 Sumonto Mitra
Sumonto Mitra Homira Behbahani
Homira Behbahani Maria Eriksdotter
Maria Eriksdotter- 1Division of Clinical Geriatrics, Center for Alzheimer Research, Department of Neurobiology, Care Sciences and Society, Karolinska Institutet, Huddinge, Sweden
- 2Division of Neurogeriatrics, Center for Alzheimer Research, Department of Neurobiology, Care Sciences and Society, Karolinska Institutet, Solna, Sweden
- 3Aging Theme, Karolinska University Hospital, Karolinska Institutet, Stockholm, Sweden
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder associated with abnormal protein modification, inflammation and memory impairment. Aggregated amyloid beta (Aβ) and phosphorylated tau proteins are medical diagnostic features. Loss of memory in AD has been associated with central cholinergic dysfunction in basal forebrain, from where the cholinergic circuitry projects to cerebral cortex and hippocampus. Various reports link AD progression with declining activity of cholinergic neurons in basal forebrain. The neurotrophic molecule, nerve growth factor (NGF), plays a major role in the maintenance of cholinergic neurons integrity and function, both during development and adulthood. Numerous studies have also shown that NGF contributes to the survival and regeneration of neurons during aging and in age-related diseases such as AD. Changes in neurotrophic signaling pathways are involved in the aging process and contribute to cholinergic and cognitive decline as observed in AD. Further, gradual dysregulation of neurotrophic factors like NGF and brain derived neurotrophic factor (BDNF) have been reported during AD development thus intensifying further research in targeting these factors as disease modifying therapies against AD. Today, there is no cure available for AD and the effects of the symptomatic treatment like cholinesterase inhibitors (ChEIs) and memantine are transient and moderate. Although many AD treatment studies are being carried out, there has not been any breakthrough and new therapies are thus highly needed. Long-term effective therapy for alleviating cognitive impairment is a major unmet need. Discussion and summarizing the new advancements of using NGF as a potential therapeutic implication in AD are important. In summary, the intent of this review is describing available experimental and clinical data related to AD therapy, priming to gain additional facts associated with the importance of NGF for AD treatment, and encapsulated cell biodelivery (ECB) as an efficient tool for NGF delivery.
Introduction
The world’s population is aging at a great pace, especially in the western developed countries where mean life expectancy is usually high (>60 years) (World Health Organization [WHO], 2018). Increased life expectancy, owing to better medical and technological advancements, allows a person to gain higher achievements in life (professional, personal, social, etc.). However, increased age also brings in several age-associated health issues like sarcopenia, cancer, dementia, diabetes, cardiovascular diseases, hypertension, osteoporosis, and others, which become major reasons of sufferings (emotional and economical) for the affected patients and associated families (Franceschi et al., 2018). Table 1 summarizes the mean age of population and the percent of population above 60 years of age in the current top 10 countries in the world (U.S. News & World Report; CIA fact book; World Report on Ageing and Health 2015). Although several other countries have both lower mean age and percent of aged population (especially in Africa where typically less than 9% of the population is above 60 years old and forecasted to remain the same until 2050), but the usual life expectancy in those countries is also low due to several confounding factors (poverty, unmet medical situations, geo-political issues, etc.). This puts focus on developed countries and demands a well-planned approach to deal with the health burden of the aging population.
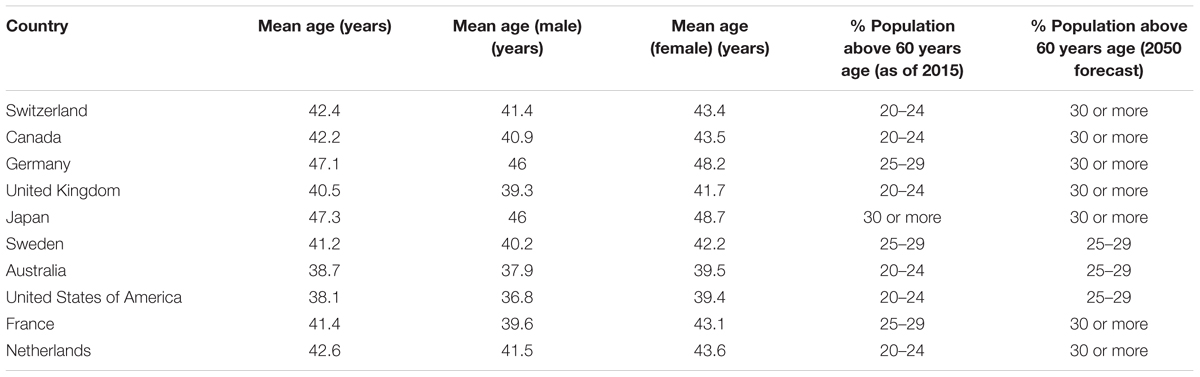
Table 1. Summary of population statistics in the current top 10 countries in the world.
The World Health Organization (WHO) identifies Alzheimer’s disease (AD) (including other dementia’s) as the fifth leading cause of death worldwide in the year 2016 (∼2 million deaths), only after ischemic heart disease, stroke, chronic obstructive pulmonary disease (COPD) and lower respiratory infections (World Health Organization [WHO], 2016). The incidence of dementia related death has doubled since 2000 (ranked at the 14th spot), which is an important aspect of concern and can be attributed to higher life expectancy. Among the upper-middle-income and high-income countries, Alzheimer’s and dementia associated deaths were ranked at fifth and third spots, respectively. By 2050, the world’s population above the age of 60 years will nearly double (from 900 million in 2015 to 2 billion), among which 80% of them will reside in low- and middle-income-countries (World Health Organization [WHO], 2018). Therefore, immediate attention should be provided in developing strategies to prevent the occurrence of such devastating health conditions. However, until such measures are developed, palliative methods will remain of utmost importance to relieve the suffering and enhance quality of life for both the affected individual and the caregiver.
Currently almost 47 million people live with dementia, which is likely to become 131 million by 2050 (World Alzheimer Report, 2016). AD alone constitutes about 60–80% of dementia cases in North America and Europe, and by itself is the single largest cause of dementia related death worldwide (Puzzo et al., 2015). The global expense for the overall direct and indirect costs are estimated to be around 1.09% and 0.65% of the global gross domestic product (GDP), respectively, whereas in the United States, the cost of patients care is in the excess of United States $100 billion (Puzzo et al., 2015; World Alzheimer Report, 2016).
Alzheimer’s disease is a neuropathological condition in which the affected individual displays a varied constellation of cognitive impairments, which progressively hamper normal daily life activities. Pathologically, AD is identified by the presence of aggregated form of amyloid-β (Aβ) proteins (produced by β-cleavage of amyloid precursor protein, APP) as extracellular plaques and hyper-phosphorylated forms of tau protein as intra-cellular tangles. AD can be classified into two major groups, familial (early onset, Mendelian-inheritance) which has an occurrence rate of ∼5% and sporadic (late onset) with an occurrence rate of 95%, respectively (Lista et al., 2015). Apart from establishing some genetic basis of the disease as predisposing factors (ApoE4, APP, presenilin 1, presenilin 2) in case of familial AD, the overall cause of the sporadic AD remains multimodal and phenotypically heterogeneous. Recently several genome wide association (GWAS) studies resulted in the identification of more than 20 genetic loci as AD associated risk factors, which were related to various physiological processes including the immune system, inflammation, lipid metabolism, and vesicle recycling (Van Cauwenberghe et al., 2016). Among non-genetic determinants, several lifestyle factors (diabetes, smoking, physical inactivity, depression, diet, low education) and early life vascular diseases were identified as confounding factors in developing AD later in life (Qiu et al., 2010; Scheltens et al., 2016).
Among several factors, Aβ is one of the highly researched targets in the field of early onset AD neuropathology and is believed to be the causal factor of the disease. It has been more than 25 years since the proposal of the ‘amyloid hypothesis’ and many clinical trials have been directed toward this hypothesis to reduce Aβ levels in search of curing AD itself (Selkoe and Hardy, 2016). However, recent failures of several clinical trials targeting Aβ clearance have initiated interest in the search of alternative targets (De Strooper and Karran, 2016; Makin, 2018). Unless further scientific progress can be achieved to find an effective strategy to cure or halt the disease progression in AD, the current state of palliative care will remain as an important aspect in treating the present and future AD affected individuals.
Presently, the only available treatment option is the use of drugs called cholinesterase inhibitors (ChEIs, which includes donepezil, rivastigmine, and galantamine) and glutamate receptor (NMDA) antagonist (memantine). Activity of acetylcholinesterase in central nerve cells was observed a few decades back leading to large research efforts to therapeutically target them for modifications of the cholinergic pathways in AD (Giacobini and Zajicek, 1956; Marchi et al., 1980; De Sarno et al., 1989; Giacobini et al., 1989; Messamore et al., 1993a,b; Zhu and Giacobini, 1995; Giacobini, 1998, 2004; Taylor, 1998). These drugs provide symptomatic relief and temporarily stabilize cognitive functions, without any impact on disease progression or severity (Rodda and Carter, 2012; Deardorff et al., 2015). However, some studies have shown that ChEIs can modulate Aβ pathways by affecting Aβ generation and fibrillation properties (Inestrosa et al., 1996; Nordberg, 2006). The ChEIs were developed to halt the activity of the acetylcholine (ACh) degrading enzyme called acetylcholinesterase (AChE), which in turn is part of the cholinergic pathway, and was inherently based on the ‘cholinergic hypotheses’ of AD development (Bartus et al., 1982; Bartus, 2000; Mufson et al., 2008). The cholinergic hypothesis highlights the importance of the basal forebrain, which contains the cholinergic neurons that are important for normal cognition, and is found degenerated in AD (Bartus et al., 1982).
AD and the Cholinergic System
The cholinergic system of the human brain has been attributed to be the master regulator of executive and mnemonic functions, and its loss is associated with cognitive decline (Ballinger et al., 2016; Gielow and Zaborszky, 2017). During the initial studies on AD related cognitive impairments in the late 1970s and early 1980s, several groups demonstrated the association between cholinergic neurons depletion in the basal forebrain and simultaneous cognitive decline (Drachman and Leavitt, 1974; Davies and Maloney, 1976; Smith and Swash, 1979; Whitehouse et al., 1982). These studies along with many other supporting observations led to the proposal of the cholinergic hypothesis, which for the first time proposed the importance of cholinergic neuron degeneration in AD associated cognitive impairment (Bartus et al., 1982). Several studies have reported discrepancies in this hypothesis, including the observations that the levels of the ACh-synthesizing enzyme (choline acetyltransferase, ChAT) actually increase during early stages of AD. Nevertheless, proponents of the theory attribute this change as a compensatory event linked to the early cholinergic deficit during AD initiation (Bartus, 2000; DeKosky et al., 2002; Mesulam, 2004; Mufson et al., 2008). Noteworthy, ChAT activity is not the rate-limiting step in ACh synthesis (Haubrich and Chippendale, 1977; Brandon et al., 2004), and the levels of ACh are maintained even when ChAT levels are depleted by 90% (Jenden et al., 1976). Widespread loss of cholinergic neurons in the basal forebrain during advanced stages of AD associated with incremental loss in cognition strengthened the cholinergic hypothesis and established the involvement of cholinergic loss in AD (Bierer et al., 1995; Bartus, 2000; Ferreira-Vieira et al., 2016; Hampel et al., 2018).
The reports on cholinergic deficits associated with early cognitive decline are supported by recent studies, where a long preclinical phase followed by mild cognitive impairment are stages in the AD development (Jack et al., 2012). Recent reports demonstrate a direct correlation between reduced basal forebrain volume and cognitive decline, which were found to be correlated with Aβ burden (Grothe et al., 2010; Kerbler et al., 2015). Interestingly, the decrement in the volume of basal forebrain precedes any major impact on hippocampal volume and predicts the cortical spread of AD pathology, thereby implying the early susceptibility of basal forebrain during AD development (Schmitz et al., 2016; Teipel et al., 2018). The cholinergic neurons are essentially dependent on the neurotrophic factor, nerve growth factor (NGF), for their survival and plasticity, and NGF metabolism is reported to be hampered in AD (Madziar et al., 2005; Iulita and Cuello, 2014; Isaev et al., 2017). Impaired signaling of NGF through its receptors is another important factor contributing to the development of AD (Latina et al., 2017).
Physiological Role of NGF
Nerve growth factor is the founding member of the ‘neurotrophin family,’ primarily produced from the GABAergic neurons in the adult rat brain cortex among many other cell types across the brain (Lauterborn et al., 1995; Biane et al., 2014). NGF regulates differentiation, growth, survival and plasticity of certain cell types, including the cholinergic neurons, in the central and peripheral systems (Levi-Montalcini, 2004; Niewiadomska et al., 2011). NGF is tonically secreted in a precursor form (proNGF), which following maturation by the extracellular protease plasmin is converted to the mature form (mNGF). Both proNGF and mNGF are biologically active (Iulita and Cuello, 2014). The cholinergic neurons require mNGF for their growth and plasticity, and expresses the NGF receptors, tropomyosin receptor kinase A (TrkA, high affinity receptor) and p75 (also known as low-affinity NGF receptor, LNGFR; or p75 neurotrophin receptor, p75NTR), respectively (Niewiadomska et al., 2011; Isaev et al., 2017). Although both proNGF and mNGF can induce neurotrophic effects through TrkA (mNGF more potent than proNGF), but proNGF alone is also capable of inducing apoptotic signaling through its interaction with p75 (with cooperation from another receptor called sortilin) (Al-Shawi et al., 2007; Bradshaw et al., 2015; Ioannou and Fahnestock, 2017). Mature NGF is finally degraded by the matrix metalloproteinase 9 (MMP9) whose expression is upregulated in AD (Mroczko et al., 2013; Iulita and Cuello, 2014).
During normal physiological conditions, the cholinergic neurons in the basal forebrain play an important role in cognition through ACh innervation to the cortex and hippocampus. NGF is released by the postsynaptic cortical and hippocampal neurons, and is taken up by the pre-synaptic cholinergic neuronal projections through TrkA/p75 receptors (Segal, 2003; Biane et al., 2014). These receptor-bound-NGF molecules are then internalized and retrogradely transported to cholinergic cell bodies in the basal forebrain to initiate further signaling cascades that includes cell survival, growth and release of ACh through the cortico-hippocampal projections (DiStefano et al., 1992; Ure and Campenot, 1994; Campenot and MacInnis, 2004). During the pathological conditions of AD, metabolism of NGF has been found to be altered, resulting in an accumulation of proNGF levels and reduction of mNGF, which in rats have been shown to induce loss of cortical synapses and atrophy of the basal forebrain cholinergic neurons (Mufson et al., 1995; Niewiadomska et al., 2011; Iulita and Cuello, 2014). Widespread cholinergic cell loss is observed during severe stages of AD. However, in the earlier stages of AD, intact albeit functionally altered cell bodies along with a mild reduction in synaptic density have been found (Mesulam, 2004). Since these cholinergic cell bodies retain their sensitivity to NGF, delivery of NGF has been targeted over the years as a potent method to revive cholinergic signaling in cortex and hippocampus (discussed later).
What Goes Wrong in AD in Relation to NGF?
In AD, the cholinergic system is affected due to various processes including the following: (1) Altered NGF maturation, (2) Skewed TrkA/p75 receptor ratio, (3) Inefficient axonal transport and signaling, (4) Aβ induced modulation of NGF receptors, (5) Suboptimal ACh innervation induced inflammatory response, and (6) Aβ cytotoxicity. The collective interplay of these factors are depicted in Figure 1.
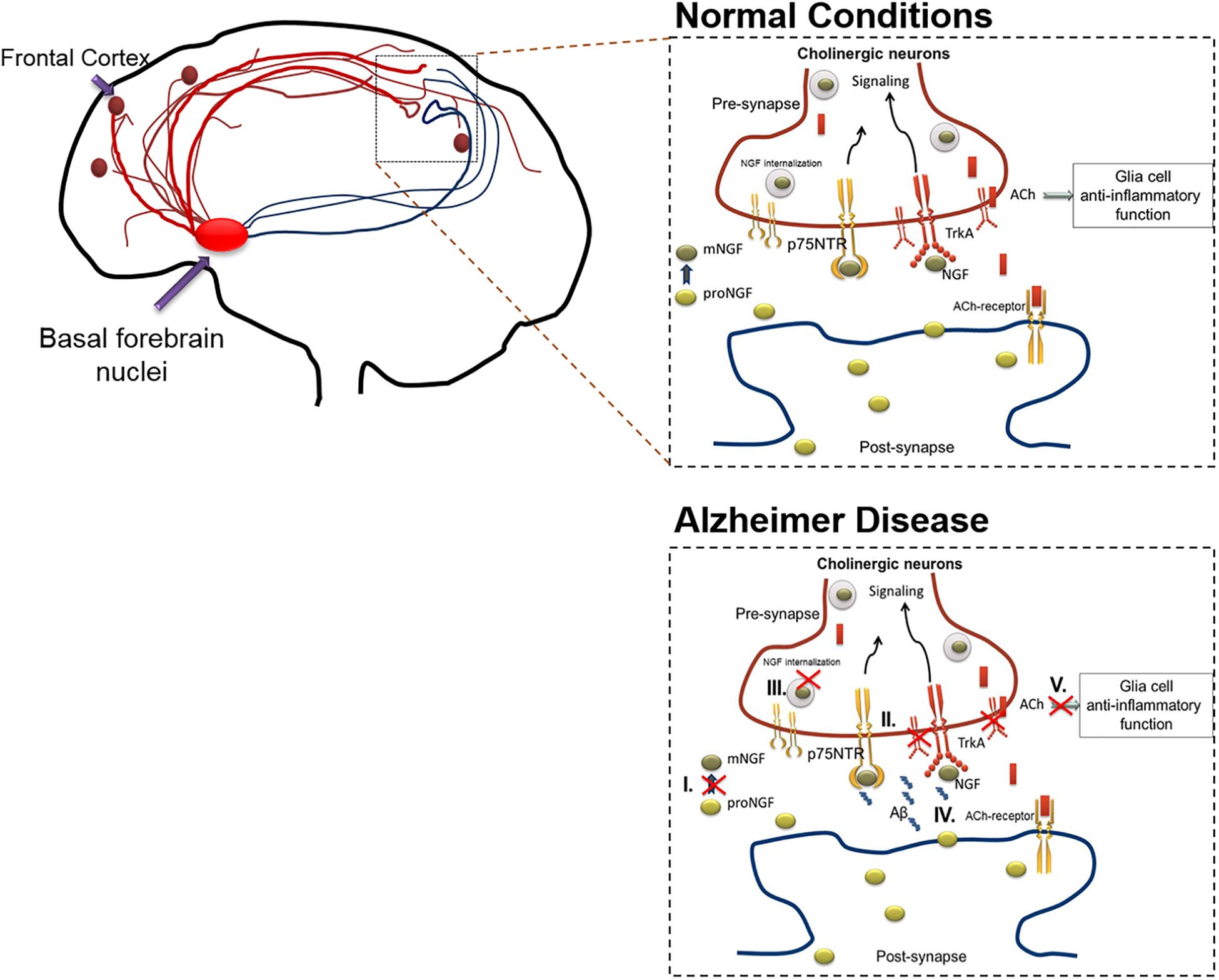
Figure 1. The action of cholinergic basal forebrain neurons in normal and in AD conditions. Cholinergic neurons are known to innervate the cortex and hippocampal regions of the brain. Under normal condition, postsynaptic resident neurons of cortical and hippocampal regions release proNGF, which is processed extracellularly (proNGF to mNGF) and taken up by the presynaptic cholinergic neurons (through TrkA, predominantly). The mNGF is then retrogradely transported within the cholinergic neurons, to the cell nuclei situated in the basal forebrain, to initiate gene expression and signaling cascades. These signaling cascades facilitate the release of acetylcholine from the cholinergic terminals present in cortical or hippocampal regions, thereby initiating cholinergic signaling in these brain regions, which includes anti-inflammatory effects mediated through alpha-7-nicotinic receptors in the glial populations. As mentioned below, this cross-talk is altered during AD in various ways which results in cholinergic degeneration. The figure illustrates that the cholinergic system is affected due to various pathways including the following: (I) Altered NGF maturation, (II) Skewed TrkA/p75 receptor ratio, (III) Inefficient axonal transport and signaling, (IV) Aβ induced modulation of NGF receptors. Apart from inducing death, increased Aβ levels can interact with several NGF receptor types including, alpha-7-nicotinic, and metabotropic receptors, leading to increased inflammation and signaling impairments. (V) Sub-optimal release of acetylcholine leading to glial activation and subsequent inflammatory response. These alterations compromise the availability of mNGF to the basal forebrain cholinergic neurons, which in-turn culminates in hampered cholinergic innervation to cortex and hippocampal regions of the brain. The red crossed out areas represent the affected pathways. Aβ, amyloid β; ACh, acetylcholine; AD, Alzheimer’s disease; NGF, nerve growth factor; TrkA, tyrosine receptor kinase A.
During the initiation and progression of AD pathology, the declining function of cholinergic innervation to cortex and hippocampus occurs due to inefficient maturation of NGF. This is attributed to reduced production and elevated clearance of mNGF (Cuello and Bruno, 2007; Allard et al., 2012) as well as reduction in levels of TrkA-receptors in neurons in nucleus basalis and cortex (Mufson et al., 1997) affecting the retrograde transport of NGF (Mufson et al., 1995). Simultaneous evaluation demonstrated hampered plasmin machinery along with increased MMP9 activity, culminating in elevated levels of proNGF in the brain tissue (Fahnestock et al., 2001; Peng et al., 2004; Fabbro and Seeds, 2009; Mroczko et al., 2013). Higher proNGF levels not only induce pro-apoptotic signaling, but are also found to affect the receptor binding of mNGF and its axonal trafficking and result in retrograde atrophy of cholinergic neurons in the basal forebrain (Sobottka et al., 2008; Ioannou and Fahnestock, 2017; Allard et al., 2018). The apoptotic signaling induced by pro-NGF is dependent on the relative expression of TrkA and p75, and is favored by a decreased ratio of TrkA/p75 that is also evident in AD (Counts et al., 2004; Ginsberg et al., 2006; Masoudi et al., 2009). These alterations have been shown in AD-like mice models (Tiveron et al., 2013). Interestingly, using the AD11 mice model that expresses anti-mNGF antibodies specifically in the brain, it was observed that lack of mNGF leads to early inflammation and Alzheimer’s neurodegeneration (Capsoni et al., 2011). NGF was found to have a direct role in modulating microglial cells toward a non-inflammatory phenotype (Rizzi et al., 2018).
In this regard, a significant cross talk between NGF and the Aβ generation machinery is present where Aβ is reported to hamper NGF maturation and NGF in turn can reduce Aβ generation, while plasmin is known to degrade Aβ (Tucker et al., 2000; Bruno et al., 2009; Yang et al., 2014). Similarly, NGF can affect APP phosphorylation levels thereby dictating Aβ generation whereas APP can modulate NGF receptor trafficking and axonal transport thereby affecting NGF downstream signaling (Zhang et al., 2013; Triaca and Calissano, 2016; Xu et al., 2016). It has been speculated that during aging, alteration in NGF maturation and signaling pathways can lead to increased Aβ generation and reduced clearance (Gutierrez-Fernandez et al., 2007; Matrone et al., 2008; Triaca and Calissano, 2016; Canu et al., 2017). Apart from inducing death, increased Aβ levels can interact with several receptor types including p75 (Yaar et al., 1997; Chakravarthy et al., 2010; May et al., 2017), alpha-7-nicotinic (Khan et al., 2010; Ni et al., 2013; Liu et al., 2017), and metabotropic receptors (Joseph and Fisher, 2003; Janickova et al., 2013), leading to increased inflammation and signaling impairments (Conejero-Goldberg et al., 2008; Mura et al., 2012; Ardura-Fabregat et al., 2017). Aβ can also impair choline re-uptake from the synaptic cleft thereby affecting ACh production in presynaptic neurons and alter ACh release leading to attentional deficits (Fu and Liu, 1997; Parikh et al., 2014). Since ACh modulates the cholinergic anti-inflammatory pathways in the glial population, hampered ACh release ensues initiation of inflammation (Wang et al., 2003; de Jonge and Ulloa, 2007; Treinin et al., 2017; Bagdas et al., 2018). Thus, it is important that mNGF levels are maintained to affect cholinergic signaling which in turn is responsible to maintain an anti-inflammatory tone in the neurological system (Pavlov et al., 2003).
In line with these findings obtained from the last three decades, NGF has been targeted to modify the associated signaling pathways and envisaged as a therapeutic strategy in AD.
Therapeutic Interventions of NGF in AD
Various Methods of NGF Delivery and Their Outcome
As a therapeutic factor, NGF has been utilized in clinical trials of varied diseases including peripheral neuropathies, human immunodeficiency virus infections, e.g., HIV and central nervous system’s (CNS) diseases like AD (Aloe et al., 2012). One of the most clinically relevant characteristic of the small neurotrophic factors like NGF is that this protein does not normally pass through the blood–brain barrier (BBB) (Liu and Chen, 2005; Wu, 2005). Furthermore, it is known that a direct delivery of neurotrophic factors can have serious peripheral side effects (Thoenen and Sendtner, 2002). These characteristics have resulted in shortcomings of systemic delivery of neurotrophic factors such as NGF in clinical trials for AD and neurological disorders.
Direct Intra-Cerebral Infusion of NGF
As mentioned above, the essential problem for many molecules and compounds is insufficient diffusion across the BBB. Therefore, it becomes imperative for these molecules, or other therapeutic targets, to be delivered locally into the brain. One of the possible routes of local delivery methods is the local cerebral injection. The challenge of this method is to find effective and compatible doses of physiologically related amounts/volumes of every individual compound, which are needed to be tested. Additionally, the infusion may not remain in the target location long enough to elicit an effect, revealing another disadvantage of this method of drug delivery. Nevertheless, the local delivery of many compounds into the brain might be the only way to generate therapy for many neurodegenerative disorders.
Researchers and companies have developed the intra-cerebral infusion platform and the devices are commercially available for many research applications. The ALZET osmotic pumps are small infusion pumps that ensure constant delivery of compounds or proteins into the brain regions of interest. The pump is connected to a metal catheter, with a plastic platform that can be customized for targeting different brain regions or be adjusted in size for different animal models. The pumps exist in different sizes suitable for both animal and human CNS and can be utilized from a few hours to 28 days. The disadvantage of the pump infusion is its short duration of compound delivery when considered for a clinical setting. To minimize the risk for infection, the pump can be placed under the skin. At the end of the infusion period, the test reagent can be retrieved from the pump to determine the remaining biological activity. Intra-cerebral infusion has been a challenging route which has been perused in clinical trials involving AD patients (Hagg, 2007). As mentioned previously, AD patients show extensive cholinergic neuron loss that may be sensitive to the levels of mature NGF. Several experimental studies in aging-related animal models have found that NGF treatment improved cognitive ability in those aged animals showing cholinergic loss and dysfunction (Fischer et al., 1987; Hefti et al., 1989).
Our research group has previously performed a small clinical study in three AD patients infusing mouse 2.5S NGF into the cerebral lateral ventricle (Olson et al., 1992; Eriksdotter Jonhagen et al., 1998). The results from this study showed an increase in nicotinic receptors expression and regional cerebral blood flow in the neocortex following NGF-infusion, as analyzed by positron emission tomography (PET) imaging. This effect was maintained for several months until the NGF infusion was stopped. However, due to induced neuropathic pain and weight loss, the intracerebroventricular NGF infusion was terminated prematurely (Eriksdotter Jonhagen et al., 1998). When we used NGF infusion into the cerebral lateral ventricle, we were aiming to influence the entire area of cholinergic terminals. The lateral ventricles are the two largest cavities of the ventricular system of the human brain containing cerebrospinal fluid (CSF). Consequently, the CSF flows directly from the ventricles into the brain tissue and passes through the spaces between the cells where it eventually enters the subarachnoid space. Taking advantage of this normal feature that occurs in the brain, it is believed that the NGF infusion into the brain can be absorbed by tissues in the brain regions like cerebral cortex (Greitz and Hannerz, 1996). This feature provides a direct administration of NGF into the lateral-ventricle-containing CSF to reach the cortical sites where the improved function of cholinergic neurons is essential to counter cognitive decline.
However, the direct intracerebroventricular NGF administration caused pain in animals by affecting the dorsal root ganglion and hypothalamus function that is correlated to the pain response, and showed weight loss among treated animals (McKelvey et al., 2013). NGF delivery also indirectly affected the release of pain mediators including histamine, and prostaglandins from mast cells (Kawamoto et al., 2002). Interestingly these cells are capable to release NGF that might form a feedback loop that sensitizes adjacent nociceptive neurons (Kawamoto et al., 2002). In contrast, other animal studies have shown that when NGF is infused directly into the rat brain parenchyma (Hao et al., 2000) and primates (Tuszynski et al., 1990) no pain-related effects were observed.
Peripheral Administration of NGF Using Nasal or Intraocular Delivery
Intranasal delivery (IND) method has overcome the obstacle for systemic delivery of both oral and parenteral drug routes into the brain. The nasal delivery of drugs provides a rapid absorption and delivers the drug directly into the brain via the olfactory route. The advantage of IND is the olfactory pathway which provides a non-invasive route for drug delivery directly to the brain, but does not protect from adverse effects. This approach has been utilized to deliver NGF into rodent brain (Cattaneo et al., 2008). The efficiency of intranasal administration was demonstrated in the AD11 anti-NGF transgenic mice model, showing reversion of the cognitive loss in AD11 mice (De Rosa et al., 2005). NGF eye drops using the ocular surface route has also been used to affect cholinergic markers in the basal forebrain of rats (Lambiase et al., 2007). Furthermore, ocular NGF has been able to activate the proliferation of cells in the sub-ventricular zone by showing increased layers of ki67 positive cells (marker for proliferative cells) which also were expressing p75NTR (Tirassa, 2011). However, this method has not been used in well-established animal models of AD. The intraocular delivery of NGF appears to be less characterized compared to the IND, particularly in terms of mechanisms and anatomical delivery to the brain (Dhuria et al., 2010). There has been studies utilizing the IND of mutated human NGF (R100) in an AD animal model (Capsoni et al., 2011, 2012), where the mutated human NGF retained the potential of NGF’s neurotrophic effect without eliciting the pain-related response. Surprisingly, such a test has not yet been validated in preclinical studies for neurodegenerative diseases (Capsoni et al., 2012). There have been various strategies for the delivery of any drug via IND such as nanotechnology-enabled drug delivery (e.g., lipid nanoparticles) for many diseases like cancer, technology of microencapsulation and the use of mucoadhesive materials and devices. However, despite the improvement and development of these systems, very few IND systems targeting AD have yet shown sufficient clinical relevance.
Stem Cell-Mediated Delivery of Therapeutics
The use of stem cells for targeting tissues or organs of interest has received much attention as carriers for therapeutic agents. The definition of these cells is based on their capability of self-renewing and differentiation into specialized progeny according to their degree of potency (Mimeault et al., 2007). Stem cells can deliver neurotrophins into diseased brain, modulate endogenous synaptic plasticity, and enhance neuronal survival. High levels of neurotrophins such as brain derived neurotrophic factor (BDNF) and NGF are expressed in neural stem cells (NSCs) (Joseph and Fisher, 2003; Kamei et al., 2007; Dhuria et al., 2010). Interestingly, increased synaptic density in hippocampus, reversal of memory and neuronal loss in transgenic AD animal models after NSCs transplantation have been shown (Joseph and Fisher, 2003; Bentz et al., 2007; Kamei et al., 2007; Yamasaki et al., 2007; Blurton-Jones et al., 2009; Kim et al., 2010).
In spite of the potential clinical applications using stem cells, there is a need to understand the pathological changes in the brain of neurodegenerative disorders that might affect the transplanted stem cells. This method should be combined with the control and observation of migratory patterns of transplanted stem cells in animal models prior to clinical trials on humans. In order to use stem cells as a replacement strategy in AD, several issues have to be addressed. By in vivo and ex vivo studies, the migration of stem cells in different brain regions and areas should be tested. Detailed knowledge of the migration, differentiation and maturation of stem cells into various neuronal subtypes is needed. These neurons would then have to re-innervate the correct target and establish neuronal connections mimicking the normal brain circuitry. Because of the safety issues, the protocols for pre-clinical experiments should be carefully controlled, standardized and undergo extensive evaluation before initiation of clinical studies. Inflammation can cause and change the pathological environment in the brain, therefore there is a possibility that transplantation of stem cells may alter the inflammatory responses in the brain. A study by Lee et al. (2012), showed an influence on inflammatory response and pathogenesis in AD animal models, when they used NSCs and mesenchymal stem cells (MSCs) as a therapeutic choice. Therefore, studies are needed to understand the mechanisms involved in direct or indirect effects of stem cell transplantation in altering the inflammation caused by tissue injury or any kind of xenotransplantation. Studies of stem cell transplantation in immune-incompetent AD models would be interesting in order to elucidate this important question (Chen and Blurton-Jones, 2012). Another benefit for AD would be the NSCs mediating delivery of enzymes such as neprilysin to degrade Aβ (de Backer et al., 2010).
Survival and differentiation of NSCs may be influenced by immune responses and the pathology of the disease may affect the efficacy of stem cell mediated therapy. Thus, further studies are needed to show if AD-associated pathology can be involved in NSC survival and differentiation. Neuronal replacement has hitherto not been clinically successful for neurodegenerative disorders like AD (Chen and Blurton-Jones, 2012). Nevertheless, the positive outcome of patient-derived induced pluripotent stem cells (iPSCs) as a model of human genetic disorders (Grskovic et al., 2011), and reprogramming of the induced NSCs (iNSCs) from AD patients can be useful for such purposes. Two different reports presented the first steps of AD iPSCs as a potential route of AD therapy (Yagi et al., 2011; Israel et al., 2012). Collectively these data suggest that stem cell mediated therapy in AD could be beneficial, and further investigations on embryonic, neural and iPSCs will contribute a basis for a future therapeutic approach for AD.
NGF Delivery Using Viral Vectors
Since the cholinergic system of the human brain is involved in memory function, and its loss is associated with cognitive decline, local NGF delivery directly to the cholinergic basal forebrain would be preferred. However, it poses a clinical and technical challenge.
The essential core of regenerative medicine revolves around cell therapy. In association with cell therapy utilization, viral vector-mediated gene transfer techniques, in particular those techniques developed for lentiviruses, have demonstrated some useful features. Hohsfield et al. (2013), demonstrated that infection by a lentiviral vector, which overexpressed NGF, showed successful production of effective NGF secretion. In parallel with these findings, lentivirus NGF gene delivery to the cholinergic basal forebrain for 1-year in aged monkeys showed no systemic leakage of NGF or formation of anti-NGF antibodies, nor activation of inflammatory markers in the brain or pain or weight loss (Nagahara et al., 2009a).
The first study using gene therapy in patients with AD was published in 2005 (Tuszynski et al., 2005). In this study, NGF gene delivery was performed to individuals with a mild AD diagnosis where the transfer of the NGF gene through genetically manipulated autologous fibroblasts was implanted into the basal forebrain. Data from this 2-year, open-label study, showed a significant increase of glucose uptake in several cortical regions and cognitive tests showed scores that were stabilized or declined at a slower rate than expected (Tuszynski et al., 2005, 2015). Also adeno-associated virus (AAV)-based gene delivery has been used in the brain of AD patients for the delivery of NGF in order to treat AD symptoms and progression (Rafii et al., 2014). A phase II clinical trial using AAV vectors expressing human NGF was recently published. This study included 49 AD patients randomly assigned to receive intracerebral injections of AAV-NGF or sham surgery. AAV-NGF delivery was well-tolerated over 2 years but with no effects on clinical outcomes. However, confirmation of accurate gene targeting by pathology is needed (Rafii et al., 2018).
There are certain limitations with in vivo gene therapy, particularly the permanent genetic modification of the patient’s brain cells and the obvious inability to control and stop the targeted release. However, with the development of self-inactivating viral vectors (SIV’s), the expression of transgenes can be delivered with minimally invasive surgery and can also be switched off (Kraunus et al., 2004; Schambach et al., 2007; Thornhill et al., 2008). Conversely, considering current technological platforms, using SIVs for long-term delivery of therapeutic proteins (for months or years) would still be a challenge, which is otherwise desirable for treating neurodegenerative disorders.
Small Molecule Modulators of NGF and Its Receptors
Apart from utilizing the full-length NGF molecule as a therapeutic agent, several chemical compounds as well as peptides are exploited and developed which are termed NGF mimetics. These compounds/peptides resemble NGF epitopes and can interact with the NGF receptors to induce downstream signaling pathways or may also act as antagonists, thereby modulating neurotrophic signaling. Nevertheless, before using these kind of analogs as neurotrophic therapeutic molecules, similar varieties were initially developed to understand NGF and TrkA interaction, and also to detect and diagnose oncogenic transformation in vivo where TrkA levels were found to be overexpressed (Longo et al., 1990; LeSauteur et al., 1996). Toward the end of the 20th century, several groups reported the development of several classes of neurotrophin mimetics targeting different receptor types including TrkA modulators, p75 modulators and BDNF (LeSauteur et al., 1995; Spiegel et al., 1995; Estenne-Bouhtou et al., 1996; Longo et al., 1997; O’Leary and Hughes, 1998).
With the advent of time, several specific small molecule modulators have been developed which can selectively target specific neurotrophin receptors as shown in Table 2. Dr. Longo and his team have received success utilizing a small molecule mimetic of NGF targeting the p75 receptor, termed LM11A-31, showing reduced microglial activation after treatment in AD-like mice (James et al., 2017). The first clinical trial using small molecule mimetics with LM11A-31 is currently underway for treatment of mild to moderate AD patients (ClinicalTrials.gov Identifier: NCT03069014).
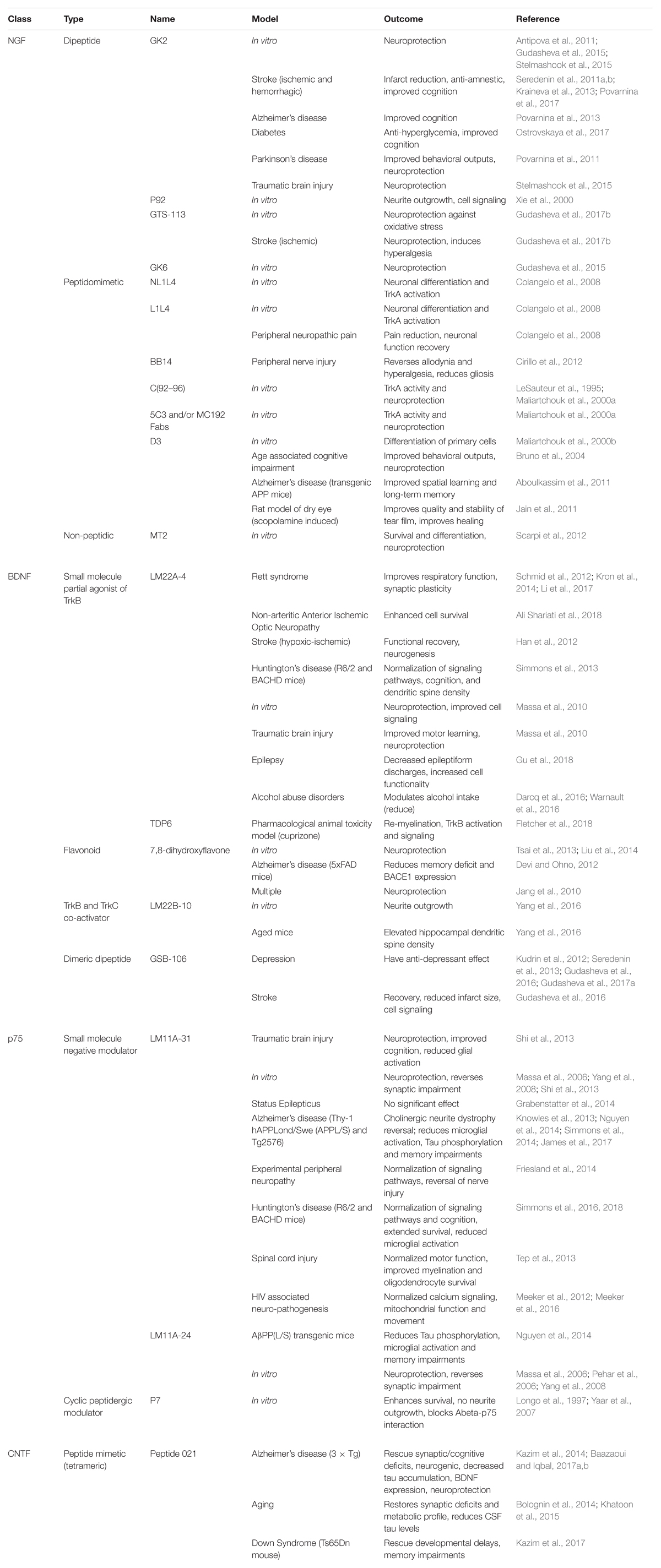
Table 2. An up-to-date comprehensive report on the type and class of different small molecule modulators evaluated so far, targeting various neurotrophin receptors in different in vitro and in vivo models.
Cell Mediated NGF Delivery
An alternative and reliable approach to deliver therapeutic molecules directly to the brain region of interest is the encapsulated cell biodelivery (ECB) system (Figure 2).
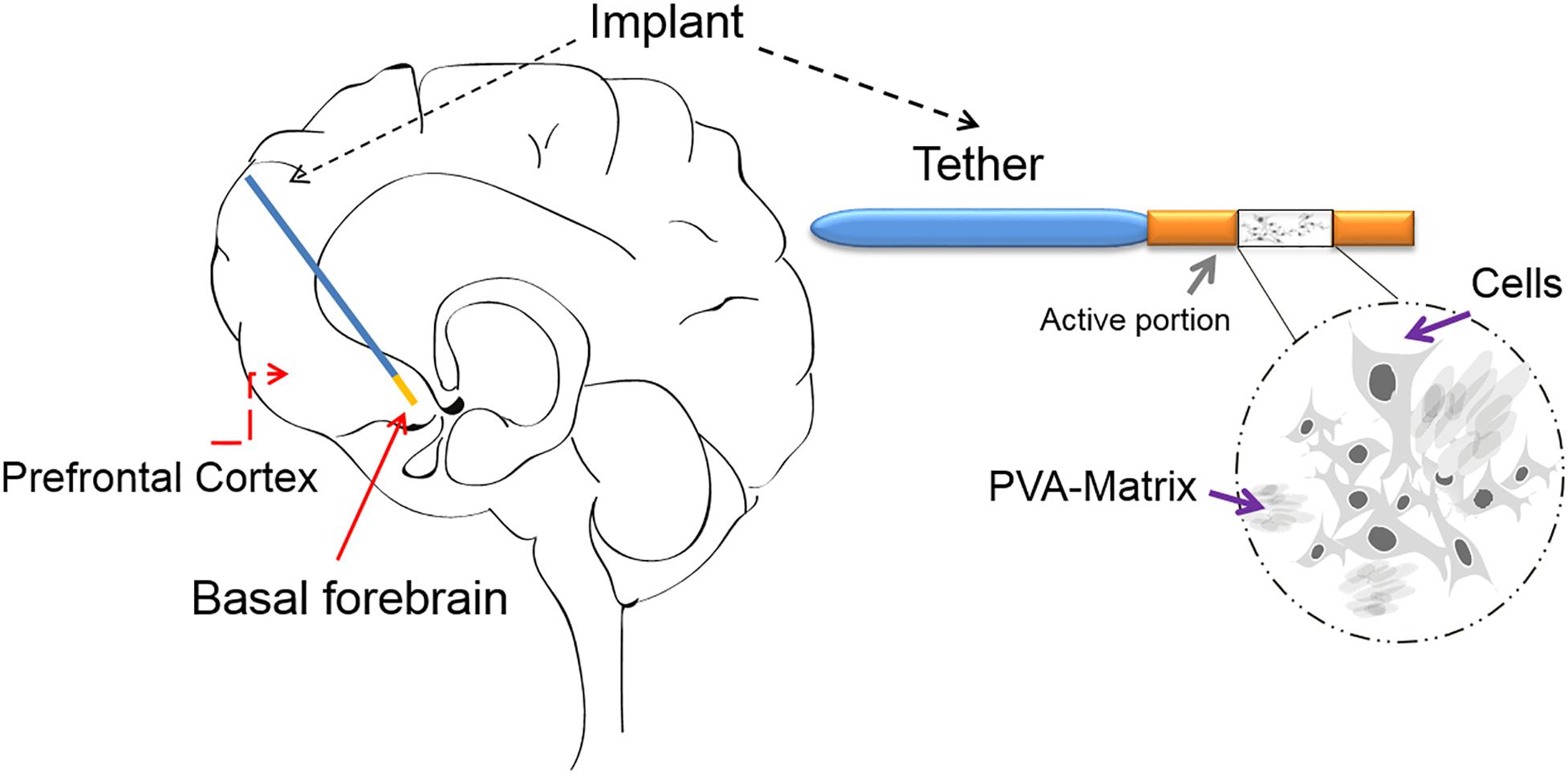
Figure 2. The ECB device: The ECB consists of a catheter-like device, which has approximately an 11 mm-long active portion and an outer diameter of 0.72 mm. In the active portion, the genetically modified human cell line (ARPE-19) is depicted growing on the spongy polyvinyl alcohol (PVA) cylindrical scaffold/matrix (a sagittal view is shown), within the semi-permeable polyethersulfone hollow fiber membrane, which allows for the influx of nutrients and the efflux of mature NGF. The membrane is in-turn linked to an inert polyurethane tether, which at its other end is finally attached to the edge of the burr hole. The device can be implanted with precision using a custom-made frame adapter, fitting a standard stereotactic frame. The schematic drawing of an implanted ECB device shows the active portion in orange and the tether in blue.
Lim and Sun (1980) achieved one of the earliest attempts to develop ECB in 1980, utilizing cross-linked alginate membranes while trying to deliver islets in rats. Subsequent studies employed ECB biodelivery of various neurotrophins including NGF, CNTF, and other sources including dopamine releasing cells and chromaffin cells (Emerich et al., 1994; Aebischer et al., 1996; Buchser et al., 1996; Winn et al., 1996; Lindner and Emerich, 1998). In the forthcoming years, several groups attempted to deliver a variety of neurotrophins like glial cell line-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF), NGF, BDNF, meteorin, galanin, and cometin. To achieve this, various encapsulation materials like alginate, poly (acrylonitrile-co-vinyl chloride) copolymer (PAN-PVC), alginate-poly-L-ornithine-alginate, poly(acrylonitrile), poly(sulfone), poly(ether sulfone) (PES), poly(amides), poly(propylene), and poly(ethylene), hydrogel, nano-carriers and agarose/poly(styrene sulfonic acid) mixed gel were employed (Orive et al., 2004, 2015; Zanin et al., 2012; Angelova et al., 2013; Emerich et al., 2014; Govender et al., 2017). More recently, 3D printing technologies are being used to construct advanced, highly defined microstructures for drug delivery (Son et al., 2017).
The ECB technology aims to integrate the therapeutic potency of gene therapy along with the precision of a safe retrievable device (Lindvall and Wahlberg, 2008). The capsule comprises a biocompatible outer membrane and an inner core comprising cells, which are engineered to secrete therapeutics, for example a biologically active compound or coding for a polypeptide or a siRNA (Lindvall and Wahlberg, 2008). The modified cells are ensheathed inside a semi-permeable polymer based capsule, which is biologically inert, thereby not eliciting xenograft responses from the surrounding cells. These retractable capsules can be implanted into specific brain locations and can be retracted later to avoid complications (if needed). The semi-permeable membrane protects the engineered cells from host immune system while simultaneously allowing bi-directional free exchange of nutrients, oxygen and therapeutic products by diffusion across the capsule walls. The advantage of this method is based on the possibility of the semi-permeable capsule to deliver therapeutic entities in a local and controlled fashion, which cannot be administered orally or intravenously. Furthermore, the therapeutic molecule can be delivered at the desired site with high concentration thereby minimizing off-target side effects and complications. The semipermeable membrane functions for; (1) the efflux of the therapeutic protein, (2) protection of cells for immunogenic reactions, and (3) influx of nutrients from the host brain (Winn et al., 1996; Lindvall and Wahlberg, 2008). Therefore, this approach tackles previous drawbacks in NGF brain delivery (BBB passage, local delivery).
Clinical Trials Using ECB-NGF Delivery and Initial Results in AD
Recently, the ECB platform was developed by NsGene Inc., with the intention to administer mature human NGF into the basal forebrain of AD patients, in an attempt to rescue basal forebrain cholinergic neurons from degeneration.
The first generation of device or implant, which is 11 mm in length, carries a genetically modified human retinal pigment epithelial cell line (ARPE-19) growing within the spongy polyvinyl alcohol scaffold, which is present inside the polyethersulphone hollow fiber membrane (Winn et al., 1996; Lindvall and Wahlberg, 2008). The semi-permeable membrane of the implant allows the influx of nutrients and the efflux of mNGF. Preclinical research from this study demonstrated that devices that were implanted in the basal forebrain of minipigs over a period up to 12 months, were retrieved without observing any associated complications (Fjord-Larsen et al., 2010). Implants showed to be intact and contained viable cells after explantation, whereas tissue in which the implant was localized showed increased levels of NGF (Fjord-Larsen et al., 2010). As compared to previous ex vivo and in vivo gene therapy studies, the ECB-NGF implant could be removed easily and thereby the treatment could be controlled or stopped whenever required.
Previously, our group utilized the ECB device in a pilot study involving ten AD patients. The study was performed in two parts in which the ECB device with NGF-producing cells were implanted into the basal forebrain of the AD patients. In the first part, six AD patients received the first generation implant having polyvinylalcohol foam scaffolding for 12 months (Eriksdotter-Jonhagen et al., 2012; Wahlberg et al., 2012). Analyses revealed that the result of this study was promising and therefore the study was continued to a second part using second generation of improved NGF releasing device containing polyethylene terephthalate (PET) yarn in additional 4 AD patients for 6 months of implantation (Eyjolfsdottir et al., 2016). The second part was carried out with improved NGF-secreting ARPE-19 cells which were transfected using the sleeping beauty transposon technology (Fjord-Larsen et al., 2010; Izsvak et al., 2010). The PET yarn improved cell viability and NGF release while also making the device manufacturing process more reproducible. In both part I and II, the devices were successfully removed after 12 and 6 months, respectively.
The outcome from both studies showed that the patients’ responses to the NGF-treatment varied and approximately half of the patients responded to the ECB-NGF-treatment with increased cholinergic markers (e.g., ChAT activity) in CSF correlating to improved cognition and brain glucose metabolism (Karami et al., 2015), less brain atrophy (Ferreira et al., 2015) and normalization of the EEG-pattern (unpublished data). In the second patient cohort in part II, we demonstrated safety and tolerability of the dose escalation from the second generation of ECB-NGF device and showed good cell survival and sustained NGF release from the encapsulated cells after explantation (Eyjolfsdottir et al., 2016).
Despite the encouraging results from ECB-NGF therapeutic approach, variations in the levels of NGF-release between capsules were observed. Furthermore, after testing for cell survival following capsule retrieval from AD patients, a few implants showed degenerating cells. Therefore, one of the challenges using this method is to understand the possible factors among all recipients that can affect secretion and survival of the encapsulated cells. Furthermore, in order to enable such approach, it is necessary to streamline and standardize the procedures. A careful characterization of the transplanted genetically engineered cells and of the quality and quantity of its produced therapeutic molecule is critical. To further develop the therapeutic platform, multifunctional approaches is needed to achieve optimal efficacy of the NGF-producing cells by investigating what factors are involved in influencing NGF-production.
Also the surgical procedure in association with the ECB implantation into the brain may cause small tissue injuries such as damaging small blood vessels, neurons and glial cells and the BBB (Hovens et al., 2012). The injured tissues surrounding the capsules might activate microglia and astrocytes which may respond by releasing various cytokines and free radicals (Feng et al., 2017; Xu et al., 2017), which in-turn can diffuse through the capsule membrane and affect the NGF-producing cell survival and NGF-release, respectively. Therefore, to some extent, the insertion of the ECB-NGF in the basal forebrain of the AD patients may lead to disruption of the normal brain environment. To test this hypothesis, the NGF-producing cells were examined by our group and was found to be negatively affected by IL-1β in a dose dependent manner suggesting that inflammation has a negative effect on the ECB cells (Eriksdotter et al., 2018). Moreover, Aβ peptides represent an obvious factor that might affect survival of the NGF-producing cells. Interestingly, at physiological concentrations, neither Aβ40 nor Aβ42 had any major impact on the NGF-release nor on the cell survival (Eriksdotter et al., 2018). To further investigate factors that could affect NGF-release or cell survival, exposure of NGF-producing cells to CSF from AD patients had a significantly reduced effect on NGF-release as compared to CSF from non-demented patients with subjective cognitive complaints (Eriksdotter et al., 2018).
Hence, the functionality of the ECB-NGF can be vulnerable to released mediators from activated glial cells, modulating the efficiency of the encapsulated NGF-releasing cells during longer duration of having devices in the brain. The variation of the patient’s clinical data, treatments and history of the diseases, could provide a milieu where the encapsulated cells might get affected. Therefore, disease conditions of the human brain play an important role in the function of encapsulated cells.
Future of ECB-Neurotrophic Factor Therapy With NGF and BDNF
The establishment of ECB-NGF delivery provides a good model for having another cell-mediated therapy of BDNF for AD and other neurodegenerative disorders. Neurotrophins, both NGF and BDNF, improve cognition and hippocampal neuron viability and are reported to be reduced in AD brain.
The hippocampus represents a region, which remains neurogenic in the adult and recent data suggest that as many as 1,500 neurons are born in the hippocampus every day (Spalding et al., 2013). Neurotrophic signaling, and in particular BDNF plays a pivotal role in hippocampal neurogenesis, synaptic plasticity, and long-term potentiation, a proposed mechanism underlying memory formation at the level of the synapse (Acheson et al., 1995; Huang and Reichardt, 2001) In addition, BDNF show important modulatory effects at the synaptic level (Simonato et al., 2006). A study by Bolton et al. (2000) demonstrated selective different regulation mechanisms by BDNF at excitatory and inhibitory synapses. Moreover, various function of BDNF, neuronal and glial development and control of short- and long-lasting synaptic interactions that influence memory and cognition are included (Sasi et al., 2017; Kowianski et al., 2018).
Several studies have shown a decrease in BDNF in the hippocampus and in CSF during AD pathogenesis, suggesting that decreased BDNF signaling may contribute to the progression of hippocampal dysfunction in AD (Allen et al., 2013). Supporting this hypothesis, direct administration of BDNF, viral overexpression of BDNF or BDNF secreted from grafted stem cells improve age related cognitive decline and also rescue cognitive decline in a preclinical mouse models developing Aβ and tau pathologies (Nagahara et al., 2009b). The latter findings were observed in absence of any changes in Aβ or tau pathology in the brain. We have recently shown an increase in CSF BDNF in mild cognitive impairment patients as compared to AD patients suggestive of a complex relationship involving preclinical and clinical stages of the disease (Hjorth et al., 2013). Collectively, these data suggest that novel therapeutic strategies aimed at stimulating key neurotrophic signaling pathways or simultaneously ameliorating the toxic insult and stimulating neurodegeneration have the potential to deliver an efficient therapy to the patient. A possibility to explore feasibility of direct CNS delivery of the BDNF (in hippocampus) is the ECB technique. However, the question remains if there is a possibility to administer not only NGF but also BDNF using encapsulated technique for AD patients. First, experiments in animal models are needed to test this hypothesis.
In the ECB-NGF clinical trials, all AD patients were also on concomitant stable long-term ChEI-treatment. Hence, combinatorial treatment with neurotrophins delivery such as NGF and BDNF along with ChEIs in AD patients would be an interesting aspect to be looked upon in future (Mehta et al., 2012; Hampel et al., 2019).
The ECB-technique may be of interest for other brain diseases besides neurodegenerative disorders. In a very recent study, BDNF has been suggested as a therapeutic for chronic epilepsy (Iughetti et al., 2018). However, the delivery of BDNF for chronic epilepsy has met comparable difficulties since like NGF; BDNF does not cross the BBB. Therefore, similar to the ECB-NGF devices used in AD clinical trials, this approach was utilized to develop devices containing genetically engineered human cells lines to release BDNF. The purpose was to gain beneficial and efficiency effects of encapsulated ECB-BDNF in chronic epilepsy treatment (Falcicchia et al., 2018). The first ECB-BDNF delivery has recently been tested in a rat model, where after insertion of the ECB-BDNF device into the hippocampus of pilocarpine-treated rats, the frequency of spontaneous seizures was decreased. Additionally, an association with improved cognitive performance was shown. Importantly, behavioral evaluation of treated rats after long-term of BDNF delivery demonstrated normal behaviors such as general activity or sleep/wake patterns. Further, quantification and evaluation of histological analyses revealed neurological improvements related to BDNF-treatment in rats together with several anatomical changes, neuronal survival, and normalization of hippocampal volume (Falcicchia et al., 2018).
ECB of Other Factors in Neurodegenerative Disorders
Besides BDNF and NGF, there are other neurotrophic factors, which can nourish, support and promote the survival and regeneration of the neurons. Among them CNTF, GDNF and neurturin have been extensively studied.
Parkinson’s disease (PD) is a neurodegenerative disorder that affects predominantly dopamine-producing (“dopaminergic”) neurons in substantia nigra. GDNF is the most efficient protective factor that promotes the survival and differentiation of dopaminergic neurons in experimental models of PD (Lin et al., 1993). Based on animal experiments and human studies, there is a need for safe, sustained and localized long-term delivery of GDNF into the basal ganglia of patients with PD (Emerich et al., 2014). Early rodent studies using encapsulated fibroblast cells and GDNF-secreting cells derived from retinal pigment epithelial cells in 6 months in rodents showed an enhanced dopaminergic function in animal models of PD (Lindner and Emerich, 1998). Compared to the practical, societal and ethical issues that have been associated with other transplantation approaches this method appeared to be promising (Sajadi et al., 2006; Huang et al., 2014; Garbayo et al., 2016). Even now, after two decades with collecting detailed knowledge of the approach of using GDNF as a therapeutic agent for PD, the main factors determining GDNF production remains obscure and the question whether GDNF can effectively be used as a therapeutic agent for PD remains unclear (d’Anglemont de Tassigny et al., 2015).
Huntington’s disease (HD) is an inherited disease that causes a progressive degeneration of nerve cells in the basal ganglia. One of the most striking physiological characteristics of HD is the loss of neurons in striatum, a component of the basal ganglia system that organizes motor movement and cognition. In recent years, there has been considerable research on neurotrophic factors and their effects on HD. Among the neurotrophic factors, CNTF has been studied in different models of HD as well as in non-human primates (Emerich et al., 1997a,b; Mittoux et al., 2000). ECB-CNTF has also been tested in one clinical trial (ClinicalTrials.gov Identifier: NCT01949324). However, in contrast to partly positive results from other neurodegenerative disorders that used ECB implants as a therapeutic approach, the clinical trial with CNTF-secreting encapsulated cells did not improve clinical symptoms. Investigation of the capsules and histopathological studies revealed that the treatment was not effective, suggesting poor distribution of CNTF from the ventricles and degeneration of the encapsulated cells (Bachoud-Levi et al., 2000; Bloch et al., 2004).
The encapsulated biodelivery technique has also been used to harbor cells that produce meteorin in animal studies, where the ECB was implanted into the rodent striatum using an animal model for HD (Tornoe et al., 2012). Meteorin is widely expressed in CNS. It is involved in glial cell differentiation and promotes axonal extension in small and intermediate neurons of basal ganglia nuclei whose degeneration is a hallmark of HD (Nishino et al., 2004). After treatment with ECB-meteorin for 4 weeks, the animals showed almost normal neurological performance and neuronal death in striatum was reduced. These results demonstrate new possibilities to further explore in neurological diseases with an excitotoxic component, such as HD (Tornoe et al., 2012). Another avenue to further investigate is the use of small molecules that can activate TrkA or p75-receptors to inhibit degeneration of neurons affected in HD (Simmons et al., 2017), as previously reported in AD (Simmons et al., 2014).
Since tau pathology correlates well with disease severity in AD and develops within the brain in anatomical connected regions, development of similar strategies using ECB is of interest. The spread of tau pathology has been suggested to occur in a prion-like manner from region to region (Jucker and Walker, 2013) suggesting that the extracellular pool of tau plays an important role in the progression of tauopathy in the brain. Indeed, high levels of anti-tau antibodies, administered intra-ventricularly via minipumps, have shown a pronounced effect on tau pathology development in an AD-like animal model, suggesting that selective targeting of extracellular tau in the brain has an impact on tau pathology development (Yanamandra et al., 2017). Proteases such as high temperature requirement A (HtrA) are involved in the protein quality control and known to degrade protein aggregations. In a recent report, human HTRA1 was shown to degrade aggregated and fibrillar tau, another pathological hallmark in AD (Poepsel et al., 2015). Interestingly, cell culture and biochemical experiments have revealed that HTRA1 can cleave both monomeric and aggregated tau. Most importantly, HTRA1 is secreted and active in the extracellular space, making it an example of a suitable candidate as an extracellular tau degrading protease. This feature of HTRA1 can be exploited, expressed and secreted using an EC biodelivery implant, to target extracellular tau.
Conclusion
Alzheimer’s disease is a devastating neurodegenerative disease, and there is currently no effective therapy available. Thus, several proposals to explore radically new concepts for a future therapeutic strategy against AD are needed. The strong supportive preclinical data on both recent clinical data with neurotrophic factor cell therapy in AD patients and in the literature indicate that stimulation of neurotrophic signaling has a therapeutic potential for AD.
With regard to the difficulties of growth factors such as NGF or BDNF that do not significantly penetrate the BBB, the clinical utility requires invasive neurosurgical procedures. Searching literature and clinical trials using neurotrophins suggest multifunctional approaches to achieve optimal efficacy of the NGF/BDNF producing cells targeted to their optimal biologically relevant brain region. However, further development of therapeutic platforms such as the ECB and its efficacy in terms of cognitive outcomes is needed in AD and other neurodegenerative diseases. As an additional option or in combination with the ECB technique, synthetic modulators of NGF or BDNF signaling can be used to stimulate neurotrophic signaling in AD. The advancement of AD pathology includes multiple and concomitant processes. These processes can be partially normalized by the administration of NGF. Therefore, a combined treatment with these neurotrophins would be an attractive therapeutic approach for AD neurodegeneration.
Author Contributions
ME initiated the manuscript and the idea, and finalized the manuscript. All authors contributed with design of manuscript and discussed content. SM and HB drafted the manuscript. All authors evaluated and reviewed the manuscript.
Funding
This review was supported by the Swedish Research Council (2016-02317), Hjärnfonden (The Brain Foundation), Gun and Bertil Stohne Foundation, Foundation for Old Servants, Lindhés Foundation, Åhlens Foundation, KI Foundation for Geriatric Research and the Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aboulkassim, T., Tong, X. K., Tse, Y. C., Wong, T. P., Woo, S. B., Neet, K. E., et al. (2011). Ligand-dependent TrkA activity in brain differentially affects spatial learning and long-term memory. Mol. Pharmacol. 80, 498–508. doi: 10.1124/mol.111.071332
Acheson, A., Conover, J. C., Fandl, J. P., DeChiara, T. M., Russell, M., Thadani, A., et al. (1995). A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 374, 450–453. doi: 10.1038/374450a0
Aebischer, P., Schluep, M., Deglon, N., Joseph, J. M., Hirt, L., Heyd, B., et al. (1996). Intrathecal delivery of CNTF using encapsulated genetically modified xenogeneic cells in amyotrophic lateral sclerosis patients. Nat. Med. 2, 696–699. doi: 10.1038/nm0696-696
Ali Shariati, M., Kumar, V., Yang, T., Chakraborty, C., Barres, B. A., Longo, F. M., et al. (2018). A small molecule TrkB neurotrophin receptor partial agonist as possible treatment for experimental nonarteritic anterior ischemic optic neuropathy. Curr. Eye Res. 43, 1489–1499. doi: 10.1080/02713683.2018.1508726
Allard, S., Jacobs, M. L., Do Carmo, S., and Cuello, A. C. (2018). Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol. Aging 67, 10–20. doi: 10.1016/j.neurobiolaging.2018.03.002
Allard, S., Leon, W. C., Pakavathkumar, P., Bruno, M. A., Ribeiro-da-Silva, A., and Cuello, A. C. (2012). Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J. Neurosci. 32, 2002–2012. doi: 10.1523/JNEUROSCI.1144-11.2012
Allen, S. J., Watson, J. J., Shoemark, D. K., Barua, N. U., and Patel, N. K. (2013). GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 138, 155–175. doi: 10.1016/j.pharmthera.2013.01.004
Aloe, L., Rocco, M. L., Bianchi, P., and Manni, L. (2012). Nerve growth factor: from the early discoveries to the potential clinical use. J. Transl. Med. 10:239. doi: 10.1186/1479-5876-10-239
Al-Shawi, R., Hafner, A., Chun, S., Raza, S., Crutcher, K., Thrasivoulou, C., et al. (2007). ProNGF, sortilin, and age-related neurodegeneration. Ann. N. Y. Acad. Sci. 1119, 208–215. doi: 10.1196/annals.1404.024
Angelova, A., Angelov, B., Drechsler, M., and Lesieur, S. (2013). Neurotrophin delivery using nanotechnology. Drug Discov. Today 18, 1263–1271. doi: 10.1016/j.drudis.2013.07.010
Antipova, T. A., Gudasheva, T. A., and Seredenin, S. B. (2011). In vitro study of neuroprotective properties of GK-2, a new original nerve growth factor mimetic. Bull. Exp. Biol. Med. 150, 607–609. doi: 10.1007/s10517-011-1202-6
Ardura-Fabregat, A., Boddeke, E., Boza-Serrano, A., Brioschi, S., Castro-Gomez, S., Ceyzeriat, K., et al. (2017). Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 31, 1057–1082. doi: 10.1007/s40263-017-0483-3
Baazaoui, N., and Iqbal, K. (2017a). Prevention of amyloid-beta and tau pathologies, associated neurodegeneration, and cognitive deficit by early treatment with a neurotrophic compound. J. Alzheimers Dis. 58, 215–230. doi: 10.3233/JAD-170075
Baazaoui, N., and Iqbal, K. (2017b). Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimers Res. Ther. 9:45. doi: 10.1186/s13195-017-0273-7
Bachoud-Levi, A. C., Deglon, N., Nguyen, J. P., Bloch, J., Bourdet, C., Winkel, L., et al. (2000). Neuroprotective gene therapy for Huntington’s disease using a polymer encapsulated BHK cell line engineered to secrete human CNTF. Hum. Gene Ther. 11, 1723–1729. doi: 10.1089/10430340050111377
Bagdas, D., Gurun, M. S., Flood, P., Papke, R. L., and Damaj, M. I. (2018). New insights on neuronal nicotinic acetylcholine receptors as targets for pain and inflammation: a focus on alpha7 nAChRs. Curr. Neuropharmacol. 16, 415–425. doi: 10.2174/1570159X15666170818102108
Ballinger, E. C., Ananth, M., Talmage, D. A., and Role, L. W. (2016). Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91, 1199–1218. doi: 10.1016/j.neuron.2016.09.006
Bartus, R. T. (2000). On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 163, 495–529. doi: 10.1006/exnr.2000.7397
Bartus, R. T., Dean, R. L. III, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. doi: 10.1126/science.7046051
Bentz, K., Molcanyi, M., Riess, P., Elbers, A., Pohl, E., Sachinidis, A., et al. (2007). Embryonic stem cells produce neurotrophins in response to cerebral tissue extract: cell line-dependent differences. J. Neurosci. Res. 85, 1057–1064. doi: 10.1002/jnr.21219
Biane, J., Conner, J. M., and Tuszynski, M. H. (2014). Nerve growth factor is primarily produced by GABAergic neurons of the adult rat cortex. Front. Cell. Neurosci. 8:220. doi: 10.3389/fncel.2014.00220
Bierer, L. M., Haroutunian, V., Gabriel, S., Knott, P. J., Carlin, L. S., Purohit, D. P., et al. (1995). Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J. Neurochem. 64, 749–760. doi: 10.1046/j.1471-4159.1995.64020749.x
Bloch, J., Bachoud-Levi, A. C., Deglon, N., Lefaucheur, J. P., Winkel, L., Palfi, S., et al. (2004). Neuroprotective gene therapy for Huntington’s disease, using polymer-encapsulated cells engineered to secrete human ciliary neurotrophic factor: results of a phase I study. Hum. Gene Ther. 15, 968–975. doi: 10.1089/hum.2004.15.968
Blurton-Jones, M., Kitazawa, M., Martinez-Coria, H., Castello, N. A., Muller, F. J., Loring, J. F., et al. (2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13594–13599. doi: 10.1073/pnas.0901402106
Bolognin, S., Buffelli, M., Puolivali, J., and Iqbal, K. (2014). Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol. Aging 35, 2134–2146. doi: 10.1016/j.neurobiolaging.2014.02.017
Bolton, M. M., Pittman, A. J., and Lo, D. C. (2000). Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J. Neurosci. 20, 3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000
Bradshaw, R. A., Pundavela, J., Biarc, J., Chalkley, R. J., Burlingame, A. L., and Hondermarck, H. (2015). NGF and ProNGF: regulation of neuronal and neoplastic responses through receptor signaling. Adv. Biol. Regul. 58, 16–27. doi: 10.1016/j.jbior.2014.11.003
Brandon, E. P., Mellott, T., Pizzo, D. P., Coufal, N., D’Amour, K. A., Gobeske, K., et al. (2004). Choline transporter 1 maintains cholinergic function in choline acetyltransferase haploinsufficiency. J. Neurosci. 24, 5459–5466. doi: 10.1523/JNEUROSCI.1106-04.2004
Bruno, M. A., Clarke, P. B., Seltzer, A., Quirion, R., Burgess, K., Cuello, A. C., et al. (2004). Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J. Neurosci. 24, 8009–8018. doi: 10.1523/JNEUROSCI.1508-04.2004
Bruno, M. A., Leon, W. C., Fragoso, G., Mushynski, W. E., Almazan, G., and Cuello, A. C. (2009). Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 857–869. doi: 10.1097/NEN.0b013e3181aed9e6
Buchser, E., Goddard, M., Heyd, B., Joseph, J. M., Favre, J., deTribolet, N., et al. (1996). Immunoisolated xenogeneic chromaffin cell therapy for chronic pain - Initial clinical experience. Anesthesiology 85, 1005–1012. doi: 10.1097/00000542-199611000-00007
Campenot, R. B., and MacInnis, B. L. (2004). Retrograde transport of neurotrophins: fact and function. J. Neurobiol. 58, 217–229. doi: 10.1002/neu.10322
Canu, N., Amadoro, G., Triaca, V., Latina, V., Sposato, V., Corsetti, V., et al. (2017). The intersection of NGF/TrkA signaling and amyloid precursor protein processing in Alzheimer’s disease neuropathology. Int. J. Mol. Sci. 18:E1319. doi: 10.3390/ijms18061319
Capsoni, S., Brandi, R., Arisi, I., D’Onofrio, M., and Cattaneo, A. (2011). A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS Neurol. Disord. Drug Targets 10, 635–647. doi: 10.2174/187152711796235032
Capsoni, S., Marinelli, S., Ceci, M., Vignone, D., Amato, G., Malerba, F., et al. (2012). Intranasal “painless” human Nerve Growth Factor [corrected] slows amyloid neurodegeneration and prevents memory deficits in App X PS1 mice. PLoS One 7:e37555. doi: 10.1371/journal.pone.0037555
Cattaneo, A., Capsoni, S., and Paoletti, F. (2008). Towards non invasive nerve growth factor therapies for Alzheimer’s disease. J. Alzheimers Dis. 15, 255–283. doi: 10.3233/JAD-2008-15210
Chakravarthy, B., Gaudet, C., Menard, M., Atkinson, T., Brown, L., Laferla, F. M., et al. (2010). Amyloid-beta peptides stimulate the expression of the p75(NTR) neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J. Alzheimers Dis. 19, 915–925. doi: 10.3233/JAD-2010-1288
Chen, W. W., and Blurton-Jones, M. (2012). Concise review: can stem cells be used to treat or model Alzheimer’s disease? Stem Cells 30, 2612–2618. doi: 10.1002/stem.1240
Cirillo, G., Colangelo, A. M., Bianco, M. R., Cavaliere, C., Zaccaro, L., Sarmientos, P., et al. (2012). BB14, a Nerve Growth Factor (NGF)-like peptide shown to be effective in reducing reactive astrogliosis and restoring synaptic homeostasis in a rat model of peripheral nerve injury. Biotechnol. Adv. 30, 223–232. doi: 10.1016/j.biotechadv.2011.05.008
Colangelo, A. M., Bianco, M. R., Vitagliano, L., Cavaliere, C., Cirillo, G., De Gioia, L., et al. (2008). A new nerve growth factor-mimetic peptide active on neuropathic pain in rats. J. Neurosci. 28, 2698–2709. doi: 10.1523/JNEUROSCI.5201-07.2008
Conejero-Goldberg, C., Davies, P., and Ulloa, L. (2008). Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neurosci. Biobehav. Rev. 32, 693–706. doi: 10.1016/j.neubiorev.2007.10.007
Counts, S. E., Nadeem, M., Wuu, J., Ginsberg, S. D., Saragovi, H. U., and Mufson, E. J. (2004). Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol. 56, 520–531. doi: 10.1002/ana.20233
Cuello, A. C., and Bruno, M. A. (2007). The failure in NGF maturation and its increased degradation as the probable cause for the vulnerability of cholinergic neurons in Alzheimer’s disease. Neurochem. Res. 32, 1041–1045. doi: 10.1007/s11064-006-9270-0
d’Anglemont de Tassigny, X., Pascual, A., and Lopez-Barneo, J. (2015). GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front. Neuroanat. 9:10. doi: 10.3389/fnana.2015.00010
Darcq, E., Morisot, N., Phamluong, K., Warnault, V., Jeanblanc, J., Longo, F. M., et al. (2016). The neurotrophic factor receptor p75 in the rat dorsolateral striatum drives excessive alcohol drinking. J. Neurosci. 36, 10116–10127. doi: 10.1523/JNEUROSCI.4597-14.2016
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. doi: 10.1016/S0140-6736(76)91936-X
de Backer, M. W., Brans, M. A., Luijendijk, M. C., Garner, K. M., and Adan, R. A. (2010). Optimization of adeno-associated viral vector-mediated gene delivery to the hypothalamus. Hum. Gene Ther. 21, 673–682. doi: 10.1089/hum.2009.169
de Jonge, W. J., and Ulloa, L. (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 151, 915–929. doi: 10.1038/sj.bjp.0707264
De Rosa, R., Garcia, A. A., Braschi, C., Capsoni, S., Maffei, L., Berardi, N., et al. (2005). Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 3811–3816. doi: 10.1073/pnas.0500195102
De Sarno, P., Pomponi, M., Giacobini, E., Tang, X. C., and Williams, E. (1989). The effect of heptyl-physostigmine, a new cholinesterase inhibitor, on the central cholinergic system of the rat. Neurochem. Res. 14, 971–977. doi: 10.1007/BF00965931
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Deardorff, W. J., Feen, E., and Grossberg, G. T. (2015). The use of cholinesterase inhibitors across all stages of Alzheimer’s disease. Drugs Aging 32, 537–547. doi: 10.1007/s40266-015-0273-x
DeKosky, S. T., Ikonomovic, M. D., Styren, S. D., Beckett, L., Wisniewski, S., Bennett, D. A., et al. (2002). Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 51, 145–155. doi: 10.1002/ana.10069
Devi, L., and Ohno, M. (2012). 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. doi: 10.1038/npp.2011.191
Dhuria, S. V., Hanson, L. R., and Frey, W. H. II (2010). Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 99, 1654–1673. doi: 10.1002/jps.21924
DiStefano, P. S., Friedman, B., Radziejewski, C., Alexander, C., Boland, P., Schick, C. M., et al. (1992). The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron 8, 983–993. doi: 10.1016/0896-6273(92)90213-W
Drachman, D. A., and Leavitt, J. (1974). Human memory and the cholinergic system. A relationship to aging?. Arch. Neurol. 30, 113–121. doi: 10.1001/archneur.1974.00490320001001
Emerich, D. F., Cain, C. K., Greco, C., Saydoff, J. A., Hu, Z. Y., Liu, H., et al. (1997a). Cellular delivery of human CNTF prevents motor and cognitive dysfunction in a rodent model of Huntington’s disease. Cell Transplant. 6, 249–266.
Emerich, D. F., Winn, S. R., Hantraye, P. M., Peschanski, M., Chen, E. Y., Chu, Y., et al. (1997b). Protective effect of encapsulated cells producing neurotrophic factor CNTF in a monkey model of Huntington’s disease. Nature 386, 395–399. doi: 10.1038/386395a0
Emerich, D. F., Orive, G., Thanos, C., Tornoe, J., and Wahlberg, L. U. (2014). Encapsulated cell therapy for neurodegenerative diseases: from promise to product. Adv. Drug Deliv. Rev. 67–68, 131–141. doi: 10.1016/j.addr.2013.07.008
Emerich, D. F., Winn, S. R., Harper, J., Hammang, J. P., Baetge, E. E., and Kordower, J. H. (1994). Implants of polymer-encapsulated human NGF-secreting cells in the nonhuman primate: rescue and sprouting of degenerating cholinergic basal forebrain neurons. J. Comp. Neurol. 349, 148–164. doi: 10.1002/cne.903490110
Eriksdotter, M., Navarro-Oviedo, M., Mitra, S., Wahlberg, L., Linderoth, B., Tjernberg, L. O., et al. (2018). Cerebrospinal fluid from Alzheimer patients affects cell-mediated nerve growth factor production and cell survival in vitro. Exp. Cell Res. 371, 175–184. doi: 10.1016/j.yexcr.2018.08.007
Eriksdotter Jonhagen, M., Nordberg, A., Amberla, K., Backman, L., Ebendal, T., Meyerson, B., et al. (1998). Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 9, 246–257. doi: 10.1159/000017069
Eriksdotter-Jonhagen, M., Linderoth, B., Lind, G., Aladellie, L., Almkvist, O., Andreasen, N., et al. (2012). Encapsulated cell biodelivery of nerve growth factor to the Basal forebrain in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 33, 18–28. doi: 10.1159/000336051
Estenne-Bouhtou, G., Kullander, K., Karlsson, M., Ebendal, T., Hacksell, U., and Luthman, K. (1996). Design, synthesis, tandem mass spectrometric sequencing and biological activity of NGF mimetics. Int. J. Pept. Protein Res. 48, 337–346. doi: 10.1111/j.1399-3011.1996.tb00850.x
Eyjolfsdottir, H., Eriksdotter, M., Linderoth, B., Lind, G., Juliusson, B., Kusk, P., et al. (2016). Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: application of a second-generation encapsulated cell biodelivery device. Alzheimers Res. Ther. 8:30. doi: 10.1186/s13195-016-0195-9
Fabbro, S., and Seeds, N. W. (2009). Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 109, 303–315. doi: 10.1111/j.1471-4159.2009.05894.x
Fahnestock, M., Michalski, B., Xu, B., and Coughlin, M. D. (2001). The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 18, 210–220. doi: 10.1006/mcne.2001.1016
Falcicchia, C., Paolone, G., Emerich, D. F., Lovisari, F., Bell, W. J., Fradet, T., et al. (2018). Seizure-suppressant and neuroprotective effects of encapsulated BDNF-producing cells in a rat model of temporal lobe epilepsy. Mol. Ther. Methods Clin. Dev. 9, 211–224. doi: 10.1016/j.omtm.2018.03.001
Feng, X., Valdearcos, M., Uchida, Y., Lutrin, D., Maze, M., and Koliwad, S. K. (2017). Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI Insight 2:e91229. doi: 10.1172/jci.insight.91229
Ferreira, D., Westman, E., Eyjolfsdottir, H., Almqvist, P., Lind, G., Linderoth, B., et al. (2015). Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J. Alzheimers Dis. 43, 1059–1072. doi: 10.3233/JAD-141068
Ferreira-Vieira, T. H., Guimaraes, I. M., Silva, F. R., and Ribeiro, F. M. (2016). Alzheimer’s disease: targeting the cholinergic system. Curr. Neuropharmacol. 14, 101–115. doi: 10.2174/1570159X13666150716165726
Fischer, W., Wictorin, K., Bjorklund, A., Williams, L. R., Varon, S., and Gage, F. H. (1987). Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329, 65–68. doi: 10.1038/329065a0
Fjord-Larsen, L., Kusk, P., Tornoe, J., Juliusson, B., Torp, M., Bjarkam, C. R., et al. (2010). Long-term delivery of nerve growth factor by encapsulated cell biodelivery in the Gottingen minipig basal forebrain. Mol. Ther. 18, 2164–2172. doi: 10.1038/mt.2010.154
Fletcher, J. L., Wood, R. J., Nguyen, J., Norman, E. M. L., Jun, C. M. K., Prawdiuk, A. R., et al. (2018). Targeting TrkB with a brain-derived neurotrophic factor mimetic promotes myelin repair in the brain. J. Neurosci. 38, 7088–7099. doi: 10.1523/JNEUROSCI.0487-18.2018
Franceschi, C., Garagnani, P., Morsiani, C., Conte, M., Santoro, A., Grignolio, A., et al. (2018). The continuum of aging and age-related diseases: common mechanisms but different rates. Front. Med. 5:61. doi: 10.3389/fmed.2018.00061
Friesland, A., Weng, Z., Duenas, M., Massa, S. M., Longo, F. M., and Lu, Q. (2014). Amelioration of cisplatin-induced experimental peripheral neuropathy by a small molecule targeting p75 NTR. Neurotoxicology 45, 81–90. doi: 10.1016/j.neuro.2014.09.005
Fu, W. M., and Liu, J. J. (1997). Regulation of acetylcholine release by presynaptic nicotinic receptors at developing neuromuscular synapses. Mol. Pharmacol. 51, 390–398.
Garbayo, E., Ansorena, E., Lana, H., Carmona-Abellan, M. D., Marcilla, I., Lanciego, J. L., et al. (2016). Brain delivery of microencapsulated GDNF induces functional and structural recovery in parkinsonian monkeys. Biomaterials 110, 11–23. doi: 10.1016/j.biomaterials.2016.09.015
Giacobini, E. (1998). Invited review: cholinesterase inhibitors for Alzheimer’s disease therapy: from tacrine to future applications. Neurochem. Int. 32, 413–419. doi: 10.1016/S0197-0186(97)00124-1
Giacobini, E. (2004). Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol. Res. 50, 433–440. doi: 10.1016/j.phrs.2003.11.017
Giacobini, E., DeSarno, P., Clark, B., and McIlhany, M. (1989). The cholinergic receptor system of the human brain: neurochemical and pharmacological aspects in aging and Alzheimer. Prog. Brain Res. 79, 335–343. doi: 10.1016/S0079-6123(08)62493-0
Giacobini, E., and Zajicek, J. (1956). Quantitative determination of acetylcholinesterase activity in individual nerve cells. Nature 177, 185–186. doi: 10.1038/177185a0
Gielow, M. R., and Zaborszky, L. (2017). The input-output relationship of the cholinergic basal forebrain. Cell Rep. 18, 1817–1830. doi: 10.1016/j.celrep.2017.01.060
Ginsberg, S. D., Che, S., Wuu, J., Counts, S. E., and Mufson, E. J. (2006). Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 97, 475–487. doi: 10.1111/j.1471-4159.2006.03764.x
Govender, T., Choonara, Y. E., Kumar, P., Bijukumar, D., du Toit, L. C., Modi, G., et al. (2017). Implantable and transdermal polymeric drug delivery technologies for the treatment of central nervous system disorders. Pharm. Dev. Technol. 22, 476–486. doi: 10.1080/10837450.2016.1189937
Grabenstatter, H. L., Carlsen, J., Raol, Y. H., Yang, T., Hund, D., Cruz Del Angel, Y., et al. (2014). Acute administration of the small-molecule p75(NTR) ligand does not prevent hippocampal neuron loss or development of spontaneous seizures after pilocarpine-induced status epilepticus. J. Neurosci. Res. 92, 1307–1318. doi: 10.1002/jnr.23402
Greitz, D., and Hannerz, J. (1996). A proposed model of cerebrospinal fluid circulation: observations with radionuclide cisternography. AJNR Am. J. Neuroradiol. 17, 431–438.
Grothe, M., Zaborszky, L., Atienza, M., Gil-Neciga, E., Rodriguez-Romero, R., Teipel, S. J., et al. (2010). Reduction of basal forebrain cholinergic system parallels cognitive impairment in patients at high risk of developing Alzheimer’s disease. Cereb. Cortex 20, 1685–1695. doi: 10.1093/cercor/bhp232
Grskovic, M., Javaherian, A., Strulovici, B., and Daley, G. Q. (2011). Induced pluripotent stem cells–opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 10, 915–929. doi: 10.1038/nrd3577
Gu, F., Parada, I., Yang, T., Longo, F. M., and Prince, D. A. (2018). Partial TrkB receptor activation suppresses cortical epileptogenesis through actions on parvalbumin interneurons. Neurobiol. Dis. 113, 45–58. doi: 10.1016/j.nbd.2018.01.018
Gudasheva, T. A., Povarnina, P., Logvinov, I. O., Antipova, T. A., and Seredenin, S. B. (2016). Mimetics of brain-derived neurotrophic factor loops 1 and 4 are active in a model of ischemic stroke in rats. Drug Des. Dev. Ther. 10, 3545–3553. doi: 10.2147/DDDT.S118768
Gudasheva, T. A., Povarnina, P. Y., Antipova, T. A., Firsova, Y. N., Konstantinopolsky, M. A., and Seredenin, S. B. (2015). Dimeric dipeptide mimetics of the nerve growth factor Loop 4 and Loop 1 activate TRKA with different patterns of intracellular signal transduction. J. Biomed. Sci. 22:106. doi: 10.1186/s12929-015-0198-z
Gudasheva, T. A., Povarnina, P. Y., and Seredenin, S. B. (2017a). Dipeptide mimetic of the brain-derived neurotrophic factor prevents impairments of neurogenesis in stressed mice. Bull. Exp. Biol. Med. 162, 454–457. doi: 10.1007/s10517-017-3638-9
Gudasheva, T. A., Tarasiuk, A. V., Sazonova, N. M., Pomogaibo, S. V., Shumskiy, A. N., Logvinov, I. O., et al. (2017b). Design, synthesis, and neuroprotective effects of a dimeric dipeptide mimetic of the third loop of the nerve growth factor. Russ. J. Bioorg. Chem. 43, 235–247. doi: 10.1134/s1068162017030050
Gutierrez-Fernandez, A., Parmer, R. J., and Miles, L. A. (2007). Plasminogen gene expression is regulated by nerve growth factor. J. Thromb. Haemost. 5, 1715–1725. doi: 10.1111/j.1538-7836.2007.02636.x
Hagg, T. (2007). Intracerebral infusion of neurotrophic factors. Methods Mol. Biol. 399, 167–180. doi: 10.1007/978-1-59745-504-6_12
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Hampel, H., Mesulam, M. M., Cuello, A. C., Khachaturian, A. S., Vergallo, A., Farlow, M. R., et al. (2019). Revisiting the cholinergic hypothesis in Alzheimer’s disease: emerging evidence from translational and clinical research. J. Prev. Alzheimers Dis. 6, 2–15. doi: 10.14283/jpad.2018.43
Han, J., Pollak, J., Yang, T., Siddiqui, M. R., Doyle, K. P., Taravosh-Lahn, K., et al. (2012). Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke 43, 1918–1924. doi: 10.1161/STROKEAHA.111.641878
Hao, J., Ebendal, T., Xu, X., Wiesenfeld-Hallin, Z., and Eriksdotter Jonhagen, M. (2000). Intracerebroventricular infusion of nerve growth factor induces pain-like response in rats. Neurosci. Lett. 286, 208–212. doi: 10.1016/S0304-3940(00)01107-1
Haubrich, D. R., and Chippendale, T. J. (1977). Regulation of acetylcholine synthesis in nervous tissue. Life Sci. 20, 1465–1478. doi: 10.1016/0024-3205(77)90437-4
Hefti, F., Hartikka, J., and Knusel, B. (1989). Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol. Aging 10, 515–533. doi: 10.1016/0197-4580(89)90118-8
Hjorth, E., Zhu, M., Toro, V. C., Vedin, I., Palmblad, J., Cederholm, T., et al. (2013). Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-beta42 by human microglia and decrease inflammatory markers. J. Alzheimers Dis. 35, 697–713. doi: 10.3233/JAD-130131
Hohsfield, L. A., Geley, S., Reindl, M., and Humpel, C. (2013). The generation of NGF-secreting primary rat monocytes: a comparison of different transfer methods. J. Immunol. Methods 391, 112–124. doi: 10.1016/j.jim.2013.02.016
Hovens, I. B., Schoemaker, R. G., van der Zee, E. A., Heineman, E., Izaks, G. J., and van Leeuwen, B. L. (2012). Thinking through postoperative cognitive dysfunction: how to bridge the gap between clinical and pre-clinical perspectives. Brain Behav. Immun. 26, 1169–1179. doi: 10.1016/j.bbi.2012.06.004
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Huang, S. L., Wang, J., He, X. J., Li, Z. F., Pu, J. N., and Shi, W. (2014). Secretion of BDNF and GDNF from free and encapsulated choroid plexus epithelial cells. Neurosci. Lett. 566, 42–45. doi: 10.1016/j.neulet.2014.02.017
Inestrosa, N. C., Alvarez, A., Perez, C. A., Moreno, R. D., Vicente, M., Linker, C., et al. (1996). Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891. doi: 10.1016/S0896-6273(00)80108-7
Ioannou, M. S., and Fahnestock, M. (2017). ProNGF, but Not NGF, switches from neurotrophic to apoptotic activity in response to reductions in TrkA receptor levels. Int. J. Mol. Sci. 18:599. doi: 10.3390/ijms18030599
Isaev, N. K., Stelmashook, E. V., and Genrikhs, E. E. (2017). Role of nerve growth factor in plasticity of forebrain cholinergic neurons. Biochemistry 82, 291–300. doi: 10.1134/S0006297917030075
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Iughetti, L., Lucaccioni, L., Fugetto, F., Predieri, B., Berardi, A., and Ferrari, F. (2018). Brain-derived neurotrophic factor and epilepsy: a systematic review. Neuropeptides 72, 23–29. doi: 10.1016/j.npep.2018.09.005
Iulita, M. F., and Cuello, A. C. (2014). Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 35, 338–348. doi: 10.1016/j.tips.2014.04.010
Izsvak, Z., Hackett, P. B., Cooper, L. J., and Ivics, Z. (2010). Translating sleeping beauty transposition into cellular therapies: victories and challenges. Bioessays 32, 756–767. doi: 10.1002/bies.201000027
Jack, C.R. Jr., Knopman, D. S., Weigand, S. D., Wiste, H. J., Vemuri, P., Lowe, V., et al. (2012). An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann. Neurol. 71, 765–775. doi: 10.1002/ana.22628
Jain, P., Li, R., Lama, T., Saragovi, H. U., Cumberlidge, G., and Meerovitch, K. (2011). An NGF mimetic, MIM-D3, stimulates conjunctival cell glycoconjugate secretion and demonstrates therapeutic efficacy in a rat model of dry eye. Exp. Eye Res. 93, 503–512. doi: 10.1016/j.exer.2011.06.014
James, M. L., Belichenko, N. P., Shuhendler, A. J., Hoehne, A., Andrews, L. E., Condon, C., et al. (2017). [(18)F]GE-180 PET detects reduced microglia activation after LM11A-31 therapy in a mouse model of Alzheimer’s disease. Theranostics 7, 1422–1436. doi: 10.7150/thno.17666
Jang, S. W., Liu, X., Yepes, M., Shepherd, K. R., Miller, G. W., Liu, Y., et al. (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692. doi: 10.1073/pnas.0913572107
Janickova, H., Rudajev, V., Zimcik, P., Jakubik, J., Tanila, H., El-Fakahany, E. E., et al. (2013). Uncoupling of M1 muscarinic receptor/G-protein interaction by amyloid beta(1-42). Neuropharmacology 67, 272–283. doi: 10.1016/j.neuropharm.2012.11.014
Jenden, D. J., Jope, R. S., and Weiler, M. H. (1976). Regulation of acetylcholine synthesis: does cytoplasmic acetylcholine control high affinity choline uptake? Science 194, 635–637.
Joseph, J. A., and Fisher, D. R. (2003). Muscarinic receptor subtype determines vulnerability to amyloid beta toxicity in transfected cos-7 cells. J. Alzheimers Dis. 5, 197–208. doi: 10.3233/JAD-2003-5304
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kamei, N., Tanaka, N., Oishi, Y., Hamasaki, T., Nakanishi, K., Sakai, N., et al. (2007). BDNF, NT-3, and NGF released from transplanted neural progenitor cells promote corticospinal axon growth in organotypic cocultures. Spine 32, 1272–1278. doi: 10.1097/BRS.0b013e318059afab
Karami, A., Eyjolfsdottir, H., Vijayaraghavan, S., Lind, G., Almqvist, P., Kadir, A., et al. (2015). Changes in CSF cholinergic biomarkers in response to cell therapy with NGF in patients with Alzheimer’s disease. Alzheimers Dement. 11, 1316–1328. doi: 10.1016/j.jalz.2014.11.008
Kawamoto, K., Aoki, J., Tanaka, A., Itakura, A., Hosono, H., Arai, H., et al. (2002). Nerve growth factor activates mast cells through the collaborative interaction with lysophosphatidylserine expressed on the membrane surface of activated platelets. J. Immunol. 168, 6412–6419. doi: 10.4049/jimmunol.168.12.6412
Kazim, S. F., Blanchard, J., Bianchi, R., and Iqbal, K. (2017). Early neurotrophic pharmacotherapy rescues developmental delay and Alzheimer’s-like memory deficits in the Ts65Dn mouse model of Down syndrome. Sci. Rep. 7:45561. doi: 10.1038/srep45561
Kazim, S. F., Blanchard, J., Dai, C. L., Tung, Y. C., LaFerla, F. M., Iqbal, I. G., et al. (2014). Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 71, 110–130. doi: 10.1016/j.nbd.2014.07.001
Kerbler, G. M., Fripp, J., Rowe, C. C., Villemagne, V. L., Salvado, O., Rose, S., et al. (2015). Basal forebrain atrophy correlates with amyloid beta burden in Alzheimer’s disease. Neuroimage Clin. 7, 105–113. doi: 10.1016/j.nicl.2014.11.015
Khan, G. M., Tong, M., Jhun, M., Arora, K., and Nichols, R. A. (2010). beta-Amyloid activates presynaptic alpha7 nicotinic acetylcholine receptors reconstituted into a model nerve cell system: involvement of lipid rafts. Eur. J. Neurosci. 31, 788–796. doi: 10.1111/j.1460-9568.2010.07116.x
Khatoon, S., Chalbot, S., Bolognin, S., Puolivali, J., and Iqbal, K. (2015). Elevated Tau level in aged rat cerebrospinal fluid reduced by treatment with a neurotrophic compound. J. Alzheimers Dis. 47, 557–564. doi: 10.3233/JAD-142799
Kim, H. J., Lee, J. H., and Kim, S. H. (2010). Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: secretion of neurotrophic factors and inhibition of apoptosis. J. Neurotrauma 27, 131–138. doi: 10.1089/neu.2008-0818
Knowles, J. K., Simmons, D. A., Nguyen, T. V., Vander Griend, L., Xie, Y., Zhang, H., et al. (2013). Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer’s mouse model. Neurobiol. Aging 34, 2052–2063. doi: 10.1016/j.neurobiolaging.2013.02.015
Kowianski, P., Lietzau, G., Czuba, E., Waskow, M., Steliga, A., and Morys, J. (2018). BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 38, 579–593. doi: 10.1007/s10571-017-0510-4
Kraineva, V. A., Gudasheva, T. A., Kotelnikova, S. O., Antipova, T. A., and Seredenin, S. B. (2013). Original nerve growth factor mimetic dipeptide GK-2 limits the manifestations of hemorrhagic stroke in rats. Bull. Exp. Biol. Med. 154, 642–644. doi: 10.1007/s10517-013-2020-9
Kraunus, J., Schaumann, D. H., Meyer, J., Modlich, U., Fehse, B., Brandenburg, G., et al. (2004). Self-inactivating retroviral vectors with improved RNA processing. Gene Ther. 11, 1568–1578. doi: 10.1038/sj.gt.3302309
Kron, M., Lang, M., Adams, I. T., Sceniak, M., Longo, F., and Katz, D. M. (2014). A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis. Model. Mech. 7, 1047–1055. doi: 10.1242/dmm.016030
Kudrin, V. S., Klodt, P. M., Narkevich, V. B., Shipilov, V. A., Poseva, V. I., Molodavkin, G. M., et al. (2012). Behavioral and neurochemical aspects of the antidepressive action of GSB-106 dipeptide BDNF fragment. Eksp. Klin. Farmakol. 75, 3–7.
Lambiase, A., Pagani, L., Di Fausto, V., Sposato, V., Coassin, M., Bonini, S., et al. (2007). Nerve growth factor eye drop administrated on the ocular surface of rodents affects the nucleus basalis and septum: biochemical and structural evidence. Brain Res. 1127, 45–51. doi: 10.1016/j.brainres.2006.09.102
Latina, V., Caioli, S., Zona, C., Ciotti, M. T., Amadoro, G., and Calissano, P. (2017). Impaired NGF/TrkA signaling causes early AD-linked presynaptic dysfunction in cholinergic primary neurons. Front. Cell. Neurosci. 11:68. doi: 10.3389/fncel.2017.00068
Lauterborn, J. C., Bizon, J. L., Tran, T. M., and Gall, C. M. (1995). NGF mRNA is expressed by GABAergic but not cholinergic neurons in rat basal forebrain. J. Comp. Neurol. 360, 454–462. doi: 10.1002/cne.903600307
Lee, H. J., Lee, J. K., Lee, H., Carter, J. E., Chang, J. W., Oh, W., et al. (2012). Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 33, 588–602. doi: 10.1016/j.neurobiolaging.2010.03.024
LeSauteur, L., Cheung, N. K., Lisbona, R., and Saragovi, H. U. (1996). Small molecule nerve growth factor analogs image receptors in vivo. Nat. Biotechnol. 14, 1120–1122. doi: 10.1038/nbt0996-1120
LeSauteur, L., Wei, L., Gibbs, B. F., and Saragovi, H. U. (1995). Small peptide mimics of nerve growth factor bind TrkA receptors and affect biological responses. J. Biol. Chem. 270, 6564–6569. doi: 10.1074/jbc.270.12.6564
Levi-Montalcini, R. (2004). The nerve growth factor and the neuroscience chess board. Prog. Brain Res. 146, 525–527. doi: 10.1016/S0079-6123(03)46033-0
Li, W., Bellot-Saez, A., Phillips, M. L., Yang, T., Longo, F. M., and Pozzo-Miller, L. (2017). A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis. Model. Mech. 10, 837–845. doi: 10.1242/dmm.029959
Lim, F., and Sun, A. M. (1980). Microencapsulated islets as bioartificial endocrine pancreas. Science 210, 908–910. doi: 10.1126/science.6776628
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S., and Collins, F. (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260, 1130–1132. doi: 10.1126/science.8493557
Lindner, M. D., and Emerich, D. F. (1998). Therapeutic potential of a polymer-encapsulated L-DOPA and dopamine-producing cell line in rodent and primate models of Parkinson’s disease. Cell Transplant. 7, 165–174.
Lindvall, O., and Wahlberg, L. U. (2008). Encapsulated cell biodelivery of GDNF: a novel clinical strategy for neuroprotection and neuroregeneration in Parkinson’s disease? Exp. Neurol. 209, 82–88. doi: 10.1016/j.expneurol.2007.08.019
Lista, S., O’Bryant, S. E., Blennow, K., Dubois, B., Hugon, J., Zetterberg, H., et al. (2015). Biomarkers in sporadic and familial Alzheimer’s disease. J. Alzheimers Dis. 47, 291–317. doi: 10.3233/JAD-143006
Liu, L., Yu, J., Li, L., Zhang, B., Liu, L., Wu, C. H., et al. (2017). Alpha7 nicotinic acetylcholine receptor is required for amyloid pathology in brain endothelial cells induced by Glycoprotein 120, methamphetamine and nicotine. Sci. Rep. 7:40467. doi: 10.1038/srep40467
Liu, X., and Chen, C. (2005). Strategies to optimize brain penetration in drug discovery. Curr. Opin. Drug Discov. Dev. 8, 505–512.
Liu, X., Obianyo, O., Chan, C. B., Huang, J., Xue, S., Yang, J. J., et al. (2014). Biochemical and biophysical investigation of the brain-derived neurotrophic factor mimetic 7,8-dihydroxyflavone in the binding and activation of the TrkB receptor. J. Biol. Chem. 289, 27571–27584. doi: 10.1074/jbc.M114.562561
Longo, F. M., Manthorpe, M., Xie, Y. M., and Varon, S. (1997). Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J. Neurosci. Res. 48, 1–17. doi: 10.1002/(SICI)1097-4547(19970401)48:1<1::AID-JNR1>3.0.CO;2-K
Longo, F. M., Vu, T. K., and Mobley, W. C. (1990). The in vitro biological effect of nerve growth factor is inhibited by synthetic peptides. Cell Regul. 1, 189–195. doi: 10.1091/mbc.1.2.189
Madziar, B., Lopez-Coviella, I., Zemelko, V., and Berse, B. (2005). Regulation of cholinergic gene expression by nerve growth factor depends on the phosphatidylinositol-3’-kinase pathway. J. Neurochem. 92, 767–779. doi: 10.1111/j.1471-4159.2004.02908.x
Makin, S. (2018). The amyloid hypothesis on trial. Nature 559, S4–S7. doi: 10.1038/d41586-018-05719-4
Maliartchouk, S., Debeir, T., Beglova, N., Cuello, A. C., Gehring, K., and Saragovi, H. U. (2000a). Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J. Biol. Chem. 275, 9946–9956.
Maliartchouk, S., Feng, Y., Ivanisevic, L., Debeir, T., Cuello, A. C., Burgess, K., et al. (2000b). A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 57, 385–391.
Marchi, M., Hoffman, D. W., Giacobini, E., and Fredrickson, T. (1980). Development and aging of cholinergic synapses. IV. Acetylcholinesterase and choline acetyltransferase activities in autonomic ganglia and iris of the chick. Dev. Neurosci. 3, 235–247. doi: 10.1159/000112396
Masoudi, R., Ioannou, M. S., Coughlin, M. D., Pagadala, P., Neet, K. E., Clewes, O., et al. (2009). Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J. Biol. Chem. 284, 18424–18433. doi: 10.1074/jbc.M109.007104
Massa, S. M., Xie, Y., Yang, T., Harrington, A. W., Kim, M. L., Yoon, S. O., et al. (2006). Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J. Neurosci. 26, 5288–5300. doi: 10.1523/JNEUROSCI.3547-05.2006
Massa, S. M., Yang, T., Xie, Y., Shi, J., Bilgen, M., Joyce, J. N., et al. (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 120, 1774–1785. doi: 10.1172/JCI41356
Matrone, C., Di Luzio, A., Meli, G., D’Aguanno, S., Severini, C., Ciotti, M. T., et al. (2008). Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J. Alzheimers Dis. 13, 81–96. doi: 10.3233/JAD-2008-13109
May, L. M., Anggono, V., Gooch, H. M., Jang, S. E., Matusica, D., Kerbler, G. M., et al. (2017). G-Protein-coupled inwardly rectifying potassium (GIRK) channel activation by the p75 neurotrophin receptor is required for amyloid beta toxicity. Front. Neurosci. 11:455. doi: 10.3389/fnins.2017.00455
McKelvey, L., Shorten, G. D., and O’Keeffe, G. W. (2013). Nerve growth factor-mediated regulation of pain signalling and proposed new intervention strategies in clinical pain management. J. Neurochem. 124, 276–289. doi: 10.1111/jnc.12093
Meeker, R. B., Poulton, W., Clary, G., Schriver, M., and Longo, F. M. (2016). Novel p75 neurotrophin receptor ligand stabilizes neuronal calcium, preserves mitochondrial movement and protects against HIV associated neuropathogenesis. Exp. Neurol. 275(Pt 1), 182–198. doi: 10.1016/j.expneurol.2015.09.012
Meeker, R. B., Poulton, W., Feng, W. H., Hudson, L., and Longo, F. M. (2012). Suppression of immunodeficiency virus-associated neural damage by the p75 neurotrophin receptor ligand, LM11A-31, in an in vitro feline model. J. Neuroimmune Pharmacol. 7, 388–400. doi: 10.1007/s11481-011-9325-0
Mehta, M., Adem, A., and Sabbagh, M. (2012). New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012:728983. doi: 10.1155/2012/728983
Messamore, E., Ogane, N., and Giacobini, E. (1993a). Cholinesterase inhibitor effects on extracellular acetylcholine in rat striatum. Neuropharmacology 32, 291–296. doi: 10.1016/0028-3908(93)90114-I
Messamore, E., Warpman, U., Ogane, N., and Giacobini, E. (1993b). Cholinesterase inhibitor effects on extracellular acetylcholine in rat cortex. Neuropharmacology 32, 745–750. doi: 10.1016/0028-3908(93)90182-3
Mesulam, M. (2004). The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn. Mem. 11, 43–49. doi: 10.1101/lm.69204
Mimeault, M., Hauke, R., and Batra, S. K. (2007). Stem cells: a revolution in therapeutics-recent advances in stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin. Pharmacol. Ther. 82, 252–264. doi: 10.1038/sj.clpt.6100301
Mittoux, V., Joseph, J. M., Conde, F., Palfi, S., Dautry, C., Poyot, T., et al. (2000). Restoration of cognitive and motor functions by ciliary neurotrophic factor in a primate model of Huntington’s disease. Hum. Gene Ther. 11, 1177–1187. doi: 10.1089/10430340050015220
Mroczko, B., Groblewska, M., and Barcikowska, M. (2013). The role of matrix metalloproteinases and tissue inhibitors of metalloproteinases in the pathophysiology of neurodegeneration: a literature study. J. Alzheimers Dis. 37, 273–283. doi: 10.3233/JAD-130647
Mufson, E. J., Conner, J. M., and Kordower, J. H. (1995). Nerve growth factor in Alzheimer’s disease: defective retrograde transport to nucleus basalis. Neuroreport 6, 1063–1066. doi: 10.1097/00001756-199505090-00028
Mufson, E. J., Counts, S. E., Perez, S. E., and Ginsberg, S. D. (2008). Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev. Neurother. 8, 1703–1718. doi: 10.1586/14737175.8.11.1703
Mufson, E. J., Lavine, N., Jaffar, S., Kordower, J. H., Quirion, R., and Saragovi, H. U. (1997). Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp. Neurol. 146, 91–103. doi: 10.1006/exnr.1997.6504
Mura, E., Zappettini, S., Preda, S., Biundo, F., Lanni, C., Grilli, M., et al. (2012). Dual effect of beta-amyloid on alpha7 and alpha4beta2 nicotinic receptors controlling the release of glutamate, aspartate and GABA in rat hippocampus. PLoS One 7:e29661. doi: 10.1371/journal.pone.0029661
Nagahara, A. H., Bernot, T., Moseanko, R., Brignolo, L., Blesch, A., Conner, J. M., et al. (2009a). Long-term reversal of cholinergic neuronal decline in aged non-human primates by lentiviral NGF gene delivery. Exp. Neurol. 215, 153–159. doi: 10.1016/j.expneurol.2008.10.004
Nagahara, A. H., Merrill, D. A., Coppola, G., Tsukada, S., Schroeder, B. E., Shaked, G. M., et al. (2009b). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 15, 331–337. doi: 10.1038/nm.1912
Nguyen, T. V., Shen, L., Vander Griend, L., Quach, L. N., Belichenko, N. P., Saw, N., et al. (2014). Small molecule p75NTR ligands reduce pathological phosphorylation and misfolding of tau, inflammatory changes, cholinergic degeneration, and cognitive deficits in AbetaPP(L/S) transgenic mice. J. Alzheimers Dis. 42, 459–483. doi: 10.3233/JAD-140036
Ni, R., Marutle, A., and Nordberg, A. (2013). Modulation of alpha7 nicotinic acetylcholine receptor and fibrillar amyloid-beta interactions in Alzheimer’s disease brain. J. Alzheimers Dis. 33, 841–851. doi: 10.3233/JAD-2012-121447
Niewiadomska, G., Mietelska-Porowska, A., and Mazurkiewicz, M. (2011). The cholinergic system, nerve growth factor and the cytoskeleton. Behav. Brain Res. 221, 515–526. doi: 10.1016/j.bbr.2010.02.024
Nishino, J., Yamashita, K., Hashiguchi, H., Fujii, H., Shimazaki, T., and Hamada, H. (2004). Meteorin: a secreted protein that regulates glial cell differentiation and promotes axonal extension. EMBO J. 23, 1998–2008. doi: 10.1038/sj.emboj.7600202
Nordberg, A. (2006). Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 20(2 Suppl. 1), S12–S18. doi: 10.1097/01.wad.0000213804.59187.2d
O’Leary, P. D., and Hughes, R. A. (1998). Structure-activity relationships of conformationally constrained peptide analogues of loop 2 of brain-derived neurotrophic factor. J. Neurochem. 70, 1712–1721. doi: 10.1046/j.1471-4159.1998.70041712.x
Olson, L., Nordberg, A., von Holst, H., Backman, L., Ebendal, T., Alafuzoff, I., et al. (1992). Nerve growth factor affects 11C-nicotine binding, blood flow, EEG, and verbal episodic memory in an Alzheimer patient (case report). J. Neural Transm. Park. Dis. Dement. Sect. 4, 79–95. doi: 10.1007/BF02257624
Orive, G., Hernandez, R. M., Rodriguez Gascon, A., Calafiore, R., Chang, T. M., de Vos, P., et al. (2004). History, challenges and perspectives of cell microencapsulation. Trends Biotechnol. 22, 87–92. doi: 10.1016/j.tibtech.2003.11.004
Orive, G., Santos, E., Poncelet, D., Hernandez, R. M., Pedraz, J. L., Wahlberg, L. U., et al. (2015). Cell encapsulation: technical and clinical advances. Trends Pharmacol. Sci. 36, 537–546. doi: 10.1016/j.tips.2015.05.003
Ostrovskaya, R. U., Yagubova, S. S., Gudasheva, T. A., and Seredenin, S. B. (2017). Low-molecular-weight NGF mimetic corrects the cognitive deficit and depression-like behavior in experimental diabetes. Acta Nat. 9, 94–102.
Parikh, V., Bernard, C. S., Naughton, S. X., and Yegla, B. (2014). Interactions between Abeta oligomers and presynaptic cholinergic signaling: age-dependent effects on attentional capacities. Behav. Brain Res. 274, 30–42. doi: 10.1016/j.bbr.2014.07.046
Pavlov, V. A., Wang, H., Czura, C. J., Friedman, S. G., and Tracey, K. J. (2003). The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol. Med. 9, 125–134. doi: 10.1007/BF03402177
Pehar, M., Cassina, P., Vargas, M. R., Xie, Y., Beckman, J. S., Massa, S. M., et al. (2006). Modulation of p75-dependent motor neuron death by a small non-peptidyl mimetic of the neurotrophin loop 1 domain. Eur. J. Neurosci. 24, 1575–1580. doi: 10.1111/j.1460-9568.2006.05040.x
Peng, S., Wuu, J., Mufson, E. J., and Fahnestock, M. (2004). Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 63, 641–649. doi: 10.1093/jnen/63.6.641
Poepsel, S., Sprengel, A., Sacca, B., Kaschani, F., Kaiser, M., Gatsogiannis, C., et al. (2015). Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 11, 862–869. doi: 10.1038/nchembio.1931
Povarnina, P. Y., Gudasheva, T. A., Vorontsova, O. N., Bondarenko, N. A., and Seredenin, S. B. (2011). Antiparkinsonian properties of a nerve growth factor dipeptide mimetic GK-2 in in vivo experiments. Bull. Exp. Biol. Med. 151, 690–693. doi: 10.1007/s10517-011-1417-6
Povarnina, P. Y., Volkova, A. A., Gudasheva, T. A., and Seredenin, S. B. (2017). Comparison of the pharmacological effects of dimeric dipeptide nerve growth factor mimetic GK-2 and mexidol on the model of ischemic stroke in rats. Bull. Exp. Biol. Med. 164, 173–176. doi: 10.1007/s10517-017-3951-3
Povarnina, P. Y., Vorontsova, O. N., Gudasheva, T. A., Ostrovskaya, R. U., and Seredenin, S. B. (2013). Original nerve growth factor mimetic dipeptide gk-2 restores impaired cognitive functions in rat models of Alzheimer’s disease. Acta Nat. 5, 84–91.
Puzzo, D., Gulisano, W., Arancio, O., and Palmeri, A. (2015). The keystone of Alzheimer pathogenesis might be sought in Abeta physiology. Neuroscience 307, 26–36. doi: 10.1016/j.neuroscience.2015.08.039
Qiu, C., Xu, W., and Fratiglioni, L. (2010). Vascular and psychosocial factors in Alzheimer’s disease: epidemiological evidence toward intervention. J. Alzheimers Dis. 20, 689–697. doi: 10.3233/JAD-2010-091663
Rafii, M. S., Baumann, T. L., Bakay, R. A., Ostrove, J. M., Siffert, J., Fleisher, A. S., et al. (2014). A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 10, 571–581. doi: 10.1016/j.jalz.2013.09.004
Rafii, M. S., Tuszynski, M. H., Thomas, R. G., Barba, D., Brewer, J. B., Rissman, R. A., et al. (2018). Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol. 75, 834–841. doi: 10.1001/jamaneurol.2018.0233
Rizzi, C., Tiberi, A., Giustizieri, M., Marrone, M. C., Gobbo, F., Carucci, N. M., et al. (2018). NGF steers microglia toward a neuroprotective phenotype. Glia 66, 1395–1416. doi: 10.1002/glia.23312
Rodda, J., and Carter, J. (2012). Cholinesterase inhibitors and memantine for symptomatic treatment of dementia. BMJ 344:e2986. doi: 10.1136/bmj.e2986
Sajadi, A., Bensadoun, J. C., Schneider, B. L., Lo Bianco, C., and Aebischer, P. (2006). Transient striatal delivery of GDNF via encapsulated cells leads to sustained behavioral improvement in a bilateral model of Parkinson disease. Neurobiol. Dis. 22, 119–129. doi: 10.1016/j.nbd.2005.10.006
Sasi, M., Vignoli, B., Canossa, M., and Blum, R. (2017). Neurobiology of local and intercellular BDNF signaling. Pflugers Arch. 469, 593–610. doi: 10.1007/s00424-017-1964-4
Scarpi, D., Cirelli, D., Matrone, C., Castronovo, G., Rosini, P., Occhiato, E. G., et al. (2012). Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 3:e339. doi: 10.1038/cddis.2012.80
Schambach, A., Galla, M., Maetzig, T., Loew, R., and Baum, C. (2007). Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol. Ther. 15, 1167–1173. doi: 10.1038/sj.mt.6300152
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Schmid, D. A., Yang, T., Ogier, M., Adams, I., Mirakhur, Y., Wang, Q., et al. (2012). A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci. 32, 1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012
Schmitz, T. W., Nathan Spreng, R., and Alzheimer’s Disease Neuroimaging Initiative (2016). Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 7:13249. doi: 10.1038/ncomms13249
Segal, R. A. (2003). Selectivity in neurotrophin signaling: theme and variations. Annu. Rev. Neurosci. 26, 299–330. doi: 10.1146/annurev.neuro.26.041002.131421
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Seredenin, S. B., Romanova, G. A., Gudasheva, T. A., Shakova, F. M., Barskov, I. V., Stelmashuk, E. V., et al. (2011a). Neuroprotective and antiamnestic effect of nerve growth factor dipeptide mimetic GK-2 in experimental ischemic infarction of brain cortex. Bull. Exp. Biol. Med. 150, 432–435.
Seredenin, S. B., Silachev, D. N., Gudasheva, T. A., Pirogov, Y. A., and Isaev, N. K. (2011b). Neuroprotective effect of GK-2, a dipeptide mimetic of nerve growth factor, during experimental focal ischemia in middle cerebral artery basin. Bull. Exp. Biol. Med. 151, 584–587.
Seredenin, S. B., Voronina, T. A., Gudasheva, T. A., Garibova, T. L., Molodavkin, G. M., Litvinova, S. A., et al. (2013). Antidepressant effect of dimeric dipeptide GSB-106, an original low-molecular-weight mimetic of BDNF. Acta Nat. 5, 105–109.
Shi, J., Longo, F. M., and Massa, S. M. (2013). A small molecule p75(NTR) ligand protects neurogenesis after traumatic brain injury. Stem Cells 31, 2561–2574. doi: 10.1002/stem.1516
Simmons, D. A., Belichenko, N. P., Ford, E. C., Semaan, S., Monbureau, M., Aiyaswamy, S., et al. (2016). A small molecule p75NTR ligand normalizes signalling and reduces Huntington’s disease phenotypes in R6/2 and BACHD mice. Hum. Mol. Genet. 25, 4920–4938. doi: 10.1093/hmg/ddw316
Simmons, D. A., Belichenko, N. P., Yang, T., Condon, C., Monbureau, M., Shamloo, M., et al. (2013). A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J. Neurosci. 33, 18712–18727. doi: 10.1523/JNEUROSCI.1310-13.2013
Simmons, D. A., James, M. L., Belichenko, N. P., Semaan, S., Condon, C., Kuan, J., et al. (2018). TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 27, 2893–2912. doi: 10.1093/hmg/ddy202
Simmons, D. A., Knowles, J. K., Belichenko, N. P., Banerjee, G., Finkle, C., Massa, S. M., et al. (2014). A small molecule p75NTR ligand, LM11A-31, reverses cholinergic neurite dystrophy in Alzheimer’s disease mouse models with mid- to late-stage disease progression. PLoS One 9:e102136. doi: 10.1371/journal.pone.0102136
Simmons, D. A., Longo, F. M., and Massa, S. M. (2017). Neurotrophin receptor signaling as a therapeutic target for Huntington’s disease. CNS Neurol. Disord. Drug Targets 16, 291–302. doi: 10.2174/1871527315666161107093047
Simonato, M., Tongiorgi, E., and Kokaia, M. (2006). Angels and demons: neurotrophic factors and epilepsy. Trends Pharmacol. Sci. 27, 631–638. doi: 10.1016/j.tips.2006.10.002
Smith, C. M., and Swash, M. (1979). Possible biochemical basis of memory disorder in Alzheimer’s disease. Age Ageing 8, 289–293. doi: 10.1093/ageing/8.4.289
Sobottka, B., Reinhardt, D., Brockhaus, M., Jacobsen, H., and Metzger, F. (2008). ProNGF inhibits NGF-mediated TrkA activation in PC12 cells. J. Neurochem. 107, 1294–1303. doi: 10.1111/j.1471-4159.2008.05690.x
Son, A. I., Opfermann, J. D., McCue, C., Ziobro, J., Abrahams, J. H. III, Jones, K., et al. (2017). An implantable micro-caged device for direct local delivery of agents. Sci. Rep. 7:17624. doi: 10.1038/s41598-017-17912-y
Spalding, K. L., Bergmann, O., Alkass, K., Bernard, S., Salehpour, M., Huttner, H. B., et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. doi: 10.1016/j.cell.2013.05.002
Spiegel, K., Agrafiotis, D., Caprathe, B., Davis, R. E., Dickerson, M. R., Fergus, J. H., et al. (1995). PD 90780, a non peptide inhibitor of nerve growth factor’s binding to the P75 NGF receptor. Biochem. Biophys. Res. Commun. 217, 488–494. doi: 10.1006/bbrc.1995.2802
Stelmashook, E. V., Genrikhs, E. E., Novikova, S. V., Barskov, I. V., Gudasheva, T. A., Seredenin, S. B., et al. (2015). Behavioral effect of dipeptide NGF mimetic GK-2 in an in vivo model of rat traumatic brain injury and its neuroprotective and regenerative properties in vitro. Int. J. Neurosci. 125, 375–379. doi: 10.3109/00207454.2014.935376
Taylor, P. (1998). Development of acetylcholinesterase inhibitors in the therapy of Alzheimer’s disease. Neurology 51(1 Suppl. 1), S30–S35; discussion S65–S67. doi: 10.1212/WNL.51.1_Suppl_1.S30
Teipel, S. J., Cavedo, E., Hampel, H., Grothe, M. J., Alzheimer’s Disease Neuroimaging Initiative and Alzheimer Precision Medicine Initiative (2018). Basal forebrain volume, but not hippocampal volume, is a predictor of global cognitive decline in patients with Alzheimer’s disease treated with cholinesterase inhibitors. Front. Neurol. 9:642. doi: 10.3389/fneur.2018.00642
Tep, C., Lim, T. H., Ko, P. O., Getahun, S., Ryu, J. C., Goettl, V. M., et al. (2013). Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J. Neurosci. 33, 397–410. doi: 10.1523/JNEUROSCI.0399-12.2013
Thoenen, H., and Sendtner, M. (2002). Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 5(Suppl.), 1046–1050. doi: 10.1038/nn938
Thornhill, S. I., Schambach, A., Howe, S. J., Ulaganathan, M., Grassman, E., Williams, D., et al. (2008). Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 16, 590–598. doi: 10.1038/sj.mt.6300393
Tirassa, P. (2011). The nerve growth factor administrated as eye drops activates mature and precursor cells in subventricular zone of adult rats. Arch. Ital. Biol. 149, 205–213. doi: 10.4449/aib.v149i1.1359
Tiveron, C., Fasulo, L., Capsoni, S., Malerba, F., Marinelli, S., Paoletti, F., et al. (2013). ProNGF∖NGF imbalance triggers learning and memory deficits, neurodegeneration and spontaneous epileptic-like discharges in transgenic mice. Cell Death Differ. 20, 1017–1030. doi: 10.1038/cdd.2013.22
Tornoe, J., Torp, M., Jorgensen, J. R., Emerich, D. F., Thanos, C., Bintz, B., et al. (2012). Encapsulated cell-based biodelivery of meteorin is neuroprotective in the quinolinic acid rat model of neurodegenerative disease. Restor. Neurol. Neurosci. 30, 225–236. doi: 10.3233/RNN-2012-110199
Treinin, M., Papke, R. L., Nizri, E., Ben-David, Y., Mizrachi, T., and Brenner, T. (2017). Role of the alpha7 nicotinic acetylcholine receptor and RIC-3 in the cholinergic anti-inflammatory pathway. Cent. Nerv. Syst. Agents Med. Chem. 17, 90–99. doi: 10.2174/1871524916666160829114533
Triaca, V., and Calissano, P. (2016). Impairment of the nerve growth factor pathway driving amyloid accumulation in cholinergic neurons: the incipit of the Alzheimer’s disease story? Neural Regen. Res. 11, 1553–1556. doi: 10.4103/1673-5374.193224
Tsai, T., Klausmeyer, A., Conrad, R., Gottschling, C., Leo, M., Faissner, A., et al. (2013). 7,8-Dihydroxyflavone leads to survival of cultured embryonic motoneurons by activating intracellular signaling pathways. Mol. Cell Neurosci. 56, 18–28. doi: 10.1016/j.mcn.2013.02.007
Tucker, H. M., Kihiko, M., Caldwell, J. N., Wright, S., Kawarabayashi, T., Price, D., et al. (2000). The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 20, 3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000
Tuszynski, M. H., Thal, L., Pay, M., Salmon, D. P., U, H. S., Bakay, R., et al. (2005). A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551–555. doi: 10.1038/nm1239
Tuszynski, M. H., U, H. S., Amaral, D. G., and Gage, F. H. (1990). Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J. Neurosci. 10, 3604–3614. doi: 10.1523/JNEUROSCI.10-11-03604.1990
Tuszynski, M. H., Yang, J. H., Barba, D., U, H. S., Bakay, R. A., Pay, M. M., et al. (2015). Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147. doi: 10.1001/jamaneurol.2015.1807
Ure, D. R., and Campenot, R. B. (1994). Leukemia inhibitory factor and nerve growth factor are retrogradely transported and processed by cultured rat sympathetic neurons. Dev. Biol. 162, 339–347. doi: 10.1006/dbio.1994.1091
Van Cauwenberghe, C., Van Broeckhoven, C., and Sleegers, K. (2016). The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430. doi: 10.1038/gim.2015.117
Wahlberg, L. U., Lind, G., Almqvist, P. M., Kusk, P., Tornoe, J., Juliusson, B., et al. (2012). Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: a technology platform for restorative neurosurgery. J. Neurosurg. 117, 340–347. doi: 10.3171/2012.2.JNS11714
Wang, H., Yu, M., Ochani, M., Amella, C. A., Tanovic, M., Susarla, S., et al. (2003). Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384–388. doi: 10.1038/nature01339
Warnault, V., Darcq, E., Morisot, N., Phamluong, K., Wilbrecht, L., Massa, S. M., et al. (2016). The BDNF valine 68 to methionine polymorphism increases compulsive alcohol drinking in mice that is reversed by tropomyosin receptor kinase B activation. Biol. Psychiatry 79, 463–473. doi: 10.1016/j.biopsych.2015.06.007
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi: 10.1126/science.7058341
World Alzheimer Report (2016). Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future. Available at: https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf (accessed January 25, 2019).
World Health Organization [WHO] (2016). World Report on Ageing and Health. Available at: https://apps.who.int/iris/bitstream/handle/10665/186463/9789240694811_eng.pdf;jsessionid=E690207076FBD4272B201D50B860261A?sequence=
World Health Organization [WHO] (2018). Ageing and Health. Available at: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed January 25, 2019).
Winn, S. R., Lindner, M. D., Lee, A., Haggett, G., Francis, J. M., and Emerich, D. F. (1996). Polymer-encapsulated genetically modified cells continue to secrete human nerve growth factor for over one year in rat ventricles: behavioral and anatomical consequences. Exp. Neurol. 140, 126–138. doi: 10.1006/exnr.1996.0123
Wu, D. (2005). Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRx 2, 120–128. doi: 10.1602/neurorx.2.1.120
Xie, Y., Tisi, M. A., Yeo, T. T., and Longo, F. M. (2000). Nerve growth factor (NGF) loop 4 dimeric mimetics activate ERK and AKT and promote NGF-like neurotrophic effects. J. Biol. Chem. 275, 29868–29874. doi: 10.1074/jbc.M005071200
Xu, J., Dong, H., Qian, Q., Zhang, X., Wang, Y., Jin, W., et al. (2017). Astrocyte-derived CCL2 participates in surgery-induced cognitive dysfunction and neuroinflammation via evoking microglia activation. Behav. Brain Res. 332, 145–153. doi: 10.1016/j.bbr.2017.05.066
Xu, W., Weissmiller, A. M., White, J. A. II, Fang, F., Wang, X., Wu, Y., et al. (2016). Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 126, 1815–1833. doi: 10.1172/JCI82409
Yaar, M., Zhai, S., Panova, I., Fine, R. E., Eisenhauer, P. B., Blusztajn, J. K., et al. (2007). A cyclic peptide that binds p75(NTR) protects neurones from beta amyloid (1-40)-induced cell death. Neuropathol. Appl. Neurobiol. 33, 533–543. doi: 10.1111/j.1365-2990.2007.00844.x
Yaar, M., Zhai, S., Pilch, P. F., Doyle, S. M., Eisenhauer, P. B., Fine, R. E., et al. (1997). Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J. Clin. Invest. 100, 2333–2340. doi: 10.1172/JCI119772
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yamasaki, T. R., Blurton-Jones, M., Morrissette, D. A., Kitazawa, M., Oddo, S., and LaFerla, F. M. (2007). Neural stem cells improve memory in an inducible mouse model of neuronal loss. J. Neurosci. 27, 11925–11933. doi: 10.1523/JNEUROSCI.1627-07.2007
Yanamandra, K., Patel, T. K., Jiang, H., Schindler, S., Ulrich, J. D., Boxer, A. L., et al. (2017). Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci. Transl. Med. 9:eaal2029. doi: 10.1126/scitranslmed.aal2029
Yang, C., Liu, Y., Ni, X., Li, N., Zhang, B., and Fang, X. (2014). Enhancement of the nonamyloidogenic pathway by exogenous NGF in an Alzheimer transgenic mouse model. Neuropeptides 48, 233–238. doi: 10.1016/j.npep.2014.04.005
Yang, T., Knowles, J. K., Lu, Q., Zhang, H., Arancio, O., Moore, L. A., et al. (2008). Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One 3:e3604. doi: 10.1371/journal.pone.0003604
Yang, T., Massa, S. M., Tran, K. C., Simmons, D. A., Rajadas, J., Zeng, A. Y., et al. (2016). A small molecule TrkB/TrkC neurotrophin receptor co-activator with distinctive effects on neuronal survival and process outgrowth. Neuropharmacology 110(Pt A), 343–361. doi: 10.1016/j.neuropharm.2016.06.015
Zanin, M. P., Pettingill, L. N., Harvey, A. R., Emerich, D. F., Thanos, C. G., and Shepherd, R. K. (2012). The development of encapsulated cell technologies as therapies for neurological and sensory diseases. J. Control Release 160, 3–13. doi: 10.1016/j.jconrel.2012.01.021
Zhang, Y. W., Chen, Y., Liu, Y., Zhao, Y., Liao, F. F., and Xu, H. (2013). APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One 8:e80571. doi: 10.1371/journal.pone.0080571
Keywords: NGF, encapsulated cell biodelivery, Alzheimer disease, cholinergic, neurotrophins
Citation: Mitra S, Behbahani H and Eriksdotter M (2019) Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Front. Neurosci. 13:38. doi: 10.3389/fnins.2019.00038
Received: 01 November 2018; Accepted: 15 January 2019;
Published: 05 February 2019.
Edited by:
A. Claudio Cuello, McGill University, CanadaReviewed by:
Wolfgang Härtig, Leipzig University, GermanyRafael Linden, Universidade Federal do Rio de Janeiro, Brazil
Copyright © 2019 Mitra, Behbahani and Eriksdotter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Eriksdotter, bWFyaWEuZXJpa3Nkb3R0ZXJAa2kuc2U=
†Equal authorship