C/EBPα, CCAAT/enhancer-binding protein α; CHOP, C/EBP homologous protein; CIL, cellular iron load; DMT1, divalent metal transporter 1; Fe, iron; FPN, ferroportin; HAMP, hepcidin antimicrobial peptide gene; HEPC, hepcidin; IL-6, interleukin-6; JAK2, janus kinase 2; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response 88; PKA, protein kinase A; SAH, subarachnoid hemorrhage; SMAD1/5, s-mothers against decapentaplegic 1/5; STAT3, signal transducer and activator of transcription 3; TFR1, transferrin receptor 1; TLR4, toll-like receptor 4.
Recently, two other pathways have been observed as inducers of brain hepcidin expression. This includes the canonical BMP/SMAD pathway, which is the classical pathway of hepcidin regulation in liver, and CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), which has also been involved in regulating liver hepcidin expression during endoplasmic reticulum (ER) stress (Oliveira et al., 2009). BMP/SMAD pathway has been shown to be involved in hepcidin expression in activated microglia in response to LPS, albeit in a reduced manner compared to IL-6/STAT3 pathway (Qian et al., 2014; Shin et al., 2018). On the other hand, CHOP pathway is involved in hepcidin expression in neurons, especially during brain hemorrhage. Similarly to liver, data suggest that CHOP regulates hepcidin expression in neurons via C/EBPα (Zhao J. et al., 2018). In this way, the picture of hepcidin regulation in the brain is unfolding, which will help in choosing the appropriate therapeutic strategies in different brain pathologies. Indeed, experiments with brain hepcidin knockout have resulted in decreased brain iron content in models with brain hemorrhage, while BMP/SMAD pathway suppression has recently been used in animal models to treat iron overload caused by hepcidin upregulation during brain ischemia (Ding et al., 2011; Tan et al., 2016; Shin et al., 2018). Similarly, CHOP silencing with siRNA reduces brain edema and improves neuronal function (Zhao J. et al., 2018). Also, TLR4 knockdown prevents brain iron accumulation during inflammation (Xiong et al., 2016).
Dual Role of Hepcidin in Brain Pathologies; Implications for its Therapeutic Potential
It is evident that local and/or systemic hepcidin is induced during brain inflammation and iron load (Urrutia et al., 2013; Xiong et al., 2016). In vivo experiments during brain inflammation show that the use of iron chelation protects from brain iron overload, reduces microglial activation and improves cognitive functions in rodents by reducing levels of hepcidin and increasing levels of FPN in hippocampus (Li Y. et al., 2016; Pan et al., 2016). In addition, maternal diet and calorie restriction have been proposed as factors that offer neuroprotection by reducing brain iron load through suppression of brain hepcidin (Wei et al., 2014; Graf et al., 2016). But, it is interesting to notice that accumulating data suggest that hepcidin role in neuronal iron load and inflammation is ambiguous; hepcidin can protect from iron load, but also can cause iron load during inflammation. The reason for this duality seems to occur due to the timing of hepcidin action; studies show that pretreatment with hepcidin protects brain cells from iron load, while hepcidin induction during inflammation aggravates iron load (Xiong et al., 2016; Urrutia et al., 2017; Zhao Y. et al., 2018). Inflammation in the brain is a strong stimulus for the increase in iron import, which increases iron load. But, inflammation has a double effect on iron export; it blocks iron export via FPN inhibition through hepcidin-independent and hepcidin-dependent mechanisms in neurons and glial cells (Urrutia et al., 2013; Zhang et al., 2017). The double effect of inflammation in blocking iron export enhances the cellular iron load in the brain even further. Previous studies have indicated that LPS can indeed affect FPN expression independently of cytokines, though cellular specific differences do exist (Liu et al., 2005; Deschemin and Vaulont, 2013). Cellular specific reactions to inflammatory stimuli have revealed that astrocytes and microglia (compared to neurons) react more strongly to LPS by increasing their hepcidin expression and decreasing FPN expression (Urrutia et al., 2013; Ma et al., 2018). Isolated neurons do not show signs of robust increase in hepcidin levels and FPN downregulation in reaction to inflammatory stimuli (Ma et al., 2018; Puy et al., 2018). But, when neurons are stimulated with high dose of inflammatory cytokines their hepcidin expression is increased significantly (Urrutia et al., 2013). This environment is created when neurons are co-cultured with microglia and astrocytes (You et al., 2017). In this scenario, prime responders to LPS are microglia, which through IL-6 act on astrocytes to increase hepcidin expression. Then, hepcidin acts on neurons in a paracrine manner to reduce FPN expression (You et al., 2017). It has to be mentioned that microglia can also affect neuronal hepcidin expression via LPS (Qian et al., 2014; Li Y. et al., 2016). So, the present picture suggests that during inflammation astrocytes are responsible for directing iron influx from plasma into the brain, where this iron is deposited in neurons via microglial and astrocyte activity, which can increase neuronal oxidative damage and even cause neurodegeneration.
On the other hand, hepcidin pretreatment has the potential to relieve the damage caused by inflammatory signaling. This happens because hepcidin pretreatment downregulates IL-6 and tumor necrosis α (TNFα) expression in astrocytes and microglia (Urrutia et al., 2017) (Figure 2). Treatment with hepcidin in these conditions protects neurons from oxidative stress (Urrutia et al., 2017). In addition, treatment with ad-hepcidin in non-inflammatory conditions protects neurons from iron load (Du et al., 2015; Gong et al., 2016; Zhou et al., 2017). The protective role of hepcidin occurs due to its effect in inhibiting iron transport through BMVEC and through its effects in reducing iron import in neuronal cells (Du et al., 2015; Gong et al., 2016; Zhou et al., 2017). These data show that timing of hepcidin treatment and presence of inflammation dramatically influences hepcidin actions in brain cells. It seems that hepcidin pretreatment primes the brain cellular milieu against the stimulative effects of inflammatory signals in increasing brain iron load (Urrutia et al., 2017). Previous studies with cultured macrophages have revealed that hepcidin-FPN interaction induces suppressor of cytokine signaling 3 (SOCS3) activity, which as its name suggest, is a suppressor of cytokine signaling during inflammation (De Domenico et al., 2010). Also, human studies have shown that levels of hepcidin are related with the inflammatory response (Burté et al., 2013). It would be interesting for future studies to reveal if beneficial effects of pretreatment with hepcidin can be sustained with increasing levels of inflammation.
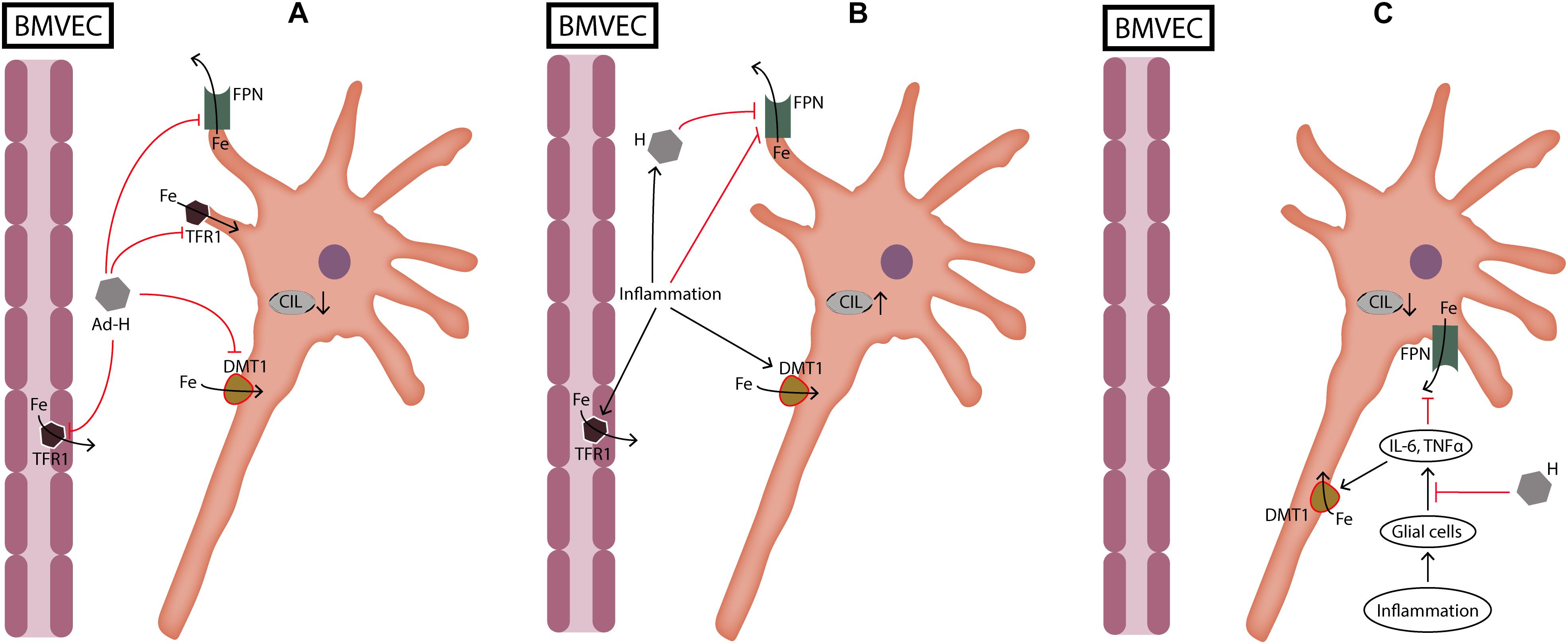
FIGURE 2. Dual role of hepcidin during neuronal cellular iron load and inflammation. (A) During cellular iron load not caused by inflammation hepcidin protects neurons by reducing iron transport through BMVEC and by reducing iron import into neurons. (B) During inflammation hepcidin production induced by cytokines is deleterious for neurons because it increases iron load by inhibiting iron export through FPN. On the other hand, inflammation increases iron import into cells which further aggravates cellular iron load. (C) When glial cells are pretreated with hepcidin, cellular iron load is decreased because hepcidin suppresses the release of cytokines from glial cells. Ad-H, ad-hepcidin; BMVEC, brain microvascular endothelial cells; CIL, cellular iron load; DMT1, divalent metal transporter 1; Fe, iron; FPN, ferroportin; IL-6, interleukin-6; TFR1, transferrin receptor 1; TNFα, tumor necrosis factor α.
Role of Inflammation and Iron Load in Neurodegenerative Diseases
Pathogenesis of neurodegenerative diseases is complex and still not entirely understood. But, in recent years accumulating evidence are linking neurodegenerative processes with iron metabolism and neuroinflammation. Iron dysmetabolism is an early feature of Alzheimer’s disease (AD) and in isolated nuclei in Parkinson’s disease (PD) (Nunomura et al., 2001; Smith et al., 2010; Guan et al., 2017). In AD, levels of ferritin in cerebrospinal fluid (CSF) are predictors of worsening cognitive performance and are also correlated with apolipoprotein E (ApoE) CSF levels and AD risk allele 𝜀4 (Ayton et al., 2015). Similarly, higher iron deposition in substantia nigra (SN) of AD patients makes them more prone to develop PD (Brar et al., 2009). Use of compounds with moderate iron-binding potency reduces levels of α-synclein and protects neurons from oxidative damage in PD (Finkelstein et al., 2017). This effect is realized by restoring the function of FPN as a cellular iron exporter (Finkelstein et al., 2017). Iron load as a risk factor in PD has been further confirmed in a meta-analysis, which has identified the presence of iron overload in SN of PD patients (Wang et al., 2016). It seems that in PD iron overload is generally more present in advanced stages of the disease compared to AD (Wang et al., 2016; Guan et al., 2017). In patients with mild and moderate PD there is also evidence of systemic perturbations of iron metabolism, with ferritin and malondialdehyde (MDA) serum levels (marker of oxidative damage) serving as significant biomarkers of PD (De Farias et al., 2017). On the other hand, it has to be mentioned that the most affected brain structure in PD, that is SN, is characterized with iron overload and significant damage in early stages of the disease (Ziegler et al., 2013; Guan et al., 2017). This would imply that iron dysmetabolism in SN in PD could occur due to a similar pathogenic mechanism observed in AD. It is interesting to notice that APP dysfunction (which is a hallmark of AD) induces dementia-like symptoms in PD (Lei et al., 2012). Importantly, APP dysfunction in AD results in its inability to stabilize the function of FPN as a cellular iron exporter, which would affect intracellular iron load (Duce et al., 2010).
Similar to iron load, inflammation is also being recognized as an important pathogenic marker of neurodegenerative diseases. It is observed early in AD and PD, often bearing evidence of peripheral (intestinal) aberrant response to different stimuli (Qin et al., 2016; Houser and Tansey, 2017; Raj et al., 2017; Gelders et al., 2018; Gurel et al., 2018; King et al., 2018; Steeland et al., 2018). This means that in AD and PD the pathogenic process is initiated and “fueled” by immunologic disturbances occurring in the gastrointestinal tract. In relation to this scenario are data coming from studies with the TNF receptor (TNFR), which have caught attention for their importance in neurodegenerative diseases. TNF and Aβ-induced inflammation is significantly reduced via TNFR1 loss (Steeland et al., 2018). It seems that TNF and Aβ oligomers act as agonists on TNFR1 to activate microglial cells. In addition, TNFR1 loss protects brain cells from propagation of peripheral inflammatory reaction by restoring blood-CSF barrier functionality (Steeland et al., 2018). In addition, direct evidence from human studies with inflammatory bowel disease (IBD) suggests that early TNF inhibition dramatically reduces the incidence of PD (Peter et al., 2018). TNF involvement in PD is further strengthened by the increased risk of early onset of PD in patients with higher expression of TNF (Lindenau et al., 2017). On the other hand, role of inflammation in AD and PD is characterized with subtle but important differences between these conditions; in vivo models with mice have shown that Aβ oligomer accumulation in AD is related to TLR4 activity, which is in contrast with α-synuclein accumulation in PD, where the role of TLR4 is low or inexistent (Noelker et al., 2013; Forloni et al., 2016; Calvo-Rodríguez et al., 2017). Also, microglial activation is somewhat more prominent with AD related oligomers than with PD related oligomers (Forloni et al., 2016). Furthermore, in PD, microglial activity is accompanied with increased number of lymphocytes in damaged brain nuclei (Wang et al., 2015). These data suggest distinct inflammatory routes of activation by pathogenic oligomers in AD and PD.
Alzheimer’s disease and PD are diseases that increase in prevalence with age. It is believed that the ability of microglia and astrocytes to secure the homeostatic equilibrium for neurons is reduced with aging. The homeostatic disequilibrium might be a result of the genetic factors that are related with increased activity of inflammatory signals, decreased ability of microglia to clear oligomer accumulation and other factors (Heppner et al., 2015; Lindenau et al., 2017; Hansen et al., 2018). “Pathogenic” microglia release cytokines which are responsible for neuronal damage, partly by inducing iron load. In experiments with cultured neurons, accumulation of pathological Aβ oligomers produces significant neurotoxic effects when neurons are co-cultured with glial cells (Urrutia et al., 2017). This shows that glial cell activity is an important factor in potentiating the toxic effect of Aβ oligomers. In order to protect themselves from cellular damage neurons increase ferritin depots, but the accumulating iron is very reactive and once it accumulates in sufficient amount it will cause oxidative damage. Iron load initiates a vicious cycle of neuronal damage, caused by the ability of excess iron to accelerate oligomer toxicity (Rottkamp et al., 2001; Wan et al., 2017).
Although evidence for the role of inflammation and resultant iron load in neurodegenerative diseases is accumulating, it is of therapeutic importance to explain how do these pathogenic factors change the dynamic of cellular biochemistry. Data from animal models show that aging creates a favorable milieu for neuronal damage via iron load. In rats, levels of DMT1 and hepcidin are increased, while levels of FPN do not change (Wang et al., 2010; Lu et al., 2017). This would indicate that cellular iron load with aging occurs due to increased iron import. Increased cellular iron stimulates hepcidin expression, which is not able to control FPN expression. It is interesting to notice that FPN is downregulated in aged rats with APP knockout (Belaidi et al., 2018). Furthermore, APP knockout increases the rate of brain iron accumulation that occurs with aging. This means that the preservation of cellular iron efflux is important for slowing age-related brain iron overload. In brains of AD patients and mice hepcidin expression is located in damaged neurons and blood vessels of amyloid plaques (Raha et al., 2013). On the other hand, similar to changes occurring with age, we observe increased DMT1 expression, but also co-localization of DMT1 with amyloid plaques in AD patients and in animal models with AD (Zheng et al., 2009). Animal models suggest that DMT1 expression is directly linked with the metabolism of pathogenic peptides that accumulate during AD (Zheng et al., 2009). Furthermore, DMT1 silencing increases cell viability in AD (Zheng et al., 2009). Similarly, in human patients, animal models and cell cultures with PD, DMT1 is also upregulated and related to oxidative stress (Salazar et al., 2008; Chen et al., 2015). Consequently, blocking DMT1 and hepcidin improves cell viability, while FPN upregulation rescues neuronal function in cultured cells and mice brain with PD (Chen et al., 2015; Xu et al., 2016; Xu Q. et al., 2017). So, at least in animal models with neurodegenerative disease, suppression of iron import and stimulation of iron export (via hepcidin manipulation) ameliorates neuronal degeneration.
In other diseases accompanied with neurodegeneration (Huntington disease, amyotrophic lateral sclerosis) iron deposition is also increased in different brain structures (Chen et al., 2013; Muller and Leavitt, 2014; Szeliga et al., 2016; Moreau et al., 2018). Changes in expression of iron protein carriers and the therapeutic potential of iron chelation evoke striking similarities with AD and PD (Salazar et al., 2008; Zheng et al., 2009; Chen et al., 2013; Belaidi and Bush, 2016; Szeliga et al., 2016; Moreau et al., 2018). It is speculative to suggest that the role of hepcidin in neurodegenerative diseases might be of primary importance, due to the dominating role of innate immune system in the pathophysiology of these diseases. Hepcidin has been evidenced quite robustly as part of our innate immune response, which is helpful in acute situations, because it controls iron availability to pathogens, but also it can regulate actions of inflammatory cytokines (De Domenico et al., 2010; Armitage et al., 2011; Burté et al., 2013). But, during chronic innate immune activity the negative feedback control between hepcidin and inflammation observed in acute situations turns into an agonistic relationship which will eventually deteriorate brain cell damage.
The Rationale for Using Hepcidin Therapeutics in Neurodegenerative Diseases
Stimulation and inhibition of hepcidin has been used in cultured cells and animal models to ameliorate brain damage. The rationale is evident; hepcidin affects FPN and iron import proteins, which means its manipulation can control cellular iron load. Furthermore, it has been shown that decreased activity or expression of FPN promotes neuronal damage (Song et al., 2010; Yang et al., 2011; Crespo et al., 2014). In animal models with PD neurodegeneration is associated with increased microglial activity and FPN downregulation (Zhang et al., 2014). In addition, human trials with iron chelators have shown promise in retarding the progress of neuronal damage (Crapper McLachlan et al., 1991; Martin-Bastida et al., 2017; Moreau et al., 2018). But, long-term results of this therapy are still unknown and pending future trials (Adlard and Bush, 2018).
In animal models with brain inflammation and increased oxidative stress, direct and indirect suppression of local and systemic hepcidin offers neuroprotection (Chen et al., 2015; Li W.-Y. et al., 2016; Pan et al., 2016; Xiong et al., 2016). But, in conditions with lack of inflammation, the use of ad-hepcidin has also beneficial effects in neuronal function, probably due to hepcidin ability to protect neurons during iron overload via suppression of cellular iron import proteins. Still, there exist practical issues concerning the use of delivery methods for hepcidin in these scenarios. In animal models this has been done via intracerebroventricular injections, but the feasibility of this method in humans is unknown (Gong et al., 2016; Urrutia et al., 2017). Recent advances suggest that nanotechnology could make an important breakthrough in treating brain diseases with targeted delivery of peptides (Goldsmith et al., 2014; Silva Adaya et al., 2017).
Based on the accumulating evidence about the role of inflammation on cellular iron content, it is reasonable to assume that neuroprotection can be achieved by blocking the inflammatory pathways through already established drugs. One such drug is acetylsalicylic acid, which has already been used in cultured cells to protect neuronal cells and microglia from inflammation-induced damage (Li W.-Y. et al., 2016; Huang et al., 2018) (Table 1). The protective effect of acetylsalicylic acid is realized through increase in iron export in neuronal cells (Huang et al., 2018), while in microglia it reduces iron import and increases iron export (Xu et al., 2015).
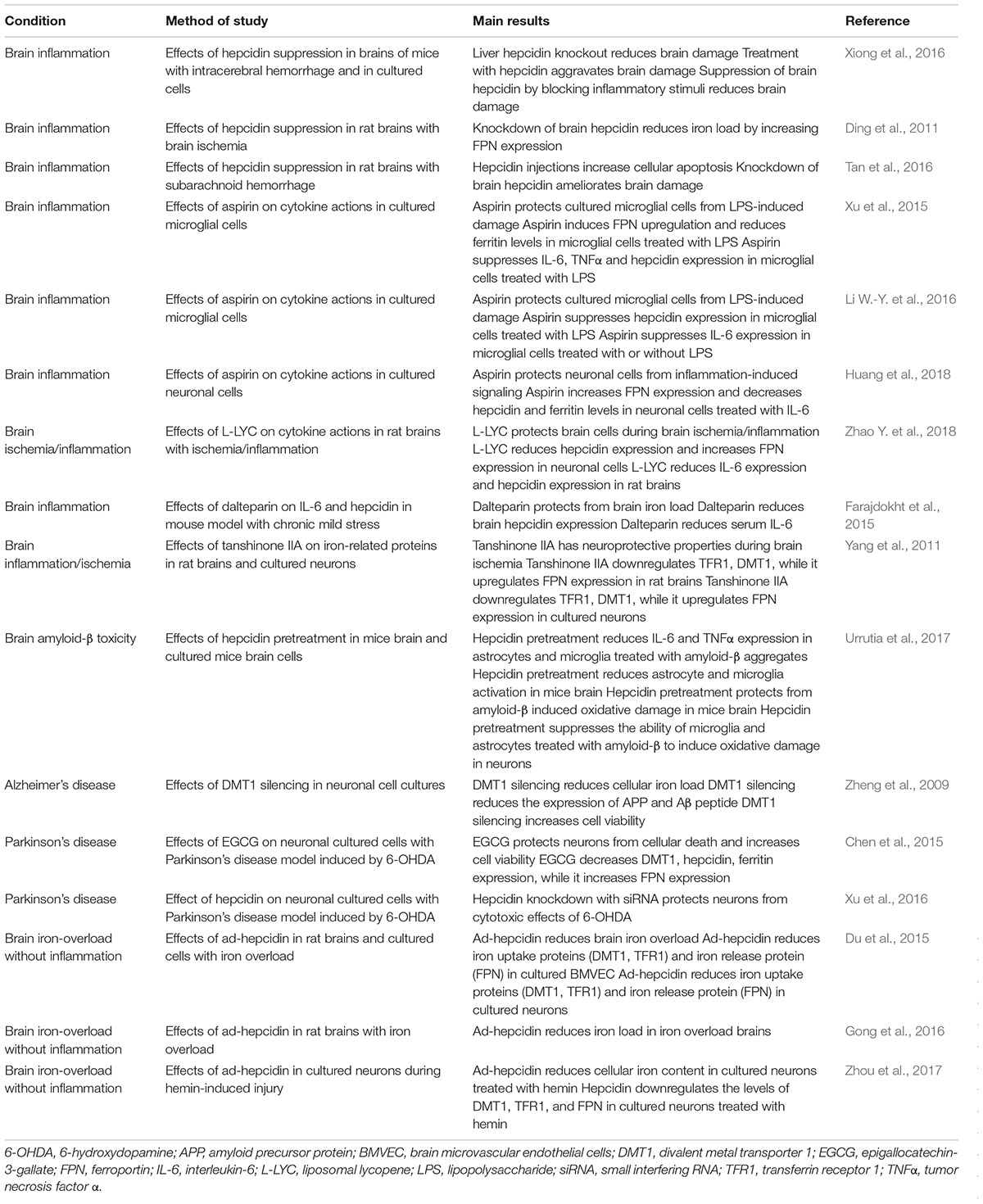
TABLE 1. Therapeutic potential of the manipulation of hepcidin and its target proteins in neurodegeneration.
As it was previously mentioned heparins can affect liver hepcidin expression. One such heparin, called dalteparin, has been administered via intraperitoneal injections in mice to suppress brain hepcidin expression during inflammatory conditions (Farajdokht et al., 2015). Dalteparin can also suppress serum IL-6 levels in these conditions, which means that its beneficial properties in reducing increased brain iron during inflammation could be due to suppression if systemic inflammation and brain hepcidin. Still the cause/effect relationship of dalteparin in the brain has not been studied thoroughly in terms of its impact on brain iron metabolism via hepcidin regulation. Future studies should also explain whether dalteparin-induced brain hepcidin suppression is dose-dependent, because of the side effects of this drug on brain homeostasis (Perry et al., 2010).
Another therapeutic option that has been used is based on methods of delivery that increase the efficiency of neuroprotective substances. One example is the use of liposomes, which serve as particles that deliver drugs to target cells, whilst keeping intact the stability of the substance that is being used for therapeutic purposes. Liposomal lycopene has been delivered in rat brains to reduce oxidative stress and ameliorate apoptosis caused by ischemia. This effect is at least partly attributed to the ability of lycopene to reduce hepcidin expression in astrocytes via its suppressive effect on IL-6 signaling (Zhao Y. et al., 2018).
Conclusion
Hepcidin is emerging as an important peptide for brain (patho)physiology. The source of hepcidin in the brain is local and probably systemic. It is believed that systemic hepcidin can pass through BBB, especially during BBB leakage (for example during inflammation). Neurons, glial cells, endothelial cells, and other brain cells may express hepcidin, although basal levels of brain hepcidin are low. They increase significantly during inflammation and most likely during brain iron load. The local molecular regulation of brain hepcidin expression is unfolding, while studies suggest that it bears resemblance with liver hepcidin regulation. Still, the details of this regulation await further clarification from future studies. Hepcidin produced by the brain cells in pathological conditions may limit iron transport through BMVEC, and thereby also limit neuronal iron load. High iron load is detrimental for neuronal function. In addition, high iron load can also affect glial cell activity by turning them into cells that promote neuronal damage. The observation of robust production of hepcidin by glial cells compared to neurons during changes in brain homeostasis is in-line with the already established role of glia as supportive, regulatory and protective cells in the brain milieu. During inflammatory conditions hepcidin promotes neuronal iron load, while the opposite occurs in non-inflammatory conditions. This observation is based on studies that have revealed the neuroprotective nature of hepcidin suppression during inflammation. This occurs due to the increase in iron import caused by inflammation, which is further exacerbated by FPN downregulation caused by hepcidin-dependent and hepcidin-independent mechanisms. On the other hand, use of ad-hepcidin during neuronal iron load (not caused by inflammation) protects these cells from oxidative stress. Intriguingly, hepcidin pretreatment also protects neurons from the deleterious effects of inflammation. It seems that pretreatment with hepcidin blocks the initiation of inflammation-induced biochemical cascade inside brain cells. This effect of hepcidin is not without precedent, because it has been observed in mice and human studies. Unfortunately, therapeutic implications of this physiological effect of hepcidin have not been studied in details, despite its potential crucial importance in brain and other pathologies. Still, pharmacologic manipulation of hepcidin is emerging as e new therapeutic territory in neurodegenerative diseases. Animal models have shown that the use of ad-hepcidin and suppression of hepcidin protects brain cells in models of neurodegenerative diseases. This has been further confirmed with the use of standard anti-inflammatory drugs such as acetylsalicylic acid or lycopene.
Finally, considering the ever increasing importance of hepcidin for brain physiology, it is paramount for future studies to examine models with different cell specific hepcidin knockout in AD and PD that will reveal the temporal changes of hepcidin, but that will also define whether hepcidin is of primary importance for the pathophysiology of neurodegenerative diseases.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adlard, P. A., and Bush, A. I. (2018). Metals and Alzheimer’s disease: how far have we come in the clinic? J. Alzheimers Dis. 62, 1369–1379. doi: 10.3233/JAD-170662
Armitage, A. E., Eddowes, L. A., Gileadi, U., Cole, S., Spottiswoode, N., Selvakumar, T. A., et al. (2011). Hepcidin regulation by innate immune and infectious stimuli. Blood 118, 4129–4139. doi: 10.1182/blood-2011-04-351957
Ayton, S., Faux, N. G., Bush, A. I., Initiative, A. D. N., Weiner, M. W., Aisen, P., et al. (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6:6760. doi: 10.1038/ncomms7760
Bacchetta, J., Zaritsky, J. J., Sea, J. L., Chun, R. F., Lisse, T. S., Zavala, K., et al. (2014). Suppression of iron-regulatory hepcidin by vitamin D. J. Am. Soc. Nephrol. 25, 564–572. doi: 10.1681/ASN.2013040355
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139, 179–197. doi: 10.1111/jnc.13425
Belaidi, A. A., Gunn, A. P., Wong, B. X., Ayton, S., Appukuttan, A. T., Roberts, B. R., et al. (2018). Marked age-related changes in brain iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics doi: 10.1007/s13311-018-0656-x
Bien-Ly, N., Yu, Y. J., Bumbaca, D., Elstrott, J., Boswell, C. A., Zhang, Y., et al. (2014). Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 211, 233–244. doi: 10.1084/jem.20131660
Bishop, G. M., Dang, T. N., Dringen, R., and Robinson, S. R. (2011). Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 19, 443–451. doi: 10.1007/s12640-010-9195-x
Boserup, M. W., Lichota, J., Haile, D., and Moos, T. (2011). Heterogenous distribution of ferroportin-containing neurons in mouse brain. Biometals 24, 357–375. doi: 10.1007/s10534-010-9405-2
Brar, S., Henderson, D., Schenck, J., and Zimmerman, E. A. (2009). Iron accumulation in the substantia nigra of patients with Alzheimer disease and parkinsonism. Arch. Neurol. 66, 371–374. doi: 10.1001/archneurol.2008.586
Burkhart, A., Skjørringe, T., Johnsen, K. B., Siupka, P., Thomsen, L. B., Nielsen, M. S., et al. (2016). Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol. Neurobiol. 53, 7237–7253. doi: 10.1007/s12035-015-9582-7
Burté, F., Brown, B. J., Orimadegun, A. E., Ajetunmobi, W. A., Afolabi, N. K., Akinkunmi, F., et al. (2013). Circulatory hepcidin is associated with the anti-inflammatory response but not with iron or anemic status in childhood malaria. Blood 121, 3016–3022. doi: 10.1182/blood-2012-10-461418
Calvo-Rodríguez, M., de la Fuente, C., García-Durillo, M., García-Rodríguez, C., Villalobos, C., and Núñez, L. (2017). Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca2+ responses, and neuron cell death in cultured rat hippocampal neurons. J. Neuroinflamm. 14:24. doi: 10.1186/s12974-017-0802-0
Canali, S., Zumbrennen-Bullough, K. B., Core, A. B., Wang, C.-Y., Nairz, M., Bouley, R., et al. (2017). Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129, 405–414. doi: 10.1182/blood-2016-06-721571
Casanovas, G., Mleczko-Sanecka, K., Altamura, S., Hentze, M. W., and Muckenthaler, M. U. (2009). Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 87, 471–480. doi: 10.1007/s00109-009-0447-2
Chen, D., Kanthasamy, A. G., and Reddy, M. B. (2015). EGCG protects against 6-OHDA-induced neurotoxicity in a cell culture model. Parkinsons. Dis. 2015:843906. doi: 10.1155/2015/843906
Chen, J., Marks, E., Lai, B., Zhang, Z., Duce, J. A., Lam, L. Q., et al. (2013). Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PLoS One 8:e77023. doi: 10.1371/journal.pone.0077023
Codazzi, F., Pelizzoni, I., Zacchetti, D., and Grohovaz, F. (2015). Iron entry in neurons and astrocytes: a link with synaptic activity. Front. Mol. Neurosci. 8:18. doi: 10.3389/fnmol.2015.00018
Crapper, McLachlan, D. R., Dalton, A. J., Kruck, T. P., Bell, M. Y., Smith, W. L., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308.
Crespo,Â. C., Silva, B., Marques, L., Marcelino, E., Maruta, C., Costa, S., et al. (2014). Genetic and biochemical markers in patients with Alzheimer’s disease support a concerted systemic iron homeostasis dysregulation. Neurobiol. Aging 35, 777–785. doi: 10.1016/j.neurobiolaging.2013.10.078
De Domenico, I., Zhang, T. Y., Koening, C. L., Branch, R. W., London, N., Lo, E., et al. (2010). Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J. Clin. Invest. 120, 2395–2405. doi: 10.1172/JCI42011
De Farias, C. C., Maes, M., Bonifacio, K. L., Matsumoto, A. K., Bortolasci, C. C., Nogueira, A. S., et al. (2017). Parkinson’s disease is accompanied by intertwined alterations in iron metabolism and activated immune-inflammatory and oxidative stress pathways. CNS Neurol. Disord. – Drug Targets 16, 484–491. doi: 10.2174/1871527316666170223161004
Deschemin, J.-C., and Vaulont, S. (2013). Role of hepcidin in the setting of hypoferremia during acute inflammation. PLoS One 8:e61050. doi: 10.1371/journal.pone.0061050
Dietrich, R. B., and Bradley, W. G. (1988). Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 168, 203–206. doi: 10.1148/radiology.168.1.3380958
Ding, H., Yan, C.-Z., Shi, H., Zhao, Y.-S., Chang, S.-Y., Yu, P., et al. (2011). Hepcidin is involved in iron regulation in the ischemic brain. PLoS One 6:e25324. doi: 10.1371/journal.pone.0025324
Du, F., Qian, Z.-M., Luo, Q., Yung, W.-H., and Ke, Y. (2015). Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol. Neurobiol. 52, 101–114. doi: 10.1007/s12035-014-8847-x
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867. doi: 10.1016/j.cell.2010.08.014
Farajdokht, F., Soleimani, M., Mehrpouya, S., Barati, M., and Nahavandi, A. (2015). The role of hepcidin in chronic mild stress-induced depression. Neurosci. Lett. 588, 120–124. doi: 10.1016/j.neulet.2015.01.008
Finkelstein, D. I., Billings, J. L., Adlard, P. A., Ayton, S., Sedjahtera, A., Masters, C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5:53. doi: 10.1186/s40478-017-0456-2
Fleming, R. E. (2009). Iron sensing as a partnership: HFE and transferrin receptor 2. Cell Metab. 9, 211–212. doi: 10.1016/J.CMET.2009.02.004
Forloni, G., Artuso, V., La Vitola, P., and Balducci, C. (2016). Oligomeropathies and pathogenesis of Alzheimer and Parkinson’s diseases. Mov. Disord. 31, 771–781. doi: 10.1002/mds.26624
Garton, T., Keep, R. F., Hua, Y., and Xi, G. (2016). Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 1, 172–184. doi: 10.1136/svn-2016-000042
Gelders, G., Baekelandt, V., and Van der Perren, A. (2018). Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 1–12. doi: 10.1155/2018/4784268
Goldsmith, M., Abramovitz, L., and Peer, D. (2014). Precision nanomedicine in neurodegenerative diseases. ACS Nano 8, 1958–1965. doi: 10.1021/nn501292z
Goldstein, E. Z., Church, J. S., Pukos, N., Gottipati, M. K., Popovich, P. G., and McTigue, D. M. (2017). Intraspinal TLR4 activation promotes iron storage but does not protect neurons or oligodendrocytes from progressive iron-mediated damage. Exp. Neurol. 298, 42–56. doi: 10.1016/j.expneurol.2017.08.015
Gong, J., Du, F., Qian, Z. M., Luo, Q. Q., Sheng, Y., Yung, W., et al. (2016). Pre-treatment of rats with ad-hepcidin prevents iron-induced oxidative stress in the brain. Free Radic. Biol. Med. 90, 126–132. doi: 10.1016/j.freeradbiomed.2015.11.016
Graf, A. E., Lallier, S. W., Waidyaratne, G., Thompson, M. D., Tipple, T. E., Hester, M. E., et al. (2016). Maternal high fat diet exposure is associated with increased hepcidin levels, decreased myelination, and neurobehavioral changes in male offspring. Brain. Behav. Immun. 58, 369–378. doi: 10.1016/j.bbi.2016.08.005
Guan, X., Xuan, M., Gu, Q., Huang, P., Liu, C., Wang, N., et al. (2017). Regionally progressive accumulation of iron in Parkinson’s disease as measured by quantitative susceptibility mapping. NMR Biomed. 30:e3489. doi: 10.1002/nbm.3489
Gurel, B., Cansev, M., Sevinc, C., Kelestemur, S., Ocalan, B., Cakir, A., et al. (2018). Early stage alterations in CA1 extracellular region proteins indicate dysregulation of IL6 and iron homeostasis in the 5XFAD Alzheimer’s disease mouse model. J. Alzheimer’s Dis. 61, 1399–1410. doi: 10.3233/JAD-170329
Hänninen, M. M., Haapasalo, J., Haapasalo, H., Fleming, R. E., Britton, R. S., Bacon, B. R., et al. (2009). Expression of iron-related genes in human brain and brain tumors. BMC Neurosci. 10:36. doi: 10.1186/1471-2202-10-36
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Hofer, T., and Perry, G. (2016). Nucleic acid oxidative damage in Alzheimer’s disease-explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J. Trace Elem. Med. Biol. 38, 1–9. doi: 10.1016/j.jtemb.2016.06.005
Houser, M. C., and Tansey, M. G. (2017). The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 3:3. doi: 10.1038/s41531-016-0002-0
Hua, Y., Nakamura, T., Keep, R. F., Wu, J., Schallert, T., Hoff, J. T., et al. (2006). Long-term effects of experimental intracerebral hemorrhage: the role of iron. J. Neurosurg. 104, 305–312. doi: 10.3171/jns.2006.104.2.305
Huang, S.-N., Ruan, H.-Z., Chen, M. Y.-J., Zhou, G., and Qian, Z. M. (2018). Aspirin increases ferroportin 1 expression by inhibiting hepcidin via the JAK/STAT3 pathway in interleukin 6-treated PC-12 cells. Neurosci. Lett. 662, 1–5. doi: 10.1016/j.neulet.2017.10.001
Hunter, H. N., Fulton, D. B., Ganz, T., and Vogel, H. J. (2002). The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J. Biol. Chem. 277, 37597–37603. doi: 10.1074/jbc.M205305200
Iacovelli, J., Mlodnicka, A. E., Veldman, P., Ying, G.-S., Dunaief, J. L., and Schumacher, A. (2009). Brain and retinal ferroportin 1 dysregulation in polycythaemia mice. Brain Res. 1289, 85–95. doi: 10.1016/j.brainres.2009.06.098
Ji, C., and Kosman, D. J. (2015). Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J. Neurochem. 133, 668–683. doi: 10.1111/jnc.13040
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 46, 678–684. doi: 10.1038/ng.2996
King, E., O’Brien, J. T., Donaghy, P., Morris, C., Barnett, N., Olsen, K., et al. (2018). Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J. Neurol. Neurosurg. Psychiatry 89, 339–345. doi: 10.1136/jnnp-2017-317134
Krause, A., Neitz, S., Mägert, H.-J., Schulz, A., Forssmann, W.-G., Schulz-Knappe, P., et al. (2000). LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 480, 147–150. doi: 10.1016/S0014-5793(00)01920-7
Kumagai, A. K., Eisenberg, J. B., and Pardridge, W. M. (1987). Absorptive-mediated endocytosis of cationized albumin and a beta-endorphin-cationized albumin chimeric peptide by isolated brain capillaries. Model system of blood-brain barrier transport. J. Biol. Chem. 262, 15214–15219.
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Li, W.-Y., Li, F.-M., Zhou, Y.-F., Wen, Z.-M., Ma, J., Ya, K., et al. (2016). Aspirin down regulates hepcidin by inhibiting NF-κB and IL6/JAK2/STAT3 pathways in BV-2 microglial cells treated with lipopolysaccharide. Int. J. Mol. Sci. 17:1921. doi: 10.3390/ijms17121921
Li, Y., Pan, K., Chen, L., Ning, J.-L., Li, X., Yang, T., et al. (2016). Deferoxamine regulates neuroinflammation and iron homeostasis in a mouse model of postoperative cognitive dysfunction. J. Neuroinflamm. 13, 268. doi: 10.1186/s12974-016-0740-2
Lindenau, J. D., Altmann, V., Schumacher-Schuh, A. F., Rieder, C. R., and Hutz, M. H. (2017). Tumor necrosis factor alpha polymorphisms are associated with Parkinson’s disease age at onset. Neurosci. Lett. 658, 133–136. doi: 10.1016/j.neulet.2017.08.049
Liu, X.-B., Nguyen, N.-B. H., Marquess, K. D., Yang, F., and Haile, D. J. (2005). Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol. Dis. 35, 47–56. doi: 10.1016/j.bcmd.2005.04.006
Liu, Y., Liu, J., Liu, H., Liao, Y., Cao, L., Ye, B., et al. (2016). Investigation of cerebral iron deposition in aged patients with ischemic cerebrovascular disease using susceptibility-weighted imaging. Ther. Clin. Risk Manage. 12, 1239–1247. doi: 10.2147/TCRM.S107783
Lu, L.-N., Qian, Z.-M., Wu, K.-C., Yung, W.-H., and Ke, Y. (2017). Expression of iron transporters and pathological hallmarks of Parkinson’s and Alzheimer’s diseases in the brain of young, adult, and aged rats. Mol. Neurobiol. 54, 5213–5224. doi: 10.1007/s12035-016-0067-0
Ma, J., Zhang, F.-L., Zhou, G., Bao, Y.-X., Shen, Y., and Qian, Z.-M. (2018). Different characteristics of hepcidin expression in IL-6 + / + and IL-6-/- neurons and astrocytes treated with lipopolysaccharides. Neurochem. Res. 43, 1624–1630. doi: 10.1007/s11064-018-2577-9
Martin-Bastida, A., Ward, R. J., Newbould, R., Piccini, P., Sharp, D., Kabba, C., et al. (2017). Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7:1398. doi: 10.1038/s41598-017-01402-2
McCarthy, R. C., and Kosman, D. J. (2014). Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PLoS One 9:e89003. doi: 10.1371/journal.pone.0089003
McCarthy, R. C., and Kosman, D. J. (2015a). Iron transport across the blood-brain barrier: development, neurovascular regulation and cerebral amyloid angiopathy. Cell. Mol. Life Sci. 72, 709–727. doi: 10.1007/s00018-014-1771-4
McCarthy, R. C., and Kosman, D. J. (2015b). Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front. Mol. Neurosci. 8:31. doi: 10.3389/fnmol.2015.00031
McCarthy, R. C., Sosa, J. C., Gardeck, A. M., Baez, A. S., Lee, C.-H., and Wessling-Resnick, M. (2018). Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 293, 7853–7863. doi: 10.1074/jbc.RA118.001949
Moos, T., and Rosengren Nielsen, T. (2006). Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin. Pediatr. Neurol. 13, 149–157. doi: 10.1016/j.spen.2006.08.003
Moreau, C., Danel, V., Devedjian, J. C., Grolez, G., Timmerman, K., Laloux, C., et al. (2018). Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 29, 742–748. doi: 10.1089/ars.2017.7493
Muller, M., and Leavitt, B. R. (2014). Iron dysregulation in Huntington’s disease. J. Neurochem. 130, 328–350. doi: 10.1111/jnc.12739
Myhre, O., Utkilen, H., Duale, N., Brunborg, G., and Hofer, T. (2013). Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: possible impact of environmental exposures. Oxid. Med. Cell. Longev. 2013:726954. doi: 10.1155/2013/726954
Noelker, C., Morel, L., Lescot, T., Osterloh, A., Alvarez-Fischer, D., Breloer, M., et al. (2013). Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci. Rep. 3:1393. doi: 10.1038/srep01393
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
Oliveira, S. J., Pinto, J. P., Picarote, G., Costa, V. M., Carvalho, F., Rangel, M., et al. (2009). ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS One 4:e6618. doi: 10.1371/journal.pone.0006618
Ostergaard, C., Sandvang, D., Frimodt-Møller, N., and Kristensen, H.-H. (2009). High cerebrospinal fluid (CSF) penetration and potent bactericidal activity in CSF of NZ2114, a novel plectasin variant, during experimental pneumococcal meningitis. Antimicrob. Agents Chemother. 53, 1581–1585. doi: 10.1128/AAC.01202-08
Pan, K., Li, X., Chen, Y., Zhu, D., Li, Y., Tao, G., et al. (2016). Deferoxamine pre-treatment protects against postoperative cognitive dysfunction of aged rats by depressing microglial activation via ameliorating iron accumulation in hippocampus. Neuropharmacology 111, 180–194. doi: 10.1016/j.neuropharm.2016.09.004
Pelizzoni, I., Zacchetti, D., Campanella, A., Grohovaz, F., and Codazzi, F. (2013). Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim. Biophys. Acta – Mol. Basis Dis. 1832, 1326–1333. doi: 10.1016/J.BBADIS.2013.04.007
Perry, J. R., Julian, J. A., Laperriere, N. J., Geerts, W., Agnelli, G., Rogers, L. R., et al. (2010). PRODIGE: a randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J. Thromb. Haemost. 8, 1959–1965. doi: 10.1111/j.1538-7836.2010.03973.x
Peter, I., Dubinsky, M., Bressman, S., Park, A., Lu, C., Chen, N., et al. (2018). Anti–tumor necrosis factor therapy and incidence of parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 75, 939–946. doi: 10.1001/jamaneurol.2018.0605
Pietrangelo, A., Dierssen, U., Valli, L., Garuti, C., Rump, A., Corradini, E., et al. (2007). STAT3 is required for IL-6-gp130–dependent activation of hepcidin in vivo. Gastroenterology 132, 294–300. doi: 10.1053/j.gastro.2006.10.018
Pigeon, C., Ilyin, G., Courselaud, B., Leroyer, P., Turlin, B., Brissot, P., et al. (2001). A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819. doi: 10.1074/jbc.M008923200
Poli, M., Girelli, D., Campostrini, N., Maccarinelli, F., Finazzi, D., Luscieti, S., et al. (2011). Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood 117, 997–1004. doi: 10.1182/blood-2010-06-289082
Puy, V., Darwiche, W., Trudel, S., Gomila, C., Lony, C., Puy, L., et al. (2018). Predominant role of microglia in brain iron retention in sanfilippo syndrome, a pediatric neurodegenerative disease. Glia 66, 1709–1723. doi: 10.1002/glia.23335
Qian, Z.-M., He, X., Liang, T., Wu, K.-C., Yan, Y.-C., Lu, L.-N., et al. (2014). Lipopolysaccharides upregulate hepcidin in neuron via microglia and the IL-6/STAT3 signaling pathway. Mol. Neurobiol. 50, 811–820. doi: 10.1007/s12035-014-8671-3
Qin, X.-Y., Zhang, S.-P., Cao, C., Loh, Y. P., and Cheng, Y. (2016). Aberrations in peripheral inflammatory cytokine levels in parkinson disease. JAMA Neurol. 73, 1316. doi: 10.1001/jamaneurol.2016.2742
Raha, A. A., Vaishnav, R. A., Friedland, R. P., Bomford, A., and Raha-Chowdhury, R. (2013). The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 1:55. doi: 10.1186/2051-5960-1-55
Raha-Chowdhury, R., Raha, A. A., Forostyak, S., Zhao, J.-W., Stott, S. R. W., and Bomford, A. (2015). Expression and cellular localization of hepcidin mRNA and protein in normal rat brain. BMC Neurosci. 16:24. doi: 10.1186/s12868-015-0161-7
Raj, D., Yin, Z., Breur, M., Doorduin, J., Holtman, I. R., Olah, M., et al. (2017). Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front. Mol. Neurosci. 10:206. doi: 10.3389/fnmol.2017.00206
Ramey, G., Deschemin, J.-C., Durel, B., Canonne-Hergaux, F., Nicolas, G., and Vaulont, S. (2010). Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 95, 501–504. doi: 10.3324/haematol.2009.014399
Rottkamp, C. A., Raina, A. K., Zhu, X., Gaier, E., Bush, A. I., Atwood, C. S., et al. (2001). Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med. 30, 447–450.
Rousselle, C., Clair, P., Lefauconnier, J. M., Kaczorek, M., Scherrmann, J. M., and Temsamani, J. (2000). New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol. Pharmacol. 57, 679–686. doi: 10.1124/mol.57.4.679
Rutgers, M. P., Pielen, A., and Gille, M. (2007). Chronic cerebellar ataxia and hereditary hemochromatosis: causal or coincidental association? J. Neurol. 254, 1296–1297. doi: 10.1007/s00415-006-0507-2
Salazar, J., Mena, N., Hunot, S., Prigent, A., Alvarez-Fischer, D., Arredondo, M., et al. (2008). Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 105, 18578–18583. doi: 10.1073/pnas.0804373105
Sansing, L. H., Harris, T. H., Welsh, F. A., Kasner, S. E., Hunter, C. A., and Kariko, K. (2011). Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 70, 646–656. doi: 10.1002/ana.22528
Schluesener, H., and Meyermann, R. (1995). Neutrophilic defensins penetrate the blood-brain barrier. J. Neurosci. Res. 42, 718–723. doi: 10.1002/jnr.490420515
Schulz, K., Kroner, A., and David, S. (2012). Iron efflux from astrocytes plays a role in remyelination. J. Neurosci. 32, 4841–4847. doi: 10.1523/JNEUROSCI.5328-11.2012
Shin, J. A., Kim, Y. A., Kim, H. W., Kim, H.-S., Lee, K.-E., Kang, J. L., et al. (2018). Iron released from reactive microglia by noggin improves myelin repair in the ischemic brain. Neuropharmacology 133, 202–215. doi: 10.1016/j.neuropharm.2018.01.038
Shinozaki, Y., Shibata, K., Yoshida, K., Shigetomi, E., Gachet, C., Ikenaka, K., et al. (2017). Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Rep. 19, 1151–1164. doi: 10.1016/j.celrep.2017.04.047
Silva Adaya, D., Aguirre-Cruz, L., Guevara, J., and Ortiz-Islas, E. (2017). Nanobiomaterials’ applications in neurodegenerative diseases. J. Biomater. Appl. 31, 953–984. doi: 10.1177/0885328216659032
Simpson, I. A., Ponnuru, P., Klinger, M. E., Myers, R. L., Devraj, K., Coe, C. L., et al. (2015). A novel model for brain iron uptake: introducing the concept of regulation. J. Cereb. Blood Flow Metab. 35, 48–57. doi: 10.1038/jcbfm.2014.168
Skjørringe, T., Burkhart, A., Johnsen, K. B., and Moos, T. (2015). Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 8:19. doi: 10.3389/fnmol.2015.00019
Smith, M. A., Zhu, X., Tabaton, M., Liu, G., McKeel, D. W., Cohen, M. L., et al. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimer’s Dis. 19, 363–372. doi: 10.3233/JAD-2010-1239
Song, N., Wang, J., Jiang, H., and Xie, J. (2010). Ferroportin 1 but not hephaestin contributes to iron accumulation in a cell model of Parkinson’s disease. Free Radic. Biol. Med. 48, 332–341. doi: 10.1016/j.freeradbiomed.2009.11.004
Song, N., Wang, J., Jiang, H., and Xie, J. (2018). Astroglial and microglial contributions to iron metabolism disturbance in Parkinson’s disease. Biochim. Biophys. Acta – Mol. Basis Dis. 1864, 967–973. doi: 10.1016/j.bbadis.2018.01.008
Steeland, S., Gorlé, N., Vandendriessche, C., Balusu, S., Brkic, M., Van Cauwenberghe, C., et al. (2018). Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 10:e8300. doi: 10.15252/emmm.201708300
Steinbicker, A. U., Bartnikas, T. B., Lohmeyer, L. K., Leyton, P., Mayeur, C., Kao, S. M., et al. (2011). Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118, 4224–4230. doi: 10.1182/blood-2011-03-339952
Sun, C., Song, N., Xie, A., Xie, J., and Jiang, H. (2012). High hepcidin level accounts for the nigral iron accumulation in acute peripheral iron intoxication rats. Toxicol. Lett. 212, 276–281. doi: 10.1016/J.TOXLET.2012.05.022
Szeliga, M., Róñycka, A., Jȩdrak, P., Barañska, S., Janik, P., Jamrozik, Z., et al. (2016). Expression of RNAs coding for metal transporters in blood of patients with Huntington’s disease. Neurochem. Res. 41, 101–106. doi: 10.1007/s11064-015-1737-4
Tan, G., Liu, L., He, Z., Sun, J., Xing, W., and Sun, X. (2016). Role of hepcidin and its downstream proteins in early brain injury after experimental subarachnoid hemorrhage in rats. Mol. Cell. Biochem. 418, 31–38. doi: 10.1007/s11010-016-2730-1
Trotta, T., Porro, C., Calvello, R., and Panaro, M. A. (2014). Biological role of Toll-like receptor-4 in the brain. J. Neuroimmunol. 268, 1–12. doi: 10.1016/j.jneuroim.2014.01.014
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Urrutia, P. J., Hirsch, E. C., González-Billault, C., and Núñez, M. T. (2017). Hepcidin attenuates amyloid beta-induced inflammatory and pro-oxidant responses in astrocytes and microglia. J. Neurochem. 142, 140–152. doi: 10.1111/jnc.14005
Wan, W., Jin, L., Wang, Z., Wang, L., Fei, G., Ye, F., et al. (2017). Iron deposition leads to neuronal α-synuclein pathology by inducing autophagy dysfunction. Front. Neurol. 8:1. doi: 10.3389/fneur.2017.00001
Wang, J.-Y., Zhuang, Q.-Q., Zhu, L.-B., Zhu, H., Li, T., Li, R., et al. (2016). Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 6:36669. doi: 10.1038/srep36669
Wang, Q., Du, F., Qian, Z.-M., Ge, X. H., Zhu, L., Yung, W. H., et al. (2008). Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology 149, 3920–3925. doi: 10.1210/en.2007-1626
Wang, Q., Liu, Y., and Zhou, J. (2015). Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4:19. doi: 10.1186/s40035-015-0042-0
Wang, S.-M., Fu, L.-J., Duan, X.-L., Crooks, D. R., Yu, P., Qian, Z.-M., et al. (2010). Role of hepcidin in murine brain iron metabolism. Cell. Mol. Life Sci. 67, 123–133. doi: 10.1007/s00018-009-0167-3
Wang, Y., Chang, C. F., Morales, M., Chou, J., Chen, H. L., Chiang, Y. H., et al. (2001). Bone morphogenetic protein-6 reduces ischemia-induced brain damage in rats. Stroke 32, 2170–2178.
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/S1474-4422(14)70117-6
Wei, S., Shi, W., Li, M., and Gao, Q. (2014). Calorie restriction down-regulates expression of the iron regulatory hormone hepcidin in normal and d -galactose-induced aging mouse brain. Rejuvenation Res. 17, 19–26. doi: 10.1089/rej.2013.1450
Welling, M. M., Nabuurs, R. J. A., and van der Weerd, L. (2015). Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimer’s Dement. 11, 51–57. doi: 10.1016/j.jalz.2013.12.020
Wu, J., Hua, Y., Keep, R. F., Nakamura, T., Hoff, J. T., and Xi, G. (2003). Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 34, 2964–2969. doi: 10.1161/01.STR.0000103140.52838.45
Xiong, X.-Y., Chen, J., Zhu, W.-Y., Zhao, T., Zhong, Q., Zhou, K., et al. (2015). Serum hepcidin concentrations correlate with serum iron level and outcome in patients with intracerebral hemorrhage. Neurol. Sci. 36, 1843–1849. doi: 10.1007/s10072-015-2266-2
Xiong, X.-Y., Liu, L., Wang, F.-X., Yang, Y.-R., Hao, J.-W., Wang, P.-F., et al. (2016). Toll-like receptor 4/MyD88–mediated signaling of hepcidin expression causing brain iron accumulation, oxidative injury, and cognitive impairment after intracerebral hemorrhage. Circulation 134, 1025–1038. doi: 10.1161/CIRCULATIONAHA.116.021881
Xu, H., Wang, Y., Song, N., Wang, J., Jiang, H., and Xie, J. (2017). New progress on the role of glia in iron metabolism and iron-induced degeneration of dopamine neurons in Parkinson’s disease. Front. Mol. Neurosci. 10:455. doi: 10.3389/fnmol.2017.00455
Xu, Q., Langley, M., Kanthasamy, A. G., and Reddy, M. B. (2017). Epigallocatechin gallate has a neurorescue effect in a mouse model of parkinson disease. J. Nutr. 147, 1926–1931. doi: 10.3945/jn.117.255034
Xu, Q., Kanthasamy, A. G., Jin, H., and Reddy, M. B. (2016). Hepcidin plays a key role in 6-OHDA induced iron overload and apoptotic cell death in a cell culture model of parkinson’s disease. Parkinsons Dis. 2016:8684130. doi: 10.1155/2016/8684130
Xu, Y. X., Du, F., Jiang, L. R., Gong, J., Zhou, Y.-F., Luo, Q. Q., et al. (2015). Effects of aspirin on expression of iron transport and storage proteins in BV-2 microglial cells. Neurochem. Int. 91, 72–77. doi: 10.1016/j.neuint.2015.10.014
Yang, L., Zhang, B., Yin, L., Cai, B., Shan, H., Zhang, L., et al. (2011). Tanshinone IIA prevented brain iron dyshomeostasis in cerebral ischemic rats. Cell. Physiol. Biochem. 27, 23–30. doi: 10.1159/000325202
You, L.-H., Yan, C.-Z., Zheng, B.-J., Ci, Y.-Z., Chang, S.-Y., Yu, P., et al. (2017). Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 8:e2676. doi: 10.1038/cddis.2017.93
Zechel, S., Huber-Wittmer, K., and von Bohlen und Halbach, O. (2006). Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J. Neurosci. Res. 84, 790–800. doi: 10.1002/jnr.20991
Zhang, D.-L., Senecal, T., Ghosh, M. C., Ollivierre-Wilson, H., Tu, T., and Rouault, T. A. (2011). Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood 118, 2868–2877. doi: 10.1182/blood-2011-01-330241
Zhang, F.-L., Hou, H.-M., Yin, Z.-N., Chang, L., Li, F.-M., Chen, Y.-J., et al. (2017). Impairment of hepcidin upregulation by lipopolysaccharide in the interleukin-6 knockout mouse brain. Front. Mol. Neurosci. 10:367. doi: 10.3389/fnmol.2017.00367
Zhang, Z., Hou, L., Song, J.-L., Song, N., Sun, Y.-J., Lin, X., et al. (2014). Pro-inflammatory cytokine-mediated ferroportin down-regulation contributes to the nigral iron accumulation in lipopolysaccharide-induced Parkinsonian models. Neuroscience 257, 20–30. doi: 10.1016/J.NEUROSCIENCE.2013.09.037
Zhao, J., Xiang, X., Zhang, H., Jiang, D., Liang, Y., Qing, W., et al. (2018). CHOP induces apoptosis by affecting brain iron metabolism in rats with subarachnoid hemorrhage. Exp. Neurol. 302, 22–33. doi: 10.1016/j.expneurol.2017.12.015
Zhao, Y., Xin, Z., Li, N., Chang, S., Chen, Y., Geng, L., et al. (2018). Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic. Biol. Med. 124, 1–11. doi: 10.1016/j.freeradbiomed.2018.05.082
Zheng, W., Xin, N., Chi, Z.-H., Zhao, B.-L., Zhang, J., Li, J.-Y., et al. (2009). Divalent metal transporter 1 is involved in amyloid precursor protein processing and Aβ generation. FASEB J. 23, 4207–4217. doi: 10.1096/fj.09-135749
Zhou, Y., Wang, Y., Wang, J., Anne Stetler, R., and Yang, Q.-W. (2014). Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog. Neurobiol. 115, 25–44. doi: 10.1016/j.pneurobio.2013.11.003
Zhou, Y.-F., Zhang, C., Yang, G., Qian, Z.-M., Zhang, M.-W., Ma, J., et al. (2017). Hepcidin protects neuron from hemin-mediated injury by reducing iron. Front. Physiol. 8:332. doi: 10.3389/fphys.2017.00332
Keywords: hepcidin, brain, neurodegenerative diseases, glial cells, inflammation, iron load
Citation: Vela D (2018) The Dual Role of Hepcidin in Brain Iron Load and Inflammation. Front. Neurosci. 12:740. doi: 10.3389/fnins.2018.00740
Received: 25 July 2018; Accepted: 26 September 2018;
Published: 15 October 2018.
Edited by:
Isabella Zanella, Università degli Studi di Brescia, ItalyReviewed by:
Torben Moos, Aalborg University, DenmarkAndrzej Friedman, Medical University of Warsaw, Poland
Copyright © 2018 Vela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Driton Vela, ZHJpdG9uLnZlbGFAdW5pLXByLmVkdQ==