 Lesha Pretorius
Lesha Pretorius Douglas B. Kell
Douglas B. Kell Etheresia Pretorius
Etheresia Pretorius- 1Department of Physiological Sciences, Stellenbosch University, Stellenbosch, South Africa
- 2School of Chemistry, The University of Manchester, Manchester, United Kingdom
- 3The Manchester Institute of Biotechnology, The University of Manchester, Manchester, United Kingdom
Alzheimer’s disease and other similar dementias are debilitating neurodegenerative disorders whose etiology and pathogenesis remain largely unknown, even after decades of research. With the anticipated increase in prevalence of Alzheimer’s type dementias among the more susceptible aging population, the need for disease-modifying treatments is urgent. While various hypotheses have been put forward over the last few decades, we suggest that Alzheimer’s type dementias are triggered by external environmental factors, co-expressing in individuals with specific genetic susceptibilities. These external stressors are defined in the Iron Dysregulation and Dormant Microbes (IDDM) hypothesis, previously put forward. This hypothesis is consistent with current literature in which serum ferritin levels of individuals diagnosed with Alzheimer’s disease are significantly higher compared those of age- and gender-matched controls. While iron dysregulation contributes to oxidative stress, it also causes microbial reactivation and virulence of the so-called dormant blood (and tissue) microbiome. Dysbiosis (changes in the microbiome) or previous infections can contribute to the dormant blood microbiome (atopobiosis1), and also directly promotes systemic inflammation via the amyloidogenic formation and shedding of potent inflammagens such as lipopolysaccharides. The simultaneous iron dysregulation and microbial aberrations affect the hematological system, promoting fibrin amylodiogenesis, and pathological clotting. Systemic inflammation and oxidative stress can contribute to blood brain barrier permeability and the ensuing neuro-inflammation, characteristic of Alzheimer’s type dementias. While large inter-individual variability exists, especially concerning disease pathogenesis, the IDDM hypothesis acknowledges primary causative factors which can be targeted for early diagnosis and/or for prevention of disease progression.
Introduction
Alzheimer’s type dementia (AD) is the most prevalent example of dementia, accounting for 75% of all cases (Qui et al., 2009), with an estimated global economic impact of $604 billion per year (Langa, 2015). In 2013 this neurodegenerative disorder was diagnosed in 44.4 million individuals worldwide. This number is expected to increase to 75.6 million by 2030 (Vradenburg, 2015; Pretorius et al., 2016a). The increasing aging population and anticipated increase in prevalence of AD has severe socio-economic implications (Prince et al., 2014). Familial AD can be promoted by mutations of the amyloid precursor protein (APP), presenilin (Bu et al., 2015) and apolipoprotein E (Ayton et al., 2015) genes, however, most AD patients suffer from the sporadic form, and even after decades of research, the etiology and pathogenesis of this disease remains largely unknown (Makin, 2018), and with essentially no existing disease-modifying treatments (Vogt et al., 2017). Despite the differences between familial and sporadic AD, the resultant neuropathology is shared by both (Pritchard et al., 2017).
Vascular dementia (VaD), also known as multi-infarct dementia, is caused by obstructions in the supply of blood to the brain, which over time results in stepwise neurodegeneration (Karantzoulis and Galvin, 2011). The pathology of VaD is characterized by atheromas of primarily cerebral arteries and arterioles. Vascular lesions characteristic of VaD often co-exist with AD, with 25–80% of individuals with dementia showing both AD symptoms and cerebrovascular lesions (Jellinger, 2008), a condition termed mixed dementia (MD). In early AD, critically located small vascular lesions in subcortical regions may promote cognitive decline. This suggests a synergistic relation between disorders (Jellinger and Attems, 2007). The distinction between isolated AD, VaD, and MD, where both pathologies coexist in the same individual, remains a controversial issue and one of the most difficult diagnostic challenges (Zekry et al., 2002; Jellinger and Attems, 2007). Additionally the population-based prevalence of MD in prospective and retrospective autopsy studies, ranges from 2 to 58%, emphasizing the coexistence of AD with multiple cerebrovascular lesions in patients with cognitive impairments. Moreover inflammation and alterations of inflammatory biomarkers are common to both AD and VaD (Schmidt et al., 2002). For these reasons, VaD and the pathogenesis thereof, fall under the AD umbrella of this review.
For the individual, AD shortens life expectancy significantly, with the cognitive deficit often causing institutionalization, physical disability and reduced quality of life (Qui et al., 2009; Pritchard et al., 2017). Hallmark features of AD pathology in the brain often include extracellular plaques composed of amyloid-β (Aβ), intracellular neurofibrillary tangles which are composed of hyperphosphorylated tau, basal forebrain cholinergic insufficiency and extensive neuronal and synaptic loss in the cortex and hippocampus (Law et al., 2001; Vogt et al., 2017). Aβ42 peptides either as monomers, dimers or intermediate amyloids stimulate arrays of inflammatory gene expression characteristic of the innate immune system (Zhao et al., 2015). These Aβ42 peptides are not only highly immunogenic, but they may self-aggregate into structures that interact with the plasma membrane and cause uncontrolled seepage of ions into and/or out of the neurons (Zhao and Lukiw, 2015), disrupting their normal physiological functioning.
Another hallmark of AD is activated neuroglial cells which produce significant amounts of inflammatory molecules. At low levels this may have a protective function but the increased expression in AD could induce neurodegeneration (Kamer et al., 2008). This said, cerebral inflammation caused by the activation of glial cells is generally asymptomatic as this occurs in the normal aging brain. According to Barrientos et al. (2016) prolonged production of pro-inflammatory cytokines as well as increased neuroinflammatory responses are common in the normal aging brain. The primary source of this exaggerated response is sensitized microglia. Other causes of this phenomenon may include neuroendocrine system dysregulation and potentiation of neuroinflammatory responses following an immune challenge, due to persistently elevated glucocorticoid levels in older individuals (Barrientos et al., 2016). Excessive age-related glial priming, present in AD, could also be due to microbial infiltration from disturbed microbiomes across a permeabilized blood brain barrier, providing slow inflammatory damage (Pritchard et al., 2017).
Recent research (Dorey et al., 2014; Filiou et al., 2014; Steardo et al., 2015) also suggests that neuro-inflammation may play a central role in the pathological progression of AD. Several neuro-inflammatory mediators including reactive oxygen species, chemokines, cytokines and activated complement are produced and secreted by microglia and astrocytes in the AD brain (Pretorius et al., 2016a). While neuro-inflammation is a part of the normal aging brain, and is a hallmark of acute injury (Lucas et al., 2006) and also of infection (McColl et al., 2009), in AD excessive systemic inflammation also occurs (Pretorius et al., 2016a), simultaneously exacerbating and perpetuating neuro-inflammation. Previous studies have verified that peripheral inflammatory markers such as interferon-γ, tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and IL-6 are related to the advancement of AD (Rubio-Perez and Morillas-Ruiz, 2012; Bu et al., 2015).
Some individuals who develop AD seem to possess genetic susceptibilities that are influenced by co-occurring environmental factors (Pritchard et al., 2017). In this review, we rehearse the Iron Dysregulation and Dormant Microbes (IDDM) hypothesis, and its ability to relate events which trigger and promote the pathogenesis of inflammatory conditions such as AD. The IDDM hypothesis emphasizes two main external triggers, namely (1) stress-induced iron dysregulation, and (2) its ability to reactivate dormant or non-replicating microbes which the host had acquired via previous infections or dysbiotic communities. We believe the primary origin of such microbes comes from a dysbiotic gut (Kell and Pretorius, 2015a). These microbes are capable of sloughing off functionally significant inflammagens such as lipopolysaccharide(s) (LPS) from Gram-negative bacteria and lipoteichoic acid (LTA) from Gram-positive bacteria. The consequences of this include significant coagulopathies, for example the amyloidogenic clotting of blood, which increases cell mortality (Kell and Pretorius, 2018). The contents of this manuscript are summarized in Figure 1.
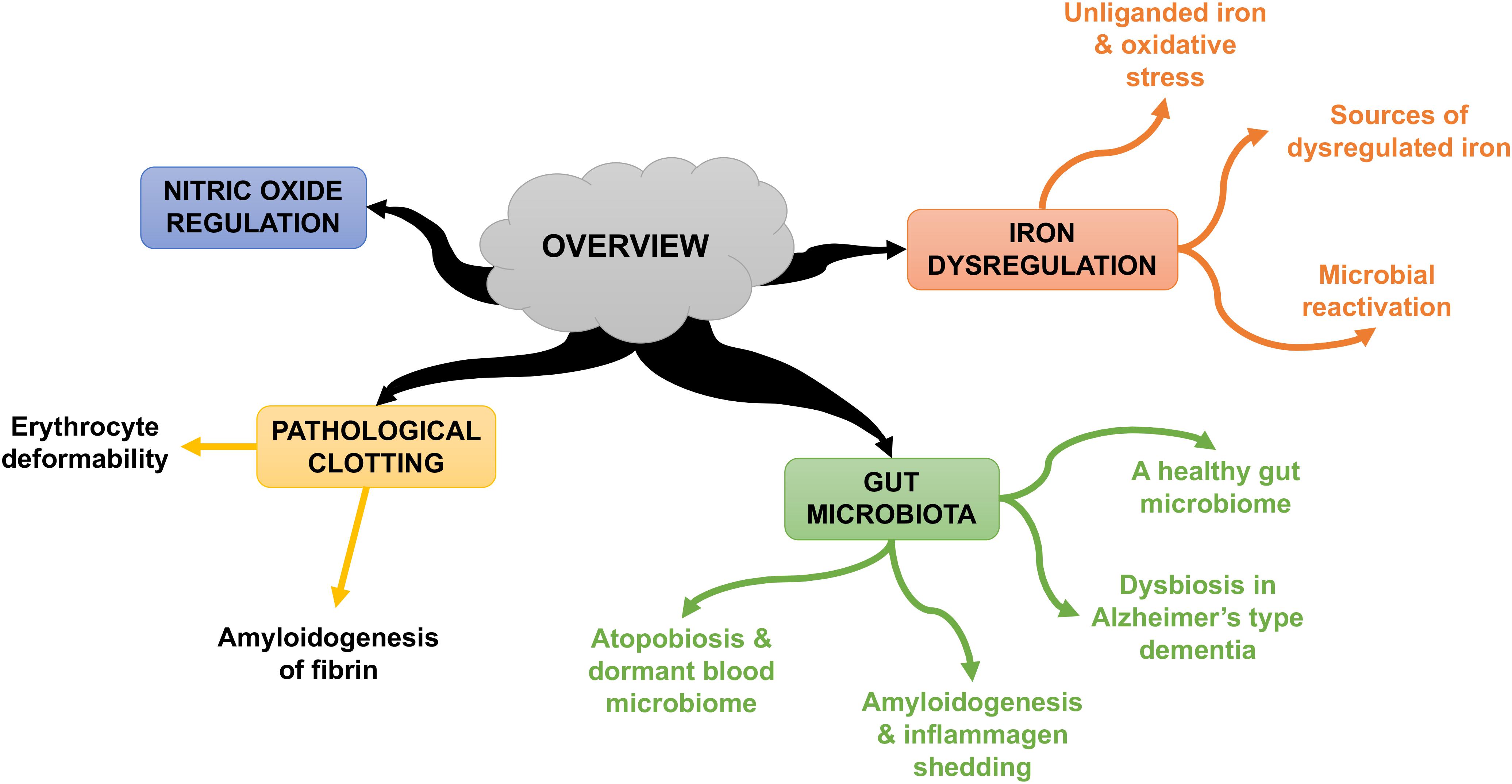
FIGURE 1. An overview of the contents of this manuscript.
Iron Dysregulation
Iron dysregulation is any form of deviation from normal, homeostatic iron metabolism; in particular it includes cerebrospinal fluid ferritin and serum ferritin levels as implicated in Alzheimer’s type dementias (Ayton et al., 2015). According to the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort study, increased ferritin levels, measured in cerebrospinal fluid, were negatively correlated to cognitive performance (Mueller et al., 2005), and these levels predicted conversion of mild cognitive impairment to Alzheimer’s disease in 144 individuals over 7 years (Ayton et al., 2015). Furthermore, a strong association between ferritin levels and cerebrospinal fluid apolipoprotein E levels was observed in this study, with the major risk allele, APOE-𝜀4, inducing 22% higher levels of cerebrospinal fluid ferritin levels when compared to non-𝜀4 carriers (Ayton et al., 2015). In the same study a modest association between cerebrospinal fluid ferritin and plasma ferritin levels was noted (p = 0.0002). Thus, there may be great clinical relevance for the use of systemically elevated serum ferritin (SF) levels as cognitive performance markers (Kell and Pretorius, 2014; Pretorius et al., 2016a).
Causes of Iron Dysregulation
Major sources of iron dysregulation stem from externally induced stressors (Kell and Pretorius, 2018). This form of iron dysregulation can be initiated by several factors that contribute to or cause cell death, such as mechanical damage (Zhang et al., 2013), nutritional stress (Schaffer, 2016), pharmacological stress (Primohamed et al., 2004), and of course oxidative stress (Kerley et al., 2018). Another source of free iron is via heme metabolism, due to the functioning of heme oxygenase-1 (HO-1), which catalyzes the degradation of heme (Pretorius and Kell, 2014). Since upregulation of HO-1 activity occurs in systemic inflammatory disorders in which erythrocytes are lysed, it may also be an important marker of inflammation and iron dysregulation.
Additionally, hepcidin, produced by the liver, is a key regulator of iron metabolism (Michels et al., 2015; Reichert et al., 2017). Decreases in hepcidin levels enhance surface exposure of ferroportin (Ganz and Nemeth, 2012) on enterocytes, macrophages and hepatocytes to increase serum ferritin levels (illustrated by Figure 2). Hepcidin expression is induced by inflammatory markers such as LPS, IL-1β, and IL-6, while increases in 1,25(OH)2D3 (calcitriol) levels cause hepcidin levels to decrease (Kell and Pretorius, 2018). According to a report by Bacchetta et al. (2014), decreases in hepcidin levels by 1,25(OH)2D3 are due to suppression of the HAMP gene by the vitamin D receptor (VDR). Chromatin immunoprecipitation assays confirmed the binding of VDR to the vitamin D response element within the proximal promotor region of the HAMP gene (Bacchetta et al., 2014). While this process is intricate, it appears that alterations in vitamin D metabolism could potentially instigate iron dysregulation.
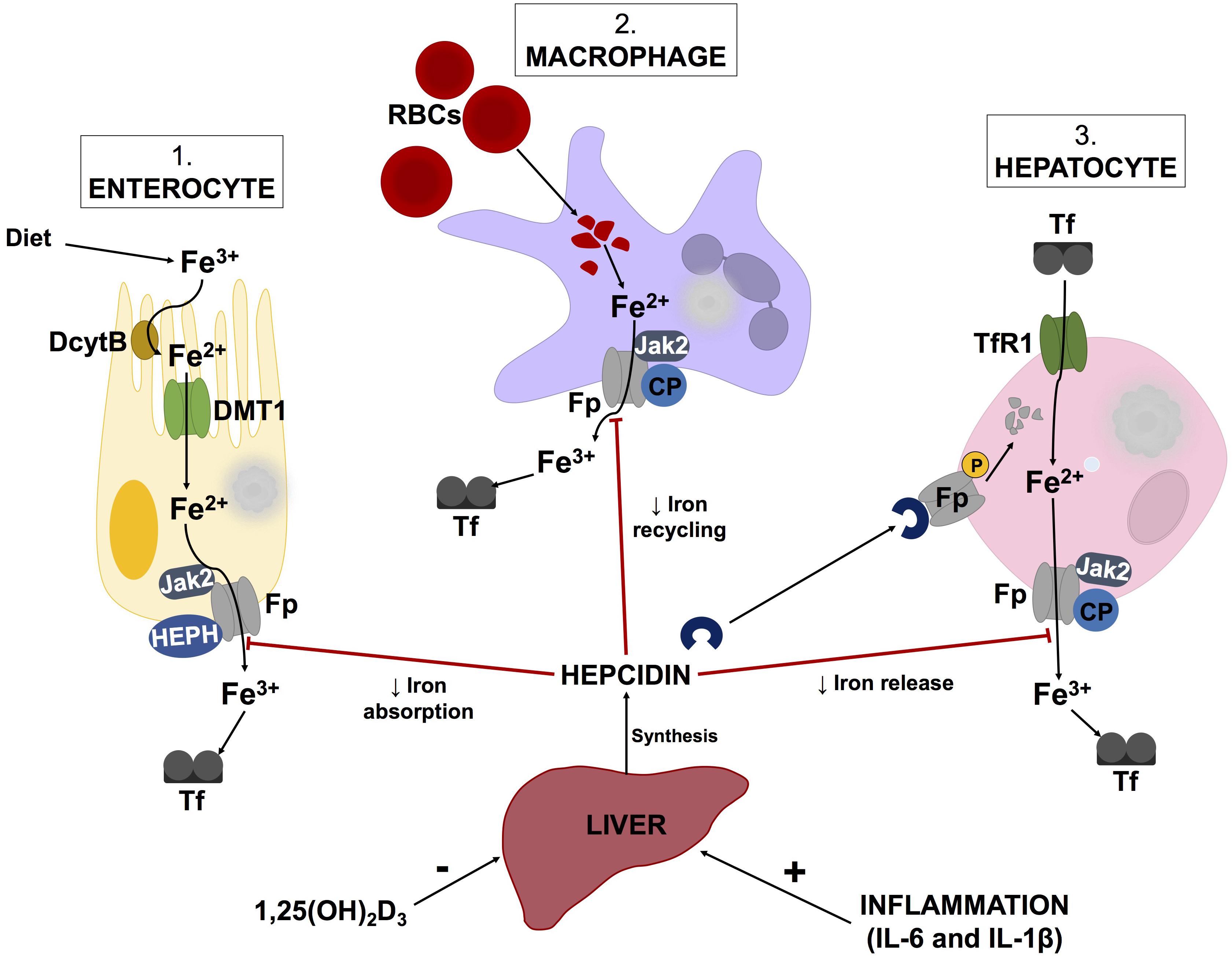
FIGURE 2. Schematic illustration of the hepcidin-ferroportin axis and its regulation of systemic iron homeostasis. Hepcidin synthesis is regulated at transcriptional level by various stimuli such as inflammatory markers and vitamin D levels. Serum ferritin concentrations are regulated by hepcidin, which causes phosphorylation, internalization and subsequent lysosomal degradation of ferroporitin (Fp), thereby reducing its expression on iron exporting cells. Adapted from Cui et al. (2009) and Mariani et al. (2009) Abbreviations: Fe3+, ferric iron; Fe2+, ferrous cation; DcytB, duodenal cytochrome B; DMT1, divalent metal transporter 1; Jak2, Janus kinase 2; HEPH, hepaestin; Tf, transferritin; RBCs, red blood cells; CP, ceruloplasmin; TfR1, transferritin receptor 1; 1,25(OH)2D3, calcitriol; IL-6, interleukin-6; IL-1β, interleukin-1beta.
Intestinal inflammation caused by gut dysbiosis can impact iron homeostasis within the GI tract (Cherayil et al., 2011), however, whether these findings have been extrapolated to serum iron homeostasis has not yet been elucidated. While iron dysregulation within the GI tract and gut dysbiosis potentially exacerbate one another, Constante et al. (2017) concluded that luminal heme originating from gastrointestinal bleeding or dietary components more likely contributes to dysbiosis of the gut microbiota in mice than vice versa. However, dietary non-heme iron intake from food has been associated with a 30% increased risk of Parkinson’s disease (p = 0.02) (Logroscino et al., 2008). In the same study authors also observed that supplemental iron intake was associated with a borderline increase in Parkinson’s disease among men (Logroscino et al., 2008). Nonetheless, the most prominent cause of iron dysregulation in the form of elevated serum ferritin levels is cell death (Kell and Pretorius, 2018).
Unliganded Iron and Oxidative Damage
In AD, iron dysregulation and the benefits of its chelation have been known for decades (Crapper McLachlan et al., 1991), and many reviews point to the role of poorly liganded iron in AD development (Exley, 2006; Castellani et al., 2007, 2012; Kell, 2009, 2010; Sandro and Muckenthaler, 2009; Grunblatt et al., 2011; Weinreb et al., 2011; Peters et al., 2015; Wood, 2015; Belaidi and Bush, 2016). Unliganded iron and the accompanying oxidative damage is causatively related to neuro-inflammation (Bester et al., 2013; Hongkuan et al., 2015). According to Nunomura et al. (2001), neurons with neurofibrillary tangles show a 40–56% decrease in relative 8-hydroxyguanosine (8-OHG) levels when compared to neurons without neurofibrillary tangles in AD patients. 8-OHG is a marker of hydroxyl radical formation (Kell, 2009, 2010), so this finding indicates that oxidative damage is an early event in AD progression, which dissipates with lesion formation (Nunomura et al., 2001). This is further supported by (Moreira et al., 2005) who found one of the earliest pathological events in AD to be an imbalance between free radical scavenging and generation. The relationship between poorly liganded iron and oxidative stress involves iron’s catalytic contribution to the Fenton and Haber-Weiss reactions. These reactions are involved in the formation of the highly reactive hydroxyl radical (OH-), the most biologically detrimental free radical species, specifically with regards to macromolecules such as lipids, proteins and nucleic acids (Moreira et al., 2005; Kell, 2009, 2010; Lipinski and Pretorius, 2013; Pretorius et al., 2016a). Notably, iron chelation can reverse tau protein hyperphosphorylation in mice brains (Guo et al., 2013), thereby reducing the formation of intracellular neurofibrillary tangles.
Microbial Reactivation
Dormancy is extremely common in microbiology, even among non-sporulating bacteria (Kaprelyants et al., 1993; Lewis, 2007; Kell et al., 2015; Kell and Kenny, 2016), for sound evolutionary reasons (Mukamolova et al., 2003). Our major hypothesis involves shared symptoms across numerous chronic inflammatory diseases caused by dormant microbes (Pretorius et al., 2016a). As reviewed in 2016 (Pretorius et al., 2016a), microbial reactivation or resuscitation, which may be autocatalytic (Mukamolova et al., 1998), indicates that these so-called dormant microbes which are non-growing and appear operationally dead (Kell et al., 1998), can recover culturability. Metals, although explicitly involved in production of free radicals and potentially oxidative stress in AD, are also involved in the process of dormant microbe reactivation (Bester et al., 2015; Kell et al., 2015). All microbes (with the possible exception of Borrelia) require free available iron to grow. This follows from the in vivo limitation of microbial growth in the absence or decreased availability of free iron (Markel et al., 2007; Reid et al., 2009; Chu et al., 2010; Nairz et al., 2010, 2014; Skaar, 2010; Haley and Skaar, 2012; Armitage and Drakesmith, 2014; Subashchandrabose and Mobley, 2015; Damron et al., 2016; Carver, 2018). It is its absence in normal metabolism that causes dormant bacteria to remain in that state. With this in mind, iron dysregulation specifically in the form of high SF levels (Kell and Pretorius, 2014), accelerates AD pathology in two ways (Pretorius et al., 2016a) which suggests the opportunity of its use as a biomarker for early diagnosis and/or a target for slowing down disease progression (Bester et al., 2013).
Gut Microbiota
The human gut microbiome contains approximately 10–100 trillion microbes, outnumbering human genes by 100-fold (Zhu et al., 2010). The gut microbiome forms the largest diffuse organ system in the body, and is more metabolically active than is the liver (Zhao et al., 2015). Through various systemic effects, the well-being and health of the human host is largely impacted by these microbes (Zhao and Lukiw, 2015). However, the human microbiome displays a high degree of variation at inter-individual levels (Costello et al., 2009). For example, even as infants the composition of the gut microbiota is idiosyncratic with significant inter-individual variation being evident from the 1st day after birth (Chong et al., 2018). As a consequence it is has been impossible to define a healthy microbiome (Dave et al., 2012), but, it has become apparent that certain compositions of microbial communities may be able to promote both health and disease (Blum, 2017).
Intriguingly the human gastrointestinal (GI) tract has co-evolved with two major phyla: (1) Firmicutes which constitutes 80% and, (2) Bacteriodetes which constitutes 20% of all GI tract bacteria (Pritchard et al., 2017; Zhao et al., 2017). There is increasing evidence implicating host–microbiome interactions at all stages of complexity including the central nervous system (Stilling et al., 2014). According to Stilling et al. (2014) gut-microbial products can distress chromatin plasticity, leading to changes in neuronal transcription and host conduct. This is due to the fact that the microbiota is an essential mediator of gene–environment interactions and may themselves be regarded as an epigenetic entity. Current research which supports this view includes reports which have characterized small non-coding RNA secreted from microbial cells in the GI tract (Ghosal et al., 2015; Zhao et al., 2017).
Alterations of the gut microbiota can promote pro-inflammatory cytokine release and increase intestinal permeability, which has also been associated with AD (Pistollato et al., 2016). In particular, changes of gut microbial diversity and density can result in systemic and neuro-inflammation as well as dysfunction of the cerebellum and hippocampus (Pistollato et al., 2016). Considering the multitudes of LPSs and amyloids in the GI tract, it is plausible that the microbiota is involved in the pathogenesis of neurological disorders hallmarked by amyloidogenic features (Tran and Greenwood-Van Meerveld, 2013; Shoemark and Allen, 2015; Zhao and Lukiw, 2015) such as AD.
Dysbiosis
Dysbiosis refers to alteration in the composition of the microbiota and has been implicated in the etiology of various disease states (de Oliveira et al., 2017; Kho and Lal, 2018). Perturbations of the gut microbiota can occur as consequences of antibiotic treatment (especially during infancy), dietary changes, sedentary behavior, food additives, non-steroidal anti-inflammatory and other drugs (Wallace, 2013; Ticinesi et al., 2017; Maier et al., 2018) and various other conditions (Pistollato et al., 2016). Dietary changes, for example, may explain up to 57% of the total structural variation the gut microbiome (Brown et al., 2012). Using a humanized mouse model in which adult human fecal microbiota were transplanted into germ-free mice, Brown et al. (2012) reported a shift in the composition of microbiota after switching the mice from a low-fat, plant polysaccharide-rich diet to a Western diet. While diets rich in refined sugars result in different changes to the microbiota from diets lacking fiber, due to the intricate balance that exists between microbial species, alterations in one species can disrupt the entire microbial community (Brown et al., 2012). Thus, alterations in diet, although modifiable, could potentially be a major hurdle in treatment dysbiosis-related conditions if not adequately addressed. Furthermore, physical activity and its relative intensity can induce or prevent gut dysbiosis by influencing splanchnic blood flow, intestinal permeability and inflammatory cytokine expression (Monda et al., 2017). For example, in mice, even in combination with a high-fat diet, exercise may reduce inflammatory infiltrate into epithelial cells, protecting the integrity of the GI wall (Campbell et al., 2016).
Broad-scale fluctuations in the gut microbiota play significant roles in disease advancement through immune and systemic activation (Vogt et al., 2017). There is considerable evidence for the existence of the gut-brain axis which allows bi-directional communication through various pathways such as neural, endocrine and immune mechanisms (Gareau, 2014; Lyte, 2014; Bienenstock et al., 2015; Köhler et al., 2016; Fung et al., 2017; Sandhu et al., 2017). Within this context, changes in microbial communities could lead to pathophysiological changes in the brain of individuals with AD. Dysbiosis is often associated with increased anxiety and memory impairment due to decreased secretion of neurotrophic factors such as brain-derived neurotrophic factor (Stilling et al., 2014).
Dysbiosis in Alzheimer’s Type Dementia
The recognition of dysbiosis and its possible link to neurodegenerative diseases are increasing as our understanding about the gut-brain axis improves (Miklossy and McGeer, 2016). A recent study in which transgenic AD mice were raised under germ-free conditions, indicated that these mice, compared to conventionally-raised AD mice, had less cerebral amyloid deposition (Harach et al., 2017). This indicates that a dysbiotic gut may influence progression of amyloid pathologies.
The gut microbiome diversity of AD patients is notably decreased and compositionally distinct from age- and gender-matched controls (Vogt et al., 2017). In this study Vogt et al. (2017), the authors reported decreases in Firmicutes and Bifidobacterium spp. and increased Bacteriodetes in AD patients. Bifidobacterium spp. are important in gut health and their beneficial effects are well-documented (Arboleya et al., 2016). For instance, particular Bifidobacterium species, such as B. bifidum, B. cantenulatum, B. breve, and B. adolescentis, have anti-inflammatory properties such as inhibiting LPS-induced IL-8 production and TNFα expression (Khokhlova et al., 2012) Bifidobacteria are also known to reduce intestinal permeability (Underwood et al., 2015). Moreover, Bifidobacterium supplementation has shown to decrease intestinal LPS levels and mend the GI-mucosal barrier in mice (Griffiths et al., 2004; Wang et al., 2006). The phylum Bacteriodetes encompasses an abundant and diverse group of Gram-negative bacteria, which have been detected as being increased in patients with Parkinson’s disease (Keshavarzian et al., 2015). The shedding of LPS and subsequent induction of inflammation is also associated with the increase in numbers of fraction of these bacteria. Considering these findings, individuals with AD may present with a gut microbial phenotype that has an increased propensity for the translocation of inflammatory bacterial components (Vogt et al., 2017).
Amyloidogenesis
Amyloid by definition is protein that is deposited as an insoluble fibril as a result of successive alterations in the protein folding process, known as amyloidosis. Similarly to prions, there is no change in the primary amino acid sequence of the proteins when they adopt an insoluble amyloid form (Kell and Pretorius, 2018). Interestingly, self-associating amyloidogenic lipoproteins are secreted by most microbial species (Schwartz et al., 2016), and amyloid is one of the main secretory products of the gut microbiome (Zhao et al., 2017). The role of the gut in dissemination of amyloid proteins has been reviewed by Pistollato et al. (2016), in which the authors adopted the unifying prion concept to explain transmission of these prion-like proteins. The cumulative life-long contribution of microbial amyloid to neurodegenerative pathophysiology is little recognized. Progressive production and aggregation of amyloids contributes to the pathogenesis of diseases in which amyloids accumulate, which is often mediated by microglial cells (Zhao et al., 2017). Thus, bacterial amyloids originating from the gut microbiome may enhance inflammatory responses to cerebral accumulation of Aβ and play a role in the pathogenesis of AD (Pistollato et al., 2016).
For example, functional amyloid from Klebsiella pneumoniae can depolymerise from its fibrillary state to release oligomers which induce cytotoxicity equal to that caused by pathological Aβ in AD (Shahnawaz and Soto, 2012). The molecular mimicry theory (Ebringer and Rashid, 2009; Proal et al., 2013, 2017) includes microbial curli fibers2 and Aβ as a protein–protein interaction which could result in cross-seeding, even if these proteins are essentially unalike (Friedland, 2015; Zhao et al., 2015). These extrinsic curli proteins act as pathogen-associated molecular patterns with increased β-pleated sheet structures, and can cross-react with antibodies to Aβ plaques (Miklossy, 2016). It is plausible that extracellular senile plaques, due to their morphological appearance and density, may in fact be constituted of curli-like Aβ compositions. In theory, this gives bacterial phylotypes a more blatant role in AD causality (Pritchard et al., 2017). The influence of gut microbiota on amyloid formation and propagation is more significant throughout aging as both the epithelium of the GI tract and the blood-brain barrier (BBB) increase in permeability to smaller molecules (Tran and Greenwood-Van Meerveld, 2013; Pistollato et al., 2016).
Shedding of Inflammagens
Some of the foremost bacterial species of the GI tract, such as the Gram-negative bacilli Bacteroides fragilis and Escherichia coli, secrete a multifarious selection of pro-inflammatory neurotoxins which are not only pathogenic but also detrimental to the homeostatic functioning of neurons (Zhao et al., 2017). The shedding of inflammagens such as LPS from gut-microbiota-related dysbiosis causes systemic and neuro-inflammation and promotes gut permeability. Shedding can occur in response to varying physiological and environmental signals with the most extreme example of microbial shedding known as the Jarisch-Herxheimer reaction. This is basically an uninhibited cytokine storm, which is caused by prompt release of inflammagenic material, often following antibiotic treatment of syphilis (Kell and Pretorius, 2018), or other diseases caused by spirochetes. As mentioned earlier, this may promote neuronal dysfunction and apoptosis, impairment of synaptic plasticity and induced vulnerability to cognitive decline (Daulatzai, 2014). Bacterial LPS has also been found in AD hippocampal brain lysates where the mean LPS levels were three-fold higher in the hippocampus of AD patients than in age-matched controls. This was said to increase to 26-fold in advanced AD hippocampal cases (Zhao et al., 2017).
LPSs are sparingly soluble but over a period of time characteristically form large heterogenous aggregates which are exceptionally immunogenic (Zhao et al., 2015). Interestingly the glycosylphosphatidyl-inositol-anchored LPS and microbe-detecting CD41 receptor, which is crucial in the neutralization of invading microbes, is similarly, stimulated by Aβ fibrils (Dowhan, 2014), relating innate-immune signaling with amyloidogenesis in AD. It has also been noted that previous bacterial infections which resulted in the formation of antibodies to amyloids or bacterial inflammagens may predispose central nervous system amyloids to ensuing attack by antibodies, which results in upregulation of neuro-inflammation (Via et al., 2015; Zhao et al., 2015). The inflammagenic potency of LPS is so pronounced that it is frequently used to stimulate in vivo AD models (Lee et al., 2008; Catorce and Gevorkian, 2016; Zakaria et al., 2017).
LTA is the cell wall equivalent of LPS in Gram-positive bacteria and is equally capable of inducing an inflammatory response through its interactions with toll-like receptor (TLR) 2 (Kell and Pretorius, 2018). LTA has not been as well studied as LPS with regards to inflammagenesis, however, recent work suggests that LTAs may in fact be more potent than LPSs (Pretorius et al., 2018). The stimulation of inflammatory cytokine production by both LPS and LTA are mediated through TLR binding (reviewed by Kell and Pretorius, 2018). In response to inflammagens, concentrations of acute phase biomarkers and inflammatory cytokines such as TNF-α, IL-1β, IL-6, IL-8, C-reactive protein (CRP), serum amyloid A (SAA), and fibrinogen increase significantly (deRosset and Strutz, 2015; De Buck et al., 2016; Pindjakova et al., 2017).
Additionally, increased BBB permeability typically observed in individuals with AD can be caused by shed bacterial inflammagens. The BBB plays a fundamental role in the initiation and continuance of chronic inflammation during AD (Zenaro et al., 2017). While Aβ accumulation in vasculature results in inflammatory events that increase BBB permeability in AD (Erickson and Banks, 2013), proteolytic enzymes such as carbonic anhydrases, peptidyl deiminases and gingipains, and appendages such as curli fibers, fimbriae and other amyloid-like proteins, which are carried with LPS (Pritchard et al., 2017) and contribute significantly to BBB permeabilization. BBB dysfunction during AD effects Aβ clearance, endothelial cell function, tight junction integrity and may activate glial cells which accelerates migration of leukocytes to the brain (Zenaro et al., 2017). This could stimulate the beginning of chronic neuro-inflammation in AD.
Atopobiosis
Atopobiosis refers to the appearance of some of the gut microbes in the wrong place (Potgieter et al., 2015). The sterility of blood is seemingly a controversial topic. The presence of a “physiological” blood microbiota has been reported by several studies using 16S ribosomal DNA quantitative polymerase chain reactions (Amar et al., 2013; Lelouvier et al., 2016; Martel et al., 2017; Proal and Marshall, 2018). Yet, in these studies the plausible source of these microbes from gut permeability was not investigated. Authors of the same studies have reported that changes in the composition of this blood microbiota can be associated with disease. However, based on immunological and clinical understanding (Ochei and Kolhatkar, 2000) the detection of bacteria in blood is always abnormal (Kinnby and Chavez de Paz, 2016; Andreadou et al., 2017) and thus referred to as atopobiosis (Potgieter et al., 2015). As seen in Figure 3, atopobiosis is caused by bacterial translocation in which bacteria move from the GI tract to normally sterile blood and tissues (Potgieter et al., 2015). Once translocation has taken place, these bacteria can cause direct infection and inflammation. The process of bacterial translocation has been reviewed by Potgieter et al. (2015) but can briefly be described by the simultaneous dysfunction of three role players: (1) dendritic cells (2) GI epithelium and, (3) M cells which overlay Peyer’s patches. It is a combination of various compromised mucosal defenses that leads to bacterial translocation. When the influx of gut microbes and toxins is particularly abundant, it is often referred to as a “leaky gut” (Maes, 2009; Fasano, 2012; Quigley, 2016; Mu et al., 2017; Kell and Pretorius, 2018) and is due to severely compromised epithelial tight junctions (Fasano, 2012).
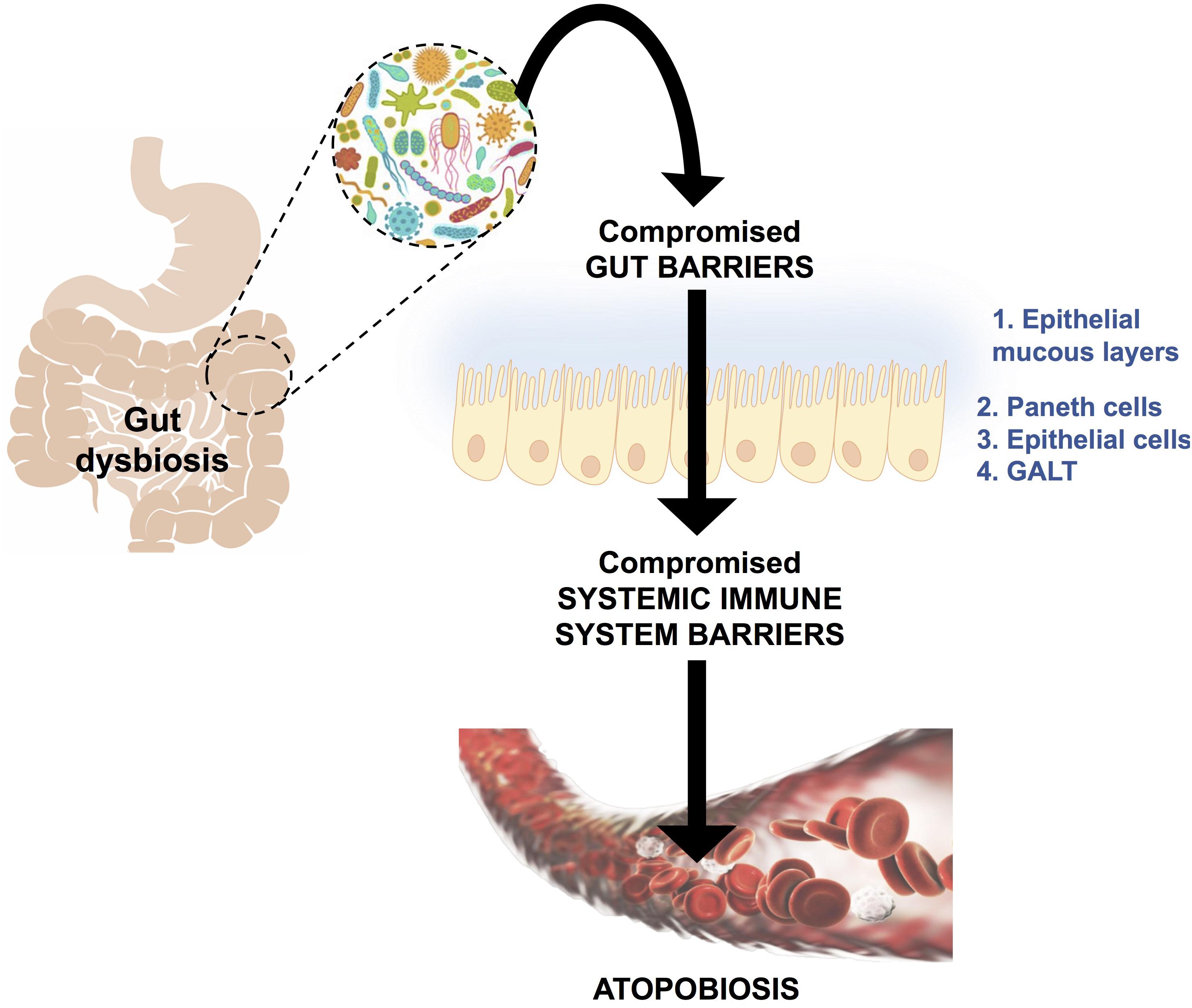
FIGURE 3. A schematic representation of bacterial translocation from a dysbiotic gastrointestinal tract to normally sterile tissues, such as blood. This occurs due to compromised gut barriers such as the epithelial mucous layer, Paneth cells (which secreted antimicrobial peptides) and epithelial cells and gut associated lymphoid tissue (GALT) as mechanical barriers. Simultaneously, there will be compromised systemic immune system function, which enables microbes to enter into normally sterile tissues via dendritic cells, injured epithelial, M-cells and Peyer’s patches. This phenomenon is known as atopobiosis and is implicated in both communicable and non-communicable diseases. Figure adapted from Potgieter et al. (2015).
Dormant Blood Microbiome
Bacteria that have successfully translocated to the blood have the ability to become dormant and reactivate upon appropriate stimuli, such as iron dysregulation in the form of high SF levels as discussed in section Causes of Iron Dysregulation. In contrast to conventional infections, these bacteria do not multiply but enter dormant states (Kell and Kenny, 2016; Pretorius et al., 2016a; Kell and Pretorius, 2018). This is especially relevant in clinical settings when toxic concentrations of antibiotics promote bacterial persistence or adoption of a dormant state that permits their survival (Kell and Pretorius, 2018). Even though the major source of this dormant blood microbiome has been reviewed here as a dysbiotic and permeabilized GI tract, we also recognize periodontitis, gingivitis, urinary tract infections and even insemination as possible points of microbial infection and subsequent translocation (Kamer et al., 2008; Flores-Mireles et al., 2015; Kenny and Kell, 2017; Pretorius et al., 2017; Pritchard et al., 2017). Current research has drifted toward the correlation between an individual’s infectious burden and risk of developing AD, with particular emphasis on herpes simplex virus 1 (HSV1) and human herpesvirus 6 and 7 infections (Honjo et al., 2009; Itzhaki, 2014, 2017; Bu et al., 2015; Itzhaki et al., 2016; Itzhaki and Tabet, 2017; Eimer et al., 2018; Itzhaki and Lathe, 2018; Readhead et al., 2018; Tzeng et al., 2018). Dormant bacteria can survive in leukocytes (Thwaites and Gant, 2011; Liehl et al., 2015) and erythrocytes (Potgieter et al., 2015) to establish the dormant blood microbiome. In Potgieter et al. (2015) transmission electron micrographs of cellular inclusions thought to be L-forms of bacteria inside erythrocytes of individuals diagnosed with AD can be seen (see also Mattman, 2001). Figure 4 illustrates the association of bacteria with pathological coagulations within the blood of individuals diagnosed with AD. This dormant blood microbiome affects the integrity of the hematological system, promoting pathological coagulation.
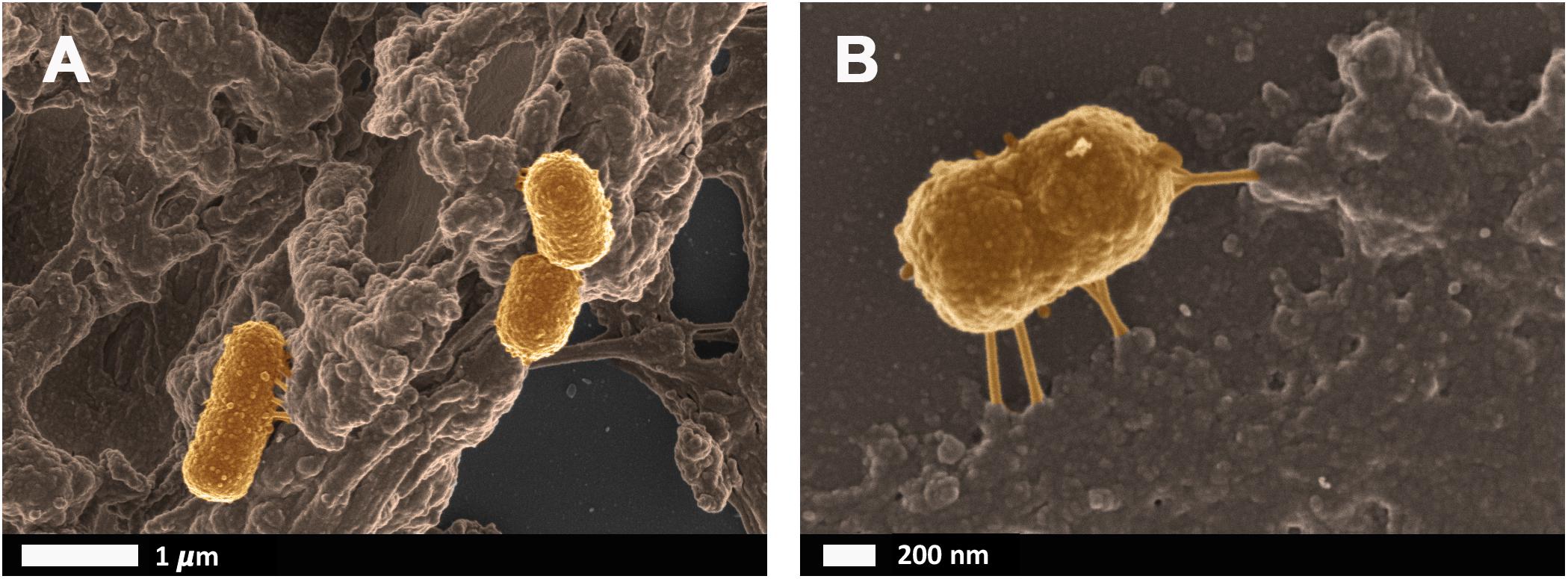
FIGURE 4. Scanning electron micrographs of bacteria associated with pathological coagulations found within the blood of individuals diagnosed with Alzheimer’s disease. Unpublished data from Potgieter et al. (2015). (A) low magnification, (B) higher magnification.
Pathological Coagulation
LPS is well known to cause inflammatory cytokine production which can cause hypercoagulation, often referred to as endotoxin-mediated hypercoagulation (Slotta et al., 2008; Kell and Pretorius, 2017). LPS may activate the coagulation pathway via upregulation of tissue factor (TF), which leads to activation of pro-thrombin (Landsem et al., 2015). Two main components of the hematological system namely fibrinogen and erythrocytes and the changes induced during inflammation and AD, will be discussed below.
Fibrinogen
Fibrinogen is the precursor of fibrin and thus plays a major role in thrombosis and hemostasis (Pretorius and Kell, 2014). During inflammation, circulating fibrinogen levels are increased, while fibrinolysis is impaired. Amyloidogenesis of the fibrin fibers occurs, resulting in the formation of dense matted deposits which trap other blood cells and distort their shape. The combination of the increased propensity to form a stiffer fibrin network and reduced fibrinolysis is a feature of numerous inflammatory diseases such as AD (Ahn et al., 2010; Zamolodchikov and Strickland, 2012). Figure 5 illustrates some of the morphological changes of fibrin fibers due to the addition of LPS.
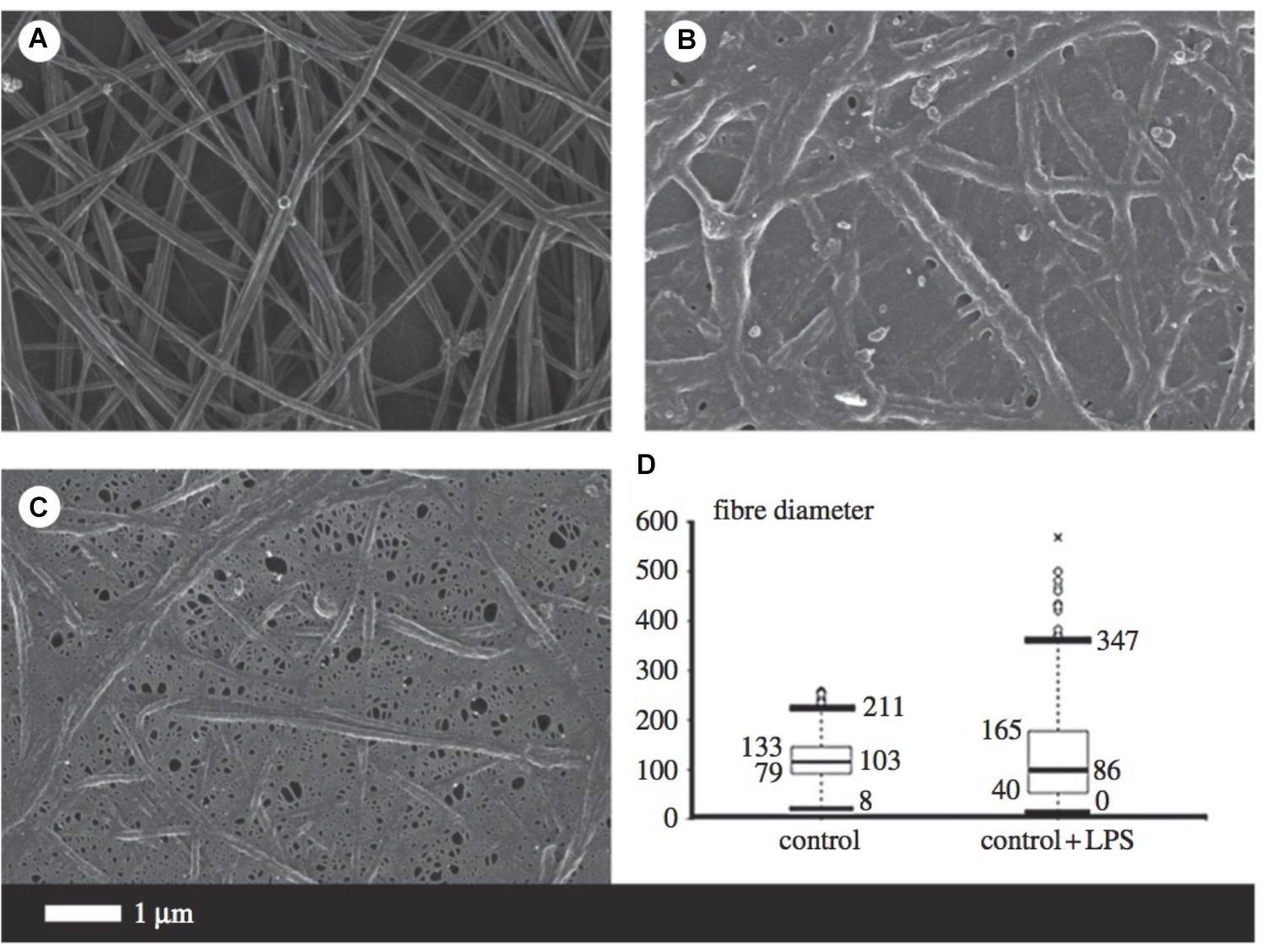
FIGURE 5. Micrographs from scanning electron microscopy (A–C) as well as a box and whisker plot indicating fiber distribution (D) taken from Pretorius et al. (2016b) (open access publication). (A) Is indicative of healthy fibrin fibers during coagulation (with the addition of thrombin), while (B,C) demonstrate the large effect of 0.2 ng/l-1 0111:B4 LPS on fibrin morphology. LPS, lipopolysaccharide. ∗, outlier measurement.
The presence of excessive (unliganded) iron and the consequential production of hydroxyl radicals is one of the leading causes of altered fibrin cross-linking (Pretorius and Kell, 2014; Kell and Pretorius, 2015b). Dense matted deposits produced in the presence of ferric ions are abnormally resistant to chemical and proteolytic degradation, hypothesized to be due to the existence of intermolecular hydrophobic bonds (Lipinski and Pretorius, 2012). As reviewed, it seems that iron dysregulation may serve as a causative factor for multiple pathologies involved in AD progression. This altered fibrinogen structure, with excessive cross-linking and increased β-pleated sheet structure, has been implicated in the development of neuro-inflammation and memory impairments. Increased fibrinogen levels are a strong indicator of cerebrovascular risk since fibrinogen binds to Aβ which further delays clot degradation (Bester et al., 2013; Pretorius et al., 2016a). In addition, pharmacological inhibition of the fibrinogen–Aβ interaction with Ru-505 altered thrombus structure and paused cognitive decline in mice (Ahn et al., 2014), providing a strong association between pathological clotting and neurodegenerative diseases.
Erythrocytes
The dynamics of erythrocytes represent an important but understudied feature of the cardiovascular system. Central to this is their rheology, viscosity, aggregation and deformability (Baskurt et al., 2011; Pretorius and Kell, 2014). It is thought that alterations in erythrocytes and their rheology can contribute to the pathogenesis of AD by hindering oxygen delivery (Tripathy et al., 2013). Dense matted deposits may also trap erythrocytes, impairing their effective delivery of oxygen to the brain. Proficient oxygen delivery is vital for normal functioning of the brain, underpinned by that fact that 20% of an individual’s total oxygen intake is used by the brain. Neurons are particularly susceptible to hypoxic periods, which can cause irreversible neurological consequences (Lipinski and Pretorius, 2013) after only a few minutes. Altered functioning of erythrocytes is also strongly suggested by changes in their size distribution (Ozturk et al., 2013; Weuve et al., 2014; Pilling et al., 2017; Winchester et al., 2018).
As reviewed by Pretorius and Kell (2014), erythrocyte deformability is a complex process which is significantly altered by pathophysiological conditions. Briefly, reduced erythrocyte deformability is an important feature of inflammation in which reactive oxygen species cause degradation of spectrin and band 3, for example, which are important membrane proteins. Since the plasma membrane and the cytoskeleton are responsible for the preservation of erythrocyte shape and stability, modifications of the phospholipid bilayer can affect deformability. In 2010, Mohanty et al. (2010) reported that 15% of erythrocytes in AD were elongated and suggested that a potential link between changes in the erythrocyte proteome could contribute to AD pathology. Deformability and impaired reformability, particularly after erythrocytes have moved through capillaries, reduces their oxygen carrying capacity, potentiating transient states of hypoxia. Figure 6 illustrates erythrocyte deformability as well as loss of membrane integrity in typical blood samples from individual’s diagnosed with AD. It is clear to see how the loss of the typical bi-concave structure of these erythrocytes would impair their physiological functioning.
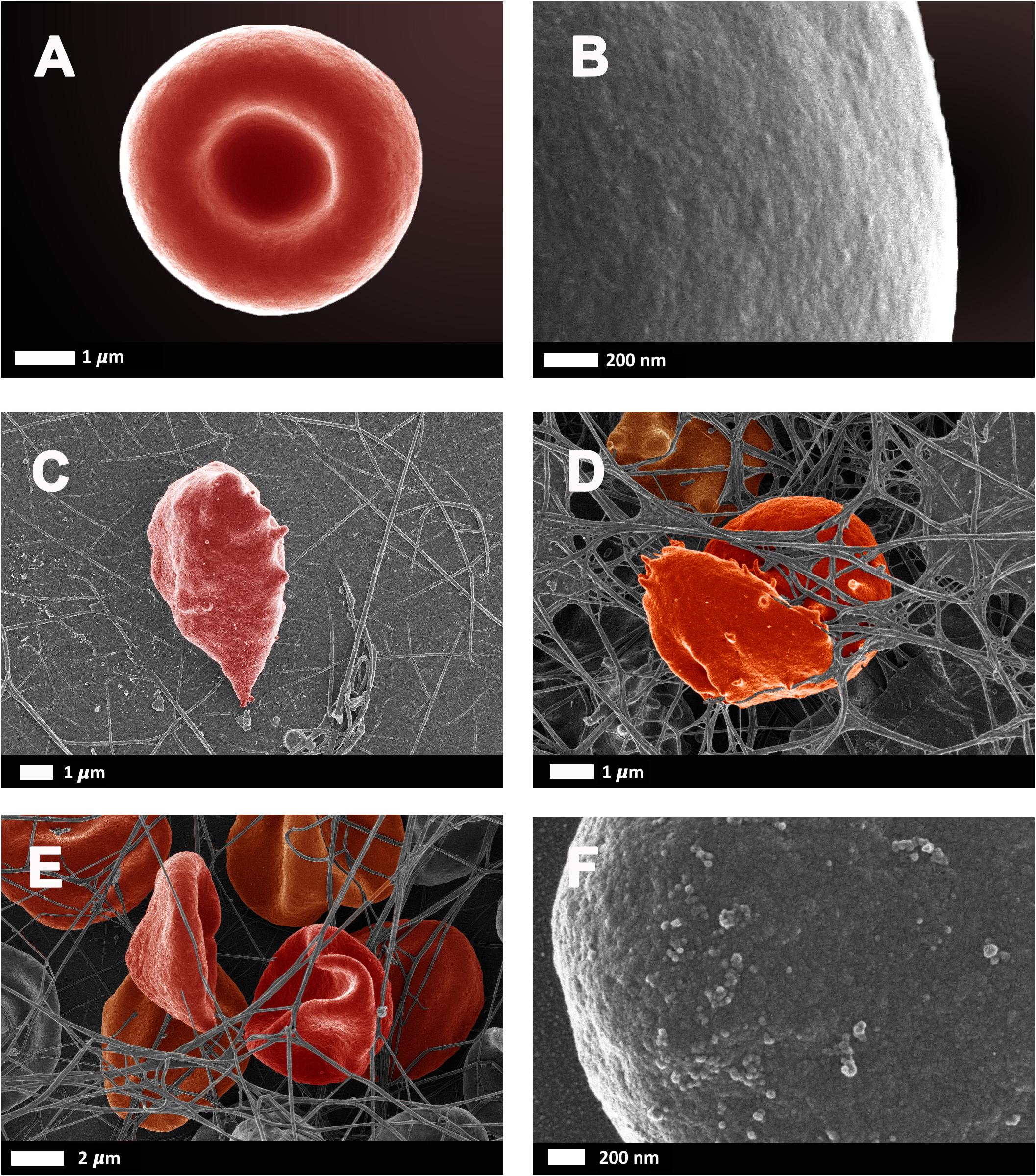
FIGURE 6. Erythrocytes from healthy individuals (A–B) and individuals diagnosed with Alzheimer’s type dementia (C–F). (C) Serum ferritin: 57 ng.L-1 (D) Serum ferritin: 256 ng.L-1 (E) Serum ferritin: 302 ng.L-1 (F) Serum ferritin: 302 ng.L-1 (100,000× machine magnification of erythrocyte membrane). Unpublished data from Bester et al. (2013).
Nitric Oxide Regulation
The dysregulation of nitric oxide (NO) synthesis is prevalent in many diseased conditions (Cooke and Dzau, 1997; Kolios et al., 2004; Naseem, 2005; Karpuzoglu and Ahmed, 2006; Pacher et al., 2007). Recently, the physiological role of erythrocytes in regulating nitric oxide levels has also been investigated (Saldanha, 2016). Due to the consequences of chronic systemic inflammation and pathological coagulation, such as erythrocyte deformability and lysis, it is plausible that aberrant NO regulation could occur and causatively contribute to AD pathogenesis.
NO is synthesized by three isoforms of nitric oxide synthases. Although NO has vasoactive, immunological and neurophysiological functions, it can be neurotoxic at high concentrations and has been implicated in neurodegenerative diseases (Law et al., 2001). In this regard, erythrocytes have an important role in vascular function. Normally, erythrocytes regulate hemostasis by balancing oxygen delivery and NO scavenging and production. More specifically, hemoglobin (Hb) compartmentalization and encapsulation reduces an erythrocyte’s ability to scavenge NO. However, in various diseases hemolysis introduces cell-free Hb into circulation, increasing NO scavenging and inducing hypertension (Helms et al., 2018). A decrease in NO may lead to a reduced ability to learn and memorize due to impairment of long-term potentiation, since NO is responsible for the synaptic efficiency of pre-synaptic glutamatergic neurons (Panthi et al., 2018). Hypoperfusion of the brain and increased oxidative stress within the vasculature are common phenomena in AD (Edwards and Rickard, 2007) and some researchers have reported abnormalities in NO synthase expression as early symptoms of cognitive impairment and AD (Lüth et al., 2002; Malinski, 2007).
Furthermore, NO can exert its neurotoxic effects through three main mechanisms. Firstly, NO is a free radical which can react with superoxide anions to produce another damaging radical, peroxynitrite (Eliasson et al., 1999). This can produce significant oxidative stress which leads to oxidative damage and eventually neuronal death. Secondly, NO causes nitrosylation in a variety of proteins such as glyceraldehyde-3 phosphate dehydrogenase (GADPH) and protein kinase C (PKC), inhibiting their functioning. Lastly, NO can impair glycolysis and the latter energy manufacturing by potentiating ADP-ribosylation of GADPH (Law et al., 2001) as well as binding aconitase and thereby inhibiting electron transfer in the electron transport chain.
Together, pathological coagulation and altered nitric oxide regulation can enhance the propensity of spontaneous clot formation in cerebral arteries and arterioles. These clots are more resistant to degradation, due to amylodiogenesis of fibrin fibers in the presence of ferric irons, and may thus potentiate transient periods of hypoxia in the brain, promoting the occurrence of MD.
Conclusion
While numerous hypotheses for the pathogenesis of AD exist (extensively reviewed by Law et al., 2001; Hardy and Selkoe, 2002; Swerdlow, 2007; Armstrong, 2011;
Dong et al., 2012; Barage and Sonawane, 2015; Selkoe and Hardy, 2016; Area-Gomez and Schon, 2017; Makin, 2018), the IDDM hypothesis (illustrated in Figure 7) recognizes that a combination of a dormant blood or tissue microbiome can be re-activated by unliganded iron. This in turn can induce shedding of highly inflammagenic LPS and LTA molecules, from which all known sequlae of AD can be seen to follow.
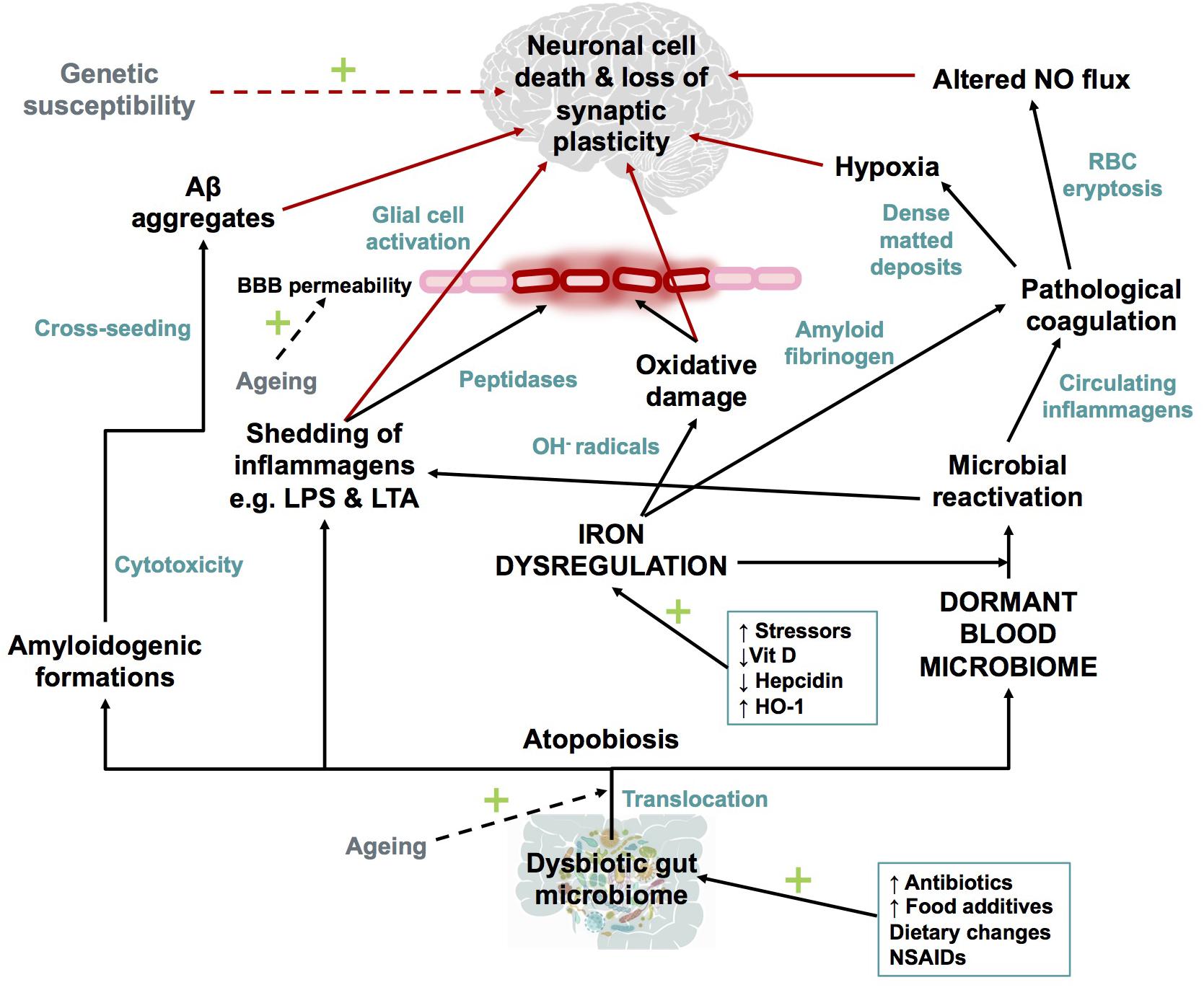
FIGURE 7. Schematic representation of The Iron Dysregulation and Dormant Microbes hypothesis. With reference to the scheme it is plausible that systemic inflammation caused by stress induced iron dysregulation and subsequent microbial reactivation can lead to increased permeability of the blood brain barrier as well as a neuro-inflammatory environment which promotes neuronal cell death. The red arrows indicate factors which could contribute to synaptic loss and neuronal death. Dashed arrows have not been discussed in detail in this review but are recognized as contributors to the pathogenesis of Alzheimer’s-type dementias. Adapted from Kell and Pretorius (2018). Abbreviations: Aβ, amyloid beta; NO, nitric oxide; BBB, blood brain barrier; RBC, red blood cell; OH-, hydroxyl; LPS, lipopolysaccharide; LTA, lipoteichoic acid; HO-1, heme oxygenase-1 and NSAIDs, non-steroidal anti-inflammatory drugs.
As reviewed here, dysbiotic bacterial communities from the GI tract or microbes from other sources of infection, translocate into normally sterile tissues and blood to establish a dormant blood microbiome. Characteristic of AD (and many other chronic, inflammatory diseases; Kell, 2009) is iron dysregulation, that amongst many other affects, causes microbial reactivation. Iron dysregulation, specifically in the form of elevated SF levels, which can causatively modulate oxidative damage, the formation of amyloidogenic fibrinogen and the aforementioned microbial reactivation as part of AD pathogenesis. Microbes contribute to amyloidogenic formations, inflammagen shedding, pathological clotting and systemic as well as neuro-inflammation. Various changes to the hematological system ensue, increasing the risk of pathological coagulation and transient hypoxic events in the AD brain. Increasing BBB permeability and glial cell activation initiate the slow inflammatory progression of AD, in combination with loss of synaptic plasticity and neuronal death. While AD is a disease too complex to fit to any particular model, the IDDM hypothesis highlights several causative role players which can be targeted for early diagnosis and/or for prevention of disease progression.
Consent for Publication
All authors have read the paper and agree that it can be published.
Author Contributions
LP wrote the paper. DK co-wrote the paper and edited the paper. EP co-wrote the paper and is the study leader.
Funding
Funders include the Biotechnology and Biological Sciences Research Council (Grant No. BB/L025752/1) as well as the National Research Foundation (NRF) of South Africa (91548: Competitive Program) and the Medical Research Council of South Africa (MRC) (Self-Initiated Research Program) for supporting this collaboration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This is paper 18 in the series “A dormant blood microbiome in chronic, inflammatory disease”. We thank the Biotechnology and Biological Sciences Research Council, the National Research Foundation (NRF) of South Africa and the Medical Research Council (MRC) of South Africa, for financially supporting this collaboration.
Footnotes
- ^ Presence of microbes in the wrong place, i.e., sterile environments such as blood.
- ^ Extracellular bacterial amyloids.
References
Ahn, H. J., Glickman, J. F., Poon, K. L., Zamolodchikov, D., Jno-Charles, O. C., Norris, E. H., et al. (2014). A novel Abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J. Exp. Med. 211, 1049–1062. doi: 10.1084/jem.20131751
Ahn, H. J., Zamolodchikov, D., Cortes-Canteli, M., Norris, E. H., Glickman, J. F., and Strickland, S. (2010). Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U.S.A. 107, 21812–21817. doi: 10.1073/pnas.1010373107
Amar, J., Lange, C., Payros, G., Garret, C., Chabo, C., Lantieri, O., et al. (2013). Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the D.E.S.I.R. study. PLoS One 8:e54461. doi: 10.1371/journal.pone.0054461
Andreadou, E., Pantazaki, A. A., Daniilidou, M., and Magba, T. (2017). Rhamnolipids, microbial virulence factors, in Alzheimer’s disease. J. Alzheimers Dis. 59, 209–222. doi: 10.3233/JAD-161020
Arboleya, S., Watkins, C., Stanton, C., and Ross, R. P. (2016). Gut bifidobacteria populations in human health and aging. Front. Microbiol. 7:1204. doi: 10.3389/fmicb.2016.01204
Area-Gomez, E., and Schon, E. A. (2017). On the pathogenesis of Alzheimer’s disease: the MAM hypothesis. FASEB J. 31, 864–867. doi: 10.1096/fj.201601309
Armitage, A. E., and Drakesmith, H. (2014). The battle for iron. Science 346, 1299–1300. doi: 10.1126/science.aaa2468
Armstrong, R. A. (2011). The pathogenesis of Alzheimer’s disease: a reevaluation of the “amyloid cascade hypothesis”. Int. J. Alzheimers Dis. 2011:630865. doi: 10.4061/2011/630865
Ayton, S., Faux, N. G., Bush, A. I., Alzheimer’s Disease, and Neuroimaging Initiative. (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6:6760. doi: 10.1038/ncomms7760
Bacchetta, J., Zaritsky, J. J., Sea, J. L., Chun, R. F., Lisse, T. S., Zavala, K., et al. (2014). Suppression of iron-regulatory hepcidin by vitamin D. J. Am. Soc. Nephrol. 25, 564–572. doi: 10.1681/ASN.2013040355
Barage, S. H., and Sonawane, K. D. (2015). Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52, 1–18. doi: 10.1016/j.npep.2015.06.008
Barrientos, R. M., Kitt, M. M., Watkins, L. R., and Maier, S. F. (2016). Neuroinflammation in the normal aging hippocampus. Neuroscience 309, 84–99. doi: 10.1016/j.neuroscience.2015.03.007
Baskurt, O., Neu, B., and Meiselman, H. J. (2011). Red Blood Cell Aggregation, 1st Edn. Boca Raton, FL: CRC Press. doi: 10.1201/b11221
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl. 1), 179–197. doi: 10.1111/jnc.13425
Bester, J., Buys, A. V., Lipinski, B., Kell, D. B., and Pretorius, E. (2013). High ferritin levels have major effects on the morphology of erythrocytes in Alzheimer’s disease. Front. Aging Neurosci. 5:88. doi: 10.3389/fnagi.2013.00088
Bester, J., Soma, P., Kell, D. B., and Pretorius, E. (2015). Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 6, 35285–35303. doi: 10.18632/oncotarget.6074
Bienenstock, J., Kunze, W., and Forsythe, P. (2015). Microbiota and the gut-brain axis. Nutr. Rev. 73(Suppl. 1), 28–31. doi: 10.1093/nutrit/nuv019
Blum, H. E. (2017). The human microbiome. Adv. Med. Sci. 62, 414–420. doi: 10.1016/j.advms.2017.04.005
Brown, K., DeCoffe, D., Molcan, E., and Gibson, D. L. (2012). Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 4, 1095–1119. doi: 10.3390/nu4081095
Bu, X. L., Yao, X. Q., Jiao, S. S., Zeng, F., Liu, Y. H., Xiang, Y., et al. (2015). A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 22, 1519–1525. doi: 10.1111/ene.12477
Campbell, S. C., Wisniewski, P. J., Noji, M., McGuinness, L. R., Haggblom, M. M., Lightfoot, S. A., et al. (2016). The effect of diet and exercise on intestinal integrity and microbial diversity in mice. PLoS One 11:e0150502. doi: 10.1371/journal.pone.0150502
Carver, P. L. (2018). The battle for iron between humans and microbes. Curr. Med. Chem. 25, 25–36. doi: 10.2174/0929867324666170720110049
Castellani, R. J., Moreira, P. I., Liu, G., Dobson, J., Perry, G., Smith, M. A., et al. (2007). Iron: the Redox-active center of oxidative stress in Alzheimer disease. Neurochem. Res. 32, 1640–1645. doi: 10.1007/s11064-007-9360-7
Castellani, R. J., Moreira, P. I., Perry, G., and Zhu, X. (2012). The role of iron as a mediator of oxidative stress in Alzheimer disease. Biofactors 38, 133–138. doi: 10.1002/biof.1010
Catorce, M. N., and Gevorkian, G. (2016). LPS-induced murine neuroinflammation model: main features and suitability for pre-clinical assessment of nutraceuticals. Curr. Neuropharmacol. 14, 155–164. doi: 10.2174/1570159X14666151204122017
Cherayil, B. J., Ellenbogen, S., and Shanmugam, N. N. (2011). Iron and intestinal immunity. Curr. Opin. Gastroenterol. 27, 523–528. doi: 10.1097/MOG.0b013e32834a4cd1
Chong, C. Y. L., Bloomfield, F. H., and O’Sullivan, J. M. (2018). Factors affecting gastrointestinal microbiome development in neonates. Nutrients 10;E274. doi: 10.3390/nu10030274
Chu, B. C., Garcia-Herrero, A., Johanson, T. H., Krewulak, K. D., Lau, C. K., Peacock, R. S., et al. (2010). Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals 23, 601–611. doi: 10.1007/s10534-010-9361-x
Constante, M., Fragoso, G., Calvé, A., Samba-Mondonga, M., and Santos, M. M. (2017). Dietary heme induces gut dysbiosis, aggravates colitis, and potentiates the development of adenomas in mice. Front. Microbiol. 8:1809. doi: 10.3389/fmicb.2017.01809
Cooke, J. P., and Dzau, V. J. (1997). NITRIC OXIDE SYNTHASE: role in the genesis of vascular disease. Annu. Rev. Med. 48, 489–509. doi: 10.1146/annurev.med.48.1.489
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Crapper McLachlan, D. R., Dalton, A. J., Kruck, T. P. A., Bell, M. Y., Smith, W. L., Kalow, W., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-B
Cui, Y., Wu, Q., and Zhou, Y. (2009). Iron-refractory iron deficiency anemia: new molecular mechanisms. Kidney Int. 76, 1137–1141. doi: 10.1038/ki.2009.357
Damron, F. H., Oglesby-Sherrouse, A. G., Wilks, A., and Barbier, M. (2016). Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 6:39172. doi: 10.1038/srep39172
Daulatzai, M. A. (2014). Role of stress, depression, and aging in cognitive decline and Alzheimer’s disease. Curr. Top. Behav. Neurosci. 18, 265–296. doi: 10.1007/7854_2014_350
Dave, M., Higgins, P. D., Middha, S., and Rioux, K. P. (2012). The human gut microbiome: current knowledge, challenges, and future directions. Transl. Res. 160, 246–257. doi: 10.1016/j.trsl.2012.05.003
De Buck, M., Gouwy, M., Wang, J. M., Van Snick, J., Proost, P., Struyf, S., et al. (2016). The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. 30, 55–69. doi: 10.1016/j.cytogfr.2015.12.010
de Oliveira, G. L. V., Leite, A. Z., Higuchi, B. S., Gonzaga, M. I., and Mariano, V. S. (2017). Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 152, 1–12. doi: 10.1111/imm.12765
deRosset, L., and Strutz, K. L. (2015). Developmental origins of chronic inflammation: a review of the relationship between birth weight and C-reactive protein. Ann. Epidemiol. 25, 539–543. doi: 10.1016/j.annepidem.2015.01.003
Dong, S., Duan, Y., Hu, Y., and Zhao, Z. (2012). Advances in the pathogenesis of Alzheimer’s disease: a re-evaluation of amyloid cascade hypothesis. Transl. Neurodegener. 1:18. doi: 10.1186/2047-9158-1-18
Dorey, E., Chang, N., Liu, Q. Y., Yang, Z., and Zhang, W. (2014). Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci. Bull. 30, 317–330. doi: 10.1007/s12264-013-1422-z
Dowhan, W. (2014). Lipids and extracellular materials. Annu. Rev. Biochem. 83, 45–49. doi: 10.1146/annurev-biochem-010314-112017
Ebringer, A., and Rashid, T. (2009). Rheumatoid arthritis is caused by Proteus- the molecular mimicry theory and Karl Popper. Front. Biosci. 1, 577–586. doi: 10.2741/e56
Edwards, T. M., and Rickard, N. S. (2007). New perspectives on the mechanisms through which nitric oxide may affect learning and memory processes. Neurosci. Biobehav. Rev. 31, 413–425. doi: 10.1016/j.neubiorev.2006.11.001
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Washicosky, K. J., Rodriguez, A. S., Gyorgy, B., et al. (2018). Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56–63.e3. doi: 10.1016/j.neuron.2018.06.030
Eliasson, M. J. L., Haung, Z., Ferrante, R. J., Sasamata, M., Molliver, M. E., and Snyder, S. H. (1999). Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J. Neurosci. 19, 5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999
Erickson, M. A., and Banks, W. A. (2013). Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 33, 1500–1513. doi: 10.1038/jcbfm.2013.135
Exley, C. (2006). Aluminium and iron, but neither copper nor zinc, are key to the precipitation of beta-sheets of Abeta42 in senile plaque cores in Alzheimer’s disease. J. Alzheimers Dis. 10, 173–177. doi: 10.3233/JAD-2006-102-305
Fasano, A. (2012). Leaky gut and autoimmune diseases. Clin. Rev. Allergy Immunol. 42, 71–78. doi: 10.1007/s12016-011-8291-x
Filiou, M. D., Arefin, A. S., Moscato, P., and Graeber, M. B. (2014). ’Neuroinflammation’ differs categorically from inflammation: transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 15, 201–212. doi: 10.1007/s10048-014-0409-x
Flores-Mireles, A. L., Walker, J. N., Caparon, M., and Hultgren, S. J. (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284. doi: 10.1038/nrmicro3432
Friedland, R. P. (2015). Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheimers Dis. 45, 349–362. doi: 10.3233/JAD-142841
Fung, T. C., Olson, C. A., and Hsiao, E. Y. (2017). Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155. doi: 10.1038/nn.4476
Ganz, T., and Nemeth, E. (2012). Hepcidin and iron homeostasis. Biochim. Biophys. Acta 1823, 1434–1443. doi: 10.1016/j.bbamcr.2012.01.014
Gareau, M. G. (2014). Microbiota-gut-brain axis and cognitive function. Adv. Exp. Med. Biol. 817, 357–371. doi: 10.1007/978-1-4939-0897-4_16
Ghosal, A., Upadhyaya, B. B., Fritz, J. V., Heintz-Buschart, A., Desai, M. S., Yusuf, D., et al. (2015). The extracellular RNA complement of Escherichia coli. Microbiologyopen 4, 252–266. doi: 10.1002/mbo3.235
Griffiths, E. A., Duffy, L. C., Schanbacher, F. L., Qiao, H., Dryja, D., Leavens, A., et al. (2004). In vivo effects of bifidobacteria and lactoferrin on gut endotoxin concentration and mucosal immunity in Balb/c mice. Dig. Dis. Sci. 49, 579–589. doi: 10.1023/B:DDAS.0000026302.92898.ae
Grunblatt, E., Bartl, J., and Riederer, P. (2011). The link between iron, metabolic syndrome, and Alzheimer’s disease. J. Neural. Transm. 118, 371–379. doi: 10.1007/s00702-010-0426-3
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Haley, K. P., and Skaar, E. P. (2012). A battle for iron: host sequestration and Staphylococcus aureus acquisition. Microbes Infect. 14, 217–227. doi: 10.1016/j.micinf.2011.11.001
Harach, T., Marungruang, N., Duthilleul, N., Cheatham, V., Mc Coy, K. D., Frisoni, G., et al. (2017). Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7:1802. doi: 10.1038/srep41802
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Helms, C. C., Gladwin, M. T., and Kim-Shapiro, D. B. (2018). Erythrocytes and vascular function: oxygen and nitric oxide. Front. Physiol. 9:125. doi: 10.3389/fphys.2018.00125
Hongkuan, Y., Hongpeng, G., Mingchun, Y., Ziyi, L., Shigeko, T., Daijiro, Y., et al. (2015). Upregulation of mitochondrial ferritin by proinflammatory cytokines: implications for a role in Alzheimer’s disease. J. Alzheimers Dis. 45, 797–811. doi: 10.3233/JAD-142595
Honjo, K., van Reekum, R., and Verhoeff, N. P. (2009). Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 5, 348–360. doi: 10.1016/j.jalz.2008.12.001
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front. Aging Neurosci. 6:202. doi: 10.3389/fnagi.2014.00202
Itzhaki, R. F. (2017). Herpes simplex virus type 1 and Alzheimer’s disease: possible mechanisms and signposts. FASEB J. 31, 3216–3226. doi: 10.1096/fj.201700360
Itzhaki, R. F., and Lathe, R. (2018). Herpes viruses and senile dementia: first population evidence for a causal link. J. Alzheimers Dis. 64, 363–366. doi: 10.3233/JAD-180266
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer’s disease. J. Alzheimers Dis. 51, 979–984. doi: 10.3233/JAD-160152
Itzhaki, R. F., and Tabet, N. (2017). Herpes simplex encephalitis and Alzheimer’s disease: is there a link? J. Neurol. Sci. 380, 20–21. doi: 10.1016/j.jns.2017.06.046
Jellinger, K. A. (2008). Morphologic diagnosis of “vascular dementia” - a critical update. J. Neurol. Sci. 270, 1–12. doi: 10.1016/j.jns.2008.03.006
Jellinger, K. A., and Attems, J. (2007). Neuropathological evaluation of mixed dementia. J. Neurol. Sci. 257, 80–87. doi: 10.1016/j.jns.2007.01.045
Kamer, A. R., Craig, R. G., Dasanayake, A. P., Brys, M., Glodzik-Sobanska, L., and de Leon, M. J. (2008). Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. 4, 242–250. doi: 10.1016/j.jalz.2007.08.004
Kaprelyants, A. S., Gottschal, J. C., and Kell, D. B. (1993). Dormancy in non-sporulating bacteria. FEMS Microbiol. Rev. 104, 271–286. doi: 10.1111/j.1574-6968.1993.tb05871.x
Karantzoulis, S., and Galvin, J. E. (2011). Distinguishing Alzheimer’s disease from other major forms of dementia. Expert Rev. Neurother. 11, 1579–1591. doi: 10.1586/ern.11.155
Karpuzoglu, E., and Ahmed, S. A. (2006). Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide 15, 177–186. doi: 10.1016/j.niox.2006.03.009
Kell, D., Potgieter, M., and Pretorius, E. (2015). Individuality, phenotypic differentiation, dormancy and ’persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Res. 4:179. doi: 10.12688/f1000research.6709.2
Kell, D. B. (2009). Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genomics 2:2. doi: 10.1186/1755-8794-2-2
Kell, D. B. (2010). Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 84, 825–889. doi: 10.1007/s00204-010-0577-x
Kell, D. B., Kaprelyants, A. S., Weichart, D. H., Harwood, C. R., and Barer, M. R. (1998). Viability and activity in readily culturable bacteria- a review and discussion of the practical issues. Antonie Van Leeuwenhoek 73, 169–187. doi: 10.1023/A:1000664013047
Kell, D. B., and Kenny, L. C. (2016). A dormant microbial component in the development of preeclampsia. Front. Med. 3:60. doi: 10.3389/fmed.2016.00060
Kell, D. B., and Pretorius, E. (2014). Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 6, 748–773. doi: 10.1039/c3mt00347g
Kell, D. B., and Pretorius, E. (2015a). On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr. Biol. 7, 1339–1377. doi: 10.1039/c5ib00158g
Kell, D. B., and Pretorius, E. (2015b). The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr. Biol. 7, 24–52. doi: 10.1039/c4ib00173g
Kell, D. B., and Pretorius, E. (2017). Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 123, 16–41. doi: 10.1016/j.pbiomolbio.2016.08.006
Kell, D. B., and Pretorius, E. (2018). No effects without causes: the Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol. Rev. Camb. Philos. Soc. 93, 1518–1557. doi: 10.1111/brv.12407
Kenny, L. C., and Kell, D. B. (2017). Immunological tolerance, pregnancy, and preeclampsia: the roles of semen microbes and the father. Front. Med. 4:239. doi: 10.3389/fmed.2017.00239
Kerley, R. N., McCarthy, C., Kell, D. B., and Kenny, L. C. (2018). The potential therapeutic effects of ergothioneine in pre-eclampsia. Free Radic. Biol. Med. 117, 145–157. doi: 10.1016/j.freeradbiomed.2017.12.030
Keshavarzian, A., Green, S. J., Engen, P. A., Voigt, R. M., Naqib, A., Forsyth, C. B., et al. (2015). Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 30, 1351–1360. doi: 10.1002/mds.26307
Kho, Z. Y., and Lal, S. K. (2018). The human gut microbiome - a potential controller of wellness and disease. Front. Microbiol. 9:1835. doi: 10.3389/fmicb.2018.01835
Khokhlova, E. V., Smeianov, V. V., Efimov, B. A., Kafarskaia, L. I., Pavlova, S. I., and Shkoporov, A. N. (2012). Anti-inflammatory properties of intestinal Bifidobacterium strains isolated from healthy infants. Microbiol. Immunol. 56, 27–39. doi: 10.1111/j.1348-0421.2011.00398.x
Kinnby, B., and Chavez de Paz, L. E. (2016). Plasminogen coating increases initial adhesion of oral bacteria in vitro. Microb. Pathog. 100, 10–16. doi: 10.1016/j.micpath.2016.08.002
Köhler, C. A., Maes, M., Slyepchenko, A., Berk, M., Solmi, M., Lanctôt, K. L., et al. (2016). The gut-brain axis, including the microbiome, leaky gut and bacterial translocation: mechanisms and pathophysiological role in Alzheimer’s disease. Curr. Pharm. Des. 22, 6152–6166. doi: 10.2174/1381612822666160907093807
Kolios, G., Valatas, V., and Ward, S. G. (2004). Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology 113, 427–437. doi: 10.1111/j.1365-2567.2004.01984.x
Landsem, A., Fure, H., Christiansen, D., Nielsen, E. W., Osterud, B., Mollnes, T. E., et al. (2015). The key roles of complement and tissue factor in Escherichia coli-induced coagulation in human whole blood. Clin. Exp. Immunol. 182, 81–89. doi: 10.1111/cei.12663
Langa, K. M. (2015). Is the risk of Alzheimer’s disease and dementia declining? Alzheimers Res. Ther. 7:34. doi: 10.1186/s13195-015-0118-1
Law, A., Gauthier, S., and Quirion, R. (2001). Say NO to Alzheimer’s disease: the putative links between nitric oxide and dementia of the Alzheimer’s type. Brain Res. Rev. 35, 73–96. doi: 10.1016/S0165-0173(00)00051-5
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
Lelouvier, B., Servant, F., Paisse, S., Brunet, A. C., Benyahya, S., Serino, M., et al. (2016). Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology 64, 2015–2027. doi: 10.1002/hep.28829
Lewis, K. (2007). Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56. doi: 10.1038/nrmicro1557
Liehl, P., Zuzarte-Luis, V., and Mota, M. M. (2015). Unveiling the pathogen behind the vacuole. Nat. Rev. Microbiol. 13, 589–598. doi: 10.1038/nrmicro3504
Lipinski, B., and Pretorius, E. (2012). Novel pathway of iron-induced blood coagulation: implications for diabetes mellitus and its complications. Pol. Arch. Med. Wewn. 122, 115–122. doi: 10.20452/pamw.1201
Lipinski, B., and Pretorius, E. (2013). The role of iron-induced fibrin in the pathogenesis of Alzheimer’s disease and the protective role of magnesium. Front. Hum. Neurosci. 7:735. doi: 10.3389/fnhum.2013.00735
Logroscino, G., Gao, X., Chen, H., Wing, A., and Ascherio, A. (2008). Dietary iron intake and risk of Parkinson’s disease. Am. J. Epidemiol. 168, 1381–1388. doi: 10.1093/aje/kwn273
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147(Suppl. 1), S232–S240. doi: 10.1038/sj.bjp.0706400
Lüth, H., Münch, G., and Arendt, T. (2002). Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 953, 135–143. doi: 10.1016/S0006-8993(02)03280-8
Lyte, M. (2014). Microbial endocrinology and the microbiota-gut-brain axis. Adv. Exp. Med. Biol. 817, 3–24. doi: 10.1007/978-1-4939-0897-4_1
Maes, M. (2009). Leaky gut in chronic fatigue syndrome: a review. Act. Nerv. Super. Rediviva 51, 21–28.
Maier, L., Pruteanu, M., Kuhn, M., Zeller, G., Telzerow, A., Anderson, E. E., et al. (2018). Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628. doi: 10.1038/nature25979
Makin, S. (2018). The amyloid hypothesis on trial. Nature 559, S4–S7. doi: 10.1038/d41586-018-05719-4
Malinski, T. (2007). Nitric oxide and nitroxidative stress in Alzheimer’s disease. J. Alzheimers Dis. 11, 207–218. doi: 10.3233/JAD-2007-11208
Mariani, R., Trombini, P., Pozzi, M., and Piperno, A. (2009). Iron metabolism in thalassemia and sickle cell disease. Mediterr. J. Hematol. Infect. Dis. 1:e2009006. doi: 10.4084/MJHID.2009.006
Markel, T. A., Crisostomo, P. R., Wang, M., Herring, C. M., Meldrum, K. K., Lillemoe, K. D., et al. (2007). The struggle for iron: gastrointestinal microbes modulate the host immune response during infection. J. Leukoc. Biol. 81, 393–400. doi: 10.1189/jlb.0906579
Martel, J., Wu, C. Y., Huang, P. R., Cheng, W. Y., and Young, J. D. (2017). Pleomorphic bacteria-like structures in human blood represent non-living membrane vesicles and protein particles. Sci. Rep. 7:10650. doi: 10.1038/s41598-017-10479-8
Mattman, L. (2001). Cell Wall Deficient forms: Stealth Pathogens, 3rd Edn. Boca Raton, FL: CRC Press.
McColl, B. W., Allan, S. M., and Rothwell, N. J. (2009). Systemic infection, inflammation and acute ischemic stroke. Neuroscience 158, 1049–1061. doi: 10.1016/j.neuroscience.2008.08.019
Michels, K., Nemeth, E., Ganz, T., and Mehrad, B. (2015). Hepcidin and host defense against infectious diseases. PLoS Pathog. 11:e1004998. doi: 10.1371/journal.ppat.1004998
Miklossy, J. (2016). Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimers Dis. 53, 1459–1473. doi: 10.3233/JAD-160451
Miklossy, J., and McGeer, P. L. (2016). Common mechanisms involved in Alzheimer’s disease and type 2 diabetes: a key role of chronic bacterial infection and inflammation. Aging 8, 575–588. doi: 10.18632/aging.100921
Mohanty, J. G., Shukla, H. D., Williamson, J. D., Launer, L. J., Saxena, S., and Rifkind, J. M. (2010). Alterations in the red blood cell membrane proteome in Alzheimer’s subjects reflect disease-related changes and provide insight into altered cell morphology. Proteome Sci. 8:11. doi: 10.1186/1477-5956-8-11
Monda, V., Villano, I., Messina, A., Valenzano, A., Esposito, T., Moscatelli, F., et al. (2017). Exercise modifies the gut microbiota with positive health effects. Oxid. Med. Cell. Longev. 2017:3831972. doi: 10.1155/2017/3831972
Moreira, P. I., Honda, K., Liu, Q., Santos, M. S., Oliveira, C. R., Gjumrakch, A., et al. (2005). Oxidative stress: the old enemy in Alzheimer’s disease pathophysiology. Curr. Alzheimer Res. 2, 430–438. doi: 10.2174/156720505774330537
Mu, Q., Kirby, J., Reily, C. M., and Luo, X. M. (2017). Leaky gut as a danger signal for autoimmune diseases. Front. Immunol. 8:598. doi: 10.3389/fimmu.2017.00598
Mueller, S. G., Weiner, M. W., Thal, L. J., Petersen, R. C., Jack, C., Jagust, W., et al. (2005). The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin. N. Am. 15, 869–881. doi: 10.1016/j.nic.2005.09.008
Mukamolova, G. V., Kaprelyants, A. S., Kell, D. B., and Young, M. (2003). Adoption of the transiently non-culturable state - a bacterial survival strategy? Adv. Microb. Physiol. 47, 65–129. doi: 10.1016/S0065-2911(03)47002-1
Mukamolova, G. V., Kaprelyants, A. S., Young, D. I., Young, M., and Kell, D. B. (1998). A bacterial cytokine. Proc. Natl. Acad. Sci. U.S.A. 95, 8916–8921. doi: 10.1073/pnas.95.15.8916
Nairz, M., Haschka, D., Demetz, E., and Weiss, G. (2014). Iron at the interface of immunity and infection. Front. Pharmacol. 5:152. doi: 10.3389/fphar.2014.00152
Nairz, M., Schroll, A., Sonnweber, T., and Weiss, G. (2010). The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol. 12, 1691–1702. doi: 10.1111/j.1462-5822.2010.01529.x
Naseem, K. M. (2005). The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 26, 33–65. doi: 10.1016/j.mam.2004.09.003
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Ochei, J., and Kolhatkar, A. (eds). (2000). “Pus abscess and wound drain,” in Medical Laboratory Science: Theory and Practice, (New Delhi: Tata McGraw-Hill Publishing Company Limited), 622.
Ozturk, Z. A., Unal, A., Yigiter, R., Yesil, Y., Kuyumcu, M. E., Neyal, M., et al. (2013). Is increased red cell distribution width (RDW) indicating the inflammation in Alzheimer’s disease (AD)? Arch. Gerontol. Geriatr. 56, 50–54. doi: 10.1016/j.archger.2012.10.002
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. doi: 10.1152/physrev.00029.2006
Panthi, S., Manandhar, S., and Gautam, K. (2018). Hydrogen sulfide, nitric oxide, and neurodegenerative disorders. Transl. Neurodegener. 7:3. doi: 10.1186/s40035-018-0108-x
Peters, D. G., Connor, J. R., and Meadowcroft, M. D. (2015). The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: two sides of the same coin. Neurobiol. Dis. 81, 49–65. doi: 10.1016/j.nbd.2015.08.007
Pilling, L. C., Atkins, J. L., Duff, M. O., Beaumont, R. N., Jones, S. E., Tyrrell, J., et al. (2017). Red blood cell distribution width: genetic evidence for aging pathways in 116,666 volunteers. PLoS One 12:e0185083. doi: 10.1371/journal.pone.0185083
Pindjakova, J., Sartini, C., Lo Re, O., Rappa, F., Coupe, B., Lelouvier, B., et al. (2017). Gut dysbiosis and adaptive immune response in diet-induced obesity vs. Systemic inflammation. Front. Microbiol. 8:1157. doi: 10.3389/fmicb.2017.01157
Pistollato, F., Sumalla Cano, S., Elio, I., Masias Vergara, M., Giampieri, F., and Battino, M. (2016). Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 74, 624–634. doi: 10.1093/nutrit/nuw023
Potgieter, M., Bester, J., Kell, D. B., and Pretorius, E. (2015). The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 39, 567–591. doi: 10.1093/femsre/fuv013
Pretorius, E., Akeredolu, O. O., Soma, P., and Kell, D. B. (2017). Major involvement of bacterial components in rheumatoid arthritis and its accompanying oxidative stress, systemic inflammation and hypercoagulability. Exp. Biol. Med. 242, 355–373. doi: 10.1177/1535370216681549
Pretorius, E., Bester, J., and Kell, D. B. (2016a). A bacterial component to Alzheimer’s-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J. Alzheimers Dis. 53, 1237–1256. doi: 10.3233/JAD-160318
Pretorius, E., Mbotwe, S., Bester, J., Robinson, C. J., and Kell, D. B. (2016b). Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J. R. Soc. Interface 13:20160539. doi: 10.1098/rsif.2016.0539
Pretorius, E., and Kell, D. B. (2014). Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integr. Biol. 6, 486–510. doi: 10.1039/c4ib00025k
Pretorius, E., Page, M. J., Hendricks, L., Nkosi, N. B., Benson, S. R., and Kell, D. B. (2018). Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: assessment with novel Amytracker stains. J. R. Soc. Interface 15:20170941. doi: 10.1098/rsif.2017.0941
Primohamed, M., James, S., Meakin, S., Green, C., Scott, A. K., Walley, T. J., et al. (2004). Adverse drug reactions as cause of admission to hospital- prospective analysis of 18 820 patients. BMJ 329, 15–19. doi: 10.1136/bmj.329.7456.15
Prince, M., Albanese, E., Guerchet, M., and Prina, M. (2014). World Alzheimer report 2014. Alzheimers Dis. Int. 2014, 104.
Pritchard, A. B., Crean, S., Olsen, I., and Singhrao, S. K. (2017). Periodontitis, microbiomes and their role in Alzheimer’s disease. Front. Aging Neurosci. 9:336. doi: 10.3389/fnagi.2017.00336
Proal, A. D., Albert, P. J., and Marshall, T. G. (2013). The human microbiome and autoimmunity. Curr. Opin. Rheumatol. 25, 234–240. doi: 10.1097/BOR.0b013e32835cedbf
Proal, A. D., Lindseth, I. A., and Marshall, T. G. (2017). Microbe-microbe and host-microbe interactions drive microbiome dysbiosis and inflammatory processes. Discov. Med. 23, 51–60.
Proal, A. D., and Marshall, T. G. (2018). Re-framing the theory of autoimmunity in the era of the microbiome: persistent pathogens, autoantibodies, and molecular mimicry. Discov. Med. 140, 299–308.
Qui, C., Kivipelto, M., and von Strauss, E. (2009). Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 11, 111–128.
Quigley, E. M. (2016). Leaky gut - concept or clinical entity? Curr. Opin. Gastroenterol. 32, 74–79. doi: 10.1097/MOG.0000000000000243
Readhead, B., Haure-Mirande, J. V., Funk, C. C., Richards, M. A., Shannon, P., Haroutunian, V., et al. (2018). Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human Herpesvirus. Neuron 99, 64–82.e7. doi: 10.1016/j.neuron.2018.05.023
Reichert, C. O., da Cunha, J., Levy, D., Bydlowski, S. P., and Spada, C. (2017). Hepcidin: homeostasis and diseases related to iron metabolism. Acta Haematol. 137, 220–236. doi: 10.1159/000471838
Reid, D. W., Anderson, G. J., and Lamont, I. L. (2009). Role of lung iron in determining the bacterial and host struggle in cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L795–L802. doi: 10.1152/ajplung.00132.2009
Rubio-Perez, J. M., and Morillas-Ruiz, J. M. (2012). A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012:756357. doi: 10.1100/2012/756357
Saldanha, C. (2016). Physiological role of erythrocyte nitric oxide. Clin. Hemorheol. Microcirc. 64, 517–520. doi: 10.3233/CH-168028
Sandhu, K. V., Sherwin, E., Schellekens, H., Stanton, C., Dinan, T. G., and Cryan, J. F. (2017). Feeding the microbiota-gut-brain axis: diet, microbiome, and neuropsychiatry. Transl. Res. 179, 223–244. doi: 10.1016/j.trsl.2016.10.002
Sandro, A., and Muckenthaler, M. U. (2009). Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimers Dis. 14, 879–895. doi: 10.3233/JAD-2009-1010
Schaffer, J. E. (2016). Lipotoxicity: many roads to cell dysfunction and cell death: introduction to a thematic review series. J. Lipid Res. 57, 1327–1328. doi: 10.1194/jlr.E069880
Schmidt, R., Schmidt, H., Curb, J. D., Masaki, K., White, L. R., and Launer, L. J. (2002). Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann. Neurol. 52, 168–174. doi: 10.1002/ana.10265
Schwartz, K., Ganesan, M., Payne, D. E., Solomon, M. J., and Boles, B. R. (2016). Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol. Microbiol. 99, 123–134. doi: 10.1111/mmi.13219
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shahnawaz, M., and Soto, C. (2012). Microcin amyloid fibrils A are reservoir of toxic oligomeric species. J. Biol. Chem. 287, 11665–11676. doi: 10.1074/jbc.M111.282533
Shoemark, D. K., and Allen, S. J. (2015). The microbiome and disease: reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J. Alzheimers Dis. 43, 725–738. doi: 10.3233/JAD-141170
Skaar, E. P. (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Slotta, J. E., Braun, O. O., Menger, M. D., and Thorlacius, H. (2008). Central role of Rho kinase in lipopolysaccharide-induced platelet capture on venous endothelium. J. Investig. Med. 56, 720–725. doi: 10.2310/JIM.0b013e31816c3e81
Steardo, L. Jr., Bronzuoli, M. R., Iacomino, A., Esposito, G., Steardo, L., and Scuderi, C. (2015). Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 9:259. doi: 10.3389/fnins.2015.00259
Stilling, R. M., Dinan, T. G., and Cryan, J. F. (2014). Microbial genes, brain & behaviour - epigenetic regulation of the gut-brain axis. Genes Brain Behav. 13, 69–86. doi: 10.1111/gbb.12109
Subashchandrabose, S., and Mobley, H. L. (2015). Back to the metal age: battle for metals at the host-pathogen interface during urinary tract infection. Metallomics 7, 935–942. doi: 10.1039/c4mt00329b
Thwaites, G. E., and Gant, V. (2011). Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9, 215–222. doi: 10.1038/nrmicro2508
Ticinesi, A., Milani, C., Lauretani, F., Nouvenne, A., Mancabelli, L., Lugli, G. A., et al. (2017). Gut microbiota composition is associated with polypharmacy in elderly hospitalized patients. Sci. Rep. 7:11102. doi: 10.1038/s41598-017-10734-y
Tran, L., and Greenwood-Van Meerveld, B. (2013). Age-associated remodeling of the intestinal epithelial barrier. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1045–1056. doi: 10.1093/gerona/glt106
Tripathy, D., Sanchez, A., Yin, X., Luo, J., Martinez, J., and Grammas, P. (2013). Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 5:19. doi: 10.3389/fnagi.2013.00019
Tzeng, N. S., Chung, C. H., Lin, F. H., Chiang, C. P., Yeh, C. B., Lu, R. B., et al. (2018). Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics 15, 417–429. doi: 10.1007/s13311-018-0611-x
Underwood, M. A., German, J. B., Lebrilla, C. B., and Mills, D. A. (2015). Bifidobacterium longum subspecies infantis: champion colonizer of the infant gut. Pediatr. Res. 77, 229–235. doi: 10.1038/pr.2014.156
Via, A., Uyar, B., Brun, C., and Zanzoni, A. (2015). How pathogens use linear motifs to perturb host cell networks. Trends Biochem. Sci. 40, 36–48. doi: 10.1016/j.tibs.2014.11.001
Vogt, N. M., Kerby, R. L., Dill-McFarland, K. A., Harding, S. J., Merluzzi, A. P., Johnson, S. C., et al. (2017). Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7:13537. doi: 10.1038/s41598-017-13601-y
Vradenburg, G. (2015). A pivotal moment in Alzheimer’s disease and dementia: how global unity of purpose and action can beat the disease by 2025. Expert Rev. Neurother. 15, 73–82. doi: 10.1586/14737175.2015.995638
Wallace, J. L. (2013). Polypharmacy of osteoarthritis: the perfect intestinal storm. Dig. Dis. Sci. 58, 3088–3093. doi: 10.1007/s10620-013-2777-8
Wang, Z., Xiao, G., Yao, Y., Guo, S., Lu, K., and Sheng, Z. (2006). The role of bifidobacteria in gut barrier function after thermal injury in rats. J. Trauma 61, 650–657. doi: 10.1097/01.ta.0000196574.70614.27
Weinreb, O., Mandel, S., Bar-Am, O., and Amit, T. (2011). Iron-chelating backbone coupled with monoamine oxidase inhibitory moiety as novel pluripotential therapeutic agents for Alzheimer’s disease: a tribute to Moussa Youdim. J. Neural Transm. 118, 479–492. doi: 10.1007/s00702-011-0597-6
Weuve, J., Mendes de Leon, C. F., Bennett, D. A., Dong, X., and Evans, D. A. (2014). The red cell distribution width and anemia in association with prevalent dementia. Alzheimer Dis. Assoc. Disord. 28, 99–105. doi: 10.1097/WAD.0b013e318299673c
Winchester, L. M., Powell, J., Lovestone, S., and Nevado-Holgado, A. J. (2018). Red blood cell indices and anaemia as causative factors for cognitive function deficits and for Alzheimer’s disease. Genome Med. 10:51. doi: 10.1186/s13073-018-0556-z
Wood, H. (2015). Alzheimer disease: iron–the missing link between ApoE and Alzheimer disease? Nat. Rev. Neurol. 11:369. doi: 10.1038/nrneurol.2015.96
Zakaria, R., Yaacob, W. M. W., Othman, Z., Long, I., Ahmad, A. H., and Al-Rahbi, B. (2017). Lipopolysaccharide-induced memory impairment in rats: a model of Alzheimer’s disease. Physiol. Res. 66, 553–565.
Zamolodchikov, D., and Strickland, S. (2012). Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 119, 3342–3351. doi: 10.1182/blood-2011-11-389668
Zekry, D., Hauw, J., and Gold, G. (2002). Mixed dementia: epidemiology, diagnosis, and treatment. J. Am. Geriatr. Soc. 50, 1431–1438. doi: 10.1046/j.1532-5415.2002.50367.x
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zhang, L., Hu, R., Li, M., Li, F., Meng, H., Zhu, G., et al. (2013). Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol. Sci. 34, 639–645. doi: 10.1007/s10072-012-1090-1
Zhao, Y., Dua, P., and Lukiw, W. J. (2015). Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD). J. Alzheimers Dis. Parkinsonism 5:177.
Zhao, Y., Jaber, V., and Lukiw, W. J. (2017). Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front. Cell. Infect. Microbiol. 7:318. doi: 10.3389/fcimb.2017.00318
Zhao, Y., and Lukiw, W. J. (2015). Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J. Nat. Sci. 1:e138.
Keywords: Alzheimer’s type dementia, iron, gut dysbiosis, iron dysregulation and dormant microbes hypothesis, inflammation
Citation: Pretorius L, Kell DB and Pretorius E (2018) Iron Dysregulation and Dormant Microbes as Causative Agents for Impaired Blood Rheology and Pathological Clotting in Alzheimer’s Type Dementia. Front. Neurosci. 12:851. doi: 10.3389/fnins.2018.00851
Received: 04 September 2018; Accepted: 30 October 2018;
Published: 16 November 2018.
Edited by:
Isabella Zanella, Università degli Studi di Brescia, ItalyReviewed by:
Zhou Liu, Guangdong Medical University, ChinaAndrea Ticinesi, Università degli Studi di Parma, Italy
Copyright © 2018 Pretorius, Kell and Pretorius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas B. Kell, ZGJrQG1hbmNoZXN0ZXIuYWMudWs= orcid.org/0000-0001-5838-7963 Etheresia Pretorius, cmVzaWFwQHN1bi5hYy56YQ== orcid.org/0000-0002-9108-2384
† Present address: Douglas B. Kell, Dept of Biochemistry, Institute of Integrative Biology, University of Liverpool, Liverpool, United Kingdom