 Ebrahim M. Yimer
Ebrahim M. Yimer Hailemichael Zeru Hishe
Hailemichael Zeru Hishe Kald Beshir Tuem
Kald Beshir Tuem- Department of Pharmacology and Toxicology, School of Pharmacy, College of Health Sciences, Mekelle University, Mekelle, Ethiopia
To date, there is no cure or disease-modifying agents available for most well-known neurological disorders. Current therapy is typically focused on relieving symptoms and supportive care in improving the quality of life of affected patients. Furthermore, the traditional de novo drug discovery technique is more challenging, particularly for neurological disorders. Therefore, the repurposing of existing drugs for these conditions is believed to be an efficient and dynamic approach that can substantially reduce the investments spent on drug development. Currently, there is emerging evidence that suggests the potential effect of a beta-lactam antibiotic, ceftriaxone (CEF), to alleviate the symptoms of different experimentally-induced neurological disorders: Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, epileptic-seizure, brain ischemia, traumatic brain injuries, and neuropathic pain. CEF also affects the markers of oxidative status and neuroinflammation, glutamatergic systems as well as various aggregated toxic proteins involved in the pathogenesis of different neurological disorders. Moreover, it was found that CEF administration to drug dependent animal models improved the withdrawal symptoms upon drug discontinuation. Thus, this review aimed to describe the effects of CEF against multiple models of neurological illnesses, drug dependency, and withdrawal. It also emphasizes the possible mechanisms of neuroprotective actions of CEF with respective neurological maladies.
Introduction
The conventional target-based approach for new drug discovery brings paramount challenges as well as massive economic burden, lengthy processes, and might even expose study participants to unexpected adverse events (Lee and Kim, 2016). One alternative paradigm for novel drug discovery is drug repurposing which has recently emerged as a potential strategy of off-target therapeutic actions of existing drugs (Kim, 2015; Parsons, 2018). Currently, drug repurposing is considered a promising tool for novel drug discovery as it is relatively rapid, less costly, and poses a minimal risk of adverse outcomes to study participants. These advantages would possibly overcome the challenges of the conventional de novo discovery of new pharmacological agents (Lee and Kim, 2016; Corsello et al., 2017).
Mental and neurological illnesses are multifarious and have a substantial influence on patients as well as being a significant economic burden for nations (Hurd et al., 2013; Gooch et al., 2017). The etiological aspects of such disorders are diverse, including pathological protein accumulation causing mostly neurodegeneration and dysregulation of the normal developmental and functional process (Di Luca et al., 2018). In 2015, neurological disorders were placed as the foremost global cause of disability and the 2nd leading cause of mortality (~17% of global deaths), while Alzheimer's disease (AD) and other dementias were found to be the 4th leading cause of mortality and morbidity worldwide, and is steadily among the top three causes of disability in most nations. During the period of 1990–2015, the overall mortality from neurological illnesses was increased by 37% (Feigin et al., 2017).
Moreover, there is no pharmacological agent currently available for the curative or disease-modifying actions of most of the neurological disorders. The present therapeutic approach therefore provides symptomatic management and supportive care in order to improve the longevity and quality of life of patients (Xie et al., 2014; Cummings, 2017; Dorst et al., 2018).
Thus, the repurposing of existing drugs for central nervous system (CNS) related disorders is an attractive, efficient, and dynamic drug development approach that can substantially reduce the investment spent during drug development in terms of both time and money (Caban et al., 2017; Hernandez et al., 2017).
Ceftriaxone (CEF) is a third generation cephalosporin, under the group of β-lactam antibiotics, and is the most frequently used drug for local (skin and soft tissue infections) as well as systemic community and hospital-acquired infections (Pinto Pereira et al., 2004). Recently, emerging evidence, mainly from preclinical studies, have highlighted the therapeutic efficacy of CEF against various neurological disorders, drug dependency, and withdrawal syndrome as well as its neuroprotective potential against various neurotoxic chemicals (Tables 1–3).
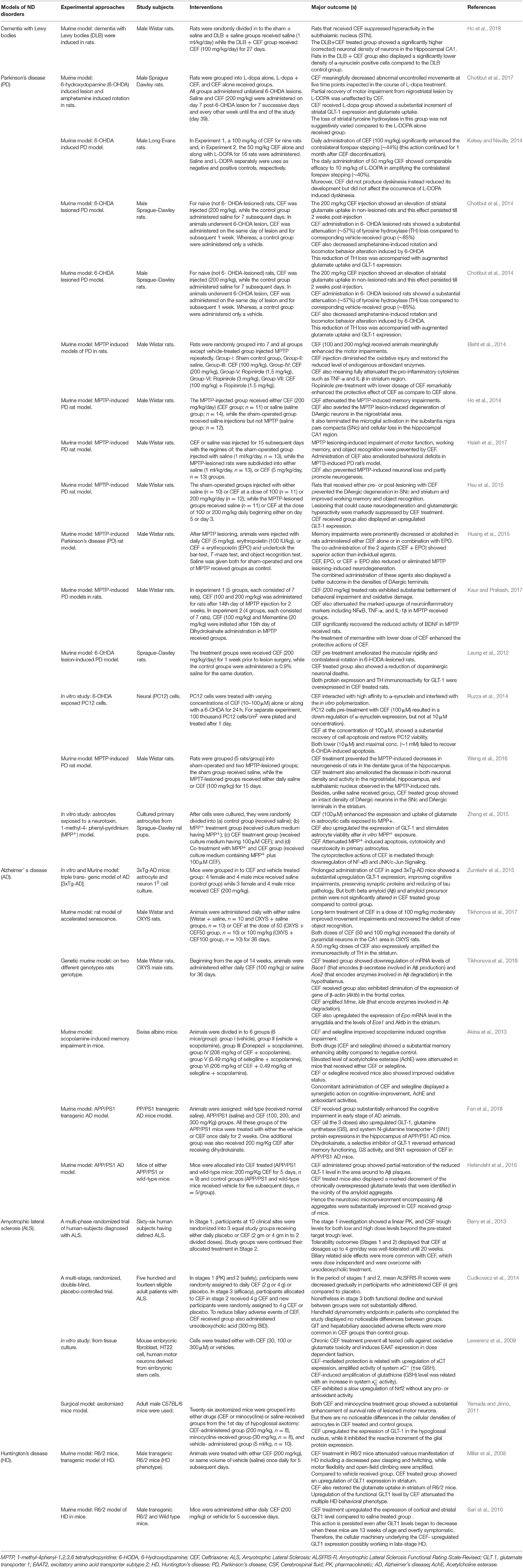
Table 1. Effects of CEF against neurodegenerative (ND) disorders.
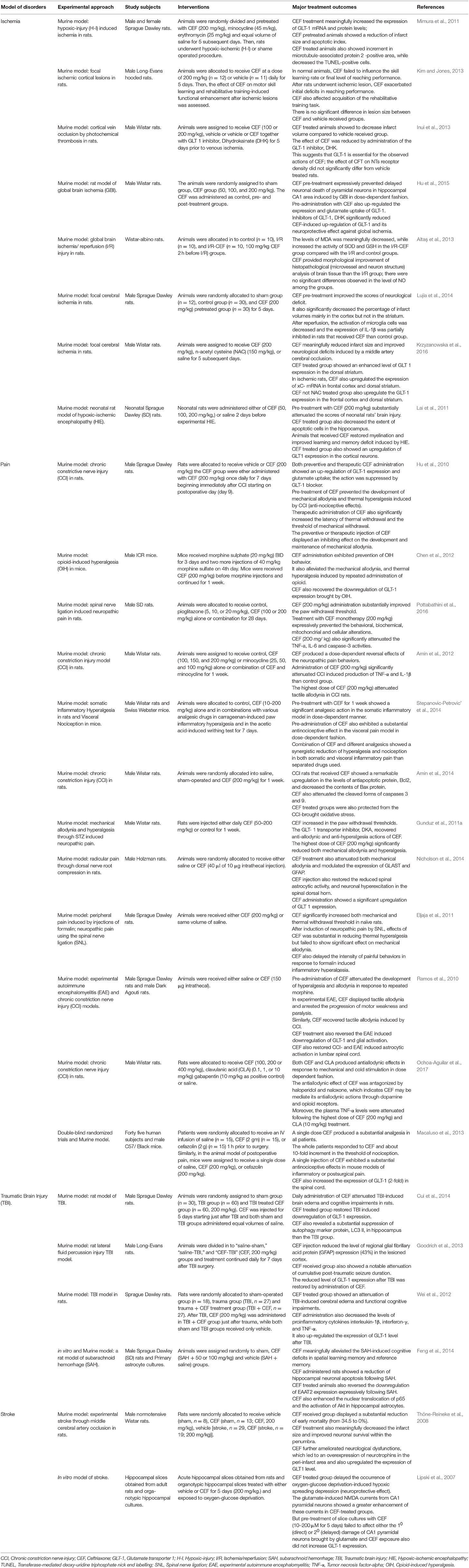
Table 2. Effect of CEF against ischemia, pain, traumatic brain injury, and stroke.
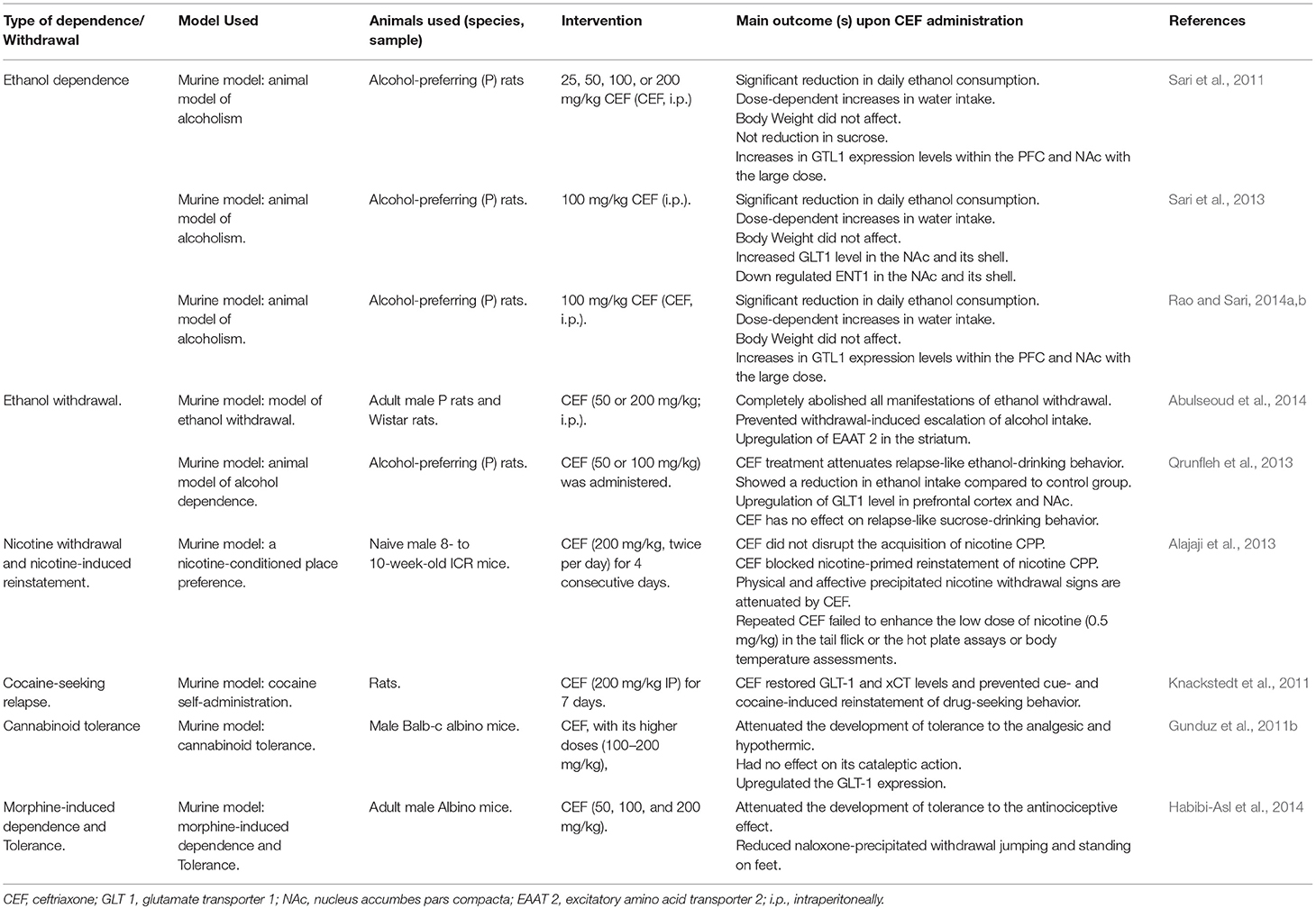
Table 3. Effect of CEF on different types drug/alcohol of dependencies and withdrawals.
Therefore, this review is intended to describe the pharmacological effects of CEF in various CNS related disorders conducted in preclinical and clinical studies along with its respective suggested neuroprotective mechanisms.
Effects of CEF Against Neurodegenerative Disorders
Neurodegenerative diseases are a diverse set of disorders characterized by a progressive loss of neuronal structure and function in distinct sections of the central nervous system. The neurological sequel of neurodegeneration results in a devastating outcome on mental as well as physical functioning of patients (Gao and Hong, 2008; Cannon and Greenamyre, 2011). Among many neurodegenerative disorders, Alzheimer's disease (AD) and Parkinson's disease (PD) are the most commonly encountered disorders (Xie et al., 2014). Huntington's disease (HD) and amyotrophic lateral sclerosis (ALS) are still devastating neurological disorders, though less prevalent (Siddique and Siddique, 2008; Franco-Iborra et al., 2018). To date, there is no cure or disease-modifying agents available for most of these neurodegenerative disorders and therapy is currently focused on symptomatic management and supportive care so as to improve the quality of life of the patients (Xie et al., 2014; Cummings, 2017).
A beta-lactam antibiotic, CEF is currently attracting the scientific community due to its multiple mechanisms to relieve symptoms and modify the natural history of various neurodegenerative diseases. Of the different possible mechanisms, upregulation of GLT-1 expression, attenuation of oxidative stress, and neuroinflammation are among the common suggested mechanisms for its neuroprotective actions (Table 1 and Figure 1). For instance, in animal models of Parkinson's disease, CEF exhibited a recovery of memory deficits (Huang et al., 2015), ameliorated abnormal uncontrolled movements (Chotibut et al., 2017), attenuated oxidative damage and restored the reduced levels of endogenous antioxidant enzymes (Bisht et al., 2014; Kaur and Prakash, 2017). Additionally, CEF modulated the expression of tyrosine hydroxylase (Chotibut et al., 2014), α-synuclein expression (Ho et al., 2018), and neuroinflammation (Kaur and Prakash, 2017) as well as prevented dopaminergic degeneration (Ho et al., 2014), while upregulating the levels of GLT-1 expression and glutamate uptake (Chotibut et al., 2014).
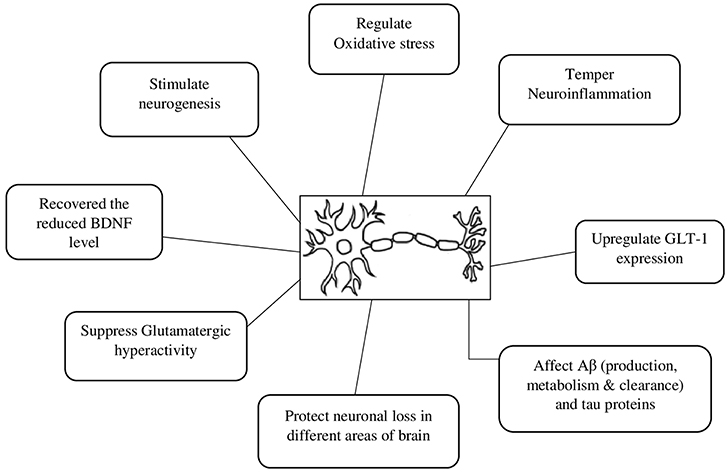
Figure 1. Possible neuroprotective mechanisms of ceftriaxone. Aβ, beta amyloid protein; BDNF, brain-derived neurotrophic factor; GLT 1, Glutamate transporter 1.
Alzheimer's disease (AD) is one of the most prevalent forms of dementia (Sadigh-Eteghad et al., 2015). Different animal models of Alzheimer's disease were affected by the administration of CEF (Table 1). Despite the most commonly underlining protein implicated in the pathology of AD, beta-amyloid (Aβ) protein was not directly affected (Zumkehr et al., 2015), CEF downregulated the messenger RNA (mRNA) expression of Bace1 (a gene that encodes β-secretase involved in Aβ formation), Ace2 (a gene that encodes enzymes play a part in Aβ metabolism), and the expression of gene of β-actin. Furthermore, CEF amplified the gene expression of Mme and Ide (genes that encode enzymes involved in Aβ degradation) as well as the expression of EPO gene (a gene that encodes erythropoietin correlated to endothelial function and removal of Aβ) (Tikhonova et al., 2018). CEF-treated animals also showed improvements in memory impairments and restoration of cognitive function and neuronal density (Tikhonova et al., 2017). CEF also attenuated increased levels of acetylcholine esterase enzyme and oxidative stress (Akina et al., 2013). In addition, CEF administration to animal models of Alzheimer's disease displayed an upregulation of GLT-1 expression, preservation of synaptic proteins and downregulation of tau proteins (Zumkehr et al., 2015). Furthermore, CEF showed neuroprotective actions in various models of amyotrophic lateral sclerosis (Lewerenz et al., 2009; Yamada and Jinno, 2011; Cudkowicz et al., 2014) and Huntington's disease (Miller et al., 2008; Sari et al., 2010).
Analgesic Effect of CEF
Analgesic effects of the beta-lactam antibiotic CEF, in different preclinical animal models (nociceptive and neuropathic pain models) have also been reported (Table 2). In a neuropathic pain model in rats, CEF-treated groups showed a meaningful overexpression of GLT-1 level and glutamate uptake in the spinal dorsal horn (Hu et al., 2010). Another study on a similar model showed that CEF significantly attenuated the production of TNF-α and IL-1β (Amin et al., 2012). CEF mitigated the increased levels of Bax and cleaved forms of caspases 3 and 9, while the expression of Bcl2 was markedly amplified (Amin et al., 2014). CEF inhibited opioid-induced hyperalgesia (OIH) in mice. Further, the beta-lactam antibiotic, CEF reversed the OIH induced downregulation of GLT-1 expression (Chen et al., 2012). Finally, CEF produced marked dose-dependent antihyperalgesic effects in the somatic inflammatory and visceral pain models (Stepanovic-Petrovic' et al., 2014).
Protective Effect of CEF Against Brain Ischemia
Various studies reported the protective role of CEF against different models of brain ischemia (Table 2). CEF pre-treatment reduced infarct volume and apoptotic index and CEF treatment significantly increased GLT-1 mRNA and protein levels (Mimura et al., 2011). In a rat model of global brain ischemia, it was found that pre-treatment with CEF substantially prevented the delayed neuronal death in the hippocampal CA1 area commonly induced by global brain ischemia (Hu et al., 2015; Table 2).
Protective Effect of CEF Against Stroke and Traumatic Brain Injury
CEF also showed protective activity on animal models of stroke (Table 2). CEF strongly reduced infarct size, marked improvement of neuronal survival within the penumbra, and diminished neurological impairment (Thöne-Reineke et al., 2008). Furthermore, CEF was found to produce promising protective effects against different models of brain injury (Cui et al., 2014; Table 2).
Effect of CEF on Alcohol Dependence and Withdrawal
Studies evidenced that glutamate transmission is implicated in various features of drug addiction (Sari et al., 2011). The foremost transporter of glutamate, GLT1, in the nucleus accumbens (NAc) and prefrontal cortex (PFC) plays a notable role in alcohol dependence behavior attenuation (Rao and Sari, 2014a). CEF is found to be the most effective β-lactam antibiotic in increasing the expression of GLT1 in the brain (Rothstein et al., 2005). Though CEF attenuated ethanol consumption in P rats, only the relatively higher doses (100 and 200 mg/kg) were associated with marked upregulation of GLT1 expression in the PFC and NAc. This increases in the expression of GLT1 appear to be inversely associated with a post treatment attenuation of ethanol intake (Sari et al., 2011). According to Lee et al. CEF induced indirect upregulation of GLT-1 expression and GSH via the activation of NF-κB, specifically at the NF-κB binding site (272 position) of the GLT-1 promoter (Lee et al., 2008). Another study has also revealed that CEF treatment improved GSH and xCT (a catalytic subunit of xC−) levels in PFC, NAc, and associated brain regions (Lewerenz et al., 2009). This CEF-induced increase in the xCT level was found to be correlated with downregulation of extracellular levels of glutamate (Sari et al., 2011), which is the very reason for alcohol dependence. On the other hand, upregulation in the levels of xCT and subsequent upsurges of GSH level may be one possible way to reverse the GLT impairments caused by free radical overproduction. Ethanol withdrawal is related to an increment of oxygen-derived free radicals (Labra Ruiz et al., 2018), which in turn have been shown to inhibit glutamate uptake by oxidation of thiol groups (Volterra et al., 1994).
During CEF treatment in ethanol dependence, an increase in water intake was observed that could be due to the compensatory effect of water to the decrease in ethanol intake (Sari et al., 2011; Table 3). On the other side, GLT1 and ENT1 were inversely affected as a consequence of ethanol consumption and this suggests that the neuroadaptative mechanisms are involving these proteins in both NAc core and shell (Sari et al., 2013).
Alcohol withdrawal syndrome is a medical emergency which is related to significant mortality rates (Campos et al., 2011). CEF administration was found to reduce or almost completely abolish all manifestations of ethanol withdrawal in P and Wistar rat variants and prevented withdrawal-induced escalation of alcohol intake. CEF treatment was also associated with long-term upregulation of excitatory amino acid transporter subtype 2 (EAAT2) in the striatum that was downregulated by ethanol withdrawal (Abulseoud et al., 2014). CEF might therefore be used as a potential therapeutic treatment for the attenuation of relapse-like ethanol-drinking behavior (Alajaji et al., 2013; Qrunfleh et al., 2013; Table 3).
Effect of CEF on Nicotine Withdrawal and Nicotine-Induced Reinstatement
Tobacco use is the foremost cause of premature mortality in the United States and around the globe. Nicotine is the primarily responsible component for addiction and related behaviors (Castane et al., 2005). Different studies suggested that nicotine-induced adaptations in glutamatergic neurotransmission play an important role in the development of nicotine dependence (Liechti and Markou, 2008). With this regard, CEF administration reversed nicotine withdrawal manifestations and mitigated nicotine-primed reinstatement of nicotine-conditioned place preference (CPP) without affecting acquisition (Alajaji et al., 2013; Table 3).
Effect of CEF on Acute Cocaine-Evoked Dopaminergic Neurotransmission
CEF exerts its prominent effects on the dopaminergic system in addition to its effects on the glutamatergic system. It decreased the negative effects of cocaine on locomotor and dopamine-activities. This effect was not responsive to a glutamate re-uptake blocker but rather affected some of the principal regulatory components of dopamine transmission in the NAc (Barr et al., 2015; Table 3).
Effect of CEF on Cannabinoid Tolerance
CEF prevented the development of tolerance to the analgesic and hypothermic actions of WIN 55,212-2 (cannabinoid receptor agonist) by increasing glutamate uptake. Even though the mechanism is not clear, stimulation of GLT-1 by CEF is one of the suggested mechanisms in preventing the development of tolerance to cannabinoids (Gunduz et al., 2011b; Table 3).
Effect of CEF on Morphine-Induced Dependence
Morphine-induced tolerance and dependence were diminished when mice were pretreated with amitriptyline and CEF alone or in combination. Moreover, the co-administration of low doses of CEF (50 mg/kg) and amitriptyline (5 mg/kg) significantly attenuated morphine-induced dependence compared to the administration of CEF or amitriptyline alone. This enhanced effect could be due to the additive or synergistic effect of CEF and amitriptyline on morphine-induced drug dependence (Habibi-Asl et al., 2014; Table 3).
Antidepressant Effects of CEF
For a long time, depressive disorder (DD) was thought to be attributed mainly to biochemical alterations of the monoamines and their receptors (Labra Ruiz et al., 2018). A majority of the currently available antidepressants were developed based on this hypothesis and act by increasing the availability of the monoamines including norepinephrine, dopamine, and serotonin. This is effected either by preventing the metabolism of these neurotransmitters or by blocking the transporter mediated reuptake of the these neurotransmitters (Delgado, 2000; Fiedorowicz and Swartz, 2004).
Moreover, DD is thought to be related to excessive glutamatergic neurotransmission. In such cases increased extracellular concentrations of glutamate were observed in several brain regions (Lowy et al., 2002). In another way, GLT1 expression is elevated as a compensatory mechanism to overcome the increased stress-induced glutamate release (Delgado, 2000). Recent evidence revealed that the β-lactam antibiotic, CEF enhanced the uptake of glutamate via the up-regulation of GLT1, suggesting its antidepressant effect. CEF treated animals also showed a marked decrement of immobility in the forced swim and tail suspension tests. Even though not statistically significant, a similar trend was noted in novelty-suppressed feeding (Mineur et al., 2007; Borah et al., 2018). A more recent study also showed that the antidepressant effect of CEF was comparable to that of fluoxetine in the tail suspension test in a dose-dependent manner (Borah et al., 2018).
Effects of CEF in Seizure Modulation
It was assumed that oxidative stress and reactive oxygen species (ROS) produced an important role in the progression of epileptic-seizures because they gradually disrupt the intracellular calcium homeostasis, which leads to neuronal loss (Chang and Yu, 2010). A study revealed that pre-treatment of animals with CEF provided meaningful protective actions against pentylenetetrazole (PTZ)-induced generalized clonic seizure, generalized tonic-clonic seizure, and convulsion-associated deaths (Jelenkovic et al., 2008).
A similar study also showed that CEF treatment substantially improved PTZ-induced convulsions and caused a noticeable modulation of oxidative stress indicators and Connexin 43 expression in the CA3 region of the hippocampal area. CEF also prominently reduced tonic-clonic convulsions and duration of these convulsions and prolonged the latency time in the PTZ-kindling model (Hussein et al., 2016).
The Possible Mechanism of Actions of CEF in Neurological Disorders
After induction of different models of neurological illness by lesioning or chemicals, it is known that animals manifest behavioral alterations in early onset and in the long-term they might also exhibit memory deficits (Van Dam and De Deyn, 2011; Savio et al., 2012; More et al., 2016). Numerous studies evidenced that CEF substantially improved such chemical-induced behavioral alteration and memory impairments in different rodent models of neurological conditions (Lai et al., 2011; Wei et al., 2012; Akina et al., 2013; Chotibut et al., 2014; Feng et al., 2014; Hsieh et al., 2017; Kaur and Prakash, 2017; Fan et al., 2018).
It is well-known that an abnormally increased level of glutamate (Glu) in the brain can induce neuronal damage and excitotoxicity that contributes to the pathogenesis of various neurological disorders including epilepsy, ALS, cerebral ischemia, parkinsonism, and AD (Massie et al., 2010; Annweiler et al., 2014; Kleteckova et al., 2014). There are different glutamate transporters involved in terminating glutamatergic transmission and physiological actions (Kanai et al., 2013; Divito and Underhill, 2014). The pre-synaptic glutamate transporter, GLT-1, clears most of the glutamate released in the cortex and hippocampus (Scofield and Kalivas, 2014). Furthermore, GLT-1 is one of the most common transporters and consists of about 80% of the glutamate transporters expressed in the hippocampus (Mookherjee et al., 2011). There is also evolving evidence suggesting a blockade of certain Glu receptors and/ or enhancing the expression of GLT-1 shown to improve neurological outcomes in various experimental models of neurological illnesses (Zlotnik et al., 2008, 2009; Wang et al., 2014; Bai et al., 2016). Among different agents tested, CEF is one of the beta-lactam antibiotics reported to have neuroprotective actions.
Several studies have revealed that the mechanism of action behind the beneficial effect of CEF in neurodegenerative diseases like PD (Leung et al., 2012; Chotibut et al., 2014, 2017; Hsu et al., 2015; Zhang et al., 2015), AD (Zumkehr et al., 2015), HD (Miller et al., 2008; Sari et al., 2010), and ALS (Yamada and Jinno, 2011) is mediated by enhancing the expression of GLT-1 mRNA levels. Similarly, the mechanism behind CEF's analgesic potential has been associated with the upregulation of the pre-synaptic GLT expression (Ramos et al., 2010; Gunduz et al., 2011a; Chen et al., 2012; Macaluso et al., 2013; Nicholson et al., 2014).
The upregulation of GLT-1 expression is associated with the significant attenuation of the neuronal damage caused by brain ischemia, and the overexpression of GLT-1 in the ischemic cortex is associated with a reduction of the size of the lesion and improves behavioral and cognitive recovery (Harvey et al., 2011). Studies also showed the neuroprotective actions of CEF in different models of ischemia (Lai et al., 2011; Mimura et al., 2011; Inui et al., 2013; Hu et al., 2015; Krzyzanowska et al., 2016), TBI (Goodrich et al., 2013; Cui et al., 2014; Feng et al., 2014), and stroke (Lipski et al., 2007; Thöne-Reineke et al., 2008) supposedly via an increased expression of GLT-1.
Abnormal regulation of glutamatergic neurotransmission, due to an excessive amount of Glu in brain reward circuitry, has been involved in both initiation and expression of addiction to the drug of abuse related behavior (Kalivas et al., 2009; D'Souza, 2015; Liu et al., 2017). It has also been demonstrated that the expression of the catalytic subunit of xC−, xCT, and GLT-1 are reduced in the NAc following substance of abuse (Massie et al., 2015; Roberts-Wolfe and Kalivas, 2015). From various preclinical studies, it has been reported that CEF markedly attenuates alcohol/ drug-seeking behavior and drug clued-up recall while restoring the reduced levels of both xCT as well as GLT-1 (Knackstedt et al., 2011; Sari et al., 2011, 2013; Qrunfleh et al., 2013; Abulseoud et al., 2014).
Long-lasting neuroinflammation and oxidative stress are the other pathological processes involved in brain aging and neurodegeneration (Lee et al., 2008; Mishra et al., 2016; Shah et al., 2016). It has been reported that neuroinflammation is a key player in various neurological disorders, including neurodegenerative illnesses and CNS injury (Chen et al., 2016; Yang and Zhou, 2018). Hence, controlling of the neuroinflammation and excitotoxicity are supposed to limit the abnormal alterations and progression of different neurological disorders (Zhou and Hu, 2013; Yimer et al., 2019), which is further supported by evidence reported, where the use of anti-inflammatory agents or targeting inflammatory markers prominently prevented or at least reduced the progression of neurological disorders (Gagne and Power, 2010; Decourt et al., 2016). Fortunately, pre-treatment of CEF showed attenuation of proinflammatory mediators including NF-κB, IL-1β, INF-γ, and/ or TNF-α in various models of neurological disorders such as PD (Kaur and Prakash, 2017), TBI (Wei et al., 2012), neuropathic pain (Pottabathini et al., 2016), and cerebral ischemia (Lujia et al., 2014), which might contribute its own share of reported neuroprotective actions of CEF.
In addition to neuroinflammation, oxidative stress, and mitochondrial dysfunction have also been involved in the development and progression of a wide range of neurodegenerative and mental disorders (Tobe, 2013; Rossignol and Frye, 2014; Yimer et al., 2019). There were also some reports suggesting that antioxidant therapy moderated the production of reactive oxygen species and prevented downstream pathologies of certain neurological diseases (Uttara et al., 2009; Du et al., 2017). Similarly, there are reports that animal models of different neurological disorder received CEF, which attenuated oxidative stress by either reducing oxidative markers such as MDA or by improving the endogenous antioxidant actions such as GSH and SOD (Lewerenz et al., 2009; Altaş et al., 2013).
BDNF is a neurotrophic factor which plays a crucial role in neuronal survival, neurogenesis, and plasticity. It has also been reported that abnormal BDNF expression is involved in several neurological illnesses. There are few studies that report CEF caused upregulation and restoration of reduced BDNF levels in certain animal models (Lee et al., 2008; Kaur and Prakash, 2017).
Besides, CEF might exert its neuroprotective actions through other mechanisms such as by affecting Aβ and tau protein metabolism and clearance in an AD model, prevent polymerization of α-synuclein in DLB (Ho et al., 2018) and PD (Ruzza et al., 2014; Tikhonova et al., 2018) models, which is of course, a call for further research to substantiate that the neuroprotective actions of CEF is also mediated via these important pathological proteins.
Generally, it appears that the shared mechanism of actions of CEF in the range of various neurologic disorders is through the upregulation of GLT-1 expression and the reduction of proinflammatory mediators and oxidative stress. However, this requires further investigation in order to verify the suggested and possible other newer neuroprotective mechanisms of CEF for each neurological ailment.
Conclusion
This review revealed that the beta-lactam antibiotic, CEF might have neuroprotective actions, which can affect a wide array of neurological disorders including AD, PD, HD, stroke, brain ischemia, seizure, and drug/alcohol dependency and withdrawal. It is particularly interesting as CEF affects the wider pathological states of CNS disorders including glutamatergic system, oxidative stress, neuroinflammation, apoptotic index, and various toxic protein aggregations. Since most of the studies conducted so far are preclinical studies, clinical studies such as randomized clinical trials and further mechanistic studies are required to assure its neuroprotective actions in real clinical scenarios.
Author Contributions
EY developed the research concept and took initiatives of the work by drafting the manuscript while KT and HH provided greater contributions toward collecting, extracting, and organizing relevant data and also revising the review paper and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CEF, Ceftriaxone; AD, Alzheimer's disease; HD, Huntington's disease; PD, Parkinson's disease; CNS, central nervous system; TBI, Traumatic brain injury; GLT 1, Glutamate transporter 1; NAc, Nucleus accumbens; PFC, Prefrontal cortex; EAAT2, excitatory amino acid transporter subtype 2
References
Abulseoud, O. A., Camsari, U. M., Ruby, C. L., Kasasbeh, A., Choi, S., and Choi, D. S. (2014). Attenuation of ethanol withdrawal by ceftriaxone-induced upregulation of glutamate transporter EAAT2. Neuropsychopharmacology 39, 1674–1684. doi: 10.1038/npp.2014.14
Akina, S., Thati, M., and Puchchakayala, G. (2013). Neuroprotective effect of ceftriaxone and selegiline on scopolamine induced cognitive impairment in mice. Adv. Biol. Res. 7, 266–275. doi: 10.5829/idosi.abr.2013.7.6.75119
Alajaji, M., Bowers, M. S., Knackstedt, L., and Damaj, M. I. (2013). Effects of the beta-lactam antibiotic ceftriaxone on nicotine withdrawal and nicotine-induced reinstatement of preference in mice. Psychopharmacology 228, 419–426. doi: 10.1007/s00213-013-3047-3
Altaş, M., Meydan, S., Aras, M., Yilmaz, N., Ulutaş, K. T., Okuyan, H. M., et al. (2013). Effects of ceftriaxone on ischemia/reperfusion injury in rat brain. J. Clin. Neurosci. 20, 457–461. doi: 10.1016/j.jocn.2012.05.030
Amin, B., Hajhashemi, V., Abnous, K., and Hosseinzadeh, H. (2014). Ceftriaxone, a Beta-Lactam antibiotic, modulates apoptosis pathways and oxidative stress in a rat model of neuropathic pain. Biomed Res. Int. 2014:937568. doi: 10.1155/2014/937568
Amin, B., Hajhashemi, V., Hosseinzadeh, H., and Abnous, K. (2012). Antinociceptive evaluation of ceftriaxone and minocycline alone and in combination in a neuropathic pain model in rat. Neuroscience 224, 15–25. doi: 10.1016/j.neuroscience.2012.07.058
Annweiler, C., Brugg, B., Peyrin, J. M., Bartha, R., and Beauchet, O. (2014). Combination of memantine and vitamin D prevents axon degeneration induced by amyloid-beta and glutamate. Neurobiol. Aging 35, 331–335. doi: 10.1016/j.neurobiolaging.2013.07.029
Bai, X., Zhang, C., Chen, A., Liu, W., Li, J., Sun, Q., et al. (2016). Protective effect of edaravone on glutamate-induced neurotoxicity in spiral ganglion neurons. Neural Plast. 2016:4034218. doi: 10.1155/2016/4034218
Barr, J. L., Rasmussen, B. A., Tallarida, C. S., Scholl, J. L., Forster, G. L., Unterwald, E. M., et al. (2015). Ceftriaxone attenuates acute cocaine-evoked dopaminergic neurotransmission in the nucleus accumbens of the rat. Br. J. Pharmacol. 172, 5414–5424. doi: 10.1111/bph.13330
Berry, J. D., Shefner, J. M., Conwit, R., Schoenfeld, D., Keroack, M., Felsenstein, D., et al. (2013). Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS ONE 8:61177. doi: 10.1371/journal.pone.0061177
Bisht, R., Kaur, B., Gupta, H., and Prakash, A. (2014). Ceftriaxone mediated rescue of nigral oxidative damage and motor deficits in MPTP model of Parkinson's disease in rats. Neurotoxicology 44, 71–79. doi: 10.1016/j.neuro.2014.05.009
Borah, A., Singha, B., and Phukan, S. (2018). Anti-depressant effect of ceftriaxone in forced swimming test and in tail suspension test in mice. Int. J. Pharm. Sci. 75, 191–194. doi: 10.22159/ijpps.2016v8i11.14466
Caban, A., Pisarczyk, K., Kopacz, K., Kapuśniak, A., Toumi, M., Rémuzat, C., et al. (2017). Filling the gap in CNS drug development: evaluation of the role of drug repurposing. J. Mark. Access Heal. Policy 5:1299833. doi: 10.1080/20016689.2017.1299833
Campos, J., Roca, L., and Gude, F. G.-Q. A (2011). Long-term mortality of patients admitted to the hospital with alcohol withdrawal syndrome. Alcohol. Clin. Exp. Res. 35, 1180–1186. doi: 10.1111/j.1530-0277.2011.01451.x
Cannon, J. R., and Greenamyre, J. T. (2011). The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 124, 225–250. doi: 10.1093/toxsci/kfr239
Castane, A., Berrendero, F., and Maldonado, R. (2005). The role of the cannabinoid system in nicotine addiction. Pharm. Biochem Behav 81, 381–386. doi: 10.1016/j.pbb.2005.01.025
Chang, S., and Yu, B. (2010). Mitochondrial matters of the brain: mitochondrial dysfunction and oxidative status in epilepsy. J. Bioenerg. Biomembr. 41, 457–459. doi: 10.1007/s10863-010-9317-4
Chen, W. W., Zhang, X., and Huang, W. J. (2016). Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 13, 3391–3396. doi: 10.3892/mmr.2016.4948
Chen, Z., He, Y., and Wang, Z. J. (2012). The beta-lactam antibiotic, ceftriaxone, inhibits the development of opioid-induced hyperalgesia in mice. Neurosci. Lett. 509, 69–71. doi: 10.1016/j.neulet.2011.12.029
Chotibut, T., Davis, R. W., Arnold, J. C., Frenchek, Z., Gurwara, S., Bondada, V., et al. (2014). Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson's model. Mol. Neurobiol. 49, 1282–1292. doi: 10.1007/s12035-013-8598-0
Chotibut, T., Meadows, S., Kasanga, E. A., McInnis, T., Cantu, M. A., Bishop, C., et al. (2017). Ceftriaxone reduces L-dopa–induced dyskinesia severity in 6-hydroxydopamine parkinson's disease model. Mov. Disord. 32, 1547–1556. doi: 10.1002/mds.27077
Corsello, S. M., Bittker, J. A., Liu, Z., Gould, J., McCarren, P., Hirschman, J. E., et al. (2017). The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 23, 405–408. doi: 10.1038/nm.4306
Cudkowicz, M. E., Titus, S., Kearney, M., Yu, H., Sherman, A., Schoenfeld, D., et al. (2014). Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 13, 1083–1091. doi: 10.1016/S1474-4422(14)70222-4
Cui, C., Cui, Y., Gao, J., Sun, L., Wang, Y., Wang, K., et al. (2014). Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol. Sci. 35, 695–700. doi: 10.1007/s10072-013-1585-4
Cummings, J. (2017). Disease modification and neuroprotection in neurodegenerative disorders. Transl. Neurodegener. 6:25. doi: 10.1186/s40035-017-0096-2
Decourt, B., Lahiri, D., and Sabbagh, M. (2016). Targeting tumor necrosis factor alpha for alzheimer's disease. Curr. Alzheimer Res. 13, 1–1. doi: 10.2174/1567205013666160930110551
Delgado, P. L. (2000). Depression: the case for a monoamine deficiency. J. Clin. Psychiatry (61 Suppl. 6), 7–11. doi: 10.1073/pnas.0307294101
Di Luca, M., Nutt, D., Oertel, W., Boyer, P., Jaarsma, J., Destrebecq, F., et al. (2018). Towards earlier diagnosis and treatment of disorders of the brain. Bull. World Health Organ. 96, 298–298A. doi: 10.2471/BLT.17.206599
Divito, C. B., and Underhill, S. M. (2014). Excitatory amino acid transporters: roles in glutamatergic neurotransmission. Neurochem. Int. 73, 172–180. doi: 10.1016/j.neuint.2013.12.008
Dorst, J., Ludolph, A. C., and Huebers, A. (2018). Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 11:175628561773473. doi: 10.1177/1756285617734734
D'Souza, M. S. (2015). Glutamatergic transmission in drug reward: implications for drug addiction. Front. Neurosci. 9:404. doi: 10.3389/fnins.2015.00404
Du, X., West, M. B., Cai, Q., Cheng, W., Ewert, D. L., Li, W., et al. (2017). Antioxidants reduce neurodegeneration and accumulation of pathologic Tau proteins in the auditory system after blast exposure. Free Radic. Biol. Med. 108, 627–643. doi: 10.1016/j.freeradbiomed.2017.04.343
Eljaja, L., Bjerrum, O. J., Honoré, P. H., and Abrahamsen, B. (2011). Effects of the excitatory amino acid transporter subtype 2 (EAAT-2) inducer ceftriaxone on different pain modalities in rat. Scand. J. Pain 2, 132–136. doi: 10.1016/j.sjpain.2011.03.003
Fan, S., Xian, X., Li, L., Yao, X., Hu, Y., Zhang, M., et al. (2018). Ceftriaxone improves cognitive function and upregulates glt-1-related glutamate-glutamine cycle in APP/PS1 mice. J. Alzheimers Dis. 66, 1731–1743. doi: 10.3233/JAD-180708
Feigin, V. L., Abajobir, A. A., Abate, K. H., Abd-Allah, F., Abdulle, A. M., Abera, S. F., et al. (2017). Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16, 877–897. doi: 10.1016/S1474-4422(17)30299-5
Feng, D., Wang, W., Dong, Y., Wu, L., Huang, J., Ma, Y., et al. (2014). Ceftriaxone alleviates early brain injury after subarachnoid hemorrhage by increasing excitatory amino acid transporter 2 expression via the PI3K/Akt/NF-κB signaling pathway. Neuroscience 268, 21–32. doi: 10.1016/j.neuroscience.2014.02.053
Fiedorowicz, J. G., and Swartz, K. L. (2004). The role of monoamine oxidase inhibitors in current psychiatric practice. J. Psychiatr. Pract. 10, 239–248. doi: 10.1016/j.neuron.2013.04.027
Franco-Iborra, S., Vila, M., and Perier, C. (2018). Mitochondrial Quality control in neurodegenerative diseases: focus on parkinson's disease and huntington's disease. Front. Neurosci. 12:342. doi: 10.3389/fnins.2018.00342
Gagne, J. J., and Power, M. C. (2010). Anti-inflammatory drugs and risk of parkinson disease: a meta-analysis. Neurology 74, 995–1002. doi: 10.1212/WNL.0b013e3181d5a4a3
Gao, H.-M., and Hong, J.-S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
Gooch, C. L., Pracht, E., and Borenstein, A. R. (2017). The burden of neurological disease in the United States: a summary report and call to action. Ann. Neurol. 81, 479–484. doi: 10.1002/ana.24897
Goodrich, G. S., Kabakov, A. Y., Hameed, M. Q., Dhamne, S. C., Rosenberg, P. A., and Rotenberg, A. (2013). Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 30, 1434–1441. doi: 10.1089/neu.2012.2712
Gunduz, O., Oltulu, C., Buldum, D., Guven, R., and Ulugol, A. (2011a). Anti-allodynic and anti-hyperalgesic effects of ceftriaxone in streptozocin-induced diabetic rats. Neurosci. Lett. 491, 23–25. doi: 10.1016/j.neulet.2010.12.063
Gunduz, O., Oltulu, C., and Ulugol, A. (2011b). Role of GLT-1 transporter activation in prevention of cannabinoid tolerance by the beta-lactam antibiotic, ceftriaxone, in mice. Pharmacol. Biochem. Behav. 99, 100–103. doi: 10.1016/j.pbb.2011.04.012
Habibi-Asl, B., Vaez, H., Najafi, M., Bidaghi, A., and Ghanbarzadeh, S. (2014). Attenuation of morphine-induced dependence and tolerance by ceftriaxone and amitriptyline in mice. Acta Anaesthesiol. Taiwanica 52, 163–168. doi: 10.1016/j.aat.2014.11.001
Harvey, B. K., Airavaara, M., Hinzman, J., Wires, E. M., Chiocco, M. J., Howard, D. B., et al. (2011). Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 6:22135. doi: 10.1371/journal.pone.0022135
Hefendehl, J. K., LeDue, J., Ko, R. W. Y., Mahler, J., Murphy, T. H., and MacVicar, B. A. (2016). Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun. 7:13441. doi: 10.1038/ncomms13441
Hernandez, J. J., Pryszlak, M., Smith, L., Yanchus, C., Kurji, N., Shahani, V. M., et al. (2017). Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as cancer therapeutics. Front. Oncol. 7:273. doi: 10.3389/fonc.2017.00273
Ho, S. C., Hsu, C. C., Pawlak, C. R., Tikhonova, M. A., Lai, T. J., Amstislavskaya, T. G., et al. (2014). Effects of ceftriaxone on the behavioral and neuronal changes in an MPTP-induced Parkinson's disease rat model. Behav. Brain Res. 268, 177–184. doi: 10.1016/j.bbr.2014.04.022
Ho, Y., Weng, J., Lin, C., Shen, M., Li, H., Liao, W., et al. (2018). Ceftriaxone treatment for neuronal deficits: a histological and MEMRI study in a rat model of dementia with lewy bodies. Behav. Neurol. 2018:4618716. doi: 10.1155/2018/4618716
Hsieh, M. H., Meng, W. Y., Liao, W. C., Weng, J. C., Li, H. H., Su, H. L., et al. (2017). Ceftriaxone reverses deficits of behavior and neurogenesis in an MPTP-induced rat model of Parkinson's disease dementia. Brain Res. Bull. 132, 129–138. doi: 10.1016/j.brainresbull.2017.05.015
Hsu, C.-Y., Hung, C.-S., Chang, H.-M., Liao, W.-C., Ho, S.-C., and Ho, Y.-J. (2015). Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson's disease dementia. Neuropharmacology 91, 43–56. doi: 10.1016/j.neuropharm.2014.11.023
Hu, Y., Li, W., Lu, L., Cai, J., Xian, X., Zhang, M., et al. (2010). An anti-nociceptive role for ceftriaxone in chronic neuropathic pain in rats. Pain 148, 284–301. doi: 10.1016/j.pain.2009.11.014
Hu, Y. Y., Xu, J., Zhang, M., Wang, D., Li, L., Li, W., and Bin (2015). Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J. Neurochem. 132, 194–205. doi: 10.1111/jnc.12958
Huang, C. K., Chang, Y. T., Amstislavskaya, T. G., Tikhonova, M. A., Lin, C. L., Hung, C. S., et al. (2015). Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson's disease dementia. Behav. Brain Res. 294, 198–207. doi: 10.1016/j.bbr.2015.08.011
Hurd, M. D., Martorell, P., Delavande, A., Mullen, K. J., and Langa, K. M. (2013). Monetary costs of dementia in the united States. N. Engl. J. Med. 368, 1326–1334. doi: 10.1056/NEJMsa1204629
Hussein, A. M., Ghalwash, M., Magdy, K., and Abulseoud, O. A. (2016). Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J. Epilepsy Res. 6, 8–15. doi: 10.14581/jer.16002
Inui, T., Alessandri, B., Heimann, A., Nishimura, F., Frauenknecht, K., Sommer, C., et al. (2013). Neuroprotective effect of ceftriaxone on the penumbra in a rat venous ischemia model. Neuroscience 242, 1–10. doi: 10.1016/j.neuroscience.2013.03.018
Jelenkovic, A. V., Jovanovic, M. D., Stanimirovic, D. D., Bokonjic, D. D., Ocic, G. G., and Boskovic, S. (2008). Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions. Exp. Biol. Med. 233, 1389–1394. doi: 10.3181/0803-RM-83
Kalivas, P. W., LaLumiere, R. T., Knackstedt, L., and Shen, H. (2009). Glutamate transmission in addiction. Neuropharmacology 56, 169–173. doi: 10.1016/j.neuropharm.2008.07.011
Kanai, Y., Clémençon, B., Simonin, A., Leuenberger, M., Lochner, M., Weisstanner, M., et al. (2013). The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Aspects Med. 34, 108–120. doi: 10.1016/j.mam.2013.01.001
Kaur, B., and Prakash, A. (2017). Ceftriaxone attenuates glutamate-mediated neuro-inflammation and restores BDNF in MPTP model of Parkinson's disease in rats. Pathophysiology 24, 71–79. doi: 10.1016/j.pathophys.2017.02.001
Kelsey, J. E., and Neville, C. (2014). The effects of the β-lactam antibiotic, ceftriaxone, on forepaw stepping and l-DOPA-induced dyskinesia in a rodent model of Parkinson's disease. Psychopharmacology 231, 2405–2415. doi: 10.1007/s00213-013-3400-6
Kim, S. Y., and Jones, T. A. (2013). The effects of ceftriaxone on skill learning and motor functional outcome after ischemic cortical damage in rats. Restor. Neurol. Neurosci. 31, 87–97. doi: 10.3233/RNN-2012-120245
Kim, T.-W. (2015). Drug repositioning approaches for the discovery of new therapeutics for alzheimer's disease. Neurotherapeutics 12, 132–142. doi: 10.1007/s13311-014-0325-7
Kleteckova, L., Tsenov, G., Kubova, H., Stuchlik, A., and Vales, K. (2014). Neuroprotective effect of the 3α5β-pregnanolone glutamate treatment in the model of focal cerebral ischemia in immature rats. Neurosci. Lett. 564, 11–15. doi: 10.1016/j.neulet.2014.01.057
Knackstedt, A. L., Melendez, R. I., and Kalivas, P. W. (2011). Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine-seeking. Biol. Psychiatry 67, 81–84. doi: 10.1016/j.biopsych.2009.07.018.Ceftriaxone
Krzyzanowska, W., Pomierny, B., Budziszewska, B., Filip, M., and Pera, J. (2016). N-acetylcysteine and ceftriaxone as preconditioning strategies in focal brain ischemia: influence on glutamate transporters expression. Neurotox. Res. 29, 539–550. doi: 10.1007/s12640-016-9602-z
Labra Ruiz, N. A., Santamaría del Ángel, D., Juárez Olguín, H., and Lindoro Silva, M. (2018). Neuroprogression: the hidden mechanism of depression. Neuropsychiatr. Dis. Treat. 14, 2837–2845. Available online at: http://www.ijp-online.com/text.asp?2010/42/5/332/70404
Lai, P., Huang, Y., Wu, C., Lai, C. J., Wang, P., and Chiu, T. H. (2011). Ceftriaxone attenuates hypoxic-ischemic brain injury in neonatal rats. J. Biomed. Sci. 18:69. doi: 10.1186/1423-0127-18-69
Lee, H.-M., and Kim, Y. (2016). Drug repurposing is a new opportunity for developing drugs against neuropsychiatric disorders. Schizophr. Res. Treat. 2016, 1–12. doi: 10.1155/2016/6378137
Lee, S.-G., Su, Z.-Z., Emdad, L., Gupta, P., Sarkar, D., Borjabad, A., et al. (2008). Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 283, 13116–13123. doi: 10.1074/jbc.M707697200
Leung, T. C. H., Lui, C. N. P., Chen, L. W., Yung, W. H., Chan, Y. S., and Yung, K. K. L. (2012). Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem. Neurosci. 3, 22–30. doi: 10.1021/cn200072h
Lewerenz, J., Albrecht, P., Tien, M. L. T., Henke, N., Karumbayaram, S., Kornblum, H. I., et al. (2009). Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem. 111, 332–343. doi: 10.1111/j.1471-4159.2009.06347.x
Liechti, M., and Markou, A. (2008). Role of the glutamatergic system in nicotine dependence: implications for the discovery and development of new pharmacological smoking cessation therapies. CNS Drugs 22, 705–724. doi: 10.2165/00023210-200822090-00001
Lipski, J., Wan, C. K., Bai, J. Z., Pi, R., Li, D., and Donnelly, D. (2007). Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience 146, 617–629. doi: 10.1016/j.neuroscience.2007.02.003
Liu, X.-L., Li, L., Li, J.-N., Tang, J.-H., Rong, J.-H., Liu, B., et al. (2017). Quantifying absolute glutamate concentrations in nucleus accumbens of prescription opioid addicts by using 1 H MRS. Brain Behav. 7:e00769. doi: 10.1002/brb3.769
Lowy, M. T., Wittenberg, L., and Yamamoto, B. K. (2002). Effect of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats. J. Neurochem. 65, 268–274. doi: 10.1046/j.1471-4159.1995.65010268.x
Lujia, Y., Xin, L., Shiquan, W., Yu, C., Shuzhuo, Z., and Hong, Z. (2014). Ceftriaxone pretreatment protects rats against cerebral ischemic injury by attenuating microglial activation-induced IL-1β expression. Int. J. Neurosci. 124, 657–665. doi: 10.3109/00207454.2013.856009
Macaluso, A., Bernabucci, M., Trabucco, A., Ciolli, L., Troisi, F., Baldini, R., et al. (2013). Analgesic effect of a single preoperative dose of the antibiotic ceftriaxone in humans. J. Pain 14, 604–612. doi: 10.1016/j.jpain.2013.01.774
Massie, A., Boillée, S., Hewett, S., Knackstedt, L., and Lewerenz, J. (2015). Main path and byways: non-vesicular glutamate release by system x c – as an important modifier of glutamatergic neurotransmission. J. Neurochem. 135, 1062–1079. doi: 10.1111/jnc.13348
Massie, A., Goursaud, S., Schallier, A., Vermoesen, K., Meshul, C. K., Hermans, E., et al. (2010). Time-dependent changes in GLT-1 functioning in striatum of hemi-Parkinson rats. Neurochem. Int. 57, 572–578. doi: 10.1016/j.neuint.2010.07.004
Miller, B. R., Dorner, J. L., Shou, M., Sari, Y., Barton, S. J., Sengelaub, D. R., et al. (2008). Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience 153, 329–337. doi: 10.1016/j.neuroscience.2008.02.004
Mimura, K., Tomimatsu, T., Minato, K., Jugder, O., Kinugasa-Taniguchi, Y., Kanagawa, T., et al. (2011). Ceftriaxone preconditioning confers neuroprotection in neonatal rats through glutamate transporter 1 upregulation. Reprod. Sci. 18, 1193–1201. doi: 10.1177/1933719111410710
Mineur, Y. S., Picciotto, M. R., and Sanacora, G. (2007). Antidepressant-like effects of ceftriaxone in male. Biol. Psychiatry 61, 205–252. doi: 10.1016/j.biopsych.2006.04.037
Mishra, V., Shuai, B., Kodali, M., Shetty, G. A., Hattiangady, B., Rao, X., et al. (2016). Resveratrol treatment after status epilepticus restrains neurodegeneration and abnormal neurogenesis with suppression of oxidative stress and inflammation. Sci. Rep. 5:17807. doi: 10.1038/srep17807
Mookherjee, P., Green, P. S., Watson, G. S., Marques, M. A., Tanaka, K., Meeker, K. D., et al. (2011). GLT-1 loss accelerates cognitive deficit onset in an Alzheimer's disease animal model. J. Alzheimers Dis. 26, 447–455. doi: 10.3233/JAD-2011-110503
More, S., Kumar, H., Cho, D.-Y., Yun, Y.-S., and Choi, D.-K. (2016). Toxin-induced experimental models of learning and memory impairment. Int. J. Mol. Sci. 17:E1447. doi: 10.3390/ijms17091447
Nicholson, K. J., Gilliland, T. M., and Winkelstein, B. A. (2014). Upregulation of GLT-1 by treatment with ceftriaxone alleviates radicular pain by reducing spinal astrocyte activation and neuronal hyperexcitability. J. Neurosci. Res. 92, 116–129. doi: 10.1002/jnr.23295
Ochoa-Aguilar, A., Sotomayor-Sobrino, M. A., Jaimez, R., Rodríguez, R., Plancarte-Sánchez, R., and Ventura-Martinez, R. (2017). Antiallodynic activity of ceftriaxone and clavulanic acid in acute administration is associated with serum tnf-α modulation and activation of dopaminergic and opioidergic systems. Drug Dev. Res. 78, 105–115. doi: 10.1002/ddr.21381
Parsons, C. G. (2018). CNS repurposing–potential new uses for old drugs: examples of screens for alzheimer's disease, parkinson's disease and spasticity. Neuropharmacology. 147, 4–10. doi: 10.1016/j.neuropharm.2018.08.027
Pinto Pereira, L. M., Phillips, M., Ramlal, H., Teemul, K., and Prabhakar, P. (2004). Third generation cephalosporin use in a tertiary hospital in Port of Spain, trinidad: need for an antibiotic policy. BMC Infect. Dis. 4:59. doi: 10.1186/1471-2334-4-59
Pottabathini, R., Kumar, A., Bhatnagar, A., Garg, S., and Ekavali (2016). Ameliorative potential of pioglitazone and ceftriaxone alone and in combination in rat model of neuropathic pain: targeting PPARγ and GLT-1 pathways. Pharmacol. Reports 68, 85–94. doi: 10.1016/j.pharep.2015.06.010
Qrunfleh, A. M., Alazizi, A., and Sari, Y. (2013). Ceftriaxone, a beta-lactam antibiotic, attenuates relapse-like ethanol-drinking behavior in alcohol-preferring rats. J. Psychopharmacology 27, 541–549. doi: 10.1177/0269881113482529
Ramos, K. M., Lewis, M. T., Morgan, K. N., Crysdale, N. Y., Kroll, J. L., Taylor, F. R., et al. (2010). Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169, 1888–1900. doi: 10.1016/j.neuroscience.2010.06.014
Rao, P. S. S., and Sari, Y. (2014a). Effectiveness of Ceftriaxone Treatment in preventing relapse-like drinking behavior following long-term ethanol dependence in p rats addiction research and therapy. Addict. Res. Ther. 5:1000183. doi: 10.4172/2155-6105.1000183
Rao, P. S. S., and Sari, Y. (2014b). Effects of ceftriaxone on chronic ethanol consumption: a potential role for xCT and GLT1 modulation of glutamate levels in male P rats. J. Mol. Neurosci. 54, 71–77. doi: 10.1007/s12031-014-0251-5
Roberts-Wolfe, D., and Kalivas, P. (2015). Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol. Disord. Drug Targets 14, 745–756. doi: 10.2174/1871527314666150529144655
Rossignol, D. A., and Frye, R. E. (2014). Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 5:150. doi: 10.3389/fphys.2014.00150
Rothstein, J. D., Patel, S., Regan, M. R., Haenggeli, C., Huang, Y. H., Bergles, D. E., et al. (2005). β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77. doi: 10.1038/nature03180
Ruzza, P., Siligardi, G., Hussain, R., Marchiani, A., Islami, M., Bubacco, L., et al. (2014). Ceftriaxone blocks the polymerization of α-synuclein and exerts neuroprotective effects in vitro. ACS Chem. Neurosci. 5, 30–38. doi: 10.1021/cn400149k
Sadigh-Eteghad, S., Sabermarouf, B., Majdi, A., Talebi, M., Farhoudi, M., and Mahmoudi, J. (2015). Amyloid-beta: a crucial factor in alzheimer's disease. Med. Princ. Pract. 24, 1–10. doi: 10.1159/000369101
Sari, Y., Prieto, A. L., Barton, S. J., Miller, B. R., and Rebec, G. V. (2010). Ceftriaxone-induced up-regulation of cortical and striatal GLT1 in the R6/2 model of Huntington's disease. J. Biomed. Sci. 17:62. doi: 10.1186/1423-0127-17-62
Sari, Y., Sakai, M., Weedman, J. M., Rebec, G. V., and Bell, R. L. (2011). Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol. 46, 239–246. doi: 10.1093/alcalc/agr023
Sari, Y., Sreemantula, S. N., Lee, M. R., and Choi, D. S. (2013). Ceftriaxone treatment affects the levels of GLT1 and ENT1 as well as ethanol intake in alcohol-preferring rats. J. Mol. Neurosci. 51, 779–787. doi: 10.1007/s12031-013-0064-y
Savio, L. E. B., Vuaden, F. C., Piato, A. L., Bonan, C. D., and Wyse, A. T. S. (2012). Behavioral changes induced by long-term proline exposure are reversed by antipsychotics in zebrafish. Prog. Neuro Psychopharmacol. Biol. Psychiatry 36, 258–263. doi: 10.1016/j.pnpbp.2011.10.002
Scofield, M. D., and Kalivas, P. W. (2014). Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist 20, 610–622. doi: 10.1177/1073858413520347
Shah, S. A., Amin, F. U., Khan, M., Abid, M. N., Rehman, S. U., Kim, T. H., et al. (2016). Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflammation 13:286. doi: 10.1186/s12974-016-0752-y
Siddique, N., and Siddique, T. (2008). Genetics of amyotrophic lateral sclerosis. Phys. Med. Rehabil. Clin. N. Am. 19, 429–439. doi: 10.1016/j.pmr.2008.05.001
Stepanovic-Petrovic', R. M., Micov, A. M., Tomic', M. A., Kovacevic', J. M., and Boškovic', B. D. (2014). Antihyperalgesic/Antinociceptive effects of ceftriaxone and its synergistic interactions with different analgesics in inflammatory pain in rodents. Anesthesiology 120, 737–750. doi: 10.1097/ALN.0000435833.33515.ba
Thöne-Reineke, C., Neumann, C., Namsolleck, P., Schmerbach, K., Krikov, M., Schefe, J. H., et al. (2008). The β-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J. Hypertens. 26, 2426–2435. doi: 10.1097/HJH.0b013e328313e403
Tikhonova, M. A., Amstislavskaya, T. G., Belichenko, V. M., Fedoseeva, L. A., Kovalenko, S. P., Pisareva, E. E., et al. (2018). Modulation of the expression of genes related to the system of amyloid-beta metabolism in the brain as a novel mechanism of ceftriaxone neuroprotective properties. BMC Neurosci. 19(Suppl. 1):13. doi: 10.1186/s12868-018-0412-5
Tikhonova, M. A., Ho, S. C., Akopyan, A. A., Kolosova, N. G., Weng, J. C., Meng, W. Y., et al. (2017). Neuroprotective effects of ceftriaxone treatment on cognitive and neuronal deficits in a rat model of accelerated senescence. Behav. Brain Res. 330, 8–16. doi: 10.1016/j.bbr.2017.05.002
Tobe, E. (2013). Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr. Dis. Treat. 9, 567–573. doi: 10.2147/NDT.S44282
Uttara, B., Singh, A., Zamboni, P., and Mahajan, R. (2009). Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 7, 65–74. doi: 10.2174/157015909787602823
Van Dam, D., and De Deyn, P. P. (2011). Animal models in the drug discovery pipeline for Alzheimer's disease. Br. J. Pharmacol. 164, 1285–1300. doi: 10.1111/j.1476-5381.2011.01299.x
Volterra, A., Trotti, D., Tromba, C., Floridi, S., and Racagni, G. (1994). Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 14, 2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994
Wang, J., Pang, T., Hafko, R., Benicky, J., Sanchez-Lemus, E., and Saavedra, J. M. (2014). Telmisartan ameliorates glutamate-induced neurotoxicity: roles of AT1 receptor blockade and PPARγ activation. Neuropharmacology 79, 249–261. doi: 10.1016/j.neuropharm.2013.11.022
Wei, J., Pan, X., Pei, Z., Wang, W., Qiu, W., Shi, Z., et al. (2012). The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J. Trauma Acute Care Surg. 73, 654–660. doi: 10.1097/TA.0b013e31825133c0
Weng, J. C., Tikhonova, M. A., Chen, J. H., Shen, M. S., Meng, W. Y., Chang, Y. T., et al. (2016). Ceftriaxone prevents the neurodegeneration and decreased neurogenesis seen in a Parkinson's disease rat model: an immunohistochemical and MRI study. Behav. Brain Res. 305, 126–139. doi: 10.1016/j.bbr.2016.02.034
Xie, A., Gao, J., Xu, L., and Meng, D. (2014). Shared mechanisms of neurodegeneration in alzheimer's disease and parkinson's disease. Biomed. Res. Int. 2014:648740. doi: 10.1155/2014/648740
Yamada, J., and Jinno, S. (2011). Alterations in neuronal survival and glial reactions after axotomy by ceftriaxone and minocycline in the mouse hypoglossal nucleus. Neurosci. Lett. 504, 295–300. doi: 10.1016/j.neulet.2011.09.051
Yang, Q., and Zhou, J. (2018). Neuroinflammation in the central nervous system: symphony of glial cells. Glia. doi: 10.1002/glia.23571. [Epub ahead of print].
Yimer, E. M., Surur, A., Wondafrash, D. Z., and Gebre, A. K. (2019). The effect of metformin in experimentally induced animal models of epileptic seizure. Behav. Neurol. 2019:6234758. doi: 10.1155/2019/6234758
Zhang, Y., Zhang, X., and Qu, S. (2015). Ceftriaxone protects astrocytes from MPP+via suppression of NF-κB/JNK/c-jun signaling. Mol. Neurobiol. 52, 78–92. doi: 10.1007/s12035-014-8845-z
Zhou, W., and Hu, W. (2013). Anti-neuroinflammatory agents for the treatment of Alzheimer's disease. Future Med. Chem. 5, 1559–1571. doi: 10.4155/fmc.13.125
Zlotnik, A., Gruenbaum, S. E., Artru, A. A., Rozet, I., Dubilet, M., Tkachov, S., et al. (2009). The neuroprotective effects of oxaloacetate in closed head injury in rats is mediated by its blood glutamate scavenging activity. J. Neurosurg. Anesthesiol. 21, 235–241. doi: 10.1097/ANA.0b013e3181a2bf0b
Zlotnik, A., Gurevich, B., Cherniavsky, E., Tkachov, S., Matuzani-Ruban, A., Leon, A., et al. (2008). The contribution of the blood glutamate scavenging activity of pyruvate to its neuroprotective properties in a rat model of closed head injury. Neurochem. Res. 33, 1044–1050. doi: 10.1007/s11064-007-9548-x
Zumkehr, J., Rodriguez-Ortiz, C. J., Cheng, D., Kieu, Z., Wai, T., Hawkins, C., et al. (2015). Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer's disease. Neurobiol. Aging 36, 2260–2271. doi: 10.1016/j.neurobiolaging.2015.04.005
Keywords: ceftriaxone, drug repurposing, neurodegenerative disorders, Alzheimer's disease, Parkinson's disease, stroke, pain, brain ischemia
Citation: Yimer EM, Hishe HZ and Tuem KB (2019) Repurposing of the β-Lactam Antibiotic, Ceftriaxone for Neurological Disorders: A Review. Front. Neurosci. 13:236. doi: 10.3389/fnins.2019.00236
Received: 23 November 2018; Accepted: 27 February 2019;
Published: 26 March 2019.
Edited by:
Francisco Lopez-Munoz, Universidad Camilo José Cela, SpainReviewed by:
Ying-Jui Ho, Chung Shan Medical University, TaiwanMaria A. Tikhonova, State Scientific-Research Institute of Physiology and Basic Medicine, Russia
Copyright © 2019 Yimer, Hishe and Tuem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ebrahim M. Yimer, ZWJyYWhpbTk5bXVoYW1tZWRAZ21haWwuY29t orcid.org/0000-0003-3140-4967